
TALTZ injektioneste, liuos, esitäytetty kynä 80 mg, injektioneste, liuos, esitäytetty ruisku 80 mg
Vaikuttavat aineet ja niiden määrät
Iksekitsumabi on valmistettu kiinanhamsterin munasarjasoluissa yhdistelmä-DNA-tekniikalla.
Taltz 80 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kynä sisältää 80 mg iksekitsumabia 1 millilitrassa.
Apuaine(et), joiden vaikutus tunnetaan:
Yksi millilitra liuosta sisältää 0,30 mg polysorbaatti 80:tä.
Taltz 40 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 40 mg iksekitsumabia 0,5 millilitrassa.
Apuaine(et), joiden vaikutus tunnetaan:
Yksi millilitra liuosta sisältää 0,30 mg polysorbaatti 80:tä.
Taltz 80 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 80 mg iksekitsumabia 1 millilitrassa.
Apuaine(et), joiden vaikutus tunnetaan:
Yksi millilitra liuosta sisältää 0,30 mg polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
Läiskäpsoriaasi
Taltz on tarkoitettu keskivaikean tai vaikean läiskäpsoriaasin hoitoon aikuisille, joille harkitaan systeemistä hoitoa.
Läiskäpsoriaasi lapsilla
Taltz on tarkoitettu keskivaikean tai vaikean läiskäpsoriaasin hoitoon vähintään 6-vuotiaille lapsille, jotka painavat vähintään 25 kg, sekä nuorille, joille harkitaan systeemistä hoitoa.
Nivelpsoriaasi
Taltz on tarkoitettu yksinään tai yhdistelmänä metotreksaatin kanssa aktiivisen nivelpsoriaasin hoitoon aikuisille, joilla yksi tai useampi tautiprosessiin vaikuttava reumalääke (DMARD) ei ole tuottanut riittävää vastetta tai on huonosti siedetty (ks. kohta Farmakodynamiikka).
Aksiaalinen spondylartriitti
Selkärankareuma (röntgenpositiivinen aksiaalinen spondylartriitti)
Taltz on tarkoitettu aktiivisen selkärankareuman hoitoon aikuisille, jotka eivät ole saaneet riittävää vastetta tavanomaisesta hoidosta.
Aksiaalinen spondylartriitti ilman radiografista näyttöä selkärankareumasta (röntgennegatiivinen aksiaalinen spondylartriitti)
Taltz on tarkoitettu aktiivisen röntgennegatiivisen aksiaalisen spondylartriitin hoitoon aikuisille, joilla on objektiivisina tulehduksen merkkeinä suurentunut C-reaktiivisen proteiinin (CRP) pitoisuus ja/tai löydöksiä magneettikuvissa (MRI), silloin kun steroideihin kuulumattomilla tulehduskipulääkkeillä (NSAID) ei ole saatu riittävää vastetta.
Lastenreuma (juveniili idiopaattinen artriitti)
Lasten nivelpsoriaasi (juveniili nivelpsoriaasi)
Taltz on tarkoitettu yksinään tai yhdistelmänä metotreksaatin kanssa aktiivisen lasten nivelpsoriaasin hoitoon vähintään 6-vuotiaille potilaille, jotka painavat vähintään 25 kg, ja joilla tavanomainen hoito on tuottanut riittämättömän vasteen tai ollut huonosti siedetty.
Entesiitteihin liittyvä artriitti
Taltz on tarkoitettu yksinään tai yhdistelmänä metotreksaatin kanssa aktiivisen entesiitteihin liittyvän artriitin hoitoon vähintään 6-vuotiaille potilaille, jotka painavat vähintään 25 kg, ja joilla tavanomainen hoito on tuottanut riittämättömän vasteen tai ollut huonosti siedetty.
Ehto
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Tämä lääkevalmiste on tarkoitettu käytettäväksi sen käyttöaiheina olevien sairauksien diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Taltz 80 mg injektioneste, liuos, esitäytetty kynä
Annostus
Läiskäpsoriaasi aikuisilla
Suositeltu aloitusannos on 160 mg injektiona ihon alle (kaksi 80 mg injektiota) viikolla 0, minkä jälkeen annos on 80 mg (yksi injektio) viikoilla 2, 4, 6, 8, 10 ja 12. Tämän jälkeen käytetään ylläpitoannostelua 80 mg (yksi injektio) 4 viikon välein.
Läiskäpsoriaasi lapsilla (vähintään 6-vuotiaat)
Tietoja tehosta ja turvallisuudesta alle 6-vuotiailla lapsilla ei ole saatavilla (ks. kohta Farmakodynamiikka).
Saatavilla olevat tiedot eivät puolla annostelua alle 25 kg painaville.
Lapsille suositeltu annos, joka annetaan injektiona ihon alle, perustuu seuraaviin painoluokkiin:
| Lapsen paino | Suositeltu aloitusannos (viikko 0) | Suositeltu annos 4 viikon välein aloitusannoksen jälkeen |
| Yli 50 kg | 160 mg (kaksi 80 mg injektiota) | 80 mg |
| 25–50 kg | 80 mg | 40 mg |
Jos 40 mg:n valmistetta ei ole saatavilla, asianmukaisen pätevyyden omaavan terveydenhuollon ammattilaisen on valmisteltava ja annosteltava 40 mg iksekitsumabiannokset käyttämällä Taltz 80 mg esitäytettyä ruiskua.
Käytä Taltz 80 mg esitäytettyä kynää vain niillä lapsilla, joiden annos on 80 mg ja joille annosta ei tarvitse valmistella.
Taltz-valmistetta ei suositella käytettäväksi lapsilla, jotka painavat alle 25 kg. Lapsen paino on kirjattava ylös ja tarkistettava säännöllisesti uudelleen ennen annostelua.
Nivelpsoriaasi
Suositeltu annos on 160 mg injektiona ihon alle (kaksi 80 mg injektiota) viikolla 0, minkä jälkeen annetaan 80 mg (yksi injektio) 4 viikon välein. Jos nivelpsoriaasipotilaalla on samanaikaisesti keskivaikea tai vaikea läiskäpsoriaasi, suositusannostus on sama kuin läiskäpsoriaasin yhteydessä.
Aksiaalinen spondylartriitti (röntgenpositiivinen ja röntgennegatiivinen)
Suositeltu annos on 160 mg (kaksi 80 mg injektiota) injektiona ihon alle viikolla 0, minkä jälkeen annos on 80 mg 4 viikon välein (ks. lisätietoja kohta Farmakodynamiikka).
Lastenreuma (vähintään 6-vuotiaat)
Lasten nivelpsoriaasi tai entesiitteihin liittyvä artriitti
Tietoja tehosta ja turvallisuudesta alle 6-vuotiailla lapsilla ei ole saatavilla (ks. kohta Farmakodynamiikka).
Saatavilla olevat tiedot eivät puolla annostelua alle 25 kg painaville.
Lapsille suositeltu annos, joka annetaan injektiona ihon alle, perustuu seuraaviin painoluokkiin:
| Lapsen paino | Suositeltu aloitusannos (viikko 0) | Suositeltu annos 4 viikon välein aloitusannoksen jälkeen |
| Yli 50 kg | 160 mg (kaksi 80 mg injektiota) | 80 mg |
| 25–50 kg | 80 mg | 40 mg |
Jos 40 mg:n valmistetta ei ole saatavilla, asianmukaisen pätevyyden omaavan terveydenhuollon ammattilaisen on valmisteltava ja annosteltava 40 mg iksekitsumabiannokset käyttämällä Taltz 80 mg esitäytettyä ruiskua.
Käytä Taltz 80 mg esitäytettyä kynää vain niillä lapsilla, joiden annos on 80 mg ja joille annosta ei tarvitse valmistella.
Taltz-valmistetta ei suositella käytettäväksi lapsilla, jotka painavat alle 25 kg. Lapsen paino on kirjattava ylös ja tarkistettava uudelleen säännöllisesti ennen annostelua.
Jos potilaalla ei ole todettu vastetta 16–20 hoitoviikon jälkeen, hoidon lopettamista on harkittava kaikissa käyttöaiheissa (läiskäpsoriaasi aikuisilla ja lapsilla, nivelpsoriaasi, aksiaalinen spondylartriitti, lastenreuma mukaan lukien lasten nivelpsoriaasi ja entesiitteihin liittyvä artriitti). Joillakin potilailla aluksi saavutettu osittainen vaste saattaa myöhemmin parantua, kun hoitoa jatketaan yli 20 viikon ajan.
Erityisryhmät
Iäkkäät
Annosta ei tarvitse muuttaa ≥ 65 vuotiailla (ks. kohta Farmakokinetiikka).
75 vuotta täyttäneiden hoidosta on rajallisesti tietoa.
Munuaisten tai maksan vajaatoiminta
Taltz-valmistetta ei ole tutkittu näissä potilasryhmissä. Annossuosituksia ei voida tehdä.
Pediatriset potilaat
Läiskäpsoriaasi lapsilla ja lastenreuma (lasten nivelpsoriaasi ja entesiitteihin liittyvä artriitti) (alle 25 kg painavat ja alle 6-vuotiaat lapset)
Ei ole asianmukaista käyttää Taltz-valmistetta alle 25 kg painavien ja alle 6-vuotiaiden lasten keskivaikean tai vaikean läiskäpsoriaasin ja lastenreuman, mukaan lukien lasten nivelpsoriaasi ja entesiitteihin liittyvä artriitti, hoitoon.
Antotapa
Ihon alle.
Taltz on tarkoitettu annettavaksi injektiona ihon alle. Injektiokohtia voidaan vaihdella. Lääkkeen pistämistä ihoalueille, joilla näkyy psoriaasimuutoksia, tulee mahdollisuuksien mukaan välttää. Liuosta/kynää ei saa ravistaa.
Kun potilaalle on opetettu ihon alle pistämisessä käytettävä asianmukainen pistostekniikka, hän voi pistää Taltz-valmisteen itse, mikäli terveydenhuollon ammattilainen katsoo sen olevan tarkoituksenmukaista. Lääkärin on kuitenkin huolehdittava potilaiden asianmukaisesta seurannasta. Pakkausselosteessa ja käyttöohjeessa on valmisteen antoa koskevat kattavat ohjeet.
Taltz 40 mg ja 80 mg injektioneste, liuos, esitäytetty ruisku
Annostus
Läiskäpsoriaasi aikuisilla
Suositeltu aloitusannos on 160 mg injektiona ihon alle viikolla 0, minkä jälkeen annos on 80 mg viikoilla 2, 4, 6, 8, 10 ja 12. Tämän jälkeen käytetään ylläpitoannostelua 80 mg 4 viikon välein.
Läiskäpsoriaasi lapsilla (vähintään 6-vuotiaat)
Tietoja tehosta ja turvallisuudesta alle 6-vuotiailla lapsilla ei ole saatavilla (ks. kohta Farmakodynamiikka).
Saatavilla olevat tiedot eivät puolla annostelua alle 25 kg painaville.
Lapsille suositeltu annos, joka annetaan injektiona ihon alle, perustuu seuraaviin painoluokkiin:
| Lapsen paino | Suositeltu aloitusannos (viikko 0) | Suositeltu annos 4 viikon välein aloitusannoksen jälkeen |
| Yli 50 kg | 160 mg (kaksi 80 mg injektiota) | 80 mg |
| 25–50 kg | 80 mg | 40 mg |
Lapsille, joille on määrätty annokseksi 80 mg, Taltz voidaan annostella suoraan esitäytetyllä ruiskulla.
Jos 40 mg:n esitäytettyä ruiskua ei ole saatavilla, terveydenhuollon ammattilaisen on valmisteltava alle 80 mg:n annokset. Iksekitsumabi 40 mg -annosten valmisteluohjeet, ks. kohta Käyttö- ja käsittelyohjeet.
Taltz-valmistetta ei suositella käytettäväksi lapsilla, jotka painavat alle 25 kg. Lapsen paino on kirjattava ylös ja tarkistettava säännöllisesti uudelleen ennen annostelua.
Nivelpsoriaasi
Suositeltu annos on 160 mg injektiona ihon alle viikolla 0, minkä jälkeen annetaan 80 mg 4 viikon välein. Jos nivelpsoriaasipotilaalla on samanaikaisesti keskivaikea tai vaikea läiskäpsoriaasi, suositusannostus on sama kuin läiskäpsoriaasin yhteydessä.
Aksiaalinen spondylartriitti (röntgenpositiivinen ja röntgennegatiivinen)
Suositeltu annos on 160 mg injektiona ihon alle viikolla 0, minkä jälkeen annos on 80 mg 4 viikon välein (ks. lisätietoja kohta Farmakodynamiikka).
Lastenreuma (vähintään 6-vuotiaat)
Lasten nivelpsoriaasi tai entesiitteihin liittyvä artriitti
Tietoja tehosta ja turvallisuudesta alle 6-vuotiailla lapsilla ei ole saatavilla (ks. kohta Farmakodynamiikka).
Saatavilla olevat tiedot eivät puolla annostelua alle 25 kg painaville.
Lapsille suositeltu annos, joka annetaan injektiona ihon alle, perustuu seuraaviin painoluokkiin:
| Lapsen paino | Suositeltu aloitusannos (viikko 0) | Suositeltu annos 4 viikon välein aloitusannoksen jälkeen |
| Yli 50 kg | 160 mg (kaksi 80 mg injektiota) | 80 mg |
| 25–50 kg | 80 mg | 40 mg |
Lapsille, joille on määrätty 80 mg annos, Taltz voidaan annostella suoraan esitäytetyllä ruiskulla.
Jos 40 mg:n esitäytettyä ruiskua ei ole saatavilla, terveydenhuollon ammattilaisen on valmisteltava alle 80 mg annokset. Taltz 40 mg annoksen valmisteluohjeet, ks. kohta Käyttö- ja käsittelyohjeet.
Taltz-valmistetta ei suositella käytettäväksi lapsilla, jotka painavat alle 25 kg. Lapsen paino on kirjattava ylös ja tarkistettava uudelleen säännöllisesti ennen annostelua.
Jos potilaalla ei ole todettu vastetta 16–20 hoitoviikon jälkeen, hoidon lopettamista on harkittava kaikissa käyttöaiheissa (läiskäpsoriaasi aikuisilla ja lapsilla, nivelpsoriaasi, aksiaalinen spondylartriitti, lastenreuma mukaan lukien lasten nivelpsoriaasi ja entesiitteihin liittyvä artriitti). Joillakin potilailla aluksi saavutettu osittainen vaste saattaa myöhemmin parantua, kun hoitoa jatketaan yli 20 viikon ajan.
Erityisryhmät
Iäkkäät
Annosta ei tarvitse muuttaa ≥ 65 vuotiailla (ks. kohta Farmakokinetiikka).
75 vuotta täyttäneiden hoidosta on rajallisesti tietoa.
Munuaisten tai maksan vajaatoiminta
Taltz-valmistetta ei ole tutkittu näissä potilasryhmissä. Annossuosituksia ei voida tehdä.
Pediatriset potilaat
Läiskäpsoriaasi lapsilla ja lastenreuma (lasten nivelpsoriaasi ja entesiitteihin liittyvä artriitti) (alle 25 kg painavat ja alle 6-vuotiaat lapset)
Ei ole asianmukaista käyttää Taltz-valmistetta alle 25 kg painavien ja alle 6-vuotiaiden lasten keskivaikean tai vaikean läiskäpsoriaasin ja lastenreuman, mukaan lukien lasten nivelpsoriaasi ja entesiitteihin liittyvä artriitti, hoitoon.
Antotapa
Ihon alle.
Taltz on tarkoitettu annettavaksi injektiona ihon alle. Injektiokohtia voidaan vaihdella. Lääkkeen pistämistä ihoalueille, joilla näkyy psoriaasimuutoksia, tulee mahdollisuuksien mukaan välttää. Liuosta/ruiskua ei saa ravistaa.
Kun potilaalle on opetettu ihon alle pistämisessä käytettävä asianmukainen pistostekniikka, hän voi pistää Taltz-valmisteen itse, mikäli terveydenhuollon ammattilainen katsoo sen olevan tarkoituksenmukaista. Lääkärin on kuitenkin huolehdittava potilaiden asianmukaisesta seurannasta. Pakkausselosteessa ja käyttöohjeessa on valmisteen antoa koskevat kattavat ohjeet.
Jos 40 mg:n esitäytettyä ruiskua ei ole saatavilla, annoksen valmistelua vaativat alle 80 mg:n annokset voi antaa vain terveydenhuollon ammattilainen.
Ohjeet lääkevalmisteen valmisteluun ennen lääkkeen antoa, ks. kohta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Vakava yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Kliinisesti merkittävä aktiivinen infektio (esim. aktiivinen tuberkuloosi, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Taltz-hoitoon liittyy infektioiden kuten ylähengitystieinfektioiden, suun hiivainfektioiden, sidekalvotulehdusten ja silsainfektioiden esiintymistiheyden suurenemista (ks. kohta Haittavaikutukset).
Taltz-valmisteen käytössä on noudatettava varovaisuutta potilailla, joilla on kliinisesti merkittävä krooninen infektio tai taustalla toistuvia infektioita. Potilaita on ohjattava olemaan yhteydessä lääkäriin mikäli infektion merkkejä tai oireita ilmenee. Jos potilaalle kehittyy infektio, hänen vointiaan on seurattava tarkoin ja Taltz-hoito on lopetettava, mikäli potilas ei reagoi tavanomaiseen hoitoon tai mikäli infektio muuttuu vakavaksi. Taltz-hoitoa ei saa aloittaa uudelleen ennen kuin infektio on parantunut.
Taltz-valmistetta ei saa antaa potilaille, joilla on aktiivinen tuberkuloosi. Jos potilaalla on latentti tuberkuloosi, tuberkuloosilääkitystä on harkittava ennen Taltz-hoidon aloittamista.
Yliherkkyys
Vakavia yliherkkyysreaktioita on ilmoitettu, mm. joitakin anafylaksia-, angioedeema-, nokkosihottuma-tapauksia ja harvinaisissa tapauksissa myöhäisiä (10–14 päivän kuluttua injektiosta ilmenneitä) vakavia yliherkkyysreaktioita, joihin kuuluu laaja-alaista nokkosihottumaa, hengenahdistusta ja suuret vasta-ainetitterit. Jos potilaalle kehittyy vakava yliherkkyysreaktio, Taltz-valmisteen anto on lopetettava heti ja asianmukainen hoito on aloitettava.
Tulehduksellinen suolistosairaus (mukaan lukien Crohnin tauti ja haavainen paksusuolitulehdus)
Tulehduksellisen suolistosairauden kehittymistä ja pahenemista on ilmoitettu iksekitsumabihoidon yhteydessä (ks. kohta Haittavaikutukset). Iksekitsumabia ei suositella potilaille, jotka sairastavat tulehduksellista suolistosairautta. Mikäli potilaalle kehittyy tulehduksellisen suolistosairauden merkkejä tai oireita tai hänellä ilmenee olemassa olevan tulehduksellisen suolistosairauden pahenemista, iksekitsumabi on lopetettava ja asianmukainen hoito aloitettava.
Rokotukset
Eläviä rokotteita ei saa antaa Taltz-hoidon aikana. Tietoja vasteesta eläviin rokotteisiin ei ole saatavilla; vasteesta inaktivoituja patogeeneja sisältäviin rokotteisiin ei ole riittävästi tietoa (ks. kohta Farmakodynamiikka).
Apuaineet, joiden vaikutus tunnetaan
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 40 mg annos ja per 80 mg annos eli sen voidaan sanoa olevan ”natriumiton”.
Polysorbaatti
Taltz 80 mg injektioneste, liuos, esitäytetty kynä:
Tämä lääkevalmiste sisältää 0,3 mg polysorbaatti 80:tä yhdessä 80 mg:n esitäytetyssä kynässä, mikä vastaa 0,30 mg/ml.
Polysorbaatit voivat aiheuttaa allergisia reaktioita.
Taltz 40 mg ja 80 mg injektioneste, liuos, esitäytetty ruisku:
Tämä lääkevalmiste sisältää 0,15 mg polysorbaatti 80:tä yhdessä 40 mg:n esitäytetyssä ruiskussa, mikä vastaa 0,30 mg/ml.
Tämä lääkevalmiste sisältää 0,3 mg polysorbaatti 80:tä yhdessä 80 mg:n esitäytetyssä ruiskussa, mikä vastaa 0,30 mg/ml.
Polysorbaatit voivat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Läiskäpsoriaasitutkimuksissa ei ole tutkittu Taltz-valmisteen turvallisuutta yhdistelmähoitona muiden immunomodulaattorien tai valohoidon kanssa.
Populaatiofarmakokineettisissä analyyseissä samanaikainen oraalisten kortikosteroidien, steroideihin kuulumattomien tulehduskipulääkkeiden (NSAID), sulfasalatsiinin tai metotreksaatin annostelu ei vaikuttanut iksekitsumabin puhdistumaan.
Sytokromi P450 substraatit
Keskivaikeaa tai vaikeaa psoriaasia sairastavilla potilailla tehdyn yhteisvaikutustutkimuksen mukaan 12 viikon iksekitsumabiannostelu yhdessä entsyymien CYP3A4 (ts. midatsolaami), CYP2C9 (ts. varfariini), CYP2C19 (ts. omepratsoli), CYP1A2 (ts. kofeiini) tai CYP2D6 (ts. dekstrometorfaani) välityksellä metaboloituvien aineiden kanssa ei vaikuta kliinisesti merkittävästi näiden aineiden farmakokinetiikkaan.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisymenetelmää hoidon aikana ja vähintään 10 viikon ajan hoidon päättymisen jälkeen.
Raskaus
Iksekitsumabin käytöstä raskaana oleville naisille on vain vähän tietoa. Eläinkokeiden perusteella ei ole saatu tietoa suorista tai epäsuorista haitallisista vaikutuksista raskauteen, alkion/sikiön kehitykseen, synnytykseen tai postnataaliseen kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta). Taltz-valmisteen käyttöä raskauden aikana on hyvä välttää varmuuden vuoksi.
Imetys
Ei tiedetä, erittyykö iksekitsumabi ihmisillä äidinmaitoon tai imeytyykö nielty lääkeaine systeemisesti. Iksekitsumabi erittyy kuitenkin pieninä määrinä makakiapinoiden maitoon. On päätettävä, lopetetaanko imetys vai lopetetaanko Taltz-hoito ottaen huomioon imetyksestä aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Iksekitsumabin vaikutusta ihmisen hedelmällisyyteen ei ole tutkittu. Eläinkokeissa ei ole havaittu suoria tai epäsuoria haitallisia vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Taltz-valmisteella ei ole haitallista vaikutusta tai on merkityksetön vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitettuja haittavaikutuksia olivat pistoskohdan reaktiot (15,5 %) ja ylähengitystieinfektiot (16,4 %) (joista yleisin oli nenänielutulehdus).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa ja markkinoille tulon jälkeen ilmoitetut haittavaikutukset (taulukko 1) esitetään MedDRA-elinjärjestelmäluokituksen mukaisesti. Haittavaikutukset on luokiteltu kussakin elinjärjestelmäluokassa yleisyyden mukaan yleisimmistä alkaen. Kunkin yleisyysluokan haittavaikutukset on esitetty vakavuusjärjestyksessä vakavimmasta alkaen. Kunkin haittavaikutuksen kohdalla mainittu yleisyysluokka perustuu seuraavaan käytäntöön: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000).
Yhteensä 8 956 potilasta on saanut Taltz-hoitoa sokkoutetuissa ja avoimissa kliinisissä tutkimuksissa läiskäpsoriaasin, nivelpsoriaasin ja aksiaalisen spondylartriitin sekä muiden autoimmuunisairauksien hoitoon. Näistä potilaista 6 385 sai Taltz-valmistetta vähintään vuoden ajan, mikä vastaa 19 833 potilasvuoden kumulatiivista altistusta aikuisilla, ja 196 lasta, mikä vastaa 343 potilasvuoden kumulatiivista altistusta.
Taulukko 1. Kliinisissä tutkimuksissa ja markkinoille tulon jälkeenilmoitetut haittavaikutukset
| Elinjärjestelmä | Yleisyys | Haittavaikutus |
Infektiot
| Hyvin yleinen | Ylähengitystieinfektio |
| Yleinen | Silsainfektio, herpes simplex (mukokutaaninen) | |
| Melko harvinainen | Influenssa, nuha, suun hiivainfektio, sidekalvotulehdus, selluliitti | |
| Harvinainen | Ruokatorven hiivainfektio | |
| Veri ja imukudos | Melko harvinainen | Neutropenia, trombosytopenia |
| Immuunijärjestelmä | Melko harvinainen | Angioedeema |
| Harvinainen | Anafylaksi | |
| Hengityselimet, rintakehä ja välikarsina | Yleinen | Suunielun kipu |
| Ruoansulatuselimistö | Yleinen | Pahoinvointi |
| Melko harvinainen | Tulehduksellinen suolistosairaus | |
| Iho ja ihonalainen kudos | Melko harvinainen | Urtikaria, ihottuma, ekseema, dyshidroottinen ekseema |
| Harvinainen | Eksfoliatiivinen dermatiitti | |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Pistoskohdan reaktiota |
a Ks. kohta tiettyjen haittavaikutusten kuvaus.
Tiettyjen haittavaikutusten kuvaus
Pistoskohdan reaktiot
Yleisimmin havaittuja pistoskohdan reaktioita olivat punoitus ja kipu. Ne olivat useimmiten lieviä tai keskivaikeita eivätkä johtaneet Taltz-hoidon lopettamiseen.
Aikuisilla tehdyissä läiskäpsoriaasitutkimuksissa pistoskohdan reaktiot olivat yleisempiä potilailla, joiden paino oli < 60 kg verrattuna ryhmään, jossa potilaiden paino oli ≥ 60 kg (25 % vs. 14 % yhdistetyille kahden ja neljän viikon välein tutkimuslääkettä saaneille ryhmille).
Nivelpsoriaasitutkimuksissa pistoskohdan reaktiot olivat yleisempiä potilailla, joiden paino oli < 100 kg verrattuna ryhmään, jossa potilaiden paino oli ≥ 100 kg (24 % vs. 13 % yhdistetyille kahden ja neljän viikon välein tutkimuslääkettä saaneille ryhmille). Aksiaalisen spondylartriitin tutkimuksissa pistoskohdan reaktiot olivat samankaltaisia potilailla, joiden paino oli < 100 kg verrattuna ryhmään, jossa potilaiden paino oli ≥ 100 kg (14 % vs. 9 % yhdistetyille kahden ja neljän viikon välein tutkimuslääkettä saaneille ryhmille). Pistoskohdan reaktioiden yleisyyden lisääntyminen yhdistetyissä kahden ja neljän viikon välein tutkimuslääkettä saaneissa ryhmissä ei johtanut keskeytysten lisääntymiseen läiskäpsoriaasi-, nivelpsoriaasi- tai aksiaalisen spondylartriitin tutkimuksissa.
Edellä kuvatut tulokset saatiin Taltz-valmisteen alkuperäisellä formulaatiolla. Yksisokkoutetussa, satunnaistetussa ristikkäistutkimuksessa 45 terveellä koehenkilöllä, jossa verrattiin alkuperäistä formulaatiota tarkistettuun sitraattivapaaseen formulaatioon, saatiin tilastollisesti merkitsevästi alhaisemmat VAS (visuaalinen analoginen asteikko, Visual Analogue Scale) -kipupisteet sitraattivapaalla valmisteella verrattuna alkuperäiseen formulaatioon injektion aikana (ero pienimmän neliösumman keskiarvossa (LSM) VAS-mittarilla mitattuna -21,69) ja 10 minuuttia injektion jälkeen (ero pienimmän neliösumman keskiarvossa (LSM) VAS-mittarilla mitattuna -4,47).
Infektiot
Vaiheen III kliinisten, aikuisilla tehtyjen läiskäpsoriaasitutkimusten lumekontrolloidussa osassa infektioita ilmoitettiin ensimmäisten 12 viikon aikana 27,2 %:lla Taltz-hoitoa saaneista potilaista ja 22,9 %:lla lumehoitoa saaneista potilaista.
Valtaosa infektioista oli ei-vakavia ja vaikeusasteeltaan lieviä tai keskivaikeita, eivätkä ne useimmiten edellyttäneet hoidon lopettamista. Vakavia infektioita esiintyi 13:lla (0,6 %) Taltz-hoitoa saaneista potilaista ja 3:lla (0,4 %) lumehoitoa saaneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Koko hoitojakson aikana infektioita ilmoitettiin 52,8 %:lla Taltz-hoitoa saaneista potilaista (46,9 tapausta 100 potilasvuotta kohden). Vakavia infektioita ilmoitettiin 1,6 %:lla Taltz-hoitoa saaneista (1,5 tapausta 100 potilasvuotta kohden).
Kliinisissä nivelpsoriaasin ja aksiaalisen spondylartriitin tutkimuksissa todetut infektioprosentit vastasivat läiskäpsoriaasitutkimuksissa todettuja. Poikkeuksena olivat influenssan ja sidekalvotulehduksen esiintymistiheydet. Influenssa ja sidekalvotulehdus olivat yleisiä nivelpsoriaasipotilailla.
Neutropenian ja trombosytopenian laboratorioarvioinnit
Läiskäpsoriaasitutkimuksissa 9 %:lle Taltz-hoitoa saaneista potilaista kehittyi neutropenia. Useimmiten veren neutrofiilimäärä oli ≥ 1 000 solua/mm3. Tämän tasoinen neutropenia voi pitkittyä, fluktuoida tai olla ohimenevää. 0,1 %:lla Taltz-hoitoa saaneista potilaista neutrofiilimäärä laski tasolle < 1 000 solua/mm3. Neutropenia ei yleensä edellyttänyt Taltz-hoidon lopettamista.
3 %:lla Taltz-hoitoa saaneista potilaista lähtötasoltaan normaali trombosyyttimäärä laski tasolle < 150 000/mm3 – ≥ 75 000/mm3. Trombosytopenia voi pitkittyä, fluktuoida tai olla ohimenevää.
Neutropenian ja trombosytopenian esiintymistiheydet kliinisissä nivelpsoriaasin ja aksiaalisen spondylartriitin tutkimuksissa vastasivat läiskäpsoriaasitutkimusten havaintoja.
Immunogeenisuus
Noin 9–17 %:lle aikuisista läiskäpsoriaasipotilaista, jotka saivat Taltz-valmistetta annossuositusten mukaisesti, kehittyi vasta-aineita lääkettä kohtaan. Useimmiten vasta-aineiden titterit olivat matalat eikä niihin liittynyt kliinisen vasteen huononemista 60 hoitoviikkoon mennessä. Noin 1 %:lla Taltz-hoitoa saaneista potilaista todettiin kuitenkin neutraloivia vasta-aineita, joilla oli yhteys pieniin lääkepitoisuuksiin ja kliinisen vasteen huononemiseen.
Kun nivelpsoriaasipotilaat saivat Taltz-hoitoa suositusannostuksella 52 viikon ajan, noin 11 %:lle kehittyi lääkevasta-aineita, joiden titterit olivat useimmiten matalat. Noin 8 %:lla todettiin neutraloivia vasta-aineita. Neutraloivilla vasta-aineilla ei todettu olevan ilmeistä yhteyttä lääkkeen pitoisuuteen tai tehoon.
Kun pediatrista psoriaasia sairastavat potilaat saivat Taltz-hoitoa suositusannoksella 12 viikon ajan, 21 potilaalle (18 %) kehittyi lääkevasta-aineita, joista noin puolen titterit olivat matalat. 5 potilaalla (4 %) todettiin neutraloivia vasta-aineita, joilla oli yhteys pieniin lääkepitoisuuksiin. Yhteyttä kliiniseen vasteeseen tai haittatapahtumiin ei havaittu.
Kun röntgenpositiivista aksiaalista spondylartriittia sairastavat potilaat saivat Taltz-valmistetta annossuositusten mukaisesti 16 viikon ajan, 5,2 %:lle kehittyi lääkevasta-aineita, joiden titterit olivat useimmiten matalat. 1,5 %:lla (3 potilaalla) oli neutraloivia vasta-aineita (NAb). Näillä kolmella potilaalla positiivisissa NAb-näytteissä oli matala iksekitsumabipitoisuus, ja yksikään näistä potilaista ei saavuttanut ASAS40-vastetta. Röntgennegatiivista aksiaalista spondylartriittia sairastavilla potilailla, jotka saivat Taltz-valmistetta annossuositusten mukaisesti 52 viikon ajan, 8,9 %:lle kehittyi lääkevasta-aineita, joiden kaikkien titterit olivat matalat. Yhdelläkään potilaalla ei ollut neutraloivia vasta-aineita, eikä lääkevasta-aineiden ja lääkkeen pitoisuuden, tehon tai turvallisuuden välillä havaittu ilmeistä yhteyttä.
Kun lastenreumaa sairastavat potilaat (lasten nivelpsoriaasi ja entesiitteihin liittyvä artriitti) saivat iksekitsumabihoitoa suositusannoksella 104 viikon ajan, 18 potilaalle (22,8 %) kehittyi lääkevasta-aineita, joiden kaikkien titterit olivat matalat tai kohtalaiset. Lääkevasta-aineiden esiintymisen ja lääkkeen pitoisuuden, tehon tai turvallisuuden välillä ei havaittu ilmeistä yhteyttä.
Minkään käyttöaiheen osalta immunogeenisuuden mahdollista yhteyttä hoidon aikana ilmenneisiin haittatapahtumiin ei ole vahvistettu selvästi.
Pediatriset potilaat
Läiskäpsoriaasia sairastavilla lapsilla, jotka saivat Taltz-hoitoa 4 viikon välein, havaittu turvallisuusprofiili on muuten yhdenmukainen läiskäpsoriaasia sairastavien aikuispotilaiden turvallisuusprofiilin kanssa, mutta sidekalvotulehduksen, influenssan ja urtikarian yleisyys oli ”yleinen”. Tulehduksellinen suolistosairaus oli myös yleisempää pediatrisilla potilailla, vaikka sen yleisyys olikin ”melko harvinainen”. Pediatrisilla potilailla tehdyssä kliinisessä tutkimuksessa Crohnin tautia esiintyi 0,9 %:lla Taltz-hoitoa saaneista potilaista ja 0 %:lla lumelääkettä saaneista potilaista 12 viikkoa kestäneessä lumelääkekontrolloidussa vaiheessa. Yhteensä Crohnin tautia esiintyi 4 Taltz-hoitoa saaneella potilaalla (2,0 %) pediatrisen kliinisen tutkimuksen lumelääkekontrolloidun vaiheen ja ylläpitohoidon aikana.
Haittavaikutukset pediatrisilla potilailla, jotka saivat avoimessa kliinisessä lääketutkimuksessa iksekitsumabia suositusannoksella ihonalaisena injektiona lastenreuman (lasten nivelpsoriaasi tai entesiitteihin liittyvä artriitti) hoitoon, olivat yhdenmukaisia iksekitsumabin tunnetun turvallisuusprofiilin kanssa integroidussa turvallisuusaineistossa lasten läiskäpsoriaasin indikaation osalta sekä aikuisten keskivaikean tai vaikean läiskäpsoriaasin, nivelpsoriaasin ja aksiaalisen spondylartriitin indikaatioiden osalta, lukuun ottamatta influenssan (yleinen), nuhan (yleinen) ja sidekalvotulehduksen (yleinen) esiintymistiheyksiä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa lääkevalmisteen myyntiluvan myöntämisen jälkeisistä epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 Fimea
Yliannostus
Kliinisissä tutkimuksissa ihon alle on annettu enintään 180 mg annoksia ilman annosta rajoittavaa toksisuutta. Kliinisissä tutkimuksissa on ilmoitettu enintään 240 mg yliannosten kertaluonteista antoa ihon alle ilman vakavia haittatapahtumia.
Yliannostustapauksissa on suositeltavaa seurata potilaan vointia haittavaikutusten oireiden ja löydösten varalta ja aloittaa asianmukainen oireenmukainen hoito viipymättä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC13
Vaikutusmekanismi
Iksekitsumabi on monoklonaalinen IgG4-vasta-aine, joka sitoutuu suurella affiniteetilla (< 3 pM) ja erittäin spesifisesti interleukiini 17A:han (sekä IL‑17A että IL‑17A/F). Suurentuneiden IL‑17A-pitoisuuksien on arveltu osallistuvan psoriaasin patogeneesiin edistämällä keratinosyyttien proliferaatiota ja aktivoitumista sekä osallistuvan nivelpsoriaasin ja aksiaalisen spondylartriitin patogeneesiin edistämällä erosiivisiin luuvaurioihin ja patologiseen uuden luun muodostumiseen johtavaa tulehdusta. Iksekitsumabi neutraloi IL‑17A:ta, mikä estää nämä vaikutukset. Iksekitsumabi ei sitoudu IL‑17B-, IL‑17C-, IL‑17D-, IL‑17E- eikä IL‑17F-ligandeihin.
In vitro ‑sitoutumistutkimukset ovat vahvistaneet, että iksekitsumabi ei sitoudu ihmisen Fcγ-reseptoreihin I, IIa ja IIIa eikä komplementin C1q-komponenttiin.
Farmakodynaamiset vaikutukset
Iksekitsumabi moduloi IL‑17A:n indusoimia tai säätelemiä biologisia vasteita. Vaiheen I tutkimuksessa otettujen psoriaasi-ihobiopsioiden tietojen perusteella todettiin, että orvaskeden paksuus oheni, proliferoituvien keratinosyyttien, T-solujen ja dendriittisolujen määrät vähenivät ja paikalliset tulehdusmerkkiaineet vähenivät annosriippuvaisesti lähtötilanteesta päivään 43 mennessä. Tämän välittömänä seurauksena iksekitsumabihoito vähentää läiskäpsoriaasimuutoksissa esiintyvää punoitusta, kovettumista ja hilseilyä.
Iksekitsumabihoidon on todettu pienentävän (1 viikon kuluessa hoidosta) C-reaktiivisen proteiinin pitoisuuksia. Kyseinen proteiini on tulehduksen merkkiaine.
Kliininen teho ja turvallisuus
Läiskäpsoriaasi aikuisilla
Iksekitsumabin tehoa ja turvallisuutta arvioitiin kolmessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen III tutkimuksessa keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavilla aikuispotilailla (N = 3 866), joille harkittiin valohoitoa tai systeemistä hoitoa (UNCOVER-1, UNCOVER-2 ja UNCOVER-3). Lisäksi iksekitsumabin tehoa ja turvallisuutta verrattiin etanerseptiin (UNCOVER-2 ja UNCOVER-3). Iksekitsumabihoitoon satunnaistetut potilaat, jotka olivat saavuttaneet sPGA-vasteen (0,1) (sPGA = Static Physicians Global Assessment) viikolla 12, satunnaistettiin uudelleen saamaan joko lume- tai iksekitsumabihoitoa vielä 48 viikon ajan (UNCOVER-1 ja UNCOVER-2); lume-, etanersepti- tai iksekitsumabihoitoon satunnaistetut potilaat, jotka eivät olleet saavuttaneet sPGA-vastetta (0,1), saivat iksekitsumabia enintään 48 viikon ajan. Lisäksi pitkäaikaista tehoa ja turvallisuutta arvioitiin kaikissa kolmessa tutkimuksessa yhteensä viiden vuoden ajan potilailla, jotka osallistuivat tutkimukseen koko tutkimuksen ajan.
64 % potilaista oli saanut aiemmin systeemistä hoitoa (biologiset lääkkeet, tavanomaiset systeemiset lääkkeet tai psoraleeni- ja ultravioletti A ‑hoito [PUVA]), 43,5 % valohoitoa, 49,3 % tavanomaista systeemistä hoitoa ja 26,4 % biologista hoitoa. 14,9 % oli saanut vähintään yhtä TNF-alfan estäjää ja 8,7 % IL-12:n/IL-23:n estäjää. 23,4 %:lla potilaista oli lähtötilanteessa anamneesissa nivelpsoriaasi.
Kaikkien kolmen tutkimuksen rinnakkaisina ensisijaisina päätetapahtumina olivat PASI 75 ‑vasteen (PASI = Psoriasis Area and Severity Index -mittari) ja sPGA-vasteen "0" (”ei ihomuutoksia”) tai "1" (”minimaaliset ihomuutokset”) saavuttaneiden potilaiden osuudet viikolla 12 lumelääkkeeseen verrattuna. Lähtötilanteen PASI-pisteiden mediaani vaihteli välillä 17,4–18,3; 48,3–51,2 %:lla potilaista lähtötilanteen sPGA-arvo oli ”vaikea” tai ”hyvin vaikea”, ja kutinan NRS-arvo (Itch Numeric Rating Scale, Itch NRS) oli lähtötilanteessa keskimäärin 6,3–7,1.
Kliininen vaste viikolla 12
UNCOVER-1-tutkimukseen satunnaistettiin 1 296 potilasta (1:1:1) saamaan joko lumelääkettä tai iksekitsumabia (80 mg 2 viikon (Q2) tai 4 viikon (Q4) välein, alussa 160 mg aloitusannos) 12 viikon ajan.
Taulukko 2. Tehotulokset UNCOVER-1-tutkimuksen viikolla 12
| Päätetapahtumat | Potilaat (%) | Vasteprosentin ero lumehoitoon verrattuna (95 % lv) | |||
Lume (N = 431) | Iksekitsumabi 80 mg 4 viikon välein (N = 432) | Iksekitsumabi 80 mg 2 viikon välein (N = 433) | Iksekitsumabi 80 mg 4 viikon välein | Iksekitsumabi 80 mg 2 viikon välein | |
| sPGA ”0” (ei ihomuutoksia) tai ”1” (minimaaliset ihomuutokset) | 14 (3,2) | 330 (76,4)a | 354 (81,8)a | 73,1 (68,8, 77,5) | 78,5 (74,5, 82,5) |
| sPGA ”0” (ei ihomuutoksia) | 0 | 149 (34,5)a | 160 (37,0)a | 34,5 (30,0, 39,0) | 37,0 (32,4, 41,5) |
| PASI 75 | 17 (3,9) | 357 (82,6)a | 386 (89,1)a | 78,7 (74,7, 82,7) | 85,2 (81,7, 88,7) |
| PASI 90 | 2 (0,5) | 279 (64,6)a | 307 (70,9)a | 64,1 (59,6, 68,7) | 70,4 (66,1, 74,8) |
| PASI 100 | 0 | 145 (33,6)a | 153 (35,3)a | 33,6 (29,1, 38,0) | 35,3 (30,8, 39,8) |
| Kutinan NRS-arvo pieneni ≥ 4b | 58 (15,5) | 305 (80,5)a | 336 (85,9)a | 65,0 (59,5, 70,4) | 70,4 (65,4, 75,5) |
Lyhenteet: N = hoitoaikomuspopulaation (intent-to-treat) potilasmäärä, Iv = luottamusväli
Huom. Jos potilaalta puuttui tietoja, katsottiin, että hän ei ollut saavuttanut vastetta.
a p < 0,001 verrattuna lumeeseen.
b Potilaat, joilla kutinan NRS-arvo lähtötilanteessa ≥ 4: lumeryhmässä N = 374, iksekitsumabi 80 mg 4 viikon välein ‑ryhmässä N = 379, iksekitsumabi 80 mg 2 viikon välein ‑ryhmässä N = 391.
UNCOVER-2-tutkimukseen satunnaistettiin 1 224 potilasta (1:2:2:2) saamaan joko lumelääkettä tai iksekitsumabia (80 mg 2 viikon tai 4 viikon välein, alussa 160 mg aloitusannos) tai etanerseptia (50 mg kahdesti viikossa) 12 viikon ajan.
Taulukko 3. Tehotulokset UNCOVER-2-tutkimuksen viikolla 12
| Päätetapahtumat | Potilaat (%) | Vasteprosentin ero lumehoitoon verrattuna (95 % Iv) | ||||
Lume (N = 168) | Iksekitsumabi (N = 347) | Iksekitsumabi 80 mg 2 viikon välein (N = 351) | Etanersepti 50 mg kahdesti viikossa (N = 358) | Iksekitsumabi 80 mg 4 viikon välein | Iksekitsumabi 80 mg 2 viikon välein | |
| sPGA ”0” (ei ihomuutoksia) tai ”1” (minimaaliset ihomuutokset) | 4 (2,4) | 253 (72,9)a,b | 292 (83,2)a,b | 129 (36,0)a | 70,5 (65,3, 75,7) | 80,8 (76,3, 85,4) |
| sPGA ”0” (ei ihomuutoksia) | 1 (0,6) | 112 (32,3)a,b | 147 (41,9)a,b | 21 (5,9)c | 31,7 (26,6, 36,7) | 41,3 (36,0, 46,6) |
| PASI 75 | 4 (2,4) | 269 (77,5)a,b | 315 (89,7)a,b | 149 (41,6)a | 75,1 (70,2, 80,1) | 87,4 (83,4, 91,3) |
| PASI 90 | 1 (0,6) | 207 (59,7)a,b | 248 (70,7)a,b | 67 (18,7)a | 59,1 (53,8, 64,4) | 70,1 (65,2, 75,0) |
| PASI 100 | 1 (0,6) | 107 (30,8)a,b | 142 (40,5)a,b | 19 (5,3)c | 30,2 (25,2, 35,2) | 39,9 (34,6, 45,1) |
| Kutinan NRS-arvo pieneni ≥ 4d | 19 (14,1) | 225 (76,8)a,b | 258 (85,1)a,b | 177 (57,8)a | 62,7 (55,1, 70,3) | 71,1 (64,0, 78,2) |
Lyhenteet: N = hoitoaikomuspopulaation (intent-to-treat) potilasmäärä, Iv = luottamusväli
Huom. Jos potilaalta puuttui tietoja, katsottiin, että hän ei ollut saavuttanut vastetta.
a p < 0,001 verrattuna lumeeseen.
b p < 0,001 verrattuna etanerseptiin.
c p < 0,01 verrattuna lumeeseen.
d Potilaat, joilla kutinan NRS-arvo lähtötilanteessa ≥ 4: lumeryhmässä N = 135, iksekitsumabi 80 mg 4 viikon välein ‑ryhmässä N = 293, iksekitsumabi 80 mg 2 viikon välein ‑ryhmässä N = 303, etanerseptiryhmässä N = 306.
UNCOVER-3-tutkimukseen satunnaistettiin 1346 potilasta (1:2:2:2) saamaan joko lumelääkettä tai iksekitsumabia (80 mg 2 viikon tai 4 viikon välein, alussa 160 mg aloitusannos) tai etanerseptia (50 mg kahdesti viikossa) 12 viikon ajan.
Taulukko 4. Tehotulokset UNCOVER-3-tutkimuksen viikolla 12
| Päätetapahtumat | Potilaat (%) | Vasteprosentin ero lumehoitoon verrattuna (95 % Iv) | ||||
Lume (N = 193) | Iksekitsumabi 80 mg 4 viikon välein (N = 386) | Iksekitsumabi 80 mg 2 viikon välein (N = 385) | Etanersepti 50 mg kahdesti viikossa (N = 382) | Iksekitsumabi 80 mg 4 viikon välein | Iksekitsumabi 80 mg 2 viikon välein | |
| sPGA ”0” (ei ihomuutoksia) tai ”1” (minimaaliset ihomuutokset) | 13 (6,7) | 291 (75,4)a,b | 310 (80,5)a,b | 159 (41,6)a | 68,7 (63,1, 74,2) | 73,8 (68,5, 79,1) |
| sPGA ”0” (ei ihomuutoksia) | 0 | 139 (36,0)a,b | 155 (40,3)a,b | 33 (8,6)a | 36,0 (31,2, 40,8) | 40,3 (35,4, 45,2) |
| PASI 75 | 14 (7,3) | 325 (84,2)a,b | 336 (87,3)a,b | 204 (53,4)a | 76,9 (71,8, 82,1) | 80,0 (75,1, 85,0) |
| PASI 90 | 6 (3,1) | 252 (65,3)a,b | 262 (68,1)a,b | 98 (25,7)a | 62,2 (56,8, 67,5) | 64,9 (59,7, 70,2) |
| PASI 100 | 0 | 135 (35,0)a,b | 145 (37,7)a,b | 28 (7,3)a | 35 (30,2, 39,7) | 37,7 (32,8, 42,5) |
| Kutinan NRS-arvo pieneni ≥ 4c | 33 (20,9) | 250 (79,9)a,b | 264 (82,5)a,b | 200 (64,1)a | 59,0 (51,2, 66,7) | 61,6 (54,0, 69,2) |
Lyhenteet: N = hoitoaikomuspopulaation (intent-to-treat) potilasmäärä, Iv = luottamusväli
Huom. Jos potilaalta puuttui tietoja, katsottiin, että hän ei ollut saavuttanut vastetta.
a p < 0,001 verrattuna lumeeseen.
b p < 0,001 verrattuna etanerseptiin.
c Potilaat, joilla kutinan NRS-arvo lähtötilanteessa ≥ 4: lumeryhmässä N = 158, iksekitsumabi 80 mg 4 viikon välein ‑ryhmässä N = 313, iksekitsumabi 80 mg 2 viikon välein ‑ryhmässä N = 320, etanerseptiryhmässä N = 312.
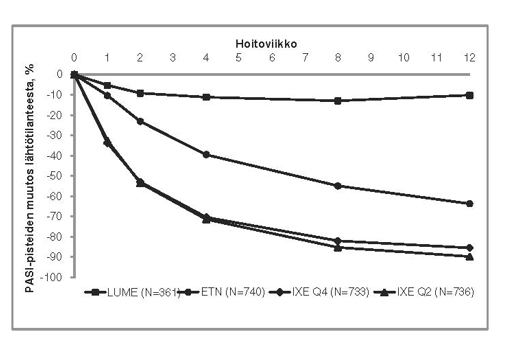
Iksekitsumabihoidolla oli yhteys nopeasti alkavaan tehoon ja PASI-pistekeskiarvon pienenemiseen > 50 % viikkoon 2 mennessä (kuva 1). PASI 75 ‑vasteen saavuttaneiden potilaiden osuus oli iksekitsumabiryhmässä merkitsevästi suurempi kuin lume- ja etanerseptiryhmissä jo viikolla 1. Noin 25 % iksekitsumabihoitoa saaneista potilaista saavutti PASI-pistemäärän < 5 viikkoon 2 mennessä, yli 55 % saavutti PASI-pistemäärän < 5 viikkoon 4 mennessä, ja tämän pistearvon saavuttaneiden osuus suureni 85 %:iin viikkoon 12 mennessä (etanerseptia saaneilla vastaavat osuudet olivat 3 %, 14 % ja 50 %). Iksekitsumabihoitoa saaneilla kutinan vaikeusaste lievittyi merkitsevästi viikolla 1.
Kuva 1. PASI-pisteiden prosentuaalinen muutos hoitoaikomuspopulaatiossa kullakin lähtötasokäynnin jälkeisellä käynnillä (mBOCF) UNCOVER-2- ja UNCOVER-3-tutkimusten induktioannosteluvaiheessa
Iksekitsumabi todettiin tehokkaaksi ja turvalliseksi potilaan iästä, sukupuolesta, etnisestä taustasta, painosta, lähtötilanteen PASI-vaikeusasteesta, läiskien sijainnista, samanaikaisesta nivelpsoriaasista ja aiemmasta biologisesta hoidosta riippumatta. Iksekitsumabi oli tehokas potilailla, jotka eivät olleet saaneet aiempaa systeemistä hoitoa tai aiempaa biologista hoitoa, sekä potilailla, jotka olivat saaneet aiempaa biologista / TNF:n estäjähoitoa tai joilla biologinen tai TNF:n estäjähoito oli epäonnistunut.
Potilaista, joilla etanersepti ei tuottanut sPGA (0,1) ‑vastetta UNCOVER-2-tutkimuksen viikolla 12 (N = 200) ja jotka siirtyivät iksekitsumabihoitoon (80 mg 4 viikon välein) 4 viikon puhdistumavaiheen jälkeen, 73 % saavutti sPGA (0,1) ‑vasteen ja 83,5 % saavutti PASI 75 ‑vasteen 12 viikon iksekitsumabihoidon jälkeen.
Kahdessa kliinisessä tutkimuksessa, joissa käytettiin aktiivista vertailuvalmistetta (UNCOVER-2 ja UNCOVER-3), vakavia haittatapahtumia esiintyi 1,9 %:lla sekä etanersepti- että iksekitsumabihoitoa saaneista potilaista ja haittatapahtumista johtuva hoidon keskeyttämisprosentti oli 1,2 % etanerseptiryhmässä ja 2,0 % iksekitsumabiryhmässä. Infektioita esiintyi 21,5 %:lla etanerseptia saaneista ja 26,0 %:lla iksekitsumabia saaneista, vakavia infektioita 0,4 %:lla etanerseptia saaneista ja 0,5 %:lla iksekitsumabia saaneista.
Vasteen säilyminen viikolla 60 ja viiden vuoden ajan
Alun perin iksekitsumabihoitoon satunnaistetut potilaat, jotka saavuttivat vasteen UNCOVER-1- ja UNCOVER-2-tutkimusten viikolla 12 (sPGA-pisteet 0, 1), satunnaistettiin uudelleen saamaan vielä 48 viikon ajan joko lumehoitoa tai iksekitsumabihoitoa (80 mg joko 4 viikon tai 12 viikon välein).
Kun potilaat, jotka saavuttivat sPGA (0,1) ‑vasteen viikolla 12, satunnaistettiin uudelleen lopettamaan hoito (ts. vaihtamaan lumehoitoon), relapsiin (sPGA ≥ 3) kulunut mediaaniaika oli UNCOVER-1- ja UNCOVER-2-tutkimuksien yhdistetyissä tiedoissa 164 päivää. Näistä potilaista 71,5 % saavutti uudelleen vähintään sPGA (0,1) ‑vasteen 12 viikon kuluessa iksekitsumabihoidon (80 mg 4 viikon välein) uudelleen aloittamisesta.
Taulukko 5. Vasteen ja tehon säilyminen viikolla 60 (UNCOVER-1- ja UNCOVER-2-tutkimukset)
| Päätetapahtumat | Potilaat (%) | Vasteprosentin ero lumehoitoon verrattuna (95 % Iv) | ||||
| 80 mg 4 viikon välein (induktio) / lume (ylläpito) (N = 191) | 80 mg 2 viikon välein (induktio) / lume (ylläpito) (N = 211) | 80 mg 4 viikon välein (induktio) / 80 mg 4 viikon välein (ylläpito) (N = 195) | 80 mg 2 viikon välein (induktio) / 80 mg 4 viikon välein (ylläpito) (N = 221) | 80 mg 4 viikon välein (induktio) / 80 mg 4 viikon välein (ylläpito) | 80 mg 2 viikon välein (induktio) / 80 mg 4 viikon välein (ylläpito) | |
| sPGA-arvo ”0” (ei ihomuutoksia) tai ”1” (minimaaliset ihomuutokset) säilyi | 12 (6,3) | 16 (7,6) | 134 (68,7)a | 173 (78,3)a | 62,4 (55,1, 69,8) | 70,7 (64,2, 77,2) |
| sPGA-arvo 0 (ei ihomuutoksia) säilyi/saavutettiin | 3 (1,6) | 6 (2,8) | 96 (49,2)a | 130 (58,8)a | 47,7 (40,4, 54,9) | 56,0 (49,1, 62,8) |
| PASI 75 ‑vaste säilyi/saavutettiin | 15 (7,9) | 19 (9,0) | 145 (74,4)a | 184 (83,3)a | 66,5 (59,3, 73,7) | 74,3 (68,0, 80,5) |
| PASI 90 ‑vaste säilyi/saavutettiin | 9 (4,7) | 10 (4,7) | 130 (66,7)a | 169 (76,5)a | 62,0 (54,7, 69,2) | 71,7 (65,4, 78,0) |
| PASI 100 ‑vaste säilyi/saavutettiin | 3 (1,6) | 6 (2,8) | 97 (49,7)a | 127 (57,5)a | 48,2 (40,9, 55,4) | 54,6 (47,7, 61,5) |
Lyhenteet: N = analyysipopulaation potilasmäärä, Iv = luottamusväli
Huom. Jos potilaalta puuttui tietoja, katsottiin, että hän ei ollut saavuttanut vastetta.
a p < 0,001 verrattuna lumeeseen.
Iksekitsumabi ylläpiti hoitovastetta potilailla, jotka eivät olleet saaneet aiempaa systeemistä hoitoa tai aiempaa biologista hoitoa, sekä potilailla, jotka olivat saaneet aiempaa biologista / TNF:n estäjähoitoa tai joilla biologinen tai TNF:n estäjähoito oli epäonnistunut.
Viikolla 12 todettiin kynsipsoriaasin (mittarina NAPSI, Nail Psoriasis Severity Index), päänahan psoriaasin (mittarina PSSI, Psoriasis Scalp Severity Index) ja palmoplantaarisen psoriaasin (mittarina PPASI, Psoriasis Palmoplantar Severity Index) lievittyneen lähtötilanteeseen nähden merkitsevästi enemmän iksekitsumabihoitoa saaneilla kuin lume- ja etanerseptihoitoa saaneilla ja tämä säilyi viikolle 60 asti iksekitsumabihoitoa saaneilla potilailla, jotka saavuttivat sPGA (0,1) ‑vasteen viikolla 12.
591 potilaasta, jotka saivat iksekitsumabia induktiovaiheessa 2 viikon välein ja tämän jälkeen 4 viikon välein tutkimuksissa UNCOVER‑1, UNCOVER‑2 ja UNCOVER‑3, 427 potilaalla iksekitsumabihoito kesti 5 vuotta. Näistä 101 potilasta tarvitsi annoksen tihentämistä. Potilaista, jotka saavuttivat viikon 264 arvioinnin (N=427), 295 potilaalla (69%) havaittiin sPGA (0,1) -vaste, 289 potilaalla (68%) PASI 90 -vaste ja 205 potilaalla (48%) PASI 100 -vaste viikolla 264. DLQI-pisteet (Dermatology Life Quality Index) kerättiin induktiovaiheen jälkeen UNCOVER‑1 ja UNCOVER‑2 tutkimuksissa. 113 potilaalla (66%) havaittiin DLQI (0,1) -vaste.
Elämänlaatu / potilaiden raportoimat hoitotulokset
Eri tutkimuksissa viikon 12 kohdalla iksekitsumabihoito oli yhteydessä terveyteen liittyvän elämänlaadun tilastollisesti merkitsevään paranemiseen, jonka arviointiperusteena oli DLQI-pisteiden (Dermatology Life Quality Index) keskipienenemän vaihteluväli verrattuna lähtötilanteeseen (iksekitsumabi 80 mg 2 viikon välein arvosta ‑10,2 arvoon ‑11,1; iksekitsumabi 80 mg 4 viikon välein arvosta ‑9,4, arvoon ‑10,7; etanersepti arvosta ‑7,7 arvoon ‑8,0; ja lume arvosta ‑1,0 arvoon ‑2,0). Iksekitsumabihoitoa saaneiden potilaiden joukossa merkitsevästi suurempi osuus potilaista saavutti DLQI-arvon 0 tai 1. Eri tutkimuksissa niiden potilaiden osuus, joilla kutinan NRS-pisteet olivat pienentyneet ≥ 4 pistettä viikolla 12, oli iksekitsumabiryhmässä merkitsevästi suurempi (84,6 % iksekitsumabi 2 viikon välein ‑ryhmässä, 79,2 % iksekitsumabi 4 viikon välein ‑ryhmässä ja 16,5 % lumeryhmässä). Hoidon hyödyt säilyivät viikkoon 60 asti niillä iksekitsumabihoitoa saaneilla potilailla, jotka saavuttivat sPGA-vasteen (sPGA-arvo 0 tai 1) viikolla 12. Enintään 60 viikkoa kestäneen iksekitsumabihoidon aikana ei todettu masennuksen pahenemista 16-kohtaisella Quick Inventory of Depressive Symptomatology Self Report ‑mittarilla mitattuna.
Markkinoilletulon jälkeiset suorat vertailututkimukset
IXORA-S: Kaksoissokkoutetussa tutkimuksessa iksekitsumabi osoittautui paremmaksi verrattuna ustekinumabiin ensisijaisen tutkimustavoitteen suhteen (PASI 90-vaste viikolla 12, taulukko 6). Vaste saavutettiin nopeammin: PASI 75-vaste jo viikolla 2 (p < 0,001) ja PASI 90- ja PASI 100-vasteet viikkoon 4 mennessä (p < 0,001). Iksekitsumabi osoittautui paremmaksi kuin ustekinumabi myös painon perusteella stratifioiduissa alaryhmissä.
Taulukko 6. Iksekitsumabin ja ustekinumabin vertailututkimuksen PASI-tulokset
| Viikko 12 | Viikko 24 | Viikko 52 | |||||||
| Iksekitsumabi* | Ustekinumabi** | Iksekitsumabi* | Ustekitsumabi** | Iksekitsumabi* | Ustekinumabi** | ||||
| Potilaita (n) | 136 | 166 | 136 | 166 | 136 | 166 | |||
| PASI 75, n (%) | 120 (88,2 %) | 114 (68,7 %) | 124 (91,2 %) | 136 (81,9 %) | 120 (88,2 %) | 126 (75,9 %) | |||
| PASI 90, n (%) | 99 (72,8 %)§ | 70 (42,2 %) | 113 (83,1 %) | 98 (59,0 %) | 104 (76,5 %) | 98 (59,0 %) | |||
| PASI 100, n (%) | 49 (36,0 %) | 24 (14,5 %) | 67 (49,3 %) | 39 (23,5 %) | 71 (52,2 %) | 59 (35,5 %) | |||
* Iksekitsumabin aloitusannos 160 mg, minkä jälkeen 80 mg viikoilla 2,4,6,8,10 ja 12 ja sitten 80 mg neljän viikon välein
** Annostelu painon perusteella: Ustekinumabiryhmän potilaat saivat 45 mg tai 90 mg annoksen viikoilla 0 ja 4 ja tämän jälkeen painon mukaisen annoksen (hyväksytyn annostuksen mukaisesti) 12 viikon välein viikolle 52 saakka
§ p < 0,001 vs. ustekinumabi (p-arvo annettu ainoastaan ensisijaiselle päätetapahtumalle)
IXORA‑R: Iksekitsumabin tehoa ja turvallisuutta arvioitiin myös 24 viikon satunnaistetussa ja kaksoissokkoutetussa rinnakkaisryhmätutkimuksessa, jossa verrattiin iksekitsumabia guselkumabiin. Iksekitsumabi osoittautui paremmaksi kuin guselkumabi jo viikolla 4, kun iho-oireet paranivat täysin ja tutkimuksen ensisijainen tutkimustavoite (PASI 100 viikolla 12) saavutettiin, ja PASI 100 viikolla 24 osoittautui vähintään yhtä hyväksi (taulukko 7).
Taulukko 7. Iksekitsumabin ja guselkumabin vertailututkimuksen tehotulokset, hoitoaikomuspopulaatioa
| Päätetapahtuma | Ajankohta | Guselkumabi (N = 507) vaste, n (%) | Iksekitsumabi (N = 520) vaste, n (%) | Ero (IXE - GUS), % (lv) | p-arvo |
| Ensisijainen tutkimustavoite | |||||
| PASI 100 | Viikko 12 | 126 (24,9) | 215 (41,3) | 16,5 (10,8, 22,2) | < 0,001 |
| Keskeisimmät toissijaiset päätetapahtumat | |||||
| PASI 75 | Viikko 2 | 26 (5,1) | 119 (22,9) | 17,8 (13,7, 21,8) | < 0,001 |
| PASI 90 | Viikko 4 | 40 (7,9) | 109 (21,0) | 13,1 (8,9, 17,3) | < 0,001 |
| PASI 100 | Viikko 4 | 7 (1,4) | 35 (6,7) | 5,4 (3,0, 7,7) | < 0,001 |
| PASI 90 | Viikko 8 | 182 (35,9) | 304 (58,5) | 22,6 (16,6, 28,5) | < 0,001 |
| sPGA (0) | Viikko 12 | 128 (25,2) | 218 (41,9) | 16,7 (11,0, 22,4) | < 0,001 |
| PASI 50 | Viikko 1 | 47 (9,3) | 143 (27,5) | 18,2 (13,6, 22,8) | < 0,001 |
| PASI 100 | Viikko 8 | 69 (13,6) | 154 (29,6) | 16,0 (11,1, 20,9) | < 0,001 |
| PASI 100 | Viikko 24 | 265 (52,3) | 260 (50,0) | -2,3 (-8,4, 3,8) | 0,414 |
Lyhenteet: Iv = luottamusväli; GUS = guselkumabi; IXE = iksekitsumabi; N = analyysipopulaation potilasmäärä; n = tietyn ryhmän potilasmäärä; PASI = psoriasis area and severity index; sPGA = static physician global assessment.
a Päätetapahtumien tilastolliset testit rajattiin tässä järjestyksessä.
Kuva 2. PASI 100 viikolla 4, 8, 12 ja 24, NRI
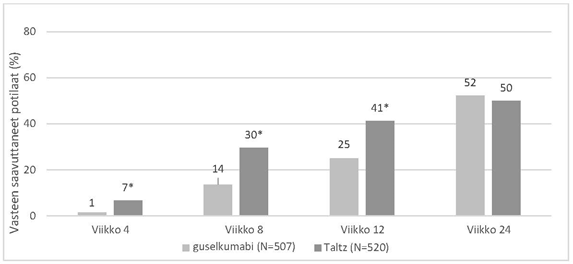
* p < 0,001 vs guselkumabi viikoilla 4, 8 ja 12
NRI = Non-responder imputation (päätetapahtumatiedon puuttuminen tulkittiin niin, että vastetta ei saavutettu)
Teho sukuelinten alueen psoriaasin hoidossa
Satunnaistettuun, kaksoissokkoutettuun, lumekontrolloituun tutkimukseen (IXORA-Q) osallistui 149 aikuista tutkittavaa (24 % naisia), joilla oli keskivaikea tai vaikea psoriaasi sukuelinten alueella (sPGA-G, static Physician Global Assessment of Genitalia ≥ 3 pistettä), BSA (Body Surface Area) oli vähintään 1 % kehon pinta-alasta (60,4 %:lla BSA oli ≥ 10 % kehon pinta-alasta) ja vähintään yksi sukuelinten alueen psoriaasin paikallishoito oli aiemmin epäonnistunut tai ollut huonosti siedetty. Potilailla oli vähintään keskivaikea läiskäpsoriaasi (sPGA-arvo ≥ 3 ja joille soveltuu valohoito ja/tai hoito systeemisillä lääkkeillä) vähintään 6 kk.
Iksekitsumabihoitoon satunnaistetut tutkittavat saivat 160 mg:n aloitusannoksen ja tämän jälkeen 80 mg 2 viikon välein 12 viikon ajan. Ensisijaisena päätetapahtumana oli niiden potilaiden osuus, joilla sPGA-G-vaste oli vähintään ”0” (”ei ihomuutoksia”) tai ”1” (”minimaaliset ihomuutokset”) (sPGA-G 0/1). Viikolla 12 merkitsevästi suurempi määrä tutkittavia saavutti sPGA-G-arvon 0/1 ja sPGA-arvon 0/1 iksekitsumabiryhmässä kuin lumeryhmässä lähtötason BSA-arvosta riippumatta. Iksekitsumabiryhmässä sPGA-G-arvon 0/1 saavutti 71 % potilaista, joiden lähtötason BSA oli välillä 1 - <10 % ja 75 % potilaista, joiden lähtötason BSA oli ≥10 %. Lumelääkeryhmässä vastaavat luvut olivat 0 % ja 13 %. Iksekitsumabihoitoa saaneessa ryhmässä oli merkitsevästi yleisempää, että potilaan täyttämillä itsearviointimittareilla (PRO, Patient reported outcome) mitattuna sukuelinten kivun ja sukuelinten kutinan vaikeusasteet lievittyivät, sukuelinten alueen psoriaasin vaikutus seksuaaliseen aktiivisuuteen väheni ja Dermatology Quality of Life Index -pistemäärä (DLQI) laski.
Taulukko 8. Tehotulokset IXORA-Q-tutkimuksen viikolla 12 aikuisilla, joilla oli sukuelinten alueen psoriaasi; NRIa
| Päätetapahtumat | Iksekitsumabi | Lume | Ero lumelääkkeeseen nähden (95 % lv) |
| Satunnaistetut potilaat (N) | N = 75 | N = 74 | |
| sPGA-G-arvo ”0” tai ”1” | 73 % | 8 % | 65 % (53 %, 77 %) |
| sPGA-arvo ”0” tai ”1” | 73 % | 3 % | 71 % (60 %, 81 %) |
| DLQI 0,1b | 45 % | 3 % | 43 % (31 %, 55 %) |
| GPSS-mittarin kutinapisteet (NRS) lähtötilanteessa ≥ 3, N | N = 62 | N = 60 | |
| GPSS-mittarin sukuelinten kutinaa kuvaavat pisteet (paranema ≥ 3 pistettä) | 60 % | 8 % | 51 % (37 %, 65 %) |
| SFQ-mittarin kohdan kaksi pisteet lähtötilanteessa ≥ 2, N | N = 37 | N = 42 | |
| SFQ-mittarin kohdan kaksi pisteet ”0” (ei rajoita koskaan) tai ”1” (rajoittaa harvoin) | 78 % | 21 % | 57 % (39 %, 75 %) |
a Lyhenteet: NRI = Non-responder imputation; sPGA = static Physician Global Assessment, lääkärin staattinen kokonaisarvio sukuelinten alueesta; GPSS = Genital Psoriasis Symptom Scale, sukuelinten psoriaasioiremittari; SFQ = Sexual Frequency Questionnaire, yhdyntätiheyskysely; DLQI = Dermatology Quality of Life Index; b DLQI-kokonaispistemäärä 0,1 tarkoittaa, että ihotauti ei vaikuttanut mitenkään potilaan elämään. sPGA-arvo ”0” tai ”1” tarkoittaa ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset”; NRS = Numeric Rating Scale, numeerinen arviointiasteikko.
Läiskäpsoriaasi lapsilla
Satunnaistettuun, kaksoissokkoutettuun, lumekontrolloituun monikeskustutkimukseen (IXORA-Peds) otettiin mukaan 201 vähintään 6- ja alle 18-vuotiasta lasta, jotka sairastivat keskivaikeaa tai vaikeaa läiskäpsoriaasia (sPGA-pistemäärä ≥ 3, ≥ 10 % kehon pinta-alasta, ja PASI-pistemäärä ≥ 12) ja joille harkittiin valohoitoa tai systeemistä hoitoa, tai joiden sairauden hallinta oli riittämätöntä paikallishoidolla.
Potilaat satunnaistettiin saamaan lumelääkettä (n = 56), etanerseptiä (n = 30) tai iksekitsumabihoitoa (n = 115), jonka annostelu oli stratifioitu painon mukaan:
< 25 kg: 40 mg viikolla 0 ja sen jälkeen 20 mg neljän viikon välein (n = 4)
25 kg−50 kg: 80 mg viikolla 0 ja sen jälkeen 40 mg neljän viikon välein (n = 50)
> 50 kg: 160 mg viikolla 0 ja sen jälkeen 80 mg neljän viikon välein (n = 147)
Potilaat, jotka oli satunnaistettu saamaan etanerseptiä (nämä potilaat sairastivat vaikeaa läiskäpsoriaasia), saivat 0,8 mg/kg, ei enempää kuin 50 mg per annos, joka viikko viikosta 0 viikkoon 11.
Alkuperäisen 12 viikon kaksoissokkoutetun induktiovaiheen jälkeen potilaat olivat soveltuvia siirtymään 48 viikon avoimeen ylläpitojaksoon (viikot 12–60), jossa käytettiin iksekitsumabia painon mukaisella annoksella. Tämän jälkeen seurasi jatkovaihe, joka kesti enimmillään viikolle 108 asti.
Kliininen vaste viikolla 12
Hoitovaste arvioitiin 12 viikon jälkeen. Rinnakkaisina ensisijaisina päätetapahtumina käytettiin niiden potilaiden osuutta, jotka saavuttivat sPGA-pisteet ”0” (ei ihomuutoksia) tai ”1” (minimaaliset ihomuutokset) ja vähintään 2 pisteen vähenemisen lähtötasosta, sekä niiden potilaiden osuutta, jotka saavuttivat vähintään 75 % PASI-pisteiden vähenemisen (PASI 75) lähtötasosta.
Muihin viikolla 12 arvioituihin tuloksiin kuuluivat niiden potilaiden osuus, jotka saavuttivat PASI 90- tai PASI 100-vasteen, sPGA-pistemäärän ”0”, sekä paranemisen kutinan vaikeudessa (määriteltynä vähintään 4 pisteen vähenemisen 11-pisteisellä kutinan numeerisella arviointiasteikolla [itch Numeric Rating Scale; NRS]).
Potilaiden lähtötason PASI-arvon mediaani oli 17 ja vaihteluväli 12−49. Lähtötason sPGA-arvo oli ”vaikea” tai ”hyvin vaikea” 49 %:lla potilaista. Kaikista potilaista 22 % oli saanut aiempaa valohoitoa ja 32 % oli saanut aiempaa tavanomaista systeemistä hoitoa psoriaasin hoitoon.
25 % potilaista (n = 43) oli alle 12-vuotiaita (14 % potilaista [n = 24] oli 6−9-vuotiaita ja 11 % [n = 19] oli 10−11-vuotiaita); 75 % (n = 128) oli vähintään 12-vuotiaita.
Tiedot kliinisestä vasteesta esitetään taulukossa 9.
Taulukko 9. Tehotulokset pediatrisilla läiskäpsoriaasipotilailla, NRI
| Päätetapahtumat | Iksekitsumabia (N = 115) n (%) | Lumelääke (N = 56) n (%) | Ero vs. lumelääke (95 % lv) | Etanerseptib (N = 30) n (%) | Ero vs. etanersepti (95 % lv)b |
| sPGA ”0” (ei ihomuutoksia) tai ”1” (minimaaliset ihomuutokset)c | |||||
| viikko 4 | 55 (48) | 4 (7) | 40,7 (29,3; 52,0)f | 0(0) | 36,8 (21,5; 52,2) |
| viikko 12c | 93 (81) | 6 (11) | 70,2 (59,3; 81,0)f | 16 (53) | 23,0 (0,6; 45,4) |
| sPGA ”0” (ei ihomuutoksia)d | 60 (52) | 1 (2) | 50,4 (40,6; 60,2)f | 5 (17) | 46,5 (26,2; 66,8) |
| PASI 75 | |||||
| viikko 4 | 62 (54) | 5 (9) | 45,0 (33,2; 56,8)f | 3 (10) | 34,7 (15,6; 53,8) |
| viikko 12c | 102 (89) | 14 (25) | 63,7 (51,0; 76,4)f | 19 (63) | 20,9 (0,1; 41,7) |
| PASI 90d | 90 (78) | 3 (5) | 72,9 (63,3; 82,5)f | 12 (40) | 36,3 (14,2; 58,5) |
| PASI 100d | 57 (50) | 1 (2) | 47,8 (38,0; 57,6)f | 5 (17) | 43,9 (23,4; 64,3) |
| Kutinan NRS-arvo (≥ 4 pisteen paraneminen)d,e | 59 (71) | 8 (20) | 51,1 (35,3; 66,9)f | Ei arvioitu | --- |
Lyhenteet: N = hoitoaikomuspopulaation (intent-to-treat) potilasmäärä; NRI = Non-Responder Imputation
a Viikolla 0 potilaat saivat 160 mg, 80 mg tai 40 mg iksekitsumabia ja sen jälkeen 80 mg, 40 mg tai 20 mg 4 viikon välein, riippuen painoluokasta, 12 viikon ajan.
b Vertailut etanerseptiin tehtiin alapopulaatiossa, johon kuuluivat Yhdysvaltojen ja Kanadan ulkopuolella asuvat potilaat, joilla oli vaikea psoriaasi (iksekitsumabihoidon N = 38).
c Rinnakkaiset ensisijaiset päätetapahtumat.
d Tulokset viikolla 12.
e Kutinan NRS-arvo (≥ 4 pisteen paraneminen) potilailla, joiden lähtötason kutinan NRS-arvo oli ≥ 4. Niiden hoitoaikomuspopulaatioon kuuluneiden potilaiden määrä, joilla lähtötason kutinan NRS-arvo oli ≥ 4: iksekitsumabi, n = 83; lumelääke, n = 40.
f p < 0,001
Kuva 3. Niiden potilaiden prosenttiosuus, jotka saavuttivat PASI 75-vasteen lasten psoriaasissa viikolle 12
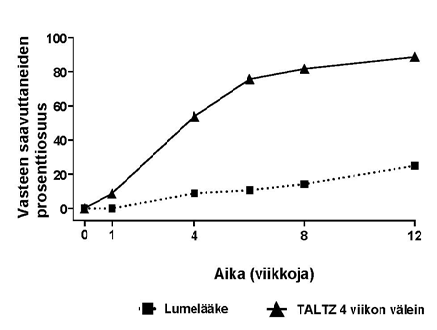
Iksekitsumabia saaneessa hoitoryhmässä saavutettiin kliinisesti merkittävästi enemmän CDLQI/DLQI (0,1)-vasteita viikolla 12 lumelääkkeeseen verrattuna. Analyysissa vastetiedon puuttuminen tulkittiin niin, että potilas ei ollut saavuttanut vastetta. Ero hoitoryhmien välillä havaittiin jo viikolla 4.
Viikolla 12 todettiin kynsipsoriaasin (mittarina Nail Psoriasis Severity Index [NAPSI=0: iksekitsumabi 18 % (6/34), lumelääke 0% (0/12)]), päänahan psoriaasin (mittarina Psoriasis Scalp Severity Index [PSSI=0: iksekitsumabi 69% (70/102), lumelääke 16% (8/50)]) sekä palmoplantaarisen psoriaasin (mittarina Psoriasis Palmoplantar Severity Index (PPASI 75: iksekitsumabi 53 % (9/17), lumelääke 11 % (1/9)]) lievittyneen lähtötilanteesta enemmän lumelääkkeeseen verrattuna.
Vasteen säilyminen viikolla 60 ja viikkoon 108 asti
Potilailla, joita hoidettiin jatkuvasti iksekitsumabilla, kliinisesti merkittävät paranemiset tehoa koskevissa päätetapahtumissa säilyivät viikolle 108 asti:
- PASI 75: 83,0 % viikolla 60; 76,6 % viikolla 108 (NRI)
- sPGA (0,1): 74,5 % viikolla 60; 68,1 % viikolla 108 (NRI)
Nivelpsoriaasi
Iksekitsumabia arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen III tutkimuksessa, joihin osallistuneilla 780 potilaalla oli aktiivinen nivelpsoriaasi (≥ 3 turvonnutta ja ≥ 3 aristavaa niveltä). Potilailla oli todettu nivelpsoriaasi (Classification Criteria for Psoriatic Arthritis -kriteerien [CASPAR] mukaisesti), taudin mediaanikesto oli 5,33 vuotta, ja heillä oli myös tämänhetkisiä läiskäpsoriaasin ihomuutoksia (94,0 %) tai anamneesissa dokumentoitu läiskäpsoriaasi, ja 12,1 %:lla potilaista oli lähtötilanteessa keskivaikea tai vaikea läiskäpsoriaasi. Yli 58,9 %:lla nivelpsoriaasipotilaista oli lähtötilanteessa entesiitti ja 22,3 %:lla daktyliitti. Molempien tutkimusten ensisijainen päätetapahtuma oli ACR 20-vaste (ACR = American College of Rheumatology) viikolla 24, jota seurasi pitkäaikainen jatkovaihe viikolta 24 viikolle 156 (3 vuotta).
Nivelpsoriaasitutkimuksessa 1 (SPIRIT-P1) aktiivista nivelpsoriaasia sairastavia potilaita, jotka eivät olleet saaneet aiemmin biologisia lääkkeitä, satunnaistettiin saamaan lumelääkettä, adalimumabia (40 mg 2 viikon välein; aktiivista kontrollivalmistetta saanut vertailuryhmä) tai iksekitsumabia (80 mg 2 viikon välein tai 80 mg 4 viikon välein). Iksekitsumabihoitoon kuului molemmissa tapauksissa myös 160 mg:n aloitusannos. 85,3 % tämän tutkimuksen potilaista oli saanut aiemmin vähintään yhtä cDMARD (conventional disease-modifying antirheumatic drug) -hoitoa. 53 % potilaista käytti samanaikaisesti metotreksaattia, jonka keskiannos viikossa oli 15,8 mg. Samanaikaista metotreksaattihoitoa saaneista potilaista 67 % käytti vähintään 15 mg:n annosta. Potilaat, joiden vaste oli riittämätön viikolla 16, saivat varahoitoa (rescue therapy: taustahoitoa muutettiin). Potilaat, jotka saivat iksekitsumabia 2 tai 4 viikon välein, jatkoivat heille alussa määrätyn iksekitsumabiannoksen käyttöä. Adalimumabia tai lumelääkettä saaneet potilaat satunnaistettiin uudelleen suhteessa 1:1 vasteesta riippuen viikolla 16 tai 24 saamaan iksekitsumabia 2 viikon tai 4 viikon välein. 243 potilasta oli tutkimuksessa iksekitsumabihoidolla 3 vuotta.
Nivelpsoriaasitutkimukseen 2 (SPIRIT-P2) otettiin potilaita, jotka olivat aiemmin saaneet TNF-estäjähoitoa ja lopettaneet sen käytön joko tehottomuuden tai siedettävyysongelmien vuoksi (anti-TNF-IR-ryhmä). Potilaat satunnaistettiin saamaan joko lumelääkettä tai iksekitsumabia, joko 80 mg 2 viikon välein tai 80 mg 4 viikon välein. Iksekitsumabihoitoon kuului molemmissa tapauksissa myös 160 mg:n aloitusannos. 56 % potilaista oli saanut riittämättömän vasteen yhdelle TNF-estäjälle ja 35 % taas ≥ 2:lle TNF-estäjälle. SPIRIT-P2-tutkimuksessa arvioiduista 363 potilaasta 41 % käytti samanaikaisesti metotreksaattia, jonka keskiannos viikossa oli 16,1 mg. Samanaikaista metotreksaattihoitoa saaneista potilaista 73,2 % käytti vähintään 15 mg:n annosta. Potilaat, joiden vaste oli riittämätön viikolla 16, saivat varahoitoa (rescue therapy: taustahoitoa muutettiin). Potilaat, jotka saivat iksekitsumabia 2 tai 4 viikon välein, jatkoivat heille alussa määrätyn iksekitsumabiannoksen käyttöä. Lumelääkettä saaneet potilaat satunnaistettiin uudelleen suhteessa 1:1 vasteesta riippuen viikolla 16 tai 24 saamaan iksekitsumabia 2 viikon tai 4 viikon välein.
168 potilasta oli tutkimuksessa iksekitsumabihoidolla 3 vuotta.
Oireet ja löydökset
Iksekitsumabihoito paransi merkitsevästi tautiaktiivisuuden mittareita verrattuna lumelääkkeeseen viikolla 24 (ks. taulukko 10).
Taulukko 10. Tehotulokset SPIRIT-P1- ja SPIRIT-P2-tutkimusten viikolla 24
| SPIRIT-P1 | SPIRIT-P2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Päätetapahtumat | Vasteprosentin ero verrattuna lumehoitoon (95 % lv) | Vasteprosentin ero verrattuna lumehoitoon (95 % lv) | |||||||||
Lume
| Iksekitsu-mabi 4 viikon välein (N = 107) | Iksekitsu-mabi 2 viikon välein (N = 103) | ADA (N = 101) | Iksekit-sumabi 4 viikon välein | Iksekit-sumabi 2 viikon välein | Lume (N = 118) | Iksekitsu-mabi 4 viikon välein (N = 122) | Iksekitsu-mabi 2 viikon välein (N = 123) | Iksekit-sumabi 4 viikon välein | Iksekit-sumabi 2 viikon välein | |
| ACR 20-vaste, n (%) | |||||||||||
| Viikko 24 | 32 (30,2) | 62 (57,9) | 64 (62,1) | 58 (57,4) | 27,8 (15,0; 40,6)c | 31,9 (19,1; 44,8)c | 23 (19,5) | 65 (53,3) | 59 (48,0) | 33,8 (22,4; 45,2)c | 28,5 (17,1; 39,8)c |
| ACR 50-vaste, n (%) | |||||||||||
| Viikko 24 | 16 (15,1) | 43 (40,2) | 48 (46,6) | 39 (38,6) | 25,1 (13,6; 36,6)c | 31,5 (19,7; 43,3)c | 6 (5,1) | 43 (35,2) | 41 (33,3) | 30,2 (20,8; 39,5)c | 28,3 (19,0; 37,5)c |
| ACR 70-vaste, n (%) | |||||||||||
| Viikko 24 | 6 (5,7) | 25 (23,4) | 35 (34,0) | 26 (25,7) | 17,7 (8,6; 26,8)c | 28,3 (18,2; 38,5)c | 0 | 27 (22,1) | 15 (12,2) | 22,1 (14,8; 29,5)c | 12,2 (6,4; 18,0)c |
| Minimaalinen tautiaktiivisuus (MDA), n (%) | |||||||||||
| Viikko 24 | 16 (15,1) | 32 (29,9) | 42 (40,8) | 32 (31,7) | 14,8 (3,8; 25,8)a | 25,7 (14,0; 37,4)c | 4 (3,4) | 34 (27,9) | 29 (23,6) | 24,5 (15,9; 33,1)c | 20,2 (12,0; 28,4)c |
| ACR 50- ja PASI 100-vasteet potilailla, joilla psoriaasi-ihomuutosten laajuus oli lähtötilanteessa ≥ 3 % kehon pinta-alasta, n (%) | |||||||||||
| Viikko 24 | 1 (1,5) | 21 (28,8) | 19 (32,2) | 9 (13,2) | 27,3 (16,5; 38,1)c | 30,7 (18,4; 43,0)b | 0 (0,0) | 12 (17,6) | 10 (14,7) | 17,6 (8,6; 26,7)c | 14,7 (6,3; 23,1)c |
Lyhenteet: ACR 20/50/70 = American College of Rheumatology -järjestön 20 %/50 %/70 % -vasteen saavuttaneiden prosenttiosuus; ADA = adalimumabi; lv = luottamusväli; N = analyysipopulaation potilasmäärä; n = kyseisen luokan potilasmäärä; NRI = Non-responder imputation; PASI100 = Psoriasis Area and Severity Index -mittarin pisteet paranivat 100 %.
Huom. Jos potilas siirtyi varahoitoon viikolla 16 tai keskeytti osallistumisensa tai häneltä puuttui tietoja, viikon 24 analyysien imputoinneissa katsottiin, että vastetta ei saavutettu.
Samanaikaisesti käytettyjä tavanomaisia reumalääkkeitä olivat metotreksaatti, leflunomidi ja sulfasalatsiini.
a p < 0,05; b p < 0,01; c p < 0,001 verrattuna lumeeseen.
Potilailla, joilla oli entuudestaan daktyliitti tai entesiitti, iksekitsumabin käyttö 4 viikon välein johti daktyliitin ja entesiitin lievittymiseen viikolla 24 verrattuna lumehoitoon (daktyliitti korjautui 78 %:lla iksekitsumabiryhmäläisistä ja 24 %:lla lumeryhmäläisistä, p < 0,001; entesiitti korjautui 39 %:lla iksekitsumabiryhmäläisistä ja 21 %:lla lumeryhmäläisistä, p < 0,01).
Jos psoriaasimuutosten laajuus oli ≥ 3 % kehon pinta-alasta, iho-oireet korjautuivat eli PASI 75-vaste saavutettiin (Psoriasis Area Severity Index -pisteet laskivat 75 %) viikkoon 12 mennessä 67 %:lla iksekitsumabia 4 viikon välein saaneista (94/141) ja 9 %:lla lumehoitoa saaneista (12/134) (p < 0,001). PASI 75-, PASI 90- tai PASI 100-vasteen saavuttaneiden osuudet viikolla 24 olivat iksekitsumabihoitoa 4 viikon välein saaneilla suuremmat kuin lumeryhmässä (p < 0,001). Potilailla, joilla oli samanaikaisesti keskivaikea tai vaikea psoriaasi ja nivelpsoriaasi, iksekitsumabihoito 2 viikon välein tuotti merkitsevästi paremmat PASI 75-, PASI 90- ja PASI 100-vasteprosentit kuin lumelääke (p < 0,001) ja osoittautui kliinisesti merkittävästi paremmaksi kuin iksekitsumabihoito 4 viikon välein.
Iksekitsumabihoito tuotti merkitsevästi lumelääkettä paremmat ACR 20-vasteprosentit jo viikolla 1, merkitsevästi paremmat ACR 50-vasteprosentit viikolla 4 ja merkitsevästi paremmat ACR 70-vasteprosentit viikolla 8. Ero säilyi viikolle 24 asti; vaikutukset pysyivät 3 vuoden ajan potilailla, jotka jatkoivat tutkimuksessa.
Kuva 4. ACR 20-vasteet SPIRIT-P1-tutkimuksen eri ajankohtina viikolle 24 asti
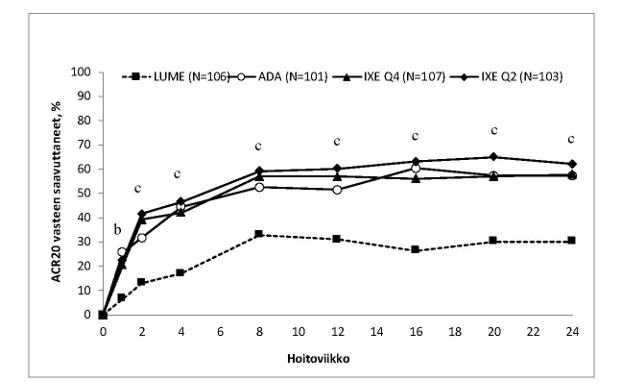
Sekä iksekitsumabi 2 viikon välein (Q2) että iksekitsumabi 4 viikon välein (Q4): b p < 0,01
ja c p < 0,001 verrattuna lumelääkkeeseen.
SPIRIT-P1- ja SPIRIT-P2-tutkimuksissa nivelpsoriaasipotilailla todettiin samankaltaiset ACR 20/50/70-vasteprosentit riippumatta siitä, saivatko he samanaikaisesti tavanomaisia reumalääkkeitä (kuten metotreksaattia) vai eivät.
SPIRIT-P1- ja SPIRIT-P2-tutkimuksissa todettiin paranemista kaikilla ACR-vastekriteerien osa-alueilla, ja mm. potilaiden arvioima kipu lievittyi. Viikolla 24 niiden potilaiden osuus, jotka saavuttivat muokattujen Psoriatic Arthritis Response Criteria -vastekriteerien (PsARC) mukaisen vasteen, oli iksekitsumabihoitoa saaneiden potilaiden joukossa suurempi kuin lumehoitoa saaneilla.
SPIRIT-P1-tutkimuksessa hoidon teho säilyi viikolle 52 asti, kun arviointiperusteina olivat ACR 20/50/70-vasteet, minimaalinen tautiaktiivisuus, entesiittien korjautuminen, daktyliittien korjautuminen ja PASI 75/90/100-vasteprosentit.
Iksekitsumabihoito osoittautui tehokkaaksi ja turvalliseksi riippumatta potilaan iästä, sukupuolesta, etnisestä taustasta, taudin kestosta, lähtötilanteen painosta, lähtötilanteen psoriaasiaffisiosta, lähtötilanteen CRP-arvosta, lähtötilanteen DAS28-CRP-arvosta, samanaikaisesta kortikosteroidihoidosta ja aiemmasta biologisesta lääkehoidosta. Iksekitsumabi oli tehokas potilailla, jotka eivät olleet saaneet aiempaa biologista hoitoa; potilailla, jotka olivat saaneet biologista lääkettä; ja potilailla, joilla biologinen lääkehoito oli epäonnistunut.
SPIRIT-P1-tutkimuksessa 63 potilasta sai iksekitsumabihoitoa 4 viikon välein 3 vuoden ajan. 107 potilaasta, jotka satunnaistettiin saamaan iksekitsumabia 4 viikon välein (NRI-analyysi hoitoaikomuspopulaatiossa), 54 potilaalla (50 %) havaittiin ACR 20-vaste, 41 potilaalla (38 %) ACR 50-vaste, 29 potilaalla (27 %) ACR 70-vaste ja 36 potilaalla (34 %) MDA-vaste viikolla 156.
SPIRIT-P2-tutkimuksessa 70 potilasta sai iksekitsumabihoitoa 4 viikon välein 3 vuoden ajan. 122 potilaasta, jotka satunnaistettiin saamaan iksekitsumabia 4 viikon välein (NRI-analyysi hoitoaikomuspopulaatiossa), 56 potilaalla (46 %) havaittiin ACR 20-vaste, 39 potilaalla (32 %) ACR 50-vaste, 24 potilaalla (20 %) ACR 70-vaste ja 33 potilaalla (27 %) MDA-vaste viikolla 156.
Radiologinen vaste
SPIRIT-P1-tutkimuksessa rakenteellisten vaurioiden etenemisen estoa arvioitiin radiologisesti ja se ilmaistiin muokattujen Sharpin kokonaispisteiden (modified Total Sharp Score, mTSS) ja tämän mittarin komponenttien eli eroosiopisteiden (Erosion Score, ES) ja nivelraon kaventumispisteiden (Joint Space Narrowing, JSN) muutoksena viikolla 24 ja 52 verrattuna lähtötilanteeseen. Viikon 24 tiedot esitetään taulukossa 11.
Taulukko 11. Muokattujen Sharpin kokonaispisteiden (mTSS-pisteet) muutos SPIRIT-P1-tutkimuksessa
| Ero lumeeseen nähden (95 % lv) | ||||||
| Lume (N = 106) | Iksekitsumabi 4 viikon välein (N = 107) | Iksekitsumabi 2 viikon välein (N = 103) | ADA (N = 101) | Iksekitsumabi 4 viikon välein | Iksekitsumabi 2 viikon välein | |
| Lähtötasopisteet, keskiarvo (SD) | 17,6 (28,62) | 19,2 (32,68) | 15,2 (28,86) | 15,9 (27,37) | Ei saatavilla | Ei saatavilla |
| Muutos lähtötilanteesta viikolle 24, LSM (SE) | 0,51 (0,092) | 0,18 (0,090) | 0,09 (0,091) | 0,13 (0,093) | -0,33 (-0,57; -0,09)b | -0,42 (-0,66; -0,19)c |
Lyhenteet: ADA = adalimumabi; lv = luottamusväli; LSM = pienimmän neliösumman keskiarvo; N = analyysipopulaation potilasmäärä; SE = keskivirhe; SD = keskihajonta.
b p < 0,01; c p < 0,001 verrattuna lumeeseen.
Iksekitsumabi esti radiologisesti todettavaa nivelvaurioiden etenemistä (taulukko 11) viikolla 24, ja niiden potilaiden prosenttiosuus, joilla nivelvauriot eivät edenneet radiologisesti (määritelmä: mTSS-arvon muutos lähtötilanteesta ≤ 0,5) satunnaistamisesta viikolle 24, oli 94,8 % iksekitsumabi 2 viikon välein ‑ryhmässä (p < 0,001), 89,0 % iksekitsumabi 4 viikon välein -ryhmässä (p = 0,026) ja 95,8 % adalimumabiryhmässä (p < 0,001), kun näitä kaikkia verrattiin lumeryhmän tuloksiin (77,4 %). mTSS-arvon keskimuutos lähtötilanteesta viikolle 52 oli lume / iksekitsumabi 4 viikon välein ‑ryhmässä 0,27; iksekitsumabi 4 viikon välein / iksekitsumabi 4 viikon välein ‑ryhmässä 0,54; ja adalimumabi / iksekitsumabi 4 viikon välein ‑ryhmässä 0,32. Niiden potilaiden prosenttiosuus, joilla ei todettu nivelvaurioiden radiologista etenemistä satunnaistamishetkeltä viikolle 52, oli lume / iksekitsumabi 4 viikon välein ‑ryhmässä 90,9 %; iksekitsumabi 4 viikon välein / iksekitsumabi 4 viikon välein ‑ryhmässä 85,6 %; ja adalimumabi / iksekitsumabi 4 viikon välein ‑ryhmässä 89,4 %. Potilailla ei ollut rakenteellisten vaurioiden etenemistä lähtötilanteesta (määritelty mTSS≤0,5) eri hoitohaaroissa seuraavien osuuksien mukaisesti: Lume/iksekitsumabi 4 viikon välein 81,5 % (N=22/27), iksekitsumabi 4 viikon välein / iksekitsumabi 4 viikon välein 73,6 % (N = 53/72), ja adalimumabi / iksekitsumabi 4 viikon välein 88,2 % (N = 30/34).
Fyysinen toimintakyky ja terveyteen liittyvä elämänlaatu
Sekä SPIRIT-P1- että SPIRIT-P2-tutkimuksessa iksekitsumabihoitoa 2 viikon välein (p < 0,001) ja 4 viikon välein (p < 0,001) saaneiden potilaiden fyysinen toimintakyky parani merkitsevästi verrattuna lumehoitoa saaneisiin, kun tilannetta arvioitiin Health Assessment Questionnaire-Disability Index ‑mittarilla (HAQ-DI) viikolla 24. SPIRIT-P1-tutkimuksessa ero säilyi viikolle 52.
Iksekitsumabihoitoa saaneet potilaat ilmoittivat terveyteen liittyneen elämänlaadun parantuneen Short Form-36 Health Survey ‑kyselyn fyysisen osion yhteispisteillä (SF-36 PCS) mitattuna (p < 0,001). Myös uupumus lievittyi uupumuksen vaikeusasteen numeerisella NRS-mittarilla mitattuna (p < 0,001).
Markkinoilletulon jälkeinen, vaiheen 4 suora vertailututkimus
Iksekitsumabin tehoa ja turvallisuutta tutkittiin avoimessa, satunnaistetussa monikeskustutkimuksessa, jossa arvioijat olivat sokkoutettuja. Tutkimus oli rinnakkaisryhmätutkimus (SPIRIT-H2H), jossa iksekitsumabia verrattiin adalimumabiin (ADA) 566 nivelpsoriaasia sairastavalla potilaalla, jotka eivät olleet käyttäneet biologisia tautiprosessiin vaikuttavia reumalääkkeitä (bDMARD). Potilaat stratifioitiin lähtötilanteessa samanaikaisen cDMARD-lääkityksen ja keskivaikean tai vaikean psoriaasin (PASI ≥ 12, BSA ≥ 10 ja sPGA ≥ 3) perusteella.
Iksekitsumabi osoittautui paremmaksi kuin ADA ensisijaisen tutkimustavoitteen suhteen: samanaikainen ACR 50-vasteen sekä PASI 100-vasteen saavuttaminen viikon 24 kohdalla (iksekitsumabi 36,0 % vs. ADA 27,9 %; p = 0,036; 95 % luottamusväli [0,5 %; 15,8 %]). Iksekitsumabi osoittautui myös vähintään yhtä hyväksi (etukäteen määritetty -12 % marginaali) kuin ADA ACR 50-vasteen suhteen (ITT-analyysi: iksekitsumabi 50,5 % vs. ADA 46,6 %; 3,9 % ero vs. ADA; 95 % luottamusväli [-4,3 %; 12,1 %]; PPS-analyysi iksekitsumabi: 52,3 %, ADA: 53,1 %, ero -0,8 % [luottamusväli -10,3 %; 8,7 %]), ja iksekitsumabi osoittautui paremmaksi PASI 100-vasteen suhteen viikon 24 kohdalla (60,1 % iksekitsumabin kanssa vs. 46,6 % ADA:n kanssa, p = 0,001). ACR 50- ja PASI 100-vasteet viikolla 24 olivat tutkimuksen keskeisimmät toissijaiset päätetapahtumat. Viikolla 52 suurempi osa iksekitsumabihoitoa saaneista potilaista saavutti samanaikaisesti ACR 50- ja PASI 100-vasteen [39 % (111/283)] verrattuna ADA-hoitoa saaneisiin potilaisiin [26 % (74/283)]. PASI 100-vasteen saavutti iksekitsumabihoitoa saaneista potilaista 64 % (182/283) ja ADA-hoitoa saaneista 41 % (117/283). Iksekitsumabi- ja ADA-hoitojen ACR 50-vasteet olivat samanlaiset [iksekitsumabi 49,8 % (141/283) ja ADA 49,8 % (141/283)]. Vasteet iksekitsumabihoitoon olivat yhdenmukaisia monoterapiana tai yhdessä metotreksaatin kanssa käytettynä.
Kuva 5. Ensisijainen päätetapahtuma (samanaikainen ACR 50 ja PASI 100) sekä keskeisimmät toissijaiset päätetapahtumat (ACR 50; PASI 100), vasteprosentit viikoilla 0–24 [ITT-populaatio, NRI]**
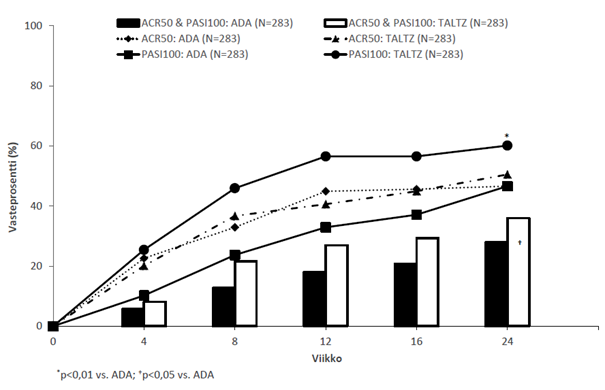
** Iksekitsumabi 160 mg viikon 0 kohdalla, sitten 80 mg kahden viikon välein viikkoon 12 asti ja sen jälkeen joka 4. viikko potilaille, joilla oli keskivaikea tai vaikea läiskäpsoriaasi, tai 160 mg viikon 0 kohdalla, sitten 80 mg joka 4. viikko muille potilaille, ADA 80 mg viikon 0 kohdalla, sitten 40 mg kahden viikon välein viikosta 1 alkaen potilaille, joilla oli keskivaikea tai vaikea läiskäpsoriaasi, tai 40 mg viikon 0 kohdalla, sitten 40 mg kahden viikon välein muille potilaille.Tilastollisen merkitsevyyden taso on merkitty vain päätetapahtumalle, joka oli etukäteen määritetty ja jonka monitestaus oli suoritettu.
Aksiaalinen spondylartriitti
Iksekitsumabia arvioitiin yhteensä 960 aikuispotilaalla, jotka sairastivat aksiaalista spondylartriittia, kolmessa satunnaistetussa, lumekontrolloidussa tutkimuksessa (kaksi röntgenpositiivisen ja yksi röntgennegatiivisen aksiaalisen spondylartriitin tutkimusta).
Röntgenpositiivinen aksiaalinen spondylartriitti
Iksekitsumabia arvioitiin yhteensä 657 potilaalla kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa (COAST-V ja COAST-W) aikuispotilailla, joilla oli BASDAI-indeksin (Bath Ankylosing Spondylitis Disease Activity Index) pistemäärän ≥ 4 perusteella aktiivinen sairaus sekä yhteenlaskettu selkäkipu ≥ 4 numeerisella arviointiasteikolla steroideihin kuulumattomien tulehduskipulääkkeiden (NSAID) käytöstä huolimatta. Kummankin tutkimuksen alkaessa potilailla oli ollut oireita keskimäärin 17 vuoden ajan (mediaani 16 vuotta). Lähtötilanteessa noin 32 %:lla potilaista oli käytössä samanaikainen cDMARD-lääkitys.
COAST-V -tutkimuksessa arvioitiin 341 potilasta, jotka eivät olleet ennen saaneet biologisia lääkkeitä. Potilaat saivat joko iksekitsumabia (80 mg tai 160 mg aloitusannos viikolla 0 ja sen jälkeen 80 mg kahden tai neljän viikon välein), adalimumabia (40 mg kahden viikon välein), tai lumelääkettä. Lumelääkettä saaneet potilaat satunnaistettiin uudestaan viikon 16 kohdalla saamaan iksekitsumabihoitoa (160 mg aloitusannos, minkä jälkeen 80 mg kahden tai neljän viikon välein). Adalimumabia saaneet potilaat satunnaistettiin uudestaan viikon 16 kohdalla saamaan iksekitsumabihoitoa (80 mg kahden tai neljän viikon välein).
COAST-W -tutkimuksessa arvioitiin 316 potilasta, jotka olivat aiemmin käyttäneet yhtä tai kahta TNF-estäjää (90 % oli saanut riittämättömän vasteen ja 10 % ei ollut sietänyt TNF-estäjiä). Kaikki potilaat saivat joko iksekitsumabia (80 mg tai 160 mg aloitusannos viikolla 0 ja sen jälkeen 80 mg kahden tai neljän viikon välein), tai lumelääkettä. Lumelääkettä saaneet potilaat satunnaistettiin uudelleen viikon 16 kohdalla saamaan iksekitsumabia (160 mg aloitusannos, minkä jälkeen 80 mg kahden tai neljän viikon välein).
Ensisijainen päätetapahtuma molemmissa tutkimuksissa oli niiden potilaiden osuus, jotka saavuttivat ASAS40-vasteen (Assessment of Spondyloarthritis International Society 40) viikolla 16.
Kliininen vaste
Kummassakin tutkimuksessa niiden potilaiden, jotka saivat iksekitsumabihoitoa 80 mg kahden viikon välein tai 80 mg neljän viikon välein, ASAS40 ja ASAS20 -vasteet paranivat enemmän verrattuna lumelääkkeeseen viikon 16 kohdalla (taulukko 12). Vasteet olivat potilailla samanlaisia riippumatta muista samanaikaisista hoidoista. COAST-W-tutkimuksessa vasteet havaittiin riippumatta aiempien käytössä olleiden TNF-estäjien määrästä.
Taulukko 12. Tehotulokset tutkimuksissa COAST-V ja COAST-W viikolla 16
COAST-V, ei aiempaa hoitoa biologisilla lääkkeillä | COAST-W, aiemmin TNF-estäjä käytössä | ||||||
Iksekitsumabi 80 mg 4 viikon väleina (N = 81) | Lumelääke
| Ero lume-lääkkeeseeng | Adalimumabi 40 mg 2 viikon välein (N = 90) | Iksekitsumabi 80 mg 4 viikon väleinc (N = 114) | Lumelääke
| Ero lume-lääkkeeseeng | |
| ASAS20-vasteb, n (%), NRI | 52 (64,2%) | 35 (40,2 %) | 24,0 (9,3; 38,6)** | 53 (58,9 %) | 55 (48,2 %) | 31 (29,8 %) | 18,4 (5,7; 31,1)** |
| ASAS40-vasteb,c, n (%), NRI | 39 (48,1%) | 16 (18,4 %) | 29,8 (16,2; 43,3)*** | 32 (35,6 %) | 29 (25,4 %) | 13 (12,5 %) | 12,9 (2,7; 23,2)* |
| ASDAS | |||||||
| Muutos lähtötilanteesta | -1,4 | -0,5 | -1,0 (-1,3; -0,7)*** | -1,3*** | ‑1,2 | -0,1 | -1,1 (-1,3; -0,8)*** |
| Lähtötilanne | 3,7 | 3,9 | 3,7 | 4,2 | 4,1 | ||
| BASDAI-pisteet | |||||||
| Muutos lähtötilanteesta | -2,9 | -1,4 | -1,5 (-2,1; -0,9)*** | -2,5*** | ‑2,2 | -0,9 | -1,2 (-1,8; -0,7)*** |
| Lähtötilanne | 6,8i | 6,8i | 6,7i | 7,5 | 7,3 | ||
| MRI Spine SPARCCd | |||||||
| Muutos lähtötilanteesta | ‑11,0 | -1,5 | -9,5 (-12,6; -6,4) *** | -11,6*** | ‑3,0 | 3,3 | -6,3 (-10,0; -2,5)** |
| Lähtötilanne | 14,5 | 15,8 | 20,0 | 8,3 | 6,4 | ||
| BASDAI50e n (%), NRI | 34 (42,0 %) | 15 (17,2 %) | 24,7 (11,4; 38,1)*** | 29 (32,2 %)* | 25 (21,9 %)i | 10 (9,6 %)i | 12,3 (2,8; 21,8)* |
| ASDAS < 2,1, n (%) (alhainen tautiaktiivisuus), NRI | 35 (43,2 %)h | 11 (12,6 %)h | 30,6 (17,7; 43,4)*** | 34 (37,8 %)*** h | 20 (17,5 %) | 5 (4,8 %) | 12,7 ( 4,6; 20,8)** |
| ASDAS < 1,3, n (%) (inaktiivinen tauti), NRI | 13 (16,0 %) | 2 (2,3 %) | 13,8 (5,2; 22,3)** | 14 (15,6 %)** | 4 (3,5 %)i | 1 (1,0 %)i | 2,5 (-1,3; 6,4) |
| ASAS HIf Muutos lähtötilanteesta |
-2,4 |
-1,3 |
-1,1 (-2,0; -0,3)* |
-2,3* |
‑1,9 |
-0,9 |
-1,0 (-1,9; -0,1)* |
| Lähtötilanne | 7,5 | 8,1 | 8,2 | 10,0 | 9,0 | ||
| SF-36 PCS Muutos lähtötilanteesta |
7,7 |
3,6 |
4,1 (1,9; 6,2)*** |
6,9** |
6,6 |
1,4 |
5,2 (3,0; 7,4)*** |
| Lähtötilanne | 34,0 | 32,0 | 33,5 | 27,5 | 30,6 | ||
Lyhenteet: N = hoitoaikomuspopulaation (intent-to-treat) potilasmäärä, NRI = Non-responder Imputation; ASAS HI = Assessment of SpondyloArthritis International Society Health Index;ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CFB = pienimmän neliösumman keskiarvo, muutos lähtötilanteesta viikolla 16;MRI Spine SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the Spine (23 nikamavälilevy-yksikön asteikko)
a Viikon 0 kohdalla potilaat saivat 80 mg tai 160 mg iksekitsumabia.
b ASAS20-vasteeksi määritellään ≥ 20 % pareneminen ja ≥ 1 yksikön (asteikko 0−10) absoluuttinen paraneminen lähtötilanteesta vähintään kolmessa neljästä arvioitavasta osa-alueesta (potilaan kokonaisarvio, selkärankakipu, toimintakyky ja tulehdus), eikä jäljellä olevassa osa-alueesta ≥ 20 % ja ≥ 1 yksikön (asteikko 0−10) huononemista. ASAS40-vasteeksi määritellään ≥ 40 % paraneminen ja ≥ 2 yksikön absoluuttinen paraneminen lähtötilanteesta vähintään kolmessa neljästä arvioitavasta osa-alueesta ilman yhtään huononemista jäljelle jäävässä osa-alueessa.
c Ensisijainen päätetapahtuma.
d Niiden hoitoaikomuspopulaatioon kuuluvien potilaiden määrä, joiden MRI-tiedot saatavilla lähtötilanteessa: COAST-V: iksekitsumabi, n = 81; lumelääke, n = 82; ADA, n = 85. COAST-W: iksekitsumabi, n = 58; lumelääke, n = 51.
e BASDAI50-vasteen määritelmä on ≥ 50 % paraneminen BASDAI-pisteissä verrattuna lähtötilanteeseen.
f ASAS HI: Assessment of SpondyloArthritis International Society Health Index (ASAS HI) kaikkien arvioitavien osa-alueiden perusteella.
g Luokka-arvoisille muuttujille on ilmoitettu ero prosenteissa (95 %:n luottamusväli), jatkuville muuttujille on ilmoitettu ero pienimmän neliösumman keskiarvossa (95 %:n luottamusväli).
h post hoc -analyysi, ei korjattu kertautuvuuden suhteen.
i ennalta määritetty, mutta ei huomioitu kertautuvuutta.
* p < 0,05; ** p < 0,01; *** p < 0,001 verrattuna lumelääkkeeseen.
ASAS40-vasteen keskeisissä osatekijöissä (selkärangan kipu, BASFI, potilaan kokonaisarvio, jäykkyys) ja muissa sairauden aktiivisuuden mittareissa, mukaan lukien CRP:ssä, havaittiin paranemista viikolla 16.
Kuva 6. ASAS40-vasteen COAST-V ja COAST-W -tutkimuksissa saavuttaneiden potilaiden prosenttiosuudet viikkoon 16 asti, NRIa
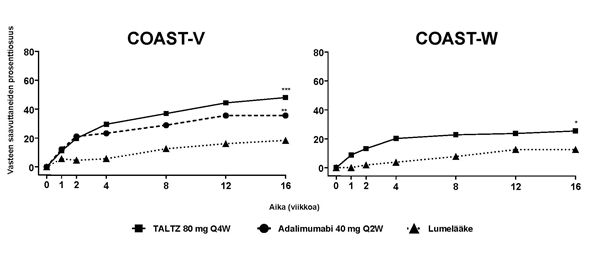
a Jos potilaalta puuttui tietoja, katsottiin, että hän ei ollut saavuttanut vastetta.
* p < 0,05; ** p < 0,01; *** p < 0,001 lumelääkkeeseen verrattuna.
Potilailla havaittiin samankaltainen ASAS40-vaste riippumatta lähtötilanteen CRP-tasoista, lähtötilanteen ASDAS-pisteistä ja selkärangan magneettikuvan SPARCC -pisteistä. ASAS40-vaste ei riippunut iästä, sukupuolesta, etnisestä taustasta, taudin kestosta, kehon painosta lähtötilanteessa, lähtötilanteen BASDAI-pisteistä tai aiemmasta hoidosta biologisella valmisteella.
Tutkimuksissa COAST-V ja COAST-W teho säilyi viikolle 52 asti taulukossa 12 esitettyjen päätetapahtumien, mukaan lukien ASAS20-, ASAS40-, ASDAS-, BASDAI- ja ASAS HI-vasteosuudet, perusteella arvioituna.
Terveyteen liittyvät tulokset
Selkärankakivussa havaittiin paranemista lumelääkkeeseen verrattuna jo viikon 1 kohdalla, ja paraneminen säilyi viikkoon 16 asti [iksekitsumabi vs. lumelääke: COAST-V -3,2 vs. -1,7; COAST-W -2,4 vs. -1,0]; väsymyksessä ja selkärangan liikkuvuudessa havaittiin paranemista lumelääkkeeseen verrattuna viikon 16 kohdalla. Selkärankakivun, väsymyksen ja selkärangan liikkuvuuden paraneminen säilyivät viikolle 52 asti.
Röntgennegatiivinen aksiaalinen spondylartriitti
Iksekitsumabia arvioitiin satunnaistetussa, kaksoissokkoutetussa tutkimuksessa (COAST-X), jossa oli 52 viikon lumelääkekontrolloitu jakso. Tutkimuksessa oli 303 aikuista potilasta, joilla oli vähintään 3 kuukauden ajan aktiivinen aksiaalinen spondylartriitti. Potilailla oli oltava objektiivisina tulehduksen merkkeinä kohonnut C-reaktiivisen proteiinin (CRP) pitoisuus ja/tai magneettikuvassa sakroiliitti, eikä ehdotonta radiografista näyttöä risti-suoliluunivelten rakenteellisesta vauriosta. Potilailla oli BASDAI-indeksin (Bath Ankylosing Spondylitis Disease Activity Index) pistemäärän ≥ 4 perusteella aktiivinen sairaus ja selkärankakipu ≥ 4 numeerisella arviointiasteikolla 0−10 steroideihin kuulumattomien tulehduskipulääkkeiden (NSAID) käytöstä huolimatta. Potilaat saivat joko iksekitsumabia (80 mg tai 160 mg aloitusannos viikolla 0 ja sen jälkeen 80 mg kahden tai neljän viikon välein) tai lumelääkettä. Samanaikaisesti käytössä olleiden muiden lääkkeiden (NSAID, cDMARD, kortikosteroidit, kipulääkkeet) annosmuutokset ja/tai hoidon aloitukset sallittiin viikosta 16 alkaen.
Lähtötilanteessa potilailla oli ollut röntgennegatiivisen aksiaalisen spondylartriitin oireita keskimäärin 11 vuoden ajan. Noin 39 %:lla potilaista oli samanaikaisesti käytössä cDMARD-lääkitys.
Ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat Assessment of Spondyloarthritis International Society 40 (ASAS40) -vasteen viikolla 16.
Kliininen vaste
Suurempi osuus iksekitsumabihoitoa 80 mg neljän viikon välein saaneista potilaista saavutti ASAS40-vasteen verrattuna lumelääkettä saaneisiin potilaisiin viikon 16 kohdalla (taulukko 13). Vasteet olivat samankaltaisia muista samanaikaisessa käytössä olleista lääkkeistä riippumatta.
Taulukko 13. Tehotulokset viikon 16 kohdalla tutkimuksessa COAST-X, NRIa,b
Iksekitsumabi 80 mg (N = 96) | Lumelääke (N = 105) | Ero lumelääkkeeseenh | |
| ASAS20-vasted, n (%), NRI | 52 (54,2 %) | 41 (39,0 %) | 15,1 (1,5; 28,8)* |
| ASAS40-vasted,e, n (%), NRI | 34 (35,4 %) | 20 (19,0 %) | 16,4 (4,2; 28,5)** |
| ASDAS | |||
| Muutos lähtötilanteesta | -1,1 | -0,6 | -0,5 (-0,8; -0,3)*** |
| Lähtötilanne | 3,8 | 3,8 | |
| BASDAI-pisteet | |||
| Muutos lähtötilanteesta | -2,2 | -1,5 | -0,7 (-1,3; -0,1)* |
| Lähtötilanne | 7,0 | 7,2 | |
| MRI SIJ SPARCCf | |||
| Muutos lähtötilanteesta | -3,4 | -0,3 | -3,1 (-4,6; -1,6)*** |
| Lähtötilanne | 5,1 | 6,3 | |
| ASDAS < 2,1, n (%) (alhainen tautiaktiivisuus), NRIg | 26 (27,7 %) | 13 (12,4 %) | 15,3 (4,3; 26,3)** |
| SF-36 PCS | |||
| Muutos lähtötilanteesta | 8,1 | 5,2 | 2,9 (0,6; 5,1)* |
| Lähtötilanne | 33,5 | 32,6 | |
a Lyhenteet: N = hoitoaikomuspopulaation (intent-to-treat) potilasmäärä, NRI = Non-responder Imputation; ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; Muutos lähtötilanteesta = pienimmän neliösumman keskiarvo, muutos lähtötilanteesta viikolla 16;MRI SIJ SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the sacroiliac joint.
b Jos potilaalta puuttui tietoja, katsottiin, että hän ei ollut saavuttanut vastetta.
c Viikon 0 kohdalla potilaat saivat 80 mg tai 160 mg iksekitsumabia.
d ASAS20-vasteeksi määritellään ≥ 20 % pareneminen ja ≥ 1 yksikön (asteikko 0−10) absoluuttinen paraneminen lähtötilanteesta vähintään kolmessa neljästä arvioitavasta osa-alueesta (potilaan kokonaisarvio, selkärankakipu, toimintakyky ja tulehdus), eikä huononemista jäljellä olevassa osa-alueessa ≥ 20 % ja ≥ 1 yksikön (asteikko 0−10) huononemista. ASAS40-vasteeksi määritellään ≥ 40 % paraneminen ja ≥ 2 yksikön absoluuttinen paraneminen lähtötilanteesta vähintään kolmessa neljästä arvioitavasta osa-alueesta ilman yhtään huononemista jäljelle jäävässä osa-alueessa.
e Ensisijainen päätetapahtuma viikolla 16.
f Niiden hoitoaikomuspopulaatioon kuuluvien potilaiden määrä, joiden MRI-tiedot saatavilla lähtötilanteessa ja viikolla 16: iksekitsumabi, n = 85; lumelääke, n = 90.
g Jos potilaalta puuttui tietoja, katsottiin, että hän ei ollut saavuttanut vastetta. Prosenttiosuudet perustuvat niihin hoitoaikomuspopulaatioon kuuluviin potilaisiin, joiden lähtötilanteen ASDAS-pisteet olivat ≥ 2,1.
h Luokka-arvoisille muuttujille on ilmoitettu ero prosenteissa (95 %:n luottamusväli), jatkuville muuttujille on ilmoitettu ero pienimmän neliösumman keskiarvossa (95 %:n luottamusväli).
* p < 0,05; ** p < 0,01; *** p < 0,001 verrattuna lumelääkkeeseen.
ASAS40-vasteen keskeisissä osatekijöissä (selkärangan kipu, BASFI, potilaan kokonaisarvio, jäykkyys) ja muissa sairauden aktiivisuuden mittareissa havaittiin merkitsevä kliininen paraneminen viikolla 16.
Kuva 7. ASAS40-vasteen tutkimuksessa COAST-X saavuttaneiden potilaiden prosenttiosuudetviikolle 16 asti, NRIa
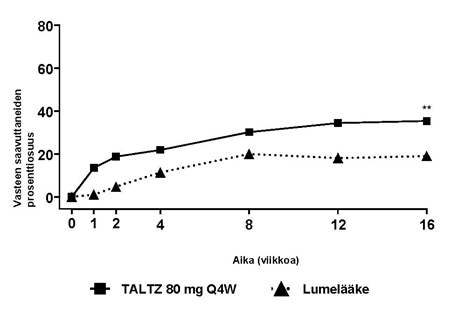
a Jos potilaalta puuttui tietoja, katsottiin, että hän ei ollut saavuttanut vastetta.
** p < 0,01 lumelääkkeeseen verrattuna.
Teho säilyi viikolle 52 asti taulukossa 13 esitettyjen päätetapahtumien perusteella arvioituna.
Terveyteen liittyvät tulokset
Selkärankakivussa havaittiin paranemista lumelääkkeeseen verrattuna jo viikon 1 kohdalla, ja paraneminen säilyi viikkoon 16 asti [iksekitsumabi vs. lumelääke: COAST-X: -2,4 vs. -1,5]. Lisäksi useampi iksekitsumabihoitoa saanut potilas saavutti hyvän terveydentilan (ASAS HI ≤5) lumelääkkeeseen verrattuna viikoilla 16 ja 52.
Pitkäaikaistulokset aksiaalisessa spondylartriitissa
Potilaille, jotka olivat mukana loppuun asti yhdessä kolmesta keskeisestä tutkimuksesta COAST‑V/W/X (52 viikkoa) tarjottiin mahdollisuutta osallistua pitkäaikaiseen jatkotutkimukseen, jossa osa potilaista satunnaistettiin keskeyttämään hoito (COAST‑Y, 350 potilasta otettiin iksekitsumabi neljän viikon välein ja 423 potilasta iksekitsumabi kahden viikon välein -ryhmään). Niiden potilaiden joukossa, jotka saavuttivat remission 157/773 (20,3 %) (Ankylosing Spondylitis Disease Activity Score [ASDAS] < 1,3 ainakin kerran ja ASDAS-pistemäärä ei ollut ≥ 2,1, viikoilla 16 ja 20), 155 potilasta altistui iksekitsumabille 76 viikon ajan. Viikolla 24 heidät satunnaistettiin COAST‑Y tutkimukseen (lumelääke, N = 53; iksekitsumabi neljän viikon välein, N=48; ja iksekitsumabi kahden viikon välein, N = 54); näistä 148 (95,5 %) saavutti viikon 64 käynnin (lumelääke, N = 50; iksekitsumabi neljän viikon välein, N = 47; iksekitsumabi kahden viikon välein, N = 51). Ensisijainen päätetapahtuma oli niiden potilaiden osuus satunnaistetussa hoidon keskeyttäneiden ryhmässä, joiden oireet eivät pahentuneet viikkojen 24‑64 aikana (yhdistetyssä iksekitsumabi kahden viikon välein ja iksekitsumabi neljän viikon välein -ryhmässä verrattuna lumelääkkeeseen). Merkitsevästi suuremmalla osuudella potilaista (NRI) yhdistetyssä iksekitsumabiryhmässä (83,3% (85/102), p < 0,001) ja iksekitsumabi neljän viikon välein -ryhmässä (83,3 % (40/48), p = 0,003) ei ollut oireiden pahenemista viikkojen 24‑64 aikana verrattuna niihin, jotka siirtyivät iksekitsumabista lumelääkkeeseen (54,7 % (29/53)). Iksekitsumabi (sekä yhdistetyssä iksekitsumabiryhmässä että iksekitsumabi neljän viikon välein -ryhmässä) viivästytti merkitsevästi oireiden pahenemista (Log‑Rank Test p < 0,001 yhdistetyssä iksekitsumabiryhmässä ja p < 0,01 iksekitsumabi neljän viikon välein -ryhmässä) verrattuna lumelääkkeeseen.
Potilailla, jotka saivat koko ajan iksekitsumabia neljän viikon välein (N = 157), ASAS40, ASDAS < 2,1 ja BASDAI50 -vasteet säilyivät viikolle 116.
Lastenreuma (juveniili idiopaattinen artriitti)
Lasten nivelpsoriaasi ja entesiitteihin liittyvä artriitti
Tehoa, turvallisuutta, siedettävyyttä ja farmakokinetiikkaa tarkastelleessa avoimessa monikeskustutkimuksessa (COSPIRIT-JIA), jossa oli adalimumabi-vertailuhaara, ihonalaisesti annostellun iksekitsumabin turvallisuutta ja tehoa arvioitiin 16 viikon ajan hoidon aloittamisen jälkeen vähintään 2- ja alle 18-vuotiailla lapsilla, joilla oli lasten nivelpsoriaasi tai entesiitteihin liittyvä artriitti. Tutkimuksen ensisijainen päätetapahtuma oli niiden potilaiden osuus (prosentteina), jotka täyttivät lastenreuman ACR 30 (30 % parannus American College of Rheumatology -kriteereissä) vastekriteerit viikolla 16.
Iksekitsumabille satunnaistettiin 20 potilasta ja adalimumabille 20 potilasta. Satunnaistaminen stratifioitiin lastenreuman luokan mukaan (lasten nivelpsoriaasi tai entesiitteihin liittyvä artriitti). Jäljellä olevat potilaat, jotka olivat tai eivät olleet saaneet bDMARD-hoitoa, saivat iksekitsumabia. Tutkimukseen tulevilta potilailta ei vaadittu dokumentoitua riittämätöntä vastetta aiempaan hoitoon.
Iksekitsumabiryhmässä (N = 81) lastenreumaa sairastavien potilaiden alaluokat tutkimukseen tullessa olivat: lasten nivelpsoriaasi 33,3 % ja entesiitteihin liittyvä artriitti 66,7 %. Potilaista 74,1 % (60/81) ei ollut saanut bDMARD-hoitoa ja 33,3 % (27/81) ei ollut saanut cDMARD-hoitoa. Kaiken kaikkiaan 72,8 % iksekitsumabilla hoidetuista potilaista sai vähintään yhtä samanaikaista hoitoa lastenreumaan avoimen hoitojakson (OLT) aikana. Lähtötilanteessa samanaikaista metotreksaatin käyttöä ilmoitettiin 40,7 %:lla potilaista, sulfasalatsiinin käyttöä 4,9 %:lla potilaista, steroideihin kuulumattomien tulehduskipulääkkeiden (NSAID) käyttöä 49,4 %:lla potilaista ja kortikosteroidien käyttöä 11,1 %:lla potilaista.
Iksekitsumabivalmistetta saaneet potilaat (N = 81) saivat painon mukaan stratifioidun annostuksen seuraavasti:
10 kg − < 25 kg: 40 mg viikolla 0 ja sen jälkeen 20 mg neljän viikon välein (n = 6)
25 kg−50 kg: 80 mg viikolla 0 ja sen jälkeen 40 mg neljän viikon välein (n = 20)
> 50 kg: 160 mg viikolla 0 ja sen jälkeen 80 mg neljän viikon välein (n = 55)
Satunnaistetuilla potilailla, jotka eivät olleet saaneet bDMARD-hoitoa, lastenreuman ACR 30 vastekriteerit viikolla 16 olivat iksekitsumabiryhmässä 18/20 (90 %) ja adalimumabiryhmässä 19/20 (95 %). Kaiken kaikkiaan iksekitsumabilla hoidetuilla potilailla (n = 81) lastenreuman ACR 30 vastekriteerit olivat 54/60 (90 %) potilailla, jotka eivät olleet saaneet bDMARD-hoitoa ja 18/21 (85,7 %) potilailla, jotka olivat saaneet bDMARD-hoitoa.
Lastenreuman ACR 30 -vasteosuudet viikolla 16 olivat myös yhdenmukaiset molemmissa alaluokissa, lasten nivelpsoriaasi (24/27, 88,9 %) ja entesiitteihin liittyvä artriitti (48/54, 88,9 %).
Lisäksi arvioitiin niiden potilaiden osuudet (prosentteina), jotka saavuttivat lastenreuman ACR 30/50/70/90/100 -vasteen viikolla 16. Tiedot kliinisestä vasteesta esitetään kuvassa 8.
Kuva 8. Lastenreuman ACR 30/50/70/90/100 -vasteosuudet iksekitsumabiryhmässä 16 viikon aikana – ITT-populaatio (NRI-menetelmä)
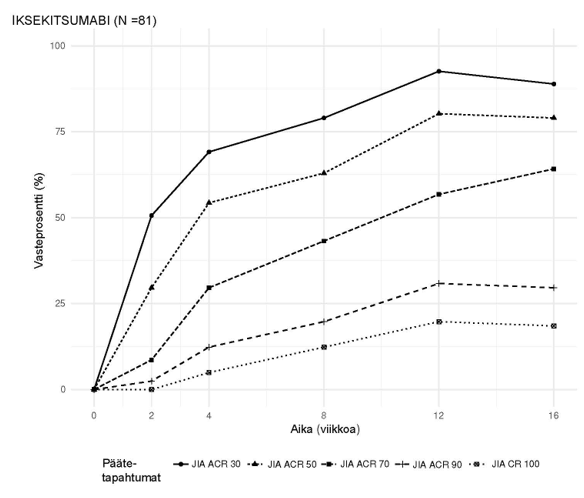
Lyhenteet: ACR 30/50/70/90/100 = 30 %/50 %/70 %/90 %/100 % parannus American College of Rheumatology -kriteereillä; ITT = hoitoaikomuspopulaatio (intent-to-treat); JIA = lastenreuma (juveniili idiopaattinen artriitti); N = analyysipopulaation potilasmäärä; NRI = Non-responder Imputation.
Rokotukset
Terveillä henkilöillä tehdyssä tutkimuksessa ei todettu turvallisuusriskejä, kun tutkimushenkilöille annetun kahden iksekitsumabiannoksen (160 mg annos ja 2 viikon kuluttua 80 mg annos) jälkeen heille annettiin kahta inaktivoitua rokotetta (jäykkäkouristus- ja pneumokokkirokotteet). Rokotuksia koskevat tiedot eivät kuitenkaan riitä selvittämään, tuottavatko kyseiset rokotteet asianmukaisen immuunivasteen iksekitsumabin annon jälkeen.
Pediatriset potilaat
Euroopan lääkevirasto on luopunut vaatimuksesta toimittaa tulokset tutkimuksista koskien iksekitsumabin käyttöä yhdessä tai useammassa lapsipotilaiden osajoukossa läiskäpsoriaasin ja nivelpsoriaasin/aksiaalisen spondylartriitin hoidossa (ks. kohta Annostus ja antotapa pediatrista käyttöä koskevien tietojen osalta).
Farmakokinetiikka
Imeytyminen
Kun iksekitsumabia annettiin kerta-annos psoriaasipotilaiden ihon alle, huippupitoisuuden keskiarvot saavutettiin 4–7 päivässä 5–160 mg annoksilla. Iksekitsumabin maksimipitoisuuden keskiarvo plasmassa (Cmax) oli 160 mg aloitusannoksen jälkeen 19,9 (keskihajonta [SD] 8,15) µg/ml.
160 mg aloitusannoksen jälkeen vakaa tila saavutettiin viikkoon 8 mennessä, kun lääkettä otettiin 80 mg 2 viikon välein. Cmax,ss-pitoisuuden keskiarvo (SD) on arviolta 21,5 (9,16) µg/ml ja Ctrough,ss-pitoisuuden keskiarvo (SD) vastaavasti 5,23 (3,19) µg/ml.
Kun potilaat siirtyvät 80 mg annoksista 2 viikon välein 80 mg annoksiin 4 viikon välein viikolla 12, vakaa tila saavutetaan noin 10 viikon kuluttua. Cmax,ss-pitoisuuden keskiarvo (SD) on arviolta 14,6 (6,04) µg/ml ja Ctrough,ss-pitoisuuden keskiarvo (SD) vastaavasti 1,87 (1,30) µg/ml.
Ihon alle annetun iksekitsumabin biologisen hyötyosuuden keskiarvo oli eri analyyseissä 54–90 %.
Jakautuminen
Populaatiofarmakokineettisten analyysien perusteella kokonaisjakautumistilavuuden keskiarvo oli vakaassa tilassa 7,11 l.
Biotransformaatio
Iksekitsumabi on monoklonaalinen vasta-aine, jonka oletetaan hajoavan pieniksi peptideiksi ja aminohapoiksi kataboliareittien välityksellä samaan tapaan kuin endogeeniset immunoglobuliinit.
Eliminaatio
Populaatiofarmakokineettisessä analyysissä lääkkeen puhdistuma seerumista oli keskimäärin 0,0161 l/h. Puhdistuma ei riipu annoksesta. Populaatiofarmakokineettisen analyysin perusteella arvioitu eliminaation puoliintumisajan keskiarvo on läiskäpsoriaasipotilailla 13 päivää.
Lineaarisuus/ei-lineaarisuus
Altistus (käyrän alle jäävä pinta-ala, AUC) suureni suhteessa annokseen, kun 5–160 mg annoksia annettiin injektiona ihon alle.
Farmakokineettiset ominaisuudet eri käyttöaiheissa
Iksekitsumabin farmakokinetiikka oli samankaltainen käyttöaiheissa läiskäpsoriaasi, nivelpsoriaasi, röntgenpositiivinen aksiaalinen spondylartriitti sekä röntgennegatiivinen aksiaalinen spondylartriitti.
Iäkkäät potilaat
Kliinisissä tutkimuksissa iksekitsumabia on saanut 4 204 läiskäpsoriaasipotilasta. Heistä yhteensä 301 oli vähintään 65-vuotiaita ja 36 oli vähintään 75-vuotiaita. 1 118 nivelpsoriaasipotilasta altistui iksekitsumabille kliinisissä tutkimuksissa. Heistä yhteensä 122 oli vähintään 65-vuotiaita ja 6 oli vähintään 75-vuotiaita.
Populaatiofarmakokineettisessä analyysissä, jossa oli mukana rajallinen määrä iäkkäitä potilaita (≥ 65-vuotiaiden n = 94 ja ≥ 75-vuotiaiden n = 12), puhdistuma oli iäkkäillä potilailla samankaltainen kuin alle 65-vuotiailla potilailla.
Munuaisten tai maksan vajaatoiminta
Munuaisten ja maksan vajaatoiminnan vaikutusta iksekitsumabin farmakokinetiikkaan ei ole arvioitu spesifisissä kliinisen farmakologian tutkimuksissa. Muuttumattoman iksekitsumabin (monoklonaalinen IgG-vasta-aine) munuaisteitse tapahtuvan eliminaation oletetaan olevan niukkaa ja merkitykseltään vähäistä; monoklonaaliset IgG-vasta-aineet eliminoituvat lähinnä solunsisäisen katabolian kautta, ja myöskään maksan vajaatoiminnan ei oleteta vaikuttavan iksekitsumabin puhdistumaan.
Pediatriset potilaat
Pediatrisille psoriaasipotilaille (iältään 6-vuotiaasta alle 18-vuotiaaseen) annosteltiin iksekitsumabia lapsille suositellun annoksen mukaisesti 12 viikon ajan. Yli 50 kg painavien potilaiden keskimääräinen vakaan tilan jäännöspitoisuus ± keskihajonta oli 3,8 ± 2,2 µg/ml ja 25−50 kg painavien potilaiden 3,9 ± 2,4 µg/ml viikolla 12.
Pediatrisille lastenreumapotilaille (iältään 6-vuotiaasta alle 18-vuotiaaseen) annettin iksekitsumabia lapsille suositellun annoksen mukaisesti 16 viikon ajan. Yli 50 kg painavien potilaiden keskimääräinen vakaan tilan jäännöspitoisuus ± keskihajonta oli 3,9 ± 1,8 µg/ml ja 25−50 kg painavien potilaiden 3,5 ± 1,3 µg/ml viikolla 16. Tietoja farmakokinetiikasta alle 25 kg painavilla potilailla oli rajoitetusti.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien tutkimusten, farmakologisen turvallisuuden arviointien sekä lisääntymis- ja kehitystoksisuutta koskevien ei-kliinisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Iksekitsumabin anto makakiapinoille 39 viikon ajan (enintään 50 mg/kg viikossa ihon alle) ei aiheuttanut elimiin kohdistuvaa toksisuutta eikä vaikuttanut haitallisesti immuunijärjestelmän toimintaan (esim. T-soluista riippuvaiseen vasta-ainevasteeseen tai NK-solujen toimintaan). Apinoille annettu 50 mg/kg annos viikossa ihon alle on noin 19 kertaa iksekitsumabin 160 mg aloitusannoksen suuruinen ja tuottaa apinoilla altistuksen (AUC), joka on vähintään 61 kertaa suurempi kuin ennustettu vakaan tilan keskialtistus ihmisillä, jotka saavat annostussuosituksen mukaisia annoksia.
Iksekitsumabin karsinogeenisuutta ja mutageenisuutta ei ole arvioitu ei-kliinisissä tutkimuksissa.
Kun sukukypsille makakiapinoille annettiin iksekitsumabia 13 viikon ajan (50 mg/kg viikossa ihon alle), sukupuolielimiin, kiimakiertoon tai siittiöihin kohdistuvia vaikutuksia ei havaittu.
Kehitystoksisuustutkimuksissa iksekitsumabin todettiin läpäisevän istukan, ja sitä esiintyi jälkeläisten veressä jopa 6 kuukauden ajan. Iksekitsumabia saaneiden apinoiden jälkeläisten postnataalinen kuolleisuus oli suurempaa kuin verrokkien jälkeläisten. Tämä liittyi lähinnä varhaisiin synnytyksiin tai siihen, että emo ei huolehtinut jälkeläisistä. Nämä löydökset ovat yleisiä kädellistutkimuksissa, eikä niillä katsota olevan kliinistä merkitystä.
Farmaseuttiset tiedot
Apuaineet
Sakkaroosi
Polysorbaatti 80 (E 433)
Injektionesteisiin käytettävä vesi
Natriumhydroksidi (pH:n säätämiseen)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
2 vuotta.
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Taltz-valmistetta voidaan säilyttää huoneenlämmössä enintään 5 päivän ajan korkeintaan 30 °C:n lämpötilassa.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TALTZ injektioneste, liuos, esitäytetty kynä
80 mg (L:ei) 1 kpl (1 ml) (937,29 €)
TALTZ injektioneste, liuos, esitäytetty ruisku
80 mg (L:ei) 1 kpl (1 ml) (937,29 €)
PF-selosteen tieto
Taltz 80 mg injektioneste, liuos, esitäytetty kynä
1 ml liuosta tyypin I kirkkaassa lasiruiskussa.
Ruisku on pakattu kertakäyttöiseen kerta-annoskynään.
Pakkauskoko: 1 esitäytetty kynä.
Taltz 40 mg injektioneste, liuos, esitäytetty ruisku
0,5 ml liuosta tyypin I kirkkaassa lasiruiskussa.
Pakkauskoko: 1 esitäytetty ruisku.
Taltz 80 mg injektioneste, liuos, esitäytetty ruisku
1 ml liuosta tyypin I kirkkaassa lasiruiskussa.
Pakkauskoko: 1 esitäytetty ruisku.
Valmisteen kuvaus:
Liuos on kirkas ja väritön tai hiukan kellertävä. Liuoksen pH on vähintään 5,2 ja enintään 6,2, ja osmolaliteetti vähintään 235 mOsm/kg ja enintään 360 mOsm/kg.
Käyttö- ja käsittelyohjeet
Pakkausselosteen mukana olevia kynän tai ruiskun käyttöohjeita on noudatettava tarkoin.
Esitäytetty kynä tai ruisku on tarkoitettu vain yhtä käyttökertaa varten.
Taltz-valmistetta ei saa käyttää, jos siinä on hiukkasia tai jos liuos on sameaa ja/tai selvästi ruskeaa.
Jäätynyttä Taltz-valmistetta ei saa käyttää.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Vain Taltz 80 mg injektioneste, liuos, esitäytetty ruisku:
40 mg iksekitsumabiannoksen valmistelu lapsille, jotka painavat 25−50 kg
Jos 40 mg:n esitäytettyä ruiskua ei ole saatavilla, asianmukaisen pätevyyden omaavan terveydenhuollon ammattilaisen on valmisteltava ja annosteltava 40 mg iksekitsumabiannokset. Käytä vain Taltz 80 mg, injektioneste, liuos, esitäytettyä ruiskua lapselle määrätyn 40 mg annoksen valmisteluun.
- Ruiskuta koko esitäytetyn ruiskun sisältö steriiliin, kirkkaaseen lasiseen injektiopulloon. ÄLÄ ravista tai pyöritä injektiopulloa.
- Käytä 0,5 ml tai 1 ml kertakäyttöistä ruiskua ja steriiliä neulaa, ja vedä ruiskuun määrätty annos (0,5 ml 40 mg annosta varten) injektiopullosta.
- Vaihda neula ja käytä 27 G steriiliä neulaa potilaan pistämiseen. Hävitä pulloon jäänyt käyttämätön iksekitsumabi.
Valmisteltu iksekitsumabi on annosteltava 4 tunnin sisällä steriilin injektiopullon lävistämisestä huoneenlämmössä.
Korvattavuus
TALTZ injektioneste, liuos, esitäytetty kynä
80 mg 1 kpl
TALTZ injektioneste, liuos, esitäytetty ruisku
80 mg 1 kpl
- Alempi erityiskorvaus (65 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli ja tosilitsumabi (tulehdukselliset reumasairaudet): Nivelreuman, juveniilin polyartriitin, psoriaasiin liittyvän niveltulehduksen, selkärankareuman tai edellä mainittuja niveltulehduksia läheisesti muistuttavan niveltulehduksen hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman hoito erityisin edellytyksin (281).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli, tosilitsumabi ja ustekinumabi (tulehdukselliset reumasairaudet): Eräiden reumasairauksien hoito erityisin edellytyksin / Adalimumabi: Uveiitin hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman ja jättisoluarteriitin hoito erityisin edellytyksin (313), Adalimumabi, bimekitsumabi, brodalumabi, etanersepti, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sekukinumabi, sertolitsumabipegoli, tildrakitsumabi ja ustekinumabi (ihopsoriaasi): Vaikean kroonisen ihopsoriaasin hoito erityisin edellytyksin (319).
ATC-koodi
L04AC13
Valmisteyhteenvedon muuttamispäivämäärä
26.03.2026
Yhteystiedot
 OY ELI LILLY FINLAND AB
OY ELI LILLY FINLAND AB Mannerheimintie 117
00280 Helsinki
09 854 5250
www.lilly.com/fi
medinfo_finland@lilly.com
Lääketietopalvelu puh. 0800-140 240