
FORXIGA tabletti, kalvopäällysteinen 5 mg, 10 mg
Vaikuttavat aineet ja niiden määrät
Forxiga 5 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää dapagliflotsiinipropaanidiolimonohydraattia määrän, joka vastaa 5 mg dapagliflotsiinia.
Apuaine, jonka vaikutus tunnetaan
Yksi 5 mg:n tabletti sisältää 25 mg laktoosia.
Forxiga 10 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää dapagliflotsiinipropaanidiolimonohydraattia määrän, joka vastaa 10 mg:aa dapagliflotsiinia.
Apuaine, jonka vaikutus tunnetaan
Yksi 10 mg:n tabletti sisältää 50 mg laktoosia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen
Kliiniset tiedot
Käyttöaiheet
Tyypin 2 diabetes
Forxiga on tarkoitettu aikuisille potilaille ja vähintään 10‑vuotiaille lapsipotilaille tyypin 2 diabeteksen hoitoon ruokavalion ja liikunnan lisäksi, kun sairaus ei ole riittävässä hoitotasapainossa
- monoterapiana, kun metformiinin käyttöä ei voida pitää tarkoituksenmukaisena sietokyvyttömyyden takia
- yhdistettynä muihin tyypin 2 diabeteksen hoitoon käytettäviin lääkevalmisteisiin.
Yhdistelmähoitoja, vaikutuksia glukoositasapainoon, sydän- ja verisuonitapahtumia, munuaistapahtumia sekä tutkittuja populaatioita koskevat tutkimustulokset on kuvattu kohdissa Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakodynamiikka.
Sydämen vajaatoiminta
Forxiga on tarkoitettu aikuisille potilaille sydämen oireisen kroonisen vajaatoiminnan hoitoon.
Krooninen munuaistauti
Forxiga on tarkoitettu aikuisille potilaille kroonisen munuaistaudin hoitoon.
Annostus ja antotapa
Annostus
Tyypin 2 diabetes
Suositeltu dapagliflotsiiniannos on 10 mg kerran vuorokaudessa.
Kun dapagliflotsiinia käytetään yhdessä insuliinin tai insuliinin eritystä lisäävän lääkeaineen, kuten sulfonyyliurean, kanssa, voidaan harkita insuliinin tai insuliinin eritystä lisäävän lääkeaineen annoksen pienentämistä hypoglykemian riskin pienentämiseksi (ks. kohdat Yhteisvaikutukset ja Haittavaikutukset).
Sydämen vajaatoiminta
Suositeltu dapagliflotsiiniannos on 10 mg kerran vuorokaudessa.
Krooninen munuaistauti
Suositeltu dapagliflotsiiniannos on 10 mg kerran vuorokaudessa.
Erityisryhmät
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen munuaisten toiminnan perusteella.
Vähäisen kokemuksen vuoksi dapagliflotsiinihoidon aloittamista ei suositella, jos potilaan GFR-arvo on < 25 ml/min.
Tyypin 2 diabetesta sairastavilla potilailla dapagliflotsiinin glukoosipitoisuutta pienentävä teho on alentunut, jos glomerulusten suodatusnopeus (GFR) on < 45 ml/min, ja todennäköisesti puuttuu potilailta, joilla on vaikea munuaisten vajaatoiminta. Jos GFR-arvo siis pienenee alle 45 ml/min, tyypin 2 diabetesta sairastavien potilaiden kohdalla on harkittava jotakin glukoosipitoisuutta pienentävää lisähoitoa, mikäli glukoositasapainon parantaminen edelleen on tarpeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa lievää tai kohtalaista maksan vajaatoimintaa sairastavilla potilailla. Potilaille, joilla on vaikea maksan vajaatoiminta, suositellaan 5 mg:n aloitusannosta. Jos annos on hyvin siedetty, voidaan annosta nostaa 10 mg:aan (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Iäkkäät (vähintään 65‑vuotiaat)
Annosta ei tarvitse muuttaa iän perusteella.
Pediatriset potilaat
Annosta ei tarvitse muuttaa, kun valmistetta käytetään tyypin 2 diabeteksen hoidossa vähintään 10‑vuotiailla lapsilla (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka). Tietoja ei ole saatavilla alle 10 vuoden ikäisistä lapsista
Dapagliflotsiinin turvallisuutta ja tehoa alle 18 vuoden ikäisillä lapsilla sydämen vajaatoiminnan tai kroonisen munuaistaudin hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Forxiga voidaan ottaa suun kautta kerran vuorokaudessa mihin aikaan vuorokaudesta tahansa joko ruoan kanssa tai tyhjään mahaan. Tabletit on nieltävä kokonaisina.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Yleistä
Dapagliflotsiinia ei pidä käyttää tyypin 1 diabetesta sairastavien potilaiden hoitoon (ks. ”Diabeettinen ketoasidoosi” kohdassa Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Vähäisen kokemuksen vuoksi dapagliflotsiinihoidon aloittamista ei suositella, jos potilaan GFR-arvo on < 25 ml/min.
Dapagliflotsiinin glukoosipitoisuutta pienentävä teho riippuu munuaisten toiminnasta. Teho on alentunut potilailla, joiden GFR-arvo on < 45 ml/min, ja todennäköisesti puuttuu potilailta, joilla on vaikea munuaisten vajaatoiminta (ks. kohdat Annostus ja antotapa, Farmakodynamiikka ja Farmakokinetiikka).
Yhdessä tutkimuksessa tyypin 2 diabetesta sairastavilla potilailla, joilla oli kohtalainen munuaisten vajaatoiminta (GFR <60 ml/min), kreatiniini-, fosfori- ja lisäkilpirauhashormoniarvojen suurenemista sekä hypotensiota ilmeni haittavaikutuksina enemmän dapagliflotsiiniryhmässä kuin lumelääkeryhmässä.
Maksan vajaatoiminta
Kliinisistä tutkimuksista on vähän kokemusta käytöstä potilaille, joilla on maksan vajaatoiminta. Dapagliflotsiinialtistus suurenee potilailla, joilla on vaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Käyttö nestehukan ja/tai hypotension riskiryhmään kuuluville potilaille
Vaikutusmekanisminsa vuoksi dapagliflotsiini lisää virtsaneritystä, mikä voi johtaa kliinisissä tutkimuksissa todettuun vähäiseen verenpaineen laskuun (ks. kohta Farmakodynamiikka). Lasku voi olla merkittävämpi potilailla, joilla on erittäin korkea veren glukoosipitoisuus.
Varovaisuutta on noudatettava potilailla, joille dapagliflotsiinin aiheuttama verenpaineen lasku saattaa olla riski, kuten verenpainelääkitystä käyttävillä potilailla, joilla on ollut hypotensiota, tai iäkkäillä potilailla.
Jos potilaalla on lisäksi muita sairaustiloja, jotka saattavat johtaa nestehukkaan (esim. maha-suolikanavan sairaus), suositellaan nestetasapainon huolellista seurantaa (esim. lääkärintarkastus, verenpainemittaukset, laboratoriotutkimukset mukaan lukien hematokriitti ja elektrolyyttitasapaino). Jos potilaalle kehittyy nestehukka, suositellaan dapagliflotsiinihoidon tilapäistä keskeyttämistä, kunnes nestehukka on korjaantunut (ks. kohta Haittavaikutukset).
Diabeettinen ketoasidoosi
Harvinaisia diabeettisen ketoasidoosin tapauksia, myös henkeä uhkaavia ja kuolemaan johtaneita, on ilmoitettu potilailla, joita on hoidettu natriumin- ja glukoosinkuljettajaproteiini 2:n (SGLT2) estäjillä, dapagliflotsiini mukaan lukien. Useissa tapauksissa sairaustila ilmeni epätyypillisenä sikäli, että veren glukoosipitoisuudet olivat vain jonkin verran koholla, alle 14 mmol/l (250 mg/dl).
Diabeettisen ketoasidoosin riski täytyy ottaa huomioon, jos potilaalla on epäspesifisiä oireita, kuten pahoinvointia, oksentelua, ruokahaluttomuutta, vatsakipua, epänormaalin voimakasta janoa, hengitysvaikeuksia, sekavuutta, epätavallista väsymystä tai uneliaisuutta. Jos tällaisia oireita ilmenee, potilas on tutkittava ketoasidoosin varalta välittömästi veren glukoosipitoisuudesta riippumatta.
Dapagliflotsiinihoito on lopetettava välittömästi, jos potilaalla epäillään olevan tai todetaan diabeettinen ketoasidoosi.
Hoito on keskeytettävä potilailta, jotka ovat sairaalahoidossa suuren kirurgisen toimenpiteen tai äkillisen vakavan sairauden takia. Näillä potilailla suositellaan ketonien seurantaa. Ketonipitoisuus kannattaa mitata verestä eikä virtsasta. Dapagliflotsiinihoito voidaan aloittaa uudelleen, kun ketonipitoisuus on normaali ja potilaan tila on jälleen vakaa.
Ennen dapagliflotsiinihoidon aloittamista on otettava huomioon potilaalla aiemmin ilmenneet ketoasidoosille mahdollisesti altistavat tekijät.
Dapagliflotsiinin käytön yhteydessä on havaittu pitkittynyttä ketoasidoosia ja pitkittynyttä glukosuriaa. Ketoasidoosi voi jatkua dapagliflotsiinihoidon keskeyttämisen jälkeen pidempään kuin dapagliflotsiinin puoliintumisaika plasmassa antaisi aihetta odottaa (ks. kohta Farmakokinetiikka). Dapagliflotsiinista riippumattomat tekijät, kuten insuliinin puute, voivat vaikuttaa ketoasidoosin pitkittymiseen.
Diabeettisen ketoasidoosin suurentuneen riskin ryhmään saattavat kuulua potilaat, joiden toiminnallisten beetasolujen määrä on vähentynyt (kuten tyypin 2 diabetesta sairastavat potilaat, joiden C-peptidiarvot ovat pienet, tai potilaat, joilla on aikuisiällä alkava autoimmuunityyppinen diabetes (LADA) tai joilla on aiemmin ollut haimatulehdus), potilaat, joilla on rajoittuneeseen ravinnonsaantiin tai vaikeaan nestehukkaan johtava tila, potilaat, joiden insuliiniannosta on pienennetty, ja potilaat, joiden insuliinin tarve on suurentunut akuutin sairauden, leikkauksen tai alkoholin väärinkäytön vuoksi. SGLT2:n estäjiä on käytettävä varoen näille potilaille.
Hoitoa SGLT2:n estäjällä ei suositella aloitettavan uudelleen, jos potilaalla on aiemmin ollut diabeettinen ketoasidoosi SGLT:n estäjän käytön aikana, ellei diabeettiselle ketoasidoosille löytynyt muuta selkeää selittävää syytä, joka on korjautunut.
Tyypin 1 diabetesta sairastavilla tutkittavilla tehdyissä dapagliflotsiinitutkimuksissa diabeettista ketoasidoosia ilmoitettiin esiintymistiheydellä yleinen. Dapagliflotsiinia ei pidä käyttää tyypin 1 diabetesta sairastavien potilaiden hoitoon.
Välilihan nekrotisoiva faskiitti (Fournier’n gangreeni)
Markkinoilletulon jälkeisistä välilihan nekrotisoivan faskiitin (tunnetaan myös nimellä Fournier’n gangreeni) tapauksista on ilmoitettu nais- ja miespotilailla, jotka käyttävät SGLT2:n estäjiä (ks. kohta Haittavaikutukset). Tämä on harvinainen mutta vakava ja mahdollisesti hengenvaarallinen tapahtuma, joka edellyttää kiireellistä leikkausta ja antibioottihoitoa.
Potilaita on kehotettava kääntymään lääkärin puoleen, jos heillä on kipua, aristusta, punoitusta tai turvotusta genitaali- tai perineaalialueella ja tähän liittyy kuumetta tai huonovointisuutta. Huomatkaa, että nekrotisoivaa faskiittia voi edeltää urogenitaali-infektio tai perineaaliabsessi. Jos Fournier’n gangreenia epäillään, Forxiga-valmisteen käyttö on keskeytettävä ja hoito (mukaan lukien antibioottihoito ja puhdistusleikkaus) on aloitettava viipymättä.
Virtsatieinfektiot
Glukoosin erittyminen virtsaan saattaa lisätä virtsatieinfektion riskiä. Siksi dapagliflotsiinihoidon väliaikaista keskeyttämistä on harkittava hoidettaessa pyelonefriittiä tai urosepsista.
Iäkkäät (vähintään 65‑vuotiaat)
Iäkkäillä potilailla voi olla suurempi nestehukan riski, ja heitä hoidetaan todennäköisemmin diureeteilla.
Munuaisten vajaatoiminta ja/tai munuaisten toimintaan mahdollisesti vaikuttavien verenpainelääkkeiden, kuten angiotensiinikonvertaasin estäjien (ACE-I) ja angiotensiinireseptorin salpaajien (ATR:n salpaaja), käyttö on todennäköisempää iäkkäillä potilailla. Iäkkäitä potilaita koskevat samat munuaisten toimintaan liittyvät suositukset kuin kaikkia muitakin potilaita (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset ja Farmakodynamiikka).
Sydämen vajaatoiminta
Kokemusta dapagliflotsiinin käytöstä NYHA-luokan IV potilaille on vain vähän.
Infiltratiivinen kardiomyopatia
Infiltratiivista kardiomyopatiaa sairastavia potilaita ei ole tutkittu.
Krooninen munuaistauti
Dapagliflotsiinin käytöstä kroonisen munuaistaudin hoidossa ei-diabeetikoilla joilla ei ole albuminuriaa, ei ole kokemusta. Potilaat, joilla on albuminuriaa, saattavat hyötyä enemmän dapagliflotsiinihoidosta.
Suurentunut hematokriittiarvo
Dapagliflotsiinihoidon yhteydessä on havaittu hematokriittiarvon suurenemista (ks. kohta Haittavaikutukset). Jos hematokriittiarvo on merkittävästi suurentunut, potilasta on seurattava ja hänet on tutkittava hematologisen perussairauden varalta.
Alaraajojen amputaatiot
SGLT2:n estäjillä tehdyissä pitkäaikaisissa kliinisissä tutkimuksissa tyypin 2 diabetesta sairastavilla tutkittavilla on havaittu alaraaja-amputaatioiden (pääasiassa varvasamputaatioiden) määrän lisääntymistä. Ei tiedetä, onko kyseessä luokkavaikutus. On tärkeää antaa diabetespotilaalle ohjeita rutiininomaisesta ennaltaehkäisevästä jalkojenhoidosta.
Virtsan laboratoriotutkimukset
Forxiga-valmisteen toimintamekanismin vuoksi potilaiden virtsan glukoosimääritys on positiivinen.
Laktoosi
Tabletit sisältävät laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Yhteisvaikutukset
Farmakodynaamiset yhteisvaikutukset
Diureetit
Dapagliflotsiini voi lisätä tiatsidi- ja loop-diureettien diureettisia vaikutuksia, ja nestehukan ja hypotension riski voi kohota (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Insuliini ja insuliinin eritystä lisäävät lääkeaineet
Insuliini ja insuliinin eritystä lisäävät lääkeaineet, kuten sulfonyyliureat, aiheuttavat hypoglykemiaa. Siksi insuliiniannosta tai insuliinin eritystä lisäävän lääkeaineen annosta voidaan joutua pienentämään hypoglykemiariskin pienentämiseksi yhdistelmähoidossa dapagliflotsiinin kanssa tyypin 2 diabetesta sairastavalla potilaalla (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Farmakokineettiset yhteisvaikutukset
Dapagliflotsiini metaboloituu pääasiassa UDP-glukuronosyltransferaasi 1A9 -entsyymin (UGT1A9) välittämän glukuronidikonjugaation kautta.
In vitro -tutkimuksissa dapagliflotsiini ei estänyt sytokromi P450 (CYP) 1A2-, CYP2A6-, CYP2B6‑, CYP2C8-, CYP2C9-, CYP2C19-, CYP2D6- ja CYP3A4-entsyymejä eikä indusoinut CYP1A2-, CYP2B6- tai CYP3A4-entsyymejä. Tämän vuoksi dapagliflotsiinin ei odoteta muuttavan näiden entsyymien kautta metaboloituvien, samanaikaisesti annettavien lääkevalmisteiden metabolista puhdistumaa.
Muiden lääkevalmisteiden vaikutukset dapagliflotsiiniin
Terveillä tutkittavilla pääasiassa kerta-annosta käyttämällä tehdyt yhteisvaikutustutkimukset viittaavat siihen, että metformiini, pioglitatsoni, sitagliptiini, glimepiridi, vogliboosi, hydroklorotiatsidi, bumetanidi, valsartaani tai simvastatiini eivät muuta dapagliflotsiinin farmakokinetiikkaa.
Kun rifampisiinia (useiden aktiivisten kuljettajaproteiinien ja lääkkeitä metaboloivien entsyymien induktori) annettiin samanaikaisesti dapagliflotsiinin kanssa, dapagliflotsiinin systeemisessä altistuksessa (AUC) havaittiin rifampisiinin annon jälkeen 22 %:n pieneneminen, mutta kliinisesti merkittävää vaikutusta glukoosin erittymiseen virtsaan 24 tunnin aikana ei todettu. Annoksen muuttamista ei suositella. Kliinisesti merkittävää vaikutusta ei odoteta muiden induktorien (esim. karbamatsepiini, fenytoiini, fenobarbitaali) samanaikaisen annon yhteydessä.
Dapagliflotsiinin ja mefenaamihapon (UGT1A9:n estäjä) samanaikaisen annon jälkeen havaittiin 55 %:n suureneminen dapagliflotsiinin systeemisessä altistuksessa, mutta ei kliinisesti merkityksellistä vaikutusta glukoosin erittymiseen virtsaan 24 tunnin aikana. Annoksen muuttamista ei suositella.
Dapagliflotsiinin vaikutukset muihin lääkevalmisteisiin
Dapagliflotsiini saattaa lisätä litiumin erittymistä munuaisten kautta, ja veren litiumpitoisuudet saattavat pienentyä. Seerumin litiumpitoisuutta on seurattava useammin dapagliflotsiinihoidon aloittamisen ja annosmuutosten jälkeen. Potilas tulee ohjata litiumia määränneen lääkärin vastaanotolle seerumin litiumpitoisuuden seurantaa varten.
Terveillä tutkittavilla pääasiassa kerta-annoksia käyttämällä tehdyissä yhteisvaikutustutkimuksissa dapagliflotsiinin ei todettu muuttavan metformiinin, pioglitatsonin, sitagliptiinin, glimepiridin, hydroklooritiatsidin, bumetanidin, valsartaanin, digoksiinin (P‑gp-substraatti) tai varfariinin (S‑varfariini, CYP2C9-substraatti) farmakokinetiikkaa tai varfariinin veren hyytymistä estäviä vaikutuksia INR-lukemilla mitattuna. Dapagliflotsiinin 20 mg:n kerta-annoksen ja simvastatiinin samanaikainen käyttö (CYP3A4-substraatti) suurensi simvastatiinin AUC-arvoa 19 % ja simvastatiinihapon AUC-arvoa 31 %. Suurentunutta simvastatiini- ja simvastatiinihappoaltistusta ei pidetä kliinisesti merkittävänä.
Häiriöt 1,5-anhydroglusitolin (1,5-AG) määrityksessä
Glukoositasapainon seurantaa 1,5-AG-määrityksellä ei suositella, koska SGLT2:n estäjiä käyttäviltä potilailta 1,5-AG-määrityksellä mitatut arvot eivät luotettavasti kuvaa glukoositasapainoa. Vaihtoehtoisten menetelmien käyttöä glukoositasapainon seurantaan suositellaan.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja dapagliflotsiinin käytöstä raskaana oleville naisille. Rotilla tehdyissä tutkimuksissa on osoitettu kehittyvään munuaiseen kohdistuvia toksisia vaikutuksia ajanjaksolla, joka vastaa ihmisen toista ja kolmatta raskauskolmannesta (ks. kohta Prekliiniset tiedot turvallisuudesta). Tämän vuoksi dapagliflotsiinin käyttöä ei pidä suositella toisen ja kolmannen raskauskolmanneksen aikana.
Dapagliflotsiinihoito on keskeytettävä raskauden havaitsemisen jälkeen.
Imetys
Ei tiedetä, erittyvätkö dapagliflotsiini ja/tai sen metaboliitit ihmisen rintamaitoon. Olemassa olevat farmakokineettiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet dapagliflotsiinin/metaboliittien erittyvän maitoon sekä farmakologisesti välittyviä vaikutuksia imeväisiin (ks. kohta Prekliiniset tiedot turvallisuudesta). Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Dapagliflotsiinia ei pidä käyttää rintaruokinnan aikana.
Hedelmällisyys
Dapagliflotsiinin vaikutusta ihmisten hedelmällisyyteen ei ole tutkittu. Millään tutkitulla dapagliflotsiiniannoksella ei ollut vaikutusta uros- ja naarasrottien hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Forxiga-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Potilaita on varoitettava hypoglykemian riskistä, kun dapagliflotsiinia käytetään samanaikaisesti sulfonyyliurean tai insuliinin kanssa.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Tyypin 2 diabetes
Tyypin 2 diabetesta koskevissa kliinisissä tutkimuksissa yli 15 000:ta potilasta on hoidettu dapagliflotsiinilla.
Ensisijainen turvallisuutta ja siedettävyyttä koskeva arviointi tehtiin ennalta määritellyssä 13 lyhyen (enintään 24 viikkoa kestäneen) lumekontrolloidun tutkimuksen yhdistetyssä analyysissa. Tutkimuksissa 2 360 tutkittavaa oli saanut dapagliflotsiinia annoksella 10 mg ja 2 295 tutkittavaa lumelääkettä.
Dapagliflotsiinilla tehdyssä sydän- ja verisuonituloksia koskevassa tutkimuksessa tyypin 2 diabetesta sairastavilla tutkittavilla (DECLARE-tutkimus, ks. kohta Farmakodynamiikka) 8 574 potilasta sai dapagliflotsiinia 10 mg:n annoksella ja 8 569 potilasta sai lumelääkettä. Altistuksen keston mediaani oli 48 kuukautta. Tutkimuksessa dapagliflotsiinialtistus oli yhteensä 30 623 potilasvuotta.
Kliinisissä tutkimuksissa yleisimmin ilmoitettuja haittavaikutuksia olivat genitaali-infektiot.
Sydämen vajaatoiminta
Dapagliflotsiinilla tehdyssä sydän- ja verisuonituloksia koskevassa tutkimuksessa sydämen vajaatoimintaa sairastavilla potilailla, joilla vajaatoimintaan liittyi pienentynyt ejektiofraktio (DAPA‑HF-tutkimus), 2 368 potilasta sai dapagliflotsiinia 10 mg:n annoksella ja 2 368 potilasta sai lumelääkettä. Altistuksen keston mediaani oli 18 kuukautta. Potilaspopulaatiossa oli mukana tyypin 2 diabetesta sairastavia potilaita, potilaita, joilla ei ollut diabetesta, ja potilaita, joiden eGFR oli ≥ 30 ml/min/1,73 m2. Dapagliflotsiinilla tehdyssä sydän- ja verisuonituloksia koskevassa tutkimuksessa sydämen vajaatoimintaa sairastavilla potilailla, joilla vasemman kammion ejektiofraktio oli > 40 % (DELIVER-tutkimus), 3 126 potilasta sai dapagliflotsiinia 10 mg:n annoksella ja 3 127 potilasta sai lumelääkettä. Altistuksen keston mediaani oli 27 kk. Potilaspopulaatiossa oli mukana tyypin 2 diabetesta sairastavia potilaita, potilaita, joilla ei ollut diabetesta, ja potilaita, joiden eGFR oli ≥ 25 ml/min/1,73 m2.
Dapagliflotsiinin kokonaisturvallisuusprofiili sydämen vajaatoimintaa sairastavilla potilailla vastasi dapagliflotsiinin tunnettua turvallisuusprofiilia.
Krooninen munuaistauti
Munuaisiin liittyviä hoitotuloksia koskevassa dapagliflotsiinitutkimuksessa, johon osallistuneilla potilailla oli krooninen munuaistauti (DAPA-CKD), 2 149 potilasta sai dapagliflotsiinia 10 mg:n annoksella ja 2 149 potilasta sai lumelääkettä. Altistuksen keston mediaani oli 27 kk. Potilaspopulaatiossa oli mukana tyypin 2 diabetesta sairastavia potilaita ja potilaita, joilla ei ollut diabetesta; potilaiden eGFR oli ≥ 25 – ≤ 75 ml/min/1,73 m2 ja potilailla oli albuminuriaa (virtsan albumiinin ja kreatiniinin suhde [U-AlbKrea] ≥ 200 – ≤ 5 000 mg/g). Hoitoa jatkettiin, jos eGFR pieneni alle tason 25 ml/min/1,73 m2.
Dapagliflotsiinin kokonaisturvallisuusprofiili kroonista munuaistautia sairastavilla potilailla vastasi dapagliflotsiinin tunnettua turvallisuusprofiilia.
Taulukoitu luettelo haittavaikutuksista
Seuraavat haittavaikutukset on havaittu lumekontrolloiduissa kliinisissä tutkimuksissa ja markkinoilletulon jälkeen tehdyssä seurannassa. Yksikään haittavaikutuksista ei ollut annosriippuvainen. Alla luetellut haittavaikutukset on luokiteltu esiintymistiheyden ja elinjärjestelmäluokituksen (SOC) mukaan. Esiintymistiheyden luokat on muodostettu seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1 000), hyvin harvinainen (< 1 / 10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1. Lumekontrolloiduissa kliinisissä tutkimuksissaa ja myyntiintulon jälkeen havaitut haittavaikutukset
| Elinjärjestelmäluokitus | Hyvin yleinen | Yleinen*
| Melko harvinainen**
| Harvinainen | Hyvin harvinainen |
| Infektiot | Vulvovaginiitti, balaniitti ja vastaavat genitaali-infektiot*,b,c Virtsatieinfektiot*,b,d | Sieni-infektio** | Välilihan nekrotisoiva faskiitti (Fournier’n gangreeni)b,i | ||
| Aineenvaihdunta ja ravitsemus | Hypoglykemia (kun käytettiin yhdessä sulfonyyliurean tai insuliinin kanssa)b | Nestehukkab,e Jano** | Diabeettinen ketoasidoosi (kun käytettiin tyypin 2 diabetesta sairastaville)b,i, k | ||
| Hermosto | Huimaus | ||||
| Ruoansulatuselimistö | Ummetus** Suun kuivuus** | ||||
| Iho ja ihonalainen kudos | Ihottumaj | Angioedeema | |||
| Luusto, lihakset ja sidekudos | Selkäkipu* | ||||
| Munuaiset ja virtsatiet | Dysuria Polyuria*,f | Nokturia**
| Tubulointerstitiaalinen nefriitti | ||
| Sukupuolielimet ja rinnat | Vulvovaginaalinen kutina** Sukupuolielinten kutina** | ||||
| Tutkimukset | Hematokriitin kohoamineng Pienentynyt kreatiniinin munuais-puhdistuma hoidon alussab Dyslipidemiah | Veren kreatiniinipitoisuuden kohoaminen hoidon alussa**,b Veren ureapitoisuuden kohoaminen** Painon lasku** |
aTaulukossa näkyvät tulokset viikolle 24 saakka (lyhyen aikavälin tulokset) glukoositasapainon palauttamisesta (glycaemic rescue) riippumatta.
bLisätietoja on vastaavassa alakohdassa.
cVulvovaginiitti, balaniitti ja vastaavat genitaali-infektiot sisältävät esim. seuraavat ennalta määritellyt suositellut termit: vulvovaginaalinen mykoottinen infektio, vaginaalinen infektio, balaniitti, genitaalialueen sieni-infektio, vulvovaginaalinen kandidiaasi, vulvovaginiitti, Candidan aiheuttama balaniitti, genitaalialueen kandidiaasi, sukupuolielinten infektio, miehen sukupuolielinten infektio, penistulehdus, vulviitti, bakteerin aiheuttama vaginiitti, vulvan absessi.
dVirtsatieinfektio sisältää seuraavat suositellut termit, jotka on lueteltu ilmoitetun esiintymistiheyden mukaisessa järjestyksessä: virtsatieinfektio, virtsarakkotulehdus, Escherichia colin aiheuttama virtsatieinfektio, virtsa- ja sukupuolielinten infektio, pyelonefriitti, trigoniitti, virtsaputkitulehdus, munuaisinfektio ja eturauhastulehdus.
eNestehukka sisältää esim. seuraavat ennalta määritellyt suositellut termit: dehydraatio, hypovolemia, hypotensio.
fPolyuria sisältää seuraavat suositellut termit: pollakisuria, polyuria, lisääntynyt virtsantuotanto.
gHematokriittiarvon keskimääräinen muutos lähtötilantesta oli 2,30 % 10 mg dapagliflotsiinia saaneiden ryhmässä ja ‑0,33 % lumelääkeryhmässä. Yli 55 %:n hematokriittiarvoja raportoitiin 1,3 %:lla tutkittavista 10 mg dapagliflotsiinia saaneiden ryhmässä ja 0,4 %:lla tutkittavista lumelääkeryhmässä.
hKeskimääräiset prosentuaaliset muutokset lähtötilanteesta 10 mg dapagliflotsiinia saaneilla verrattuna lumelääkettä saaneisiin olivat: kokonaiskolesteroli 2,5 % (lumelääke: 0,0 %); HDL‑kolesteroli 6,0 % (lumelääke: 2,7 %); LDL‑kolesteroli 2,9 % (lumelääke: ‑1,0 %); triglyseridit ‑2,7 % (lumelääke: ‑0,7 %).
iKs. kohta Varoitukset ja käyttöön liittyvät varotoimet
jHaittavaikutus havaittiin myyntiintulon jälkeisessä seurannassa. Ihottuma sisältää seuraavat suositellut termit, jotka on lueteltu kliinisisissä tutkimuksissa todetun esiintymistiheyden mukaisessa järjestyksessä: ihottuma, yleistynyt ihottuma, kutiava ihottuma, täpläinen ihottuma, täpläinen ja näppyläinen ihottuma, märkärakkulainen ihottuma, vesrirakkulainen ihottuma ja punoittava ihottuma. Aktiivi- ja lumekontrolloiduissa kliinisissä tutkimuksissa (dapagliflotsiini n = 5 936, verrokkiryhmät n = 3 403) ihottuman esiitymistiheys oli samanlainen dapagliflotsiini- (1,4 %) ja verrokkiryhmissä (1,4 %).
kRaportoitiin tyypin 2 diabetesta sairastavilla potilailla tehdyssä sydän- ja verisuonituloksia koskevassa tutkimuksessa (DECLARE). Esiintymistiheys perustuu vuosittaiseen määrään.
*Raportoitiin ≥ 2 %:lla tutkittavista ja lumelääkeryhmään verrattuna ≥ 1 % useammin ja vähintään 3 tutkittavalla enemmän niiden tutkittavien ryhmässä, jotka saivat 10 mg dapagliflotsiinia.
**Tutkija ilmoitti mahdollisesti tutkimushoitoon liittyvänä, todennäköisesti tutkimushoitoon liittyvänä tai tutkimushoitoon liittyvänä haittavaikutuksena, jota raportoitiin ≥ 0,2 %:lla tutkittavista ja lumelääkkeeseen verrattuna ≥ 0,1 % useammin ja vähintään 3 tutkittavalla enemmän niiden tutkittavien ryhmässä, jotka saivat 10 mg dapagliflotsiinia.
Valikoitujen haittavaikutusten kuvaus
Vulvovaginiitti, balaniitti ja niihin liittyvät genitaali-infektiot
13 tutkimuksen yhdistetyssä turvallisuusanalyysissä vulvovaginiittia, balaniittia ja niihin liittyviä genitaali-infektioita raportoitiin 5,5 %:lla dapagliflotsiinin 10 mg:n annosta saaneista tutkittavista ja 0,6 %:lla lumelääkettä saaneista tutkittavista. Useimmat infektiot olivat lieviä tai kohtalaisia, ja tutkittavat saivat hoitovasteen ensimmäiseen tavanomaiseen hoitoon. Infektiot johtivat harvoin dapagliflotsiinihoidon keskeyttämiseen. Nämä infektiot olivat yleisempiä naisilla (8,4 %:lla dapagliflotsiinia saaneista ja 1,2 %:lla lumelääkettä saaneista), ja aiemmin infektioita sairastaneilla infektion uusiutuminen oli todennäköisempää.
DECLARE-tutkimuksessa vakavia genitaali-infektiotapahtumia ilmeni pienellä määrällä potilaita, ja niitä ilmeni tasaisesti sekä dapagliflotsiini- että lumelääkeryhmässä: 2 potilaalla kummassakin.
DAPA‑HF-tutkimuksessa vakavia genitaali-infektiotapahtumia ei raportoitu yhdelläkään potilaalla dapagliflotsiiniryhmässä, mutta niitä raportoitiin yhdellä potilaalla lumelääkeryhmässä. Genitaali-infektioiden vuoksi hoidon keskeyttämiseen johtaneita haittatapahtumia ilmeni 7 potilaalla (0,3 %) dapagliflotsiiniryhmässä, mutta ei yhdelläkään potilaalla lumelääkeryhmässä. DELIVER-tutkimuksessa yhdellä potilaalla (< 0,1 %) kummassakin hoitoryhmässä raportoitiin vakavia genitaali-infektiotapahtumia. Genitaali-infektioiden vuoksi hoidon keskeyttämiseen johtaneita haittatapahtumia ilmeni 3 potilaalla (0,1 %) dapagliflotsiiniryhmässä, mutta ei yhdelläkään potilaalla lumelääkeryhmässä.
DAPA-CKD-tutkimuksessa todettiin vakavia genitaali-infektiotapahtumia 3 potilaalla (0,1 %) dapagliflotsiiniryhmässä eikä yhdelläkään potilaalla lumelääkeryhmässä. Genitaali-infektioiden vuoksi hoidon keskeyttämiseen johtaneita haittatapahtumia ilmeni 3 potilaalla (0,1 %) dapagliflotsiiniryhmässä, mutta ei yhdelläkään potilaalla lumelääkeryhmässä. Vakavia genitaali-infektiotapahtumia tai genitaali-infektioiden vuoksi hoidon keskeyttämiseen johtaneita haittatapahtumia ei raportoitu yhdelläkään potilaalla, jolla ei ollut diabetesta.
Fimoosia / hankinnaista fimoosia on raportoitu esiintyvän samanaikaisesti sukupuolielinten infektioiden kanssa, ja joissakin tapauksissa tarvittiin ympärileikkaus.
Välilihan nekrotisoiva faskiitti (Fournier’n gangreeni)
Fournier’n gangreenin tapauksia on ilmoitettu markkinoille-tulon jälkeen potilailla, jotka käyttävät SGLT2:n estäjiä, dapagliflotsiini mukaan lukien (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
DECLARE-tutkimuksessa, johon osallistui 17 160 potilasta, joilla oli tyypin 2 diabetes, ja jossa altistuksen keston mediaani oli 48 kuukautta, raportoitiin yhteensä kuusi Fournier’n gangreenin tapausta, yksi dapagliflotsiiniryhmässä ja viisi lumelääkeryhmässä.
Hypoglykemia
Hypoglykemian esiintyvyys riippui diabetespotilailla tehdyissä tutkimuksissa käytetyn taustahoidon tyypistä.
Tutkimuksissa, joissa dapagliflotsiinia arvioitiin monoterapiana, metformiinin lisälääkkeenä tai sitagliptiinin lisälääkkeenä (metformiinin kanssa tai ilman sitä), lievien hypoglykemiatapahtumien esiintymistiheys oli samanlainen (< 5 %) hoitoryhmien välillä, lumeryhmä mukaan lukien, kun hoitoa jatkettiin enintään 102 viikon ajan. Kaikissa tutkimuksissa merkittävät hypoglykemiatapahtumat olivat melko harvinaisia ja niitä oli saman verran dapagliflotsiini- ja lumeryhmässä. Hypoglykemiatapahtumien määrä oli korkeampi tutkimuksissa, joissa hoitoon oli yhdistetty sulfonyyliurea tai insuliini (ks. kohta Yhteisvaikutukset).
Tutkimuksessa, jossa dapagliflotsiinia annettiin glimepiridin lisänä, lieviä hypoglykemiatapahtumia raportoitiin 24 ja 48 viikon kohdalla esiintyneen useammin niillä potilailla, joita hoidettiin dapagliflotsiinin 10 mg:n annoksella yhdistettynä glimepiridiin (6,0 %:lla 24 viikon kohdalla ja 7,9 %:lla 48 viikon kohdalla), kuin niillä potilailla, joita hoidettiin lumelääkkeellä yhdistettynä glimepiridiin (2,1 %:lla 24 viikon kohdalla ja 2,1 %:lla 48 viikon kohdalla).
Yhdistelmähoidossa merkittäviä hypoglykemiatapahtumia raportoitiin viikon 24 kohdalla 0,5 %:lla ja viikon 104 kohdalla 1,0 %:lla potilaista, jotka saivat 10 mg dapagliflotsiinia yhdistettynä insuliiniin. Vastaava luku lumelääkkeen ja insuliinin yhdistelmää saaneilla oli 0,5 % viikoilla 24 ja 104. Lieviä hypoglykemiatapahtumia raportoitiin viikon 24 kohdalla 40,3 %:lla ja viikon 104 kohdalla 53,1 %:lla potilaista, jotka saivat 10 mg dapagliflotsiinia yhdistettynä insuliiniin. Vastaavat luvut lumelääkkeen ja insuliinin yhdistelmää saaneilla olivat 34,0 % viikolla 24 ja 41,6 % viikolla 104.
Tutkimuksessa, jossa dapagliflotsiinia annettiin metformiinin ja sulfonyyliurean lisänä, merkittäviä hypoglykemiatapauksia ei raportoitu 24 tutkimusviikon aikana. Lieviä hypoglykemiatapauksia raportoitiin 12,8 %:lla potilaista, jotka saivat 10 mg dapagliflotsiinia yhdistettynä metformiiniin ja sulfonyyliureaan, ja 3,7 %:lla potilaista, jotka saivat lumelääkettä yhdistettynä metformiiniin ja sulfonyyliureaan.
DECLARE-tutkimuksessa merkittävien hypoglykemiatapahtumien riskin ei havaittu suurentuneen dapagliflotsiinihoitoa saaneilla lumelääkettä saaneisiin verrattuna. Merkittäviä hypoglykemiatapahtumia ilmoitettiin 58:lla dapagliflotsiinia saaneella potilaalla (0,7 %) ja 83:lla lumelääkettä saaneella potilaalla (1,0 %).
DAPA‑HF-tutkimuksessa merkittäviä hypoglykemiatapahtumia raportoitiin 4 potilaalla (0,2 %) sekä dapagliflotsiini- että lumehoitoryhmässä. DELIVER-tutkimuksessa merkittäviä hypoglykemiatapahtumia raportoitiin 6 potilaalla (0,2 %) dapagliflotsiiniryhmässä ja 7 potilaalla (0,2 %) lumehoitoryhmässä. Merkittäviä hypoglykemiatapahtumia havaittiin vain tyypin 2 diabetesta sairastavilla potilailla.
DAPA-CKD-tutkimuksessa merkittäviä hypoglykemiatapahtumia raportoitiin 14 potilaalla (0,7 %) dapagliflotsiiniryhmässä ja 28 potilaalla (1,3 %) lumelääkeryhmässä, ja niitä havaittiin vain tyypin 2 diabetesta sairastavilla potilailla.
Nestehukka
13 tutkimuksen yhdistetyssä turvallisuusanalyysissä nestehukkaan viittaavia haittavaikutuksia (mm. dehydraatio, hypovolemia tai hypotensio) raportoitiin 1,1 %:lla tutkittavista, joita hoidettiin dapagliflotsiinin 10 mg:n annoksella ja 0,7 %:lla tutkittavista, jotka saivat lumelääkettä. Vakavia haittavaikutuksia esiintyi < 0,2 %:lla tutkittavista ja niitä todettiin saman verran dapagliflotsiinin 10 mg:n annosta ja lumelääkettä saaneilla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
DECLARE-tutkimuksessa potilaita, joilla todettiin nestehukkaan viittaavia tapahtumia, oli saman verran eri hoitoryhmissä: 213 (2,5 %) dapagliflotsiiniryhmässä ja 207 (2,4 %) lumelääkeryhmässä. Vakavia haittatapahtumia raportoitiin 81:llä dapagliflotsiiniryhmän potilaista (0,9 %:lla) ja 70:llä lumelääkeryhmän potilaista (0,8 %:lla). Tapahtumia oli yleisesti saman verran eri hoitoryhmissä ikään, diureettien käyttöön, verenpaineeseen ja angiotensiinikonvertaasin estäjien (ACE-I) tai angiotensiinireseptorin salpaajien (ATR:n salpaajien) käyttöön perustuvissa alaryhmissä. Potilailla, joiden eGFR oli lähtötilanteessa < 60 ml/min/1,73 m2, todettiin 19 nestehukkaan viittaavaa vakavaa haittatapahtumaa dapagliflotsiiniryhmässä ja 13 tapahtumaa lumelääkeryhmässä.
DAPA‑HF-tutkimuksessa niiden potilaiden määrä, joilla ilmeni nestehukkaan viittaavia tapahtumia, oli 170 (7,2 %) dapagliflotsiiniryhmässä ja 153 (6,5 %) lumelääkeryhmässä. Potilaita, joilla ilmeni vakavia tapahtumia, joiden oireet viittasivat nestehukkaan, oli dapagliflotsiiniryhmässä vähemmän (23 [1,0 %]) kuin lumelääkeryhmässä (38 [1,6 %]). Tulokset olivat samanlaisia riippumatta tutkittavien diabetesstatuksesta lähtötilanteessa ja lähtötilanteen eGFR-arvoista. DELIVER-tutkimuksessa niiden potilaiden määrä, joilla ilmeni vakavia tapahtumia, joiden oireet viittasivat nestehukkaan, oli 35 (1,1 %) dapagliflotsiiniryhmässä ja 31 (1,0 %) lumelääkeryhmässä.
DAPA-CKD-tutkimuksessa niiden potilaiden määrä, joilla ilmeni nestehukkaan viittaavia tapahtumia, oli 120 (5,6 %) dapagliflotsiiniryhmässä ja 84 (3,9 %) lumelääkeryhmässä. Potilaita, joilla ilmeni vakavia tapahtumia, joiden oireet viittasivat nestehukkaan, oli dapagliflotsiiniryhmässä 16 (0,7 %) ja lumelääkeryhmässä 15 (0,7 %).
Diabeettinen ketoasidoosi tyypin 2 diabetesta sairastavilla tutkittavilla
DECLARE-tutkimuksessa, jossa altistuksen keston mediaani oli 48 kuukautta, diabeettista ketoasidoosia raportoitiin 27 potilaalla 10 mg dapagliflotsiinia saaneiden ryhmässä ja 12 potilaalla lumelääkeryhmässä. Tapahtumat jakautuivat tasaisesti koko tutkimusjaksolle. 27:stä dapagliflotsiiniryhmän potilaasta, joilla esiintyi diabeettista ketoasidoosia, 22:ta hoidettiin tapahtumahetkellä samanaikaisesti insuliinilla. Diabeettisen ketoasidoosin taustalla olevat syyt olivat tyypin 2 diabetesta sairastavien potilaiden populaatiossa odotusten mukaisia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.).
DAPA‑HF-tutkimuksessa diabeettista ketoasidoosia raportoitiin kolmella tyypin 2 diabetesta sairastavalla potilaalla dapagliflotsiiniryhmässä, mutta ei yhdelläkään lumelääkeryhmässä. DELIVER-tutkimuksessa diabeettista ketoasidoosia raportoitiin kahdella tyypin 2 diabetesta sairastavalla potilaalla dapagliflotsiiniryhmässä, mutta ei yhdelläkään lumelääkeryhmässä.
DAPA-CKD-tutkimuksessa diabeettista ketoasidoosia ei raportoitu yhdelläkään potilaalla dapagliflotsiiniryhmässä, mutta kahdella tyypin 2 diabetesta sairastavalla potilaalla lumelääkeryhmässä.
Virtsatieinfektiot
13 tutkimuksen yhdistetyssä turvallisuusanalyysissä virtsatieinfektioita raportoitiin useammin dapagliflotsiinin 10 mg:n annoksella hoidetuilla (4,7 %) kuin lumelääkettä saaneilla (3,5 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Useimmat infektiot olivat luonteeltaan lieviä tai kohtalaisia, ja tutkittavat saivat hoitovasteen ensimmäiseen tavanomaiseen hoitoon. Infektiot johtivat harvoin dapagliflotsiinihoidon keskeyttämiseen. Nämä infektiot olivat yleisempiä naisilla, ja aiemmin infektioita sairastaneilla infektion uusiutuminen oli todennäköisempää.
DECLARE-tutkimuksessa vakavia virtsatieinfektioita ilmoitettiin harvemmin 10 mg dapagliflotsiinia saaneiden ryhmässä (79, 0,9 %) verrattuna lumelääkeryhmään (109, 1,3 %).
DAPA‑HF-tutkimuksessa ilmeni vakavina haittatapahtumina virtsatieinfektioita 14 potilaalla (0,6 %) dapagliflotsiiniryhmässä ja 17 potilaalla (0,7 %) lumelääkeryhmässä. Sekä dapagliflotsiini- että lumelääkeryhmässä 5 potilaalla (0,2 %) ilmeni virtsatieinfektioiden vuoksi hoidon keskeyttämiseen johtaneita haittatapahtumia. DELIVER-tutkimuksessa ilmeni vakavina haittatapahtumina virtsatieinfektioita 41 potilaalla (1,3 %) dapagliflotsiiniryhmässä ja 37 potilaalla (1,2 %) lumelääkeryhmässä. Dapagliflotsiiniryhmässä 13 potilaalla (0,4 %) ja lumelääkeryhmässä 9 potilaalla (0,3 %) ilmeni virtsatieinfektioiden vuoksi hoidon keskeyttämiseen johtaneita haittatapahtumia.
DAPA-CKD-tutkimuksessa ilmeni vakavina haittatapahtumina virtsatieinfektioita 29 potilaalla (1,3 %) dapagliflotsiiniryhmässä ja 18 potilaalla (0,8 %) lumelääkeryhmässä. Dapagliflotsiiniryhmässä 8 potilaalla (0,4 %) ja lumelääkeryhmässä 3 potilaalla (0,1 %) ilmeni virtsatieinfektioiden vuoksi hoidon keskeyttämiseen johtaneita haittatapahtumia. Niiden potilaiden määrä, joilla ei ollut diabetesta ja jotka ilmoittivat vakavia virtsatieinfektiotapahtumia tai virtsatieinfektioiden vuoksi hoidon keskeyttämiseen johtaneita haittatapahtumia, oli samankaltainen molemmissa hoitoryhmissä (6 vakavaa haittatapahtumaa [0,9 %] dapagliflotsiiniryhmässä ja 4 vakavaa haittatapahtumaa [0,6 %] lumelääkeryhmässä;1 hoidon keskeyttämiseen johtanut haittatapahtuma [0,1 %] dapagliflotsiiniryhmässä eikä yhtään tällaista tapahtumaa lumelääkeryhmässä).
Suurentuneet kreatiniiniarvot
Suurentuneisiin kreatiniiniarvoihin liittyvät haittavaikutukset koottiin ryhmäksi (esim. pienentynyt kreatiniinin munuaispuhdistuma, munuaisten vajaatoiminta, kohonneet kreatiniiniarvot veressä ja pienentynyt glomerulusten suodatusnopeus). 13 tutkimuksen yhdistetyssä turvallisuusanalyysissä tähän ryhmään kuuluvia haittavaikutuksia ilmoitettiin 3,2 %:lla potilaista, jotka saivat 10 mg dapagliflotsiinia, ja 1,8 %:lla potilaista, jotka saivat lumelääkettä. Potilailla, joiden munuaisten toiminta oli normaali tai joilla oli lievä munuaisten vajaatoiminta (lähtötilanteen eGFR ≥ 60 ml/min/1,73 m2), tähän ryhmään kuuluvia haittavaikutuksia ilmoitettiin 1,3 %:lla potilaista, jotka saivat 10 mg dapagliflotsiinia, ja 0,8 %:lla potilaista, jotka saivat lumelääkettä. Nämä haittavaikutukset olivat yleisempiä potilailla, joiden lähtötilanteen eGFR on ≥ 30 ja < 60 ml/min/1,73 m2 (10 mg dapagliflotsiinia saaneiden ryhmässä 18,5 % ja lumelääkeryhmässä 9,3 %).
Munuaisiin liittyneitä haittavaikutuksia saaneiden potilaiden tarkempi arviointi osoitti, että useimmilla seerumin kreatiniiniarvot olivat muuttuneet ≤ 44 mikromoolia/l (≤ 0,5 mg/dl) lähtötilanteesta. Kreatiniiniarvojen suureneminen oli yleensä tilapäistä jatkuvan hoidon aikana tai palautuvaa hoidon lopettamisen jälkeen.
DECLARE-tutkimuksessa, johon osallistui myös iäkkäitä potilaita ja potilaita, joilla oli munuaisten vajaatoiminta (eGFR alle 60 ml/min/1,73 m2), eGFR pieneni ajan myötä molemmissa hoitoryhmissä. 1 vuoden kohdalla keskimääräinen eGFR oli dapagliflotsiiniryhmässä hieman pienempi ja 4 vuoden kohdalla hieman suurempi verrattuna lumelääkeryhmään.
DAPA‑HF- ja DELIVER-tutkimuksissa eGFR pieneni ajan myötä sekä dapagliflotsiiniryhmässä että lumelääkeryhmässä. DAPA‑HF-tutkimuksessa eGFR pieneni aluksi keskimäärin 4,3 ml/min/1,73 m2 dapagliflotsiiniryhmässä ja 1,1 ml/min/1,73 m2 lumelääkeryhmässä. 20 kuukauden kohdalla eGFR oli muuttunut lähtötilanteesta saman verran hoitoryhmissä: ‑5,3 ml/min/1,73 m2 dapagliflotsiiniryhmässä ja ‑4,5 ml/min/1,73 m2 lumelääkeryhmässä. DELIVER-tutkimuksessa eGFR-arvo oli pienentynyt yhden kuukauden kohdalla keskimäärin 3,7 ml/min/1,73 m2 dapagliflotsiiniryhmässä ja 0,4 ml/min/1,73 m2 lumelääkeryhmässä. 24 kuukauden kohdalla eGFR oli muuttunut lähtötilanteesta saman verran hoitoryhmissä: ‑4,2 ml/min/1,73 m2 dapagliflotsiiniryhmässä ja ‑3,2 ml/min/1,73 m2 lumelääkeryhmässä.
DAPA-CKD-tutkimuksessa eGFR pieneni ajan myötä sekä dapagliflotsiiniryhmässä että lumelääkeryhmässä. Keskimääräinen eGFR pieneni aluksi (päivään 14 mennessä) niin, että muutos oli ‑4,0 ml/min/1,73 m2 dapagliflotsiiniryhmässä ja ‑0,8 ml/min/1,73 m2 lumelääkeryhmässä. 28 kuukauden kohdalla eGFR:n muutos lähtötilanteesta oli ‑7,4 ml/min/1,73 m2 dapagliflotsiiniryhmässä ja ‑8,6 ml/min/1,73 m2 lumelääkeryhmässä.
Pediatriset potilaat
Kliinisessä tutkimuksessa vähintään 10‑vuotiailla, tyypin 2 diabetesta sairastavilla lapsilla havaittu dapagliflotsiinin turvallisuusprofiili (ks. kohta Farmakodynamiikka) oli samankaltainen kuin aikuisilla tehdyissä tutkimuksissa on todettu.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Dapagliflotsiinin ei osoittettu aiheuttavan minkäänlaista toksisuutta terveillä tutkittavilla suun kautta otettuina, enintään 500 mg:n kerta-annoksina (50‑kertainen ihmiselle suositeltuun enimmäisannokseen nähden). Näillä tutkittavilla havaittiin glukoosia virtsassa annoksen suuruuteen suhteessa olevan ajan (vähintään 5 päivän ajan 500 mg:n annoksella) ilman nestehukkaa, hypotensiota tai elektrolyyttihäiriötä tai kliinisesti merkittävää vaikutusta QTc-aikaan. Hypoglykemian esiintyvyys oli samankaltaista kuin lumeryhmässä. Kliinisissä tutkimuksissa, joissa terveille tutkittaville ja tyypin 2 diabetesta sairastaville tutkittaville annettiin kerran päivässä 2 viikon ajan enintään 100 mg:n annoksia (10‑kertainen ihmisen suositeltuun enimmäisannokseen nähden), hypoglykemiaa ilmaantui hieman useammin kuin lumeryhmässä, eikä ilmaantuvuus ollut annoksesta riippuvainen. Haittavaikutusten, mukaan lukien nestehukan tai hypotension, määrät olivat samankaltaiset kuin lumeryhmässä. Laboratorioparametreissa, mukaan lukien seerumin elektrolyytit ja munuaistoiminnan biomarkkerit, ei ollut kliinisesti merkittäviä annoksesta riippuvia muutoksia.
Yliannostustapauksessa on aloitettava asianmukainen tukihoito potilaan kliinisen tilan mukaan. Dapagliflotsiinin poistamista hemodialyysilla ei ole tutkittu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Diabeteslääkkeet, natrium-glukoosi-kuljettajaproteiini 2:n (SGLT2) estäjät, ATC-koodi: A10BK01
Vaikutusmekanismi
Dapagliflotsiini on erittäin voimakas (Ki: 0,55 nM), selektiivinen ja reversiibeli SGLT2:n estäjä.
Dapagliflotsiini estää SGLT2:ta, mikä vähentää glukoosin takaisinimeytymistä glomerulussuodoksesta proksimaaliseen munuaistiehyeen ja samanaikaisesti vähentää natriumin takaisinimeytymistä, mikä johtaa glukoosin erittymiseen virtsaan ja osmoottiseen diureesiin. Siten dapagliflotsiini lisää natriumin kulkeutumista distaaliseen tiehyeen, mikä lisää tubuloglomerulaarista palautetta ja pienentää intraglomerulaarista painetta. Tämä yhdessä osmoottisen diureesin kanssa johtaa tilavuusylikuormituksen vähenemiseen, alentaa verenpainetta ja pienentää sydämen esikuormitusta ja jälkikuormitusta, millä saattaa olla hyödyllisiä vaikutuksia sydämen remodellaatioon ja diastoliseen toimintaan ja mikä saattaa ylläpitää munuaisten toimintaa. DAPA‑HF-, DELIVER ja DAPA‑CKD-tutkimuksissa osoitettiin, että dapagliflotsiinin sydämeen ja munuaisiin liittyvät hyödyt eivät perustu pelkästään verensokeria alentavaan vaikutukseen eivätkä rajoitu diabetespotilaisiin. Muita vaikutuksia ovat hematokriitin suureneminen ja painon lasku.
Dapagliflotsiini parantaa sekä plasman paastoglukoosiarvoa että aterian jälkeistä glukoosiarvoa vähentämällä glukoosin takaisinimeytymistä munuaisissa, mikä johtaa glukoosin erittymiseen virtsaan. Tämä glukoosin erittyminen (glukosuurinen vaikutus) havaitaan ensimmäisen annoksen jälkeen, jatkuu 24 tunnin annosvälien aikana ja kestää koko hoitoajan. Munuaisten tällä mekanismilla poistaman glukoosin määrä riippuu veren glukoosipitoisuudesta ja glomerulusten suodatusnopeudesta. Siksi tutkittavilla, joilla veren glukoosipitoisuus on normaali, dapagliflotsiinin taipumus aiheuttaa hypoglykemiaa on pieni. Dapagliflotsiini ei heikennä normaalia hypoglykemian aiheuttamaa endogeenista glukoosin tuotantoa. Dapagliflotsiini toimii insuliinin erityksestä ja toiminnasta riippumatta. Beetasolujen toiminnan on havaittu parantuneen homeostaasimallimäärityksessä (HOMA beta cell) dapagliflotsiinilla tehdyissä kliinisissä tutkimuksissa.
SGLT2 ilmentyy selektiivisesti munuaisissa. Dapagliflotsiini ei estä muita glukoosin kuljettajaproteiineja, jotka ovat tärkeitä glukoosin kuljettamisessa perifeerisiin kudoksiin. Se on > 1400 kertaa selektiivisempi SGLT2-kuljettajaproteiinille kuin SGLT1-kuljettajaproteiinille, joka on suolessa tärkein glukoosin imeytymisestä vastaava kuljettajaproteiini.
Farmakodynaamiset vaikutukset
Virtsaan erittyvän glukoosin määrän havaittiin lisääntyneen terveillä tutkittavilla ja tyypin 2 diabetesta sairastavilla tutkittavilla dapagliflotsiinin antamisen jälkeen. Tyypin 2 diabetesta sairastavilla tutkittavilla, jotka saivat dapagliflotsiinia 10 mg:n vuorokausiannoksella 12 viikon ajan, virtsaan erittyi noin 70 grammaa glukoosia vuorokaudessa (mikä vastaa 280 kcal:ia/vrk). Jatkuvaa glukoosin erittymistä havaittiin tyypin 2 diabetesta sairastavilla tutkittavilla, joille annettiin 10 mg dapagliflotsiinia vuorokaudessa 2 vuoden ajan.
Tämä dapagliflotsiiniin aikaansaama glukoosin erittyminen virtsaan johtaa lisäksi osmoottiseen diureesiin ja virtsamäärän lisääntymiseen tyypin 2 diabetesta sairastavilla henkilöillä. Virtsamäärän lisääntyminen tyypin 2 diabetesta sairastavilla tutkittavilla, joita hoidettiin 10 mg:n dapagliflotsiiniannoksella, oli pysyvää 12 viikon ajan, ja virtsamäärä lisääntyi n. 375 ml vuorokaudessa. Virtsamäärän lisääntyminen lisäsi vähän ja ohimenevästi natriumin erittymistä virtsaan, mihin ei liittynyt seerumin natriumpitoisuuksien muutoksia.
Myös uraatin erittyminen virtsaan lisääntyi hetkellisesti (3‑7 päivän ajaksi), mitä seurasi pysyvä lasku seerumin uraattipitoisuudessa. Viikon 24 kohdalla seerumin uraattipitoisuuden väheneminen vaihteli välillä ‑48,3 ja -18,3 mikromoolia/l (‑0,87 ja ‑0,33 mg/dl).
Kliininen teho ja turvallisuus
Tyypin 2 diabetes
Glukoositasapainon parantaminen ja sydämeen, verisuoniin ja munuaisiin liittyvän sairastuvuuden ja kuolleisuuden vähentäminen ovat olennaisia osia tyypin 2 diabeteksen hoitoa.
Forxiga-valmisteen turvallisuutta ja glykeemistä tehoa tutkittiin neljässätoista kaksoissokkoutetussa, satunnaistetussa, kontrolloidussa kliinisessä tutkimuksessa, joihin osallistui 7 056 tyypin 2 diabetesta sairastavaa aikuista tutkittavaa. Näissä tutkimuksissa 4 737 henkilöä hoidettiin dapagliflotsiinilla. Hoitoaika oli 24 viikkoa kahdessatoista tutkimuksessa, joista kahdeksassa oli 24–80 viikon pitkäaikainen jatkovaihe (tutkimuksen kokonaiskestoaika enintään 104 viikkoa), yhdessä tutkimuksessa hoitoaika oli 28 viikkoa, ja yhden tutkimuksen kesto oli 52 viikkoa pidennettynä pitkäaikaisilla 52 ja 104 viikon pituisilla jatkovaiheilla (tutkimuksen kokonaiskestoaika 208 viikkoa). Lähtötilanteessa keskimääräinen aika, jonka tutkittavat olivat sairastaneet diabetesta, oli 1,4–16,9 vuotta. Lievää munuaisten vajaatoimintaa havaittiin 50 %:lla ja kohtalaista munuaisten vajaatoimintaa 11 %:lla tutkittavista. 51 % tutkittavista oli miehiä, 84 % oli valkoisia, 8 % oli aasialaisia, 4 % oli mustia ja 4 % muista etnisistä ryhmistä. Tutkittavista 81 %:lla painoindeksi (BMI) oli ≥ 27. Lisäksi tehtiin kaksi 12 viikon pituista lumelääkekontrolloitua tutkimusta potilailla, joiden tyypin 2 diabeteksen hoitotasapaino oli riittämätön ja joilla oli verenpainetauti.
Sydän- ja verisuonituloksia koskevassa tutkimuksessa (DECLARE) arvioitiin dapagliflotsiinin vaikutusta sydän-, verisuoni- ja munuaistapahtumiin vertaamalla 10 mg:n dapagliflotsiiniannosta lumelääkkeeseen tyypin 2 diabetesta sairastavalla 17 160 potilaalla, joilla joko oli tai ei ollut todettua sydän- ja verisuonisairautta.
Glukoositasapaino
Monoterapia
Kaksoissokkoutetussa, lumekontrolloidussa 24 viikkoa kestäneessä tutkimuksessa (jossa oli jatkovaihe) arvioitiin Forxiga-monoterapian turvallisuutta ja tehoa tutkittavilla, joiden tyypin 2 diabetes ei ollut hoitotasapainossa. Kerran vuorokaudessa annetun dapagliflotsiinihoidon tuloksena HbA1c-arvot laskivat tilastollisesti merkittävästi (p < 0,0001) lumelääkkeeseen verrattuna (taulukko 2).
Tutkimuksen jatkovaiheessa HbA1c-arvon laskut säilyivät viikkoon102 asti: HbA1c-arvon korjatut keskimääräiset muutokset lähtötilanteesta olivat ‑0,61 % (dapagliflotsiini 10 mg) ja ‑0,17 % (lumelääke).
Taulukko 2. Viikon 24 tulokset (LOCFa) lumekontrolloidusta dapagliflotsiini-monoterapiatutkimuksesta
| Monoterapiana | ||
Dapagliflotsiini 10 mg | Lumelääke | |
| Nb | 70 | 75 |
| HbA1c (%) | ||
| Lähtötilanne (keskiarvo) | 8,01 | 7,79 |
| Muutos lähtötilanteestac | ‑0,89 | ‑0,23 |
| Ero lumelääkkeeseen verrattunac | ‑0,66* | |
| (95 %:n luottamusväli) | (‑0,96, ‑0,36) | |
Tutkittavat (%), jotka saavuttivat seuraavat arvot: HbA1c < 7 % | ||
| Korjattuna suhteessa lähtötilanteeseen | 50,8§ | 31,6 |
| Paino (kg) | ||
| Lähtötilanne (keskiarvo) | 94,13 | 88,77 |
| Muutos lähtötilanteestac | ‑3,16 | ‑2,19 |
| Ero lumelääkkeeseen verrattunac | ‑0,97 | |
| (95 %:n luottamusväli) | (‑2,20, 0,25) | |
| aLOCF: Viimeisimmästä havainnosta laskettu arvio (last observation carried forward) (ennen glukoositason korjaamista [glycaemic rescue] sitä edellyttäneillä tutkittavilla) bKaikki satunnaistetut tutkittavat, jotka saivat vähintään yhden annoksen kaksoissokkoutettua tutkimuslääkevalmistetta lyhytkestoisen kaksoissokkoutetun tutkimusjakson aikana cPienimmän neliösumman keskiarvo suhteutettuna lähtötilanteen arvoon *p‑arvo < 0,0001 verrattuna lumelääkkeeseen §Tilastollista merkitsevyyttä ei ole arvioitu toissijaisten päätetapahtumien peräkkäisen testimenetelmän takia | ||
Yhdistelmähoito
Viisikymmentäkaksi viikkoa kestäneessä aktiivikontrolloidussa tutkimuksessa (mukaan lukien 52 ja 104 viikon pituiset jatkovaiheet), jossa pyrittiin osoittamaan dapagliflotsiini vähintään samanveroiseksi kuin vertailulääke (non-inferiority), verrattiin Forxiga-valmistetta metformiinin lisähoitona sulfonyyliureaan (glipitsidi) tutkittavilla, joiden glukoositasapaino oli riittämätön (HbA1c > 6,5 % ja ≤ 10 %). Tulokset osoittivat HbA1c-arvon keskimääräisen pienenemisen lähtötilanteesta viikolle 52 olevan samankaltainen verrattuna glipitsidiin, mikä osoittaa samanveroisuutta (non-inferiority) (taulukko 3). HbA1c-arvon korjattu keskimääräinen muutos lähtötilanteesta viikolla 104 oli -0,32 % dapagliflotsiinia saaneiden ryhmässä ja -0,14 % glipitsidia saaneiden ryhmässä. HbA1c-arvon korjattu keskimääräinen muutos lähtötilanteesta viikolla 208 oli ‑0,10 % dapagliflotsiinia saaneiden ryhmässä ja 0,20 % glipitsidia saaneiden ryhmässä. Dapagliflotsiinilla hoidetun ryhmän tutkittavista huomattavasti pienemmällä osuudella (3,5 % viikolla 52, 4,3 % viikolla 104 ja 5,0 % viikolla 208) ilmeni vähintään yksi hypoglykemiatapahtuma verrattuna glipitsidillä hoidettuun ryhmään (40,8 % viikolla 52, 47,0 % viikolla 104 ja 50,0 % viikolla 208). Niiden tutkittavien osuus, jotka olivat jatkaneet tutkimuksessa viikkoon 104 asti, oli 56,2 % dapagliflotsiinilla hoidettujen ryhmässä ja 50,0 % glipitsidillä hoidettujen ryhmässä, ja niiden tutkittavien osuus, jotka olivat jatkaneet tutkimuksessa viikkoon 208 asti, oli 39,7 % dapagliflotsiinilla hoidettujen ryhmässä ja 34,6 % glipitsidilla hoidettujen ryhmässä.
Taulukko 3. Tulokset viikolla 52 (LOCFa) vertailututkimuksessa, jossa verrattiin dapagliflotsiinia glipitsidiin metformiiniin lisähoitona
| Parametri | Dapagliflotsiini + metformiini | Glipitsidi + metformiini |
| Nb | 400 | 401 |
| HbA1c (%) | ||
| Lähtötilanne (keskiarvo) | 7,69 | 7,74 |
| Muutos lähtötilanteestac | ‑0,52 | ‑0,52 |
| Ero glipitsidiin + metformiiniin verrattunac | 0,00d | |
| (95 %:n luottamusväli) | (‑0,11, 0,11) | |
| Paino (kg) | ||
| Lähtötilanne (keskiarvo) | 88,44 | 87,60 |
| Muutos lähtötilanteestac | ‑3,22 | 1,44 |
| Ero glipitsidiin + metformiiniin verrattunac | ‑4,65* | |
| (95 %:n luottamusväli) | (‑5,14, ‑4,17) | |
| aLOCF: Viimeisimmästä havainnosta laskettu arvio (last observation carried forward) bSatunnaistetut ja hoidetut tutkittavat, joilla lääkkeen teho oli mitattu lähtötilanteessa ja vähintään yhden kerran lähtötilanteen jälkeen cPienimmän neliösumman keskiarvo suhteutettuna lähtötilanteen arvoon dSamanveroinen glipitsidin + metformiinin kanssa *p‑arvo < 0,0001 | ||
Dapagliflotsiini yhdistettynä metformiiniin, glimepiridiin, metformiiniin ja sulfonyyliureaan, sitagliptiiniin (metformiinin kanssa tai ilman sitä) tai insuliiniin johti tilastollisesti merkitsevään HbA1c-arvon alenemiseen 24 viikon kohdalla verrattuna lumelääkettä saaneisiin tutkittaviin (p < 0,0001, taulukot 4, 5 ja 6).
Viikolla 24 havaitut HbA1c-arvojen paranemiset säilyivät niissä yhdistelmähoitotutkimuksissa (glimepiridi ja insuliini), joissa seuranta-aika jatkui 48 viikkoon asti (glimepiridi) ja viikkoon 104 asti (insuliini). Kun dapagliflotsiinia annettiin sitagliptiinin lisälääkkeenä (metformiinin kanssa tai ilman sitä), keskimääräinen muutos lähtötilanteesta viikkoon 48 oli 10 mg dapagliflotsiinia saaneilla ‑0,30 % ja lumelääkettä saaneilla 0,38 %. Tutkimuksessa, jossa dapagliflotsiinia annettiin yhdistettynä metformiinin HbA1c-arvojen parantuminen säilyi viikolle 102 saakka (korjattu HbA1c-arvon muutos lähtötilanteesta 10 mg:n dapagliflotsiiniannoksella ‑0,78 % ja lumelääkkeellä 0,02 %). Insuliinia (yhdessä suun kautta lisäksi annettujen glukoosipitoisuutta alentavien lääkkeiden kanssa tai ilman niitä) saaneilla HbA1c-arvon korjattu keskimääräinen muutos lähtötilanteesta viikolla 104 oli ‑0,71 % dapagliflotsiiniryhmässä ja ‑0,06 % lumelääkeryhmässä. Viikoilla 48 ja 104 insuliiniannos oli pysynyt vakaana (keskimäärin 76 IU/vrk) lähtötilanteeseen nähden potilailla, joita hoidettiin 10 mg:n dapagliflotsiiniannoksella. Lumelääkettä saaneiden ryhmässä annos oli suurentunut lähtötilanteesta keskimäärin 10,5 IU/vrk viikolla 48 (keskimääräinen annos 84 IU/vrk) ja 18,3 IU/vrk viikolla 104 (keskimääräinen annos 92 IU/vrk). Niiden potilaiden osuus, jotka olivat jatkaneet tutkimuksessa viikkoon 104, oli 72,4 % dapagliflotsiinia 10 mg saaneiden ryhmässä ja 54,8 % lumelääkettä saaneiden ryhmässä.
Taulukko 4. Tulokset 24 viikkoa kestäneistä (LOCFa) lumevertailututkimuksista, joissa dapagliflotsiini oli yhdistettynä metformiiniin tai sitagliptiiniin (metformiinin kanssa tai ilman sitä)
| Yhdistelmähoito | ||||
| Metformiini1 | DPP-4:n estäjä (sitagliptiini2) ± metformiini1 | |||
Dapagliflotsiini 10 mg | Lumelääke | Dapagliflotsiini 10 mg | Lumelääke | |
| Nb | 135 | 137 | 223 | 224 |
| HbA1c (%) | ||||
| Lähtötilanne (keskiarvo) | 7,92 | 8,11 | 7,90 | 7,97 |
| Muutos lähtötilanteestac | ‑0,84 | ‑0,30 | -0,45 | 0,04 |
| Ero lumelääkkeeseen verrattunac | ‑0,54* | -0,48* | ||
| (95 %:n luottamusväli) | (‑0,74, ‑0,34) | (-0,62, ‑0,34) | ||
Tutkittavat (%), jotka saavuttivat seuraavat arvot: HbA1c < 7 % | ||||
| Korjattuna suhteessa lähtötilanteeseen | 40,6** | 25,9* | ||
| Paino (kg) | ||||
| Lähtötilanne (keskiarvo) | 86,28 | 87,74 | 91,02 | 89,23 |
Muutos lähtötilanteestac | ‑2,86 | ‑0,89 | -2,14 | -0,26 |
Ero lumelääkkeeseen verrattunac | ‑1,97* | -1,89* | ||
| (95 %:n luottamusväli) | (‑2,63, ‑1,31) | (-2,37, ‑1,40) | ||
| 1Metformiini ≥ 1 500 mg/vrk; 2sitagliptiini 100 mg/vrk aLOCF: Viimeisimmästä havainnosta (ennen glukoositason korjaamista [glycaemic rescue] sitä edellyttäneillä tutkittavilla) laskettu arvio (last observation carried forward) bKaikki satunnaistetut tutkittavat, jotka saivat vähintään yhden annoksen kaksoissokkoutettua tutkimuslääkevalmistetta lyhytkestoisen kaksoissokkoutetun hoitojakson aikana cPienimmän neliösumman keskiarvo suhteutettuna lähtötilanteen arvoon *p‑arvo < 0,0001 verrattuna lumelääkkeeseen + suun kautta otettavaan verensokeria alentavaan lääkevalmisteeseen **p‑arvo < 0,05 verrattuna lumelääkkeeseen + suun kautta otettavaan verensokeria alentavaan lääkevalmisteeseen | ||||
Taulukko 5. Tulokset 24 viikkoa kestäneestä lumevertailututkimuksesta, jossa tutkittiin dapagliflotsiinia yhdistettynä sulfonyyliureaan (glimepiridi) tai metformiiniin ja sulfonyyliureaan
| Yhdistelmähoito | ||||
Sulfonyyliurea (glimepiridi1) | Sulfonyyliurea + metformiini2 | |||
Dapagliflotsiini 10 mg | Lumelääke | Dapagliflotsiini 10 mg | Lumelääke | |
| Na | 151 | 145 | 108 | 108 |
| HbA1c (%)b | ||||
| Lähtötilanne (keskiarvo) | 8,07 | 8,15 | 8,08 | 8,24 |
| Muutos lähtötilanteestac | -0,82 | -0,13 | -0,86 | -0,17 |
| Ero lumelääkkeeseenc | -0,68* | -0,69* | ||
| (95 %:n luottamusväli) | (-0,86, -0,51) | (-0,89, -0,49) | ||
Tutkittavat (%), jotka saavuttivat seuraavat arvot: HbA1c < 7 % (LOCF)d Korjattuna suhteessa lähtötilanteeseen | 31,7* | 13,0 | 31,8* | 11,1 |
| Paino (kg) (LOCF)d | ||||
| Lähtötilanne (keskiarvo) | 80,56 | 80,94 | 88,57 | 90,07 |
| Muutos lähtötilanteesta | -2,26 | -0,72 | -2,65 | -0,58 |
| Ero lumelääkkeeseenc | -1,54* | -2,07* | ||
| (95 %:n luottamusväli) | (-2,17, -0,92) | (-2,79, -1,35) | ||
| 1glimepiridi 4 mg/vrk 2metformiini (välittömästi vaikuttavat tai depotlääkemuodot) ≥ 1 500 mg/vrk yhdistettynä sulfonyyliurean suurimpaan siedettyyn annokseen, jonka täytyy olla vähintään puolet enimmäisannoksesta, vähintään 8 viikon ajan ennen tutkimukseen osallistumista. aSatunnaistetut lääkettä saaneet potilaat, joilta oli mitattu lääkkeen teho lähtötilanteessa ja ainakin kerran sen jälkeen. bSarakkeet 1 ja 2: HbA1c analysoitiin käyttämällä LOCF:ää (katso alaviite d); sarakkeet 3 ja 4: HbA1c analysoitiin käyttämällä LRM:ää (katso alaviite e) cPienimmän neliösumman keskiarvo suhteutettuna lähtötilanteen arvoon dLOCF: Viimeisimmästä havainnosta (ennen glukoositason korjaamista [glycaemic rescue] sitä edellyttäneillä tutkittavilla) laskettu arvio (last observation carried forward) eLRM: Toistettujen mittausten pitkittäisanalyysi (longitudinal repeated measures analysis) *p‑arvo < 0,0001 verrattuna lumelääkkeeseen + suun kautta otettavaan verensokeria alentavaan lääkevalmisteeseen | ||||
Taulukko 6. Tulokset viikolla 24 (LOCFa) lumevertailututkimuksessa, jossa tutkittiin dapagliflotsiinin ja insuliinin yhdistelmää (yksin tai yhdessä suun kautta annettavien verensokeria alentavien lääkevalmisteiden kanssa)
| Parametri | Dapagliflotsiini 10 mg + insuliini ± suun kautta otettavat verensokeria alentavat lääkevalmisteet2 | Lumelääke + insuliini ± suun kautta otettavat verensokeria alentavat lääkevalmisteet2 |
| Nb | 194 | 193 |
| HbA1c (%) | ||
| Lähtötilanne (keskiarvo) | 8,58 | 8,46 |
| Muutos lähtötilanteestac | ‑0,90 | ‑0,30 |
| Ero lumelääkkeeseen verrattunac | ‑0,60* | |
| (95 %:n luottamusväli) | (‑0,74, ‑0,45) | |
| Paino (kg) | ||
| Lähtötilanne (keskiarvo) | 94,63 | 94,21 |
| Muutos lähtötilanteestac | ‑1,67 | 0,02 |
| Ero lumelääkkeeseen verrattunac | ‑1,68* | |
| (95 %:n luottamusväli) | (‑2,19, ‑1,18) | |
| Insuliinin keskimääräinen vuorokausiannos (IU)1 | ||
| Lähtötilanne (keskiarvo) | 77,96 | 73,96 |
| Muutos lähtötilanteestac | ‑1,16 | 5,08 |
| Ero lumelääkkeeseen verrattunac | ‑6,23* | |
| (95 %:n luottamusväli) | (‑8,84, ‑3,63) | |
| Tutkittavat, joiden insuliinin vuorokausiannos on keskimäärin alentunut vähintään 10 % (%) | 19,7** | 11,0 |
| aLOCF: Viimeisestä havainnosta (ennen sitä päivää tai sinä päivänä, kun insuliiniannosta on ensimmäinen kerran nostettu, mikäli tarpeen) laskettu arvo (last observation carried forward) bKaikki satunnaistetut tutkittavat, jotka ottivat vähintään yhden annoksen kaksoissokkoutettua tutkimuslääkevalmistetta lyhytkestoisen kaksoissokkoutetun hoitojakson aikana cPienimmän neliösumman keskiarvo suhteutettuna lähtötilanteen arvoon, kun käytössä on suun kautta otettava verensokeria alentava lääkevalmiste *p‑arvo < 0,0001 verrattuna lumelääkkeeseen + insuliiniin ± suun kautta otettavaan verensokeria alentavaan lääkevalmisteeseen **p‑arvo < 0,05 verrattuna lumelääkkeeseen + insuliiniin ± suun kautta otettavaan verensokeria alentavaan lääkevalmisteeseen 1Insuliinihoitojen (lyhytvaikutteisen, keskipitkävaikutteisen ja perusinsuliinin) titraus ylöspäin sallittiin vain tutkittavilla, jotka täyttivät paastoverensokeria koskevat ennalta asetetut kriteerit. 2Tutkittavista 50 % sai insuliinia monoterapiana lähtötilanteessa; 50 % sai yhtä tai kahta suun kautta otettavaa verensokeria alentavaa lääkevalmistetta insuliinin lisäksi. Jälkimmäisestä ryhmästä 80 % sai pelkästään metformiinia, 12 % metformiinia ja sulfonyyliureaa ja loput muita suun kautta otettavia verensokeria alentavia lääkevalmisteita. | ||
Yhdistelmähoito metformiinin kanssa, kun potilaat eivät ole aiemmin saaneet lääkehoitoa
Kahteen aktiivikontrolloituun 24 viikkoa kestäneeseen tutkimukseen osallistui 1 236 aiemmin hoitamatonta potilasta, joilla oli tyypin 2 diabetes, jonka hoitotasapaino ei ollut riittävä (HbA1c ≥ 7,5 % ja ≤ 12 %). Tutkimuksissa arvioitiin yhdessä metformiinin kanssa annetun dapagliflotsiinin (5 mg tai 10 mg) tehoa ja turvallisuutta potilailla, jotka eivät olleet aiemmin saaneet lääkehoitoa, verrattuna hoitoon yksittäisillä aineilla.
Hoito dapagliflotsiinin 10 mg:n annoksen ja metformiinin (enintään 2 000 mg vuorokaudessa) yhdistelmällä paransi HbA1c-arvoja selvästi verrattuna yksittäisiin aineisiin (taulukko 8) ja pienensi plasman paastoglukoosiarvoja (enemmän kuin yksittäiset aineet) ja painoa (enemmän kuin metformiini).
Taulukko 7. Tulokset viikolla 24 (LOCFa) aktiivikontrolloidussa tutkimuksessa, jossa tutkittiin dapagliflotsiinin ja metformiinin yhdistelmää potilailla, jotka eivät olleet aiemmin saaneet lääkehoitoa
| Parametri | Dapagliflotsiini 10 mg + metformiini | Dapagliflotsiini 10 mg | Metformiini |
| Nb | 211b | 219b | 208b |
| HbA1c (%) | |||
| Lähtötilanne (keskiarvo) | 9,10 | 9,03 | 9,03 |
| Muutos lähtötilanteestac | ‑1,98 | ‑1,45 | ‑1,44 |
| Ero dapagliflotsiiniin verrattunac | ‑0,53* | ||
| (95 %:n luottamusväli) | (‑0,74, ‑0,32) | ||
| Ero metformiiniin verrattunac | ‑0,54* | ‑0,01 | |
| (95 %:n luottamusväli) | (‑0,75, ‑0,33) | (‑0,22, 0,20) | |
| aLOCF: viimeisestä havainnosta (ennen glukoositason korjaamista [glycaemic rescue] sitä edellyttäneillä potilailla) laskettu arvio. bKaikki satunnaistetut potilaat, jotka saivat vähintään yhden annoksen kaksoissokkoutettua tutkimuslääkevalmistetta lyhytkestoisen kaksoissokkoutetun hoitojakson aikana. cPienimmän neliösumman keskiarvo suhteutettuna lähtötilanteen arvoon. *p-arvo < 0,0001. | |||
Yhdistelmähoito depotmuotoisen eksenatidin kanssa
28 viikkoa kestäneessä kaksoissokkoutetussa, aktiivisella vertailuvalmisteella kontrolloidussa tutkimuksessa dapagliflotsiinin ja depotmuotoisen eksenatidin (GLP‑1‑reseptoriagonisti) yhdistelmää verrattiin pelkkään dapagliflotsiiniin ja pelkkään depotmuotoiseen eksenatidiin pelkästään metformiinia saaneilla tutkittavilla, joiden glukoositasapaino oli riittämätön (HbA1c ≥ 8 % ja ≤ 12 %). HbA1c-arvo pieneni kaikissa hoitoryhmissä lähtötilanteeseen verrattuna. Yhdistelmähoito dapagliflotsiinin 10 mg:n annoksella ja depotmuotoisella eksenatidilla pienensi HbA1c‑arvoa lähtötilanteesta enemmän kuin pelkkä dapagliflotsiini tai pelkkä depotmuotoinen eksenatidi (taulukko 8).
Taulukko 8. Tulokset 28 viikkoa kestäneestä tutkimuksesta, jossa verrattiin dapagliflotsiinia ja depotmuotoista eksenatidia pelkkään dapagliflotsiiniin ja pelkkään depotmuotoiseen eksenatidiin yhdistelmänä metformiinin kanssa (intent to treat -potilaat)
| Parametri | Dapagliflotsiini 10 mg kerran vuorokaudessa + depotmuotoinen eksenatidi 2 mg kerran viikossa | Dapagliflotsiini 10 mg kerran vuorokaudessa + lumelääke kerran viikossa | Depotmuotoinen eksenatidi 2 mg kerran viikossa + lumelääke kerran vuorokaudessa |
| N | 228 | 230 | 227 |
| HbA1c (%) | |||
| Lähtötilanne (keskiarvo) | 9,29 | 9,25 | 9,26 |
| Muutos lähtötilanteestaa | ‑1,98 | ‑1,39 | ‑1,60 |
| Keskimääräinen ero muutoksessa lähtötilanteesta yhdistelmän ja yksittäisen lääkevalmisteen välillä (95 %:n luottamusväli) | ‑0,59* (‑0,84, ‑0,34) | ‑0,38** (‑0,63, ‑0,13) | |
| Tutkittavat (%), jotka saavuttivat alle 7 %:n HbA1c-arvon | 44,7 | 19,1 | 26,9 |
| Paino (kg) | |||
| Lähtötilanne (keskiarvo) | 92,13 | 90,87 | 89,12 |
| Muutos lähtötilanteestaa | ‑3,55 | ‑2,22 | ‑1,56 |
| Keskimääräinen ero muutoksessa lähtötilanteesta yhdistelmän ja yksittäisen lääkevalmisteen välillä (95 %:n luottamusväli) | ‑1,33* (‑2,12, ‑0,55) | ‑2,00* (‑2,79, ‑1,20) | |
| N = potilaiden määrä a Korjatut pienimmän neliösumman keskiarvot ja tutkimusryhmien ero(t) lähtötilanteessa todetuista arvoista tapahtuneen muutoksen suhteen viikolla 28 mallinnettiin käyttämällä toistettujen mittausten sekamallia. Malli sisälsi kiinteinä tekijöinä hoidon, alueen, lähtötilanteen HbA1c-ositteen (< 9,0 % tai ≥ 9,0 %), viikon sekä hoidon ja viikon yhteisvaikutuksen. Kovariaattina oli lähtötilanteessa mitattu arvo. *p < 0,001, **p < 0,01 Kaikki p-arvot ovat kerrannaisuuden suhteen korjattuja p-arvoja. Analyysit eivät sisällä mittauksia, jotka on tehty hätälääkityksen käytön jälkeen tai tutkimuslääkkeen käytön ennenaikaisen lopettamisen jälkeen. | |||
Plasman paastoglukoosi
Dapagliflotsiini 10 mg monoterapiana tai yhdistettynä joko metformiiniin, glimepiridiin, metformiiniin ja sulfonyyliureaan, sitagliptiiniin (metformiinin kanssa tai ilman sitä) tai insuliiniin laski plasman paastoglukoosiarvoa tilastollisesti merkitsevästi (‑1,90 – ‑1,20 mmol/l [‑34,2 –‑21,7 mg/dl]) verrattuna lumelääkkeeseen (-0,33 – 0,21 mmol/l [-6,0 – 3,8 mg/dl]). Tämä vaikutus havaittiin hoitoviikolla 1, ja vaikutus säilyi niissä tutkimuksissa, joita jatkettiin viikolle 104.
Yhdistelmähoito dapagliflotsiinin 10 mg:n annoksella ja depotmuotoisella eksenatidilla oli pienentänyt huomattavasti enemmän plasman paastoglukoosiarvoa viikolla 28: (‑3,66 mmol/l [‑65,8 mg/dl]) verrattuna pelkkään dapagliflotsiiniin (‑2,73 mmol/l [‑49,2 mg/dl]), p < 0,001, tai pelkkään eksenatidiin (‑2,54 mmol/l [‑45,8 mg/dl]), p < 0,001.
Tutkimuksessa, joka tehtiin nimenomaan diabetespotilailla, joiden eGFR oli ≥ 45 − < 60 ml/min/1,73 m2, dapagliflotsiinihoito oli pienentänyt paastoglukoosiarvoa viikolla 24 (‑1,19 mmol/l [‑21,46 mg/dl]), verrattuna lumelääkkeeseen (‑0,27 mmol/l [‑4,87 mg/dl]) (p = 0,001).
Aterianjälkeinen glukoosi
Dapagliflotsiini 10 mg yhdistettynä glimepiridiin oli alentanut kaksi tuntia aterian jälkeen mitattua glukoosipitoisuutta tilastollisesti merkitsevästi viikon 24 kohdalla, ja tämä tulos säilyi viikkoon 48 saakka.
Dapagliflotsiini 10 mg sitaglipitiinin lisälääkkeenä (metformiinin kanssa tai ilman sitä) oli alentanut kaksi tuntia aterian jälkeen mitattua glukoosipitoisuutta viikon 24 kohdalla, ja tämä tulos säilyi viikkoon 48 saakka.
Yhdistelmähoito dapagliflotsiinin 10 mg:n annoksella ja depotmuotoisella eksenatidilla oli pienentänyt kaksi tuntia aterian jälkeen mitattua glukoosipitoisuutta viikon 28 kohdalla huomattavasti enemmän kuin kumpikaan lääkevalmiste yksinään.
Paino
Dapagliflotsiini 10 mg yhdistettynä joko metformiiniin, glimepiridiin, metformiiniin ja sulfonyyliureaan, sitagliptiiniin (metformiinin kanssa tai ilman sitä) tai insuliiniin laski tutkittavien painoa tilastollisesti merkitsevästi viikkoon 24 mennessä (p < 0,0001, taulukot 4 ja 5). Nämä vaikutukset säilyivät pitkäaikaistutkimuksissa. Viikolla 48 ero lumelääkkeeseen verrattuna oli dapagliflotsiinia sitagliptiinin lisälääkkeenä (metformiinin kanssa tai ilman sitä) saaneilla ‑2,22 kg. Viikolla 102 ero lumelääkkeeseen verrattuna oli dapagliflotsiinia metformiinin lisälääkkeenä saaneilla ‑2,14 kg ja insuliinin lisälääkkeenä saaneilla ‑2,88 kg.
Lääkevertailututkimuksessa, jossa pyrittiin osoittamaan dapagliflotsiini vähintään samanveroiseksi kuin vertailulääke (non-inferiority), glipitsidiin verrattuna dapagliflotsiini metformiinin lisälääkkeenä oli laskenut painoa tilastollisesti merkitsevästi -4,65 kg viikkoon 52 mennessä (p < 0,0001, taulukko 3) ja tämä vaikutus oli säilynyt viikolla 104 (-5,06 kg) ja viikolla 208 (‑4,38 kg).
Dapagliflotsiinin 10 mg:n annoksen ja depotmuotoisen eksenatidin yhdistelmän käyttö laski painoa huomattavasti enemmän kuin kumpikaan lääkevalmiste yksinään (taulukko 8).
24 viikon kestoisessa tutkimuksessa, johon osallistui 182 diabetesta sairastavaa potilasta, kehon koostumusta tutkittiin kaksienergisellä röntgenabsorptiometrialla (DEXA). Tutkimus osoitti, että dapagliflotsiini 10 mg yhdistettynä metformiiniin vähensi kehon painoa ja kehon rasvakudosta DEXA:lla mitattuna verrattuna lumelääkkeen ja metformiinin yhdistelmään; vähentämättä rasvatonta kehon painoa (lean body mass) tai nestettä. Magneettikuvauksella tehty alatutkimus osoitti Forxiga + metformiini-yhdistelmähoidon vähentävän numeraalisesti viskeraalisen rasvakudoksen määrää verrattuna lumelääke + metformiini-yhdistelmähoitoon.
Verenpaine
13 lumevertailututkimuksen ennalta määritellyssä yhteisanalyysissä dapagliflotsiini annoksella 10 mg aiheutti ‑3,7 mmHg:n systolisen verenpaineen muutoksen lähtötilanteesta ja ‑1,8 mmHg:n ja diastolisen verenpaineen muutoksen viikkoon 24 mennessä. Vastaavat lukemat lumeryhmässä olivat ‑0,5 mmHg ja ‑0,5 mmHg. Samanlaista verenpaineen alenemista todettiin viikolle 104 asti.
Yhdistelmähoito dapagliflotsiinin 10 mg:n annoksella ja depotmuotoisella eksenatidilla oli laskenut systolista verenpainetta huomattavasti enemmän viikon 28 kohdalla (‑4,3 mmHg) kuin pelkkä dapagliflotsiini (‑1,8 mmHg, p < 0,05) tai pelkkä depotmuotoinen eksenatidi (‑1,2 mmHg, p < 0,01).
Kahdessa 12 viikon pituisessa lumelääkekontrolloidussa tutkimuksessa, joihin osallistui yhteensä 1 062 potilasta, joiden tyypin 2 diabeteksen hoitotasapaino oli riittämätön ja joilla oli verenpainetauti (siitä huolimatta, että he toisessa tutkimuksessa käyttivät jo tutkimuksen aloittaessaan pysyvänä lääkityksenä angiotensiinikonvertaasin estäjää (ACE:n estäjää) tai angiotensiinireseptorin salpaajaa (ATR‑salpaajaa) ja toisessa tutkimuksessa angiotensiinikonvertaasin estäjää tai angiotensiinireseptorin salpaajaa sekä lisäksi yhtä verenpainelääkettä), potilaille annettiin dapagliflotsiinia 10 mg:n annoksella tai lumelääkettä.Viikon 12 kohdalla 10 mg:n dapagliflotsiiniannos yhdessä tavallisen diabeteshoidon kanssa oli parantanut HbA1c‑arvoja ja pienentänyt lumelääkkeen suhteen korjattua systolista verenpainetta toisessa tutkimuksessa keskimäärin 3,1 mmHg ja toisessa tutkimuksessa 4,3 mmHg.
Tutkimuksessa, joka tehtiin nimenomaan diabetespotilailla, joiden eGFR oli ≥ 45 − < 60 ml/min/1,73 m2, dapagliflotsiinihoito oli pienentänyt potilaan istuessa mitattua systolista verenpainetta viikon 24 kohdalla (‑4,8 mmHg) verrattuna lumelääkkeeseen (‑1,7 mmHg) (p < 0,05).
Glukoositasapaino potilailla, joilla oli keskivaikea munuaisten vajaatoiminta CKD-luokka 3A (eGFR ≥ 45 – < 60 ml/min/1,73 m2)
Dapagliflotsiinin tehoa arvioitiin tutkimuksessa, joka tehtiin nimenomaan diabetespotilailla, joiden eGFR oli ≥ 45 − < 60 ml/min/1,73 m2 ja joiden glukoositasapaino oli tavanomaisella hoidolla riittämätön. Dapagliflotsiinihoito pienensi HbA1c-arvoja ja painoa lumelääkkeeseen verrattuna (taulukko 9).
Taulukko 9. Tulokset viikolla 24 lumevertailututkimuksessa, jossa tutkittiin dapagliflotsiinia diabetespotilailla, joiden eGFR oli ≥ 45 − < 60 ml/min/1,73 m2
Dapagliflotsiinia 10 mg | Lumelääkea | |
| Nb | 159 | 161 |
| HbA1c (%) | ||
| Lähtötilanne (keskiarvo) | 8,35 | 8,03 |
| Muutos lähtötilanteestab | ‑0,37 | ‑0,03 |
Ero lumelääkkeeseen verrattunab (95 %:n luottamusväli) | ‑0,34* (‑0,53, ‑0,15) | |
| Paino (kg) | ||
| Lähtötilanne (keskiarvo) | 92,51 | 88,30 |
| Prosentuaalinen muutos lähtötilanteestac | -3,42 | -2,02 |
Prosentuaalisen muutoksen ero lumelääkkeeseen verrattunac (95 %:n luottamusväli) | -1,43* (-2,15, -0,69) | |
a Metformiini tai metformiinihydrokloridi olivat osa tavanomaista hoitoa 69,4 %:lla dapagliflotsiiniryhmän potilaista ja 64,0 %:lla lumeryhmän potilaista.
b Pienimmän neliösumman keskiarvo suhteutettuna lähtötilanteen arvoon
c Johdettu pienimmän neliösumman keskiarvosta suhteutettuna lähtötilanteen arvoon
* p < 0,001
Potilaat, joiden HbA1c-arvo lähtötilanteessa oli ≥ 9 %
Ennalta määritellyssä analyysissä tutkimuspotilaista, joiden HbA1c-arvo lähtötilanteessa oli ≥ 9 %, todettiin, että dapagliflotsiinihoito 10 mg:n annoksella sai aikaan tilastollisesti merkitsevän aleneman HbA1c-arvossa viikkoon 24 mennessä ainoana lääkkeenä (vakioitu keskimääräinen muutos lähtötilanteesta: ‑2,04 % dapagliflotsiinia 10 mg:n saaneiden ryhmässä ja 0,19 % lumelääkeryhmässä) ja yhdistelmänä metformiinin kanssa (vakioitu keskimääräinen muutos lähtötilanteesta: ‑1,32 % dapagliflotsiiniryhmässä ja ‑0,53 % lumelääkeryhmässä).
Sydän-, verisuoni- ja munuaistulokset
Dapagliflotsiinin vaikutusta sydän- ja verisuonitapahtumiin arvioinut tutkimus (Dapagliflozin Effect on Cardiovascular Events, DECLARE) oli kansainvälinen, satunnaistettu, kaksoissokkoutettu, lumekontrolloitu kliininen monikeskustutkimus, jossa verrattiin käytössä olevaan taustahoitoon lisätyn dapagliflotsiinin ja lumelääkkeen vaikutusta sydän- ja verisuonituloksiin. Kaikilla potilailla oli tyypin 2 diabetes ja joko vähintään kaksi muuta sydän- ja verisuonisairauksien riskitekijää (ikä ≥ 55 vuotta miehillä tai ≥ 60 vuotta naisilla ja vähintään yksi seuraavista: dyslipidemia, kohonnut verenpaine tai tupakointi) tai todettu sydän- ja verisuonisairaus.
17 160 satunnaistetusta potilaasta 6 974:llä (40,6 %:lla) oli todettu sydän- ja verisuonisairaus ja 10 186:lla (59,4 %:lla) ei ollut todettua sydän- ja verisuonisairautta. 8 582 potilasta satunnaistettiin saamaan 10 mg dapagliflotsiinia ja 8 578 saamaan lumelääkettä, ja seurannan keston mediaani oli 4,2 vuotta.
Tutkimuspopulaation keski-ikä oli 63,9 vuotta, ja 37,4 % potilaista oli naisia. Yhteensä 22,4 %:lla oli ollut diabetes ≤ 5 vuotta, ja diabeteksen keskimääräinen kesto oli 11,9 vuotta. Keskimääräinen HbA1c oli 8,3 % ja keskimääräinen painoindeksi oli 32,1 kg/m2.
Lähtötilanteessa 10,0 %:lla potilaista oli aiemmin todettu sydämen vajaatoiminta. Keskimääräinen eGFR oli 85,2 ml/min/1,73 m2, potilaista 7,4 %:lla eGFR oli < 60 ml/min/1,73 m2, ja potilaista 30,3 %:lla oli mikroalbuminuria (U-AlbKrea ≥ 30− ≤ 300 mg/g) tai makroalbuminuria (U-AlbKrea > 300 mg/g).
Suurin osa potilaista (98 %) käytti lähtötilanteessa vähintään yhtä diabeteslääkevalmistetta, kuten metformiinia (82 %), insuliinia (41 %) tai sulfonyyliureaa (43 %).
Ensisijaiset päätetapahtumat olivat aika sydän- ja verisuonikuoleman, sydäninfarktin ja iskeemisen aivohalvauksen muodostaman yhdistelmäpäätetapahtuman (merkittävät sydänperäiset haittatapahtumat, MACE) ensimmäiseen tapahtumaan ja aika sydämen vajaatoiminnasta johtuvan sairaalahoitojakson ja sydän- ja verisuonikuoleman muodostaman yhdistelmäpäätetapahtuman ensimmäiseen tapahtumaan. Toissijaiset päätetapahtumat olivat munuaisiin liittyvä yhdistelmäpäätetapahtuma ja kaikista syistä johtuva kuolleisuus.
Merkittävät sydänperäiset haittatapahtumat
10 mg:n dapagliflotsiiniannos osoitettiin vähintään samanveroiseksi kuin lumelääke sydän- ja verisuonikuoleman, sydäninfarktin ja iskeemisen aivohalvauksen muodostaman yhdistelmäpäätetapahtuman suhteen (yksitahoinen p < 0,001).
Sydämen vajaatoiminta tai sydän- ja verisuonikuolema
10 mg:n dapagliflotsiiniannoksen osoitettiin ehkäisevän sydämen vajaatoiminnasta johtuvan sairaalahoitojakson ja sydän- ja verisuonikuoleman yhdistelmäpäätetapahtumaa paremmin kuin lumelääke (kuva 1). Ero hoitovaikutuksessa liittyi sydämen vajaatoiminnasta johtuviin sairaalahoitojaksoihin, eikä sydän- ja verisuonikuolemissa ollut eroa (kuva 2).
Dapagliflotsiinihoidon hyöty lumelääkkeeseen verrattuna havaittiin riippumatta siitä, oliko potilailla todettu sydän- ja verisuonisairaus ja oliko potilailla lähtötilanteessa sydämen vajaatoiminta. Hyöty oli johdonmukainen kaikissa keskeisissä alaryhmissä, mukaan lukien ikä, sukupuoli, munuaisten toiminta (eGFR) ja maantieteellinen alue.
Kuva 1: Aika ensimmäiseen sydämen vajaatoiminnasta johtuvaan sairaalahoitojaksoon tai sydän- ja verisuonikuolemaan
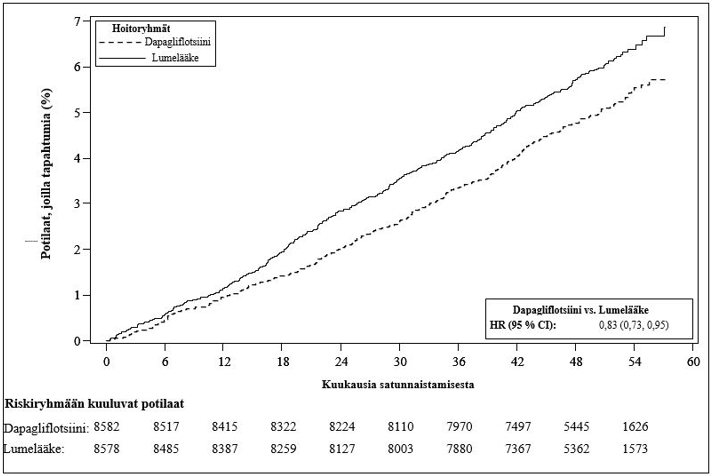
Riskiryhmään kuuluvien potilaiden määrä on riskiryhmään kuuluvien potilaiden määrä jakson alussa.
HR = riskisuhde CI = luottamusväli
Ensisijaisia ja toissijaisia päätemuuttujia koskevat tulokset on esitetty kuvassa 2. Dapagliflotsiinin paremmuutta lumelääkkeeseen nähden ei osoitettu MACE:n suhteen (p = 0,172). Munuaisia koskevaa yhdistelmäpäätetapahtumaa ja kaikista syistä johtuvaa kuolleisuutta ei näin ollen testattu osana tulokset vahvistavaa testaustoimenpidettä.
Kuva 2: Hoitovaikutukset ensisijaisten yhdistelmäpäätetapahtumien ja niiden komponenttien sekä toissijaisten päätetapahtumien ja niiden komponenttien suhteen
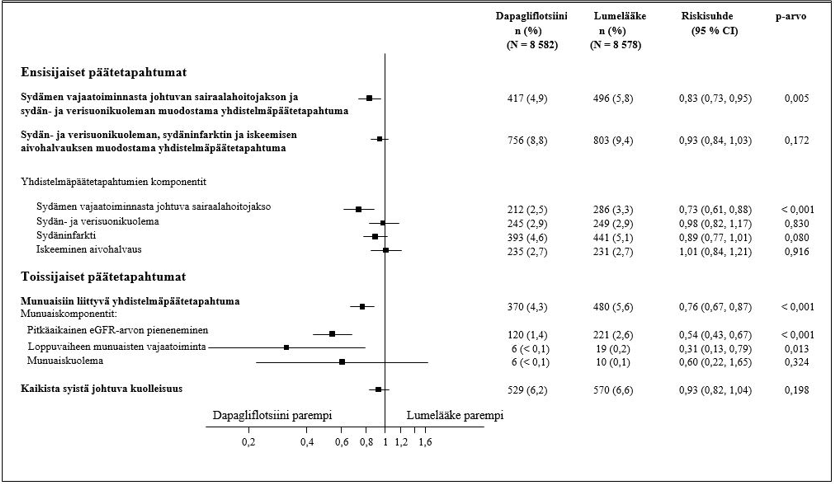
Munuaisiin liittyvän yhdistelmäpäätetapahtuman määritelmä: pitkäaikainen vahvistettu eGFR-arvon ≥ 40 %:n pieneneminen arvoon eGFR < 60 ml/min/1,73 m2 ja/tai loppuvaiheen munuaisten vajaatoiminta (≥ 90 päivää dialyysihoitoa tai munuaisensiirto, pitkäaikainen vahvistettu eGFR < 15 ml/min/1,73 m2) ja/tai munuais- tai sydän- ja verisuonikuolema.
p-arvot ovat kaksitahoisia. Toissijaisten päätetapahtumien ja yksittäisten komponenttien osalta p-arvot ovat nimellisiä. Aika ensimmäiseen tapahtumaan analysoitiin käyttämällä Coxin suhteellisten riskitiheyksien mallia. Yksittäisten komponenttien ensimmäisten tapahtumien lukumäärät ovat kunkin komponentin ensimmäisten tapahtumien todelliset lukumäärät, eikä niiden yhteenlaskettu summa vastaa yhdistelmäpäätetapahtuman tapahtumien lukumäärää.
CI = luottamusväli.
Nefropatia
Dapagliflotsiini vähensi vahvistetun pitkäaikaisen eGFR-arvon pienenemisen, loppuvaiheen munuaisten vajaatoiminnan ja munuais- tai sydän- ja verisuonikuoleman muodostaman yhdistelmäpäätetapahtuman tapahtumien ilmaantuvuutta. Ryhmien välinen ero liittyi munuaiskomponenttien (pitkäaikaisen eGFR-arvon pienenemisen, loppuvaiheen munuaisten vajaatoiminnan ja munuaiskuoleman) tapahtumien vähenemiseen (kuva 2).
Dapagliflotsiinin ja lumelääkkeen välinen riskisuhde (HR) nefropatian (pitkäaikainen eGFR:n pieneneminen, loppuvaiheen munuaissairaus ja munuaiskuolema) ilmenemiseen kuluneen ajan suhteen oli 0,53 (95 %:n luottamusväli 0,43, 0,66).
Lisäksi dapagliflotsiini pienensi pitkäkestoisen albuminurian ilmaantuvuutta (HR 0,79 [95 %:n luottamusväli 0,72, 0,87]) ja johti makroalbuminurian suurempaan regressioon (HR 1,82 [95 %:n luottamusväli 1,51, 2,20]) verrattuna lumelääkkeeseen.
Sydämen vajaatoiminta
DAPA‑HF-tutkimus: Sydämen vajaatoiminta, johon liittyi pienentynyt ejektiofraktio (LVEF ≤ 40 %)
DAPA‑HF-tutkimus (Dapagliflozin And Prevention of Adverse outcomes in Heart Failure) oli kansainvälinen, satunnaistettu, kaksoissokkoutettu, lumekontrolloitu monikeskustutkimus, joka tehtiin sydämen vajaatoimintaa sairastavilla potilailla (NYHA [New York Heart Association] ‑toimintakykyluokat II–IV), joilla vajaatoimintaan liittyi pienentynyt ejektiofraktio (vasemman kammion ejektiofraktio [LVEF] ≤ 40 %). Tutkimuksessa selvitettiin tavanomaisen taustahoidon lisänä käytetyn dapagliflotsiinin vaikutuksia sydän- ja verisuonikuoleman sekä sydämen vajaatoiminnan pahenemisen ilmaantuvuuteen lumelääkkeeseen verrattuna.
4 744 potilaasta 2 373 satunnaistettiin saamaan dapagliflotsiinia 10 mg:n annoksella ja 2 371 lumelääkettä. Seurannan keston mediaani oli 18 kuukautta. Tutkimuspopulaation keskimääräinen ikä oli 66 vuotta, ja 77 % potilaista oli miehiä.
Lähtötilanteessa 67,5 % potilaista kuului NYHA-luokkaan II, 31,6 % luokkaan III ja 0,9 % luokkaan IV, LVEF:n mediaani oli 32 %, 56 %:lla potilaista sydämen vajaatoiminta oli iskeeminen, 36 %:lla ei-iskeeminen ja 8 %:lla etiologia oli tuntematon. Kummassakin hoitoryhmässä 42 %:lla potilaista oli aiemmin todettu tyypin 2 diabetes, ja lisäksi 3 % kummankin ryhmän potilaista luokiteltiin tyypin 2 diabetesta sairastaviksi potilaiksi sillä perusteella, että HbA1c oli ≥ 6,5 % sekä tutkimuksen sisäänottovaiheessa että satunnaistamishetkellä. Potilaat saivat tavanomaista hoitoa; 94 % potilaista sai angiotensiinikonvertaasin estäjää (ACE:n estäjää), angiotensiinireseptorin salpaajaa (ATR-salpaajaa) tai angiotensiinireseptorin salpaajan ja neprilysiinin estäjän yhdistelmää (ARNI, 11 %), 96 % sai beetasalpaajaa, 71 % sai mineralokortikoidireseptorin salpaajaa (MRA), 93 % sai diureettia ja 26 %:lla potilaista oli implantoitu laite (jossa oli defibrillointitoiminto).
Tutkimukseen otettiin potilaita, joiden eGFR oli ≥ 30 ml/min/1,73 m2 tutkimuksen sisäänottovaiheessa. Keskimääräinen eGFR oli 66 ml/min/1,73 m2, 41 %:lla potilaista eGFR oli < 60 ml/min/1,73 m2 ja 15 %:lla eGFR oli < 45 ml/min/1,73 m2.
Sydän- ja verisuonikuolemat ja sydämen vajaatoiminnan paheneminen
Dapagliflotsiini ennaltaehkäisi lumelääkettä paremmin primaarista yhdistelmäpäätetapahtumaa, jonka muodostivat sydän- ja verisuonikuolema, sydämen vajaatoiminnasta johtuva sairaalahoitojakso ja sydämen vajaatoiminnasta johtuva kiireellinen vastaanottokäynti (HR 0,74 [95 %:n luottamusväli 0,65, 0,85], p < 0,0001). Vaikutus havaittiin varhain ja se säilyi koko tutkimuksen ajan (kuva 3).
Kuva 3: Aika ensimmäiseen yhdistelmätapahtumaan, jonka muodostivat sydän- ja verisuonikuolema, sydämen vajaatoiminnasta johtuva sairaalahoitojakso ja sydämen vajaatoiminnasta johtuva kiireellinen vastaanottokäynti
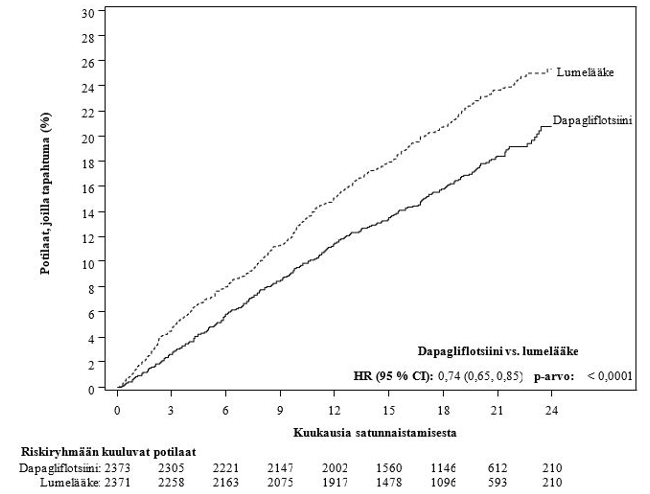
Sydämen vajaatoiminnasta johtuva kiireellinen vastaanottokäynti määriteltiin lääkärin esimerkiksi päivystysyksikössä tekemäksi kiireelliseksi, ennalta suunnittelemattomaksi arvioinniksi, jonka perusteella sydämen vajaatoiminnan pahenemisen katsottiin edellyttävän hoitoa (muuta kuin vain suun kautta otettavien diureettien annoksen suurentamista).
Riskiryhmään kuuluvien potilaiden määrä on riskiryhmään kuuluvien potilaiden määrä jakson alussa.
Ensisijaisen yhdistelmäpäätetapahtuman kaikki kolme komponenttia olivat erikseen osallisina hoitovaikutuksessa (kuva 4). Sydämen vajaatoiminnasta johtuvia kiireellisiä vastaanottokäyntejä oli vain vähän.
Kuva 4: Hoitovaikutukset ensisijaisen yhdistelmäpäätetapahtuman, sen komponenttien ja kaikista syistä johtuvan kuolleisuuden suhteen
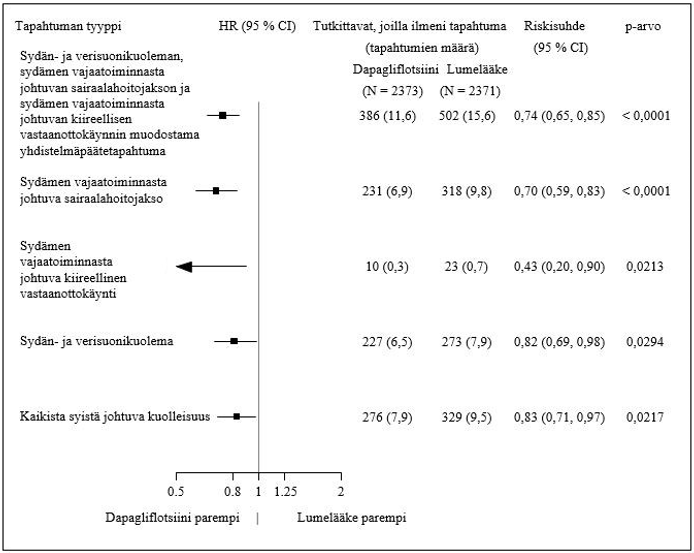
Sydämen vajaatoiminnasta johtuva kiireellinen vastaanottokäynti määriteltiin lääkärin esimerkiksi päivystysyksikössä tekemäksi kiireelliseksi, ennalta suunnittelemattomaksi arvioinniksi, jonka perusteella sydämen vajaatoiminnan pahenemisen katsottiin edellyttävän hoitoa (muuta kuin vain suun kautta otettavien diureettien annoksen suurentamista).
Yksittäisten komponenttien ensimmäisten tapahtumien lukumäärät ovat kunkin komponentin ensimmäisten tapahtumien todelliset lukumäärät, eikä niiden yhteenlaskettu summa vastaa yhdistelmäpäätetapahtuman tapahtumien lukumäärää.
Tapahtumien määrät on ilmoitettu niiden tutkittavien määränä, joilla ilmeni tapahtuma, seurannassa 100:aa potilasvuotta kohti.
Yksittäisten komponenttien ja kaikista syistä johtuvan kuolleisuuden osalta p-arvot ovat nimellisiä.
Dapagliflotsiini pienensi myös sydämen vajaatoiminnasta johtuvien sairaalahoitojaksojen (ensimmäisten ja uusintahoitojaksojen) ja sydän- ja verisuonikuoleman tapahtumien kokonaismäärää; dapagliflotsiiniryhmässä ilmeni 567 tapahtumaa ja lumelääkeryhmässä 742 tapahtumaa (esiintymistiheyksien suhde 0,75 [95 %:n luottamusväli 0,65, 0,88]; p = 0,0002).
Dapagliflotsiinihoidon hyöty havaittiin sekä sydämen vajaatoimintaa sairastavilla potilailla, joilla oli tyypin 2 diabetes, että sydämen vajaatoimintaa sairastavilla potilailla, joilla ei ollut diabetesta. Dapagliflotsiini vähensi ensisijaista yhdistelmäpäätetapahtumaa, sydän- ja verisuonikuoleman tai sydämen vajaatoiminnan pahenemisen ilmaantuvuutta, riskisuhteella 0,75 (95 %:n luottamusväli 0,63, 0,90) diabetespotilailla ja 0,73 (95 %:n luottamusväli 0,60, 0,88) potilailla, joilla ei ollut diabetesta.
Dapagliflotsiinihoidon hyöty ensisijaisen päätetapahtuman suhteen lumelääkkeeseen verrattuna oli johdonmukainen kaikissa keskeisissä alaryhmissä, mukaan lukien samanaikainen sydämen vajaatoiminnan lääkehoito, munuaisten toiminta (eGFR), ikä, sukupuoli ja maantieteellinen alue.
Potilaiden ilmoittamat hoitotulokset – sydämen vajaatoiminnan oireet
Dapagliflotsiinihoidon vaikutukset sydämen vajaatoiminnan oireisiin arvioitiin KCCQ-mittarin kokonaispistemäärällä (Total Symptom Score of the Kansas City Cardiomyopathy Questionnaire, KCCQ-TSS), jolla määritetään kvantitatiivisesti sydämen vajaatoiminnan oireiden, kuten väsymyksen, perifeerisen turvotuksen, hengenahdistuksen ja ortopnean, esiintymistiheys ja vaikeusaste. Pistemäärä arvioidaan asteikolla 0–100, niin että suurempi pistemäärä tarkoittaa parempaa terveydentilaa.
Lumelääkkeeseen verrattuna dapagliflotsiinihoito johti sydämen vajaatoiminnan oireisiin liittyvään tilastollisesti merkitsevään ja kliinisesti merkitykselliseen hyötyyn mitattuna KCCQ-kokonaispistemäärän muutoksena lähtötilanteesta kuukauden 8 kohdalla (voittosuhde 1,18 [95 %:n luottamusväli 1,11, 1,26]; p < 0,0001). Tuloksiin vaikuttivat sekä oireiden esiintymistiheys että oiretaakka. Hyöty havaittiin sekä sydämen vajaatoiminnan oireiden lievittymisessä että sydämen vajaatoiminnan oireiden pahenemisen ennaltaehkäisyssä.
Vasteanalyyseissä niiden potilaiden osuus, joilla todettiin 8 kuukauden kohdalla lähtötilanteeseen nähden KCCQ-kokonaispistemäärän kliinisesti merkityksellinen, vähintään 5 pisteeksi määritelty paraneminen oli suurempi dapagliflotsiinihoitoryhmässä kuin lumelääkeryhmässä. Niiden potilaiden osuus, joilla ilmeni kliinisesti merkityksellinen, vähintään 5 pisteeksi määritelty huononeminen oli pienempi dapagliflotsiinihoitoryhmässä kuin lumelääkeryhmässä. Dapagliflotsiinin käytön yhteydessä havaitut hyödyt säilyivät, kun käytettiin konservatiivisempia raja-arvoja kliinisesti merkityksellisemmälle muutokselle (taulukko 10).
Taulukko 10. Niiden potilaiden määrät ja prosentuaaliset osuudet, joilla todettiin KCCQ-kokonaispistemäärän kliinisesti merkityksellinen paraneminen tai huononeminen 8 kuukauden kohdalla
| Muutos lähtötilanteesta 8 kuukauden kohdalla: | Dapagliflotsiini 10 mg na = 2 086 | Lumelääke na = 2 062 | ||
| Paraneminen | n (%), parantunutb | n (%), parantunutb | Kerroinsuhdec (95 %:n luottamusväli) | p-arvof |
| ≥ 5 pistettä | 933 (44,7) | 794 (38,5) | 1,14 (1,06, 1,22) | 0,0002 |
| ≥ 10 pistettä | 689 (33,0) | 579 (28,1) | 1,13 (1,05, 1,22) | 0,0018 |
| ≥ 15 pistettä | 474 (22,7) | 406 (19,7) | 1,10 (1,01, 1,19) | 0,0300 |
| Huononeminen | n (%), huonontunutd | n (%), huonontunutd | Kerroinsuhdee (95 %:n luottamusväli) | p-arvof |
| ≥ 5 pistettä | 537 (25,7) | 693 (33,6) | 0,84 (0,78, 0,89) | < 0,0001 |
| ≥ 10 pistettä | 395 (18,9) | 506 (24,5) | 0,85 (0,79, 0,92) | < 0,0001 |
| a Niiden potilaiden määrä, joilla todettiin KCCQ-kokonaispistemäärä tai jotka kuolivat ennen 8 kuukauden ajankohtaa. b Niiden potilaiden määrä, joilla havaittiin vähintään 5, 10 tai 15 pisteen paraneminen lähtötilanteesta. Potilaat, jotka kuolivat ennen mainittua ajankohtaa, laskettiin potilaiksi, joilla pistemäärä ei ollut parantunut. c Paranemisen kohdalla kerroinsuhde > 1 suosii dapagliflotsiinin 10 mg:n vahvuutta. d Niiden potilaiden määrä, joilla havaittiin vähintään 5 tai 10 pisteen huononeminen lähtötilanteesta. Potilaat, jotka kuolivat ennen mainittua ajankohtaa, laskettiin potilaiksi, joilla pistemäärä oli huonontunut. e Huononemisen kohdalla kerroinsuhde < 1 suosii dapagliflotsiinin 10 mg:n vahvuutta. f p‑arvot ovat nimellisiä. | ||||
Nefropatia
Munuaisiin liittyvän yhdistelmäpäätetapahtuman tapahtumia (vahvistettu pitkäaikainen eGFR-arvon ≥ 50 %:n pieneneminen, loppuvaiheen munuaissairaus tai munuaiskuolema) oli vain vähän; niiden ilmaantuvuus oli dapagliflotsiiniryhmässä 1,2 % ja lumelääkeryhmässä 1,6 %.
DELIVER-tutkimus: Sydämen vajaatoiminta, jossa vasemman kammion ejektiofraktio oli > 40 %
DELIVER-tutkimus (Dapagliflozin Evaluation to Improve the LIVEs of Patients with PReserved Ejection Fraction Heart Failure) oli kansainvälinen, satunnaistettu, kaksoissokkoutettu, lumekontrolloitu monikeskustutkimus, joka tehtiin ≥ 40‑vuotiailla sydämen vajaatoimintaa sairastavilla potilailla (NYHA-luokat II–IV), joilla LVEF oli > 40 % ja joiden kohdalla oli näyttöä rakenteellisesta sydänsairaudesta. Tutkimuksessa selvitettiin dapagliflotsiinin vaikutuksia sydän- ja verisuonikuoleman sekä sydämen vajaatoiminnan pahenemisen ilmaantuvuuteen lumelääkkeeseen verrattuna.
6 263 potilaasta 3 131 satunnaistettiin saamaan dapagliflotsiinia 10 mg:n annoksella ja 3 132 lumelääkettä. Seurannan keston mediaani oli 28 kuukautta. Tutkimukseen osallistuneista potilaista 654 (10 %) sairasti subakuuttia sydämen vajaatoimintaa (määritelmä: satunnaistaminen sydämen vajaatoiminnasta johtuvan sairaalahoitojakson aikana tai 30 päivän kuluessa kotiutumisesta). Tutkimuspopulaation keskimääräinen ikä oli 72 vuotta, ja 56 % potilaista oli miehiä.
Lähtötilanteessa 75 % potilaista kuului NYHA-luokkaan II, 24 % luokkaan III ja 0,3 % luokkaan IV. LVEF:n mediaani oli 54 %, 34 %:lla potilaista LVEF oli ≤ 49 %, 36 %:lla LVEF oli 50–59 % ja 30 %:lla LVEF oli ≥ 60 %. Kummassakin hoitoryhmässä 45 %:lla potilaista oli aiemmin todettu tyypin 2 diabetes. Lähtötilanteessa hoitoon kuului angiotensiinikonvertaasin estäjä (ACE:n estäjä), angiotensiinireseptorin salpaaja (ATR-salpaaja) tai angiotensiinireseptorin salpaajan ja neprilysiinin estäjän yhdistelmä (ARNI) (77 %), beetasalpaaja (83 %), diureetti (98 %) tai mineralokortikoidireseptorin salpaaja (MRA; 43 %).
Keskimääräinen eGFR oli 61 ml/min/1,73 m2, 49 %:lla potilaista eGFR oli < 60 ml/min/1,73 m2, 23 %:lla eGFR oli < 45 ml/min/1,73 m2 ja 3 %:lla eGFR oli < 30 ml/min/1,73 m2.
Dapagliflotsiini pienensi lumelääkettä paremmin ensisijaisen yhdistelmäpäätetapahtuman (sydän- ja verisuonikuolema, sydämen vajaatoiminnasta johtuva sairaalahoitojakso ja sydämen vajaatoiminnasta johtuva kiireellinen vastaanottokäynti) ilmaantuvuutta (HR 0,82 [95 %:n luottamusväli 0,73, 0,92], p = 0,0008) (kuva 5).
Kuva 5: Aika ensimmäiseen yhdistelmätapahtumaan, jonka muodostivat sydän- ja verisuonikuolema, sydämen vajaatoiminnasta johtuva sairaalahoitojakso ja sydämen vajaatoiminnasta johtuva kiireellinen vastaanottokäynti
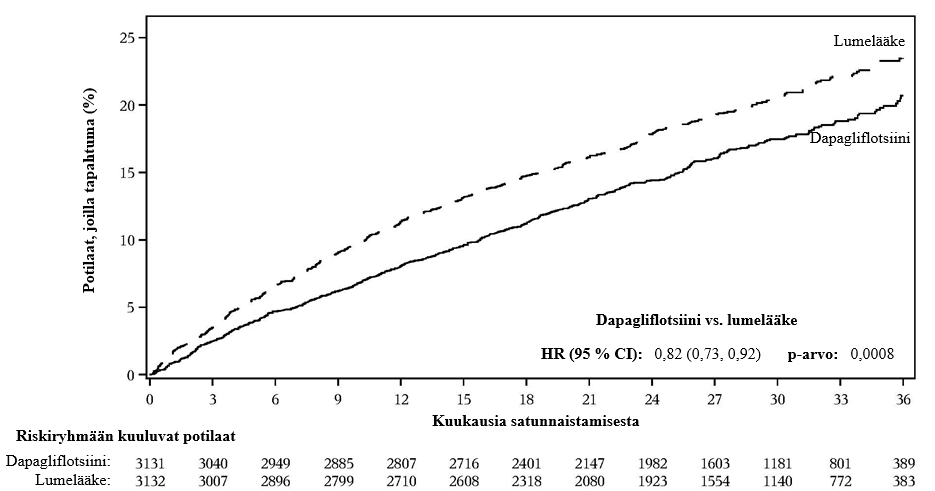
Sydämen vajaatoiminnasta johtuva kiireellinen vastaanottokäynti määriteltiin lääkärin esimerkiksi päivystysyksikössä tekemäksi kiireelliseksi, ennalta suunnittelemattomaksi arvioinniksi, jonka perusteella sydämen vajaatoiminnan pahenemisen katsottiin edellyttävän hoitoa (muuta kuin vain suun kautta otettavien diureettien annoksen suurentamista).
Riskiryhmään kuuluvien potilaiden määrä on riskiryhmään kuuluvien potilaiden määrä jakson alussa.
Kuvassa 6 esitetään ensisijaisen yhdistelmäpäätetapahtuman kolmen eri komponentin osallisuus hoitovaikutukseen.
Kuva 6: Hoitovaikutukset ensisijaisen yhdistelmäpäätetapahtuman ja sen komponenttien suhteen
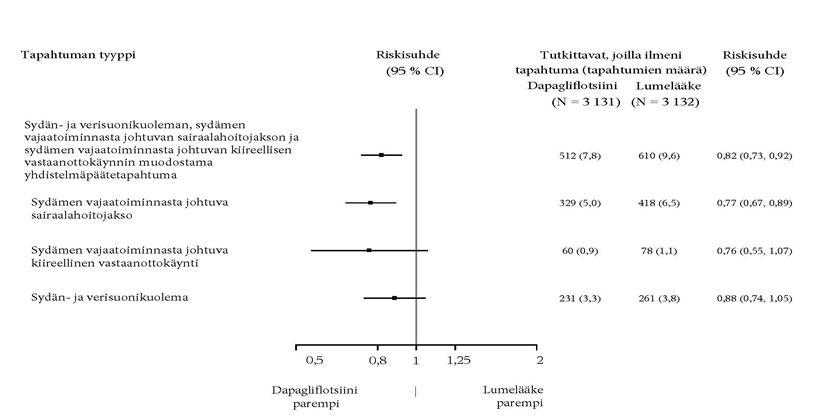
Sydämen vajaatoiminnasta johtuva kiireellinen vastaanottokäynti määriteltiin lääkärin esimerkiksi päivystysyksikössä tekemäksi kiireelliseksi, ennalta suunnittelemattomaksi arvioinniksi, jonka perusteella sydämen vajaatoiminnan pahenemisen katsottiin edellyttävän hoitoa (muuta kuin vain suun kautta otettavien diureettien annoksen suurentamista).
Yksittäisten komponenttien ensimmäisten tapahtumien lukumäärät ovat kunkin komponentin ensimmäisten tapahtumien todelliset lukumäärät, eikä niiden yhteenlaskettu summa vastaa yhdistelmäpäätetapahtuman tapahtumien lukumäärää.
Tapahtumien määrät on ilmoitettu niiden tutkittavien määränä, joilla ilmeni tapahtuma, seurannassa 100:aa potilasvuotta kohti.
Sydän- ja verisuonikuolema, joka esitetään tässä ensisijaisen päätetapahtuman komponenttina, testattiin myös toissijaisena päätetapahtumana tyypin 1 virheen suhteen muodollisesti kontrolloituna.
Dapagliflotsiini pienensi lumelääkettä paremmin sydämen vajaatoiminnan tapahtumien (määritelmänä ensimmäinen sairaalahoitojakso tai uusintahoitojakso sydämen vajaatoiminnan vuoksi tai sydämen vajaatoiminnasta johtuva ensimmäinen kiireellinen vastaanottokäynti tai kiireellinen uusintakäynti) ja sydän- ja verisuonikuolemien kokonaismäärää; dapagliflotsiiniryhmässä ilmeni 815 tapahtumaa ja lumelääkeryhmässä 1 057 tapahtumaa (esiintymistiheyksien suhde 0,77 [95 %:n luottamusväli 0,67, 0,89]; p = 0,0003).
Dapagliflotsiinihoidon hyöty ensisijaisen päätetapahtuman suhteen lumelääkkeeseen verrattuna havaittiin kaikissa alaryhmissä, joissa potilaiden LVEF oli ≤ 49 %, 50–59 % tai ≥ 60 %. Vaikutukset olivat johdonmukaiset myös muissa keskeisissä alaryhmissä, joissa ryhmittelyperusteena oli esimerkiksi ikä, sukupuoli, NYHA-luokka, NT‑proBNP-pitoisuus, vajaatoiminnan subakuutti luonne ja potilaan status tyypin 2 diabeteksen suhteen.
Potilaiden ilmoittamat hoitotulokset – sydämen vajaatoiminnan oireet
Lumelääkkeeseen verrattuna dapagliflotsiinihoito johti sydämen vajaatoiminnan oireisiin liittyvään tilastollisesti merkitsevään hyötyyn mitattuna KCCQ-kokonaispistemäärän muutoksena lähtötilanteesta kuukauden 8 kohdalla (voittosuhde 1,11 [95 %:n luottamusväli 1,03, 1,21]; p = 0,0086). Tuloksiin vaikuttivat sekä oireiden esiintymistiheys että oiretaakka.
Vasteanalyyseissä niiden potilaiden osuus, joilla todettiin 8 kuukauden kohdalla lähtötilanteeseen nähden KCCQ-kokonaispistemäärän kohtalainen huononeminen (≥ 5 pistettä) tai suuri huononeminen (≥ 14 pistettä), oli dapagliflotsiinihoitoryhmässä pienempi; dapagliflotsiiniryhmässä 24,1 %:lla ja lumelääkeryhmässä 29,1 %:lla todettiin pistemäärän kohtalainen huononeminen (kerroinsuhde 0,78 [95 %:n luottamusväli 0,64, 0,95]), ja dapagliflotsiiniryhmässä 13,5 %:lla ja lumelääkeryhmässä 18,4 %:lla todettiin suuri huononeminen (kerroinsuhde 0,70 [95 %:n luottamusväli 0,55, 0,88]). Hoitoryhmien välillä ei ollut eroja niiden potilaiden osuudessa, joilla ilmeni pieni tai kohtalainen paraneminen (≥ 13 pistettä) tai suuri paraneminen (≥ 17 pistettä).
Sydämen vajaatoiminta DAPA‑HF- ja DELIVER-tutkimuksissa
DAPA‑HF- ja DELIVER-tutkimusten yhdistetyssä analyysissä dapagliflotsiinin ja lumelääkkeen välinen riskisuhde sydän- ja verisuonikuoleman, sydämen vajaatoiminnasta johtuvan sairaalahoitojakson ja sydämen vajaatoiminnasta johtuvan kiireellisen vastaanottokäynnin muodostaman yhdistelmäpäätetapahtuman suhteen oli 0,78 (95 %:n luottamusväli 0,72, 0,85), p < 0,0001. Hoitovaikutus oli johdonmukainen koko LVEF-alueella, eikä vaikutus heikentynyt LVEF-arvon muuttuessa.
DAPA‑HF- ja DELIVER-tutkimusten ennalta määritellyssä potilaskohtaisten tietojen yhdistetyssä analyysissä dapagliflotsiini pienensi sydän- ja verisuonikuoleman riskiä verrattuna lumelääkkeeseen (HR 0,85 [95 %:n luottamusväli 0,75, 0,96], p = 0,0115). Molemmat tutkimukset olivat osallisina vaikutuksessa.
Krooninen munuaistauti
Tutkimus, jossa arvioitiin dapagliflotsiinin vaikutusta munuaistapahtumiin ja sydän- ja verisuonitauteihin liittyvään kuolleisuuteen kroonista munuaistautia sairastavilla potilailla (The Study to Evaluate the Effect of Dapagliflozin on Renal Outcomes and Cardiovascular Mortality in Patients with Chronic Kidney Disease, DAPA-CKD), oli kansainvälinen, satunnaistettu, kaksoissokkoutettu, lumekontrolloitu monikeskustutkimus. Potilailla oli krooninen munuaistauti, eGFR-arvo oli ≥ 25 – ≤ 75 ml/min/1,73 m2 ja heillä oli albuminuriaa (U-AlbKrea ≥ 200 – ≤ 5 000 mg/g). Tutkimuksessa verrattiin käytössä olevaan tavanomaiseen taustahoitoon lisätyn dapagliflotsiinin tai lumelääkkeen vaikutusta yhdistetyn päätetapahtuman, pitkäaikainen eGFR-arvon ≥ 50 %:n pieneneminen, loppuvaiheen munuaisten vajaatoiminta (määriteltiin tilanteeksi, jossa eGFR oli pitkäaikaisesti < 15 ml/min/1,73 m2 tai potilas sai pitkäkestoista dialyysihoitoa tai munuaisensiirron), sydän- ja verisuonikuolema tai munuaiskuolema, ilmaantuvuuteen.
4 304 potilaan ryhmästä 2 152 potilasta satunnaistettiin saamaan dapagliflotsiinia 10 mg:n annoksella ja 2 152 satunnaistettiin saamaan lumelääkettä, ja seurannan keston mediaani oli 28,5 kuukautta. Hoitoa jatkettiin, jos eGFR pieneni tutkimuksen aikana alle tason 25 ml/min/1,73 m2, ja sitä voitiin jatkaa, jos dialyysi oli tarpeen.
Tutkimuspopulaation keski-ikä oli 61,8 vuotta, ja 66,9 % potilaista oli miehiä. Lähtötilanteessa keskimääräinen eGFR oli 43,1 ml/min/1,73 m2 ja U-AlbKrea-arvon mediaani oli 949,3 mg/g. 44,1 %:lla potilaista eGFR oli 30 – < 45 ml/min/1,73 m2 ja 14,5 %:lla eGFR oli < 30 ml/min/1,73 m2. 67,5 %:lla potilaista oli tyypin 2 diabetes. Potilaat saivat tavanomaista hoitoa; 97,0 % potilaista sai angiotensiinikonvertaasin estäjää (ACE:n estäjää) tai angiotensiinireseptorin salpaajaa (ATR-salpaajaa).
Tutkimus keskeytettiin tehosyistä etuajassa ennen suunniteltua analyysiä, koska riippumaton arviointikomitea suositteli sen keskeyttämistä. Dapagliflotsiini ennaltaehkäisi lumelääkettä paremmin ensisijaista yhdistelmäpäätetapahtumaa, pitkäaikainen eGFR-arvon ≥ 50 %:n pieneneminen, loppuvaiheen munuaisten vajaatoiminnan kehittyminen, sydän- ja verisuonikuolema tai munuaiskuolema. Kaplan–Meier-kuvaaja ajasta ensimmäiseen ensisijaiseen yhdistelmäpäätetapahtumaan osoittaa, että hoitovaikutus oli ilmeinen 4 kuukaudesta alkaen ja säilyi tutkimuksen loppuun asti (kuva 7).
Kuva 7: Aika ensimmäiseen ensisijaiseen yhdistelmäpäätetapahtumaan, pitkäaikainen eGFR-arvon ≥ 50 %:n pieneneminen, loppuvaiheen munuaisten vajaatoiminta, sydän- ja verisuonikuolema tai munuaiskuolema
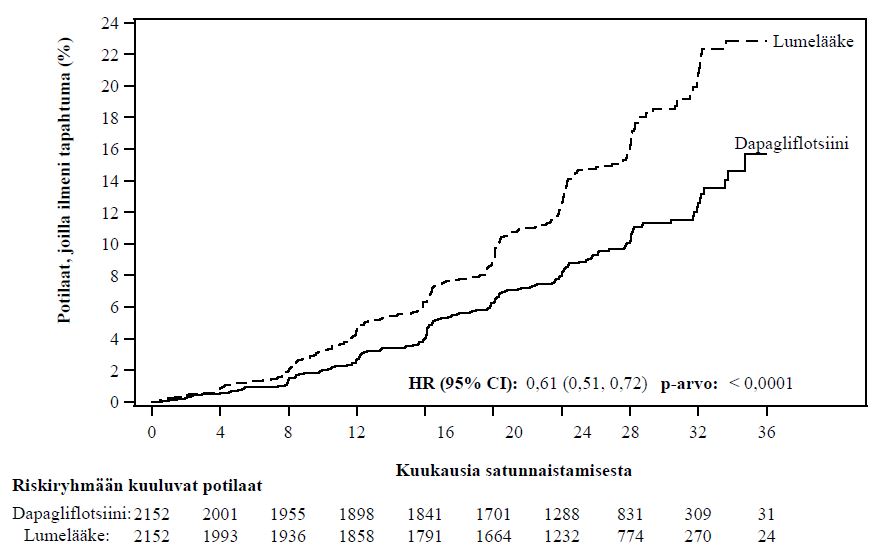
Riskiryhmään kuuluvien potilaiden määrä on riskiryhmään kuuluvien potilaiden määrä jakson alussa.
Ensisijaisen yhdistelmäpäätetapahtuman kaikki neljä komponenttia vaikuttivat itsenäisesti hoitovasteeseen. Dapagliflotsiini myös pienensi yhdistelmäpäätetapahtuman pitkäaikainen eGFR-arvon ≥ 50 %:n pieneneminen, loppuvaiheen munuaisten vajaatoiminta tai munuaiskuolema, ja yhdistelmäpäätetapahtuman sydän- ja verisuonikuolema tai sydämen vajaatoiminnasta johtuva sairaalahoitojakso, ilmaantuvuutta. Dapagliflotsiinihoito paransi kokonaiselossaoloaikaa kroonista munuaistautia sairastavilla potilailla ja vähensi kaikista syistä johtuvaa kuolleisuutta merkitsevästi (kuva 8).
Kuva 8: Hoitovaikutukset ensisijaisiin ja toissijaisiin yhdistelmäpäätetapahtumiin, niiden yksittäisiin komponentteihin ja kaikista syistä johtuvaan kuolleisuuteen
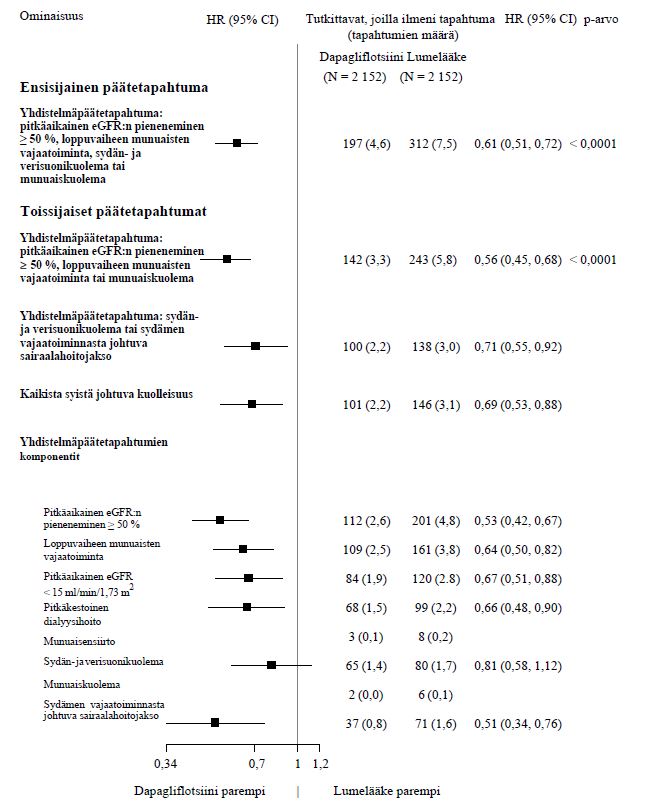
Yksittäisten komponenttien ensimmäisten tapahtumien lukumäärät ovat kunkin komponentin ensimmäisten tapahtumien todelliset lukumäärät, eikä niiden yhteenlaskettu summa vastaa yhdistelmäpäätetapahtuman tapahtumien lukumäärää.
Tapahtumien määrät on ilmoitettu niiden tutkittavien määränä, joilla ilmeni tapahtuma, seurannassa 100 potilasvuotta kohti.
Riskisuhteen (HR) estimaatteja ei esitetä alaryhmistä, joissa oli yhteensä alle 15 tapahtumaa, kun molemmat ryhmät yhdistettiin.
Dapagliflotsiinihoidon hyöty oli johdonmukainen kroonista munuaistautia sairastavilla potilailla, joilla oli tyypin 2 diabetes ja joilla ei ollut diabetesta. Dapagliflotsiini vähensi ensisijaista yhdistelmäpäätetapahtumaa (pitkäaikainen eGFR-arvon ≥ 50 %:n pieneneminen, loppuvaiheen munuaisten vajaatoiminnan kehittyminen, sydän- ja verisuonikuolema tai munuaiskuolema) riskisuhteella (HR) 0,64 (95 %:n luottamusväli 0,52, 0,79) potilailla, joilla oli tyypin 2 diabetes, ja riskisuhteella (HR) 0,50 (95 %:n luottamusväli 0,35, 0,72) potilailla, joilla ei ollut diabetesta.
Dapagliflotsiinihoidon hyöty ensisijaisen päätetapahtuman suhteen lumelääkkeeseen verrattuna oli johdonmukainen myös muissa keskeisissä alaryhmissä, mukaan lukien eGFR, ikä, sukupuoli ja maantieteellinen alue.
Pediatriset potilaat
Tyypin 2 diabetes
Kliinisessä tutkimuksessa 10–24‑vuotiailla lapsilla ja nuorilla, joilla oli tyypin 2 diabetes, 39 potilasta satunnaistettiin saamaan dapagliflotsiinia 10 mg:n annoksella ja 33 satunnaistettiin saamaan lumelääkettä metformiinin, insuliinin tai metformiinin ja insuliinin yhdistelmän lisälääkkeenä. Satunnaistamishetkellä 74 % potilaista oli alle 18‑vuotiaita. HbA1c-arvon korjattu keskimääräinen muutos lähtötilanteesta viikkoon 24 oli dapagliflotsiinia saaneilla verrattuna lumelääkettä saaneisiin ‑0,75 % (95 %:n luottamusväli ‑1,65, 0,15). Alle 18‑vuotiaiden ikäryhmässä HbA1c-arvon korjattu keskimääräinen muutos oli dapagliflotsiinia saaneilla verrattuna lumelääkettä saaneisiin ‑0,59 % (95 %:n luottamusväli ‑1,66, 0,48). Vähintään 18-vuotiaiden ikäryhmässä HbA1c-arvon keskimääräinen muutos lähtötilanteesta oli ‑1,52 % dapagliflotsiinia saaneiden ryhmässä (n = 9) ja 0,17 % lumelääkeryhmässä (n = 6). Teho ja turvallisuus olivat samankaltaiset kuin dapagliflotsiinia saaneilla aikuispotilailla on todettu. Turvallisuus ja siedettävyys vahvistettiin tarkemmin tutkimuksen 28 viikon pituisessa turvallisuutta koskevassa jatkovaiheessa.
Sydämen vajaatoiminta ja krooninen munuaistauti
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset dapagliflotsiinin käytöstä sydän- ja verisuonitapahtumien ehkäisyssä sydämen kroonista vajaatoimintaa sairastavilla potilailla ja kroonisen munuaistaudin hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Dapagliflotsiini imeytyi nopeasti ja tehokkaasti suun kautta annettuna. Dapagliflotsiinin huippupitoisuus plasmassa (Cmax) saavutettiin paastotilassa yleensä kahdessa tunnissa lääkkeen antamisen jälkeen. Dapagliflotsiinin geometrinen vakaan tilan Cmax-keskiarvo oli 158 ng/ml ja AUCτ-keskiarvo oli 628 ng h/ml kerran vuorokaudessa otetun 10 mg dapagliflotsiiniannoksen jälkeen. Suun kautta otetun 10 mg:n dapagliflotsiiniannoksen absoluuttinen hyötyosuus oli 78 %. Runsasrasvaisen aterian nauttiminen vähensi dapagliflotsiinin Cmax-arvoa enintään 50 % ja pidensi Tmax-arvoa noin yhdellä tunnilla, mutta vaikutus AUC-arvoon ei eronnut paastotilassa saadusta arvosta. Näitä muutoksia ei pidetä kliinisesti merkitsevinä. Forxiga voidaan siten ottaa joko ruoan kanssa tai ilman.
Jakautuminen
Dapagliflotsiini sitoutuu proteiineihin noin 91-prosenttisesti. Sitoutuminen proteiiniin ei vaihdellut eri sairaustiloissa (esim. munuaisen tai maksan vajaatoiminta). Dapagliflotsiinin keskimääräinen jakautumistilavuus vakaassa tilassa oli 118 litraa.
Biotransformaatio
Dapagliflotsiini metaboloituu suuressa määrin, ja sen päämetaboliitti on inaktiivinen dapagliflotsiini-3‑O‑glukuronidi. Dapagliflotsiini-3‑O‑glukuronidilla tai muilla metaboliiteilla ei ole glukoosia alentavaa vaikutusta. Dapagliflotsiini-3‑O‑glukuronidin muodostumiseen vaikuttaa UGT1A9-entsyymi, jota esiintyy maksassa ja munuaisissa. CYP‑välitteinen metabolia oli ihmisillä vähäisempi puhdistumareitti.
Eliminaatio
Dapagliflotsiinin keskimääräinen terminaalinen puoliintumisaika (t1/2) plasmassa oli terveillä tutkittavilla 12,9 tuntia yhden suun kautta otetun dapagliflotsiinin 10 mg:n annoksen jälkeen. Laskimoon annetun dapagliflotsiinin keskimääräinen systeeminen kokonaispuhdistuma oli 207 ml/min. Dapagliflotsiini ja sen metaboliitit poistuvat pääasiassa erittymällä virtsaan, jolloin alle 2 % erittyneestä lääkeaineesta on metaboloitumatonta dapagliflotsiinia. 50 mg:n [14C]‑dapagliflotsiiniannoksesta 96 % voitiin jäljittää: 75 % virtsasta ja 21 % ulosteista. Noin 15 % annoksesta erittyi ulosteeseen metaboloitumattomana lääkkeenä.
Lineaarisuus
Dapagliflotsiinialtistus suureni dapagliflotsiiniannoksen mukaisesti alueella 0,1–500 mg, eikä sen farmakokinetiikka muuttunut ajan myötä, kun toistuvia vuorokausiannoksia jatkettiin viikolle 24.
Erityisryhmät
Munuaisten vajaatoiminta
Vakaassa tilassa (20 mg dapagliflotsiinia kerran päivässä 7 päivän ajan) olevilla tutkittavilla, jotka sairastivat tyypin 2 diabetesta ja lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa (plasman ioheksolipuhdistumalla määritettynä), vastaava keskimääräinen systeeminen dapagliflotsiinialtistuma oli 32 %, 60 % ja 87 % suurempi kuin tutkittavilla, joilla oli tyypin 2 diabetes ja normaali munuaisten toiminta. Vakaassa tilassa glukoosin eritys virtsaan 24 tunnin aikana riippui voimakkaasti munuaisten toiminnasta: tyypin 2 diabetesta sairastavilla tutkittavilla glukoosia erittyi päivässä 85 g normaalissa munuaisten toiminnassa, 52 g lievässä vajaatoiminnassa, 18 g keskivaikeassa vajaatoiminnassa ja 11 g vaikeassa vajaatoiminnassa. Hemodialyysin vaikutusta dapagliflotsiinialtistukseen ei tunneta. Heikentyneen munuaisten toiminnan vaikutusta systeemiseen altistukseen arvioitiin populaatiofarmakokineettisessa mallissa. Aiempien tulosten mukaisesti mallin ennustama AUC-arvo oli suurempi potilailla, joilla oli krooninen munuaistauti, kuin potilailla, joilla oli normaali munuaisten toiminta. Mallin ennustama AUC-arvo ei eronnut merkittävästi kroonista munuaistautia ja tyypin 2 diabetesta sairastavilla potilailla ja kroonista munuaistautia sairastavilla potilailla, joilla ei ollut diabetesta.
Maksan vajaatoiminta
Tutkittavilla, joilla oli lievä tai keskivaikea maksan vajaatoiminta (Child-Pugh-luokat A ja B), dapagliflotsiinin Cmax-keskiarvo oli enintään 12 % suurempi ja AUC-keskiarvo oli enintään 36 % suurempi kuin terveillä verrokkihenkilöillä. Näitä muutoksia ei pidetty kliinisesti merkitsevinä. Vaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla (Child‑Pugh-luokka C) dapagliflotsiinin Cmax-keskiarvo oli 40 % suurempi ja AUC-keskiarvo oli 67 % suurempi kuin terveillä verrokkihenkilöillä.
Iäkkäät (vähintään 65‑vuotiaat)
Alle 70‑vuotiailla tutkittavilla altistus ei lisääntynyt kliinisesti merkittävästi pelkästään iän perusteella. Altistumisen voidaan kuitenkin olettaa lisääntyvän ikään liittyvän munuaistoiminnan heikkenemisen vuoksi. Tutkimustuloksia ei ole riittävästi, jotta voitaisiin tehdä johtopäätöksiä yli 70‑vuotiaiden potilaiden altistuksesta.
Pediatriset potilaat
Tyypin 2 diabetesta sairastavilla 10–17‑vuotiailla lapsilla farmakokinetiikka ja farmakodynamiikka (glukosuria) olivat samankaltaiset kuin tyypin 2 diabetesta sairastavilla aikuisilla on todettu.
Sukupuoli
Dapagliflotsiinin AUCss-keskiarvon arvioitiin olevan naisilla noin 22 % korkeampi kuin miehillä.
Rotu
Systeemisessä altistumisessa ei ollut kliinisesti merkitseviä eroja valkoisten, mustien ja aasialaisten välillä.
Paino
Dapagliflotsiinialtistuksen havaittiin vähenevän painon lisääntyessä. Täten hoikkien potilaiden altistuminen saattaa olla hieman lisääntynyt ja tavallista painavampien potilaiden altistuminen hieman vähentynyt. Näitä eroja ei kuitenkaan pidetty kliinisesti merkitsevinä.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, karsinogeenisuutta sekä hedelmällisyyttä koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Dapagliflotsiini ei aiheuttanut kasvaimia hiirille tai rotille millään tutkitulla annoksella kahden vuoden kestoisessa karsinogeenisuustutkimuksessa.
Lisääntymis- ja kehitystoksisuus
Dapagliflotsiinin suora antaminen vastavieroitetuille rotanpoikasille ja epäsuora altistus myöhäisen tiineyden (aikana, joka vastaa raskauden toista ja kolmatta kolmannesta ihmisen munuaisten kehittymisen suhteen) ja imetyksen aikana johtivat jälkeläisten munuaisaltaan ja munuaistiehyeiden laajenemisen esiintymistiheyden ja/tai vaikeusasteeseen kasvamiseen.
Nuorten eläinten toksisuustutkimuksessa dapagliflotsiinia annettiin suoraan rotanpoikasille syntymän jälkeisestä päivästä 21 päivään 90 saakka. Munuaisaltaan ja munuaistiehyeiden laajenemista raportoitiin kaikilla annoksilla. Poikasten altistukset pienimmällä testatulla annoksella olivat ≥ 15 kertaa ihmisille suositeltuja maksimiannoksia isommat. Näihin löydöksiin liittyi annosriippuvaista munuaisen painon kasvua ja makroskooppista munuaisten suurenemista, jota havaittiin kaikilla annoksilla. Nuorilla eläimillä havaitut munuaisaltaan ja -tiehyeiden laajenemiset eivät täysin palautuneet noin yhden kuukauden mittaisen palautumisjakson aikana.
Erillisessä syntymää edeltävää ja syntymän jälkeistä kehitystä arvioineessa tutkimuksessa emorotille annettiin tutkimuslääkettä kuudennesta tiineyspäivästä 21. synnytyksen jälkeiseen päivään, ja poikaset altistuivat epäsuorasti in utero ja koko imetyksen ajan. (Satelliittitutkimuksessa arvioitiin dapagliflotsiinialtistusta maidossa ja poikasissa.) Munuaisaltaan laajenemisen esiintymistiheyden tai vaikeusasteen kasvua havaittiin tutkimuslääkettä saaneiden emojen aikuisissa jälkeläisissä, kuitenkin vain suurimmilla testatuilla annoksilla. (Emon dapagliflotsiinialtistukset olivat 1 415 kertaa ja poikasten altistukset 137 kertaa suurempia kuin ihmisen altistus ihmiselle suositellulla maksimiannoksella. Kehitykseen kohdistunut lisätoksisuus rajoittui poikasten annoksesta riippuvaiseen painon laskuun, ja sitä havaittiin vain annoksilla ≥ 15 mg/kg/vrk (poikasten altistuksen ollessa ≥ 29 kertaa ihmisen altistus ihmiselle suositellulla maksimiannoksella). Maternaalinen toksisuus oli ilmeistä vain suurimmalla testatulla annoksella ja rajoittui ohimenevään painonlaskuun ja ruoankulutuksen vähenemiseen. Kehitystoksisuuden suurin haitaton vaikutustaso (NOAEL), pienin testattu annos, liittyy emon systeemiseen altistukseen, joka on noin 19-kertainen verrattuna ihmisen altistukseen ihmiselle suositellulla maksimiannoksella.
Rotilla ja kaniineilla tehdyissä alkion ja sikiönkehitystä selvittäneissä lisätutkimuksissa dapagliflotsiinia annettiin kyseisten lajien organogeneesin tärkeimpinä kehitysjaksoina. Kaniineilla ei havaittu maternaalista toksisuutta tai kehitystoksisuutta millään testatulla annoksella; suurimpaan testattuun annokseen liittyy systeeminen altistus, joka on noin 1 191 kertaa ihmiselle suositeltu maksimiannos. Rotilla dapagliflotsiini ei ollut alkioletaalinen eikä teratogeeninen altistuksella, joka oli 1 441 kertaa ihmiselle suositeltava maksimiannos.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa (E460i)
Laktoosi
Krospovidoni (E1202)
Piidioksidi (E551)
Magnesiumstearaatti (E470b)
Kalvopäällyste
Polyvinyylialkoholi (E1203)
Titaanidioksidi (E171)
Makrogoli 3350 (E1521)
Talkki (E553b)
Keltainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
FORXIGA tabletti, kalvopäällysteinen
5 mg (L:kyllä) 28 fol (51,19 €), 98 fol (151,74 €)
10 mg (L:kyllä) 28 fol (51,19 €), 98 fol (151,74 €)
PF-selosteen tieto
Alumiini/Alumiini-läpipainopakkaus.
Forxiga 5 mg kalvopäällysteiset tabletit
14, 28 tai 98 kalvopäällysteistä tablettia repäisyviivattomissa kalenteriläpipainopakkauksissa.
30 x 1 tai 90 x 1 kalvopäällysteistä tablettia repäisyviivallisissa kerta-annosläpipainopakkauksissa.
Forxiga 10 mg kalvopäällysteiset tabletit
14, 28 tai 98 kalvopäällysteistä tablettia repäisyviivattomissa kalenteriläpipainopakkauksissa.
10 x 1, 30 x 1 tai 90 x 1 kalvopäällysteistä tablettia repäisyviivallisissa kerta-annosläpipainopakkauksissa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Forxiga 5 mg kalvopäällysteiset tabletit
Keltainen, kaksoiskupera, halkaisijaltaan 0,7 cm:n suuruinen pyöreä kalvopäällysteinen tabletti, jonka toisella puolella on merkintä ”5” ja toisella puolella merkintä ”1427”.
Forxiga 10 mg kalvopäällysteiset tabletit
Keltainen, kaksoiskupera, suunnilleen 1,1 x 0,8 cm:n suuruinen viistosti vinoneliönmuotoinen kalvopäällysteinen tabletti, jonka toisella puolella on merkintä ”10” ja toisella puolella merkintä ”1428”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
FORXIGA tabletti, kalvopäällysteinen
10 mg 28 fol, 98 fol
- Alempi erityiskorvaus (65 %). Diabetes, muu kuin insuliinihoito (215), Dapagliflotsiini ja empagliflotsiini: Kroonisen sydämen vajaatoiminnan hoito erityisin edellytyksin (250).
- Peruskorvaus (40 %).
FORXIGA tabletti, kalvopäällysteinen
5 mg 28 fol, 98 fol
- Alempi erityiskorvaus (65 %). Diabetes, muu kuin insuliinihoito (215).
- Peruskorvaus (40 %).
ATC-koodi
A10BK01
Valmisteyhteenvedon muuttamispäivämäärä
12.03.2026
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi