
PROCREN DEPOT PDS pulver och vätska till injektionsvätska, suspension i förfylld spruta 3,75 mg, 11,25 mg, 30 mg
Kvalitativ och kvantitativ sammansättning
Leuprorelinacetat 3,75 mg, 11,25 mg respektive 30 mg
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Pulver och vätska till injektionsvätska, suspension i förfylld spruta.
Kliniska uppgifter
Terapeutiska indikationer
Män 3,75 mg, 11,25 mg och 30 mg: Långt framskriden prostatacancer, när orkiektomi inte är indicerad.
Kvinnor 3,75 mg och 11,25 mg: Behandling av svår endometrios. Behandling av bröstcancer hos pre- och perimenopausala kvinnor, hos vilka hormonbehandling är indicerad. 3,75 mg: Minskning av storleken på uterusmyom (leiomyoma uteri).
Barn 3,75 mg och 11,25 mg: Behandling av central pubertas praecox (flickor under 9 år, pojkar under 10 år).
Dosering och administreringssätt
Dosering
Prostatacancer: Den rekommenderade dosen för subkutan injektion är en enkeldos om 3,75 mg var 4:e vecka, 11,25 mg var 12:e vecka eller 30 mg var 6:e månad. Behandlingen bör inte avbrytas vid remission eller förbättrat terapisvar.
Hos patienter som behandlas med GnRH‑analoger för prostatacancer brukar behandling fortgå efter utveckling av kastrationsresistent prostatacancer. Hänvisning bör göras till relevanta riktlinjer.
Endometrios: Den rekommenderade dosen för subkutan injektion är en enkeldos om 3,75 mg var 4:e vecka eller 11,25 mg var 12:e vecka i högst sex (6) månader.
Tillägg av komplementär hormonbehandling (5 mg noretisteronacetat per dag) till behandlingen med Procren Depot PDS vid endometrios har visat sig minska osteoporos och vasomotoriska symtom. Om komplementär hormonbehandling sätts in tillsammans med Procren Depot PDS bör nyttan och riskerna med respektive behandling beaktas separat. Denna kombinationsbehandling kan fortgå i högst sex (6) månader.
Uterusmyom: Den rekommenderade dosen för subkutan injektion är en enkeldos om 3,75 mg var 4:e vecka i högst tre (3) månader.
Bröstcancer: Den rekommenderade dosen är en enkeldos om 3,75 mg var 4:e vecka eller 11,25 mg var 12:e vecka.
Pediatrisk population
Behandling med leuprorelinacetat hos barn ska i sin helhet ske under överinseende av en barnendokrinolog.
Doseringen bör anpassas individuellt.
Den rekommenderade startdosen beror på kroppsvikten.
Barn med en kroppsvikt ≥ 20 kg
1 ml (3,75 mg leuprorelinacetat) suspension, innehållande 44,1 mg mikrokapslar med långsam frisättning av läkemedlet i 1 ml vehikellösning, administreras som en enkeldos en gång i månaden som subkutan injektion.
1 ml (11,25 mg leuprorelinacetat) suspension, innehållande 130,0 mg mikrokapslar med långsam frisättning av läkemedlet i 1 ml vehikellösning, administreras som en enkeldos var 3:e månad som subkutan injektion.
Barn med en kroppsvikt < 20 kg
I dessa sällsynta fall används följande dosering utgående från den kliniska aktiviteten vid central pubertas praecox:
0,5 ml (1,88 mg leuprorelinacetat) administreras som en enkeldos en gång i månaden som subkutan injektion eller
0,5 ml (5,625 mg leuprorelinacetat) administreras som en enkeldos var 3:e månad som subkutan injektion.
Överbliven suspension bör kasseras. Barnets viktökning bör följas.
Beroende på den kliniska aktiviteten avseende central pubertas praecox kan det vara nödvändigt att öka dosen vid otillräcklig suppression (klinisk evidens t.ex. stänkblödningar eller otillräcklig hämning av gonadotropinutsöndring i LHRH‑test). Den lägsta effektiva dosen som ska administreras en gång i månaden eller var 3:e månad bör då bestämmas med hjälp av LHRH‑testet.
Vid injektionsstället uppkom ofta sterila abscesser när leuprorelinacetat administrerades intramuskulärt i högre doser än de rekommenderade. Därför bör läkemedlet i sådana här fall administreras subkutant (se avsnitt Varningar och försiktighet).
Minsta möjliga injektionsvolym rekommenderas vid administrering intramuskulärt/subkutant hos barn för att minska obehaget relaterat till injektionen.
Behandlingslängden beror på kliniska parametrar vid behandlingsstart eller under behandlingen (prognos på slutlängd, tillväxthastighet, skelettålder och/eller accelererad skelettålder) och beslutas av behandlande barnläkare tillsammans med vårdnadshavaren och, om möjligt, det behandlade barnet. Skelettåldern bör följas under behandlingen med 6–12 månaders intervall. För flickor vars skelettålder är över 12 år och pojkar vars skelettålder är över 13 år bör utsättande av behandlingen övervägas med hänsyn till de kliniska parametrarna.
Hos flickor bör graviditet uteslutas innan behandlingen inleds. Förekomst av graviditet under behandlingen kan generellt inte uteslutas. I sådana fall ska läkare kontaktas.
Observera:
För att förhindra återfall av symtom på pubertas praecox bör administreringsintervallet vid användning av Procren Depot PDS 3,75 mg vara 30 ± 2 dagar.
För att förhindra återfall av symtom på pubertas praecox bör administreringsintervallet vid användning av Procren Depot PDS 11,25 mg vara 90 ± 2 dagar.
Administreringssätt
Procren Depot PDS bör förberedas, beredas och administreras endast av hälso- och sjukvårdspersonal som är förtrogna med dessa rutiner.
Kontraindikationer
Överkänslighet mot leuprorelinacetat, liknande nonapeptider eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen (se avsnitt Varningar och försiktighet).
Leuprorelinacetat är kontraindicerat för kvinnor som är gravida eller kan bli gravida under behandlingen (se avsnitten Fertilitet, graviditet och amning och Prekliniska säkerhetsuppgifter).
Leuprorelinacetat ska inte ges till kvinnor med odiagnostiserad vaginalblödning.
Hos flickor med central pubertas praecox:
- Graviditet och amning
- Odiagnostiserad vaginalblödning.
Varningar och försiktighet
Alla patientgrupper
Enstaka fall av anafylaxi har rapporterats i samband med användning av leuprorelinacetatpreparat som administreras månatligen. Patienternas tillstånd bör därför observeras noga efter administrering av läkemedlet med avseende på eventuella överkänslighetsreaktioner. Om en anafylaktisk reaktion eller annan allvarlig allergisk reaktion uppkommer bör användningen av Procren Depot PDS avbrytas omedelbart och lämplig behandling sättas in (se avsnitten Kontraindikationer och Biverkningar).
I början av behandlingen ökar koncentrationerna av gonadotropin och könssteroider så att de överstiger koncentrationerna vid utgångsläget, vilket beror på läkemedlets naturliga stimulerande effekt. Därför kan en ökning av kliniska fynd och symtom iakttas (se avsnitt Farmakodynamiska egenskaper). Förvärringen av symtomen kan bidra till uppkomst av paralys. Den kan eventuellt vara associerad med komplikationer som leder till dödsfall (se även avsnitt Biverkningar).
Det finns en ökad risk för depression (som kan vara allvarlig) hos patienter som får behandling med GnRH‑agonister, som leuprorelin. Patienterna bör informeras om detta och få lämplig behandling om symtom uppkommer.
Bentäthet
Det kan förekomma förändringar i bentätheten i samband med vilka hypoöstrogena tillstånd som helst hos kvinnor samt när preparatet används under lång tid för behandling av prostatacancer hos män. Det finns inga data om huruvida tillståndet är reversibelt hos män efter att behandlingen med leuprorelinacetat avslutas. Hos kvinnor kan minskningen av bentätheten vara reversibel efter att behandlingen med leuprorelinacetat avslutats.
Konvulsioner
Efter godkännandet för försäljning har det förekommit rapporter om konvulsioner i samband med användning av GnRH‑agonister (inklusive leuprorelinacetat). Sådana har förekommit hos kvinnor och barn, hos patienter med krampanfall, epilepsi, rubbningar i hjärncirkulationen och avvikelser eller tumörer i CNS i anamnesen samt hos patienter med samtidig medicinering som kan disponera för konvulsioner, t.ex. bupropion eller SSRI‑läkemedel. Konvulsioner har även rapporterats hos patienter som inte har någon av ovannämnda faktorer.
Idiopatisk intrakraniell hypertension
Idiopatisk intrakraniell hypertension (pseudotumor cerebri) har rapporterats hos patienter som fått leuprorelin. Patienterna ska varnas om tecken och symtom på idiopatisk intrakraniell hypertension, däribland svår eller återkommande huvudvärk, synstörningar och tinnitus. Om idiopatisk intrakraniell hypertension uppstår ska utsättning av leuprorelin övervägas.
Mycket allvarliga hudbiverkningar
Mycket allvarliga hudbiverkningar (SCARs), inklusive Stevens-Johnsons syndrom (SJS) och toxisk epidermal nekrolys (TEN) som kan vara livshotande eller dödliga, har rapporterats i samband med behandling med leuprorelin. Vid förskrivningstillfället ska patienten informeras om tecken och symtom och ska övervakas noga för allvarliga hudreaktioner. Om tecken och symtom som tyder på dessa reaktioner uppstår ska leuprorelin omedelbart sättas ut och en alternativ behandling ska övervägas (enligt vad som är lämpligt).
Män
”Flare reaktion”
Under den första behandlingsveckan höjer leuprorelinacetat, liksom andra LHRH‑agonister, serumtestosteron med ca 50 % från utgångsläget. Övergående förvärring eller ökning av symtomen kan ibland förekomma under de första behandlingsveckorna, vilket kan förebyggas genom samtidig behandling med en antiandrogen. Ett litet antal patienter kan uppleva en temporär förvärring av osteodyni, som kan behandlas symtomatiskt. Liksom med andra LHRH‑analoger har enstaka fall av urinvägsobstruktion samt enstaka fall av kompression av ryggradskanalen som kunnat leda till paralys rapporterats med leuprorelinacetat. Därför ska patienter med urinvägsobstruktion eller metastaser i ryggraden följas noggrant under de första behandlingsveckorna.
Metabola förändringar
Användning av androgen deprivationsterapi, inklusive GnRH-agonister, kan vara associerad med en ökad risk för metabola förändringar såsom hyperglykemi, diabetes, hyperlipidemi och fettlever. Hyperglykemi kan bero på utveckling av diabetes eller försämrad glykemisk kontroll hos patienter med diabetes. Patienter med ökad risk bör övervakas med avseende på tecken och symtom på metabolt syndrom inklusive lipider, blodglukos och/eller glykosylerat hemoglobin (HbA1c) ska kontrolleras, och behandlas enligt gällande kliniska riktlinjer (se avsnitt Biverkningar).
Kardiovaskulär sjukdom
Ökad risk för hjärtinfarkt, plötslig hjärtdöd och stroke har rapporterats hos män i samband med användning av GnRH‑agonister. Risken verkar vara låg baserat på rapporterade oddskvoter, och den ska i likhet med kardiovaskulära riskfaktorer utvärderas noggrant i samband med valet av behandling för patienter med prostatacancer. Patienter som får GnRH‑agonister ska kontrolleras för symtom och fynd som tyder på utvecklande av kardiovaskulär sjukdom. I behandlingen ska gällande klinisk praxis följas.
Androgendeprivationsterapi kan förlänga QT‑intervallet
För patienter med konstaterad QT‑förlängning eller riskfaktorer för QT‑förlängning samt för patienter som samtidigt får behandling med andra läkemedel som kan förlänga QT‑intervallet (se avsnitt Interaktioner) bör förskrivare bedöma nytta‑riskbalansen inklusive risken för torsades de pointes innan behandling med Procren Depot PDS inleds.
Laboratorieprover
Responsen på behandlingen med leuprorelinacetat bör följas genom bestämning av testosteronnivån och PSA (prostataspecifikt antigen) i serum. Hos de flesta patienterna ökade testosteronnivåerna så att de översteg nivåerna vid utgångsläget under den första veckan och minskade sedan till utgångsnivån eller lägre före slutet av den andra behandlingsveckan. Koncentrationen nådde kastrationsnivå inom 2–4 veckor, och när denna koncentration hade nåtts kvarstod den så länge patienterna fick injektionerna enligt tidtabellen.
En övergående ökning av nivåerna av enzymet surt fosfatas kan förekomma hos vissa patienter i ett tidigt skede av behandlingen, men värdena återgår vanligen till normal eller nästan normal nivå inom en månad efter att behandlingen inletts.
I slutet av den sex månader långa behandlingsperioden är det bra att mäta testosteronnivån hos de patienter som inte tidigare fått behandling med LHRH‑analoger.
Kvinnor
I likhet med andra LHRH‑analoger orsakar Procren Depot PDS en reversibel ökning av östradiolnivåerna i början av behandlingen. Detta kan hos vissa patienter vara förenat med symtom som vanligen försvinner när behandlingen fortgår. I samband med submukös leiomyoma uteri har det rapporterats om fall av kraftig vaginalblödning där det krävts ett kirurgiskt ingrepp eller läkemedelsbehandling för att stoppa blödningen.
På grund av låg östrogennivå kan det ske förändringar i bentätheten, vilka kan gå tillbaka efter att behandlingen med leuprorelin avslutats. Vid behandling av endometrios med Procren Depot PDS som monoterapi eller i kombination med komplementär hormonell behandling bör behandlingsperiodens längd begränsas till sex (6) månader. Eftersom leuprorelin kan främja osteoporos bör behandlingen av endometrios upprepas endast efter noggrant övervägande.
Barn
Innan behandlingsstart bör en exakt diagnos av idiopatisk och/eller neurogen central pubertas praecox ställas.
Detta är en långtidsbehandling som anpassas individuellt. Procren Depot PDS 3,75 mg ges så exakt och regelbundet som möjligt en gång i månaden och Procren Depot PDS 11,25 mg ges så exakt och regelbundet som möjligt var 3:e månad. Fördröjning av injektionsdatum i undantagsfall och med några dagar (Procren Depot PDS 3,75 mg: 30 ± 2 dagar eller Procren Depot PDS 11,25 mg: 90 ± 2 dagar) påverkar inte behandlingsresultatet.
I händelse av att sterila abscesser uppstår vid injektionsstället (oftast rapporterat efter intramuskulär injektion med högre doser än rekommenderat) kan absorptionen av leuprorelinacetat från depotpreparatet minska. I dessa fall bör hormonella parametrar (testosteron, östradiol) kontrolleras varannan vecka (se avsnitt Dosering och administreringssätt).
Om barnet har en progressiv hjärntumör bör behandling sättas in först efter en noggrann individuell bedömning av risker och nytta.
Hos flickor kan vaginala blödningar, stänkblödningar och flytningar förekomma efter den första injektionen som ett tecken på hormonnedreglering. Vaginal blödning efter den första/andra månaden av behandlingen behöver utredas.
Bentätheten kan minska under GnRH‑behandling av central pubertas praecox. Tillväxten av benmassa återställs emellertid efter avslutad behandling, och maximal benmassa i senare tonåren verkar inte påverkas av behandlingen.
Efter avslutad GnRH‑behandling kan förskjuten femurepifys förekomma. Förklaringen kan vara att de låga koncentrationerna av östrogen under behandling med GnRH‑agonister försvagar epifysbrosket. Ökningen i tillväxthastighet som sker efter avslutad behandling resulterar i en minskning av den skjuvkraft som krävs för att förskjuta epifysen.
Om läkemedlet inte används i enlighet med vårdprogrammet eller om för låga doser används kan den pubertala utvecklingsprocessen eventuellt inte hållas tillräckligt väl under kontroll. Då kan följderna bl.a. vara att tecknen på pubertet återvänder, t.ex. i form av menstruation, bröstutveckling och testikeltillväxt. De långvariga följderna av otillräcklig reglering av könssteroidutsöndringen är okända, men de kan bl.a. innefatta en risk för att slutlängden blir kortare än förväntat.
Laboratorieprover
Skelettåldern bör mätas med 6–12 månaders intervall för att följa med utvecklingen.
Koncentrationen av könssteroider kan öka eller nå nivåer som överstiger prepubertala koncentrationer om dosen inte är tillräckligt hög. När behandlingsdosen har fastställts minskar koncentrationerna av gonadotropin och könssteroider till prepubertal nivå.
Interaktioner
Inga interaktionsstudier har utförts. Eftersom leuprorelinacetat är en peptid, i huvudsak bryts ner genom inverkan av peptidas och har en bindningsgrad till plasmaproteiner på ca 46 % förväntas inga interaktioner med andra läkemedel uppkomma.
Prostatacancer
Eftersom androgendeprivationsterapi kan förlänga QT‑intervallet bör en noggrann övervägning göras före samtidig användning av Procren Depot PDS och läkemedel som förlänger QT‑intervallet eller kan framkalla torsades de pointes, såsom klass IA antiarytmika (t.ex. kinidin, disopyramid) eller klass III antiarytmika (t.ex. amiodaron, sotalol, dofetilid, ibutilid), metadon, moxifloxacin, antipsykotika m.fl. (se avsnitt Varningar och försiktighet, Män, Androgendeprivationsterapi kan förlänga QT-intervallet).
Hos kvinnor orsakar administrering av depotformen av leuprorelinacetat suppression av hypofysen. Funktionen återgår till tidigare nivå inom tre månader efter att behandlingen avslutats. Därför kan testresultaten vid mätning av gonadotropinutsöndringen från hypofysen samt hormonkoncentrationerna vara missvisande ännu tre månader efter att behandlingen med leuprorelinacetat avslutats.
Fertilitet, graviditet och amning
Graviditet
Procren Depot PDS ska inte användas under graviditet eller amning (se avsnitt Kontraindikationer). Säkerheten av leuprorelinacetat under graviditet har inte påvisats kliniskt. Innan behandling med leuprorelinacetat inleds bör graviditet uteslutas. Leuprorelinacetat är inte ett preventivmedel. Om antikonception behövs ska en icke‑hormonell metod användas.
Fertilitet
I studier med leuprorelinacetat och andra motsvarande analoger har fertiliteten visats återgå till det normala efter utsättning av läkemedlet då behandlingen pågått i upp till 24 månader.
Pediatrisk population:
Se avsnitt Kontraindikationer.
Effekter på förmågan att framföra fordon och använda maskiner
Ingen effekt på förmågan att framföra fordon eller använda maskiner har observerats.
Biverkningar
Vanliga (≥ 1/100, < 1/10)
Mindre vanliga (≥ 1/ 1 000, < 1/100)
Sällsynta (≥ 1/10 000, < 1/1 000)
Mycket sällsynta (< 1/10 000)
Ingen känd frekvens (kan inte beräknas från tillgängliga data).
| Sammandrag av biverkningar relaterade till läkemedlet hos kvinnor | ||
| Organsystem | Frekvens | Biverkningar |
| Infektioner och infestationer | Vanliga | Vaginit |
| Psykiska störningar | Vanliga | Depression Nedsatt libido |
| Reproduktionsorgan och bröstkörtel | Vanliga | Smärta i brösten Ömhet i brösten Minskad bröststorlek Vaginal torrhet |
| Sällsynta | Dyspareuni | |
| Allmänna symtom och/eller symtom vid administreringsstället | Sällsynta | Irritation |
Allvarliga fall av venösa och arteriella tromboembolier, inklusive djup ventrombos, lungemboli, hjärtinfarkt, stroke och transitorisk ischemisk attack (TIA), har rapporterats. I vissa fall har ett tidsmässigt samband mellan händelsen och behandlingen rapporterats. Bedömningen av de flesta fallen försvåras dock av vilseledande riskfaktorer eller samtidig användning av andra läkemedel. Det är oklart om det finns ett orsakssamband mellan användning av GnRH‑agonister och dessa händelser.
I uppföljningen efter godkännandet för försäljning har allvarlig leverskada konstaterats hos kvinnor. Det finns inga data om prevalensen av dessa fall.
| Sammandrag av biverkningar relaterade till läkemedlet hos män | ||
| Organsystem | Frekvens | Biverkningar |
| Psykiska störningar | Sällsynta | Nedsatt libido |
| Lever och gallvägar | Ingen känd frekvens | Fettlever |
| Reproduktionsorgan och bröstkörtel | Vanliga | Testisatrofi Erektil dysfunktion |
| Sällsynta | Gynekomasti | |
| Undersökningar | Ingen känd frekvens | QT‑förlängning (se avsnitten Varningar och försiktighet och Interaktioner) |
I uppföljningen efter godkännandet för försäljning har allvarlig leverskada konstaterats hos män. Det finns inga data om prevalensen av dessa fall.
| Sammandrag av biverkningar relaterade till läkemedlet hos barn | ||
| Organsystem | Frekvens | Biverkningar |
| Immunsystemet | Mycket sällsynta | Omfattande allergiska reaktioner (feber, utslag, t.ex. pruritus, anafylaktisk reaktion) |
| Psykiska störningar | Vanliga | Humörsvängningar |
| Centrala och perifera nervsystemet | Vanliga | Huvudvärk* |
| Magtarmkanalen | Vanliga | Buksmärta/bukkramper Illamående/kräkningar |
| Hud och subkutan vävnad | Vanliga | Akne |
| Reproduktionsorgan och bröstkörtel | Vanliga | Vaginalblödning** Stänkblödning Flytningar |
| Allmänna symtom och/eller symtom vid administreringsstället | Vanliga | Reaktioner vid injektionsstället |
*Liksom vid användning av andra läkemedel i denna grupp har hypofysär apoplexi rapporterats i mycket sällsynta fall efter initial administrering till patienter med hypofysadenom.
**Observera: I allmänhet bör förekomst av stänkblödningar vid fortsatt behandling (efter eventuell bortfallsblödning under den första behandlingsmånaden) bedömas som ett tecken på potentiell underdosering. Suppressionen av hypofysen bör i sådana fall bestämmas genom ett LHRH‑test.
I början av behandlingen ökar koncentrationerna av könshormoner kortvarigt, varefter de minskar till prepubertal nivå. På grund av denna farmakologiska effekt kan biverkningar uppträda särskilt i början av behandlingen.
Psykiska biverkningar
Psykiska biverkningar har rapporterats hos patienter som använder GnRH‑agonister. Efter godkännandet för försäljning av denna läkemedelsgrupp har symtom relaterade till humörsvängningar rapporterats, t.ex. gråt, irritabilitet, otålighet, ilska och aggressivitet. Ett säkert orsakssamband mellan behandling med GnRH‑agonister och förekomsten av ovannämnda biverkningar har inte kunnat påvisas. Utveckling eller förvärring av psykiska symtom under behandling med leuprorelinacetat bör övervakas.
| Sammandrag av biverkningar relaterade till läkemedlet – Allmänt | ||
| Organsystem | Frekvens | Biverkningar |
| Blodet och lymfsystemet | Sällsynta | Leukopeni Trombocytopeni Anemi Rikligt antal blodkroppar |
| Immunsystemet | Sällsynta | Anafylaktisk reaktion |
| Metabolism och nutrition | Sällsynta | Diabetes mellitus Hyperkalemi Aptitlöshet Ätstörning |
| Psykiska störningar | Vanliga | Humörsvängningar, depression (långvarig användning) |
| Mindre vanliga | Humörsvängningar, depression (kortvarig användning)* | |
| Sällsynta | Sömnlöshet | |
| Centrala och perifera nervsystemet | Vanliga | Huvudvärk Yrsel |
| Sällsynta | Parestesi Försämrat minne Konvulsioner | |
| Ingen känd frekvens | Idiopatisk intrakraniell hypertension (pseudotumor cerebri) (se avsnitt Varningar och försiktighet) | |
| Ögon | Sällsynta | Synrubbningar |
| Öron och balansorgan | Sällsynta | Hörselrubbningar Tinnitus |
| Hjärtat | Sällsynta | Angina pectoris Takykardi Rytmrubbningar |
| Blodkärl | Vanliga | Vallningar |
| Sällsynta | Ischemi Hypertension | |
| Andningsvägar, bröstkorg och mediastinum | Vanliga | Dyspné |
| Sällsynta | Pneumoni | |
| Ingen känd frekvens | Interstitiell lungsjukdom | |
| Magtarmkanalen | Vanliga | Kräkningar Illamående |
| Sällsynta | Diarré Förstoppning Stomatit Muntorrhet | |
| Lever och gallvägar | Sällsynta | Ikterus |
| Hud och subkutan vävnad | Vanliga | Utslag Akne Kraftiga svettningar |
| Sällsynta | Dermatit Pruritus Hirsutism Alopeci Nagelförändringar | |
| Ingen känd frekvens | Steven-Johnsons syndrom/toxisk epidermal nekrolys (SJS/TEN) (se avsnitt Varningar och försiktighet) Toxiskt hudutslag Erythema multiforme Blåsdermatos Exfoliativ dermatit | |
| Muskuloskeletala systemet och bindväv | Vanliga | Myalgi Artropati |
| Sällsynta | Muskuloskeletal smärta Dorsalgi Osteodyni Artralgi Muskelstelhet | |
| Njurar och urinvägar | Sällsynta | Hematuri Pollakisuri Urineringsstörningar |
| Allmänna symtom och/eller symtom vid administreringsstället | Vanliga | Smärta Ödem Kraftlöshet Trötthet Smärta vid injektionsstället, hematom, inflammation, steril abscess, förhårdnad och reaktion vid injektionsstället |
| Sällsynta | Tryckkänsla över bröstet Feber Frossa Törst | |
| Ingen känd frekvens | Nekros vid injektionsstället | |
| Undersökningar | Sällsynta | Förändringar i EKG Avvikelser i leverfunktionstest Förhöjda transaminasvärden Förhöjd nivå av alkaliskt fosfatas (AFOS) i blodet Förhöjda bilirubinnivåer i blodet Förhöjda ureanivåer i blodet Förhöjda urinsyranivåer i blodet Förhöjda nivåer av triglycerider i blodet Viktförändringar |
*Frekvensen kan vara högre med vissa preparat baserat på preparat- och indikationsspecifika kliniska prövningar.
Liksom med andra läkemedel i denna grupp har hypofysinfarkt rapporterats i mycket sällsynta fall i samband med initial administrering till patienter med hypofysadenom.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta‑riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till:
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Subkutana doser som, ställda i proportion till kroppsvikten, var ca 133 gånger högre än den rekommenderade dosen för människa orsakade dyspné, minskad vakenhetsgrad och lokal irritation vid injektionsstället hos råtta. För närvarande finns inga bevis på att detta också skulle gälla hos människa. I tidiga kliniska prövningar observerades ingen avvikande biverkningsprofil vid administrering av en hög daglig subkutan dos – upp till 20 mg/dygn – under högst två år jämfört med dosen 1 mg/dygn.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Endokrin terapi, gonadotropinfrisättande hormonanaloger, ATC‑kod: L02AE02
Procren Depot PDS innehåller leuprorelinacetat, som är en syntetisk nonapeptid, superanalog till det naturligt förekommande gonadotropinfrisättande hormonet LHRH. Leuprorelin är ca 70 gånger mer aktivt än naturligt LHRH. Vid tillförsel av LHRH‑analoger erhålls initialt en ökad frisättning av FSH och LH och därmed även av testosteron och dihydrotestosteron hos män samt östron och östradiol hos premenopausala kvinnor. Efter ca 2 veckor blockeras denna frisättning, med sänkt östrogen- och testosteronproduktion som följd. Då många prostatatumörer är androgenberoende hämmar denna behandling vanligen tumörtillväxt och orsakar atrofi av reproduktionsorganen. Suppression av spermatogenesen är en följdverkan av detta, men den är dock reversibel vid utsättning.
Hos kvinnor är tillväxten av vävnad vid de benigna gynekologiska sjukdomarna endometrios och uterusmyom östrogenberoende, och den minskade östrogenproduktionen leder till att sjukdomsförändringarna blir mindre och att symtomen minskar. Behandlingen leder till att menstruationsblödningarna uteblir hos de flesta patienterna så länge behandlingen pågår.
I kliniska prövningar har man observerat att patienter med metastaserande kastrationsresistent prostatacancer har nytta av att läkemedel såsom hämmare av den androgena hormoneffekten (abirateronacetat och enzalutamid), taxaner (docetaxel och kabazitaxel) och det radioaktiva läkemedlet Ra‑223 kombineras med GnRH‑agonister som leuprorelin.
Barn
Reversibel suppression av hypofysens utsöndring av gonadotropin sker, med en efterföljande minskning av östradiol- (E2) eller testosteronnivåerna till prepubertala nivåer.
Initial uppblossande stimulering av gonaderna (kortvarig ökning av koncentrationerna) kan orsaka vaginal blödning hos flickor vars menstruation börjat redan före behandlingsstart. Bortfallsblödning kan uppträda i början av behandlingen. Blödningen upphör normalt när behandlingen fortsätter.
Följande terapeutiska effekter kan påvisas:
- suppression av basala och stimulerade gonadotropinnivåer till prepubertala nivåer
- suppression av prematurt förhöjda könshormonnivåer till prepubertala nivåer samt avstannande av prematur menstruation
- avstannande/involution av somatisk pubertetsutveckling (Tannerstadier)
- förbättring/normalisering av förhållandet mellan kronologisk ålder och skelettålder
- förebyggande av progressiv accelererande skelettålder
- minskning och normalisering av tillväxthastighet
- ökning av slutlängd.
Behandlingsresultatet är hämning av den patologiskt, prematurt aktiverade hypotalamus‑hypofys‑gonadaxeln till en nivå som är i enlighet med den prepubertala åldern.
När barn i en klinisk långtidsstudie fick behandling med leuprorelin i doser om högst 15 mg per månad i > 4 år fortsatte utvecklingen av de pubertala dragen efter avslutad behandling. När 20 kvinnliga prövningsdeltagare följdes upp till vuxen ålder var menstruationscykeln normal hos 80 %, och hos 7 av 20 prövningsdeltagare konstaterades totalt 12 graviditeter, inklusive flerbördsgraviditeter hos 4 prövningsdeltagare.
Farmakokinetiska egenskaper
Biotillgängligheten för leuprorelinacetat efter subkutan administrering är jämförbar med den vid intramuskulär administrering. Den absoluta biotillgängligheten efter administrering av en dos på 7,5 mg har beräknats vara 90 %.
Hos patienter med prostatacancer var plasmakoncentrationen av leuprorelinacetat 0,7 ng/ml respektive 1,0 ng/ml en månad efter administrering av subkutana och intramuskulära enkeldoser om 3,75 mg respektive 7,5 mg. Det framkom inget som tydde på kumulation av läkemedlet. I en studie med kastrerade män bibehölls plasmakoncentrationen av leuprorelin i över en månad efter intramuskulär injektion med depotformen (7,5 mg).
Serumkoncentrationen av leuprorelinacetat mättes under 12 veckor efter administrering av en dos om 3,75 mg hos 11 premenopausala patienter med bröstcancer. Efter fyra veckor var koncentrationen över 0,1 ng/ml och den förblev stabil efter upprepade injektioner vid vecka 8 och 12. Det framkom inget som tydde på kumulation av läkemedlet.
Distributionsvolymen för leuprorelin efter en intravenös bolusinjektion hos manliga friska frivilliga var 27 l vid steady state. Bindningsgraden till plasmaproteiner in vitro varierade mellan 43 % och 49 %.
I en studie med manliga friska frivilliga var clearance 7,6 l/h efter administrering av en intravenös bolusinjektion på 1 mg, och den terminala halveringstiden var ca tre timmar i en tvåkompartmentmodell.
Djurstudier har visat att leuprorelinacetat metaboliseras till små inaktiva peptider – en pentapeptid (metabolit I), två tripeptider (metaboliterna II och III) och en dipeptid (metabolit IV) – som därefter genomgår ytterligare metabolism.
Plasmakoncentrationen av den största metaboliten (metabolit I) nådde sin högsta nivå (ca 6 % av det ursprungliga läkemedlets maximala koncentration) två till sex timmar efter administrering mätt hos fem patienter med prostatacancer efter administrering av depotformen av leuprorelinacetat. En vecka efter administrering var koncentrationen av metabolit I ca 20 % av medelkoncentrationen för leuprorelin.
Mindre än 5 % av en leuprorelinacetatdos på 3,75 mg som gavs till tre patienter utsöndrades i urinen som leuprorelinacetat och metabolit I under 27 dygn. Farmakokinetiken för leuprorelinacetat har inte undersökts hos patienter med lever- eller njursvikt.
Barn
Bild 1 visar serumnivåerna av leuprorelin efter administrering av en subkutan enkeldos (leuprorelinacetat i depotform, dosering på 30 mikrogram/kg). Maximala serumnivåer uppnås 60 minuter efter administrering (7,81 ± 3,59 ng/ml). AUC0‑672 är 105,78 ± 52,40 ng • timme/ml.
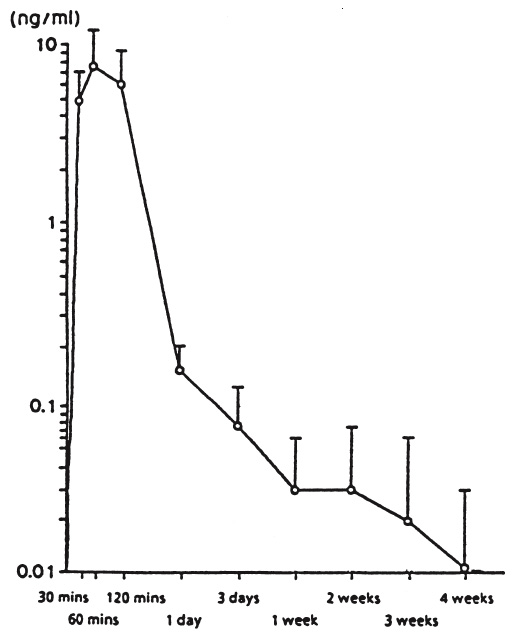
Bild 1: Serumnivåer av leuprorelin efter subkutan administrering av en enkeldos om 30 mikrogram/kg leuprorelinacetat i depotform (n = 6) (medelvärde ± standardavvikelse).
Bild 2 visar serumnivåerna av leuprorelin hos barn under de första 6 månaderna av behandlingen, när leuprorelinacetat i depotform administrerades subkutant med 3 månaders intervall (2 injektioner). Efter den första injektionen ökar serumnivåerna av leuprorelin, och maximala serumnivåer uppnås vid månad 4 (294,79 ± 105,42 pg/ml). Nivåerna minskar något fram till månad 6 (229,02 ± 103,33 pg/ml).
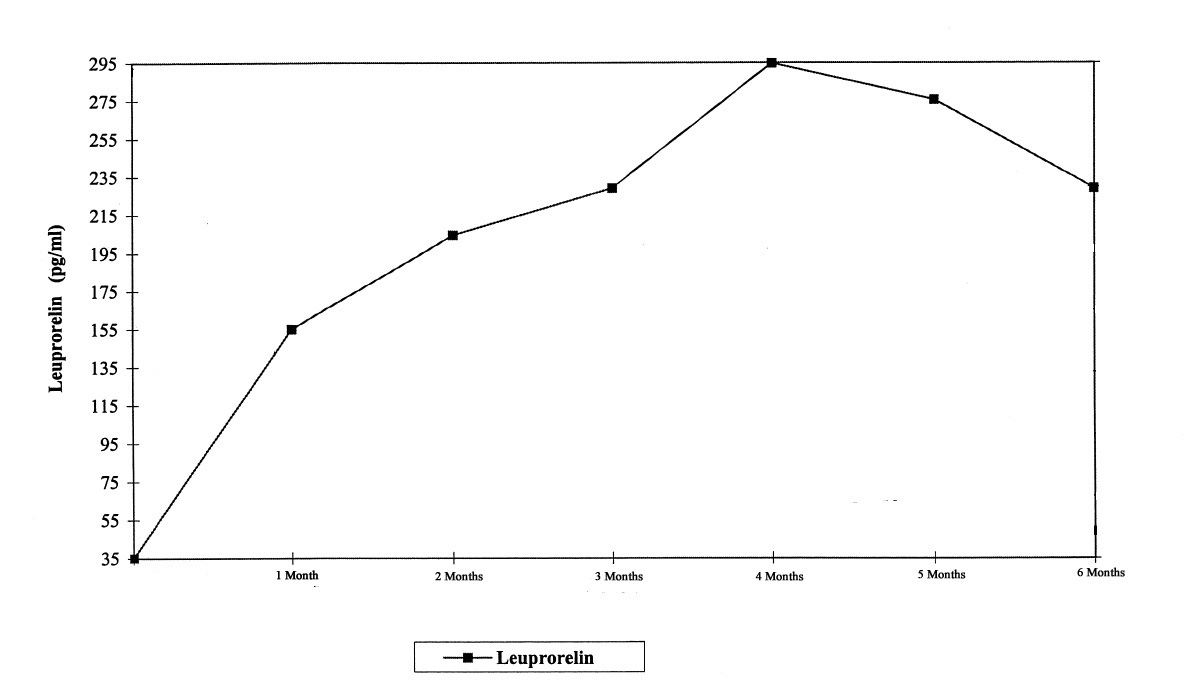
Bild 2: Serumnivåer av leuprorelin under de första 6 månaderna av behandlingen, när leuprorelinacetat i depotform administrerades med 3 månaders intervall (två subkutana injektioner, n = 42–43).
Prekliniska säkerhetsuppgifter
I en studie på kaniner administrerades leuprorelinacetat som en månatlig enkeldos på 0,00024, 0,0024 respektive 0,024 mg/kg (1/300–1/3 av den högsta dosen för människa) given på dag sex av dräktigheten. Detta orsakade en dosberoende ökning av de mest betydande utvecklingsstörningarna hos fostren. I motsvarande studier på råtta kunde en ökning av malformationer hos fostren inte påvisas. Fosterdödligheten ökade och fostervikten minskade när de två större doserna gavs till kaniner och när den största dosen gavs till råttor. Effekterna på fosterdödlighet var en logisk följd av de förändringar i hormonnivåer som läkemedlet orsakade. Därför finns risk för utvecklingsstörningar hos fostret samt spontan abort, om läkemedlet används under graviditet.
Leuprorelin kan minska fertiliteten hos båda könen. Administrering av leuprorelinacetat till han- och honråttor i doser om 0,024, 0,24 och 2,4 mg/kg per månad under 3 månader (endast 1/300 av den beräknade månadsdosen för människa) orsakade atrofi av reproduktionsorganen och försämring av den reproduktiva funktionen.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
3,75 mg:
Pulver: Poly(mjölksyra/glykolsyra) sampolymer, mannitol och gelatin.
Vätska: Karmellosnatrium, mannitol, polysorbat 80, koncentrerad ättiksyra (för pH‑justering) och vatten för injektionsvätskor.
11,25 mg och 30 mg:
Pulver: Polymjölksyra och mannitol.
Vätska: Karmellosnatrium, mannitol, polysorbat 80, koncentrerad ättiksyra (för pH‑justering) och vatten för injektionsvätskor.
Inkompatibiliteter
Då blandbarhetsstudier saknas skall detta läkemedel inte blandas med andra läkemedel.
Hållbarhet
3,75 mg: 3 år. Den färdigberedda suspensionen är hållbar i 24 timmar.
11,25 mg: 3 år. Den färdigberedda suspensionen är hållbar i 12 timmar.
30 mg: 3 år. Den färdigberedda suspensionen är hållbar i 24 timmar.
Särskilda förvaringsanvisningar
Förvaras vid högst 25 ºC. Förvaras i skydd mot kyla. Får ej frysas.
Förvaras i originalförpackningen.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
PROCREN DEPOT PDS injektiokuiva-aine ja liuotin suspensiota varten, esitäytetty ruisku
3,75 mg (L:ei) 3,75 mg (136,43 €)
11,25 mg (L:ei) 11,25 mg (346,32 €)
30 mg (L:ei) 30 mg (573,50 €)
PF-selosteen tieto
Förpackningen innehåller:
1 tvåkammarspruta (glas, Ph. Eur. typ I) 3,75 mg med en 25 G‑nål samt 11,25 mg och 30 mg med en 23 G‑nål (rostfritt stål).
Proppen är gjord av silikoniserat klorbutylgummi, kolven av propylen. Kolvens ända är försedd med en packning av vattenabsorberande etylenvinylacetat eller polyvinylalkohol. 1 spritsudd.
Läkemedlets utseende:
Beskrivning av preparatet: vitt pulver, lösningsmedel: klar, färglös vätska.
Särskilda anvisningar för destruktion och övrig hantering
Procren Depot PDS utgörs av en tvåkammarspruta med nål. Den ena kammaren innehåller antingen 3,75 mg, 11,25 mg eller 30 mg leuprorelinacetat i pulverform, och den andra kammaren innehåller 1 ml lösningsmedel. Sprutan ska hållas i upprätt ställning när injektionsvätskan bereds. Blandandet av pulvret och vätskan ska ske på ett lugnt sätt, så att läkemedlet löser sig jämnt i lösningen. I sprutans sida finns en inbuktning som underlättar upplösningen. Det rekommenderas att det färdiga läkemedlet injiceras omedelbart. Bruksanvisning finns i förpackningen.
Ersättning
PROCREN DEPOT PDS injektiokuiva-aine ja liuotin suspensiota varten, esitäytetty ruisku
30 mg 30 mg
- Ylempi erityiskorvaus (100 %). Eturauhassyöpä (116).
- Peruskorvaus (40 %).
PROCREN DEPOT PDS injektiokuiva-aine ja liuotin suspensiota varten, esitäytetty ruisku
3,75 mg 3,75 mg
11,25 mg 11,25 mg
- Ylempi erityiskorvaus (100 %). Rintasyöpä (115), Eturauhassyöpä (116).
- Peruskorvaus (40 %).
Atc-kod
L02AE02
Datum för översyn av produktresumén
17.02.2025
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi