
COFACT pulver och vätska till injektionsvätska, lösning 250 IU, 500 IU
Kvalitativ och kvantitativ sammansättning
Cofact (koncentrat av fyra koagulationsfaktorer) tillhandahålls som ett pulver och vätska till injektionsvätska, lösning och innehåller humant protrombinkomplexkoncentrat. Läkemedlet innehåller nominellt följande mängd IU av humana koagulationsfaktorer:
Cofact 250 IU (faktor IX) | Cofact 500 IU (faktor IX) | Efter beredning (IU/ml)* | |
Aktiva substanser | |||
Koagulationsfaktor II | 140–350 | 280–700 | 14–35 |
Koagulationsfaktor VII | 70–200 | 140–400 | 7–20 |
Koagulationsfaktor IX | 250 | 500 | 25 |
Koagulationsfaktor X | 140–350 | 280–700 | 14–35 |
Övriga aktiva substanser | |||
Protein C | 111–390 | 222–780 | 11–39 |
Protein S | 10–80 | 20–160 | 1–8 |
*Efter beredning med 10 ml (för Cofact 250 IU) eller 20 ml (för Cofact 500 IU) vatten för injektionsvätskor.
Det totala proteininnehållet per injektionsflaska är 130–350 mg (Cofact 250 IU) eller 260–700 mg (Cofact 500 IU). Produktens specifika aktivitet är ≥ 0,6 IU/mg, utryckt som faktor IX-aktivitet.
Aktiviteterna hos samtliga koagulationsfaktorer såväl som hos protein C och S (antigen) har testats enligt gällande standarder för WHO eller Europafarmakopén.
Hjälpämne(n) med känd effekt
Efter beredning innehåller detta läkemedel 125–195 mmol natrium/l, upp till 44,8 mg natrium per 10 ml.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Pulver och vätska till injektionsvätska, lösning.
Kliniska uppgifter
Terapeutiska indikationer
- Behandling av blödning och perioperativ blödningsprofylax vid förvärvad brist på koagulationsfaktorer i protrombinkomplexet, såsom brist till följd av behandling med vitamin K-antagonister, eller vid överdosering av vitamin K-antagonister, när snabb korrigering av bristen krävs.
- Behandling av blödning och perioperativ profylax vid medfödd brist på någon av de vitamin K-beroende koagulationsfaktorerna, när renade och specifika koagulationsfaktorprodukter inte finns att tillgå.
Villkor
Hoito tulee aloittaa hyytymishäiriöihin perehtyneen lääkärin valvonnassa.
Dosering och administreringssätt
Dosering
Endast allmänna riktlinjer för dosering ges nedan. Behandling bör sättas in under överinseende av läkare med erfarenhet av behandling av koagulationsrubbningar. Substitutionsbehandlingens dosering och duration avgörs av rubbningens svårighetsgrad, blödningens lokalisation och omfattning samt patientens kliniska tillstånd.
Dos och doseringsintervall ska beräknas individuellt för varje patient. Doseringsintervallet måste anpassas till att olika koagulationsfaktorer i protrombinkomplexet har olika halveringstid i plasma (se avsnitt Farmakokinetiska egenskaper). Individuella doseringsbehov kan bara bestämmas utifrån regelbundna mätningar av individuella plasmanivåer för koagulationsfaktorn i fråga, eller genom globala test på protrombinkomplexnivåerna (PK, INR) och kontinuerlig övervakning av patientens kliniska tillstånd.
Vid större kirurgiska ingrepp är noggrann övervakning av substitutionsbehandlingen med hjälp av koagulationsanalyser nödvändig (specifika koagulationsfaktoranalyser och/eller globala test på protrombinkomplexnivåerna).
Blödning och perioperativ blödningsprofylax under behandling med vitamin K-antagonist:
Dosen avgörs av INR före behandling, målvärdet för INR och kroppsvikten. I nedanstående tabell anges de ungefärliga doser som krävs för korrigering av INR vid olika initiala INR-nivåer.
Doseringstabellerna ska endast användas som allmänna riktlinjer och de kan inte ersätta den individuella doseringsberäkningen för varje enskild patient samt en noggrann övervakning av INR och andra koagulationsparametrar vid behandling.
Rekommenderade doser av Cofact i ml för att uppnå målvärdet för INR på ≤ 2,1
Initialt INR-värde | 7,5 | 5,9 | 4,8 | 4,2 | 3,6 | 3,3 | 3,0 | 2,8 | 2,6 | 2,5 | 2,3 | 2,2 |
| Kroppsvikt | ||||||||||||
50 kg | 40 | 40 | 40 | 30 | 30 | 30 | 20 | 20 | X | X | X | X |
60 kg | 50 | 50 | 40 | 40 | 30 | 30 | 30 | 20 | X | X | X | X |
70 kg | 60 | 50 | 50 | 50 | 40 | 40 | 30 | 30 | X | X | X | X |
80 kg | 60 | 60 | 60 | 50 | 50 | 40 | 40 | 30 | X | X | X | X |
90 kg | 60 | 60 | 60 | 60 | 50 | 50 | 40 | 30 | X | X | X | X |
100 kg | 60 | 60 | 60 | 60 | 60 | 50 | 40 | 40 | X | X | X | X |
Rekommenderade doser av Cofact i ml för att uppnå målvärdet för INR på ≤ 1,5
Initialt INR-värde | 7,5 | 5,9 | 4,8 | 4,2 | 3,6 | 3,3 | 3,0 | 2,8 | 2,6 | 2,5 | 2,3 | 2,2 |
| Kroppsvikt | ||||||||||||
50 kg | 60 | 60 | 60 | 50 | 50 | 50 | 40 | 40 | 30 | 30 | 30 | 30 |
60 kg | 80 | 70 | 70 | 60 | 60 | 60 | 50 | 50 | 40 | 40 | 40 | 30 |
70 kg | 90 | 80 | 80 | 70 | 70 | 70 | 60 | 60 | 50 | 40 | 40 | 40 |
80 kg | 100 | 100 | 90 | 90 | 90 | 80 | 80 | 70 | 60 | 50 | 50 | 40 |
90 kg | 100 | 100 | 100 | 90 | 90 | 90 | 80 | 80 | 70 | 60 | 50 | 40 |
100 kg | 100 | 100 | 100 | 100 | 100 | 90 | 90 | 80 | 70 | 70 | 60 | 50 |
Doserna beräknas baserat på koncentrationen av faktor IX i Cofact, eftersom den har relativt kort halveringstid och lågt utbyte efter infusion jämfört med övriga koagulationsfaktorer i protrombinkomplexet. Det antas att en genomsnittlig plasmakoncentration av faktor IX på ≥ 30 % är tillräcklig för att uppnå ett INR-värde på ≤ 2,1 och ≥ 60 % för att uppnå ett INR-värde på ≤ 1,5. Beräknade mängder avrundas till multiplar om 10 ml och en övre gräns på totalt 60 eller 100 ml har fastställts (se tabellerna ovan). Målvärdena för INR rekommenderas av Federation of Dutch Thrombosis Services och de är liknande som rekommendationerna i England och Tyskland.
Justeringen av en vitamin K-antagonistinducerad rubbning av hemostasen kvarstår i ungefär 6–8 timmar. Dock inträder effekterna av vitamin K, om detta givits samtidigt, vanligtvis inom 4–6 timmar. Därför krävs vanligen inte upprepad behandling med humant protrombinkomplexkoncentrat när vitamin K har givits.
Då dessa rekommendationer är empiriskt grundade, och recovery och effektduration kan variera, krävs övervakning av behandlingen med INR-bestämningar.
Blödning och perioperativ profylax vid medfödd brist på någon av de vitamin K-beroende koagulationsfaktorerna, när specifika koagulationsfaktorprodukter inte finns att tillgå:
Den beräknade dosering som krävs för behandling baseras på det empiriska fyndet att ungefär 1 IU av faktor VII eller faktor IX per kg kroppsvikt höjer plasmaaktiviteten för faktor VII respektive IX med 0,01 IU/ml, medan 1 IU av faktor II eller X per kg kroppsvikt höjer plasmaaktiviteten för faktor II eller X med 0,02 respektive 0,017 IU/ml.
Dosen för en specifik faktor uttrycks i internationella enheter (IU), vilka relateras till den aktuella WHO-standarden för varje faktor. Aktiviteten i plasma för en specifik koagulationsfaktor uttrycks antingen som en procentandel (i förhållande till normal plasma) eller i internationella enheter (i förhållande till den internationella standarden för den specifika koagulationsfaktorn).
En internationell enhet (IU) av koagulationsfaktoraktivitet motsvarar mängden i 1 ml normal humanplasma.
Exempelvis baseras beräkningen av den erforderliga doseringen av faktor X på det empiriska fyndet att 1 internationell enhet (IU) av faktor X per kg kroppsvikt höjer aktiviteten av faktor X i plasma med 0,017 IU/ml. Den erforderliga dosen beräknas med hjälp av följande formel:
Erforderligt antal enheter = kroppsvikt (kg) x önskad ökning av faktor X (IU/ml) x 60
Där 60 (ml/kg) är det reciproka värdet för uppskattad recovery.
Om individuell recovery är känd ska detta värde användas vid beräkning.
Pediatrisk population
Säkerhet och effekt för Cofact för pediatriska patienter har inte fastställts.
Administreringssätt
För anvisningar om beredning av läkemedlet före administrering, se avsnitt Särskilda anvisningar för destruktion och övrig hantering. Cofact ska administreras intravenöst.
Det rekommenderas att den färdigberedda produkten administreras med en hastighet av ungefär 2 ml/minut.
Kontraindikationer
Överkänslighet mot de aktiva substanserna eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Råd bör inhämtas från specialist med erfarenhet av behandling av koagulationsrubbningar.
Hos patienter med förvärvad brist på vitamin K-beroende koagulationsfaktorer (t.ex. inducerad av behandling med vitamin K-antagonister) ska Cofact endast användas när snabb korrigering av protrombinkomplexnivåerna är nödvändig, exempelvis vid större blödning eller akutkirurgi. I andra fall är i regel dosreduktion av vitamin K-antagonisten och/eller administrering av vitamin K tillräcklig.
Patienter som får vitamin K-antagonist kan ha ett bakomliggande tillstånd av hyperkoagulabilitet vilket kan förvärras av infusion av ett humant protrombinkomplex.
Vid medfödd brist på någon av de vitamin K-beroende faktorerna ska specifik koagulationsfaktorprodukt användas, om sådan finns att tillgå.
Om allergiska reaktioner eller reaktioner av anafylaktisk typ skulle inträffa ska injektionen/infusionen avbrytas omedelbart. Vid chock ska medicinsk standardbehandling för chock sättas in.
Standardåtgärder för att förhindra att infektioner överförs från läkemedel tillverkade av humant blod eller plasma inkluderar urval av givare, test av individuella donationer och plasmapooler för specifika infektionsmarkörer och samt att effektiva tillverkningssteg för inaktivering/eliminering av virus är en del av tillverkningsprocessen. Trots detta kan risken för överföring av infektiösa agens inte helt uteslutas när läkemedel som tillverkats av humant blod eller plasma ges. Detta gäller även okända eller nya virus eller andra patogener.
De åtgärder som vidtagits anses effektiva mot höljeförsedda virus som humant immunbristvirus (HIV), hepatit B-virus (HBV) och hepatit C-virus (HCV) samt för det icke höljeförsedda viruset hepatit A (HAV). De åtgärder som vidtagits kan ha begränsad effekt mot andra, icke höljeförsedda virus såsom parvovirus B19. Parvovirus B19-infektion kan vara allvarlig för gravida kvinnor (infektion av foster) och för patienter med immunbrist eller förkortad livslängd produktion av röda blodkroppar (t.ex. vid hemolytisk anemi).
Lämplig vaccinering (hepatit A och B) ska övervägas för patienter som fått regelbunden/upprepad behandling med protrombinkomplexprodukter framställda ur human plasma.
Det finns risk för trombos eller disseminerad intravasal koagulation när patienter, med antingen medfödd eller förvärvad brist, behandlas med humant protrombinkomplex, i synnerhet vid upprepad dosering. Risken kan vara högre vid behandling av isolerad brist på faktor VII eftersom övriga vitamin K-beroende koagulationsfaktorer, med längre halveringstid, kan nå nivåer som är betydligt högre än normalt.
Patienter som ges humant protrombinkomplex ska observeras noga med avseende på tecken eller symtom på intravasal koagulation eller trombos. På grund av risken för tromboemboliska komplikationer ska noggrann övervakning ske när man administrerar humant protrombinkomplex till patienter med anamnes på kranskärlssjukdom, till patienter med leversjukdom, till peri- eller postoperativa patienter, till nyfödda eller till patienter med risk för tromboemboliska händelser eller disseminerad intravasal koagulation. I var och en av dessa situationer ska de potentiella fördelarna av behandlingen vägas mot risken för dessa komplikationer.
Data saknas vad gäller användning av Cofact vid perinatal blödning orsakad av vitamin K-brist hos nyfödda.
Hjälpämnen
Cofact innehåller upp till 448 mg natrium per 100 ml, motsvarande 22 % av WHOs högsta rekommenderat dagligt intag (2 gram natrium för vuxna). Detta bör beaktas hos patienter som ordinerats saltfattig kost.
Pediatrisk population
Det finns inte tillräckligt med data för att rekommendera användning av Cofact till barn och ungdomar.
Interaktioner
Humana protrombinkomplexpreparat neutraliserar effekten av behandling med vitamin K-antagonist, men det finns inte några kända interaktioner med andra läkemedel.
Fertilitet, graviditet och amning
Säkerheten vid användning av humant protrombinkomplex under graviditet och amning hos människa har inte fastställts.
Djurstudier är inte lämpliga för bedömning av säkerheten med avseende på graviditet, embryonal/fetal utveckling, förlossning eller postnatal utveckling. Därför bör humant protrombinkomplex användas under graviditet och amning, endast om klar indikation föreligger. Se avsnitt Varningar och försiktighet för information om risken för parvovirus B19-infektion hos gravida kvinnor.
Effekter på förmågan att framföra fordon och använda maskiner
Inga studier har utförts om förmågan att framföra fordon och använda maskiner.
Biverkningar
Tabell över biverkningar av Cofact
De biverkningar som presenteras har rapporterats under kliniska prövningar och vid användning av Cofact efter marknadsintroduktion. Tabellen nedan anger biverkningar enligt MedDRA-organklassificering (SOC and Preferred Term Level). Frekvensen av biverkningarna definieras enligt följande konvention: mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100), sällsynta (≥ 1/10 000, < 1/1 000), mycket sällsynta (< 1/10 000), ingen känd frekvens (kan inte beräknas från tillgängliga data).
Biverkningarna presenteras inom varje frekvensgrupp efter fallande allvarlighetsgrad.
Biverkningsfrekvenser
| Organklass enligt MedDRA (SOC) | Biverkning | Frekvens |
Immunsystemsjukdomar | Anafylaktisk reaktion, överkänslighet | Ingen känd frekvens |
Centrala och perifera nervsystemet | Cerebrovaskulär händelse, yrsel | Ingen känd frekvens |
Hjärtsjukdomar | Akut hjärtinfarkt | Ingen känd frekvens |
Vaskulära sjukdomar | Tromboemboliska händelser (emboli, djup ventrombos); se avsnitt Varningar och försiktighet | Vanliga |
Hypotension | Mindre vanliga | |
Respiratoriska, torakala och mediastinala sjukdomar | Lungemboli, andningssvikt | Ingen känd frekvens |
Magtarmkanalen | Illamående, kräkningar | Ingen känd frekvens |
Sjukdomar i hud och subkutan vävnad | Hyperhidros, klåda, urtikaria | Ingen känd frekvens |
Allmänna sjukdomar och tillstånd på administreringsställe | Rodnad vid infusionsstället, irritation vid infusionsstället, svullnad vid infusionsstället
Sjukdomskänsla | Ingen känd frekvens |
Utredningar | Onormal leverfunktion | Ingen känd frekvens |
Substitutionsbehandling kan leda till bildning av cirkulerande antikroppar som hämmar en eller flera av de humana protrombinkomplexfaktorerna. Om sådan hämning uppträder kommer tillståndet att manifestera sig som dålig klinisk respons, t.ex. pågående blödning.
För information om smittsamma ämnen, se avsnitt Varningar och försiktighet.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Användning av höga doser av humana protrombinkomplexpreparat har kunnat sättas i samband med fall av hjärtinfarkt, disseminerad intravasal koagulation, ventrombos och lungemboli. Vid överdosering ökar därför risken för utveckling av tromboemboliska komplikationer eller disseminerad intravaskulär koagulation.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: hemostatika, koagulationsfaktor IX, II, VII och X i kombination, ATC-kod: B02BD01
Koagulationsfaktorerna II, VII, IX och X, vilka syntetiseras i levern med hjälp av vitamin K, brukar kallas protrombinkomplexet. Förutom koagulationsfaktorerna innehåller Cofact vitamin K-beroende koagulationshämmande protein C och protein S.
Faktor VII är zymogen till det aktiva serinproteaset faktor VIIa, med vilket den externa koagulationsvägen initieras. Vävnadsfaktor-faktor VIIa-komplexet aktiverar koagulationsfaktorerna X och IX, varigenom faktor IXa och Xa bildas. I takt med att koagulationskaskaden fortgår, aktiveras protrombin (faktor II) och omvandlas till trombin. Genom trombinets verkan konverteras fibrinogen till fibrin vilket resulterar i att ett koagel bildas. Den normala produktionen av trombin är också av avgörande betydelse för trombocytfunktionen, som ett led i den primära hemostasen.
Isolerad svår brist på faktor VII leder till minskad trombinbildning och blödningsbenägenhet beroende på försämrad fibrinbildning och försämrad primär hemostas. Isolerad brist på faktor IX är en av de klassiska hemofilisjukdomarna (hemofili B). Isolerad brist på faktor II eller faktor X är mycket sällsynt men i svår form orsakar detta en blödningsbenägenhet som liknar den som ses vid klassisk hemofili.
Övriga aktiva substanser, det koagulationshämmande protein C och protein S, syntetiseras också i levern. Den biologiska aktiviteten för protein C förstärks av kofaktorn; protein S.
Aktiverat protein C hämmar koagulationen genom att inaktivera koagulationsfaktorerna Va och VIIIa. Protein S som kofaktor till protein C stödjer inaktiveringen av koagulationen. Brist på protein C innebär en ökad risk för trombos.
Förvärvad brist på de vitamin K-beroende koagulationsfaktorerna kan uppstå under behandling med vitamin K-antagonister. Om bristen blir allvarlig resulterar det i svår blödningsbenägenhet, som karakteriseras av retroperitoneala och cerebrala blödningar, snarare än muskel- och ledblödningar. Svår leversvikt resulterar också i markant sänkta nivåer av de vitamin K-beroende koagulationsfaktorerna och en klinisk blödningsbenägenhet, vilken emellertid ofta är komplex, till följd av en samtidigt pågående låggradig intravasal koagulation, låga trombocytvärden, brist på koagulationshämmare och störd fibrinolys.
Administrering av humant protrombinkomplex ger en ökning av plasmanivåerna för de vitamin K-beroende koagulationsfaktorerna och kan temporärt korrigera koagulationsrubbningen hos patienter med brist på en eller flera av dessa faktorer.
Farmakokinetiska egenskaper
Följande information om halveringstid för de fyra koagulationsfaktorerna som Cofact innehåller beskrivs i litteraturen:
Koagulationsfaktor | Halveringstid |
Faktor II | 40–60 timmar |
Faktor VII | 4–6 timmar |
Faktor IX | 18–25 timmar |
Faktor X | 30–60 timmar |
Prekliniska säkerhetsuppgifter
Inga studier på djur har genomförts med Cofact förutom en studie med råttor på en eventuell hypotensiv effekt (det fastställdes att detta inte förekommer).
Toxikologiska studier har genomförts på försöksdjur med TNBP och Tween 80. Cofact innehåller högst 0,4 mikrog TNBP per IU av faktor IX och högst 4 mikrog Tween 80 per IU av faktor IX. När Cofact används vid den rekommenderade doseringen är mängden TNBP och Tween 80 som patienten får långt under de nivåer som visat sig skadliga i djurförsök.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Pulver: natriumcitrat, natriumklorid, antitrombin ≤ 0,6 IU/ml.
Vätska: vatten för injektionsvätskor.
Inkompatibiliteter
Detta läkemedel får inte blandas med andra läkemedel.
Cofact är kompatibelt med polypropenmaterial. Behandlingsfel kan inträffa till följd av adsorption av koagulationsfaktorn till de inre ytorna av annan injektions-/infusionsutrustning.
Hållbarhet
3 år.
Kemisk och fysikalisk stabilitet efter beredning har visats i 3 timmar vid 15 °C–25 °C. Ur mikrobiologiskt perspektiv ska produkten användas omedelbart efter beredning. Om den inte används omedelbart är förvaringstiden och förhållandena före användning användarens ansvar.
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 °C–8 °C). Får ej frysas. Förvara injektionsflaskan i ytterkartongen. Ljuskänsligt.
Inom hållbarhetstiden kan produkten förvaras vid eller under 25 °C i upp till 6 månader före användning. Om den inte används under den här tidsperioden måste den kasseras. När produkten har tagits ut ur kylskåp får den inte ställas tillbaka i kylskåp. Det datum då produkten placeras i rumstemperatur ska noteras på förpackningen.
Förvaringsanvisningar efter beredning av läkemedlet finns i avsnitt Hållbarhet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
COFACT injektiokuiva-aine ja liuotin, liuosta varten
250 IU (L:ei) 250 IU + 10 ml (237,27 €)
500 IU (L:ei) 500 IU + 20 ml (458,57 €)
PF-selosteen tieto
250 IU av pulver i en injektionsflaska (typ I glas) med en propp (brombutyl) + 10 ml vätska i en injektionsflaska (typ I glas) med en propp (brombutyl) + en överföringskanyl – i enkelförpackning.
500 IU av pulver i en injektionsflaska (typ II glas) med en propp (brombutyl) + 20 ml vätska i en injektionsflaska (typ I glas) med en propp (klorbutyl med Fluro Tec-beläggning) + en överföringskanyl – i enkelförpackning.
Läkemedlets utseende:
Pulvret har en blåaktig färg. Vätskan är en klar, färglös lösning, fri från synliga partiklar.
Särskilda anvisningar för destruktion och övrig hantering
Upplösning
Den torkade proteinfraktionen ska lösas i den föreskrivna volymen vatten för injektionsvätskor. Vid förvaring vid 2 °C–8 °C måste injektionsflaskorna med Cofact och vatten för injektionsvätskor ha nått rumstemperatur (15 °C–25 °C) innan upplösningen genomförs.
Procedur med hjälp av en överföringskanyl
- Ta bort det skyddande plastlocket från flaskan som innehåller vatten för injektionsvätskor och flaskan som innehåller produkten.
- Desinficera flaskornas gummiproppar med en kompress fuktad med alkohol (70 %).
- Ta bort det skyddande locket från den ena änden av överföringskanylen och stick in kanylen i flaskan som innehåller vatten för injektionsvätskor (A).
- Ta sedan bort det skyddande locket från den andra änden av överföringskanylen, vänd flaskan med överföringskanylen upp och ned och stick omedelbart in den fria kanylen i flaskan som innehåller produkten (B).
Undertrycket i den flaska som innehåller produkten kommer att göra att vattnet för injektionsvätskor sugs in i flaskan. Rekommendation: när vattnet för injektionsvätskor rinner mellan flaskorna, ska den flaska som innehåller produkten hållas lutad så att vattnet rinner utmed flaskans vägg. Det gör att produkten löser upp sig snabbare. Så snart vattnet har runnit över i den andra flaskan ska den tomma flaskan och överföringskanylen tas bort i en enda rörelse.
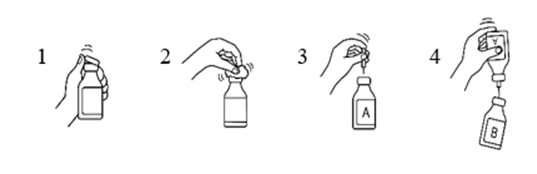
För att påskynda upplösningen kan man snurra flaskan försiktigt och, om det är nödvändigt, värma den till 30 °C. Flaskan får inte skakas och temperaturen får inte överstiga 37 °C. Om flaskan värms i vattenbad, ska försiktighet iakttas så att vattnet inte kommer i kontakt med det skyddande locket och/eller gummiproppen.
Den torkade substansen ska i regel ha löst upp sig fullständigt inom 10 minuter och bildat en blåfärgad lösning. Den blå färgen orsakas av plasmaproteinet ceruloplasmin.
Lösningen ska vara klar eller något opaliserande. Använd inte lösningar som är grumliga eller som innehåller fällning. Färdigberedd produkt ska granskas visuellt med avseende på partiklar och missfärgning före administrering.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
COFACT injektiokuiva-aine ja liuotin, liuosta varten
250 IU 250 IU + 10 ml
500 IU 500 IU + 20 ml
- Ei korvausta.
Atc-kod
B02BD01
Datum för översyn av produktresumén
22.10.2022
Yhteystiedot
 PROTHYA BIOSOLUTIONS FINLAND OY
PROTHYA BIOSOLUTIONS FINLAND OY Lars Sonckin kaari 14
02600 Espoo
09 6120 9122
www.prothya.com
info@prothya.fi