
FIBRYGA pulver och vätska till injektions-/infusionsvätska, lösning 1 g
Kvalitativ och kvantitativ sammansättning
Humant fibrinogen
Varje flaska med FIBRYGA innehåller 1 g humant fibrinogen. Efter beredning med 50 ml vatten för injektionsvätskor innehåller FIBRYGA cirka 20 mg/ml humant fibrinogen.
Innehållet koagulerbart protein fastställs enligt europeiska farmakopéns riktlinjer för humant fibrinogen.
Framställt från plasma från humana givare.
Hjälpämnen med känd effekt
Natrium upp till 132 mg (5,8 mmol) per flaska.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Pulver och vätska till injektions-/infusionsvätska, lösning.
Kliniska uppgifter
Terapeutiska indikationer
Behandling av blödningsepisoder samt perioperativ profylax hos patienter med medfödd hypo- eller afibrinogenemi med känd blödningsbenägenhet.
Som kompletterande behandling vid hantering av okontrollerad svår blödning hos patienter med förvärvad hypofibrinogenemi under pågående kirurgiska ingrepp.
Villkor
Hoito tulee aloittaa hyytymishäiriöihin perehtyneen lääkärin valvonnassa.
Dosering och administreringssätt
Behandlingen ska initieras under överinseende av en läkare med erfarenhet av att behandla koagulationssjukdomar.
Dosering
Dosering och behandlingsduration beror på sjukdomens svårighetsgrad, blödningens lokalisering och omfattning, samt patientens kliniska tillstånd.
För att kunna beräkna individuell dos bör funktionell fibrinogennivå fastställas. Mängd och administreringsfrekvens anpassas till varje enskild patient utifrån regelbundna mätningar av fibrinogennivåerna i plasma och kontinuerlig övervakning av patientens kliniska tillstånd, samt med hänsyn till andra substitutionsbehandlingar som använts.
Vid större kirurgiska ingrepp är det mycket viktigt att man noggrannt övervakar ersättningssbehandlingen via koagulationsanalyser.
1. Profylax hos patienter med medfödd hypo- eller afibrinogenemi och känd blödningsbenägenhet.
För att förebygga kraftiga blödningar under kirurgiska ingrepp rekommenderas profylaktisk behandling som höjer fibrinogenvärdet till 1 g/l, upprätthåller detta fibrinogenvärde tills hemostas är under kontroll samt håller fibrinogenvärdet över 0,5 g/l tills såret är helt läkt.
Vid kirurgiskt ingrepp eller vid behandling av en blödning ska dosen beräknas enligt följande:
Dos (mg/kg kroppsvikt) = [Målvärde (g/l) - uppmätt värde (g/l)]
0,018 (g/l per mg/kg kroppsvikt)
Efterföljande dosering (doser och injektionsfrekvens) ska anpassas efter patientens kliniska tillstånd samt laboratorieresultat.
Den biologiska halveringstiden för fibrinogen är 3–4 dagar. Om fibrinogen inte förbrukas krävs därför oftast inte någon upprepad behandling med humant fibrinogen. Med hänsyn till den ackumulering som sker vid upprepad, profylaktisk administrering, ska dosen och frekvensen fastställas enligt läkarens behandlingsmål för en specifik patient.
Pediatrisk population
Vid kirurgiskt ingrepp eller vid behandling av en blödningsepisod ska dosen hos ungdomar beräknas enligt formeln som beskrivs ovan för vuxna, medan dosen till barn < 12 år ska beräknas enligt följande:
Dos (mg/kg kroppsvikt) = [Målvärde (g/l) - uppmätt värde (g/l)]
0,014 (g/l per mg/kg kroppsvikt)
Efterföljande dosering ska anpassas efter patientens kliniska status och laboratorieresultat.
Äldre patienter
Kliniska studier av FIBRYGA inkluderade inte patienter i åldern 65 år och äldre. Det är därför oklart om denna patientgrupp uppvisar annorlunda respons än yngre patienter.
2. Behandling av blödningar
Blödning hos patienter med medfödd hypo- eller afibrinogenemi
Blödningsepisoder ska behandlas enligt ovanstående formeln för vuxna/ungdomar och barn, för att uppnå ett rekommenderat målvärde för fibrinogen i plasma på 1 g/l. Detta värde ska upprätthållas tills hemostasen är under kontroll.
Blödning hos patienter med fibrinogenbrist
Vuxna
Generellt administreras till en början 1–2 g med påföljande infusioner vid behov. I fall av svår blödning, t.ex. vid omfattande kirurgi, kan en större mängd (4–8 g) fibrinogen krävas.
Pediatrisk population
Doseringen bör fastställas på grund av kroppsvikt och kliniska behov men är vanligen 20–30 mg/kg.
Administreringssätt
Intravenös infusion eller injektion.
FIBRYGA ska administreras genom långsam intravenös infusion med en rekommenderad maximal hastighet på 5 ml per minut för patienter med medfödd hypo- eller afibrinogenemi och med en rekommenderad maxhastighet på 10 ml per minut för patienter med förvärvad fibrinogenbrist.
Anvisningar om beredning av läkemedlet före administrering finns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Tromboembolism
Det finns risk för trombos när patienter med antigen medfödd eller förvärvad brist behandlas med humant fibrinogen, i synnerhet vid hög dos eller upprepad dosering. Patienter som får humant fibrinogen ska övervakas noga med avseende på tecken eller symptom på trombos.
Hos patienter med kranskärlssjukdom eller hjärtinfarkt i anamnesen, patienter med leversjukdom, peri- eller postoperativa patienter, nyfödda eller patienter som löper risk för tromboemboliska händelser eller disseminerad intravaskulär koagulation, ska nyttan av behandling med humant plasmafibrinogen vägas mot risken för tromboemboliska komplikationer. Försiktighet ska iakttas och noggrann övervakning upprätthållas.
Förvärvad hypofibrinogenemi är förknippad med låga plasmakoncentrationer av alla koagulationsfaktorer (inte enbart fibrinogen) och inhibitorer, så behandling med blodprodukter innehållande koagulationsfaktorer bör övervägas. Det krävs noggrann monitorering av koagulationssystemet.
Allergiska reaktioner eller reaktioner av anafylaktisk typ
Om allergiska reaktioner eller reaktioner av anafylaktisk typ förekommer ska injektionen/infusionen avbrytas omedelbart. Vid anafylaktisk chock ska standardbehandling för chock sättas in.
Natriumhalt
Detta läkemedel innehåller upp till 132 mg natrium per flaska, motsvarande 6,6 % av WHOs högsta rekommenderat dagligt intag (2 gram natrium för vuxna). Detta ska beaktas för patienter som ordinerats natriumreducerad kost.
Virussäkerhet
Standardåtgärder för att förhindra infektioner till följd av användning av läkemedel som är framställda av humant blod eller plasma inkluderar urval av givare, test av individuella donationer och plasmapooler för specifika infektionsmarkörer samt effektiv inaktivering/eliminering av virus under tillverkningsprocessen. Trots detta kan risken för överföring av infektiösa agens inte helt uteslutas när läkemedel som framställts från humant blod eller plasma administreras. Detta gäller även nya, hittills okända virus och andra patogener.
Vidtagna åtgärder anses vara effektiva mot höljeförsedda virus, såsom HIV, HBV och HCV samt mot det icke-höljeförsedda viruset HAV. Vidtagna åtgärder kan vara av begränsat värde mot icke-höljeförsedda virus såsom parvovirus B19. Infektion av parvovirus B19 kan vara allvarligt för gravida kvinnor (infektion av foster) och för personer med immunbrist eller ökad erytropoes (t.ex. hemolytisk anemi).
Lämplig vaccinering (hepatit A och B) bör övervägas för patienter som regelbundet/upprepat får humana plasmaderiverade produkter.
Immunogenicitet
Vid substitutionsterapi med koagulationsfaktorer vid andra medfödda brister har antikroppsreaktioner observerats, men det finns för närvarande inga data för fibrinogenkoncentrat.
Interaktioner
Det finns inga kända interaktioner mellan humana fibrinogenprodukter och andra läkemedel.
Fertilitet, graviditet och amning
Graviditet
Säkerheten vid användning av FIBRYGA under graviditet har inte fastställts i kontrollerade kliniska studier. Klinisk erfarenhet av fibrinogenprodukter för behandling av obstetriska komplikationer tyder inte på någon skadlig inverkan på graviditetsförloppet eller på fostrets eller det nyfödda barnets hälsa. Reproduktionsstudier hos djur har inte genomförts med FIBRYGA (se avsnitt Prekliniska säkerhetsuppgifter). Eftersom den aktiva substansen är av humant ursprung kataboliseras den på samma sätt som patientens egna protein. Dessa fysiologiska humana blodkomponenter förväntas inte påverka reproduktion eller foster.
Fördelen med FIBRYGA under graviditet måste utvärderas med beaktande av att klinisk erfarenhet av fibrinogenkoncentrat finns tillgänglig men data från kontrollerade kliniska studier saknas.
Amning
Det är okänt om FIBRYGA utsöndras i bröstmjölk. Men på grund av aktiv substans natur, förväntas inga effekter på ammade nyfödda/spädbarn.
Således måste ett beslut fattas om FIBRYGA-behandling är indicerad under amning med beaktande av nyttan med amning för barnet och fördelen med terapi för kvinnan.
Fertilitet
Det finns inga tillgängliga data angående fertilitet.
Effekter på förmågan att framföra fordon och använda maskiner
FIBRYGA har ingen eller försumbar effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
Det finns inga tillförlitliga data om frekvensen av biverkningar från kliniska studier med denna produkt.
I kliniska studier har följande biverkningar rapporterats: pyrexi, läkemedelsutslag, flebit och trombos.
Följande biverkningar har rapporterats för FIBRYGA och andra fibrinogenkoncentrat:
Organsystem enligt MedDRA | Biverkningar | Frekvens* |
Immunsystemet: | Allergiska reaktioner eller reaktioner av anafylaktisk typ Hudreaktioner | Okänt |
Blodkärl: | Tromboemboliska händelser (inklusive hjärtinfarkt och lungemboli) (se avsnitt Varningar och försiktighet) Tromboflebit | Okänt |
Allmänna symtom och/eller symtom vid administreringsstället: | Ökad kroppstemperatur (pyrexi) | Okänt |
*Frekvens okänd eftersom den kan inte beräknas från tillgängliga data. Mild feber och hudreaktion var enstaka händelser i kliniska studier. Allergiska reaktioner eller anafylaktisk typ reaktioner, tromboemboliska händelser (inklusive hjärtinfarkt och lungemboli) och tromboflebit är klasseffekter. | ||
Beträffande säkerhet avseende överförbara agens, se avsnitt Varningar och försiktighet.
Pediatrisk population:
Tjugosex patienter, i åldern 1– < 18 år, ingick i säkerhetsanalysen av medfödd fibrinogenbrist, av vilka 12 ungdomar var i åldern 12–< 18 år, 8 barni åldern 6–< 12 år och 6 barn i åldern 1–< 6 år gamla.
Den övergripande säkerhetsprofilen skiljer sig inte åt mellan vuxna patienter, ungdomar och barn.
Det finns inga data om användning av FIBRYGA hos pediatriska patienter med förvärvad fibrinogenbrist.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till (se detaljer nedan).
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
För att undvika överdosering är regelbunden övervakning av fibrinogennivån i plasma indicerad (se avsnitt Dosering och administreringssätt).
Vid överdosering är risken att utveckla tromboemboliska komplikationer förhöjd.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: hemostatika, fibrinogen, ATC-kod: B02BB01
Humant fibrinogen (koagulationsfaktor I) konverteras, i närvaro av trombin, aktiverad koagulationsfaktor XIII (FXIIIa) och kalciumjoner, till ett stabilt och elastiskt tredimensionellt hemostatiskt fibrinkoagel.
Administrering av humant fibrinogen ökar fibrinogenvärdet i plasma och kan tillfälligt korrigera koagulationsdefekten hos patienter med fibrinogenbrist.
En öppen, prospektiv, randomiserad, kontrollerad, tvåarmad cross-over enkeldos farmakokinetisk fas II-studie med 22 patienter med medfödd fibrinogenbrist (afibrinogenemi) (se avsnitt Farmakokinetiska egenskaper) utvärderade även den maximala koagelfastheten (MCF - Maximal Clot Firmness) som en surrogatmarkör för hemostatisk effekt (FORMA-01). MCF bestämdes med tromboelastometri (ROTEM). För varje patient bestämdes MCF före (baslinjen) och en timme efter administrering av en enkeldos FIBRYGA. MCF-värdena var signifikant högre efter administrering av FIBRYGA än vid baslinjen (se tabellen nedan).
Tabell 1: Maximal koagelfasthet MCF [mm] (ITT population) n=22
| Tidpunkt | Medelvärde ± SD | Median (intervall) |
| Före infusion | 0 ± 0 | 0 (0-0) |
| 1 timme efter infusion | 9,7 ± 3,0 | 10,0 (4,0-16,0) |
| Genomsnittlig förändring (primäranalys)* | 9,7 ± 3,0 | 10,0 (4,0-16,0) |
| MCF = maximum clot firmness (maximal koagelfasthet); ITT = intention-to-treat (avsikt att behandla). *p < 0,0001 (95 % konfidensintervall 8,37; 10,99) | ||
En prospektiv, öppen, okontrollerad, multicenter, fas 3-studie (FORMA-02) utfördes på 25 patienter med medfödd fibrinogenbrist (afibrinogenemi och hypofibrinogenemi), i ett åldersintervall från 12 till 54 år (6 ungdomar, 19 vuxna). Detta inkluderade behandling av 89 blödningar och 12 kirurgiska ingrepp. Det var en signifikant förändring av MCF från baslinjen vilket uppmättes med ROTEM och fibrinogenvärde i plasma. Mediandosen FIBRYGA per infusion för behandling av blödningsepisoder var 57,5 mg/kg och den median totala dosen var 59,4 mg/kg. Den genomsnittliga totala dosen FIBRYGA per operation var 85,8 mg/kg. Övergripande hemostatisk effekt bedömdes som lyckade (graderade som bra eller utmärkt effekt) för 98,9 % av de behandlade blödningsepisoderna och för 100 % av operationerna av en oberoende bedömningskommitté som använde ett objektivt poängsystem.
En annan potentiell, öppen, okontrollerad, multicenter, fas 3-studie (FORMA-04) utfördes på 14 barn med medfödd fibrinogenbrist (afibrinogenemi och hypofibrinogenemi), i ett åldersintervall från 1 till 10 år (6 < 6 år gamla och 8 mellan 6 och < 12 år gamla). Detta inkluderade behandling av 10 blödningsepisoder och tre kirurgiska ingrepp, samt farmakokinetik för en dos. Det fanns en signifikant förändring från baslinjen i maximal koagelfasthet (MCF), mätt med ROTEM- och fibrinogenvärde i plasma. Mediandosen FIBRYGA per infusion för behandling av blödningsepisoder var 70,2 mg/kg och den median totala dosen var 73,9 mg/kg. Den genomsnittliga totala dosen FIBRYGA per operation var 108 mg/kg. Övergripande hemostatisk effekt bedömdes som lyckade (graderad som bra eller utmärkt effekt) för 100 % av de behandlade blödningsepisoderna och av operationerna av en oberoende bedömningskommitté som använde ett objektivt poängsystem.
Vid den prospektiva randomiserade kontrollerade studien FORMA-05 undersöktes den hemostatiska effekten och säkerheten hos FIBRYGA i jämförelse med kryoprecipitat som källa till fibrinogensupplementering hos patienter som utvecklar förvärvad fibrinogenbrist under cytoreduktiv kirurgi för den extensiva bukmaligniteten pseudomyxoma peritonei. I studien ingick 43 vuxna patienter i analysgruppen Per protokoll (PP), varav 21 patienter behandlades med FIBRYGA och 22 patienter behandlades med kryoprecipitat. Intraoperativ fibrinogensupplementering utfördes i förebyggande syfte (dvs. efter 60–90 minuters kirurgi, när excessiv blodförlust hade observerats, men innan 2 liter blod hade förlorats) med doser på 4 g FIBRYGA eller 2 pooler med 5 enheter av kryoprecipitat, med upprepning efter behov. Under kirurgin på 7,8 ± 1,7 timmar användes 6,5 ± 3 g FIBRYGA (89 ± 39 mg/kg kroppsvikt) respektive 4,1 ± 2,2 pooler med 5 enheter av kryoprecipitat. I medianvärde administrerades 1 enhet respektive 0,5 enheter RBC intraoperativt till patienter som behandlades med FIBRYGA och kryoprecipitat, med i medianvärde 0 enheter RBC under det första dygnet postoperativt i båda grupperna (se tabellen nedan). Varken färskfrusen plasma eller trombocytkoncentrat transfunderades under studien. Hemostatisk terapi baserad på fibrinogensupplementering bedömdes som framgångsrik vid 100 % av de kirurgiska ingreppen i båda grupperna enligt en oberoende bedömningskommitté som använde ett objektivt poängsystem.
Tabell 2: Transfusion av RBC* [enheter] intraoperativt och under första dygnet postoperativt (PP-population)
| Tidsram | FIBRYGA-grupp (n=21) | Kryoprecipitat-grupp (n=22) |
|---|---|---|
| Intraoperativt | 1 (0–4) | 0,5 (0–5) |
| Första dygnet postoperativt | 0 (0–2) | 0 (0–2) |
| RBC = koncentrat av röda blodkroppar; PP = per protokoll. *ingen transfusion skedde av andra allogena blodprodukter, såsom färskfrusen plasma eller trombocytkoncentrat | ||
Pediatrisk population
Vid medfödd fibrinogenbrist, FIBRYGA administrerades i två kliniska studier (FORMA-02 och FORMA-04) till 20 patienter i åldern 1‑< 18 år, varav 6 ungdomar 12‑<18 år, 8 barn 6‑< 12 år och 6 barn 1‑< 6 år gamla. Hemostatisk effekt bedömdes som lyckade av en oberoende bedömningskommitté för alla behandlade blödningsepisoder (10 blödningsepisoder hos ungdomar, 5 hos barn 6‑< 12 år och 5 hos barn 1‑< 6 år) och profylax bedömdes också lyckade för de fyra operationerna som utförts hos dessa patienter (1 hos ungdomar och 3 hos barn 1‑< 6 år).
Farmakokinetiska egenskaper
Humant fibrinogen är en normal beståndsdel i humanplasma och fungerar som endogent fibrinogen. Den biologiska halveringstiden för fibrinogen i plasma är 3–4 dagar. FIBRYGA administreras intravenöst och är omedelbart tillgänglig i en plasmakoncentration som motsvarar den administrerade dosen.
En öppen, prospektiv, randomiserad, kontrollerad, tvåarmad cross-over fas 2-studie med 22 patienter med medfödd fibrinogenbrist (afibrinogenemi) i åldersintervallet 12 till 53 år (6 ungdomar, 16 vuxna), jämförde de farmakokinetiska egenskaperna av en enkeldos FIBRYGA med egenskaperna hos ett annat kommersiellt tillgängligt fibrinogenkoncentrat hos samma patienter (FORMA-01). Varje patient fick en enkel intravenös dos FIBRYGA 70 mg/kg och av jämförelseprodukten. Blodprover togs för att bestämma fibrinogenaktiviteten vid baslinjen och upp till 14 dagar efter infusionen. FIBRYGA:s farmakokinetiska parametrar i per protokoll-analysen (PP) (n = 21) sammanfattas i tabellen nedan.
Tabell 3: Farmakokinetiska parametrar (n=21) för fibrinogenaktivitet (PP-population*)
Parametrar | Medelvärde ± SD | Intervall |
Halveringstid [tim] | 75,9 ± 23,8 | 40,0 - 157,0 |
Cmax [mg/dl] | 139,0 ± 36,9 | 83,0 - 216,0 |
AUCnorm för en dos på 70 mg/kg [mg*h/ml] | 113,7 ± 31,5 | 59,7 - 175,5 |
Clearance [ml/h/kg] | 0,67 ± 0,2 | 0,4 - 1,2 |
Genomsnittlig residenstid [tim] | 106,3 ± 30,9 | 58,7 - 205,5 |
Distributionsvolym vid steady state [ml/kg] | 70,2 ± 29,9 | 36,9 - 149,1 |
* En patient uteslöts från PP-populationen på grund av att han/hon fick <90 % av den planerade dosen FIBRYGA och jämförelseprodukt | ||
Den inkrementella in vivo-återhämtningen (IVR - in vivo recovery) fastställdes från värden som erhölls upp till 4 timmar efter infusion. Median inkrementell IVR var 1,8 mg/dl (intervall 1,08 - 2,62 mg/dl) ökning per mg/kg. Median IVR indikerar att en dos på 70 mg/kg ökar patientens fibrinogenkoncentration i plasma med cirka 125 mg/dl.
Farmakokinetik i särskilda patientgrupper
Ingen statistiskt relevant skillnad i fibrinogenaktivitet observerades mellan manliga och kvinnliga studiedeltagare.
Pediatrisk population
Farmakokinetiska data från ungdomar från 12 till 18 år erhölls i FORMA-02-studien. I PP-analysen observerades en liten skillnad mellan halveringstiden för ungdomar (n = 5) och för vuxna (n = 16), med 72,8 ± 16,5 timmar jämfört med 76,9 ± 26,1 timmar. Clearance var nästan identisk i de två åldersgrupperna, dvs. 0,68 ± 0,18 ml/h/kg respektive 0,66 ± 0,21 ml/h/kg.
De farmakokinetiska egenskaperna hos FIBRYGA undersöktes ytterligare i FORMA-04-studien på 13 barn under 12 år med medfödd fibrinogenbrist (afibrinogenemi). Varje patient fick en engångsdos på 70 mg/kg FIBRYGA intravenöst. De farmakokinetiska parametrarna för FIBRYGA sammanfattas i tabellen nedan. Median inkrementell IVR var 1,4 mg/dl (intervall, 1,3‑2,1 mg/dl) ökning per mg/kg.
Tabell 4: Farmakokinetiska parametrar (n=13) för fibrinogenaktivitet
Parametrar | Medelvärde ± SD | Intervall |
Halveringstid [tim] | 63,3 ± 12,0 | 45,6–91,6 |
Cmax [mg/dl] | 107,2 ± 16,8 | 93,0–154,0 |
AUCnorm för en dos på 70 mg/kg [mg*h/ml] | 92,0 ± 20,0 | 69,7–134,2 |
Clearance [ml/h/kg] | 0,8 ± 0,2 | 0,5–1,0 |
Genomsnittlig residenstid [tim] | 88,0 ± 16,8 | 63,6–126,7 |
Distributionsvolym vid steady state [ml/kg] | 67,6 ± 7,1 | 52,8–76,8 |
* Beräknat hos 10 av 13 patienter på grund av otillräckligt antal kvantifierbara värden hos 3 patienter. | ||
Prekliniska säkerhetsuppgifter
Säkerhet för FIBRYGA har visats i flera prekliniska säkerhetsfarmakologiska studier (kardiovaskulära effekter, trombogen potential) och toxikologiska studier (akut toxicitet, lokal tolerans). Prekliniska data visar ingen speciell risk för människa baserat på dessa studier. I ett venöst stastest (Wessler-test) visades FIBRYGA vara icke-trombogent vid doser upp till 400 mg/kg kroppsvikt.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Pulver
L-argininhydroklorid
Glycin
Natriumklorid
Natriumcitrat (dihydrat)
Vätska
Vatten för injektionsvätskor
Inkompatibiliteter
Detta läkemedel får inte blandas med andra läkemedel.
Hållbarhet
3 år.
Kemisk och fysikalisk stabilitet för den färdigberedda lösningen har visats i 24 timmar vid rumstemperatur (max 25 °C). Ur mikrobiologisk synvinkel bör produkten användas omedelbart efter beredning. Om den inte används omedelbart är förvaringstid och förhållanden vid användning användarens ansvar. Den färdigberedda lösningen får inte frysas eller förvaras i kylskåp. Delvis använda flaskor ska kasseras.
Särskilda förvaringsanvisningar
Förvaras vid högst 25 °C. Får ej frysas. Förvara flaskan i ytterkartongen. Ljuskänsligt.
Förvaringsanvisningar för läkemedlet efter beredning finns i avsnitt Hållbarhet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
FIBRYGA injektio-/infuusiokuiva-aine ja liuotin, liuosta varten
1 g (L:ei) 1 g (Octajet siirtolaite) (687,95 €)
PF-selosteen tieto
Varje förpackning innehåller:
- 1 g humant fibrinogen i en 100 ml färglös glasflaska av typ II-glas, förseglad med en infusionspropp (brombutylgummi) och ett flip-off-lock av aluminium.
- 50 ml vätska (vatten för injektionsvätskor) i en 50 ml färglöst glas injektionsflaska av typ II-glas, förseglad med en infusionspropp (halobutylgummi) och ett och ett flip-off-lock av aluminium.
- 1 nextaro överföringsset.
Läkemedlets utseende:
Pulvret är vitt eller blekgult och fuktabsorberande (hygroskopiskt), som även framstår som en söndersmulad massa.
Vätskan är klar och färglös.
Särskilda anvisningar för destruktion och övrig hantering
Allmänna instruktioner
- Den färdigberedda lösningen ska vara nästan färglös och lätt opaliserande. Använd inte lösningar som är grumliga eller som innehåller partiklar.
- FIBRYGA är endast avsett för engångsbruk. Återanvänd inte någon av komponenterna.
- Med tanke på den mikrobiologiska säkerheten ska lösningen administreras omedelbart efter beredning. Kemisk och fysikalisk stabilitet av den färdigberedda lösningen har visats i 24 timmar vid rumstemperatur (max. 25° C). Färdigberedd FIBRYGA-lösning ska inte kylas eller frysas.
Beredning
| 1. Kontrollera att flaskan med pulver (FIBRYGA) och injektionsflaskan med spädningsvätska håller rumstemperatur. Samma temperatur ska bibehållas under beredningen. Om ett vattenbad används för att värma läkemedlet, är det viktigt att se till att vatten inte kommer i kontakt med behållarnas gummiproppar eller lock. Vattenbadets temperatur ska inte överstiga 37 °C. | ||
| 2. Ta av locket från flaskan med pulver (FIBRYGA) och locket från flaskan med spädningsvätska så att infusionsproppens centrala del exponeras. Rengör gummipropparna med en alkoholservett och låt gummipropparna torka. | ||
| 3. Öppna förpackningen med överföringssetet (nextaro) genom att dra av locket (fig. 1). Bevara sterilteten genom att låta den genomskinliga blisterförpackningen sitta kvar över överföringssetet. Vidrör inte spetsen. | 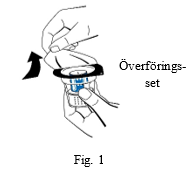 | |
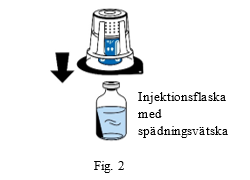
4. Placera injektionsflaskan med spädningsvätska på en slät, ren yta och håll den i ett stadigt grepp. Låt blisterförpackningen sitta kvar över överföringssetet och placera överföringssetets blå del ovanpå injektionsflaskan med spädningsvätska. Tryck stadigt rakt ned tills överföringssetet snäpper på plats (fig. 2). Vrid inte när du fäster.
Obs! |
| |
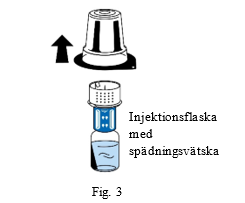
| 5. Håll kvar greppet om injektionsflaskan med spädningsvätska och ta varsamt bort blisterförpackningen från överföringssetet (nextaro) genom att dra det rakt upp. Låt överföringssetet sitta kvar stadigt ansluten till injektionsflaskan med spädningsvätska (fig. 3). |
| |
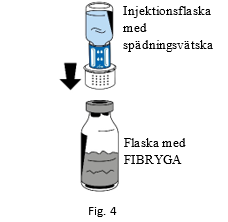
| 6. Placera flaskan med pulver (FIBRYGA) på en slät, ren yta och håll den i ett stadigt grepp. Fatta injektionsflaskan med spädningsvätska med överföringssetet anslutet och vänd upp och ned. Placera kopplingen på överföringssetets vita del ovanpå flaskan med pulver (FIBRYGA). Tryck, utan att vrida, stadigt ned tills överföringssetet snäpper på plats (fig. 4). Spädningsvätskan rinner nu automatiskt ner i flaskan med pulver (FIBRYGA). |
| |
| 7. Låt injektionsflaskan med spädningsvätska sitta kvar och snurra varsamt flaskan med FIBRYGA tills pulvret har lösts upp helt. Skaka inte flaskan eftersom detta kan orsaka skumbildning. Pulvret bör lösas upp helt inom cirka 5 minuter. Det ska inte ta längre än 20 minuter att lösa upp pulvret. Om pulvret inte har lösts upp inom 20 minuter ska produkten inte användas. | ||
| 8. I det sällsynta fall att oupplöst pulver ses flyta under överföringen av spädningsvätskan, eller om det tar oväntat lång tid att lösa upp pulvret, kan upplösningen påskyndas genom att injektionsflaskan roteras kraftigare horisontellt. | ||
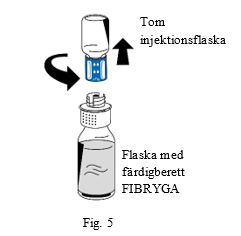
| 9. Efter avslutad beredning ska överföringssetets blå del skruvas av moturs från den vita delen (fig. 5). Vidrör inte luerlock-kopplingen på överföringssetets vita del. |
| |
| 10. Kasta den tomma injektionsflaskan tillsammans med överföringssetets blå del. | ||
Administrering
| 1. Anslut varsamt en spruta till luerlock-kopplingen på överföringssetets vita del (fig. 6) | 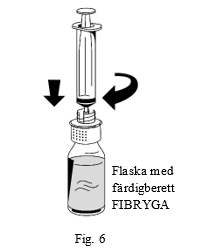 |
| 2. Vänd flaskan med FIBRYGA upp och ned och dra upp lösningen i sprutan (fig. 7). | 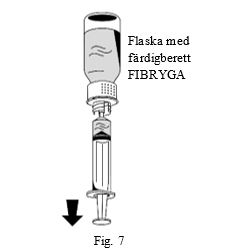 |
| 3. Håll sprutan med ett fast grepp om cylindern (med sprutans kolv pekande nedåt) och dra ut sprutan från överföringssetet när lösningen har överförts (fig. 8). | 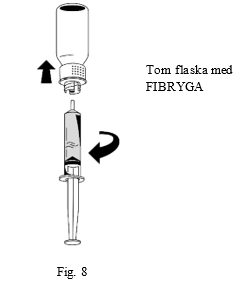 |
| 4. Kasta överföringssetets vita del tillsammans med den tomma FIBRYGA-flaskan. |
Ett standardinfusionsset rekommenderas för intravenös användning av den beredda lösningen vid rumstemperatur.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
FIBRYGA injektio-/infuusiokuiva-aine ja liuotin, liuosta varten
1 g 1 g
- Ei korvausta.
Atc-kod
B02BB01
Datum för översyn av produktresumén
17.11.2023
Yhteystiedot
Rajatorpantie 41 C
01640 Vantaa
09 8520 2710
www.octapharma.fi
info@octapharma.fi