
CABOMETYX filmdragerad tablett 20 mg, 40 mg, 60 mg
Kvalitativ och kvantitativ sammansättning
CABOMETYX 20 mg filmdragerade tabletter
Varje filmdragerad tablett innehåller kabozantinib (S)-malat motsvarande 20 mg kabozantinib.
Hjälpämnen med känd effekt
Varje filmdragerad tablett innehåller 15,54 mg laktos.
CABOMETYX 40 mg filmdragerade tabletter
Varje filmdragerad tablett innehåller kabozantinib (S)-malat motsvarande 40 mg kabozantinib.
Hjälpämnen med känd effekt
Varje filmdragerad tablett innehåller 31,07 mg laktos.
CABOMETYX 60 mg filmdragerade tabletter
Varje filmdragerad tablett innehåller kabozantinib (S)-malat motsvarande 60 mg kabozantinib.
Hjälpämnen med känd effekt
Varje filmdragerad tablett innehåller 46,61 mg laktos.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Filmdragerad tablett.
Kliniska uppgifter
Terapeutiska indikationer
Njurcellscancer (renal cell carcinoma, RCC)
CABOMETYX är indicerat som monoterapi vid avancerad njurcellscancer:
- som första linjens behandling av vuxna med intermediär eller dålig prognos (se avsnitt Farmakodynamiska egenskaper)
- hos vuxna efter tidigare vaskulär endotel tillväxtfaktor (VEGF)-riktad behandling (se avsnitt Farmakodynamiska egenskaper).
CABOMETYX i kombination med nivolumab är indicerat som första linjens behandling av vuxna med avancerad njurcellscancer (se avsnitt Farmakodynamiska egenskaper).
Hepatocellulär cancer (hepatocellular carcinoma, HCC)
CABOMETYX är avsett som monoterapi för behandling av hepatocellulär cancer (HCC) hos vuxna som tidigare behandlats med sorafenib.
Differentierad tyreoideacancer (differentiated thyroid carcinoma, DTC)
CABOMETYX är avsett som monoterapi för behandling av vuxna patienter med lokalt avancerad eller metastaserad differentierad tyreoideacancer (DTC), refraktär eller inte lämpade för radioaktivt jod (RAI) som har progredierat under eller efter systemisk behandling.
Neuroendokrina tumörer (NET)
CABOMETYX är avsett för behandling av vuxna patienter med inoperabla eller metastaserande, väldifferentierade neuroendokrina tumörer utanför pankreas (epNET) och i pankreas (pNET) som har progredierat efter minst en tidigare systemisk behandling, som inte är en somatostatinanalog.
Villkor
Hoito tulee aloittaa syövän hoitoon tarkoitettujen lääkevalmisteiden antamiseen perehtyneen lääkärin valvonnassa.
Dosering och administreringssätt
Behandling med CABOMETYX ska sättas in av en läkare med erfarenhet av administrering av läkemedel mot cancer.
Dosering
CABOMETYX tabletter och kabozantinib kapslar är inte bioekvivalenta och får inte bytas ut mot varandra (se avsnitt Farmakokinetiska egenskaper).
CABOMETYX som monoterapi
För RCC, HCC, DTC och NET är den rekommenderade dosen av CABOMETYX 60 mg en gång dagligen. Behandlingen bör fortsätta tills patienten inte längre har någon klinisk nytta av behandlingen eller tills oacceptabel toxicitet uppstår.
CABOMETYX i kombination med nivolumab vid första linjens behandling av avancerad njurcellscancer
Den rekommenderade dosen av CABOMETYX är 40 mg en gång dagligen i kombination med nivolumab infusionsvätska, lösning, som administreras intravenöst antingen 240 mg varannan vecka eller 480 mg var fjärde vecka, eller med nivolumab injektionsvätska, lösning, som administreras subkutant antingen 600 mg varannan vecka eller 1200 mg var fjärde vecka. Behandlingen bör fortsätta till sjukdomsprogress eller oacceptabel toxicitet. Behandling med nivolumab bör fortsätta till sjukdomsprogress, oacceptabel toxicitet eller upp till 24 månader hos patienter utan sjukdomsprogress (se produktresumén för dosering av nivolumab).
Dosmodifiering
Tillfällig utsättning av behandling och/eller dossänkning kan krävas för att hantera misstänkta biverkningar (se tabell 1). När dossänkning är nödvändig vid monoterapi, rekommenderas en minskning till 40 mg dagligen, och därefter till 20 mg dagligen.
När CABOMETYX administreras i kombination med nivolumab, rekommenderas dossänkning av CABOMETYX till 20 mg en gång dagligen, och därefter till 20 mg varannan dag (se nivolumabs produktresumé för rekommenderade dosjusteringar av nivolumab).
Tillfällig utsättning rekommenderas för att hantera toxiciteter av CTCAE (Common Terminology Criteria for Adverse Events) grad 3 eller högre eller intolerabel toxicitet av grad 2. Dossänkning rekommenderas för händelser som, om de kvarstår, kan bli allvarliga eller oacceptabla.
Om en patient missar en dos ska den missade dosen inte tas om det är mindre än 12 timmar kvar till nästa dos.
Tabell 1: Rekommenderade dosmodifieringar av CABOMETYX för biverkningar
| Biverkning och allvarlighetsgrad | Behandlingsmodifiering |
| Biverkningar av grad 1 och grad 2 som är tolerabla och lätta att hantera | Dosjustering krävs vanligtvis inte. Lägg till stödjande behandling om det är indicerat. |
| Biverkningar av grad 2 som är intolerabla och inte kan hanteras med en dossänkning eller stödjande behandling | Avbryt behandling tills biverkningen avtar till grad ≤ 1. Lägg till stödjande behandling om det är indicerat. Överväg återinsättning med en reducerad dos. |
| Biverkningar av grad 3 (utom kliniskt icke-relevanta laboratorieavvikelser) | Avbryt behandling tills biverkningen avtar till grad ≤ 1. Lägg till stödjande behandling om det är indicerat. Återinsättning med en reducerad dos. |
| Biverkningar av grad 4 (utom kliniskt icke-relevanta laboratorieavvikelser) | Avbryt behandling. Sätt in lämplig medicinsk behandling. Om biverkningen avtar till grad ≤ 1, återinsätt med en reducerad dos. Om biverkningen inte förbättras, sätt ut behandlingen. |
| Ökning av leverenzymer hos patienter med njurcellscancer som behandlas med CABOMETYX i kombination med nivolumab | |
| ALAT eller ASAT > 3 gånger ULN men ≤ 10 gånger ULN utan samtidig total bilirubin ≥ 2 gånger ULN | Avbryt behandlingen med CABOMETYX och nivolumab tills dessa biverkningar avtar till grad ≤ 1. Kortikosteroidbehandling kan övervägas om immunmedierade reaktioner misstänks (se nivolumabs produktresumé). Återinsätt med endast ett av läkemedlen, alternativt kan sekventiell återinsättning av båda läkemedlen övervägas efter återhämtning. Vid återinsättning av nivolumab, se nivolumabs produktresumé. |
| ALAT eller ASAT > 10 gånger ULN eller > 3 gånger ULN med samtidig total bilirubin ≥ 2 gånger ULN | Avbryt behandling med CABOMETYX och nivolumab permanent. Kortikosteroidbehandling kan övervägas om immunmedierade reaktioner misstänks (se nivolumabs produktresumé). |
Obs! Toxicitetsgrader anges i enlighet med NCI-CTCAE v4 (National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0)
Samtidig behandling med andra läkemedel
Samtidig behandling med läkemedel som är starka hämmare av CYP3A4 bör användas med försiktighet, och långvarig användning av samtidigt administrerade läkemedel som är starka inducerare av CYP3A4 bör undvikas (se avsnitt Varningar och försiktighet och Interaktioner).
Val av ett annat läkemedel för samtidig behandling som har låg eller obefintlig potential att inducera eller hämma CYP3A4 bör övervägas.
Särskilda populationer
Äldre
Ingen specifik dosjustering för användning av kabozantinib till äldre patienter (≥ 65 år) rekommenderas.
Etnicitet
Ingen dosjustering är nödvändig baserat på etnicitet (se avsnitt Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Kabozantinib ska användas med försiktighet till patienter med lätt eller måttligt nedsatt njurfunktion.
Kabozantinib rekommenderas inte till patienter med gravt nedsatt njurfunktion eftersom säkerhet och effekt inte har fastställts i denna population.
Nedsatt leverfunktion
För patienter med lindrigt nedsatt leverfunktion krävs ingen dosjustering. Eftersom endast begränsade data är tillgängliga för patienter med måttligt nedsatt leverfunktion (Child Pugh B), så kan ingen dosrekommendation ges. Noggrann övervakning av den övergripande säkerheten rekommenderas för dessa patienter (se avsnitt Varningar och försiktighet och Farmakokinetiska egenskaper). Det finns ingen klinisk erfarenhet av patienter med gravt nedsatt leverfunktion (Child Pugh C), så kabozantinib rekommenderas inte att användas till dessa patienter (se avsnitt Farmakokinetiska egenskaper).
Nedsatt hjärtfunktion
Det finns begränsade data från patienter med nedsatt hjärtfunktion. Inga specifika doseringsrekommendationer kan ges.
Pediatrisk population
Säkerhet och effekt för kabozantinib för barn och ungdomar i åldern under 18 år har ännu inte fastställts. Tillgänglig information finns i avsnitt Biverkningar, Farmakodynamiska egenskaper och Farmakokinetiska egenskaper men inga doseringsrekommendationer kan fastställas.
Administreringssätt
CABOMETYX är avsett för oral användning. Tabletterna ska sväljas hela och får inte krossas. Patienter ska instrueras att inte äta något under minst 2 timmar före och 1 timme efter intaget av CABOMETYX.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Eftersom de flesta biverkningar inträffar tidigt under behandlingen, bör läkaren övervaka patienten noga under de första åtta veckorna av behandlingen för att avgöra om det krävs några dosändringar. Biverkningar som i allmänhet uppkommer tidigt inkluderar hypokalcemi, hypokalemi, trombocytopeni, hypertoni, palmar-plantar erytrodysestesi (PPES), proteinuri och gastrointestinala biverkningar (buksmärta, slemhinneinflammation, förstoppning, diarré, kräkning).
Hantering av misstänkta biverkningar kan kräva tillfälligt behandlingsavbrott eller dosreducering av kabozantinib (se avsnitt Dosering och administreringssätt):
Dosreducering och behandlingsavbrott på grund av en biverkning förekom hos 46-67 % respektive 70-84 % av de patienter som behandlades med kabozantinib i de pivotala kliniska monoterapistudierna med RCC (METEOR, CABOSUN), HCC (CELESTIAL), DTC (COSMIC-311) och NET (CABINET). Två dosreduceringar krävdes för 9,4–33 % av patienterna. Mediantiden till första dosreduceringen var 38-106 dagar och till första behandlingsavbrottet 28-68 dagar.
När kabozantinib ges i kombination med nivolumab som första linjens behandling av avancerad RCC, förekom dosreducering samt behandlingsavbrott för kabozantinib på grund av biverkning hos 54,1 % respektive 73,4 % av patienterna i den kliniska studien (CA2099ER). Två dosreduceringar krävdes för 9,4 % av patienterna. Mediantiden till första dosreduceringen var 106 dagar och till första behandlingsavbrottet var 68 dagar.
Levertoxicitet
Avvikelser i leverfunktionstester (ökning av alaninaminotransferas [ALAT], aspartataminotransferas [ASAT] och bilirubin) har ofta observerats hos patienter som behandlas med kabozantinib. Det rekommenderas att utföra leverfunktionstester (ALAT, ASAT och bilirubin) innan behandling med kabozantinib påbörjas och noggrann övervakning under behandlingen. För patienter där försämrat resultat på leverfunktionstest anses relaterat till kabozantinibbehandling (där ingen alternativ orsak är uppenbar) bör anvisningarna för dosmodifiering i Tabell 1 följas (se avsnitt Dosering och administreringssätt).
När kabozantinib ges i kombination med nivolumab, har högre frekvenser av ALAT- och ASAT-förhöjningar av grad 3 och 4 rapporterats i förhållande till monoterapi med kabozantinib hos patienter med avancerad njurcellscancer (se avsnitt Biverkningar). Leverenzymer bör kontrolleras innan behandlingen påbörjas samt regelbundet under behandlingen. Riktlinjer för medicinsk hantering för båda läkemedlen bör följas (se avsnitt Dosering och administreringssätt samt nivolumabs produktresumé).
Sällsynta fall av vanishing bile duct syndrome (förlust av gallgångar) har rapporterats. Samtliga fall har inträffat hos patienter som har fått immuncheckpointhämmare, antingen före eller tillsammans med kabozantinibbehandling.
Kabozantinib elimineras huvudsakligen via levern. Noggrannare övervakning av den övergripande säkerheten rekommenderas för patienter med lindrigt eller måttligt nedsatt leverfunktion (se även avsnitt Dosering och administreringssätt och Farmakokinetiska egenskaper). En högre relativ andel patienter med måttligt nedsatt leverfunktion (Child-Pugh B) utvecklade hepatisk encefalopati under kabozantinibbehandling. kabozantinib rekommenderas inte till patienter med gravt nedsatt leverfunktion (Child-Pugh C, se avsnitt Dosering och administreringssätt).
Hepatisk encefalopati
I HCC-studien (CELESTIAL) rapporterades hepatisk encefalopati oftare i kabozantinib- än i placeboarmen. Kabozantinib har förknippats med diarré, kräkningar, minskad aptit och elektrolytavvikelser. Hos patienter med hepatocellulär cancer och nedsatt leverfunktion kan dessa icke-hepatiska effekter vara utlösande faktorer för utveckling av hepatisk encefalopati. Patienterna ska övervakas angående tecken och symptom på hepatisk encefalopati.
Perforeringar och fistlar
Allvarliga gastrointestinala perforeringar och fistlar, ibland med dödlig utgång, har observerats med kabozantinib. Patienter som har inflammatorisk tarmsjukdom (t.ex. Crohns sjukdom, ulcerös kolit, peritonit, divertikulit eller appendicit), har gastrointestinal tumörinfiltration eller har komplikationer från tidigare gastrointestinal kirurgi (särskilt när detta är förenat med fördröjd eller ofullständig läkning) bör utvärderas noggrant före insättning av behandling med kabozantinib och därefter kontrolleras noggrant avseende symtom på perforeringar och fistlar inklusive abscesser och sepsis. Ihållande eller återkommande diarré under behandlingen kan vara en riskfaktor för uppkomsten av analfistlar. Kabozantinib ska sättas ut för patienter som får en gastrointestinal perforering eller en fistel som inte kan hanteras på adekvat sätt.
Gastrointestinala (GI) besvär
Diarré, illamående/kräkningar, nedsatt aptit och stomatit/oral smärta var några av de vanligast rapporterade GI-händelserna (se avsnitt Biverkningar). Snabb medicinsk hantering, inklusive stödjande vård med antiemetika, antidiarroika eller antacida, bör sättas in för att förhindra uttorkning, obalans i elektrolyterna och viktminskning. Behandlingsavbrott, dosreducering eller permanent utsättning av kabozantinib bör beaktas vid bestående eller återkommande betydande GI-biverkningar (se Tabell 1).
Tromboemboliska händelser
Fall av venös tromboembolism, inklusive lungembolism, och arteriell tromboembolism, ibland dödlig, har observerats med kabozantinib. Kabozantinib bör användas med försiktighet till patienter som löper risk för eller som tidigare har haft något av detta. I HCC-studien (CELESTIAL) observerades portaventrombos med kabozantinib, inklusive ett dödsfall. Patienter med en historik av invasion i portavenen föreföll ha en högre risk att utveckla portaventrombos. Kabozantinib ska sättas ut för patienter som får en akut myokardinfarkt eller någon annan kliniskt signifikant tromboembolisk komplikation. I CABINET-studien var frekvensen av venös tromboembolism högre i pNET-kohorten (19 %) jämfört med epNET-kohorten (3,8 %) hos patienter som fick kabozantinib.
Blödning
Svår blödning, ibland dödlig, har observerats med kabozantinib. Patienter som har haft svåra blödningar innan behandlingen initieras måste utredas noggrant innan behandling med kabozantinib inleds. Kabozantinib ska inte administreras till patienter som har eller löper risk att få svåra blödningar.
I HCC-studien (CELESTIAL), rapporterades dödliga blödningar med högre incidens för kabozantinib jämfört med placebo. Riskfaktorer för svår blödning i populationen med avancerad hepatocellulär cancer kan innefatta tumörinvasion av större blodkärl och underliggande levercirros som resulterar i esofageala bråck, portahypertension och trombocytopeni. I CELESTIAL-studien uteslöts patienter som hade samtidig behandling med blodförtunnande läkemedel eller trombocytaggregationshämmare. Individer med obehandlade eller ofullständigt behandlade bråck med blödning eller hög risk för blödning uteslöts också från denna studie. I studien på kabozantinib i kombination med nivolumab vid första linjens behandling av avancerad njurcellscancer (CA2099ER) exkluderades patienter med antikoagulantia vid terapeutiska doser.
Aneurysmer och arteriella dissektioner
Användning av VEGF-hämmare till patienter med eller utan hypertoni kan främja bildningen av aneurysmer och/eller arteriella dissektioner. Denna risk ska noga övervägas innan kabozantinib sätts in hos patienter med riskfaktorer såsom hypertoni eller tidigare aneurysm.
Trombocytopeni
I HCC-studien (CELESTIAL), DTC-studien (COSMIC-311) och NET-studien (CABINET) rapporterades trombocytopeni och minskat antal blodplättar. Nivån på blodplättar bör övervakas under behandling med kabozantinib och dosen anpassas efter trombocytopenins svårighetsgrad (se Tabell 1).
Sårkomplikationer
Sårkomplikationer har observerats med kabozantinib. Om möjligt ska behandling med kabozantinib avbrytas minst 28 dagar före en planerad operation, inklusive tandkirurgiska ingrepp eller invasiva tandingrepp. Beslut om återupptagande av behandling med kabozantinib efter operation ska baseras på klinisk bedömning av adekvat sårläkning. Kabozantinib ska avbrytas för patienter med sårläkningskomplikationer som kräver medicinsk intervention.
Hypertoni
Hypertoni, inklusive hypertensiv kris har observerats med kabozantinib. Blodtrycket ska vara välkontrollerat före insättning av kabozantinib. Efter initering av kabozantinib, ska blodtrycket övervakas tidigt och regelbundet och behandlas vid behov med lämplig blodtryckssänkande behandling. Vid bestående förhöjning av blodtrycket trots blodtryckssänkande läkemedel ska behandling med kabozantinib avbrytas tills blodtrycket är kontrollerat. Därefter kan behandling med kabozantinib återupptas med reducerad dos. Kabozantinib ska sättas ut vid allvarlig och ihållande hypertoni trots blodtryckssänkande behandling och dosreducering av kabozantinib. Vid hypertonisk kris ska kabozantinib sättas ut.
Hjärtsvikt
Kabozantinib har förknippats med en ökad risk för hjärtsvikt. Denna risk kan förvärras av vanliga biverkningar av kabozantinib (till exempel hypertoni, hypotyreos och arteriella trombotiska händelser) som kan leda till hjärtsvikt. Patienterna ska övervakas för tecken och symtom på hjärtsvikt genom hela behandlingen. Dessa biverkningar ska hanteras omgående, behandlingsavbrott och/eller dosjustering bör övervägas om nödvändigt (se avsnitt Dosering och administreringssätt) och behandling med TKI ska avbrytas hos patienter som utvecklar svårt hjärtsvikt.
Osteonekros
Fall av osteonekros i käken har observerats med kabozantinib. En munundersökning bör genomföras före insättning av kabozantinib och regelbundet under behandlingen. Patienter bör instrueras i god munhygien. Behandlingen med kabozantinib bör om möjligt avbrytas minst 28 dagar före planerad tandkirurgi eller invasiva tandingrepp. Försiktighet är indicerat i patienter som får preparat förknippade med osteonekros i käken, såsom bisfosfonater. Kabozantinib bör sättas ut för patienter som får osteonekros i käken.
Palmar-plantar erytrodysestesi
Palmar-plantar erytrodysestesi (PPES) har observerats med kabozantinib. Vid allvarlig PPES bör man överväga att avbryta behandlingen med kabozantinib. Kabozantinib bör återupptas med en lägre dos när PPES har förbättrats till grad 1.
Proteinuri
Proteinuri har observerats med kabozantinib. Urinprotein bör kontrolleras regelbundet under behandling med kabozantinib. Kabozantinib bör sättas ut för patienter som får nefrotiskt syndrom.
Posteriort reversibelt encefalopatisyndrom
Posteriort reversibelt encefalopatisyndrom (PRES) har observerats med kabozantinib. Detta syndrom bör beaktas för alla patienter med multipla symtom, inklusive krampanfall, huvudvärk, synstörningar, förvirring eller förändrad mental funktion. Behandlingen med kabozantinib bör sättas ut för patienter med PRES.
Förlängning av QT-intervall
Kabozantinib bör användas med försiktighet till patienter som tidigare har haft förlängt QT-intervall, patienter som behandlas med antiarytmika och patienter med relevant tidigare hjärtsjukdom, bradykardi eller störningar i elektrolytbalansen. Vid användning av kabozantinib ska regelbunden övervakning med on-treatment EKG och elektrolyter (serumkalcium, kalium och magnesium) övervägas.
Sköldkörtelrubbingar
Laboratoriska baslinjemätningar av sköldkörtelfunktionen rekommenderas hos alla patienter. Patienter med redan förekommande hypotyreos eller hypertyreos bör behandlas enligt gällande medicinsk praxis innan behandling med kabozantinib påbörjas. Alla patienter bör hållas under noggrann övervakning för tecken och symptom på sköldkörtelrubbningar under behandlingen med kabozantinib. Sköldkörtelfunktion bör kontrolleras regelbundet under behandlingen med kabozantinib. Patienter som utvecklar sköldkörtelrubbning bör behandlas enligt gällande medicinsk praxis.
Biokemiska laboratorietestavvikelser
Kabozantinib har förknippats med ökad incidens av elektrolytavvikelser (inklusive hypo- och hyperkalemi, hypomagnesemi, hypokalcemi, hyponatremi). Hypokalcemi har observerats i en högre frekvens och/eller ökad svårighetsgrad (inklusive grad 3 och 5) med kabozantinib hos patienter med tyroideacancer i jämförelse med patienter med andra cancertyper. Det rekommenderas att biokemiska parametrar övervakas under behandling med kabozantinib och att lämplig ersättningsbehandling sätts in vid behov enligt klinisk praxis. Fall av hepatisk encefalopati hos patienter med hepatocellulär cancer kan anses bero på utvecklingen av elektrolytstörningar. Behandlingsavbrott, dosreducering eller permanent utsättning av kabozantinib bör övervägas vid bestående eller återkommande signifikanta avvikelser (se Tabell 1).
CYP3A4-inducerare och -hämmare
Kabozantinib är ett CYP3A4-substrat. Samtidig administrering av kabozantinib och den starka CYP3A4-hämmaren ketokonazol medförde en ökning av plasmaexponeringen av kabozantinib. Försiktighet krävs vid samtidig administrering av kabozantinib och medel som är starka CYP3A4-hämmare. Samtidig administrering av kabozantinib och den starka CYP3A4-induceraren rifampicin medförde en minskning av plasmaexponeringen av kabozantinib. Därför bör långvarig administrering av medel som är starka CYP3A4-inducerare tillsammans med kabozantinib undvikas (se avsnitt Dosering och administreringssätt och Interaktioner).
P-glykoproteinsubstrat
Kabozantinib var en hämmare (IC50 = 7,0 μM), men inte ett substrat för P-glykoprotein (P‑gp)-transportaktiviteter i ett dubbelriktat analyssystem med MDCK‑MDR1-celler. Därför kan kabozantinib potentiellt öka plasmakoncentrationerna av samtidigt administrerade substrat av P‑gp. Patienter ska varnas för att ta ett P‑gp-substrat (t.ex. fexofenadin, aliskiren, ambrisentan, dabigatranetexilat, digoxin, kolkicin, maravirok, posakonazol, ranolazin, saxagliptin, sitagliptin, talinolol, tolvaptan) samtidigt med kabozantinib (se avsnitt Interaktioner).
MRP2-hämmare
Administrering av MRP2-hämmare kan leda till ökning av plasmakoncentrationerna av kabozantinib. Därför bör försiktighet iakttas vid samtidig användning av MRP2-hämmare (t.ex. ciklosporin, efavirenz, emtricitabin) (se avsnitt Interaktioner).
Hjälpämnen
Laktos
Patienter med något av följande sällsynta ärftliga tillstånd bör inte använda detta läkemedel: galaktosintolerans, total laktasbrist eller glukos-galaktosmalabsorption.
Natrium
Detta läkemedel innehåller mindre än 1 mmol natrium (23 mg) per tablett, d.v.s är näst intill ”natriumfritt”.
Interaktioner
Effekt av andra läkemedel på kabozantinib
CYP3A4-hämmare och -inducerare
Administrering av den starka CYP3A4-hämmaren ketokonazol (400 mg dagligen i 27 dagar) till friska försökspersoner minskade clearance av kabozantinib (med 29 %) och ökade plasmaexponeringen vid en engångsdos av kabozantinib (AUC) med 38 %. Därför ska samtidig administrering av starka CYP3A4-hämmare (t.ex. ritonavir, itrakonazol, erytromycin, klaritromycin, grapefruktjuice) och kabozantinib ske med försiktighet.
Administrering av den starka CYP3A4-induceraren rifampicin (600 mg dagligen i 31 dagar) till friska försökspersoner ökade clearance av kabozantinib (4,3-faldigt) och minskade plasmaexponeringen vid en engångsdos av kabozantinib (AUC) med 77 %. Långvarig samtidig administrering av starka CYP3A4-inducerare (t.ex. fenytoin, karbamazepin, rifampicin, fenobarbital eller naturläkemedel som innehåller johannesört [Hypericum perforatum]) och kabozantinib ska därför undvikas.
Medel som förändrar pH i magsäcken
Samtidig administrering av protonpumpshämmaren (PPI) esomeprazol (40 mg dagligen i 6 dagar) och en engångsdos på 100 mg kabozantinib till friska försökspersoner ledde inte till någon kliniskt signifikant effekt på plasmaexponeringen av kabozantinib (AUC). Ingen dosjustering är indicerad när medel som förändrar pH i magsäcken (dvs. PPI:er, H2-receptorantagonister och antacida) ges samtidigt med kabozantinib.
MRP2-hämmare
In vitro-data visar att kabozantinib är ett substrat av MRP2. Därför kan administrering av MRP2-hämmare leda till ökningar av plasmakoncentrationerna av kabozantinib.
Sekvestreringsmedel för gallsalt
Sekvestreringsmedel för gallsalt såsom kolestyramin och kolesevelam (Cholestagel) kan interagera med kabozantinib och kan påverka absorption (eller reabsorption) vilket leder till potentiellt minskad exponering (se avsnitt Farmakokinetiska egenskaper). Den kliniska betydelsen av dessa potentiella interaktioner är okänd.
Effekten av kabozantinib på andra läkemedel
Effekten av kabozantinib på farmakokinetiken för kontraceptiva steroider har inte undersökts. Eftersom oförändrad kontraceptiv effekt inte kan garanteras, rekommenderas en ytterligare kontraceptiv metod, exempelvis en barriärmetod.
Effekten av kabozantinib på warfarins farmakokinetik har inte studerats. En interaktion med warfarin kan vara möjlig. I händelse av en sådan kombination, bör INR-värdet övervakas.
P-glykoproteinsubstrat
Kabozantinib var en hämmare (IC50 = 7,0 μM), men inte ett substrat, av P‑gp-transportaktiviteter i ett dubbelriktat analyssystem med MDCK‑MDR1-celler. Därför kan kabozantinib potentiellt öka plasmakoncentrationerna av samtidigt administrerade substrat av P‑gp. Patienter ska varnas för att ta ett P‑gp-substrat (t.ex. fexofenadin, aliskiren, ambrisentan, dabigatranetexilat, digoxin, kolkicin, maravirok, posakonazol, ranolazin, saxagliptin, sitagliptin, talinolol, tolvaptan) samtidigt med kabozantinib.
Fertilitet, graviditet och amning
Fertila kvinnor/Preventivmedel för män och kvinnor
Fertila kvinnor måste avrådas från att bli gravida medan de tar kabozantinib. Kvinnliga partners till manliga patienter som tar kabozantinib måste också undvika graviditet. Effektiva preventivmetoder bör användas av både manliga och kvinnliga patienter och deras partners under behandling och i minst 4 månader efter avslutad behandling. Eftersom p-piller möjligen inte anses vara ett ”effektivt preventivmedel” ska de användas tillsammans med en annan metod, exempelvis en barriärmetod (se avsnitt Interaktioner).
Graviditet
Det finns inga studier med gravida kvinnor som använder kabozantinib. Djurstudier har visat embryofetala och teratogena effekter (se avsnitt Prekliniska säkerhetsuppgifter). Den potentiella risken för människor är okänd. Kabozantinib ska endast användas under graviditet då tillståndet innebär att det är absolut nödvändigt att kvinnan behandlas med kabozantinib.
Amning
Det är okänt om kabozantinib och/eller dess metaboliter utsöndras i bröstmjölk. På grund av den potentiella skadan för barnet ska mödrar avstå från amning under behandling med kabozantinib och i minst 4 månader efter avslutad behandling.
Fertilitet
Det finns inga data om human fertilitet. Baserat på icke-kliniska säkerhetsdata kan manlig och kvinnlig fertilitet nedsättas av behandling med kabozantinib (se avsnitt Prekliniska säkerhetsuppgifter). Både män och kvinnor bör rådas att söka rådgivning och överväga fertilitetsbevarande åtgärder före behandling.
Effekter på förmågan att framföra fordon och använda maskiner
Kabozantinib har mindre effekt på förmågan att framföra fordon och använda maskiner. Biverkningar som trötthet och svaghet har förknippats med kabozantinib. Därför ska patienter rekommenderas att iaktta försiktighet vid framförande av fordon och användning av maskiner.
Biverkningar
Kabozantinib som monoterapi
Sammanfattning av säkerhetsprofil
De vanligaste allvarliga biverkningarna hos patienterna med njurcellscancer (incidensen ≥ 1 %) är pneumoni, buksmärta, diarré, illamående, hypertoni, blodproppar, hyponatremi, lungemboli, kräkningar, dehydrering, trötthet, asteni, minskad aptit, djup ventrombos, yrsel, hypomagnesemi och palmar-plantar erytrodysestesi (PPES).
De vanligaste allvarliga biverkningarna hos patienterna med hepatocellulär cancer (incidensen ≥ 1 %) är hepatisk encefalopati, asteni, trötthet, PPES, diarré, hyponatremi, kräkningar, buksmärta och trombocytopeni.
De vanligaste allvarliga biverkningarna hos patienterna med differentierad tyreoideacancer (incidensen ≥ 1 %) är diarré, pleurautgjutning, pneumoni, lungembolism, hypertoni, anemi, djup ventrombos, hypokalcemi, osteonekros i käke, smärta, PPES, kräkningar och nedsatt njurfunktion.
De vanligaste allvarliga biverkningarna hos patienterna med neuroendokrina tumörer (incidensen ≥ 1 %) är hypertoni, trötthet, lungembolism, kräkningar, diarré, illamående och emboli.
De vanligaste biverkningarna oavsett grad (som uppkommit hos minst 25 % av patienterna) hos patienterna med njurcellscancer, hepatocellulär cancer, differentierad tyreoideacancer och neuroendokrina tumörer var diarré, trötthet, illamående, minskad aptit, PPES och hypertoni.
Biverkningstabell
Biverkningar som rapporterats i den sammanslagna datan för patienter som behandlats med kabozantinib som monoterapi vid njurcellscancer, hepatocellulär cancer, differentierad tyreoideacancer och neuroendokrina tumörer (n = 1355) eller som rapporterats efter marknadsföring av kabozantinib listas i tabell 2. Biverkningarna listas efter MedDRAs organsystemklassificering och frekvenskategorier. Frekvenser baseras på alla grader och definieras som: mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100), ingen känd frekvens (kan inte beräknas från tillgängliga data). Inom varje frekvensområde presenteras biverkningarna efter fallande allvarlighetsgrad.
Tabell 2: Biverkningar som har rapporterats i kliniska studier eller efter marknadsföring hos patienter som behandlats med kabozantinib som monoterapi
| Infektioner och infestationer | |
| Vanliga | abscess, pneumoni |
| Blodet och lymfsystemet | |
| Mycket vanliga | anemi, trombocytopeni |
| Vanliga | neutropeni, lymfopeni |
| Endokrina systemet | |
| Mycket vanliga | hypotyreos* |
| Metabolism och nutrition | |
| Mycket vanliga | minskad aptit, hypomagnesemi, hypokalemi, hypoalbuminemi, hypokalcemi |
| Vanliga | uttorkning, hypofosfatemi, hyponatremi, hyperkalemi, hyperbilirubinemi, hyperglykemi, hypoglykemi |
| Centrala och perifera nervsystemet | |
| Mycket vanliga | dysgeusi, huvudvärk, yrsel |
| Vanliga | perifer neuropatia |
| Mindre vanliga | kramp, cerebrovaskulär händelse, reversibelt posteriort, leukoencefalopatisyndrom |
| Öron och balansorgan | |
| Vanliga | tinnitus |
| Hjärtat | |
| Mindre vanliga | akut hjärtinfarkt, hjärtsvikt |
| Blodkärl | |
| Mycket vanliga | hypertoni, blödningb* |
| Vanliga | ventrombosc, hypotoni, emboli |
| Mindre vanliga | hypertensiv kris, arteriell trombos, artäremboli |
| Ingen känd frekvens | aneurysmer och arteriella dissektioner |
| Andningsvägar, bröstkorg och mediastinum | |
| Mycket vanliga | dysfoni, dyspné, hosta |
| Vanliga | lungembolism, allergisk rinit |
| Mindre vanliga | pneumothorax |
| Magtarmkanalen | |
| Mycket vanliga | diarré*, illamående, kräkning, stomatit, förstoppning, buksmärta, dyspepsi |
| Vanliga | gastrointestinal perforation*g, pankreatit, fistel*, gastroesofageal refluxsjukdom, hemorrojder, oral smärta, muntorrhet, dysfagi, gasbildning |
| Mindre vanliga | glossodyni |
| Lever och gallvägar | |
| Vanliga | hepatisk encefalopati* |
| Mindre vanliga | kolestatisk hepatit |
| Hud och subkutan vävnad | |
| Mycket vanliga | palmar-plantar erytrodysestesi, hudutslagf |
| Vanliga | pruritus, alopeci, torr hud, förändrad hårfärg, hyperkeratos, erytem |
| Ingen känd frekvens | kutan vaskulit |
| Muskuloskeletala systemet och bindväv | |
| Mycket vanliga | smärta i extremitet, artralgi |
| Vanliga | muskelspasmer |
| Mindre vanliga | osteonekros i käken |
| Njurar och urinvägar | |
| Vanliga | proteinuri |
| Allmänna symtom och/eller symtom vid administreringsstället | |
| Mycket vanliga | trötthet, mukosal inflammation, asteni, perifera ödem |
| Undersökningard | |
| Mycket vanliga | viktnedgång, förhöjt ALAT, ASAT i serum, förhöjt alkaliskt fosfatas i blod |
| Vanliga | förhöjt GGT, förhöjt blodkreatinin, förhöjt amylas, förhöjt lipas, förhöjt blodkolesterol, förhöjda triglycerider i blodet, minskat leukocytantal |
| Skador och förgiftningar och behandlings-komplikationer | |
| Mindre vanliga | sårkomplikationere |
* Se avsnitt Biverkningar Beskrivning av utvalda biverkningar för ytterligare karaktärisering.
a Inklusive perifer neuropati; perifer neuropati är främst senorisk
b Inklusive epistaxis som den vanligaste rapporterade biverkningen
c All ventrombos inklusive djup ventrombos
d Baserat på rapporterade biverkningar
e Försämrad läkning, sårkomplikationer och sårlossning
f Utslag är en sammansatt term som inkluderar dermatit, akneiform dermatit, bullös dermatit, exfolierande utslag, erytematöst hudutslag, follikulärt hudutslag, makulärt hudutslag, makulopapulärt hudutslag, papulärt hudutslag, kliande hudutslag och läkemedelsutslag.
g Fall med dödlig utgång har rapporterats
Kabozantinib i kombination med nivolumab vid första linjens behandling av avancerad njurcellscancer
Sammanfattning av säkerhetsprofilen
När kabozantinib administreras i kombination med nivolumab, se nivolumabs produktresumé innan behandlingen påbörjas. För ytterligare information om nivolumabs säkerhetsprofil vid monoterapi, se nivolumabs produktresumé.
I ett dataset för kabozantinib 40 mg en gång dagligen i kombination med nivolumab 240 mg varannan vecka vid njurcellscancer (n = 320), med en minsta uppföljningstid på 16 månader, är de vanligaste allvarliga biverkningarna (≥ 1 % incidens) diarré, pneumonit, lungemboli, pneumoni, hyponatremi, pyrexi, binjurebarksvikt, kräkning, uttorkning.
De vanligaste biverkningarna (≥ 25 %) var diarré, trötthet, palmar-plantar erytrodysestesi, stomatit, muskuloskeletal smärta, hypertension, hudutslag, hypotyreos, minskad aptit, illamående, buksmärta. Majoriteten av biverkningarna var milda till måttliga (grad 1 eller 2).
Biverkningstabell
Biverkningar som identifierades i den kliniska studien av kabozantinib i kombination med nivolumab är listade i tabell 3 enligt MedDRA-klassificering av organsystem och frekvenskategorier. Frekvenserna baseras på alla grader och definieras som: mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100), ingen känd frekvens (kan inte beräknas från tillgängliga data). Inom varje frekvensgrupp presenteras biverkningar efter fallande allvarlighetsgrad.
Tabell 3: Biverkningar av kabozantinib i kombination med nivolumab
| Infektioner och infestationer | |
| Mycket vanliga | övre luftvägsinfektioner |
| Vanliga | pneumoni |
| Blodet och lymfsystemet | |
| Vanliga | eosinofili |
| Immunsystemet | |
| Vanliga | överkänslighet(inklusive anafylaktisk reaktion) |
| Mindre vanliga | infusionsrelaterad överkänslighetsreaktion |
| Endokrina systemet | |
| Mycket vanliga | hypotyreos, hypertyreos |
| Vanliga | binjurebarksvikt |
| Mindre vanliga | hypofysit, tyreoidit |
| Metabolism och nutrition | |
| Mycket vanliga | minskad aptit |
| Vanliga | uttorkning |
| Centrala och perifera nervsystemet | |
| Mycket vanliga | dysgeusi, yrsel, huvudvärk |
| Vanliga | perifer neuropati |
| Mindre vanliga | autoimmun encefalit, Guillain-Barrés syndrom, myasteniskt syndrom |
| Öron och balansorgan | |
| Vanliga | tinnitus |
| Ögon | |
| Vanliga | torra ögon, dimsyn |
| Mindre vanliga | uveit |
| Hjärtat | |
| Vanliga | förmaksflimmer, takykardi |
| Mindre vanliga | myokardit |
| Blodkärl | |
| Mycket vanliga | hypertension |
| Vanliga | trombosa |
| Mindre vanliga | artäremboli |
| Andningsvägar, bröstkorg och mediastinum | |
| Mycket vanliga | dysfoni, dyspné, hosta |
| Vanliga | pneumonit, lungembolism, epistaxis, pleural effusion |
| Mindre vanliga | pneumothorax |
| Magtarmkanalen | |
| Mycket vanliga | diarré, kräkning, illamående, förstoppning, stomatit, buksmärta, dyspepsi |
| Vanliga | kolit, gastrit, oral smärta, muntorrhet, hemorrojder |
| Mindre vanliga | pankreatit, tunntarmsperforeringb, glossodyni |
| Lever och gallvägar | |
| Vanliga | hepatit |
| Ingen känd frekvens | vanishing bile duct syndrome (förlust av gallgångar)c |
| Hud och subkutan vävnad | |
| Mycket vanliga | palmar-plantar erytrodysestesi, hudutslagd, pruritus |
| Vanliga | alopeci, torr hud, erytem, förändrad hårfärg |
| Mindre vanliga | psoriasis, urtikaria |
| Ingen känd frekvens | kutan vaskulit |
| Muskuloskeletala systemet och bindväv | |
| Mycket vanliga | muskuloskeletal smärtae, artralgi, muskelspasmer |
| Vanliga | artrit |
| Mindre vanliga | myopati, osteonekros i käken, fistel |
| Njurar och urinvägar | |
| Mycket vanliga | proteinuri |
| Vanliga | njursvikt, akut njurskada |
| Mindre vanliga | nefrit |
| Allmänna symtom och/eller symtom vid administreringsstället | |
| Mycket vanliga | trötthet, pyrexi, ödem |
| Vanliga | smärta, bröstsmärta |
| Undersökningarf | |
| Mycket vanliga | förhöjt ALAT, förhöjt ASAT, hypofosfatemi, hypokalcemi, hypomagnesemi, hyponatremi, hyperglykemi, lymfopeni, förhöjt alkaliskt fosfatas, förhöjt lipas, förhöjt amylas, thrombocytopeni, förhöjt kreatinin, anemi, leukopeni, hyperkalemi, neutropeni, hyperkalcemi, hypoglykemi, hypokalemi, förhöjt totalt bilirubin, hypermagnesemi, hypernatremi, viktminskning |
| Vanliga | förhöjt blodkolesterol, hypertriglyceridemi |
Biverkningsfrekvenserna som presenteras i tabell 3 beror inte nödvändigtvis enbart på kabozantinib, utan kan innefatta bidrag från underliggande sjukdom eller från nivolumab som används i kombination.
a Trombos är en sammansatt term som inkluderar portventrombos, pulmonell ventrombos, pulmonell trombos, aortatrombos, arteriell trombos, djup ventrombos, bäckenvenstrombos, vena cavatrombos, venös trombos, venös trombos i extremiteter.
b Dödsfall har rapporterats.
c Med tidigare eller samtidig exponering för immuncheckpointhämmare.
d Utslag är en sammansatt term som inkluderar dermatit, acneiform dermatit, bullös dermatit, exfoliativt utslag, erytematöst utslag, follikelutslag, makulärt utslag, makulopapulärt utslag, papulärt utslag, pruritiska utslag och läkemedelsutslag.
e Muskuloskeletal smärta är en sammansatt term som inkluderar ryggsmärta, bensmärta, muskuloskeletal bröstsmärta, muskuloskeletalt obehag, myalgi, nacksmärta, smärta i extremiteter, spinal smärta.
f Frekvenser av laboratoriska testvärden återspeglar andelen patienter som upplevde en försämring från baslinjen i laboratoriemätningar med undantag för viktminskning, ökat kolesterol i blodet och hypertriglyceridemi.
Beskrivning av utvalda biverkningar
Data för nedanstående reaktioner är baserade på patienter som fått CABOMETYX 60 mg peroralt en gång dagligen som monoterapi i den pivotala RCC-studien efter tidigare VEGF-riktad behandling och för behandlingsnaiv RCC, i HCC efter tidigare systemisk behandling, i DTC för patienter som är refraktära eller inte lämpliga för radioaktivt jod (RAI) som har progredierat under eller efter tidigare systemisk behandling, i NET som har progredierat efter tidigare systemisk behandling eller patienter som fått CABOMETYX 40 mg peroralt en gång dagligen i kombination med nivolumab vid första linjens behandling av avancerad RCC (avsnitt Farmakodynamiska egenskaper).
Gastrointestinal perforering (se avsnitt Varningar och försiktighet)
I RCC-studien (METEOR), rapporterades gastrointestinala perforeringar hos 0,9 % (3/331) av RCC-patienter som behandlats med kabozantinib. Händelserna var av grad 2 eller 3. Mediantiden till debut var 10,0 veckor.
I RCC-studien för behandlingsnaiv RCC (CABOSUN) rapporterades gastrointestinala perforeringar hos 2,6 % (2/78) av patienterna behandlade med kabozantinib. Händelserna var av grad 4 och 5.
I HCC-studien (CELESTIAL) rapporterades gastrointestinala perforeringar hos 0,9 % av patienterna som behandlats med kabozantinib (4/467). Alla händelser var av grad 3 eller 4. Mediantiden till debut var 5,9 veckor. I DTC-studien (COSMIC-311), rapporterades gastrointestinal perforering av grad 4 hos en av patienterna (0,6 %) som behandlats med kabozantinib och förekom efter 14 veckors behandling. I NET-studien (CABINET), rapporterades gastrointestinala perforeringar hos 1,3 % av patienter som behandlats med kabozantinib (3/227). Händelserna var av grad 3, 4 och 5. Mediantiden till debut var 21,6 veckor. I kombination med nivolumab vid avancerad RCC vid första linjens behandling (CA2099ER) var incidensen av GI-perforeringar 1,3 % (4/320) hos de behandlade patienterna. En händelse var av grad 3, två händelser var av grad 4 och en händelse var av grad 5 (dödlig).
Dödliga perforeringar har förekommit i det kliniska kabozantinib-programmet.
Hepatisk encefalopati (se avsnitt Varningar och försiktighet)
I HCC-studien (CELESTIAL) rapporterades hepatisk encefalopati (hepatisk encefalopati, encefalopati, hyperammonemisk encefalopati) hos 5,6 % av patienterna som behandlats med kabozantinib (26/467); varav grad 3-4-händelser var 2,8 % och en (0,2 %) grad 5-händelse. Mediantiden till debut var 5,9 veckor.
I NET-studien (CABINET), rapporterades hepatisk encefalopati hos 0,9 % av patienterna som behandlats med kabozantinib (2/227). Det var en händelse av grad 3 (0,4 %) för vilken mediantiden till debut var 14,3 veckor.
Inga fall av hepatisk encefalopati rapporterades i RCC-studierna (METEOR, CABOSUN och CA2099ER) och i DTC-studien (COSMIC-311).
Diarré (se avsnitt Varningar och försiktighet)
I RCC-studien (METEOR), rapporterades diarré hos 74 % av RCC-patienterna som behandlats med kabozantinib (245/331); varav grad 3-4-händelser var 11 %. Mediantiden till debut var 4,9 veckor.
I RCC-studien med behandlingsnaiv RCC (CABOSUN) rapporterades diarré hos 73 % av patienterna som behandlats med kabozantinib (57/78), varav grad 3-4-händelser var 10 %.
I HCC-studien (CELESTIAL) rapporterades diarré hos 54 % av patienterna som behandlats med kabozantinib (251/467), varav grad 3-4-händelser var 9,9 %. Mediantiden till debut av alla händelser var 4,1 veckor. Diarrén ledde till dosjusteringar, behandlingsavbrott och permanent utsättning hos 84/467 (18 %), 69/467 (15 %) respektive 5/467 (1 %) av patienterna.
I DTC-studien (COSMIC-311) rapporterades diarré hos 62 % av patienterna som behandlats med kabozantinib (105/170), varav grad 3-4 händelser var 7,6 %. Diarrén ledde till dosreduceringar och behandlingsavbrott hos 24/170 (1 %) respektive 36/170 (21 %) av patienterna.
I NET-studien (CABINET), rapporterades diarré hos 63 % av patienterna som behandlats med kabozantinib (144/227), varav 8,4 % var grad 3-händelser, inga grad 4 händelser. Mediantiden till debut av grad 3-händelser var 5,1 veckor.
I kombination med nivolumab vid avancerad RCC vid första linjens behandling (CA2099ER) rapporterades incidensen av diarré hos 64,7 % (207/320) av de behandlade patienterna, händelser av grad 3-4 hos 8,4 % (27/320). Mediantiden till utveckling av alla händelserna var 12,9 veckor. Dosfördröjning eller dosreducering inträffade hos 26,3 % (84/320) respektive utsättning hos 2,2 % (7/320) av patienter med diarré.
Fistlar (se avsnitt Varningar och försiktighet)
I RCC-studien (METEOR), rapporterades fistlar hos 1,2 % (4/331) av patienterna som behandlats med kabozantinib och inkluderade analfistlar hos 0,6 % (2/331) av patienterna som behandlats med kabozantinib. En händelse var av grad 3; återstoden var av grad 2. Mediantiden till debut var 30,3 veckor.
I RCC-studien med behandlingsnaiv RCC (CABOSUN) rapporterades inga fall av fistlar.
I HCC-studien (CELESTIAL) rapporterades fistlar hos 1,5 % (7/467) av HCC-patienterna. Mediantiden till debut var 14 veckor.
I DTC-studien (COSMIC-311), rapporterades fistlar (två anala och ett i svalg) hos 1,8 % (3/170) av patienter som behandlats med kabozantinib.
I NET-studien (CABINET), rapporterades fistlar (två anala och en i gallgång) hos 1,3 % (3/227) av patienterna som behandlats med kabozantinib. Analfistel var av grad 1 och 3, gallgångsfistel var av grad 2. Mediantiden till debut var 19,3 veckor.
I kombination med nivolumab vid avancerarad RCC vid första linjens behandling (CA2099ER) rapporterades incidensen av fistlar hos 0,9 % (3/320) av de behandlade patienterna och svårighetsgraden var grad 1.
Dödliga fistlar har förekommit i det kliniska kabozantinib-programmet.
Blödning (se avsnitt Varningar och försiktighet)
I RCC-studien (METEOR) var incidensen av svåra blödningshändelser (grad ≥ 3) 2,1 % (7/331) för RCC-patienter som behandlats med kabozantinib. Mediantiden till debut var 20,9 veckor.
I RCC-studien med behandlingsnaiv RCC (CABOSUN), var incidensen av svåra blödningshändelser (grad ≥ 3) för RCC-patienter som behandlats med kabozantinib 5,1 % (4/78).
I HCC-studien (CELESTIAL) var incidensen av svåra blödningshändelser (grad ≥ 3) 7,3 % för patienter som behandlats med kabozantinib (34/467). Mediantiden till debut var 9,1 veckor.
I DTC-studien (COSMIC-311), var incidensen av svåra blödninghändelser (grad ≥ 3) 2,4 % för patienter som behandlats med kabozantinib (4/170). Mediantiden till debut var 11,5 veckor.
I NET-studien (CABINET), var incidensen av svåra blödninghändelser (grad ≥ 3) 1,8 % hos patienter som behandlats med kabozantinib (4/227). Mediantiden till debut var 14,1 veckor.
I kombination med nivolumab vid avancerad RCC vid första linjens behandling (CA2099ER) var incidensen ≥ grad 3-blödningar 1,9 % (6/320) hos de behandlade patienterna.
Dödliga blödningar har förekommit i det kliniska kabozantinib-programmet.
Posteriort reversibelt encefalopatisyndrom (PRES) (se avsnitt Varningar och försiktighet)
Inget fall av PRES rapporterades i METEOR-, CABOSUN-, CA2099ER- eller CELESTIAL-studierna, men PRES har rapporterats hos en av patienterna i DTC-studien (COSMIC-311) och hos en patient i NET-studien (CABINET). PRES har i sällsynta fall rapporterats i andra kliniska studier (hos 2/4872 patienter; 0,04 %).
Förhöjda leverenzymer när kabozantinib kombineras med nivolumab vid njurcellscancer
I en klinisk studie av tidigare obehandlade patienter med RCC som fick kabozantinib i kombination med nivolumab sågs en högre incidens av förhöjt ALAT (10,1 %) och förhöjt ASAT (8,2 %) av grad 3 och 4 i förhållande till kabozantinib monoterapi hos patienter med avancerad RCC (ALAT ökade med 3,6 % och ASAT ökade med 3,3 % i METEOR-studien). Mediantiden till utveckling av förhöjt ALAT eller ASAT av grad > 2 var 10,1 veckor (intervall: 2 till 106,6 veckor; n = 85). Hos 91 % av patienterna med förhöjt ALAT eller ASAT av grad ≥ 2, gick förhöjningarna tillbaka till grad 0-1 med en mediantid för tillbakagång på 2,3 veckor (intervall: 0,4 till 108,1 veckor).
Bland de 45 patienter med förhöjt ALAT eller ASAT av grad ≥ 2 som åter behandlades med antingen kabozantinib (n = 10) eller nivolumab (n = 10) administrerat som monoterapi eller med båda (n = 25), observerades återfall av förhöjt ALAT eller ASAT grad ≥ 2 hos 4 patienter som fick kabozantinib, hos 3 patienter som fick nivolumab och hos 8 patienter som fick både kabozantinib och nivolumab.
Hypotyreos
I RCC-studien (METEOR) var indicensen av hypotyreos 21 % (68/331).
I den behandlingsnaiva RCC-studien (CABOSUN) var incidensen av hypotyreos 23 % (18/78) hos kabozantinibbehandlade RCC-patienter.
I HCC-studien (CELESTIAL) var incidensen av hypotyreos 8,1 % (38/467) hos kabozantinibbehandlade patienter och grad 3-händelser hos 0,4 % (2/467).
I DTC-studien (COSMIC-311), var incidensen av hypotyreos 2,4 % (4/170), alla av grad 1–2, utan behov av ändringar i behandlingen.
I NET-studien (CABINET), var incidensen av hypotyreos 26 % (59/227) hos patienter som behandlats med kabozantinib, alla av grad 1–2.
I kombination med nivolumab vid avancerad RCC vid första linjens behandling (CA2099ER) var incidensen av hypotyreos 35,6 % (114/320) av de behandlade patienterna.
Pediatrisk population (se avsnitt Farmakodynamiska egenskaper)
I studien ADVL1211, en begränsad doseskaleringsstudie av kabozantinib hos pediatriska och tonåriga patienter med återkommande eller refraktära solida tumörer inklusive CNS-tumörer observerades följande händelser: förhöjt ASAT (mycket vanligt, 76,9 %), förhöjt ALAT (mycket vanligt, 71,8 %), minskat antal lymfocyter (mycket vanligt, 48,7 %), minskat antal neutrofiler (mycket vanligt, 35,9 %) och förhöjt lipas (mycket vanligt, 33,3 %) vid en högre frekvens hos alla försökspersoner i alla dosgrupper som ingick i säkerhetspopulationen (N = 39), jämfört med vuxna. De ökade frekvenserna för dessa patienter gäller alla grader samt grad 3/4 av dessa biverkningar. De rapporterade biverkningarna överensstämmer kvalitativt med den kända säkerhetsprofilen för kabozantinib i vuxna populationer. Det låga antalet försökspersoner utesluter dock en slutgiltig bedömning av trender och frekvenser och ytterligare jämförelse med kabozantinibs kända säkerhetsprofil.
I studien ADVL1622 av kabozantinib hos barn och unga vuxna med följande solida tumör-strata: Ewing sarkom, rhabdomyosarkom, icke-rhabdomyosarkom mjukdelssarkom (NRSTS), osteosarkom, Wilms tumör och andra sällsynta solida tumörer (icke-statistisk kohort), säkerhetsprofilen för kabozantinibbehandlade barn och unga vuxna i alla strata var jämförbar med den som observerades hos vuxna som behandlades med kabozantinib.
Vidgning av fysen har observerats hos barn med öppna tillväxtplattor som behandlades med kabozantinib.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till
Sverige:
Läkemedelsverket
Box 26
751 03 Uppsala
www.lakemedelsverket.se
Finland:
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Det finns ingen specifik behandling för överdosering av kabozantinib och möjliga överdoseringssymtom har inte fastställts.
I händelse av misstänkt överdos ska kabozantinib sättas ut och stödjande behandling sättas in. Metabola kliniska laboratorievärden bör kontrolleras minst en gång per vecka eller när det bedöms vara kliniskt lämpligt för att utvärdera eventuella trendförändringar. Biverkningar som förknippas med överdosering ska behandlas symtomatiskt.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: antineoplastiska medel, proteinkinashämmare, ATC-kod: L01EX07.
Verkningsmekanism
Kabozantinib är en liten molekyl som hämmar flera receptortyrosinkinaser (RTK) som är inblandade i tumörtillväxt och angiogenes, patologisk benremodellering, läkemedelsresistens och metastatisk utveckling av cancer. Kabozantinib utvärderades när det gällde dess hämmande aktivitet mot flera olika kinaser och identifierades som en hämmare av receptorerna för MET (proteinet hepatocyt-tillväxtfaktorreceptor) och VEGF (vaskulär endotel tillväxtfaktor). Dessutom hämmar kabozantinib andra tyrosinkinaser inklusive GAS6-receptorn (AXL), RET, ROS1, TYRO3, MER, stamcellsfaktorreceptorn (KIT), TRKB, Fms-liknande tyrosinkinas-3 (FLT3) och TIE-2.
Farmakodynamisk effekt
Kabozantinib uppvisade dosrelaterad hämning av tumörtillväxt, tumörregression, och/eller hämmad metastasering i ett brett spektrum av prekliniska tumörmodeller.
Hjärtelektrofysiologi
En ökning från baslinjen av korrigerat QT-intervall enligt Fridericia (QTcF) på 10–15 ms på dag 29 (men inte på dag 1) efter insättning av behandling med kabozantinib (med en dos på 140 mg en gång dagligen) sågs i en kontrollerad klinisk studie av patienter med medullär tyreoideacancer. Denna effekt var inte förenad med någon förändring i hjärtats vågformsmorfologi eller nya rytmer. Inga patienter som behandlades med kabozantinib i denna studie hade ett bekräftat QTcF > 500 ms, och samma sak gällde för patienter som behandlades med kabozantinib i RCC-, HCC- eller NET-studierna (vid en dos på 60 mg).
Klinisk effekt och säkerhet
Njurcellscancer
Randomiserad studie av patienter med njurcellscancer som tidigare genomgått vaskulär endotel tillväxtfaktor (VEGF)-riktad behandling (METEOR)
Säkerheten och effekten av CABOMETYX vid behandling av njurcellscancer, efter tidigare vaskulär endotel tillväxtfaktor (VEGF)-riktad behandling, utvärderades i en randomiserad, öppen multicenterstudie i fas 3 (METEOR). Patienter (N = 658) med avancerad RCC med en klarcellskomponent som tidigare hade fått minst 1 VEGF-receptor-tyrosinkinashämmare (VEGFR TKI) randomiserades (1:1) till kabozantinib (N = 330) eller everolimus (N = 328). Patienter kunde ha fått andra behandlingar, inklusive cytokiner, och antikroppar riktade mot VEGF, PD-1 (programmerad död)-receptorn, eller dess ligander. Patienter med behandlade hjärnmetastaser fick delta. Progressionsfri överlevnad (PFS) bedömdes av en blindad, oberoende radiologisk granskningskommitté, och den primära analysen utfördes bland de första 375 randomiserade patienterna. Sekundära effektmått var objektiv svarsfrekvens (ORR) och total överlevnad (OS). Tumörbedömningar gjordes var åttonde vecka under de första 12 månaderna, och därefter var tolfte vecka.
Demografi och sjukdomsegenskaper vid baslinjen var likartade mellan kabozantinib- och everolimusarmarna. Majoriteten av patienterna var män (75 %), med en medianålder på 62 år. Sjuttioen procent (71 %) fick endast en tidigare VEGFR TKI; 41 % av patienterna fick sunitinib som sin enda tidigare VEGFR TKI. Enligt kriterierna från Memorial Sloan Kettering Cancer Center för prognostisk riskkategori, var 46 % gynnsamma (0 riskfaktorer), 42 % hade en medelrisk (1 riskfaktor) och 13 % var ogynnsamma (2 eller 3 riskfaktorer). Femtiofyra procent (54 %) av patienterna hade 3 eller flera organ med metastatisk sjukdom, inklusive lunga (63 %), lymfkörtlar (62 %), lever (29 %) och skelett (22 %). Medianvärdet för behandlingslängden var 7,6 månader (intervall 0,3–20,5) för patienter som fick kabozantinib och 4,4 månader (intervall 0,21–18,9) för patienter som fick everolimus.
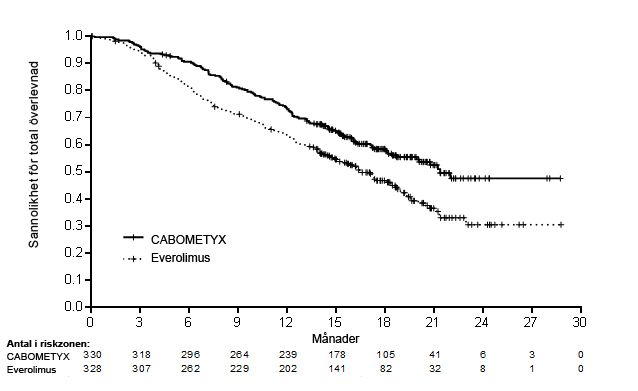
En statistiskt signifikant förbättring av PFS visades för kabozantinib jämfört med everolimus (figur 1 och tabell 4). En planerad interimsanalys av OS utfördes vid tiden för PFS-analysen och uppnådde inte interimsgränsen för statistisk signifikans (202 händelser, HR = 0,68 [0,51; 0,90], p=0,006). I en påföljande oplanerad interimsanalys av OS, visades en statistiskt signifikant förbättring för patienter som randomiserats till kabozantinib jämfört med everolimus (320 händelser, median på 21,4 månader mot 16,5 månader; HR = 0,66 [0,53; 0,83], p = 0,0003; figur 2). Jämförbara resultat för OS observerades i en uppföljande analys (beskrivande) efter 430 händelser.
Explorativa analyser av PFS och OS hos intention-to-treat (ITT)-gruppen har också visat enhetliga resultat till fördel för kabozantinib jämfört med everolimus i olika subgrupper enligt ålder (< 65 mot ≥ 65, kön, MSKCC-riskgrupp (gynnsam, medel, ogynnsam), ECOG-status (0 mot 1), tid från diagnos till randomisering (< 1 år mot ≥ 1 år), tumörens MET-status (hög jämfört med låg jämfört med okänd), skelettmetastaser (saknas jämfört med förekomst), viscerala metastaser (saknas jämfört med förekomst), viscerala och skelettmetastaser (saknas jämfört med förekomst), antal tidigare VEGFR-TKI (1 jämfört med ≥ 2), varaktighet för första VEGFR-TKI (≤ 6 månader jämfört med > 6 månader).
Objektiva svarsfrekvensfynd sammanfattas i tabell 5.
Figur 1: Kaplan-Meier-kurva för progressionsfri överlevnad enligt oberoende radiologisk granskningskommitté hos RCC-patienter efter tidigare vaskulär endotel tillväxtfaktor (VEGF)‑riktad behandling (De första 375 patienterna randomiserades) (METEOR)
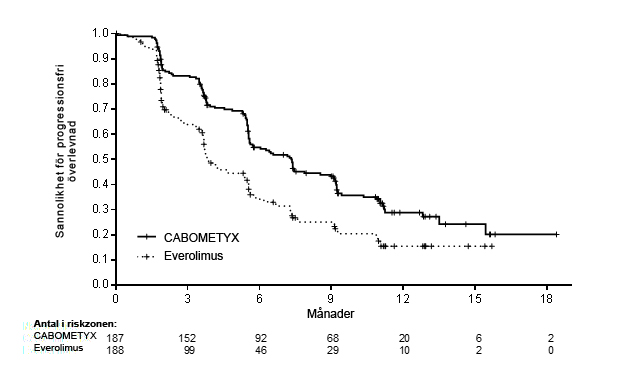
Tabell 4: Sammanfattning av PFS-fynd enligt oberoende radiologisk granskningskommitté hos RCC-patienter efter tidigare vaskulär endotel tillväxtfaktor (VEGF)-riktad behandling (METEOR)
| Primär PFS-analyspopulation | Intent-to-treat-population | |||
| Effektmått | CABOMETYX | Everolimus | CABOMETYX | Everolimus |
| N = 187 | N = 188 | N = 330 | N = 328 | |
| Medianvärde PFS (95 % KI), månader | 7,4 (5,6; 9,1) | 3,8 (3,7; 5,4) | 7,4 (6,6; 9,1) | 3,9 (3,7; 5,1) |
| HR (95 % KI), p‑värde1 | 0,58 (0,45; 0,74), p<0,0001 | 0,51 (0,41; 0,62), p<0,0001 | ||
1 stratifierat log-rank-test
Figur 2: Kaplan-Meier-kurva för total överlevnad hos RCC-patienter efter tidigare vaskulär endotel tillväxtfaktor (VEGF)-riktad behandling (METEOR)
Tabell 5: Sammanfattning av ORR-resultat per granskning av oberoende radiologisk kommitté (IRC) och prövarens granskning hos RCC-patienter efter tidigare vaskulär endotel tillväxtfaktor (VEGF)-riktad behandling
| Primär analys ORR Intent-to-treat-population (IRC) | ORR per prövarens granskning av Intent-to-treat-population | |||
| Effektmått | CABOMETYX | Everolimus | CABOMETYX | Everolimus |
| N = 330 | N = 328 | N = 330 | N = 328 | |
| ORR (endast partiella svar) (95 % KI) | 17 % (13 %, 22 %) | 3 % (2 %, 6 %) | 24 % (19 %, 29 %) | 4 % (2 %, 7 %) |
| p‑värde1 | p< 0,0001 | p< 0,0001 | ||
| Partiellt svar | 17 % | 3 % | 24 % | 4 % |
| Mediantid till första svar, månader (95 % KI) | 1,91 (1,6; 11,0) | 2,14 (1,9; 9,2) | 1,91 (1,3; 9,8) | 3,50 (1,8; 5,6) |
| Stabil sjukdom som bästa svar | 65 % | 62 % | 63 % | 63 % |
| Progressiv sjukdom som bästa svar | 12 % | 27 % | 9 % | 27 % |
1 chi-två-test
Randomiserad studie vid behandlingsnaiva njurcellscancerpatienter (CABOSUN)
Säkerheten och effekten av CABOMETYX vid behandling av behandlingsnaiv njurcellscancer utvärderades i en randomiserad, öppen multicenterstudie (CABOSUN). Patienter (N = 157) med tidigare obehandlad, lokalt avancerad eller metastaserande RCC med en klarcellskomponent randomiserades (1:1) till kabozantinib (N = 79) eller sunitinib (N=78). Patienterna var tvungna att ha intermediär eller dålig prognos som riskgruppskategori enligt International Metastatic RCC Database Consortium (IMDC). Patienterna stratifierades utifrån IMDC-riskgrupp och närvaro av benmetastaser (ja/nej). Cirka 75 % av patienterna genomgick en nefrektomi före behandlingens början.
För sjukdom med intermediär prognos uppnåddes en eller två av följande riskfaktorer, medan tre eller fler faktorer uppnåddes för dålig prognos: tid från diagnos av RCC till systemisk behandling < 1 år, Hgb < LLN, korrigerat kalcium > ULN, KPS < 80 %, antal neutrofiler > ULN och blodplättantal > ULN.
Det primära effektmåttet var PFS. Sekundära effektmått var objektiv svarsfrekvens (ORR) och total överlevnad (OS). Tumörbedömningar genomfördes var 12:e vecka.
Demografi och sjukdomsegenskaper vid baslinjen var likartade mellan kabozantinib- och sunitinibarmarna. Majoriteten av patienterna var män (78 %) med en medianålder på 62 år. Patientfördelningen enligt IMDC-riskgrupper var 81 % intermediär prognos (1-2 riskfaktorer) och 19 % dålig prognos (≥ 3 riskfaktorer). Majoriteten av patienterna (87 %) hade ECOG performance status 0 eller 1; 13 % hade ECOG performance status 2. Trettiosex procent (36 %) av patienterna hade benmetastaser.
En statistiskt signifikant förbättring av PFS, som bedömdes retrospektivt av en blindad oberoende radiologisk kommitté (IRC), visades för kabozantinib jämfört med sunitinib (Figur 3 och Tabell 6). Resultaten från prövarens och IRC:s granskning av PFS var överensstämmande.
Patienter med både positiv och negativ MET-status visade en gynnsam effekt med kabozantinib jämfört med sunitinib, med större aktivitet hos patienter med positiv MET-status jämfört med patienter med negativ MET-status (HR=0,32 (0,16, 0,63) vs 0,67 (0,37, 1,23).
Behandling med kabozantinib associerades med längre överlevnad jämfört med sunitinib (Tabell 6). Studien saknade power för OS-analys och datan var inte tillräckligt utvecklad.
Resultatet för objektiv svarsfrekvens (ORR) är sammanfattat i Tabell 6.
Figur 3: Kaplan-Meier-kurva för progressionsfri överlevnad av IRC i behandlingsnaiva RCC-patienter
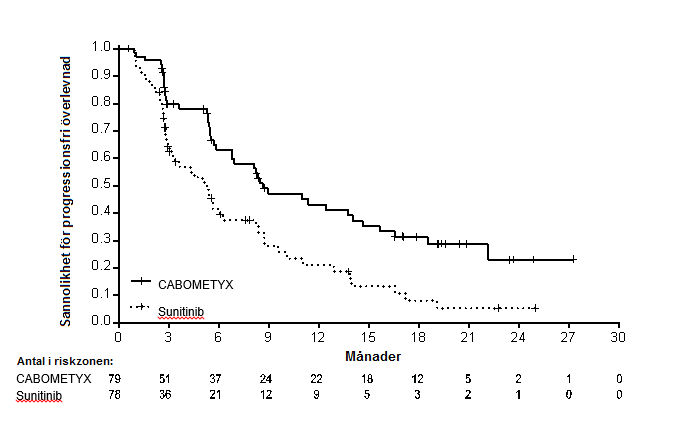
Tabell 6: Effektresultat hos behandlingsnaiva RCC-patienter (ITT population, CABOSUN)
CABOMETYX (N=79) | Sunitinib (N=78) | |
| Progressionsfri överlevnad (PFS) av IRC a | ||
| Median PFS i månader (95 % CI) | 8,6 (6,2, 14,0) | 5,3 (3,0, 8,2) |
| HR (95 % CI); stratifierat b,c | 0,48 (0,32, 0,73) | |
| Tvåsidigt log-rank p-värde: stratifierat b | p=0,0005 | |
| Progressionsfri överlevnad (PFS) av prövaren | ||
| Median PFS i månader (95 % CI) | 8,3 (6,5, 12,4) | 5,4 (3,4, 8,2) |
| HR (95 % CI); stratifierat b,c | 0,56 (0,37, 0,83) | |
| Tvåsidigt log-rank p-värde: stratifierat b | p=0,0042 | |
| Total överlevnad (OS) | ||
| Median OS i månader (95 % CI) | 30,3 (14,6, NE) | 21,0 (16,3, 27,0) |
| HR (95 % CI); stratifierat b,c | 0,74 (0,47, 1,14) | |
| Objektiv svarsfrekvens (ORR)n (%) av IRC | ||
| Fullständigt svar | 0 | 0 |
| Partiellt svar | 16 (20) | 7 (9) |
| ORR (endast partiella svar) | 16 (20) | 7 (9) |
| Stabil sjukdom | 43 (54) | 30 (38) |
| Progressiv sjukdom | 14 (18) | 23 (29) |
| Objektiv svarsfrekvens (ORR) n (%) av prövaren | ||
| Fullständiga svar | 1 (1) | 0 |
| Partiella svar | 25 (32) | 9 (12) |
| ORR (endast partiella svar) | 26 (33) | 9 (12) |
| Stabil sjukdom | 34 (43) | 29 (37) |
| Progressiv sjukdom | 14 (18) | 19 (24) |
a I enlighet med EU censoring
b Stratifieringsfaktorer per IxRS omfattar IMDC riskkategorier (intermediär prognos, dålig prognos och benmetastaser (ja, nej)
c Uppskattade med hjälp av Cox proportional hazard model justerade för stratifieringsfaktorer per IxRS. Hazard ratio < 1 indikerar progressionsfri överlevnad till förmån för kabozantinib
Randomiserad fas 3-studie av kabozantinib i kombination med nivolumab vs. sunitinib (CA2099ER)
Säkerheten och effekten av kabozantinib 40 mg peroralt dagligen i kombination med nivolumab 240 mg intravenöst varannan vecka för första linjens behandling av avancerad / metastaserad RCC utvärderades i en randomiserad, öppen fas 3-studie (CA2099ER). Studien inkluderade patienter (18 år eller äldre) med avancerad eller metastaserad RCC med en klar cellkomponent, Karnofsky Performance Status (KPS) ≥ 70 %, och mätbar sjukdom enligt RECIST v1.1 oavsett deras PD-L1-status eller IMDC-riskgrupp. Studien uteslöt patienter med autoimmun sjukdom eller andra medicinska tillstånd som kräver systemisk immunsuppression, patienter som tidigare behandlats med en anti-PD-1, anti PD-L1, anti-PD-L2, anti-CD137 eller anti-CTLA-4-antikropp, dåligt kontrollerad hypertoni trots blodtryckssänkande behandling, aktiva hjärnmetastaser och okontrollerad binjureinsufficiens. Patienterna stratifierades med IMDC-prognosvärde, PD-L1-tumöruttryck och region.
Totalt 651 patienter randomiserades till antingen kabozantinib 40 mg peroralt en gång dagligen i kombination med nivolumab 240 mg (n = 323) intravenöst varannan vecka eller sunitinib (n = 328) 50 mg peroralt dagligen i 4 veckor följt av 2 veckors uppehåll. Behandlingen fortsatte till sjukdomsprogress eller oacceptabel toxicitet med nivolumabadministrering i upp till 24 månader. Behandling efter initial prövarbedömd progression enligt RECIST version 1.1 tilläts om prövaren bedömde att patienten tolererade behandlingen och hade klinisk nytta av studieläkemedlet. Första tumörbedömningen efter baslinjen utfördes 12 veckor (± 7 dagar) efter randomisering. Efterföljande tumörbedömningar inträffade var sjätte vecka (± 7 dagar) fram till och med vecka 60, därefter var 12:e vecka (± 14 dagar) till radiologisk progress, bekräftad av Blinded Independent Central review (BICR). Den primära effektparametern var PFS vilken bestäms enligt en BICR. Ytterligare effektmått inkluderade OS och ORR som viktiga sekundära resultatmått.
Baslinjeegenskaperna var generellt likvärdiga mellan de två grupperna. Medianåldern var 61 år (intervall: 28-90) med 38,4 % ≥ 65 år och 9,5 % ≥ 75 år. Majoriteten av patienterna var män (73,9 %) och vita (81,9 %). Åtta procent av patienterna var asiatiska, 23,2 % och 76,5 % av patienterna hade en KPS på 70 till 80 % respektive 90 till 100 % vid baslinjen. Patientfördelningen enligt IMDC-riskkategorier var 22,6 % god, 57,6 % intermediär respektive 19,7 % dålig. För tumöruttryck av PD L1, hade 72,5 % av patienterna PD-L1-uttryck <1 % eller obestämbart och 24,9 % av patienterna hade PD-L1-uttryck ≥ 1 %. Sarkomatoid tumör påvisades hos 11,5 % av patienterna. Mediantiden för behandling var 14,26 månader (intervall: 0,2-27,3 månader) för patienter som behandlades med kabozantinib i kombination med nivolumab och 9,23 månader (intervall: 0,8-27,6 månader) hos sunitinibbehandlade patienter.
Studien visade en statistiskt signifikant fördel i PFS, OS och ORR för patienter randomiserade till kabozantinib i kombination med nivolumab jämfört med sunitinib.
Effektresultat från den primära analysen (minsta uppföljningstid 10,6 månader; medianuppföljningstid 18,1 månader) visas i tabell 7.
Tabell 7: Effektresultat (CA2099ER)
| kabozantinib + nivolumab (n = 323) | sunitinib (n = 328) | |
| Progressionsfri överlevnad (PFS) per BICR | ||
| Händelser | 144 (44,6 %) | 191 (58,2 %) |
| Riskkvota | 0,51 | |
| 95 % KI | (0,41, 0,64) | |
| p‑värdeb, c | < 0,0001 | |
| Median (95 % KI)d | 16,59 (12,45, 24,94) | 8,31 (6,97, 9,69) |
| Total överlevnad (OS) | ||
| Händelser | 67 (20,7 %) | 99 (30,2 %) |
| Riskkvota | 0,60 | |
| 98,89 % KI | (0,40, 0,89) | |
| p‑värdeb,c,e | 0,0010 | |
| Median (95 % KI) | N.E. | N.E. (22,6, N.E.) |
| Frekvens (95 % KI) | ||
| Vid 6 månader | 93,1 (89,7, 95,4) | 86,2 (81,9, 89,5) |
| Objektiv svarsfrekvens ORR) per BICR (CR + PR) | 180 (55,7 %) | 89 (27.1 %) |
| (95 % KI)f | (50,1, 61,2) | (22,4, 32,3) |
| Skillnad i ORR (95 % KI)g | 28,6 (21,7, 35,6) | |
| p‑värdeh | < 0,0001 | |
| Fullständigt svar (CR) | 26 (8,0 %) | 15 (4,6 %) |
| Partiellt svar (PR) | 154 (47,7 %) | 74 (22,6 %) |
| Stabil sjukdom (SD) | 104 (32,2 %) | 138 (42,1 %) |
| Medianduration för svard | ||
| Månader (intervall) | 20,17 (17,31, N.E.) | 11,47 (8,31, 18,43) |
| Mediantid till svar | ||
| Månader (intervall) | 2,83 (1,0‑19,4) | 4,17 (1,7‑12,3) |
a Stratifierad Cox proportional hazards model. Riskkvoten är kabozantinib och nivolumab över sunitinib.
b Tvåsidiga p-värden från stratifierat log-rank-test.
c Log-rank-test stratifierat av IMDC-prognostisk riskpoäng (0, 1-2, 3-6), tumöruttryck av PD-L1 (≥ 1 % jämfört med < 1 % eller obestämbar) och region (USA / Kanada / Västeuropa / Nordeuropa, övriga världen) som angetts i IRT.
d Baserat på Kaplan-Meier-uppskattningar.
e Gräns för statistisk signifikans p-värde < 0,0111.
f KI baserat på Clopper och Pearson-metoden.
g Stratajusterad skillnad i objektiv svarsfrekvens (kabozantinib + nivolumab - sunitinib) baserat på DerSimonian och Laird.
h Tvåsidigt p-värde från CMH-test.
NE = icke uppskattningsbar (non‑estimable)
Den primära analysen av PFS inkluderade censur för ny anticancerbehandling (tabell 7). Resultaten för PFS med och utan censurering för ny anticancerbehandling var konsekventa.
En fördel i PFS observerades för kabozantinib i kombination med nivolumab-armen jämfört med sunitinib oavsett tumöruttryck av PD L1. Median-PFS för tumör-PD L1-uttryck ≥ 1 % var 13,08 för kabozantinib i kombination med nivolumab och 4,67 månader i sunitinib-armen (HR = 0,45; 95 % KI: 0,29, 0,68). För tumör-PD L1-uttryck < 1 % var median-PFS 19,84 månader för kabozantinib i kombination med nivolumab och 9,26 månader i sunitinib-armen (HR = 0,50; 95 % KI: 0,38, 0,65).
En fördel i PFS observerades för kabozantinib i kombination med nivolumab-armen jämfört med sunitinib oavsett (IMDC) riskkategori. Median-PFS för riskgruppen med god prognos nåddes inte för kabozantinib i kombination med nivolumab och var 12,81 månader i sunitinib-armen (HR = 0,60; 95 % KI: 0,37, 0,98). Median-PFS för riskgruppen med intermediär prognos var 17,71 månader för kabozantinib i kombination med nivolumab och var 8,38 månader i sunitinib-armen (HR = 0,54; 95 % KI: 0,41, 0,73). Median-PFS för riskgruppen med dålig prognos var 12,29 månader för kabozantinib i kombination med nivolumab och 4,21 månader i sunitinib-armen (HR = 0,36; 95 % KI: 0,23, 0,58).
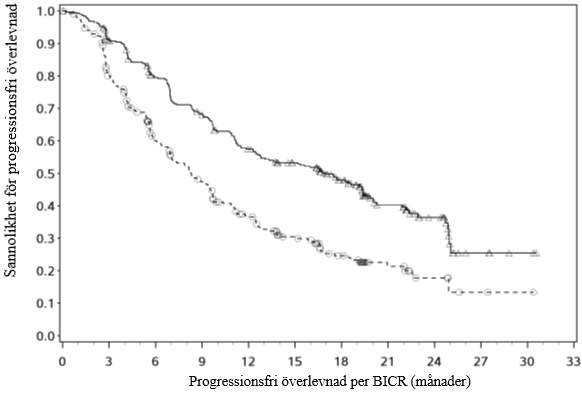
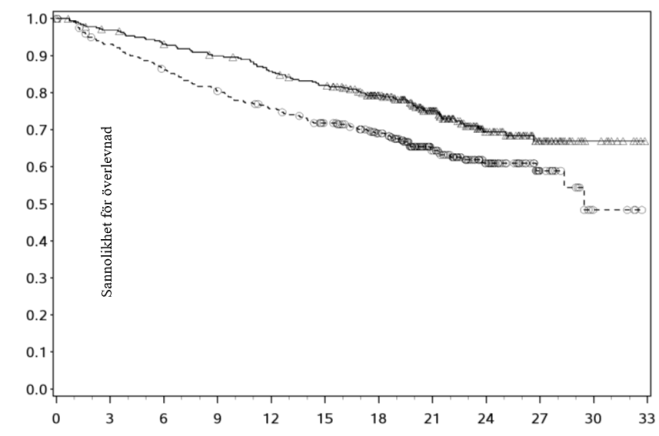
En uppdaterad PFS- och OS-analys utfördes när alla patienter haft en minsta uppföljningstid på 16 månader och en medianuppföljningstid på 23,5 månader (se figur 4 och 5). PFS-riskkvoten var 0,52 (95 % KI: 0,43; 0,64). OS-riskkvoten var 0,66 (95 % KI: 0,50; 0,87). Uppdaterade effektdata (PFS och OS) i undergrupper för IMDC-riskkategorier och PD-L1-uttrycksnivåer bekräftade de ursprungliga resultaten. Med den uppdaterade analysen uppnås median-PFS för riskgruppen med god prognos.
Figur 4: Kaplan‑Meier-kurvor för PFS (CA2099ER)
Antal individer i riskzonen
Kabozantinib + nivolumab | |||||||||||
323 | 308 | 295 | 283 | 269 | 255 | 220 | 147 | 84 | 40 | 10 | 0 |
Sunitinib | |||||||||||
328 | 295 | 272 | 254 | 236 | 217 | 189 | 118 | 62 | 22 | 4 | 0 |
Kabozantinib + nivolumab (händelser: 86/323), median och 95 % CI: NE
Sunitinib (händelser: 116/328), median och 95 % CI: 29,47 (28,35, NE)
Figur 5: Kaplan-Meier-kurvor för OS (CA2099ER)
Total överlevnad (månader)
Antal individer i riskzonen
| Kabozantinib + nivolumab | |||||||||||
| 323 | 308 | 295 | 283 | 269 | 255 | 220 | 147 | 84 | 40 | 10 | 0 |
| Sunitinib | |||||||||||
| 328 | 295 | 272 | 254 | 236 | 217 | 189 | 118 | 62 | 22 | 4 | 0 |
Kabozantinib + nivolumab (händelse: 86/323), median och 95 % CI: NE
Sunitinib (händelser: 116/328), median och 95 % CI: 29,47 (28,35, NE)
Hepatocellulär cancer
Kontrollerad studie på patienter som fått sorafenib (CELESTIAL)
Säkerheten och effekten av CABOMETYX utvärderades i en randomiserad, dubbelblind, placebokontrollerad fas 3-studie (CELESTIAL). Patienterna (N = 707) med HCC som inte var mottagliga för kurativ behandling och som tidigare fått sorafenib för avancerad sjukdom randomiserades (2:1) till att få kabozantinib (N = 470) eller placebo (N = 237). Patienterna kunde tidigare ha fått en annan systemisk behandling för avancerad sjukdom utöver sorafenib. Randomiseringen stratifierades med sjukdomens bakomliggande orsaker (hepatit B-virus [med eller utan hepatit C-virus], hepatit C-virus [utan hepatit B-virus] eller annat), geografisk region (Asien, andra regioner) och extrahepatisk spridning av sjukdom och/eller makrovaskulära invasioner (Ja, Nej).
Det primära effektmåttet var total överlevnad (OS). Sekundära effektmått var progressionsfri överlevnad (PFS) och objektiv svarsfrekvens (ORR), som utvärderades av prövaren med hjälp av Response Evaluation Criteria in Solid Tumours (RECIST) 1.1. Tumörbedömningar genomfördes var 8:e vecka. Individer fortsatte behandlingen i den blindade studien efter radiologisk sjukdomsprogression under tiden som de upplevde klinisk nytta eller tills det uppstod ett behov av påföljande systemisk eller lokal anticancerbehandling av levern. Byte från placebo till kabozantinib var inte tillåtet under den blindade behandlingsperioden.
Demografi och sjukdomsegenskaper vid baslinjen var likartade mellan kabozantinib- och placeboarmarna och visas nedan för alla 707 randomiserade patienter.
Majoriteten av patienterna (82 %) var män: medianålder var 64 år. Majoriteten av patienterna (56 %) var kaukasier och 34 % av patienterna var asiater. Femtiotre procent (53 %) av patienterna hade ECOG performance status (PS) 0 och 47 % hade ECOG PS 1. Nästan alla patienter (99 %) var Child Pugh A och 1 % var Child Pugh B. Bakomliggande orsaker till HCC inkluderade 38 % hepatit B-virus (HBV), 21 % hepatit C-virus (HCV), 40 % annat (varken HBV eller HCV). Sjuttioåtta procent (78 %) hade förekomst av makroskopisk vaskulär invasion och/eller extrahepatisk tumörspridning, 41 % hade alfa-fetoprotein (AFP)-nivåer ≥ 400 ug/l, 44 % hade fått transarteriell lokoregional embolisering eller kemoinfusion, 37 % fick strålbehandling före behandling med kabozantinib. Medianduration av behandling med sorafenib var 5,32 månader. Sjuttiotvå procent (72 %) av patienterna hade fått 1 och 28 % hade tidigare fått 2 systemiska behandlingsregimer för avancerad sjukdom.
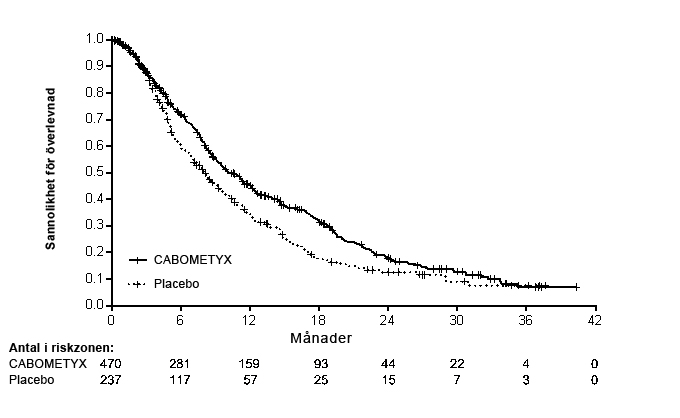
En statistiskt signifikant förbättring av OS visades för kabozantinib jämfört med placebo (Tabell 8 och Figur 6).
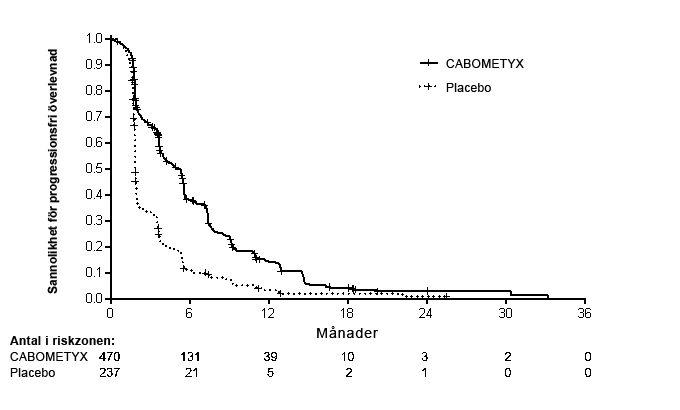
PFS- och ORR-fynden sammanfattas i Tabell 8.
Tabell 8: Effektresultat hos HCC (ITT population, CELESTIAL)
| CABOMETYX (N=470) | Placebo (N=237) | ||||
| Total överlevnad (OS) | |||||
| Median OS (95 % CI), månader | 10,2 (9,1, 12,0) | 8,0 (6,8, 9,4) | |||
| HR (95 % CI) 1,2 | 0,76 (0,63, 0,92) | ||||
| p-värde1 | p = 0,0049 | ||||
| Progressionsfri överlevnad (PFS)3 | |||||
| Median PFS i månader (95 % CI) | 5,2 (4,0, 5,5) | 1,9 (1,9, 1,9) | |||
| HR (95 % CI)1 | 0,44 (0,36, 0,52) | ||||
| p-värde1 | p < 0,0001 | ||||
| Kaplan-Meier uppskattning av individer i procent som är händelsefria vid 3 månader | |||||
| % (95 % CI) | 67,0 % (62,2 %, 71,3 %) | 33,3 % (27,1 %, 39,7 %) | |||
| Objektiv svarsfrekvens (ORR) n (%)3 | |||||
| Fullständiga svar (CR) | 0 | 0 | |||
| Partiella svar (PR) | 18 (4) | 1 (0,4) | |||
| ORR (CR+PR) | 18 (4) | 1 (0,4) | |||
| p-värde1,4 | p=0,0086 | ||||
| Stabil sjukdom | 282 (60) | 78 (33) | |||
| Progressiv sjukdom | 98 (21) | 131 (55) | |||
1 tvåsidigt stratifierat log-rank test med sjukdomens bakomliggande orsaker (HBV [med eller utan HCV], HCV [utan HBV], eller annat), geografisk region (Asien, andra regioner) och extrahepatisk spridning av sjukdom och/eller makrovaskulära invasioner (Ja, Nej) som stratifieringsfaktorer (med IVRS data)
2 uppskattat med hjälp av Cox proportional-hazardmodellen
3 bedömt av prövaren med RECIST 1.1
4 stratifierat Cochran-Mantel-Haenszel (CMH) test
Figur 6: Kaplan-Meier-kurva för total överlevnad (CELESTIAL)
Figur 7: Kaplan-Meier-kurva för progressionsfri överlevnad (CELESTIAL)
Incidensen av systemisk anticancerbehandling utanför protokollet (NPACT), utan strålning och lokalt riktad mot lever, var 26 % i kabozantinibarmen och 33 % i placeboarmen. Individer som fick dessa behandlingar var tvungna att avsluta studiebehandlingen. En explorativ analys av OS censurerad för användning av NPACT stödde den primära analysen: HR, justerad för stratifieringsfaktorer (per IxRS), var 0,66 (95 % CI: 0,52, 0,84; stratifierat logrank p-värde = 0,0005). I Kaplan-Meier uppskattas medianvärdet för OS till 11,1 månader i kabozantinibarmen jämfört med 6,9 månader i placeboarmen, en beräknad skillnad på 4,2 månader i mediantid.
Den icke-sjukdomsspecifika livskvaliteten (QoL) utvärderades med hjälp av EuroQoL EQ-5D-5L. En negativ effekt av kabozantinib kontra placebo på EQ-5D-användningsindexvärdet observerades under de första veckorna av behandlingen. Endast begränsade QoL-data finns tillgängliga efter denna period.
Differentierad tyroideacancer (DTC)
Placebokontrollerad studie på vuxna som tidigare fått systemisk behandling och är refraktär eller inte lämpliga för radioaktivt jod (COSMIC-311).
Säkerheten och effekten av CABOMETYX utvärderades i COSMIC-311, en randomiserad (2:1), dubbelblind, placebokontrollerad multicenterstudie hos vuxna patienter med lokal avancerad eller metastaserad sjukdom med differentierad tyroideacancer som har progredierat efter upp till två tidigare VEGFR-riktad behandling (inklusive, men inte begränsat till, lenvatinib eller sorafenib) och som var refraktär eller inte lämpade för radioaktivt jod. Patienter med mätbar sjukdom och dokumenterad radiografisk progression bedömt av prövare enligt RECIST 1.1, under eller efter VEGRF-riktad TKI, randomiserades (N = 258) till att få kabozantinib 60 mg peroralt en gång dagligen (N = 170) eller placebo (N = 88).
Randomiseringen stratifierades enligt tidigare mottagande av lenvantinib (ja/nej) och ålder (≤ 65 år / > 65 år). Patienter som uppfyllde kriterierna och som randomiserades till placebo fick byta till kabozantinib vid bekräftad sjukdomsprogression efter bedömning av en blindad oberoende radiologisk komitté (BIRC). I den blindade studien fortsatte individer behandlas så länge de upplevde en klinisk nytta eller fram till oacceptabel toxicitet. Det primära effektmåttet, bedömt av BIRC enligt RECIST 1.1, var progressionsfri överlevnad (PFS) hos intention-to-treat (ITT)-gruppen, och objektiv svarsfrekens (ORR) hos de första 100 randomiserade patienterna. Tumörbedömningar gjordes var åttonde vecka under de första 12 månaderna, och därefter var tolfte vecka. Ytterligare effektmått var total överlevnad (OS).
Den primära analysen av PFS inkluderade 187 randomiserade patienter, 125 till att få kabozantinib och 62 till att få placebo. Demografi och sjukdomsegenskaper vid baslinjen var generellt sett balanserad för båda behandlingsgrupper. Medianålder var 66 år (intervall 32 till 85 år), 51 % var ≥ 65 år, 13 % var ≥ 75 år. Majoriteten av patienterna var vita (70 %), 18 % av patienterna var asiatiska och 55 % var kvinnor. Femtiofem procent hade histologisk bekräftad papillär tyroideacancer, 48 % hade follikulär tyroideacancer inklusive 17 % patienter med Hürthle-cell tyroideacancer. Metastaser förekom hos 95 % av patienterna: i lungor hos 68 %, i lymfkörtlar hos 67 %, i ben hos 19 %, i lungsäck hos 18 % och i lever hos 15 %. Fem av patienterna var inte lämpade för tidigare behandling med RAI, 63 % hade tidigare fått lenvantinib, 60 % hade tidigare fått sorafenib och 23 % hade tidigare fått både sorafenib och lenvatinib. ECOG-performance-status vid baslinje var 0 (48 %) eller 1 (52 %). Mediantiden för behandling var 4,4 månader i kabozantinib-armen och 2,3 månader i placebo-armen.
Resultat från den primära analysen (med cut-off-datum den 19 augusti 2020 och medianuppföljningstid 6,2 månader för PFS), och uppdaterad analys (med cut-off-datum den 08 februari 2021 och medianuppföljningstid 10,1 månader för PFS) visas i tabell 9. Studien visade ingen statistiskt signifikant förbättring av ORR hos patienter randomiserade till att få Cabometyx (n = 67) i jämförelse med placebo (n = 33): 15 % jämfört med 0 %. Studien visade en statistiskt signifikant förbättring av PFS (medianuppföljningstid 6,2 månader) för patienter randomiserade till kabozantinib (n = 125) jämfört med placebo (n = 62).
En uppdaterad analys av PFS och OS (medianuppföljningstid 10,1 månader) utfördes för 258 randomiserade patienter, 170 till kabozantinib och 88 till placebo.
Analysen av total överlevnad kan vara missvisande då placebobehandlade patienter med bekräftad sjukdomsprogression kunde välja att byta till kabozantinib.
Tabell 9: Effektresultat från COSMIC-311
| Primär analys1 (ITT) | Uppdaterad analys2 (fullständig ITT) | ||||
| CABOMETYX (N = 125) | Placebo (N = 62) | CABOMETYX (N = 170) | Placebo (N = 88) | ||
| Progressionsfri överlevnad* | |||||
| Antal händelser, (%) | 31 (25) | 43 (69) | 62 (36) | 69 (78) | |
| Progressiv sjukdom | 25 (20) | 41 (66) | 50 (29) | 65 (74) | |
| Död | 6 (4,8) | 2 (3,2) | 12 (7,1) | 4 (4,5) | |
| Median PFS I månader (96% CI) | N.E (5,7, N.E) | 1,9 (1,8, 3,6) | 11,0 (7,4, 13,8) | 1,9 (1,9, 3,7) | |
| HR (96 % CI)3 | 0,22 (0,13, 0,36) | 0,22 (0,15, 0,32) | |||
| p‑värde4 | < 0,0001 | ||||
| Total överlevnad | |||||
| Händelser, n (%) | 17 (14) | 14 (23) | 37 (22) | 21 (24) | |
| HR3 (95 % CI) | 0,54 (0,27, 1,11) | 0,76 (0,45, 1,31) | |||
| Primär analys1 | |||||
| Objektiv svarsfrekvens (ORR)5 | |||||
| CABOMETYX (n = 67) | Placebo (n = 33) | ||||
| Total respons, (%) | 10 (15) | 0 (0) | |||
| Fullständiga svar | 0 | 0 | |||
| Partiella svar | 10 (15) | 0 | |||
| Stabil sjukdom | 46 (69) | 14 (42) | |||
| Progressiv sjukdom | 4 (6) | 18 (55) | |||
* Den primära analysen av PFS inkluderade censur för nya anticancerbehandling. Resultaten för PFS med och utan censurering av nya anticancerbehandling var konsekventa.
CI, konfidensintervall; NE = icke-uppskattningsbar (non-estimable)
1 Cut-off-datumet för den primära analysen är den 19 augusti 2020
2 Cut-off-datumet för den sekundära analysen är den 8 februari 2021
3 Uppskattade med hjälp av Cox proportional hazard
4 Log-rank-test stratifierat efter tidigare mottagande av lenvatinib (ja/nej) och ålder (≤ 65 år / > 65 år) som stratifieringsgrader (enligt IXRS-data).
5 Baserat på de 100 första inkluderade patienterna med medianuppföljningstid på 8,9 månader, n = 67 i CABOMETYX-gruppen och n = 33 i placebo-gruppen.
Figur 8: Kaplan-Meier-kurvor över progressionsfri överlevnad i COSMIC-311 (uppdaterad analys (cut-off-datum: den 8 februari 2021), N = 258)
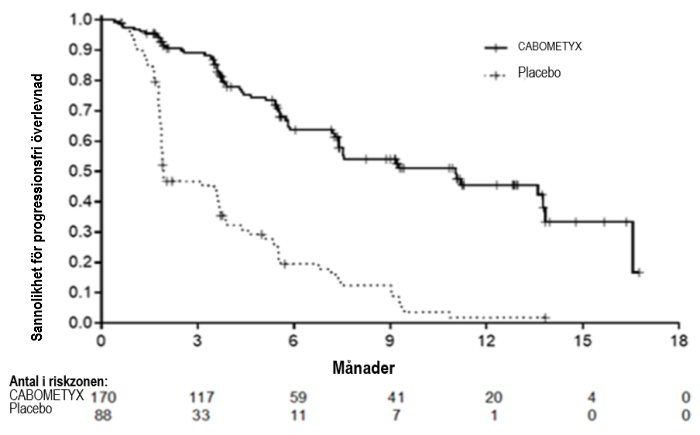
Neuroendokrina tumörer (NET)
Placebokontrollerad studie på vuxna patienter med lokal avancerad eller metastaserad epNET och pNET som har progredierat efter tidigare behandling (CABINET)
Säkerheten och effekten av CABOMETYX utvärderades i CABINET, en multicenter, randomiserad (2:1), dubbelblind, placebokontrollerad fas 3-studie hos vuxna patienter med lokal avancerad eller metastaserad väldifferentierad pNET (kabozantinib: N = 64; placebo: N = 31) och epNET (kabozantinib: N = 134; placebo: N = 69) som har progredierat efter tidigare godkänd behandling.
Patienter med epNET och pNET delades in i två separata kohorter som randomiserades och analyserades oberoende av varandra.
Patienterna fortsatte med blindad studiebehandling tills sjukdomsprogression, oacceptabel toxicitet eller återkallande av samtycke. Lämpliga patienter som randomiserats till placebo fick byta till behandling med kabozantinib vid bekräftad sjukdomsprogression genom central bedömning i realtid. Det primära effektmåttet var progressionsfri överlevnad (PFS) i ITT-populationen bedömt av en blindad oberoende radiologisk komitté (BIRC) enligt RECIST 1.1 med stratifieringsfaktorer vid randomisering enligt följande:
- epNET: Samtidig somatostatinanalog (SSA) och lokalisering av primärtumör (midgut GI/okänt vs. icke-midgut GI/lunga/annat)
- pNET: Samtidig SSA och tidigare sunitinib
Utvärdering av tumörer gjordes var 12:e vecka från behandlingsstart till sjukdomsprogression. Total överlevnad (OS) var sekundärt effektmått.
epNET-kohort:
Majoriteten av patienterna, 51,7 %, var kvinnor. Medianåldern var 66 år. Majoriteten av patienterna, 83,7 %, var vita. Dessutom hade 39,9 % av patienterna ECOG performance status 0, medan 59,1 % hade performance status 1. Ursprungslokalisering av primärtumör var oftast tunntarmen med 32,5 %, följt av lungorna med 19,2 %, annan lokalisering med 17,2 % och okänd lokalisering med 11,8 %. De flesta patienterna hade en icke-funktionell tumör och stod för 53,7 % av fallen, medan 32,5 % hade en funktionell tumör. För 13,8 % av patienterna var funktionsstatusen okänd. Den vanligaste tumörgraden var grad 2, som observerades hos 66 % av patienterna och grad 1 hos 25,6 % av patienterna. Majoriteten av patienterna, 69 %, använde SSA samtidigt och 92,6 % hade tidigare använt SSA. 45,3 % av patienterna hade endast fått en tidigare behandling utöver SSA. De flesta tumörer var väldifferentierade och representerade 93,6 % av fallen, medan 6,4 % inte specificerades. Den vanligaste lokaliseringen av metastaser var i levern, som påverkades i 89,7 % av fallen, lymfkörtlar i 70 % av fallen, skelettet i 49,3 % av fallen, annan lokalisering i 35 % av fallen och lungor i 21,2 % av fallen.
Tabell 10: Effektresultat i epNET-kohorter från CABINET-studien
| Effektmått | Kabozantinib (N = 134) | Placebo (N = 69) |
|---|---|---|
| Progressionsfri överlevnad | ||
| Antal händelser, n (%) | 71 (53) | 40 (58) |
| Dokumenterad progression, n (%) | 53 (40) | 35 (51) |
| Död, n (%) | 18 (13) | 5 (7,2) |
| Median PFS i månader1 (95 % CI) | 8,5 (7,5, 12,5) | 4,0 (3,0, 5,7) |
| HR2 (95 % CI) | 0,38 (0,25, 0,58) | |
Medianuppföljningstid var 23 månader för båda armarna. Enligt BIRC-bedömningar av progression och respons med cut-off-datum den 24 augusti 2023.
1 Baserat på Kaplan-Meier-uppskattningar
2 Uppskattat med hjälp av Cox proportional-hazard-modell. CABINET-studien avslutades m.a.p effekt vid tiden för en interimanalys som bara planerats för att utvärdera futilitet. Typ 1-fel kontrollerades inte formellt och p-värden presenteras ej. Det 95 % konfidensintervall som presenterats är deskriptiv och innebär inte att statistisk signifikans uppnåddes.
Figur 9: epNET: Kaplan-Meier-kurvor för progressionsfri överlevnad (cut-off datum: 24 augusti 2023, N = 203)
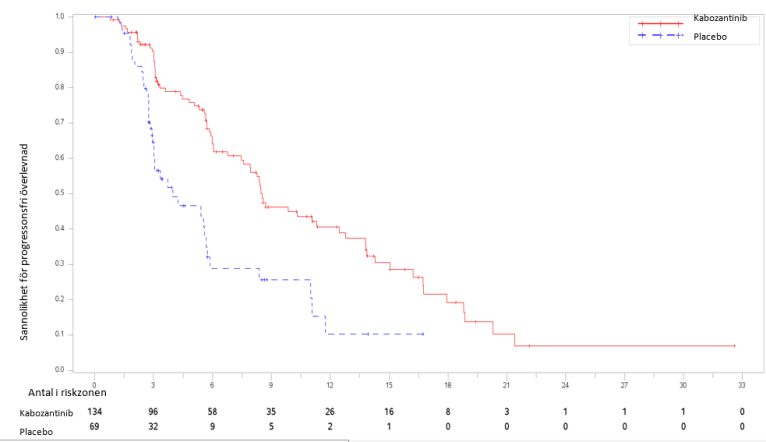
En uppdaterad explorativ OS-analys (DCO: sept 2024) med 126 OS-händelser utfördes som visade: medianöverlevnaden var 21,95 månader i kabozantinib-armen och 22,47 månader i placeboarmen, med en riskkvot (HR) på 1,04 (95 % KI: 0,71, 1,52). Vid tidpunkten för analysen växlade 28 (41 %) patienter över från placebo till kabozantinib.
pNET-kohort:
Majoriteten av patienterna, 57,9 %, var män. Medianåldern var 59,5 år i gruppen som fick kabozantinib och 64 år i placeboarmen. Majoriteten av patienterna, 83,2 %, var vita. Dessutom hade 52,6 % av patienterna ECOG performance status 0, medan 46,3 % hade performance status på 1.
De flesta patienterna hade en icke-funktionell tumör och stod för 73,7 % av fallen, medan 16,8 % hade en funktionell tumör. För 9,5 % av patienterna var funktionsstatusen okänd. Den vanligaste tumörgraden var grad 2 och observerades hos 61,1 % av patienterna. Grad 1 observerades hos 22,1 %, grad 3 hos 11,6 % av patienterna och var hos 5,3 % av patienterna okänt. Majoriteten av patienterna, 54,7 %, använde SSA samtidigt och 97,9 % hade tidigare använt SSA. 28,4 % av patienterna hade endast fått en tidigare behandling utöver SSA. De flesta tumörer var väldifferentierade och representerade 97,9 % av fallen, medan 2,1 % inte specificerades. Den vanligaste lokaliseringen av metastaser var i levern, som var påverkad i 96,8 % av fallen, lymfkörtlar i 48,4 % av fallen, skelettet i 27,4 % av fallen och annan lokalisering i 13,7 % av fallen.
Tabell 11: Effektresultat i pNET-kohorter från CABINET-studien
| Kabozantinib (N = 64) | Placebo (N = 31) | |
|---|---|---|
| Progressionsfri överlevnad | ||
| Antal händelser, n (%) | 32 (50) | 25 (81) |
| Dokumenterad progression, n (%) | 25 (39) | 21 (68) |
| Död, n (%) | 7 (11) | 4 (13) |
| Median PFS i månader1 (95 % CI) | 13,8 (8,9, 17,0) | 4,5 (3,0, 5,8) |
| HR2 (95 % CI) | 0,23 (0,12, 0,42) | |
Medianuppföljningstid var 23 månader (kabozantinib) och 25 månader (placebo). Enligt BIRC-bedömningar av progression och respons med cut-off datum den 24 augusti 2023.
1 Baserat på Kaplan-Meier-uppskattningar
2 Uppskattat med hjälp av Cox proportional-hazard-modell. CABINET-studien avslutades m.a.p effekt vid tiden för en interimanalys som bara planerats för att utvärdera futilitets. Typ 1-fel kontrollerades inte formellt och p-värden presenteras ej. Det 95 % konfidensintervall som presenterats är deskriptiv och innebär inte att statistisk signifikans uppnåddes.
Figur 10: pNET: Kaplan-Meier-kurvan för progressionsfri överlevnad i CABINET (cut-off datum: 24 augusti 2023, N = 95)
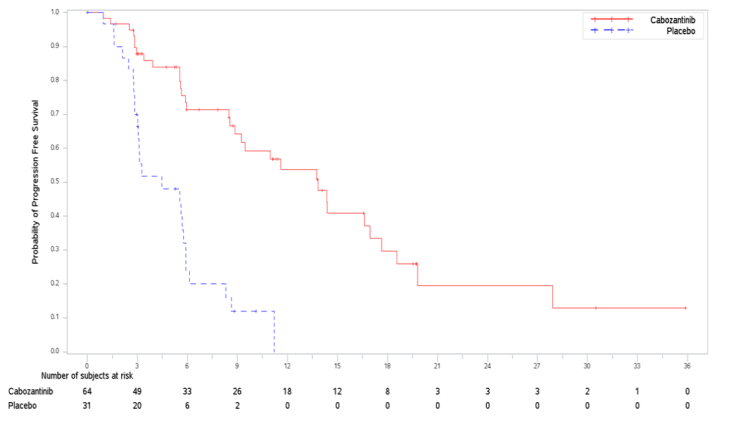
En uppdaterad explorativ OS-analys (DCO: sept 2024) med 46 OS-händelser utfördes som visade: Medianvärdet för Kaplan-Meier-uppskattningen av totalöverlevnad var 40,08 månader i kabozantinib-armen och 31,11 månader i placeboarmen, med en HR på 1,11 (0,59, 2,09). Vid tidpunkten för analysen växlade 14 (45 %) patienter över från placebo till kabozantinib.
Pediatrisk population
Europeiska läkemedelsmyndigheten har senarelagt kravet att skicka in studieresultat för vissa studier för CABOMETYX för en eller flera grupper av den pediatriska populationen för behandling av solida maligna tumörer (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
ADVL 1211
En fas 1-studie (ADVL1211) av kabozantinib på pediatriska patienter med solida tumörer har genomförts av Children Oncology Group (COG). Lämpliga patienter var ≥ 2 år och ≤ 18 år. Denna studie rekryterade patienter för 3 dosnivåer: 30 mg/m2, 40 mg/m2 och 55 mg/m2 en gång dagligen enligt ett kontinuerligt doseringsschema (veckodosering efter kroppsyta avrundat till närmaste 20 mg). Kabozantinib doserades efter kroppsyta enligt ett doseringsnomogram.
Målet var att definiera dosbegränsande toxicitet (DLT), bestämma den rekommenderade dosen för fas-2 (RP2D), erhålla preliminära farmakokinetiska data för barn och undersöka effekten i solida tumörer. Fyrtioen patienter rekryterades, varav 36 var fullständigt utvärderadbara. Patienterna hade olika solida tumörer: medullär tyreoideacancer (MTC ) (n = 5), osteosarkom (n = 2), Ewing sarkom (EWS) (n = 4), rhabdomyosarkom (RMS) (n = 2), andra mjukdelssarkom (STS) (n = 4), Wilms tumör (WT) (n = 2), hepatoblastom (n = 2), hepatocellulär cancer (HCC) (n = 2), njurcellscancer (RCC) (n = 3), tumörer i centrala nervsystemet (CNS) (n = 9) och andra (n = 6).
Av de 36 försökspersonerna i den utvärderbara populationen hade fyra försökspersoner (11,1 %) partiellt svar (PR) som bäst totalt svar och åtta försökspersoner (22,2 %) hade stabil sjukdom (SD) (som varade minst 6 cykler). Av de 12 försökspersonerna med PR eller SD större än eller lika med 6 cykler var 10 försökspersoner i grupperna kabozantinib 40 mg/m2 eller 55 mg/m2 (sju respektive tre).
Baserat på en central granskning sågs partiellt svar hos 2/5 patienter med MTC, en patient med Wilms tumör och en patient med klarcellssarkom.
ADVL1622
ADVL1622 utvärderade aktiviteten av kabozantinib i utvalda pediatriska solida tumörer. Denna multicenter, öppna tvåstegs fas 2-studie inkluderade följande solida tumör-strata: icke-osteosarkoms-strata (inklusive Ewing sarkom, rhabdomyosarkom (RMS), icke-rhabdomyosarkom mjukdelssarkom (NRSTS) och Wilms tumör), osteosarkom-strata och sällsynta solida tumörs-strata (inklusive medullär tyroideacancer (MTC), njurcellscancer (RCC), hepatocellulär cancer (HCC), hepatoblastom, binjurebarkscancer och andra solida tumörer). Kabozantinib administrerades oralt en gång dagligen enligt ett kontinuerligt doseringsschema på 28-dagarscykler med en dos på 40 mg/m2/dag (kumulativ veckodos på 280 mg/m2 med hjälp av ett doseringsnomogram). Försökspersonerna var ≥ 2 och ≤ 30 år vid tidpunkten för studiestart för alla strata förutom övre åldersgräns på ≤ 18 år för MTC, RCC och HCC.
För icke-osteosarkom och sällsynta tumör-strata var det primära effektmåttet den objektiva svarsfrekvensen (ORR). För osteosarkom-strata användes en tvåstegsdesign som inkorporerade dubbla effektmått för objektivt svar (CR + PR) baserat på responsutvärderingskriterier i solida tumörer (RECIST) version 1.1-kriterier och behandlingsframgång enligt definitionen av SD i ≥ 4 månader. Farmakokinetiken för kabozantinib hos pediatriska och tonåriga försökspersoner utvärderades (se avsnitt Farmakokinetiska egenskaper).
Sammanfattning av effektresultat
Vid cut-off datum (30 juni 2021) hade 108/109 försökspersoner fått minst en dos kabozantinib. Varje statistisk kohort i icke-osteosarkom-strata omfattade 13 försökspersoner. Inga svar observerades i dessa statistiska kohorter. Osteosarkom-strata omfattade totalt 29 försökspersoner inklusive 17 barn (ålder 9 till 17 år) och 12 vuxna (ålder 18 till 22 år).
I osteosarkom-strata hade alla försökspersoner fått tidigare systemisk behandling. En PR observerades hos en vuxen och ett barn. DCR var 34,5 % (95 % KI: 17,9, 54,3).
Farmakokinetiska egenskaper
Absorption
Efter oral administrering av kabozantinib uppnås maximala plasmakoncentrationer av kabozantinib 3 till 4 timmar efter dosering. Plasma-koncentrationstidsprofiler visar en andra absorptionstopp cirka 24 timmar efter administrering, vilket tyder på att kabozantinib kan genomgå enterohepatisk recirkulation.
Upprepad daglig dosering av kabozantinib med 140 mg i 19 dagar resulterade i en ungefär 4‑ till 5‑faldig genomsnittlig ackumulering av kabozantinib (baserat på AUC) jämfört med administrering av en engångsdos; steady state uppnås ungefär vid dag 15.
En fettrik måltid ökade måttligt värdena för Cmax och AUC (41 % respektive 57 %) jämfört med fastande tillstånd hos friska försökspersoner som fick en oral engångsdos på 140 mg kabozantinib. Det finns ingen information om den exakta effekten av födointag när detta sker 1 timme efter administrering av kabozantinib.
Bioekvivalens kunde inte visas mellan kabozantinib som kapsel respektive tablett efter en engångsdos på 140 mg till friska försökspersoner. En ökning på 19 % av Cmax för tablettformuleringen jämfört med kapselformuleringen observerades. En skillnad på mindre än 10 % i AUC observerades mellan kabozantinib formulerat som tablett och kapsel.
Distribution
Kabozantinib är i hög grad proteinbundet in vitro i human plasma (≥ 99,7 %). Baserat på den populationsfarmakokinetiska (PK) modellen har distributionsvolymen i centralt kompartment (Vc/F) uppskattats vara 212 l.
Metabolism
Kabozantinib metaboliserades in vivo. Fyra metaboliter förekom i plasma vid exponeringar (AUC) som var större än 10 % av moderföreningen: XL184‑N‑oxid, XL184 amid-spjälkningsprodukt, XL184-monohydroxisulfat och 6‑desmetylamid-spjälkningsprodukt-sulfat. Två icke-konjugerade metaboliter (XL184-N‑oxid och XL184 amid-spjälkningsprodukt), vilka innehar <1 % av den målinriktade kinashämningsförmågan hos moderföreningen kabozantinib, står var och en för <10 % av den totala läkemedelsrelaterade plasmaexponeringen.
Kabozantinib är ett substrat för CYP3A4-metabolism in vitro, i egenskap av en neutraliserande antikropp mot CYP3A4-hämmat bildande av metabolit XL184 N‑oxid med >80 % i en NADPH-katalyserad humanlevermikrosomal (HLM) inkubering. Däremot hade neutraliserande antikroppar mot CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6 och CYP2E1 ingen effekt på bildandet av kabozantinibs metaboliter. En neutraliserande antikropp mot CYP2C9 visade en minimal effekt på kabozantinibs metabolitbildning (dvs. en minskning på < 20 %).
Eliminering
I en farmakokinetisk populationsanalys av kabozantinib med användning av data som samlats in från 1883 patienter och 140 friska frivilliga efter oral administrering av ett intervall av doser från 20-140 mg, är den terminala halveringstiden i plasma för kabozantinib cirka 110 timmar. Genomsnittlig clearance (CL/F) vid steady state uppskattades till 2,48 l/timme. Inom en 48‑dagars insamlingsperiod efter en engångsdos av 14C-kabozantinib hos friska försökspersoner, återfanns cirka 81 % av den totala administrerade radioaktiviteten, varav 54 % i feces och 27 % i urin.
Farmakokinetik i speciella patientpopulationer
Nedsatt njurfunktion
I en studie på nedsatt njurfunktion som genomfördes med en enkel dos på 60 mg kabozantinib, var förhållandet mellan geometriska LS-medelvärden för total plasma-kabozantinib, Cmax och AUC0-inf 19 % och 30 % högre för patienter med lindrigt nedsatt njurfunktion (90 % KI för Cmax 91,60 % till 155,51 %; AUC0-inf 98,79 % till 171,26 %) och 2 % och 6-7 % högre (90 % KI för Cmax 78,64 % till 133,52 %; AUC0-inf 79,61 % till 140,11 %), för patienter med måttligt nedsatt njurfunktion jämfört med patienter med normal njurfunktion. Det geometriska LS-medelvärdet för obundet plasma-kabozantinib AUC0-inf var 0,2 % högre för individer med lätt nedsatt njurfunktion (90 % KI 55,9 % till 180 %) och 17 % högre (90 % KI 65,1 % till 209,7 %) för individer med måttligt nedsatt njurfunktion jämfört med individer med normal njurfunktion. Individer med gravt nedsatt njurfunktion har inte studerats.
Nedsatt leverfunktion
Baserat på en farmakokinetisk integrerad populationsanalys av kabozantinib till friska försökspersoner och cancerpatienter (inklusive HCC) observerades ingen kliniskt signifikant skillnad i den genomsnittliga plasmakoncentrationen av kabozantinib hos personer med normal leverfunktion (n = 1425) och lindrigt nedsatt leverfunktion (n = 558). Det finns begränsade data för patienter med måttligt nedsatt leverfunktion (n = 15) enligt NCI-ODWG (National Cancer Institute - Organ Dysfunction Working Group). Farmakokinetiken för kabozantinib utvärderades ej hos patienter med gravt nedsatt leverfunktion.
Etnicitet
Vid en farmakokinetisk populationsanalys identifierades inga kliniskt relevanta skillnader i PK för kabozantinib baserat på etnicitet.
Pediatrisk population
Data erhållen från simulering utförd med populationsfarmakokinetiska modeller som utvecklats i friska individer och vuxna patienter med olika typer av maligniteter, visar att patienter 12 år och äldre, som får en dos 40 mg kabozantinib en gång dagligen för patienter < 40 kg, eller en dos 60 mg en gång dagligen för patienter ≥ 40 kg resulterar i en plasmaexponering som liknar den som uppnås hos vuxna patienter som behandlats med 60 mg kabozantinib en gång dagligen (se avsnitt Dosering och administreringssätt).
I de två kliniska studier som genomfördes av COG på pediatriska patienter med solida tumörer (ADVL1211 och ADVL1622) doserades kabozantinib efter kroppsyta (BSA) enligt ett doseringsnomogram, med användning av tillgängliga 20 mg och 60 mg tabletter avsedda för vuxna. Bland de 55 patienterna var medianåldern 13 år (intervall: 4 till 18 år). En populationsfarmakokinetisk (PK) analys byggdes med hjälp av PK-data som samlats in i båda studierna. PK för kabozantinib beskrevs tillräckligt av en tvåkompartement modell med första ordningens eliminering och första ordningens absorptionsprocesser. Det fanns inga bevis för att ålder, kön, etnicitet och tumörtyp påverkade PK för kabozantinib hos barn och ungdomar. Endast BSA ansågs vara en signifikant prediktor för PK för kabozantinib. Inget dosberoende sågs i den utvecklade modellen för de tre testade dosnivåerna (30, 40 och 55 mg/m²). Exponering hos barn och ungdomar efter administrering av en BSA-baserad dos på 40 mg/m² liknar exponering hos vuxna med en fast dos på 60 mg dagligen.
Prekliniska säkerhetsuppgifter
Följande biverkningar har inte observerats i kliniska prövningar, men har setts hos djur vid exponeringsnivåer som är likartade med kliniska exponeringsnivåer och bedöms därför ha möjlig klinisk relevans:
I upp till 6 månader långa toxicitetsstudier med upprepad dosering till råttor och hundar, var målorganen för toxicitet magtarmkanal, benmärg, lymfoida vävnader, njure, binjure och vävnader i reproduktionskanal. NOAEL (nivå utan observerad skadlig effekt) för dessa fynd var lägre än kliniska exponeringsnivåer för människa vid avsedd terapeutisk dos.
Kabozantinib har inte uppvisat någon mutagen eller klastogen potential i en standarduppsättning av genotoxicitetsanalyser. Kabozantinibs cancerogena potential har utvärderats i två arter: rasH2-transgena möss och Sprague-Dawleyråttor. I den 2 år långa karcinogenicitetsstudien på råttor bestod de kabozantinibrelaterade neoplastiska fynden av en ökad incidens av godartad feokromocytom, ensamt eller i kombination med malign feokromocytom/komplex malign feokromocytom, i binjuremärgen hos båda könen vid exponeringar långt under den avsedda exponeringen hos människa. Den kliniska relevansen av de neoplastiska förändringarna som observerats i råttor är osäker, men är troligen låg. Kabozantinib var inte karcinogent i rasH2-musmodellen vid en något högre exponering än den avsedda humana terapeutiska exponeringen.
Fertilitetsstudier på råttor har visat minskad fertilitet hos hanar och honor. Dessutom observerades hypospermatogenes hos hanhundar vid exponeringsnivåer under kliniska exponeringsnivåer för människa vid avsedd terapeutisk dos.
Embryofetala utvecklingsstudier utfördes på råtta och kanin. Hos råttor orsakade kabozantinib postimplantationsförlust, fetalt ödem, gomspalt/kluven läpp, dermal aplasi och böjd eller rudimentär svans. Hos kanin orsakade kabozantinib fetala mjukdelsförändringar (minskad mjältstorlek, liten eller saknad mellanliggande lunglob) och ökad fetal incidens av totala missbildningar. NOAEL för embryofetal toxicitet och teratogena fynd understeg de kliniska exponeringsnivåerna för människa vid avsedd terapeutisk dos.
Juvenila råttor (jämförbart med en > 2 år gammal pediatrisk population) som fick kabozantinib visade ökade WBC-parametrar, minskad hematopoes, pubescent/omoget reproduktionssystem hos honor (utan fördröjd vaginal öppning), tandavvikelser, minskat benmineralinnehåll och bentäthet, leverpigmentering och lymfoid hyperplasi i lymfkörtlar. Fynd i uterus/ovarier och minskad hematopoes verkade vara övergående, medan effekter på benparametrar och leverpigmentering kvarstod. Juvenila råttor (motsvarande en < 2 år gammal pediatrisk population) visade likartade behandlingsrelaterade fynd, med ytterligare fynd i det manliga reproduktionssystemet (degeneration och/eller atrofi av sädeskanaler i testiklar, minskning av luminala spermier i epididymis), och verkade vara känsligare för kabozantinibrelaterad toxicitet vid jämförbara dosnivåer.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Tablettinnehåll
Mikrokristallin cellulosa
Laktos, vattenfri
Hydroxipropylcellulosa
Kroskarmellosnatrium
Kolloidal silikondioxid, vattenfri
Magnesiumstearat
Filmdragering
Hypromellos 2910
Titandioxid (E171)
Triacetin
Gul järnoxid (E172)
Inkompatibiliteter
Ej relevant.
Hållbarhet
4 år.
Särskilda förvaringsanvisningar
Inga särskilda förvaringsanvisningar.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
CABOMETYX tabletti, kalvopäällysteinen
20 mg (L:kyllä) 30 kpl (5121,14 €)
40 mg (L:kyllä) 30 kpl (5121,14 €)
60 mg (L:kyllä) 30 kpl (5121,14 €)
PF-selosteen tieto
HDPE-burk med en barnskyddande förslutning av polypropylen och tre behållare med torkmedel av silikongel och polyesterspiral. Varje burk innehåller 30 filmdragerade tabletter.
Läkemedlets utseende:
CABOMETYX 20 mg filmdragerade tabletter
Tabletterna är gula och runda utan skåra och präglade med ”XL” på ena sidan och ”20” på den andra sidan av tabletten.
CABOMETYX 40 mg filmdragerade tabletter
Tabletterna är gula och triangelformade utan skåra och präglade med ”XL” på ena sidan och ”40” på den andra sidan av tabletten.
CABOMETYX 60 mg filmdragerade tabletter
Tabletterna är gula och ovala utan skåra och präglade med ”XL” på ena sidan och ”60” på den andra sidan av tabletten.
Särskilda anvisningar för destruktion och övrig hantering
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
CABOMETYX tabletti, kalvopäällysteinen
20 mg 30 kpl
40 mg 30 kpl
60 mg 30 kpl
- Ylempi erityiskorvaus (100 %). Kabotsantinibi: Munuaissyövän hoito erityisin edellytyksin (1502).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Kabotsantinibi: Munuaissyövän ja kilpirauhassyövän hoito erityisin edellytyksin (3012).
Atc-kod
L01EX07
Datum för översyn av produktresumén
15.01.2026
Yhteystiedot
Kista Science Tower, Färögatan 33
SE-164 51 Kista
Sweden
+46 8 451 60 00
www.ipsen.com
info.se@ipsen.com