
BOOSTRIX injektionsvätska, suspension i förfylld spruta
Kvalitativ och kvantitativ sammansättning
1 dos (0,5 ml) innehåller:
Difteritoxoid1 minst 2 internationella enheter (IE) (2,5 Lf)
Tetanustoxoid1 minst 20 internationella enheter (IE) (5 Lf)
Bordetella pertussis-antigener
Pertussistoxoid1 | 8 mikrogram |
Filamentöst hemagglutinin1 | 8 mikrogram |
Pertaktin1 | 2,5 mikrogram |
1adsorberat på hydratiserad aluminiumhydroxid (Al(OH)3) | 0,3 milligram Al3+ |
och aluminiumfosfat (AlPO4) | 0,2 milligram Al3+ |
Vaccinet kan innehålla spår av formaldehyd som används under tillverkningsprocessen (se avsnitt Kontraindikationer).
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Injektionsvätska, suspension i förfylld spruta.
Kliniska uppgifter
Terapeutiska indikationer
Boostrix är indicerat som boostervaccination mot difteri, stelkramp (tetanus) och kikhosta (pertussis) av individer från 4 års ålder och uppåt (se avsnitt Dosering och administreringssätt).
Boostrix är också indicerat som passivt skydd mot kikhosta i tidig spädbarnsålder när modern har vaccinerats under graviditet (se avsnitt Dosering och administreringssätt, Fertilitet, graviditet och amning och Farmakodynamiska egenskaper).
Administrering av Boostrix ska ske enligt officiella rekommendationer.
Dosering och administreringssätt
Dosering
En dos om 0,5 ml av vaccinet rekommenderas.
Boostrix kan ges från och med 4 års ålder.
Administrering av Boostrix ska ske enligt officiella rekommendationer och/eller enligt lokal praxis för kombinationsvaccin som innehåller reducerad dos av difteritoxoid-, tetanustoxoid- och pertussisantigen.
Boostrix kan ges under graviditetens andra eller tredje trimester enligt officiella rekommendationer (se avsnitt Terapeutiska indikationer, Fertilitet, graviditet och amning och Farmakodynamiska egenskaper).
Boostrix kan även ges som en del av en vaccinationsserie mot difteri, stelkramp och kikhosta till ungdomar och vuxna med okänd vaccinationsstatus eller ofullständig vaccination mot difteri, stelkramp och kikhosta. Baserat på data hos vuxna rekommenderas ytterligare två doser av ett difteri- och stelkrampsvaccin, en och sex månader efter den första dosen, för att maximera vaccinsvaret mot difteri och stelkramp (se avsnitt Farmakodynamiska egenskaper).
Boostrix kan användas vid behandling av skada med risk för tetanussmitta om personen tidigare är primärvaccinerad med tetanustoxoid och om en boosterdos mot difteri och kikhosta är indicerad. Tetanusimmunglobulin kan ges samtidigt i enlighet med officiella rekommendationer.
Påfyllnadsvaccination mot difteri, stelkramp och kikhosta ska ges enligt intervall som fastslås i officiella rekommendationer (vanligtvis 10 år).
Barn
Säkerhet och effekt för Boostrix för barn under 4 års ålder har inte fastställts.
Administreringssätt
Boostrix är avsett för djup intramuskulär injektion företrädesvis i deltoideusregionen (se avsnitt Varningar och försiktighet).
Kontraindikationer
Överkänslighet mot de aktiva substanserna eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen eller formaldehyd.
Överkänslighet efter tidigare vaccination mot difteri, stelkramp eller kikhosta.
Boostrix är kontraindicerat hos personer som har fått encefalopati av okänd etiologi inom 7 dagar efter en tidigare vaccination med vaccin mot kikhosta. I sådana fall ska vaccinationen mot kikhosta avbrytas och vaccinationskuren fortsättas med vacciner mot difteri och stelkramp.
Boostrix ska inte ges till personer som upplevt övergående trombocytopeni eller neurologiska komplikationer (kramper och hypotoniska-hyporesponsiva episoder, se avsnitt Varningar och försiktighet) efter tidigare vaccination mot difteri och/eller stelkramp.
Som för andra vacciner ska vaccination med Boostrix uppskjutas vid akut svår febersjukdom. Lindrig infektion utgör inte en kontraindikation.
Varningar och försiktighet
Vaccination ska föregås av anamnes (särskilt vad det gäller tidigare vaccination och förekomst av eventuella oönskade reaktioner).
Om något av följande har inträffat i anslutning till vaccination mot kikhosta ska beslutet att ge fler doser av vaccin mot kikhosta noggrant övervägas.
- Feber ≥ 40,0 °C inom 48 timmar efter vaccination, utan annan identifierbar orsak.
- Medvetslöshet eller chockliknande tillstånd (hypotonisk-hyporesponsiv episod) inom 48 timmar efter vaccination.
- Ihållande, otröstlig gråt som varar ≥ 3 timmar och som inträffar inom 48 timmar efter vaccination.
- Kramper med eller utan feber inom 3 dagar efter vaccination.
I vissa fall, som vid hög incidens av kikhosta, överväger fördelarna riskerna.
Som för alla vaccinationer ska risk och nytta av immunisering med vaccin eller uppskjutande av vaccination noggrant övervägas hos barn som har en nydebuterande eller progressiv svår neurologisk sjukdom.
Som för alla vacciner för injektion ska lämplig medicinsk behandling och övervakning alltid finnas lätt tillgänglig i händelse av en sällsynt anafylaktisk reaktion efter administrering av vaccinet.
Boostrix ska ges med försiktighet till personer med trombocytopeni (se avsnitt Kontraindikationer) eller blödningsstörning, eftersom en intramuskulär injektion kan ge en blödning hos dessa personer. I dessa situationer kan vaccinet administreras subkutant om detta överensstämmer med lokala rekommendationer. Vid administrering via båda administreringsvägarna ska man hålla ett fast tryck (utan att gnida) på injektionsstället under minst två minuter.
Boostrix ska under inga omständigheter ges intravaskulärt.
Tidigare kramper, förekomst av kramper inom familjen och förekomst av en oönskad händelse/biverkan inom familjen efter tidigare DTP-vaccination utgör ingen kontraindikation.
Infektion med humant immunbristvirus (HIV) anses inte vara en kontraindikation. Dock uppnås eventuellt inte förväntat immunologiskt svar efter vaccination av patienter med nedsatt immunförsvar.
Svimning kan inträffa efter eller till och med före vaccination, framför allt hos ungdomar, som en psykogen reaktion på nålstick. Detta kan åtföljas av flera neurologiska symtom såsom övergående synrubbning, parestesi och rörelser av tonisk-klonisk typ i extremiteterna under återhämtning. Det är viktigt att lämplig beredskap finns på plats för att undvika skador vid svimning.
Som för alla vacciner uppnås eventuellt inte ett skyddande immunsvar hos alla vaccinerade. I detta avseende skiljer sig inte Boostrix från andra vacciner.
Hjälpämnen
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per dos, d.v.s. är näst intill ”natriumfritt”.
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Interaktioner
Användning tillsammans med andra vacciner eller immunglobuliner
Boostrix kan ges samtidigt med vaccin mot humant papillomvirus. Inga kliniskt signifikanta interaktioner har observerats mellan antikropparna som komponenterna i någotdera av dessa vacciner gett upphov till.
Boostrix kan ges samtidigt med konjugerat vaccin mot meningokockinfektion, serogrupper A, C, W‑135 och Y (MenACWY). Kliniska studier på individer i åldrarna 9 till 25 år visade att immunsvaret mot tetanus-, difteri- och meningokockantigener var opåverkat. Lägre geometriska medelkoncentrationer (GMC) observerades för pertussisantigener, dessa data tyder dock inte på någon klinisk relevant interferens.
Boostrix kan administreras samtidigt med inaktiverade säsongsinfluensavacciner som inte innehåller adjuvans. När Boostrix gavs samtidigt med ett trevalent inaktiverat influensavaccin hos personer i åldrarna 19 till 64 år visade kliniska data att immunsvaret mot stelkramps-, difteri-, pertussistoxin- (PT) och influensaantigener förblev oförändrade. Lägre geometriska medelkoncentrationer (GMC) påträffades i antigenerna hemagglutinin (FHA) och pertaktin (PRN). Dessa data tyder dock inte på någon kliniskt signifikant effekt. Inga skillnader påträffades i en predefinierad explorativ kohort när vacciner administrerades antingen samtidigt eller separat till individer som var 65 år eller äldre.
Boostrix kan administreras samtidigt med herpes zoster-vaccin som inte innehåller levande patogener. Kliniska data från individer 50 år och äldre visade att immunsvaret mot stelkramp-, difteri-, PT-, FHA- och herpes zoster-antigener förblev oförändrade. Lägre geometriska medelvärden (GMC) påträffades på PRN-antigenen, men dessa data tyder dock inte på någon kliniskt signifikant effekt.
Samtidig administrering av Boostrix och andra vacciner eller immunglobuliner har inte studerats.
Det är dock inte troligt att samtidig administrering med andra inaktiverade eller immunglobuliner har signifikant klinisk effekt på immunsvaret.
Om samtidig användning av Boostrix och andra vacciner eller immunglobuliner anses nödvändigt, ska de, enligt allmän vaccinationspraxis och rekommendationer, ges vid olika injektionsställen.
Användning tillsammans med immunsuppressiva läkemedel
Som med andra vacciner kan ett adekvat immunsvar inte alltid uppnås hos patienter som erhåller immunsuppressiv behandling.
Fertilitet, graviditet och amning
Graviditet
Boostrix kan användas under graviditetens andra eller tredje trimester enligt officiella rekommendationer.
För data om förebyggande av kikhosta hos spädbarn vars mödrar vaccinerats under graviditeten, se avsnitt Farmakodynamiska egenskaper.
I en randomiserad kontrollerad klinisk studie (342 graviditetsutfall) och en prospektiv observationsstudie (793 graviditetsutfall) gavs Boostrix till gravida kvinnor under graviditetens sista trimester. Enligt säkerhetsdata från dessa studier påvisades inga vaccinrelaterade biverkningar på graviditeten eller på fostrets/det nyfödda barnets hälsa.
Det finns inga säkerhetsdata från prospektiva kliniska studier avseende användning av Boostrix eller Boostrix Polio under graviditetens första och andra trimester.
Data från passiv övervakning där gravida kvinnor exponerades för Boostrix- eller Boostrix Polio-vaccinet under den sista eller andra trimestern har inte visat några vaccinrelaterade biverkningar på graviditeten eller på fostrets/det nyfödda barnets hälsa.
Som med andra inaktiverade vacciner, är det dock inte troligt att vaccination med Boostrix skadar fostret under någon trimester av graviditeten.
Djurstudier tyder inte på några direkta eller indirekta skadliga effekter på graviditet, embryonal/fosterutveckling, förlossning eller postnatal utveckling (se avsnitt Prekliniska säkerhetsuppgifter).
Amning
Effekten av administrering av Boostrix under amning har inte utvärderats, men eftersom Boostrix innehåller toxoider och inaktiverade antigener förväntas ingen risk för spädbarnet. Hälso- och sjukvårdspersonal ska noga väga fördelarna mot riskerna vid administrering av Boostrix till kvinnor som ammar.
Fertilitet
Det finns inga tillgängliga humandata från prospektiva kliniska studier. Djurstudier tyder inte på några direkta eller indirekta skadliga effekter på fertilitet hos hona (se avsnitt Prekliniska säkerhetsuppgifter).
Effekter på förmågan att framföra fordon och använda maskiner
Det är inte troligt att vaccinet skulle ha någon effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
Den säkerhetsprofil som presenteras nedan baseras på uppgifter från kliniska studier varvid Boostrix administrerades till 839 barn (från 4 till 8 års ålder) och 1 931 vuxna, unga och barn (från 10 till 76 års ålder) (Tabell 1).
Den vanligaste förekommande händelsen efter vaccination med Boostrix i båda grupperna var lokala reaktioner vid injektionsstället (smärta, rodnad och svullnad) vilket rapporterades hos 23,7–80,6 % av de vaccinerade i båda studierna. Vanligtvis inträffade dessa reaktioner inom 48 timmar efter vaccination och alla reaktioner försvann utan sviter.
Tabell över biverkningar
De rapporterade biverkningarna anges enligt följande frekvens:
Mycket vanlig: | > 1/10 |
Vanlig: | > 1/100, < 1/10 |
Mindre vanlig: | > 1/1 000, < 1/100 |
Sällsynt: | > 1/10 000, < 1/1 000 |
Mycket sällsynt: | < 1/10 000) |
Biverkningarna presenteras inom varje frekvensområde efter fallande allvarlighetsgrad.
- Kliniska studier
Tabell 1: Biverkningar rapporterade i kliniska studier med Boostrix
Organsystemklass | Frekvens | Biverkningar | |
Barn i åldern 4‑8 år (N = 839) | Personer i åldern 10‑76 år (N = 1931) | ||
Infektioner och infestationer | Mindre vanliga | övre luftvägsinfektion | övre luftvägsinfektion, faryngit |
Blodet och lymfsystemet | Mindre vanliga | lymfadenopati | |
Metabolism och nutrition | Vanliga | anorexi | |
Psykiska störningar | Mycket vanliga | irritabilitet | |
Centrala och perifera nervsystemet | Mycket vanliga | somnolens | huvudvärk |
Vanliga | huvudvärk | yrsel | |
Mindre vanliga | uppmärksamhetsstörningar | synkope | |
Ögon | Mindre vanliga | konjunktivit | |
Andningsvägar, bröstkorg och mediastinum | Mindre vanliga | hosta | |
Magtarmkanalen | Vanliga | diarré, kräkning, magtarmbesvär | illamående, magtarmbesvär |
Mindre vanliga | diarré, kräkning | ||
Hud och subkutan vävnad | Mindre vanliga | utslag | hyperhidros, klåda, utslag |
Muskuloskeletala systemet och bindväv | Mindre vanliga | artralgi, myalgi, ledstelhet, muskuloskeletal stelhet | |
Allmänna symtom och/eller symtom vid administreringsstället | Mycket vanliga | reaktioner vid injektionsstället (såsom rodnad och/eller svullnad), smärta vid injektionsstället, trötthet | reaktioner vid injektionsstället (såsom rodnad och/eller svullnad), allmän sjukdomskänsla, trötthet, smärta vid injektionsstället |
Vanliga | feber ≥ 37,5 °C inklusive feber > 39,0 °C, kraftig svullnad av den vaccinerade kroppsdelen (ibland även intilliggande led) | feber (≥ 37,5 °C), reaktioner vid injektionsstället (såsom induration och steril abscess vid injektionsstället) | |
Mindre vanliga | andra reaktioner vid injektionsstället (såsom induration), smärta | feber (> 39,0 °C), influensaliknande sjukdom, smärta | |
Reaktogenicitet efter upprepad dos
Data från 146 individer tyder på att en liten ökning av lokal reaktogenicitet (smärta, rodnad och svullnad) kan förekomma vid upprepad vaccination enligt ett 0, 1, 6 månaders schema till vuxna (> 40 års ålder).
Data visar på att individer som primärvaccinerats mot DTP som barn kan få en ökning av lokal reaktogenicitet efter boosterdos av Boostrix.
- Uppföljningsstudier efter lansering
Eftersom dessa händelser rapporterats spontant går det inte att med säkerhet beräkna frekvensen.
Tabell 2: Biverkningar rapporterade för Boostrix efter lansering
Organsystemklass | Frekvens | Biverkningar |
Immunsystemet | ingen känd frekvens | allergiska reaktioner, inklusive anafylaktiska och anafylaktoida reaktioner |
Centrala och perifera nervsystemet | ingen känd frekvens | hypotoniska-hyporesponsiva episoder, kramper (med eller utan feber) |
Hud och subkutan vävnad | ingen känd frekvens | urtikaria, angioödem |
Allmänna symtom och/eller symtom vid administreringsstället | ingen känd frekvens | asteni |
Efter administrering av vacciner innehållande tetanustoxoid har det förekommit mycket sällsynta rapporter om biverkningar i centrala eller perifera nervsystemet inkluderande uppåtgående paralys och även respiratorisk paralys (t.ex. Guillain-Barrés syndrom).
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till:
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Fall av överdosering har rapporterats under uppföljningsstudier efter lansering. Biverkningarna efter överdosering, när sådana rapporterades, liknade de som rapporteras vid normal vaccinadministrering.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Vacciner mot bakteriella infektioner, kombinerat, ATC-kod J07AJ52
Immunsvar
Omkring en månad efter boostervaccination med Boostrix observerades följande frekvenser för serokonversionshastigheter (Tabell 3).
Tabell 3: Immunsvar hos barn, ungdomar och vuxna
Antigen | Respons(1) | Vuxna och ungdomar från 10 års ålder och uppåt ATP(2) N = 1694 (% vaccinerade) | Barn från 4 års ålder ATP(2) N = 415 (% vaccinerade) |
Difteri | > 0,1 IE/ml | 97,2 % | 99,8 % |
Tetanus | > 0,1 IE/ml | 99,0 % | 100,0 % |
Pertussis: Pertussistoxoid Filamentöst hemagglutinin Pertaktin | > 5 ELISA-enheter/ml | 97,8 % 99,9 % 99,4 % | 99,0 % 100,0 % 99,8 % |
(1) Respons: som seroprotektion vid angiven tidpunkt ansågs en koncentration av antikroppar mot difteri och tetanus > 0,1 IE/ml, en koncentration av antikroppar mot pertussis > 5 ELISA-enheter/ml ansågs som seropositivitet.
(2) ATP: Enligt protokoll - omfattar alla individer som fått en boosterdos av Boostrix som engångsdos och för vilka immungenicitetsdata var tillgängliga för minst en antigen vid den specifika tidpunkten.
(N) Minsta antalet personer med tillgängliga data för vardera antigen.
Hos vuxna och ungdomar visade jämförande studier att difteri-antikroppstitrar en månad efter vaccination var liknande dem för Td-vacciner avsedda för vuxna med samma antigeninnehåll som Boostrix. Lägre tetanus-antikroppstitrar sågs jämfört med hos Td-vacciner avsedda för vuxna.
Som med andra Td-vacciner avsedda för vuxna inducerar Boostrix högre titrar både för anti-D- och anti-T-antikroppar hos barn och ungdomar än hos vuxna.
Varaktighet av immunsvaret
Följande seroprotektion/seropositivitet observerades 3‑3,5 år, 5‑6 år och 10 år efter första vaccinationen med Boostrix hos individer vaccinerade i enlighet med protokoll (ATP1) (Tabell 4).
Tabell 4: Varaktighet av immunsvar hos barn, ungdomar och vuxna
Antigen | Respons(2) | Vuxna och ungdomar från 10 års ålder och uppåt (% vaccinerade) | |||||
3‑3,5 års varaktighet | 5 års varaktighet | 10 års varaktighet | |||||
Vuxna(3) (N=309) | Ungdomar(3) (N=261) | Vuxna(3) (N=232) | Ungdomar(3) (N=250) | Vuxna(3) (N=158) | Ungdomar(3) (N=74) | ||
Difteri | ≥ 0,1 IE/ml | 71,2 % | 91,6 % | 84,1 % | 86,8 % | 64,6 % | 82,4 % |
≥ 0,016 IE/ml(4) | 97,4 % | 100 % | 94,4 % | 99,2 % | 89,9 % | 98,6 % | |
Tetanus | ≥ 0,1 IE/ml | 94,8 % | 100 % | 96,2 % | 100 % | 95,0 % | 97,3 % |
Pertussis Pertussistoxoid Filamentöst hemagglutinin Pertaktin | ≥ 5 ELISA-enheter/ml | 90,6 % 100 % 94,8 % | 81,6 % 100 % 99,2 % | 89,5 % 100 % 95,0 % | 76,8 % 100 % 98,1 % | 85,6 % 99,4 % 95,0 % | 61,3 % 100 % 96,0 % |
(1) ATP: Enligt protokoll - omfattar alla individer som fått en boosterdos av Boostrix, för vilka immungenicitetsdata var tillgängliga för minst ett antigen vid den specifika tidpunkten.
(2) Respons: som seroprotektion vid angiven tidpunkt ansågs en koncentration av antikroppar mot difteri och tetanus > 0,1 IE/ml, en koncentration av antikroppar mot pertussis > 5 ELISA-enheter/ml ansågs som seropositivitet.
(3) Begreppen ”vuxna” och ”ungdomar” avser åldrar där individer fick sin första vaccination med Boostrix.
(4) Andelen i procent av de vaccinerade med antikroppsnivå förknippad med skydd mot sjukdom (≥ 0,1 IE/ml med ELISA-test eller > 0,016 IE/ml med in vitro Vero-cell-neutraliseringstest).
N= Minsta antalet individer med tillgängliga data för vardera antigen.
Följande seroprotektion/seropositivitet observerades 3‑3,5 år och 5‑6 år efter första vaccinationen med Boostrix hos individer vaccinerade i enlighet med protokoll (ATP1):
Antigen | Respons(2) | Barn från 4 års ålder och uppåt (% vaccinerade) | |
3‑3,5 års varaktighet | 5‑6 års varaktighet | ||
(N=118) | (N=68) | ||
Difteri | ≥ 0,1 IE/ml | 97,5 % | 94,2 % |
≥ 0,016 IE/ml(3) | 100 % | Inte fastställt | |
Tetanus | ≥ 0,1 IE/ml | 98,4 % | 98,5 % |
Pertussis Pertussistoxoid Filamentöst hemagglutinin Pertaktin | ≥ 5 ELISA-enheter/ml | 58,7 % 100 % 99,2 % | 51,5 % 100 % 100 % |
(1) ATP: Enligt protokoll - omfattar alla individer som fått en boosterdos av Boostrix, för vilka immungenicitetsdata var tillgängliga för minst ett antigen vid den specifika tidpunkten.
(2) Respons: som seroprotektion vid angiven tidpunkt ansågs en koncentration av antikroppar mot difteri och tetanus > 0,1 IE/ml, en koncentration av antikroppar mot pertussis > 5 ELISA-enheter/ml ansågs som seropositivitet.
(3) Andelen i procent av de vaccinerade med antikroppsnivå förknippad med skydd mot sjukdom (≥ 0,1 IE/ml med ELISA-test eller > 0,016 IE/ml med in vitro Vero-cell-neutraliseringstest).
N= Minsta antalet individer med tillgängliga data för vardera antigen.
Skyddseffekt mot kikhosta
De pertussisantigener som Boostrix innehåller ingår i det kombinerade acellulära barnvaccinet (Infanrix), för vilket effekt efter primärvaccination har påvisats i en effektstudie av individer i samma hushåll. Antikroppstitrarna för alla tre pertussiskomponenterna efter vaccination med Boostrix är högre än de som påvisats i effektstudien av individer i samma hushåll. Baserat på dessa jämförelser kan Boostrix ge skydd mot pertussis, men graden och varaktigheten av skyddet har inte fastställts.
Passivt skydd mot kikhosta hos spädbarn (yngre än 3 månader) vars mödrar vaccinerats under graviditeten
I en randomiserad, placebokontrollerad överkorsningsstudie påvisades högre koncentrationer av antikroppar mot kikhosta i navelsträngsblodet hos barn vars mödrar vaccinerats med Boostrix (dTpa-grupp; N = 291) under graviditetsvecka 27‑36 jämfört med placebo (kontrollgrupp; N = 292). De geometriska medelvärdena för koncentrationerna av antikroppar i navelsträngsblodet mot kikhosteantigenerna PT, FHA och PRN var 46,9, 366,1 respektive 301,8 IE/ml i dTpa-gruppen och 5,5, 22,7 respektive 14,6 IE/ml i kontrollgruppen. Detta motsvarar antikroppstitrar som är 8, 16 respektive 21 gånger högre i navelsträngsblodet hos vaccinerade mödrars barn än barn i kontrollgruppen. I icke-experimentella effektstudier har konstaterats att dessa antikroppstitrar kan ge passivt skydd mot kikhosta.
Immunogenicitet hos spädbarn och småbarn vars mödrar har vaccinerats under graviditet
I två kliniska studier undersöktes immunogeniciteten av Infanrix hexa (difteri-, stelkramp-, kikhosta-, hepatit B-, inaktiverad poliomyelit och Haemophilus influenzae typ B-konjugatvaccin) hos spädbarn och småbarn födda till friska mödrar som vaccinerades med Boostrix under graviditetsvecka 27‑36.
Infanrix hexa administrerades i kombination med det 13-valenta pneumokockkonjugatvaccinet till spädbarn som primärvaccination (n = 268) och till samma spädbarn/barn vid 11‑18 månaders ålder som boostervaccination (n = 229).
Efter primärvaccination och boostervaccination visade immunologiska data från studierna ingen klinisk betydande interferens mellan moderns vaccination med Boostrix och spädbarnens och småbarnens svar på difteri-, stelkramps-, hepatit B-, Haemophilus influenzae typ B- eller pneumokockantigener eller inaktiverat poliovirus.
Spädbarn och småbarn vars mödrar fick Boostrix-vaccination under graviditeten visade lägre antikroppskoncentrationer mot pertussisantigener efter primärvaccination (PT, FHA och PRN) och boostervaccination (PT, FHA). Ökningen av koncentrationen av antikroppar mot kikhosta från tidpunkten före boosterdosen till en månad efter den låg vid samma nivå hos spädbarn och småbarn födda av mödrar som vaccinerats med Boostrix respektive placebo under graviditeten, vilket visar en effektiv förstärkning av immunsystemet. Eftersom det inte finns några data som tyder på ett samband mellan observationen och skyddet mot kikhosta, är den kliniska betydelsen av observationerna inte helt klarlagd. Aktuella epidemiologiska data om kikhosta efter insättande av dTpa-immunisering under graviditet tyder dock inte på att denna immunologiska interferens skulle ha någon klinisk betydelse.
Skydd mot kikhosta hos spädbarn vars mödrar vaccinerats under graviditet
Vaccineffektivitet för Boostrix eller Boostrix Polio utvärderades i tre observationsstudier, i Storbritannien, Spanien och Australien. Vaccination skedde som en del av ett maternellt vaccinationsprogram under den tredje trimestern av graviditeten för att skydda spädbarn under tre månaders ålder mot kikhosta.
Uppgifter om respektive studiedesign samt resultat finns i tabell 5.
Tabell 5: Vaccineffektivitet mot kikhosta hos spädbarn under tre månaders ålder födda av kvinnor som vaccinerats under den tredje trimestern av graviditeten med Boostrix/Boostrix Polio.
Studieland | Vaccin | Studiedesign | Vaccineffektivitet |
Storbritannien | Boostrix Polio | Retrospektiv, screeningmetod | 88 % (95 % KI: 79; 93) |
Spanien | Boostrix | Prospektiv, matchad fallkontroll | 90,9 % (95 % KI: 56,6; 98,1) |
Australien | Boostrix | Prospektiv, matchad fallkontroll | 69 % (95 % KI: 13; 89) |
KI: Konfidensintervall
Om maternell vaccination sker inom två veckor före födsel kan vaccineffektiviteten hos barnet vara lägre än det som nämns i tabellen.
Immunsvar efter en upprepad dos av Boostrix
Immunogeniciteten av Boostrix administrerat 10 år efter en tidigare boosterdos med vaccin mot difteri, stelkramp och kikhosta (acellulärt) med reducerat antigeninnehåll har utvärderats. En månad efter vaccination var > 99 % av individerna seropositiva mot kikhosta och serologiskt skyddade mot difteri och stelkramp.
Immunsvar hos individer utan tidigare eller med okänd vaccinationshistorik
Efter administrering av en dos av Boostrix till 83 ungdomar i åldrarna 11 till 18 år, utan tidigare vaccination mot kikhosta och ingen vaccination mot difteri och stelkramp under de senaste 5 åren, hade alla individer seroprotektion mot stelkramp och difteri. Seropositiviteten efter en dos varierade mellan 87 % och 100 % för de olika pertussisantigenerna.
Efter administrering av en dos av Boostrix till 139 vuxna ≥ 40 års ålder som inte fått något vaccin mot difteri och stelkramp de senaste 20 åren var mer än 98,5 % seropositiva mot alla pertussisantigenerna och 81,5 % respektive 93,4 % hade serologiskt skydd mot difteri respektive stelkramp. Efter administrering av ytterligare två doser (med 0, 1 och 6 månaders vaccinationsprogram) var graden av seropositivitet 100 % mot alla tre pertussisantigenerna och graden av seroprotektion mot difteri och stelkramp nådde 99,3 % respektive 100 %.
Immunsvar och säkerhetsprofil hos individer som står på aktiv behandling för obstruktiv luftvägssjukdom
Säkerheten och immunogeniciteten efter vaccination med Boostrix har utvärderats i en beskrivande metaanalysstudie med 222 deltagare ≥ 18 år under pågående behandling för obstruktiv luftvägssjukdom så som astma eller kronisk obstruktiv lungsjukdom (KOL). En månad efter vaccination med Boostrix uppnådde 89,0 % skyddande antikroppsnivåer (≥0,1 IE/ml) mot difteriantigen och 97,2 % mot stelkrampsantigen. Andelen som svarade mot pertussis/kikhost-antigen pertussistoxoid, filamentöst hemagglutinin och pertaktin var78,3 %, 96,1 % respektive 92,2 %. Dessa resultat överensstämmer med de resultat som erhållits i den vuxna populationen och med en liknande säkerhetsprofil.
Farmakokinetiska egenskaper
Utvärdering av farmakokinetik krävs inte för vacciner.
Prekliniska säkerhetsuppgifter
Reproduktionstoxikologi
Fertilitet
Icke-kliniska data som erhållits med Boostrix visade inte på några särskilda risker för människa baserat på konventionella fertilitetsstudier på honråtta och -kanin.
Graviditet
Icke-kliniska data som erhållits med Boostrix visade inte på några särskilda risker för människa baserat på konventionella studier av embryo-fosterutveckling som gjorts på råtta och kanin samt på råtta med avseende på förlossning och postnatal toxicitet (fram till slutet av digivningsperioden).
Djurtoxikologi och/eller farmakologi
Prekliniska data visar inga särskilda biverkningar för människa baserat på konventionella studier avseende säkerhet och toxicitet.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Natriumklorid
Vatten för injektionsvätskor
För adjuvans, se avsnitt KVALITATIV OCH KVANTITATIV SAMMANSÄTTNING.
Inkompatibiliteter
Då blandbarhetsstudier saknas ska detta läkemedel inte blandas med andra läkemedel.
Hållbarhet
4 år
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 °C–8 °C).
Stabilitetsdata indikerar att Boostrix är stabilt i 7 dagar vid temperaturer upp till 37 °C. I slutet av denna period bör Boostrix användas eller kasseras. Dessa data är endast avsedda som vägledning för hälso- och sjukvårdspersonal i händelse av tillfällig förvaring utanför kylskåpstemperatur.
Får ej frysas.
Förvaras i originalförpackningen. Ljuskänsligt.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
BOOSTRIX injektioneste, suspensio, esitäytetty ruisku
0,5 ml (34,15 €)
PF-selosteen tieto
0,5 ml suspension i en förfylld spruta (typ I glas) med en kolvring (butylgummi) och ett skyddslock.
Förpackningsstorlekar 1 och 10 med eller utan nålar.
Skyddslocket och gummikolvringen på den förfyllda sprutan är av syntetiskt gummi.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Läkemedlets utseende:
Boostrix är en grumlig, vit suspension.
Särskilda anvisningar för destruktion och övrig hantering
Före användning ska vaccinet uppnå rumstemperatur och omskakas väl till en homogen, grumlig, vit suspension. Före administrering ska vaccinet inspekteras visuellt med avseende på främmande partiklar och/eller fysikaliska förändringar. Om någotdera observeras får vaccinet inte användas.
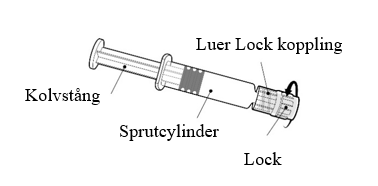
Instruktioner för den förfyllda sprutan
| Håll alltid i sprutcylindern, inte i kolvstången. Skruva av locket på sprutan genom att vrida den motsols. |
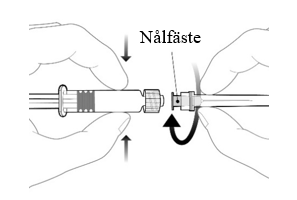
| Fäst nålen på sprutan genom att ansluta den till Luer Lock kopplingen och rotera ett kvarts varv medsols tills du känner att den låser sig. Lock Dra inte ut kolvstången ur sprutcylindern, om detta sker ska vaccinet inte administreras. |
Destruktion:
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
BOOSTRIX injektioneste, suspensio, esitäytetty ruisku
0,5 ml
- Ei korvausta.
Atc-kod
J07AJ52
Datum för översyn av produktresumén
27.06.2023
Yhteystiedot
 GLAXOSMITHKLINE OY
GLAXOSMITHKLINE OY Porkkalankatu 20 A
00180 Helsinki
010 303 030
www.glaxosmithkline.fi