
BRIVIACT oraaliliuos 10 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi millilitra sisältää 10 mg brivarasetaamia.
Apuaineet, joiden vaikutus tunnetaan
Yksi millilitra sisältää 168 mg sorbitolia (E420), 1 mg metyyliparahydroksibentsoaattia (E218) ja enintään 5,5 mg propyleeniglykolia (E1520).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Oraaliliuos
Kliiniset tiedot
Käyttöaiheet
Briviact on tarkoitettu lisälääkkeeksi paikallisalkuisten toissijaisesti yleistyvien tai yleistymättömien kohtausten hoitoon epilepsiaa sairastaville aikuisille, nuorille ja vähintään 2-vuotiaille lapsille.
Annostus ja antotapa
Annostus
Lääkäri valitsee sopivimman lääkemuodon ja vahvuuden potilaan painon ja annoksen perusteella. On suositeltavaa, että vanhemmat ja potilaan hoidosta huolehtivat henkilöt antavat Briviact-oraaliliuoksen pakkauksessa toimitetulla mittalaitteella (10 ml:n tai 5 ml:n mittaruisku).
Yhteenveto suositellusta annostuksesta aikuisille, nuorille ja vähintään 2-vuotiaille lapsille on seuraavassa taulukossa. Annos otetaan kahteen yhtä suureen annokseen jaettuna noin 12 tunnin välein.
| Suositeltu aloitusannos | Suositeltu ylläpitoannos | Terapeuttinen annosalue* |
| Nuoret ja lapset, jotka painavat vähintään 50 kg, sekä aikuiset | ||
| 50 mg/vrk (tai 100 mg/vrk)** | 100 mg/vrk | 50–200 mg/vrk |
| Nuoret ja lapset, jotka painavat vähintään 20 kg, mutta alle 50 kg | ||
| 1 mg/kg/vrk (korkeintaan 2 mg/kg/vrk)** | 2 mg/kg/vrk | 1–4 mg/kg/vrk |
| Lapset, jotka painavat vähintään 10 kg, mutta alle 20 kg | ||
| 1 mg/kg/vrk (korkeintaan 2,5 mg/kg/vrk)** | 2,5 mg/kg/vrk | 1–5 mg/kg/vrk |
* Annosta voi muuttaa potilaan yksilöllisen vasteen mukaan tällä tehokkaalla annosalueella.
** Mikäli lääkäri pitää sitä tarpeellisena kohtausten saamiseksi hallintaan.
Aikuiset
Suositeltu aloitusannos on joko 50 mg/vrk tai 100 mg/vrk sen mukaan, millaiseksi lääkäri arvioi kohtausten vähentämistarpeen verrattuna mahdollisiin haittavaikutuksiin. Annosta voi muuttaa potilaan yksilöllisen vasteen ja hoidon siedettävyyden mukaan tehokkaalla annosvälillä 50–200 mg/vrk.
Vähintään 50 kg painavat lapset ja nuoret
Suositeltu aloitusannos on 50 mg/vrk. Brivarasetaamihoito voidaan aloittaa myös annoksella 100 mg/vrk, jos lääkäri pitää sitä tarpeellisena kohtausten saamiseksi hallintaan. Suositeltu ylläpitoannos on 100 mg/vrk. Annosta voidaan säätää potilaan yksilöllisen vasteen perusteella tehokkaalla annosalueella 50–200 mg/vrk.
Vähintään 20 kg mutta alle 50 kg painavat lapset ja nuoret
Suositeltu aloitusannos on 1 mg/kg/vrk. Brivarasetaamihoito voidaan aloittaa myös annoksella, joka on korkeintaan 2 mg/kg/vrk, jos lääkäri pitää sitä tarpeellisena kohtausten saamiseksi hallintaan. Suositeltu ylläpitoannos on 2 mg/kg/vrk. Annosta voidaan säätää potilaan yksilöllisen vasteen perusteella tehokkaalla annosalueella 1–4 mg/kg/vrk.
Vähintään 10 kg, mutta alle 20 kg, painavat lapset
Suositeltu aloitusannos on 1 mg/kg/vrk. Brivarasetaamihoito voidaan aloittaa myös annoksella, joka on korkeintaan 2,5 mg/kg/vrk, jos lääkäri pitää sitä tarpeellisena kohtausten saamiseksi hallintaan. Suositeltu ylläpitoannos on 2,5 mg/kg/vrk. Annosta voidaan säätää potilaan yksilöllisen vasteen perusteella tehokkaalla annosalueella 1–5 mg/kg/vrk.
Yhden lääkkeenantokerran annos kullekin potilaalle lasketaan seuraavalla kaavalla:
Antokerralla annettava määrä (ml) = [paino (kg) x vuorokausiannos (mg/kg/vrk)] x 0,05
Briviact-oraaliliuoksen mukana toimitetaan:
- 5 ml:n ruisku (sininen mitta-asteikko), mitta-asteikon väli vastaa 0,1 ml:aa liuosta (mitta-asteikon yksi 0,1 ml:n väli vastaa 1 mg:aa brivarasetaamia). Ruiskussa on lisäksi asteikkomerkit 0,25 ml:n ja 0,75 ml:n kohdalla. Asteikkomerkit alkavat kohdasta 0,25 ml ja jatkuvat 5 ml:aan saakka.
- 10 ml:n ruisku (musta mitta-asteikko), mitta-asteikon väli vastaa 0,25 ml:aa liuosta (mitta-asteikon yksi 0,25 ml:n väli vastaa 2,5 mg:aa brivarasetaamia).
Lääkärin on neuvottava potilaalle, mikä on hänelle sopiva ruiskun koko.
Jos laskettu kerta-annos on vähintään 5 mg (0,5 ml) ja enintään 50 mg (5 ml), on käytettävä 5 ml:n oraaliruiskua.
Jos laskettu kerta-annos on yli 50 mg (5 ml), on käytettävä suurinta 10 ml:n oraaliruiskua.
Laskettu annos pyöristetään lähimpään asteikkomerkkiin. Jos laskettu annos osuu kahden asteikkomerkin keskiväliin, on käytettävä suurempaa annosta osoittavaa asteikkomerkkiä.
Seuraavassa taulukossa on esimerkkejä yhdellä antokerralla annettavista oraaliliuoksen määristä lääkärin määräämän annoksen ja potilaan painon mukaan. Oraaliliuoksen tarkka määrä on laskettava täsmälleen lapsen painon mukaan.
On huomattava, että ruiskun mitta-asteikko rajoittaa annostusta. Jos potilas tarvitsee esimerkiksi 2,15 ml:n annoksen, annettava tilavuus on pyöristettävä ylöspäin 2,2 ml:aan, sillä 5 ml:n ruiskulla voidaan antaa ainoastaan 2,1 ml:n tai 2,2 ml:n tilavuus. Vastaavasti 1,13 ml olisi pyöristettävä alaspäin 1,1 ml:n tilavuudeksi.
| Antokerralla otettava oraaliliuostilavuus vähintään 50 kg painaville nuorille ja lapsille sekä aikuisille | ||||
| Lääkärin määräämä annos | Jos annos on 50 mg/vrk 25 mg/antokerta | Jos annos on 100 mg/vrk 50 mg/antokerta | Jos annos on 150 mg/vrk 75 mg/antokerta | Jos annos on 200 mg/vrk 100 mg/antokerta |
| Suositeltu ruisku | 5 ml | 10 ml | ||
| Paino | Annettava tilavuus | Annettava tilavuus | ||
| vähintään 50 kg | 2,5 ml (25 mg) | 5 ml (50 mg) | 7,5 ml (75 mg) | 10 ml (100 mg) |
| Antokerralla otettava oraaliliuostilavuus vähintään 20 kg, mutta alle 50 kg, painaville lapsille ja nuorille | |||||
| Lääkärin määräämä annos | Jos annos on 1 mg/kg/vrk 0,05 ml/kg/antokerta (vastaa annosta 0,5 mg/kg/antokerta) | Jos annos on 2 mg/kg/vrk 0,1 ml/kg/antokerta (vastaa annosta 1 mg/kg/antokerta) | Jos annos on 3 mg/kg/vrk 0,15 ml/kg/antokerta (vastaa annosta 1,5 mg/kg/antokerta) | Jos annos on 4 mg/kg/vrk 0,2 ml/kg/antokerta (vastaa annosta 2 mg/kg/antokerta) | |
| Suositeltu ruisku | 5 ml | 5 ml tai 10 ml* | |||
| Paino | Annettava tilavuus | Annettava tilavuus | |||
| 20 kg | 1 ml (10 mg) | 2 ml (20 mg) | 3 ml (30 mg) | 4 ml (40 mg) | |
| 25 kg | 1,25 ml (12,5 mg) | 2,5 ml (25 mg) | 3,75 ml (37,5 mg) | 5 ml (50 mg) | |
| 30 kg | 1,5 ml (15 mg) | 3 ml (30 mg) | 4,5 ml (45 mg) | 6 ml* (60 mg) | |
| 35 kg | 1,75 ml (17,5 mg) | 3,5 ml (35 mg) | 5,25 ml* (52,5 mg) | 7 ml* (70 mg) | |
| 40 kg | 2 ml (20 mg) | 4 ml (40 mg) | 6 ml* (60 mg) | 8 ml* (80 mg) | |
| 45 kg | 2,25 ml (22,5 mg) | 4,5 ml (45 mg) | 6,75 ml* (67,5 mg) | 9 ml* (90 mg) | |
| * Jos tilavuus on yli 5 ml ja enintään 10 ml, potilasta neuvotaan käyttämään 10 ml:n oraaliruiskua. | |||||
| Antokerralla otettava oraaliliuostilavuus vähintään 10 kg, mutta alle 20 kg, painaville lapsille | |||||
| Lääkärin määräämä annos | Jos annos on 1 mg/kg/vrk 0,05 ml/kg/antokerta (vastaa annosta 0,5 mg/kg/antokerta) | Jos annos on 2,5 mg/kg/vrk 0,125 ml/kg/antokerta (vastaa annosta 1,25 mg/kg/antokerta) | Jos annos on 3 mg/kg/vrk 0,15 ml/kg/antokerta (vastaa annosta 1,5 mg/kg/antokerta) | Jos annos on 4 mg/kg/vrk 0,2 ml/kg/antokerta (vastaa annosta 2 mg/kg/antokerta) | Jos annos on 5 mg/kg/vrk 0,25 ml/kg/antokerta (vastaa annosta 2,5 mg/kg/antokerta) |
| Suositeltu ruisku: 5 ml | |||||
| Paino | Annettava tilavuus | ||||
| 10 kg | 0,5 ml (5 mg) | 1,25 ml (12.5 mg) | 1,5 ml (15 mg) | 2 ml (20 mg) | 2,5 ml (25 mg) |
| 12 kg | 0,6 ml (6 mg) | 1,5 ml (15 mg) | 1,8 ml (18 mg) | 2,4 ml (24 mg) | 3,0 ml (30 mg) |
| 14 kg | 0,7 ml (7 mg) | 1,75 ml (17,5 mg) | 2,1 ml (21 mg) | 2,8 ml (28 mg) | 3,5 ml (35 mg) |
| 15 kg | 0,75 ml (7,5 mg) | 1,9 ml (19,0 mg) | 2,25 ml (22,5 mg) | 3 ml (30 mg) | 3,75 ml (37,5 mg) |
Annoksen jääminen väliin
Jos potilaalta jää väliin yksi tai useampia annoksia, on suositeltavaa ottaa yksi kerta-annos heti asian muistuessa mieleen ja seuraava annos tavanomaiseen aikaan joko aamulla tai illalla. Näin saatetaan välttyä plasman brivarasetaamipitoisuuden pienenemiseltä alle tehokkaan pitoisuuden ja tästä johtuvien satunnaisten kohtausten ilmaantuminen.
Hoidon lopettaminen
Jos brivarasetaamihoito on lopetettava vähintään 16-vuotiailla potilailla, suositellaan annoksen asteittaista pienentämistä 50 mg:lla/vrk viikoittain.
Jos brivarasetaamihoito on lopetettava alle 16-vuotiailla potilailla, suositellaan annoksen pienentämistä enintään puolella annoksella viikoittain, kunnes annos on 1 mg/kg/vrk (potilaat, joiden paino on alle 50 kg) tai 50 mg/vrk (potilaat, joiden paino on 50 kg tai enemmän).
Kun hoitoa on annettu 1 viikon ajan annoksella 50 mg/vrk, suositellaan tämän jälkeen vielä viimeistä hoitoviikkoa annoksella 20 mg/vrk.
Erityisryhmät
Iäkkäät (vähintään 65‑vuotiaat)
Annosta ei tarvitse muuttaa iäkkäälle potilaalle (ks. kohta Farmakokinetiikka).
Kliininen kokemus valmisteen käytöstä vähintään 65‑vuotiaille potilaille on vähäistä.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa potilaalle, jolla on munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka). Tietojen puuttumisen vuoksi brivarasetaamia ei suositella loppuvaiheen munuaistautia sairastavalle potilaalle, joka saa dialyysihoitoa. Aikuisista saatujen tietojen perusteella annosta ei tarvitse muuttaa lapsipotilaille, joilla on munuaisten vajaatoiminta. Munuaisten vajaatoimintaa sairastavista lapsipotilaista ei ole kliinisiä tietoja saatavilla.
Maksan vajaatoiminta
Altistus brivarasetaamille suureni aikuispotilaissa, joilla oli pitkäaikainen maksasairaus.
Maksan vajaatoimintaa sairastaville potilaille suositellaan seuraavia annosmuutoksia maksan vajaatoiminnan vaikeusasteesta riippumatta. Annos jaetaan kahteen osaan ja annetaan noin 12 tunnin välein (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). Kliinisiä tietoja ei ole saatavilla maksan vajaatoimintaa sairastavista lapsipotilaista.
| Ikä ja paino | Suositeltu aloitusannos | Suurin suositeltu vuorokausiannos |
| Nuoret ja lapset, jotka painavat vähintään 50 kg, sekä aikuiset | 50 mg/vrk | 150 mg/vrk |
| Nuoret ja lapset, jotka painavat vähintään 20 kg, mutta alle 50 kg | 1 mg/kg/vrk | 3 mg/kg/vrk |
| Lapset, jotka painavat vähintään 10 kg, mutta alle 20 kg | 1 mg/kg/vrk | 4 mg/kg/vrk |
Alle 2-vuotiaat pediatriset potilaat
Brivarasetaamin tehoa alle 2-vuotiaiden pediatristen potilaiden hoidossa ei ole vielä varmistettu. Tällä hetkellä saatavilla olevat tiedot on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, mutta annostusta koskevia suosituksia ei voida antaa.
Antotapa
Brivarasetaami-oraaliliuoksen voi laimentaa veteen tai mehuun juuri ennen suun kautta ottamista. Oraaliliuoksen voi ottaa joko ruokailun yhteydessä tai tyhjään mahaan (ks. kohta Farmakokinetiikka). Brivarasetaami-oraaliliuoksen voi antaa nenä-mahaletkun tai maha-avanneletkun kautta.
Briviact-oraaliliuoksen mukana toimitetaan 5 ml:n ja 10 ml:n mittaruisku ja ruiskun sovitin.
Pakkausselosteessa on käyttöohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai muille pyrrolidonijohdoksille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Itsetuhoajatukset ja ‑käyttäytyminen
Epilepsialääkkeiden, myös brivarasetaamin, käyttäjillä on ilmoitettu itsetuhoajatuksia ja ‑käyttäytymistä lääkkeen käyttötarkoituksesta riippumatta. Satunnaistettujen, lumekontrolloitujen kliinisten epilepsialääketutkimusten meta-analyysi osoitti itsetuhoajatusten ja ‑käyttäytymisen riskin vähäistä lisääntymistä. Riskin kasvun mekanismia ei tunneta, eikä lisääntyneen riskin mahdollisuutta voida sulkea pois brivarasetaamin käyttäjillä.
Potilaita tulee seurata itsetuhoajatusten ja -käyttäytymisen varalta, ja asianmukaisen hoidon tarvetta tulee harkita. Potilaita (ja heidän omaisiaan) tulee neuvoa ottamaan yhteyttä lääkäriin, mikäli itsetuhoajatuksia tai ‑käyttäytymistä esiintyy. Ks. myös kohdasta Haittavaikutukset lapsipotilaita koskevat tiedot.
Maksan vajaatoiminta
Kliiniset tiedot brivarasetaamin käytöstä potilaalle, jolla on entuudestaan maksan vajaatoiminta, ovat vähäisiä. Annoksen muuttamista suositellaan maksan vajaatoimintapotilaalle (ks. kohta Annostus ja antotapa).
Vaikeat ihoreaktiot (SCAR)
Vaikeita ihoreaktioita (SCAR), mukaan lukien Stevens-Johnsonin oireyhtymä (SJS), jotka voivat olla hengenvaarallisia tai johtaa kuolemaan, on raportoitu brivarasetaamihoidon yhteydessä. Potilaalle on kerrottava lääkkeen määräämisen yhteydessä ihoreaktioiden oireista ja löydöksistä, ja potilasta on seurattava tarkoin niiden varalta. Jos näihin reaktioihin viittaavia oireita ja löydöksiä ilmenee, brivarasetaamihoito on lopetettava välittömästi ja sen sijaan on harkittava jotakin vaihtoehtoista hoitoa.
Apuaineet
Natriumsisältö
Briviact-oraaliliuos sisältää alle 1 mmol natriumia (23 mg) per ml eli sen voidaan sanoa olevan ”natriumiton”.
Fruktoosi-intoleranssi
Tämä lääkevalmiste sisältää 168 mg sorbitolia (E420) per millilitra. Tätä lääkevalmistetta ei pidä antaa potilaille, joilla on perinnöllinen fruktoosi-intoleranssi (HFI).
Apuaineet, jotka voivat aiheuttaa intoleranssia
Tämä oraaliliuos sisältää metyyliparahydroksibentsoaattia (E218), joka voi aiheuttaa allergisia reaktioita (mahdollisesti viivästyneitä).
Briviact-oraaliliuos sisältää propyleeniglykolia (E1520).
Yhteisvaikutukset
Virallisia yhteisvaikutustutkimuksia on tehty vain aikuisille.
Farmakodynaamiset yhteisvaikutukset
Samanaikainen levetirasetaamihoito
Kliinisissä tutkimuksissa, joihin osallistuneiden lukumäärä oli tosin pieni, brivarasetaamista ei havaittu olevan hyötyä lumelääkkeeseen verrattuna potilaille, jotka ottivat samanaikaisesti levetirasetaamia. Mitään uusia turvallisuuteen tai siedettävyyteen vaikuttavia seikkoja ei havaittu (ks. kohta Farmakodynamiikka).
Yhteisvaikutus alkoholin kanssa
Farmakokineettisessä ja farmakodynaamisessa yhteisvaikutustutkimuksessa, jossa terveille tutkittaville annettiin 200 mg brivarasetaamia kerta-annoksena ja 0,6 g/l etanolia jatkuvana infuusiona, ei ilmennyt farmakokineettistä yhteisvaikutusta, mutta brivarasetaami noin kaksinkertaisti alkoholin vaikutuksen psykomotoriseen toimintaan, tarkkaavuuteen ja muistiin. Brivarasetaamin ja alkoholin samanaikaista käyttöä ei suositella.
Farmakokineettiset yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset brivarasetaamin farmakokinetiikkaan
In vitro ‑tiedot viittaavat siihen, että brivarasetaamiin liittyvä yhteisvaikutusten mahdollisuus on pieni. Brivarasetaami metaboloituu pääasiassa hydrolysoitumalla CYP:stä riippumattomasti. Se metaboloituu myös CYP2C19-välitteisesti hydroksyloitumalla (ks. kohta Farmakokinetiikka).
Voimakkaiden CYP2C19:n estäjien (esim. flukonatsoli, fluvoksamiini) samanaikainen anto voi suurentaa plasman brivarasetaamipitoisuutta, mutta kliinisesti merkityksellisen CYP2C19-välitteisen yhteisvaikutuksen riskiä pidetään pienenä. Saatavilla olevien vähäisten kliinisten tietojen perusteella on näyttöä siitä, että samanaikainen kannabidiolin antaminen saattaa lisätä brivarasetaamin pitoisuutta plasmassa, mahdollisesti CYP2C19-eston kautta, mutta sen kliininen merkittävyys on epävarmaa.
Rifampisiini
Entsyymejä voimakkaasti indusoivan rifampisiinin samanaikainen anto (600 mg/vrk 5 päivän ajan) terveille tutkittaville pienensi brivarasetaamin AUC (plasman pitoisuus-aikakuvaajan alle jäävää pinta-alaa) ‑arvoa 45 %. Lääkettä määräävän lääkärin on harkittava brivarasetaamiannoksen muuttamista potilaalle, jolle aloitetaan rifampisiinihoito tai jonka rifampisiinihoito lopetetaan.
Entsyymejä voimakkaasti indusoivat epilepsialääkkeet
Entsyymejä voimakkaasti indusoivien epilepsialääkkeiden (karbamatsepiini, fenobarbitaali, fenytoiini) samanaikainen anto pienentää plasman brivarasetaamipitoisuutta, mutta annosta ei tarvitse muuttaa (ks. taulukko 1).
Muut entsyymejä indusoivat epilepsialääkkeet
Myös muut entsyymejä voimakkaasti indusoivat aineet (kuten mäkikuisma [Hypericum perforatum]) saattavat pienentää systeemistä brivarasetaamialtistusta. Siksi varovaisuutta on noudatettava mäkikuismahoidon aloittamisessa ja lopettamisessa.
Brivarasetaamin vaikutukset muihin lääkeaineisiin
Kun brivarasetaamiannos oli joko 50 mg/vrk tai 150 mg/vrk, se ei vaikuttanut (CYP3A4‑välitteisesti metaboloituvan) midatsolaamin AUC-arvoon. Kliinisesti merkityksellisten CYP3A4‑yhteisvaikutusten riskiä pidetään pienenä.
In vitro -tutkimusten mukaan brivarasetaami estää CYP450‑isoformeja, lukuun ottamatta CYP2C19:ää, joko vain vähän tai ei lainkaan. Brivarasetaami saattaa suurentaa CYP2C19‑välitteisesti metaboloituvien lääkeaineiden (esim. lansopratsolin, omepratsolin, diatsepaamin) pitoisuutta plasmassa. In vitro ‑tutkimuksissa brivarasetaami ei indusoinut CYP1A1/2-entsyymejä mutta indusoi CYP3A4:ää ja CYP2B6:ta. CYP3A4:n indusointia ei todettu in vivo (ks. edellä midatsolaamia koskeva tieto). CYP2B6:n indusointia ei ole tutkittu in vivo, ja brivarasetaami saattaa pienentää CYP2B6‑välitteisesti metaboloituvien lääkeaineiden (esim. efavirentsin) pitoisuutta plasmassa. In vitro ‑yhteisvaikutustutkimuksissa, joissa selvitettiin kuljettajaproteiineihin mahdollisesti kohdistuvia estovaikutuksia, todettiin, ettei kliinisesti merkityksellisiä vaikutuksia ollut, lukuun ottamatta OAT3:a. Brivarasetaamin IC50-arvo (pitoisuus, jolla puolet reaktiosta estyy) OAT3:n estossa in vitro on 42 kertaa suurempi kuin kliinisellä enimmäisannoksella saavutettava Cmax-arvo. Brivarasetaamiannos 200 mg/vrk saattaa suurentaa OAT3:n kuljettamien lääkeaineiden pitoisuutta plasmassa.
Epilepsialääkkeet
Brivarasetaamin (50–200 mg/vrk) ja muiden epilepsialääkkeiden mahdollisia yhteisvaikutuksia on selvitetty analyysissa, jossa yhdistettiin plasman lääkepitoisuusmääritysten tulokset kaikista toisen ja kolmannen vaiheen kliinisistä tutkimuksista, toisen ja kolmannen vaiheen lumekontrolloitujen tutkimusten populaatiofarmakokineettisestä analyysista sekä erityisistä lääke-lääkeyhteisvaikutuksia selvittäneistä yhteisvaikutustutkimuksista (tutkitut epilepsialääkkeet: karbamatsepiini, lamotrigiini, fenytoiini ja topiramaatti). Taulukossa 1 on tiivistelmä yhteisvaikutusten vaikutuksesta plasman pitoisuuteen (↑ = suurenee, ↓= pienenee, AUC = plasman pitoisuus-aikakuvaajan alle jäävä pinta-ala, Cmax = todettu enimmäispitoisuus).
Taulukko 1: Farmakokineettiset yhteisvaikutukset brivarasetaamin ja muiden epilepsialääkkeiden välillä
Samanaikaisesti annettu epilepsialääke | Epilepsialääkkeen vaikutus plasman brivarasetaamipitoisuuteen | Brivarasetaamin vaikutus plasman epilepsialääkepitoisuuteen |
Karbamatsepiini | AUC 29 % ↓ Cmax 13 % ↓ Annosta ei tarvitse muuttaa | Karbamatsepiini – ei vaikutusta Karbamatsepiiniepoksidi ↑ (ks. teksti jäljempänä) Annosta ei tarvitse muuttaa |
Klobatsaami | Ei tietoa saatavana | Ei vaikutusta |
Klonatsepaami | Ei tietoa saatavana | Ei vaikutusta |
Lakosamidi | Ei tietoa saatavana | Ei vaikutusta |
Lamotrigiini | Ei vaikutusta | Ei vaikutusta |
Levetirasetaami | Ei vaikutusta | Ei vaikutusta |
Okskarbatsepiini | Ei vaikutusta | Ei vaikutusta (monohydroksijohdos, MHD) |
Fenobarbitaali | AUC 19 % ↓ Annosta ei tarvitse muuttaa | Ei vaikutusta |
Fenytoiini | AUC 21 % ↓ Annosta ei tarvitse muuttaa | Ei vaikutusta aAUC 20 % ↑ aCmax 20 % ↑ |
Pregabaliini | Ei tietoa saatavana | Ei vaikutusta |
Topiramaatti | Ei vaikutusta | Ei vaikutusta |
Valproiinihappo | Ei vaikutusta | Ei vaikutusta |
Tsonisamidi | Ei tietoa saatavana | Ei vaikutusta |
aperustuu tutkimukseen, jossa brivarasetaamia annettiin supraterapeuttisena annoksena 400 mg/vrk.
Karbamatsepiini
Brivarasetaami estää epoksidihydrolaasia kohtalaisesti ja palautuvasti, jolloin karbamatsepiinin aktiivisen metaboliitin, karbamatsepiiniepoksidin, pitoisuus suurenee. Kontrolloiduissa kliinisissä tutkimuksissa plasman karbamatsepiiniepoksidipitoisuus suureni keskimäärin 37 % brivarasetaamiannoksella 50 mg/vrk, 62 % annoksella 100 mg/vrk ja 98 % annoksella 200 mg/vrk vain hieman vaihdellen. Turvallisuuteen liittyviä riskejä ei havaittu. Brivarasetaami ja valproaatti eivät vaikuttaneet karbamatsepiiniepoksidin AUC-arvoon additiivisesti.
Suun kautta otettavat ehkäisyvalmisteet
Brivarasetaamin (100 mg/vrk) samanaikainen anto etinyyliestradiolia (0,03 mg) ja levonorgestreeliä (0,15 mg) sisältävän suun kautta otettavan ehkäisyvalmisteen kanssa ei vaikuttanut kummankaan aineen farmakokinetiikkaan. Kun brivarasetaamia annettiin 400 mg/vrk (kaksinkertainen annos suositeltuun enimmäisvuorokausiannokseen verrattuna) yhdessä etinyyliestradiolia (0,03 mg) ja levonorgestreeliä (0,15 mg) sisältävän suun kautta otettavan ehkäisyvalmisteen kanssa, estrogeenin AUC-arvon todettiin pienenevän 27 % ja progestiinin 23 % ilman vaikutusta ovulaation suppressioon. Endogeenisiin merkkiaineisiin lukeutuvien estradiolin, progesteronin, luteinisoivan hormonin (LH), follikkelia stimuloivan hormonin (FSH) ja sukupuolihormoneja sitovan globuliinin (SHBG) pitoisuus-aikaprofiilit eivät yleensä muuttuneet.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Lääkärin on keskusteltava perhesuunnittelusta ja raskaudenehkäisystä sellaisen brivarasetaamia saavan naispotilaan kanssa, joka voi tulla raskaaksi (ks. Raskaus).
Jos nainen päättää tulla raskaaksi, brivarasetaamin käyttöä on arvioitava uudelleen huolellisesti.
Raskaus
Epilepsiaan ja epilepsialääkkeisiin yleisesti liittyvä riski
Kaikkien epilepsialääkkeiden osalta on osoitettu, että epämuodostumia esiintyy 2–3 kertaa enemmän hoitoa saaneiden epilepsiaa sairastavien naisten jälkeläisillä kuin yleisväestössä, jossa esiintyvyys on noin 3 %. Epämuodostumien on todettu lisääntyvän hoitoa saavassa potilasjoukossa niillä, jotka käyttävät useita lääkkeitä; ei kuitenkaan tiedetä, missä määrin tämä johtuu hoidosta ja/tai perussairaudesta. Epilepsiahoitojen lopettaminen voi pahentaa sairautta, mikä voisi olla haitallista sekä äidille että sikiölle.
Brivarasetaamiin liittyvä riski
On vain vähän tietoja brivarasetaamin käytöstä raskaana oleville naisille. Brivarasetaamin kulkeutumisesta ihmisen istukan läpi ei ole tietoa, mutta brivarasetaamin on osoitettu läpäisevän helposti rotan istukan (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmiselle ei tunneta. Brivarasetaamilla ei todettu mahdollista teratogeenisuutta eläintutkimuksissa (ks. kohta Prekliiniset tiedot turvallisuudesta).
Brivarasetaamia käytettiin kliinisissä tutkimuksissa lisälääkkeenä, ja yhdessä karbamatsepiinin kanssa se suurensi karbamatsepiinin aktiivisen metaboliitin, karbamatsepiiniepoksidin, pitoisuutta annoksen mukaan (ks. kohta Yhteisvaikutukset). Tämän vaikutuksen kliinistä merkitystä raskaudessa ei pystytä arvioimaan tietojen riittämättömyyden vuoksi.
Varotoimena brivarasetaamia saa käyttää raskauden aikana vain, jos se on kliinisesti välttämätöntä (eli jos hyöty äidille on selvästi suurempi kuin mahdollinen riski sikiölle).
Imetys
Brivarasetaami erittyy ihmisen rintamaitoon. On päätettävä lopettaa joko rintaruokinta tai brivarasetaamihoito sen mukaan, mikä on lääkevalmisteen hyöty äidille. Brivarasetaamin ja karbamatsepiinin yhteisannossa karbamatsepiiniepoksidin rintamaitoon erittyvä määrä voi suurentua. Tiedot ovat riittämättömät tämän kliinisen merkityksen arvioimiseksi.
Hedelmällisyys
Saatavilla ei ole tietoa brivarasetaamin vaikutuksesta ihmisen hedelmällisyyteen. Brivarasetaamihoito ei vaikuttanut rotan hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Brivarasetaamilla on vähäinen tai kohtalainen vaikutus ajokykyyn ja koneiden käyttökykyyn.
Mahdollisten yksilöllisten herkkyyserojen vuoksi joillakin potilailla saattaa ilmetä uneliaisuutta, heitehuimausta ja muita keskushermostoon liittyviä oireita. Potilasta on kehotettava olemaan ajamatta autoa tai käyttämättä muita mahdollisesti vaarallisia koneita, kunnes hän tietää, miten brivarasetaami vaikuttaa hänen kykyynsä suoriutua tällaisista toimista.
Haittavaikutukset
Tiivistelmä turvallisuustiedoista
Brivarasetaamihoidossa useimmiten (> 10 %) ilmoitetut haittavaikutukset olivat uneliaisuus (14,3 %) ja heitehuimaus (11,0 %). Ne olivat vaikeudeltaan tavallisesti lieviä tai keskivaikeita. Uneliaisuuden ja väsymyksen ilmaantuvuuden ilmoitettiin lisääntyvän annoksen suurenemisen myötä.
Haittavaikutusten vuoksi hoidon keskeytti brivarasetaamin eri annosryhmiin satunnaistetuista potilaista 3,5 % annosryhmässä 50 mg/vrk, 3,4 % annosryhmässä 100 mg/vrk ja 4,0 % annosryhmässä 200 mg/vrk sekä 1,7 % lumelääkeryhmään satunnaistetuista potilaista. Brivarasetaamihoidon lopettamiseen useimmiten johtaneet haittavaikutukset olivat heitehuimaus (0,8 %) ja konvulsio (0,8 %).
Haittavaikutukset taulukoituina
Seuraavassa taulukossa on lueteltu elinjärjestelmittäin ja esiintymistiheyksittäin haittavaikutukset, jotka tunnistettiin kolmen lumelääkekontrolloidun, kiinteällä annoksella tehdyn brivarasetaamitutkimuksen vähintään 16 vuoden ikäiset tutkittavat kattavaa turvallisuustietokantaa koskeneessa katsauksessa ja markkinoille tulon jälkeisistä tiedoista.
Esiintymistiheydet määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin esiintymistiheyden luokassa vakavuudeltaan alenevassa järjestyksessä.
| Elinjärjestelmä | Esiintymistiheys | Haittavaikutukset |
| Infektiot | Yleinen | Influenssa |
| Veri ja imukudos | Melko harvinainen | Neutropenia |
| Immuunijärjestelmä | Melko harvinainen | Tyypin I yliherkkyys |
| Aineenvaihdunta ja ravitsemus | Yleinen | Ruokahalun heikkeneminen |
| Psyykkiset häiriöt | Yleinen | Masennus, ahdistuneisuus, unettomuus, ärtyisyys |
| Melko harvinainen | Itsetuhoajatukset, psykoottinen häiriö, aggressiivisuus, agitaatio | |
| Hermosto | Hyvin yleinen | Heitehuimaus, uneliaisuus |
| Yleinen | Konvulsio, kiertohuimaus | |
| Hengityselimet, rintakehä ja välikarsina | Yleinen | Ylähengitystieinfektiot, yskä |
| Ruoansulatuselimistö | Yleinen | Pahoinvointi, oksentelu, ummetus |
| Iho ja ihonalainen kudos | Tuntematon | Stevens-Johnsonin oireyhtymä(1) |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Väsymys |
(1) Haittavaikutukset on raportoitu markkinoille tulon jälkeen.
Valikoitujen haittavaikutusten kuvaus
Neutropeniaa on ilmoitettu 0,5 %:lla (6/1 099) brivarasetaamia saaneista potilaista ja 0 %:lla (0/459) lumelääkettä saaneista potilaista. Näistä tutkittavista neljän neutrofiilimäärä oli pienentynyt alkutilanteessa, ja se pieneni entisestään brivarasetaamihoidon aloittamisen jälkeen. Yksikään näistä kuudesta neutropeniatapauksesta ei ollut vaikea, ei vaatinut erityistä hoitoa eikä johtanut brivarasetaamihoidon keskeyttämiseen. Yhteenkään tapaukseen ei myöskään liittynyt infektioita.
Itsetuhoajatuksia on ilmoitettu 0,3 %:lla (3/1 009) brivarasetaamia saaneista potilaista ja 0,7 %:lla (3/459) lumelääkettä saaneista potilaista. Epilepsiapotilaille tehdyissä lyhytkestoisissa kliinisissä brivarasetaamitutkimuksissa ei ilmennyt yhtään itsemurhaa eikä itsemurhayritystä; näitä molempia on kuitenkin ilmoitettu avoimissa jatkotutkimuksissa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kliinisen kehitystyön aikana pienellä joukolla brivarasetaamipotilaita (9/3022) on ilmoitettu välittömään (tyypin I) yliherkkyyteen viittaavia reaktioita.
Pediatriset potilaat
Vähintään yhden kuukauden ikäisillä lapsilla todettu brivarasetaamin turvallisuusprofiili oli yhdenmukainen aikuisilla todetun turvallisuusprofiilin kanssa. Avoimissa, kontrolloimattomissa pitkäkestoisissa tutkimuksissa itsetuhoajatuksia raportoitiin 4,7 %:lla arvioiduista vähintään 6-vuotiaista lapsipotilaista (yleisempiä nuorilla); aikuisilla niitä esiintyi 2,4 %:lla. Käyttäytymisen häiriöitä esiintyi 24,8 %:lla lapsipotilaista ja 15,1 %:lla aikuisista. Suurin osa tapahtumista oli voimakkuudeltaan lieviä tai keskivaikeita. Ne eivät olleet vakavia eivätkä johtaneet tutkimuslääkityksen lopettamiseen. Lapsilla on lisäksi ilmoitettu haittavaikutuksena psykomotorista yliaktiivisuutta (4,7 %).
Lapsilla, jotka olivat iältään yhdestä kuukaudesta alle neljään vuoteen, ei tunnistettu spesifistä haittavaikutusprofiilia vanhempiin lapsipotilaiden ikäryhmiin verrattuna. Merkittäviä turvallisuustietoja, jotka viittaisivat tietyn haittavaikutuksen suurempaan ilmaantuvuuteen tässä ikäryhmässä, ei tunnistettu. Koska tietoja on vain vähän saatavilla alle 2-vuotiaista lapsista, brivarasetaamin käyttö ei ole aiheellista tässä ikäryhmässä. Saatavilla on vain vähän kliinistä tietoa vastasyntyneistä.
Iäkkäät
Brivarasetaamin toisen ja kolmannen vaiheen kehitysohjelmaan osallistuneista 130 iäkkäästä tutkittavasta (44:llä oli epilepsia) 100 oli 65–74‑vuotiaita ja 30 oli 75–84‑vuotiaita. Iäkkäiden ja nuorten aikuispotilaiden turvallisuusprofiilit vaikuttaisivat samankaltaisilta.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta.
www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet
Brivarasetaamin yliannostuksesta ihmiselle on vain vähän kliinistä kokemusta. Terveellä tutkittavalla, joka otti brivarasetaamia 1 400 mg kerta-annoksena, ilmoitettiin uneliaisuutta ja heitehuimausta. Seuraavia haittavaikutuksia on raportoitu brivarasetaamin yliannostuksen yhteydessä markkinoilletulon jälkeisessä seurannassa: pahoinvointi, kiertohuimaus, tasapainohäiriöt, ahdistuneisuus, väsymys/uupumus, ärtyneisyys, aggressiivisuus, unettomuus, masennus ja itsetuhoajatukset. Yleisesti ottaen brivarasetaamin yliannostukseen liittyvät haittavaikutukset ovat yhdenmukaisia tunnettujen haittavaikutusten kanssa.
Yliannostuksen hoito
Brivarasetaamin yliannostukseen ei ole olemassa spesifistä vastalääkettä. Yliannostusta tulee hoitaa yleisin elintoimintoja tukevin toimin. Koska alle 10 % brivarasetaamista erittyy virtsaan, hemodialyysi ei odotettavasti suurenna brivarasetaamin puhdistumaa merkitsevästi (ks. kohta Farmakokinetiikka).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Epilepsialääkkeet, muut epilepsialääkkeet, ATC-koodi: N03AX23
Vaikutusmekanismi
Brivarasetaamin affiniteetti aivojen synaptiseen vesikkeliproteiini 2A:han (SV2A) on voimakasta ja selektiivistä. SV2A on transmembraaninen glykoproteiini, jota on presynaptisella tasolla hermosoluissa ja umpirauhassoluissa. Tämän proteiinin tarkka tehtävä on vielä selvittämättä, mutta sen on osoitettu vaikuttavan hermovälittäjäaineiden eksosytoosiin. Brivarasetaamin antikonvulsiivisen vaikutusmekanismin arvellaan perustuvan ensisijaisesti brivarasetaamin sitoutumiseen SV2A:han.
Kliininen teho ja turvallisuus
Brivarasetaamin teho paikallisalkuisten kohtausten lisälääkkeenä on osoitettu kolmessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa kliinisessä monikeskustutkimuksessa, joissa käytettiin kiinteää annosta ja joihin osallistuneet tutkittavat olivat vähintään 16‑vuotiaita. Päivittäinen brivarasetaamiannos oli näissä tutkimuksissa 5–200 mg/vrk. Kaikissa tutkimuksissa oli 8 viikon aloitusjakso ja sitten 12 viikon hoitojakso, jonka aikana annosta ei suurennettu. Tutkimuslääkettä sai 1 558 potilasta, joista 1 099 sai brivarasetaamia. Tutkimusten sisäänottoperusteisiin kuuluivat hallitsemattomat paikallisalkuiset kohtaukset huolimatta joko 1:llä tai 2 samanaikaisella epilepsialääkkeellä annetusta hoidosta. Potilailla oli oltava vähintään 8 paikallisalkuista kohtausta aloitusjakson aikana. Kolmannen vaiheen tutkimusten ensisijaiset päätetapahtumat olivat paikallisalkuisten kohtausten esiintymistiheyden prosentuaalinen väheneminen lumelääkkeeseen verrattuna ja vasteen (paikallisalkuisten kohtausten esiintymistiheyden väheneminen 50 %:lla alkutilanteesta) saavuttaminen 50 %:lla tutkittavista.
Potilaiden tavallisimmin käyttämät epilepsialääkkeet olivat tutkimukseen ottohetkellä karbamatsepiini (40,6 %), lamotrigiini (25,2 %), valproaatti (20,5 %), okskarbatsepiini (16,0 %), topiramaatti (13,5 %), fenytoiini (10,2 %) ja levetirasetaami (9,8 %). Tehdyissä kolmessa tutkimuksessa kohtaustiheyden mediaani oli alkutilanteessa 9 kohtausta 28:aa päivää kohti. Potilailla oli ollut epilepsia keskimäärin noin 23 vuotta.
Taulukossa 2 on tiivistelmä tehoa koskevista tuloksista. Brivarasetaami oli kaiken kaikkiaan tehokas vähintään 16‑vuotiaiden potilaiden paikallisalkuisten kohtausten lisälääkkeenä annoksella 50–200 mg/vrk.
Taulukko 2: Päätulokset, jotka koskevat tehoa paikallisalkuisten kohtausten esiintymistiheyteen 28 päivän aikavälillä
Tutkimus | Lume | Brivarasetaami *Tilastollisesti merkitsevä (p-arvo) | ||
50 mg/vrk | 100 mg/vrk | 200 mg/vrk | ||
Tutkimus N01253(1) | ||||
n = 96 | n = 101 | |||
Hoitoon vastanneita 50 % | 16,7 | 32,7* (p = 0,008) | ∼ | ∼ |
Prosentuaalinen väheneminen (%) lumelääkkeeseen verrattuna | ei oleellinen | 22,0* (p = 0,004) | ∼ | ∼ |
Tutkimus N01252(1) | ||||
n = 100 | n = 99 | n = 100 | ||
Hoitoon vastanneita 50 % | 20,0 | 27,3 (p = 0,372) | 36,0(2) (p = 0,023) | ∼ |
Prosentuaalinen väheneminen (%) lumelääkkeeseen verrattuna | ei oleellinen | 9.2 (p = 0,274) | 20,5(2) (p = 0,010) | ∼ |
Tutkimus N01358 | ||||
n = 259 | n = 252 | n = 249 | ||
Hoitoon vastanneita 50 % | 21,6 | ∼ | 38,9* (p < 0,001) | 37,8* (p < 0,001) |
Prosentuaalinen väheneminen (%) lumelääkkeeseen verrattuna | ei oleellinen | ∼ | 22,8* (p < 0,001) | 23,2* (p < 0,001) |
n = satunnaistetut potilaat, jotka saivat vähintään 1 annoksen tutkimuslääkettä
∼ Annosta ei tutkittu
* Tilastollisesti merkitsevä
(1) Noin 20 % potilaista sai samanaikaisesti levetirasetaamia.
(2) N01252-tutkimuksessa ei saavutettu tilastollista merkitsevyyttä ensisijaisen päätetapahtuman suhteen sekventiaalisen tutkimusmenetelmän perusteella. 100 mg:n vuorokausiannos oli nimellisesti merkitsevä.
Kohtaustiheys kliinisissä tutkimuksissa väheni lumelääkkeeseen verrattuna enemmän annoksella 100 mg/vrk kuin annoksella 50 mg/vrk. Brivarasetaamiannosten 50 mg/vrk ja 100 mg/vrk turvallisuusprofiilit olivat samankaltaiset, myös keskushermostoon liittyvien haittatapahtumien ja pitkäaikaiskäytön osalta, lukuun ottamatta uneliaisuuden ja väsymyksen ilmaantuvuuden annoksesta riippuvaista suurenemista.
Kuvassa 1 on potilaiden prosentuaaliset osuudet (pois lukien potilaat, jotka saivat samanaikaisesti levetirasetaamia) sen mukaan, kuinka paljon paikallisalkuiset kohtaukset vähenivät alkutilanteesta 28 päivän aikavälillä kaikissa kolmessa tutkimuksessa. Potilaat, joiden paikallisalkuiset kohtaukset lisääntyivät alkutilanteesta yli 25 %, on esitetty vasemmalla (”huonompi”). Potilaat, joiden paikallisalkuisten kohtausten esiintymistiheys väheni prosentuaalisesti alkutilanteesta, on esitetty neljässä äärimmäisenä oikealla olevassa luokassa. Potilaita, joiden kohtaustiheys väheni vähintään 50 %, oli 20,3 % lumelääkeryhmässä; 34,2 % annosryhmässä 50 mg/vrk; 39,5 % annosryhmässä 100 mg/vrk ja 37,8 % annosryhmässä 200 mg/vrk.
Kuva 1: Potilaiden osuudet (%) kohtaustiheyteen liittyneen vasteen mukaan luokiteltuina brivarasetaamiryhmässä ja lumelääkeryhmässä 12 viikon aikana (koskee kaikkia kolmea kaksoissokkoutettua kliinistä päätutkimusta)
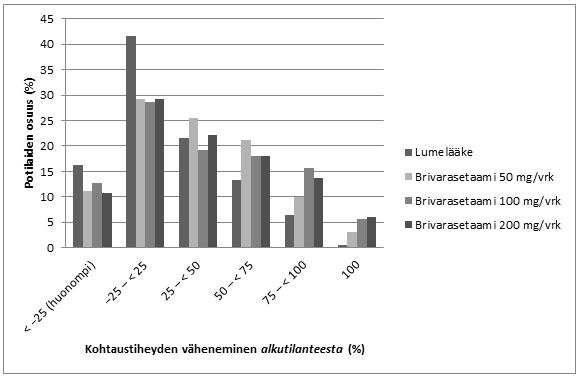
Kolmen kliinisen päätutkimuksen yhdistetyssä analyysissa ei havaittu tehoeroja (mitattuna vasteen saavuttamisena 50 %:lla tutkittavista) annosvälillä 50–200 mg/vrk, silloin kun brivarasetaami annettiin yhdessä indusoivien tai ei-indusoivien epilepsialääkkeiden kanssa. Kliinisissä tutkimuksissa kohtaukset loppuivat 12 viikon hoitojakson aikana 2,5 %:lla (4/161) potilaista brivarasetaamin annosryhmässä 50 mg/vrk, 5,1 %:lla (17/332) annosryhmässä 100 mg/vrk ja 4,0 %:lla (10/249) annosryhmässä 200 mg/vrk verrattuna 0,5 %:iin (2/418) lumelääkeryhmässä.
Havaintojen mukaan kohtaustiheyden (28 päivän aikavälillä) prosentuaalisen vähenemisen mediaani parani brivarasetaamihoidossa lumelääkkeeseen verrattuna (33,3 %, n = 115) seuraavasti potilailla, joilla oli alkutilanteessa toissijaisesti yleistyviä toonis-kloonisia kohtauksia (kohtaustyyppi IC): 66 %:lla (n = 62) brivarasetaamin annosryhmässä 50 mg/vrk, 61,2 %:lla (n = 100) annosryhmässä 100 mg/vrk ja 82,1 %:lla (n = 75) annosryhmässä 200 mg/vrk.
Brivarasetaamin tehoa yksinään käytettynä ei ole varmistettu. Brivarasetaamia ei suositella käytettäväksi yksinään.
Levetirasetaamihoito
Kahdessa kolmannen vaiheen satunnaistetussa, lumekontrolloidussa kliinisessä tutkimuksessa noin 20 % potilaista sai levetirasetaamia samanaikaisena epilepsialääkkeenä. Tutkittavia oli lukumääräisesti vähän, mutta brivarasetaamihoidosta ei lumelääkkeeseen verrattuna havaittu olevan hyötyä potilaille, jotka saivat samanaikaisesti levetirasetaamia. Tämä saattaa olla osoitus kilpailevasta sitoutumisesta synaptiseen vesikkeliproteiini 2A:han (SV2A). Mitään uusia turvallisuuteen tai siedettävyyteen liittyviä seikkoja ei havaittu.
Kolmannessa tutkimuksessa tutkimussuunnitelman mukainen analyysi osoitti, että brivarasetaamin annoksilla 100 mg/vrk ja 200 mg/vrk on lumelääkkeeseen verrattuna tehoa potilaisiin, jotka olivat altistuneet levetirasetaamille aiemmin. Brivarasetaamin teho näihin potilaisiin havaittiin vähäisemmäksi kuin potilaisiin, jotka eivät olleet altistuneet levetirasetaamille aiemmin; tämä johtui todennäköisesti siitä, että kyseiset potilaat olivat käyttäneet aiemmin useampia epilepsialääkkeitä ja heidän kohtaustiheytensä oli alkutilanteessa suurempi.
Iäkkäät (vähintään 65‑vuotiaat)
Kolmeen kaksoissokkoutettuun ja lumekontrolloituun kliiniseen päätutkimukseen osallistui 38 iältään 65–80‑vuotiasta potilasta. Vaikka tutkimuksista saadut tiedot ovat vähäisiä, teho oli verrattavissa nuoremmilla tutkittavilla saavutettuun tehoon.
Avoimet jatkotutkimukset
Pitkäkestoisiin avoimiin jatkotutkimuksiin otettiin mukaan kaikista tutkimuksista 81,7 % niistä potilaista, jotka pysyivät satunnaistetuissa tutkimuksissa mukana loppuun asti. Kohtauksettomuus saavutettiin 5,3 %:lla (n = 1 500) potilaista, joiden brivarasetaamialtistus kesti satunnaistettuun tutkimukseen mukaanottohetkestä laskettuna 6 kuukautta, verrattuna 4,6 %:iin (n = 1 188) potilaista, joiden brivarasetaamialtistus kesti 12 kuukautta, ja 3,7 %:iin (n = 847) potilaista, joiden brivarasetaamialtistus kesti 24 kuukautta. Koska suuri osa potilaista (26 %) keskeytti osallistumisensa avoimiin tutkimuksiin tehon puutteen vuoksi, kyse on voinut olla valikoitumisharhasta, koska tutkimuksessa mukana pysyneet tutkittavat vastasivat hoitoon paremmin kuin osallistumisensa ennenaikaisesti päättäneet.
Turvallisuusprofiili oli pisimmillään 8 vuoden avoimissa jatkotutkimuksissa samankaltainen kuin lumekontrolloiduissa lyhytkestoisissa kliinisissä tutkimuksissa.
Pediatriset potilaat
2-vuotiailla ja sitä vanhemmilla lapsilla paikallisalkuisten kohtausten patofysiologia on samankaltainen kuin nuorilla ja aikuisilla. Epilepsialääkkeistä kertynyt kokemus viittaa siihen, että aikuisilla tehtyjen tehokkuustutkimusten tulokset ovat sovellettavissa vähintään 2-vuotiaisiin lapsiin, kunhan annos säädetään lapsille sopivaksi ja hoidon turvallisuus on osoitettu (ks. kohdat Farmakokinetiikka ja Haittavaikutukset). Vähintään 2-vuotiaiden potilaiden annokset määritettiin muuttamalla annosta painon perusteella siten, että saavutettavat lääkeainepitoisuudet plasmassa ovat samaa luokkaa kuin tehokkaita annoksia ottavilla aikuisilla (ks. kohta Farmakokinetiikka).
Eräässä pitkäkestoisessa, kontrolloimattomassa, avoimessa turvallisuustutkimuksessa oli mukana lapsia (yhden kuukauden ikäisistä alle 16-vuotiaisiin), joiden hoitoa jatkettiin farmakokinetiikkatutkimuksen loppuun suorittamisen jälkeen (ks. kohta Farmakokinetiikka), lapsia, joiden hoitoa jatkettiin laskimoon annettavan (i.v.) valmisteen turvallisuutta koskeneen tutkimuksen päättymisen jälkeen, sekä lapsia, jotka rekrytoitiin suoraan turvallisuustutkimukseen. Suoraan tutkimukseen otetut lapset aloittivat brivarasetaamihoidon annoksella 1 mg/kg/vrk, ja vasteesta ja siedettävyydestä riippuen annos suurennettiin korkeintaan tasolle 5 mg/kg/vrk kaksinkertaistamalla se viikon välein. Yksikään lapsista ei saanut yli 200 mg:n vuorokausiannosta. Vähintään 50 kg painavien lasten brivarasetaamihoito aloitettiin annoksella 50 mg/vrk, ja vasteesta ja siedettävyydestä riippuen annosta suurennettiin viikon välein 50 mg/vrk kerrallaan korkeintaan tasolle 200 mg/vrk.
Avoimista turvallisuustutkimuksista ja liitännäishoitoa koskeneista farmakokineettisistä tutkimuksista saatujen yhdistettyjen tietojen perusteella 186 lasta, joiden ikä on 1 kuukaudesta alle 16 vuoteen ja joilla on paikallisalkuisia epilepsiakohtauksia, on saanut brivarasetaamia. Näistä 149 lasta on saanut hoitoa vähintään 3 kuukautta, 138 lasta on saanut hoitoa ≥ 6 kuukauden ajan, 123 lasta ≥ 12 kuukauden ajan, 107 lasta ≥ 24 kuukauden ajan ja 90 lasta ≥ 36 kuukauden ajan.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset brivarasetaamin käytöstä yhden tai useamman pediatrisen potilasryhmän paikallisalkuisten epileptisten kohtausten hoidossa (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Brivarasetaamin kalvopäällysteisillä tableteilla, oraaliliuoksella ja injektionesteellä laskimoon on sama AUC-arvo, sen sijaan enimmäispitoisuus plasmassa on hieman suurempi laskimoon annon kuin suun kautta annon jälkeen. Brivarasetaamin farmakokinetiikka on lineaarinen ja ajasta riippuvainen ja vaihtelee vain vähän yksilöiden sisäisesti ja välisesti. Brivarasetaami imeytyy täydellisesti, sitoutuu proteiineihin hyvin vähän, erittyy munuaisteitse laajan biotransformaation kautta, ja sen metaboliitit ovat farmakologisesti inaktiivisia.
Imeytyminen
Suun kautta otettu brivarasetaami imeytyy nopeasti ja täydellisesti, ja sen absoluuttinen biologinen hyötyosuus on noin 100 %. Tyhjään mahaan otettujen tablettien tmax saavutetaan 1 tunnin mediaaniajassa (vaihteluväli 0,25–3 h).
Brivarasetaamin ottaminen hyvin rasvaisen aterian yhteydessä hidasti imeytymistä (mediaani tmax 3 h) ja pienensi brivarasetaamin enimmäispitoisuutta plasmassa (37 % pienempi). Sen sijaan imeytymisosuus pysyi muuttumattomana.
Jakautuminen
Brivarasetaami sitoutuu heikosti (≤ 20 %) plasman proteiineihin. Jakautumistilavuus on 0,5 l/kg, mikä vastaa lähes elimistön kokonaisnestemäärää.
Rasvaliukoisuutensa vuoksi (log P) brivarasetaami läpäisee solukalvon hyvin.
Biotransformaatio
Brivarasetaami metaboloituu ensisijaisesti amidiryhmän hydrolyysin kautta vastaavaksi karboksyylihapoksi (noin 60 % eliminaatiosta) ja toissijaisesti propyylisivuketjun hydroksylaation kautta (noin 30 % eliminaatiosta). Maksassa ja muualla kuin maksassa oleva amidohydrolaasi edesauttaa amidiryhmän hydrolyysia karboksyylihappometaboliitiksi (34 % annoksesta virtsassa). Brivarasetaami hydroksyloituu in vitro ensisijaisesti CYP2C19-välitteisesti. Kumpikin metaboliitti metaboloituu edelleen muodostaen saman hydroksihapon ensisijaisesti karboksyylihappometaboliitin propyylisivuketjun hydroksylaation kautta (pääasiassa CYP2C9:n välityksellä). Yksilöillä, joilla on inefektiivisiä CYP2C19-mutaatioita, hydroksimetaboliitin muodostuminen vähenee kertoimella 10 in vivo, samalla kun brivarasetaamipitoisuus suurenee 22 % niillä, joilla on vähintään yksi mutanttialleeli, ja 42 % niillä, joilla on molemmat mutanttialleelit. Nämä kolme metaboliittia ovat farmakologisesti inaktiivisia.
Eliminaatio
Brivarasetaami eliminoituu ensisijaisesti metaboloitumalla ja erittymällä virtsaan. Yli 95 % annoksesta, myös metaboliitit, erittyvät virtsaan 72 tunnissa annoksen ottamisesta. Alle 1 % annoksesta erittyy ulosteisiin, ja alle 10 % brivarasetaamista erittyy muuttumattomana virtsaan. Loppuvaiheen puoliintumisaika (t1/2) plasmassa on noin 9 tuntia. Kokonaispuhdistuma potilaiden plasmasta oli arviolta 3,6 l/h.
Lineaarisuus
Farmakokinetiikka on annoksesta riippuvainen 10 mg:sta vähintään 600 mg:aan.
Yhteisvaikutukset lääkeaineiden kanssa
Brivarasetaami poistuu elimistöstä monia eri reittejä pitkin, mukaan lukien munuaisteitse, CYP:stä riippumattoman hydrolyysin välityksellä ja CYP‑välitteisten hapettumisreaktioiden kautta. Brivarasetaami ei ole in vitro ihmisen P‑glykoproteiinin (P‑gp) eikä monilääkeresistenssiin liittyvien proteiini 1:n (MRP1) tai proteiini 2:n (MRP2) eikä todennäköisesti orgaanisten anionien kuljettajapolypeptidien 1B1 (OATP1B1) ja OAT1B3 substraatti.
In vitro -määritysten mukaan yhdenkään CYP:n (esim. CYP1A, CYP2C8, CYP2C9, CYP2D6 ja CYP3A4) estäjistä ei pitäisi vaikuttaa merkittävästi brivarasetaamin poistumiseen elimistöstä.
Brivarasetaami ei estänyt in vitro kliinisesti merkityksellisinä pitoisuuksina CYP1A2:ta, CYP2A6:ta, CYP2B6:ta, CYP2C8:aa, CYP2C9:ää, CYP2D6:ta, CYP3A4:ää eikä P‑gp‑, BCRP‑, BSEP‑ MRP2‑, MATE-K‑, MATE-1‑, OATP1B1‑, OATP1B3‑, OAT1‑ tai OCT1‑kuljettajaproteiineja. Brivarasetaami ei indusoinut CYP1A2:ta in vitro.
Erityisryhmien farmakokinetiikka
Iäkkäät (vähintään 65‑vuotiaat)
Iäkkäille (65–79 v) tehdyssä tutkimuksessa, jossa tutkittavien kreatiniinipuhdistuma oli 53–98 ml/min/1,73 m2 ja brivarasetaamiannostus 400 mg/vrk kahdesti vuorokaudessa, brivarasetaamin puoliintumisaika plasmassa oli 7,9 tuntia 65–75‑vuotiailla ja 9,3 tuntia yli 75‑vuotiailla. Brivarasetaamin tasapainotilan puhdistuma plasmassa oli samaa luokkaa (0,76 ml/min/kg) kuin tutkituilla terveillä nuorilla miehillä (0,83 ml/min/kg) (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Tutkimus, johon osallistuneilla tutkittavilla oli vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma < 30 ml/min/1,73 m2 ilman dialyysihoidon tarvetta), osoitti, että brivarasetaamin AUC-arvo plasmassa oli kohtalaisesti suurentunut (+21 %) suhteessa terveisiin verrokkeihin, samalla kun karboksyylihappometaboliitin AUC-arvo oli suurentunut 3‑kertaiseksi, hydroksimetaboliitin 4‑kertaiseksi ja hydroksihappometaboliitin 21‑kertaiseksi. Näiden inaktiivisten metaboliittien munuaispuhdistuma pieneni kertoimella 10. Hydroksihappometaboliitilla ei todettu mitään turvallisuushuolia prekliinisissä tutkimuksissa. Brivarasetaamia ei ole tutkittu hemodialyysihoitoa saavilla potilailla (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Maksakirroosipotilaille (Child-Pugh-luokat A, B ja C) tehty farmakokineettinen tutkimus osoitti brivarasetaamialtistuksen suurenevan sairauden vaikeusasteesta riippumatta suurin piirtein saman verran (50 %, 57 % ja 59 %) kuin kaltaistetuilla terveillä verrokeilla (ks. kohta Annostus ja antotapa).
Paino
Tasapainotilassa pitoisuus plasmassa pienenee arviolta 40 % potilailla, jotka painavat 46–115 kg. Tätä eroa ei kuitenkaan pidetä kliinisesti merkityksellisenä.
Sukupuoli
Brivarasetaamin farmakokinetiikassa ei ole kliinisesti merkityksellisiä eroja sukupuolten välillä.
Rotu
Rotu (valkoihoinen, aasialainen) ei vaikuttanut brivarasetaamin farmakokinetiikkaan merkittävästi epilepsiapotilaille tehdyssä populaatiofarmakokineettisessä mallinnuksessa. Jotain muuta etnistä alkuperää olevia potilaita oli lukumääräisesti vähän.
Farmakokineettiset/farmakodynaamiset suhteet
EC50-arvo (plasman brivarasetaamipitoisuus, joka vastaa 50 %:a enimmäisvaikutuksesta) oli arviolta 0,57 mg/l. Tämä pitoisuus plasmassa on hieman suurempi kuin mediaanialtistus, joka saavutetaan brivarasetaamiannoksella 50 mg/vrk. Kohtaustiheyttä saadaan vähennettyä entisestään suurentamalla vuorokausiannos 100 mg:aan, ja tasanne saavutetaan vuorokausiannoksella 200 mg.
Pediatriset potilaat
Farmakokineettisessä tutkimuksessa, johon kuului 3 viikon pituinen arviointijakso ja jossa oraaliliuoksena annetun brivarasetaamin annosta suurennettiin aina samansuuruisina lisäyksinä kolmessa vaiheessa viikon välein, arvioitiin 99:ää tutkittavaa, joiden ikä oli 1 kk – < 16 vuotta. Brivarasetaamia annettiin viikon välein suurennettuina annoksina, jotka olivat noin 1 mg/kg/vrk, 2 mg/kg/vrk ja 4 mg/kg/vrk. Kaikki annokset säädettiin painon perusteella, eivätkä ne ylittäneet enimmäisannoksia 50 mg/vrk, 100 mg/vrk ja 200 mg/vrk. Arviointijakson lopussa tutkittavat saattoivat soveltua pitkäkestoiseen seurantatutkimukseen, jossa hoitoa jatkettiin tutkittavan viimeksi saamalla annoksella (ks. kohta Haittavaikutukset). Pitoisuuden plasmassa osoitettiin olevan suhteessa annokseen kaikissa ikäryhmissä. Populaatiofarmakokineettinen mallinnus perustui 3 viikkoa kestäneestä farmakokineettisestä tutkimuksesta sekä käynnissä olevasta pitkän aikavälin seurantatutkimuksesta harvassa näytteenotossa saatuun pitoisuutta plasmassa koskevaan aineistoon. Analyysissä oli mukana 232 pediatrista epilepsiapotilasta, jotka olivat iältään kahdesta kuukaudesta 17 vuoteen. Analyysi osoitti, että annokset 5,0 mg/kg/vrk (paino 10–20 kg) ja 4,0 mg/kg/vrk (paino 20–50 kg) tuottavat plasmaan saman keskimääräisen vakaan tilan pitoisuuden kuin aikuisten annostus 200 mg/vrk. Arvioitu plasmapuhdistuma oli 10 kg painavilla lapsilla 0,96 l/h; 20 kg painavilla lapsilla 1,61 l/h; 30 kg painavilla lapsilla 2,18 l/h; ja 50 kg painavilla lapsilla 3,19 l/h. Vertailun vuoksi aikuispotilaiden (paino 70 kg) plasmapuhdistuman arvioitiin olevan 3,58 l/h. Tällä hetkellä saatavilla ei ole kliinistä tietoa vastasyntyneistä.
Prekliiniset tiedot turvallisuudesta
Farmakologisissa turvallisuustutkimuksissa vaikutukset kohdistuivat vallitsevasti keskushermostoon (lähinnä keskushermoston toiminnan ohimenevä vaimentuminen ja spontaanin lokomotorisen aktiivisuuden väheneminen), ja niitä todettiin annoksilla, jotka olivat moninkertaisia (yli 50-kertaisia) verrattuna brivarasetaamin farmakologisesti vaikuttavaan annokseen, 2 mg/kg. Oppiminen ja muistin toiminta eivät muuttuneet.
Maksatoksiset vaikutukset (lähinnä porfyria) olivat löydös, jota ei havaittu kliinisissä tutkimuksissa mutta joka todettiin koirilla tehdyissä toistuvan annon toksisuustutkimuksissa, silloin kun kliininen altistus oli samaa luokkaa kuin kliininen altistus plasmassa AUC-arvona. Brivarasetaamista ja sen rakenteellisesta sukulaisyhdisteestä kertyneet toksikologiset tiedot osoittavat kuitenkin, että koirien maksamuutokset ovat kehittyneet sellaisten mekanismien kautta, jotka eivät ole ihmiselle merkityksellisiä. Rotilla ja apinoilla ei todettu haitallisia maksamuutoksia brivarasetaamin pitkäaikaisen annon jälkeen, kun altistus oli 5‑ ja 42‑kertainen verrattuna kliiniseen altistukseen AUC-arvona. Apinoilla keskushermostoon liittyneitä merkkejä (lamautuminen, tasapainon menetys, liikkeiden kömpelyys) ilmeni kun altistus oli 64 kertaa suurempi kuin kliininen Cmax; nämä vaikutukset lievittyivät ajan myötä.
Geenitoksisuustutkimuksissa ei todettu mutageenistä eikä klastogeenistä aktiivisuutta. Karsinogeenisuustutkimuksissa ei ilmennyt viitteitä onkogeenisuudesta rotilla. Sen sijaan uroshiirten hepatosellulaaristen kasvainten ilmaantuvuuden suurenemisen arvellaan johtuvan jostakin muusta kuin geenitoksisesta vaikutustavasta, joka liittyy fenobarbitaalityyppiseen maksaentsyymien induktioon; tämä on tunnetusti spesifisesti jyrsijöihin liittyvä ilmiö.
Brivarasetaami ei vaikuttanut rottien tai kaniinien hedelmällisyyteen kummallakaan sukupuolella, eikä sen osoitettu olevan teratogeeninen kummallekaan eläinlajille. Kaniineilla todettiin alkiotoksisuutta emolle toksisilla brivarasetaamiannoksilla, jolloin altistustaso oli 8 kertaa suurempi kuin suositellulla enimmäisannoksella saavutettava kliininen altistus AUC-arvona. Brivarasetaamin osoitettiin läpäisevän helposti rotan istukan ja erittyvän imettävän rotan maitoon pitoisuuksina, jotka vastaavat pitoisuuksia emon plasmassa.
Brivarasetaami ei aiheuttanut rotille riippuvuuden riskiä.
Nuorilla eläimillä tehdyt tutkimukset
Brivarasetaamialtistuksella, joka oli 6–15 kertaa suurempi kuin suositellulla enimmäisannoksella saavutettava kliininen altistus AUC-arvona, oli nuorten rottien kehitykseen haitallisia vaikutuksia (ts. kuolleisuus, kliiniset merkit, tavanomaista pienempi ruumiinpaino ja tavanomaista pienempi aivojen paino). Haittavaikutuksia ei todettu keskushermoston toimintakokeissa eikä neuropatologisissa tai aivojen histopatologisissa tutkimuksissa. Kun brivarasetaamialtistus oli 6 kertaa suurempi kuin suositellulla enimmäisannoksella saavutettava kliininen altistus AUC-arvona, brivarasetaamin nuorille koirille aiheuttamat muutokset olivat samankaltaisia kuin täysikasvuisilla eläimillä havaitut. Yhteenkään kehityksen tai kypsymisen tavanomaiseen päätetapahtumaan kohdistuneita haittavaikutuksia ei ilmennyt.
Farmaseuttiset tiedot
Apuaineet
Natriumsitraatti
Sitruunahappo, vedetön (pH:n säätöön)
Metyyliparahyroksibentsoaatti (E218)
Karmelloosinatrium
Sukraloosi
Sorbitoli, nestemäinen (E420)
Glyseroli (E422)
Vadelma-aromi (propyleeniglykoli (E1520) 90–98 %)
Puhdistettu vesi
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Avaamisen jälkeen: 8 kuukautta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BRIVIACT oraaliliuos
10 mg/ml (L:ei) 300 ml (146,48 €)
PF-selosteen tieto
300 ml:n meripihkanvärinen lasipullo (tyyppi III), jossa on valkoinen lapsiturvallinen suljin (polypropeeni) rasiassa, jossa on myös asteikollinen 5 ml:n (sininen mitta-asteikko) ja 10 ml:n (musta mitta-asteikko) mittaruisku (polypropeeni, polyeteeni) ja ruiskun sovitin (polyeteeni).
Valmisteen kuvaus:
Hieman viskoosinen, kirkas, väritön tai kellertävä neste.
Käyttö- ja käsittelyohjeet
Ei erityisvaatimuksia.
Käyttämätön lääkevalmiste, sekä laimentamaton että laimennettu, tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BRIVIACT oraaliliuos
10 mg/ml 300 ml
- Ylempi erityiskorvaus (100 %). Brivarasetaami, eslikarbatsepiini, gabapentiini, lakosamidi, levetirasetaami, perampaneeli, pregabaliini, tiagabiini ja tsonisamidi: Epilepsian hoito erityisin edellytyksin (182).
- Peruskorvaus (40 %).
ATC-koodi
N03AX23
Valmisteyhteenvedon muuttamispäivämäärä
19.05.2025
Yhteystiedot
Bertel Jungin aukio 5, 6 krs.
02600 Espoo
+358 9 2514 4221
etunimi.sukunimi@ucb.com