
ZOLEDRONIC ACID STADA infuusioneste, liuos 4 mg/100 ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 4 mg tsoledronihappoa, joka vastaa 4,26 mg tsoledronihappomonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
- Luustotapahtumien (patologiset murtumat, selkäydinkompressio, luuston sädehoito tai leikkaus tai kasvaimen aiheuttama hyperkalsemia) ehkäiseminen aikuispotilailla, joilla on pitkälle edennyt luustoon liittyvä syöpä.
- Kasvaimen aiheuttaman hyperkalsemian hoito aikuispotilailla (TIH).
Ehto
Valmistetta saa käyttää vain laskimoon annettavien bisfosfonaattien antoon perehtynyt lääkäri.
Annostus ja antotapa
Zoledronic acid Stada -valmistetta saa määrätä ja käyttää vain laskimoon annettavien bisfosfonaattien antoon perehtynyt terveydenhuollon ammattilainen. Pakkausseloste ja muistutuskortti potilaalle tulee antaa potilaille, joita hoidetaan Zoledronic acid Stada -valmisteella.
Annostus
Luustotapahtumien ehkäiseminen potilailla, joilla on pitkälle edennyt luustoon liittyvä syöpä
Aikuiset ja iäkkäät potilaat
Suositeltu annos luustotapahtumien ehkäisyyn potilaille, joilla on pitkälle edennyt luustoon liittyvä syöpä, on 4 mg tsoledronihappoa 3–4 viikon välein.
Potilaille annetaan myös päivittäin 500 mg kalsiumia ja 400 IU D-vitamiinia suun kautta.
Päätettäessä ehkäistä luustoon liittyviä tapahtumia potilailla, joilla on luuston etäpesäkkeitä, on otettava huomioon, että hoidon vaikutuksen alkaminen kestää 2–3 kuukautta.
Kasvaimen aiheuttaman hyperkalsemian hoito
Aikuiset ja iäkkäät potilaat
Suositeltu annos hyperkalsemiassa (albumiinilla korjattu seerumin kalsiumpitoisuus ≥ 12,0 mg/dl tai 3,0 mmol/l) on 4 mg tsoledronihappoa kerta-annoksena.
Munuaisten vajaatoiminta
Kasvaimen aiheuttaman hyperkalsemian hoito:
Kasvaimen aiheuttamaa hyperkalsemiaa sairastaville potilaille, joilla on myös vaikea munuaisten vajaatoiminta, Zoledronic acid Stada -hoitoa pitää harkita vasta hoidon riski-hyötyarvioinnin jälkeen. Potilaita, joiden seerumin kreatiniini oli > 400 mikromol/l tai > 4,5 mg/dl, ei otettu mukaan kliinisiin tutkimuksiin. Annosta ei tarvitse muuttaa potilaille, joilla on kasvaimen aiheuttama hyperkalsemia, seerumin kreatiniinin ollessa < 400 mikromol/l tai < 4,5 mg/dl (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Luustotapahtumien ehkäiseminen potilailla, joilla on pitkälle edennyt luustoon liittyvä syöpä:
Aloitettaessa Zoledronic acid Stada -hoitoa potilaille, joilla on multippeli myelooma tai kiinteiden kasvainten aiheuttamia metastaattisia luuleesioita, on määritettävä seerumin kreatiniini ja kreatiniinipuhdistuma (CLcr). CLcr lasketaan seerumin kreatiniinista Cockcroft–Gaultin kaavalla. Zoledronic acid Stada ‑valmistetta ei suositella potilaille, joilla on ennen hoidon aloittamista vaikea munuaisten vajaatoiminta. Vaikean munuaisten vajaatoiminnan määritelmä on tässä potilasryhmässä CLcr < 30 ml/min. Kliinisiin tsoledronihappotutkimuksiin ei otettu mukaan potilaita, joiden seerumin kreatiniini oli > 265 mikromol/l tai > 3,0 mg/dl.
Potilaille, joilla on normaali munuaistoiminta (CLcr > 60 ml/min), tsoledronihappo 4 mg/100 ml infuusioneste voidaan antaa suoraan ilman lisävalmisteluja. Potilaille, joilla on luuston etäpesäkkeitä ja ennen hoidon aloittamista lievä tai keskivaikea munuaisten vajaatoiminta, suositellaan pienempiä Zoledronic acid Stada -annoksia. Lievän tai keskivaikean munuaisten vajaatoiminnan määritelmä on tässä potilasryhmässä CLcr 30–60 ml/min (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet):
Lähtötason kreatiniinipuhdistuma (ml/min) | Suositeltu tsoledronihappoannos* |
> 60 | 4,0 mg tsoledronihappoa |
50–60 | 3,5 mg* tsoledronihappoa |
40–49 | 3,3 mg* tsoledronihappoa |
30–39 | 3,0 mg* tsoledronihappoa |
*Annokset on laskettu AUC-tavoitearvolle 0,66 (mg•h/l) (CLcr = 75 ml/min). Munuaisten vajaatoimintapotilaat saavuttavat oletettavasti saman AUC-arvon pienemmällä annoksella kuin potilaat, joiden kreatiniinipuhdistuma on 75 ml/min.
Hoidon aloituksen jälkeen: seerumin kreatiniini on mitattava ennen jokaista Zoledronic acid Stada -annosta ja hoito on keskeytettävä, jos munuaisten toiminta on heikentynyt. Kliinisissä tutkimuksissa munuaistoiminnan heikkeneminen määriteltiin seuraavasti:
- potilailla, joilla normaali kreatiniinipitoisuus lähtötilanteessa (< 1,4 mg/dl tai < 124 mikromol/l): 0,5 mg/dl:n tai 44 mikromol/l:n nousu
- potilailla, joilla epänormaali kreatiniinipitoisuus lähtötilanteessa (> 1,4 mg/dl tai > 124 mikromol/l): 1,0 mg/dl:n tai 88 mikromol/l:n nousu.
Kliinisissä tutkimuksissa tsoledronihappohoito aloitettiin uudestaan vain, jos kreatiniiniarvo korjaantui 10 %:n sisälle lähtöarvosta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Zoledronic acid Stada -hoito on aloitettava uudestaan samalla annoksella, jota annettiin ennen hoidon keskeyttämistä.
Pediatriset potilaat
Tsoledronihapon turvallisuutta ja tehoa 1–17-vuotiaiden lasten hoidossa ei ole varmistettu. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdassa Farmakodynamiikka, ei voida antaa suosituksia annostuksesta.
Antotapa
Laskimoon.
Zoledronic acid Stada 4 mg/100 ml infuusioneste, liuos annetaan laskimoon yhtenä vähintään 15 minuuttia kestävänä infuusiona.
Potilaille, joiden munuaiset toimivat normaalisti (CLcr > 60 ml/min), tsoledronihappo 4 mg/100 ml infuusionestettä ei saa laimentaa edelleen.
Potilaille, joilla on lievä tai keskivaikea munuaisten vajaatoiminta, suositellaan pienennettyjä tsoledronihappoannoksia (ks. kohta ”Annostus” yllä ja kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pienennettyjen annosten valmistus potilaille, joiden perustason CLcr ≤ 60 ml/min: ks. taulukko 1 alla. Ota pullosta ilmoitettu määrä tsoledronihappoliuosta ja laita tilalle vastaava määrä steriiliä natriumkloridi-injektionestettä 9 mg/ml (0,9 %) tai 5-prosenttista glukoosi-injektionestettä.
Taulukko 1. Zoledronic acid Stada 4 mg/100 ml infuusionesteen pienennettyjen annosten valmistus
Lähtötason kreatiniinipuhdistuma (ml/min) | Poista seuraava määrä Zoledronic acid Stada ‑infuusionestettä (ml) | Korvaa seuraavalla määrällä steriiliä natriumkloridi-injektionestettä 9 mg/ml (0,9 %) tai 5-prosenttista glukoosi-injektionestettä (ml) | Pienennetty annos (mg tsoledronihappoa / 100 ml) |
50–60 | 12,0 | 12,0 | 3,5 |
40–49 | 18,0 | 18,0 | 3,3 |
30–39 | 25,0 | 25,0 | 3,0 |
Zoledronic acid Stada 4 mg/100 ml infuusionestettä ei saa sekoittaa muiden infuusionesteiden kanssa, ja se on annettava eri infuusioletkulla kerta-annosliuoksena laskimoon.
Potilaat on nesteytettävä kunnolla ennen Zoledronic acid Stada -valmisteen antoa ja sen jälkeen.
Vasta-aiheet
- yliherkkyys vaikuttavalle aineelle, muille bisfosfonaateille tai kohdassa Apuaineet mainituille apuaineille
- imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Yleistä
Ennen Zoledronic acid Stada -valmisteen antoa potilaat täytyy tutkia riittävän nesteytyksen varmistamiseksi.
Liiallista nesteytystä tulee välttää, jos potilaalla on sydämen vajaatoiminnan vaara.
Hyperkalsemiaan liittyviä metabolisia peruslaboratorioarvoja, kuten seerumin kalsium-, fosfaatti- ja magnesiumpitoisuutta, on seurattava huolellisesti tsoledronihappohoidon aloittamisen jälkeen. Lyhytaikainen tukihoito voi olla tarpeen hypokalsemian, hypofosfatemian tai hypomagnesemian ilmetessä. Hoitamattomien hyperkalsemiapotilaiden munuaisten toiminta on yleensä jonkin verran heikentynyttä, minkä vuoksi on harkittava munuaisten toiminnan huolellista seurantaa.
Zoledronic acid Stada sisältää samaa vaikuttavaa ainetta kuin tsoledronihappo 5 mg/100 ml. Zoledronic acid Stada -hoitoa käyttäville potilaille ei pidä antaa samanaikaisesti tsoledronihappo 5 mg/100 ml -valmistetta tai muita bisfosfonaatteja, koska näiden aineiden yhteisvaikutuksia ei tunneta.
Munuaisten vajaatoiminta
Kasvaimen aiheuttamaa hyperkalsemiaa sairastavat potilaat, joilla on todettu heikentynyt munuaisten toiminta, on tutkittava asianmukaisesti, ja on harkittava, onko Zoledronic acid Stada ‑valmisteella saavutettava mahdollinen hyöty suurempi kuin siitä mahdollisesti aiheutuva riski.
Tehtäessä päätöstä hoidon antamisesta luustotapahtumien ehkäisemiseksi potilaille, joilla on luumetastaaseja, on otettava huomioon, että hoito alkaa tehota 2–3 kuukauden kuluttua.
Tsoledronihapon käytön yhteydessä on raportoitu munuaisten vajaatoimintaa. Tekijöihin, jotka voivat lisätä munuaisten toimintakyvyn heikkenemisen mahdollisuutta, kuuluvat dehydraatio, todettu munuaisten vajaatoiminta, useat tsoledronihappo- tai muut bisfosfonaattikäyttökerrat sekä muiden munuaistoksisten lääkkeiden käyttö. Vaikka riski on pienempi annettaessa 4 mg:n tsoledronihappoannos 15 minuutin aikana, voi munuaistoiminta silti heikentyä. Munuaisten vajaatoimintaan ja dialyysiin johtanutta munuaisten toiminnan heikkenemistä on raportoitu potilailla, jotka ovat saaneet 4 mg:n aloitusannoksen tai kerta-annoksen tsoledronihappoa. Seerumin kreatiniiniarvot ovat myös nousseet joillakin potilailla, joille on annettu pitkään tsoledronihappoa luustotapahtumien ehkäisemiseksi suositellulla annoksella, tosin harvemmin.
Potilaiden seerumin kreatiniinipitoisuudet täytyy tutkia ennen jokaista Zoledronic acid Stada -annosta. Aloitettaessa hoitoa potilaille, joilla on luumetastaaseja ja lievä tai kohtalainen munuaisten vajaatoiminta, suositellaan pienempiä tsoledronihappoannoksia. Zoledronic acid Stada -valmistetta ei saa antaa, jos potilaan munuaisten toiminnan havaitaan heikentyvän hoidon aikana. Zoledronic acid Stada aloitetaan uudestaan vasta kun seerumin kreatiniiniarvo palautuu lähtöarvoon 10 %:n tarkkuudella. Zoledronic acid Stada -hoito aloitetaan uudestaan samalla annoksella kuin annettiin ennen hoidon keskeyttämistä.
Koska tsoledronihappo voi vaikuttaa munuaisten toimintaan, eikä vaikeaa munuaisten vajaatoimintaa alkutilanteessa sairastavia potilaita (kliinisissä tutkimuksissa määrittely kasvaimen aiheuttamaa hyperkalsemiaa sairastaville potilaille: seerumin kreatiniini ≥ 400 mikromol/l tai ≥ 4,5 mg/dl, ja syöpäpotilaille, joilla on luumetastaaseja: seerumin kreatiniini ≥ 265 mikromol/l tai ≥ 3,0 mg/dl) koskevaa kliinistä turvallisuustietoa ole, ja koska farmakokinetiikasta vaikeaa munuaisten vajaatoimintaa alkutilanteessa sairastavilla potilailla (kreatiniinipuhdistuma < 30 ml/min) on vain vähän tietoa, tsoledronihapon käyttöä ei suositella potilaille, joilla on vaikea munuaisten vajaatoiminta.
Maksan vajaatoiminta
Koska on vain vähän tietoa potilaista, joilla on vaikea maksan vajaatoiminta, spesifisiä hoitosuosituksia tämän potilasryhmän osalta ei voida antaa.
Osteonekroosi
Leuan osteonekroosi
Leuan osteonekroosia (ONJ) on raportoitu melko harvoin kliinisissä tutkimuksissa ja markkinoille tulon jälkeen potilailla, jotka ovat saaneet tsoledronihappoa.
Hoidon aloittamista tai uutta hoitojaksoa on lääketieteellisiä hätätapauksia lukuun ottamatta lykättävä myöhemmäksi potilailla, joilla on suun pehmytkudoksessa avoimia vaurioita, jotka eivät ole parantuneet. Potilaille, joilla on samanaikaisia riskitekijöitä, suositellaan hammastutkimusta tarkoituksenmukaisella ehkäisevällä hoidolla ja yksilöllistä hyöty-haitta-arviota ennen bisfosfonaattihoidon aloittamista.
Seuraavat riskitekijät on huomioitava arvioitaessa potilaan riskiä leuan osteonekroosin kehittymiselle:
- bisfosfonaatin tehokkuus (mitä tehokkaampi aine, sitä suurempi riski), antoreitti (suurempi riski parenteraalisen antotavan yhteydessä) ja bisfosfonaatin kumulatiivinen annos
- syöpä, muut sairaudet (esim. anemia, hyytymishäiriö, infektio), tupakointi
- samanaikaiset hoidot: kemoterapia, angiogeneesin estäjät (ks. kohta Yhteisvaikutukset), pään ja kaulan alueen sädehoito, kortikosteroidit
- aiempi hammassairaus, huono suuhygienia, periodontaalinen sairaus, invasiiviset hammastoimenpiteet (esim. hampaan poistot) ja huonosti istuvat hammasproteesit.
Kaikkia potilaita on kehotettava tsoledronihappohoidon aikana huolehtimaan suuhygieniastaan hyvin, käymään säännöllisesti hammastarkastuksessa ja ilmoittamaan heti suun alueella esiintyvistä oireista, kuten hampaiden heilumisesta, kivusta tai turvotuksesta tai haavaumien parantumattomuudesta tai eritevuodosta. Hoidon aikana invasiiviset hammashoidot / hampaisiin liittyvät toimenpiteet on tehtävä vasta tarkan harkinnan jälkeen ja niiden ajoittamista lähelle tsoledronihappoannoksen antoa on vältettävä . Potilailla, joille kehittyy leuan osteonekroosi bisfosfonaattihoidon aikana, hammaskirurgia voi pahentaa tilaa. Ei ole tietoa, vähentääkö bisfosfonaattihoidon keskeytys leuan osteonekroosiriskiä potilailla, joille on tehtävä hampaisiin liittyviä toimenpiteitä.
Hoitavan lääkärin ja leuan osteonekroosista asiantuntemusta omaavan hammaslääkärin tai hammaskirurgin on tehtävä läheistä yhteistyötä hoitosuunnitelman laatimisessa potilaille, joille kehittyy ONJ. Tsoledronihappohoidon tilapäistä keskeyttämistäon harkittava kunnes on tila korjaantunut ja samanaikaiset riskitekijät on minimoitu tilanteissa, joissa se on mahdollista.
Muiden anatomisten osien osteonekroosi
Korvakäytävän osteonekroosia on ilmoitettu bisfosfonaattien käytön ja lähinnä pitkäaikaisen hoidon yhteydessä. Korvakäytävän osteonekroosin mahdollisia riskitekijöitä ovat steroidien käyttö ja kemoterapia ja/tai paikalliset riskitekijät, kuten infektio tai trauma. Korvakäytävän osteonekroosin mahdollisuus on huomioitava, jos bisfosfonaatteja saavalla potilaalla ilmenee korvaoireita, krooniset korvatulehdukset mukaan lukien.
Lisäksi on raportoitu lähinnä aikuisilla tsoledronihappohoitoa saavilla syöpäpotilailla satunnaisia osteonekroositapauksia muissa elimistön osissa, kuten lonkassa ja reisiluussa.
Luusto- ja lihaskipu
Lääkkeen markkinoille tulon jälkeisen kokemuksen perusteella vakavia ja ajoittaisia toimintakykyä rajoittavia luu-, nivel- ja/tai lihaskipuja on raportoitu tsoledronihappoa käyttävillä potilailla. Kyseisiä tapahtumia on kuitenkin raportoitu harvoin. Oireiden ilmaantumista edeltävä aika vaihteli yhdestä vuorokaudesta useaan kuukauteen hoidon aloittamisesta. Useimmilla potilailla oireet lievenivät hoidon lopettamisen jälkeen. Osalla potilaista oireet palasivat, kun hoito aloitettiin uudelleen tsoledronihapolla tai toisella bisfosfonaatilla.
Epätyypilliset reisiluun murtumat
Epätyypillisiä subtrokanteerisia ja diafyseaalisia reisiluun murtumia on raportoitu bisfosfonaattihoidon yhteydessä, ensisijaisesti niillä potilailla, jotka ovat saaneet pitkäaikaista bisfosfonaattihoitoa osteoporoosiin. Tällaisia poikittaisia tai lyhyitä viistomurtumia voi ilmetä missä tahansa reisiluun pienen sarvennoisen alapuolen ja nivelnastan yläpuolisen alueen välissä. Näitä murtumia ilmenee hyvin pienten traumojen yhteydessä tai ilman traumaa, ja jotkut potilaat voivat kokea kipua reidessä tai nivusissa. Usein murtumat muistuttavat ensin rasitusmurtumia ennen kuin viikkojen ja kuukausien kuluessa ne muuttuvat täydellisiksi reisiluun murtumiksi. Murtumat ovat usein molemminpuolisia, joten toinenkin reisiluu on tutkittava, jos bisfosfonaattihoitoa saavalla potilaalla todetaan reisiluun varsiosan murtuma. Näiden murtumien viivästynyttä paranemista on myös raportoitu. Potilailla, joilla epäillään epätyypillistä reisiluun murtumaa, tulee harkita bisfosfonaattihoidon keskeyttämistä potilaan tilan arvioinnin ajaksi, ja keskeyttämispäätöksen on perustuttava yksilölliseen riski-hyötysuhteen arvioon.
Potilaita on ohjeistettava ilmoittamaan kaikista mahdollisista bisfosfonaattihoidon aikana ilmenevistä reisi-, lonkka- tai nivuskivuista, ja tällaisista oireista kertovat potilaat on tutkittava mahdollisen reisiluun epätäydellisen murtuman varalta.
Hypokalsemia
Hypokalsemiaa on raportoitu tsoledronihappoa saaneilla potilailla. Sydämen rytmihäiriöitä ja neurologisia haittavaikutuksia (mukaan lukien kouristuskohtaukset, heikentynyt tuntoherkkyys ja tetania) on raportoitu vaikea-asteisten hypokalsemiatapausten seurauksena. Sairaalahoitoa vaatineita vaikea-asteisia hypokalsemiatapauksia on raportoitu. Joissakin tapauksissa hypokalsemia voi olla potilaan henkeä uhkaava (ks. kohta Haittavaikutukset). Varovaisuutta on noudatettava kun tsoledronihappoa annetaan yhdessä lääkevalmisteiden kanssa, joiden tiedetään aiheuttavan hypokalsemiaa, koska näillä valmisteilla saattaa olla synergistinen vaikutus, joka aikaansaa vakavan hypokalsemian (ks. kohta Yhteisvaikutukset). Seerumin kalsium tulee mitata ja hypokalsemia korjata ennen tsoledronihappohoidon aloittamista. Potilaille tulee antaa riittävä määrä kalsium- ja D-vitamiinilisiä.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per infuusio eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Kliinisissä tutkimuksissa tsoledronihappoa on annettu samaan aikaan yleisesti käytössä olevien syöpälääkkeiden, diureettien, antibioottien ja analgeettien kanssa ilman ilmeisiä kliinisiä yhteisvaikutuksia. Tsoledronihappo ei sitoudu merkittävästi plasman proteiineihin eikä estä ihmisen P450-entsyymejä in vitro (ks. kohta Farmakokinetiikka), mutta muodollisia kliinisiä yhteisvaikutustutkimuksia ei ole tehty.
Varovaisuutta on noudatettava, kun bisfosfonaatteja annetaan aminoglykosidien, kalsitoniinin tai loopdiureettien kanssa, koska näillä lääkkeillä voi olla additiivinen vaikutus, jolloin seerumin kalsiumpitoisuus voi pysyä pienenä pitempään kuin olisi tarpeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varovaisuutta on noudatettava annettaessa tsoledronihappoa yhdessä muiden mahdollisesti munuaistoksisten lääkkeiden kanssa. Myös hypomagnesemian kehittymisen mahdollisuus on otettava huomioon hoidon aikana.
Multippelia myeloomaa sairastavilla potilailla munuaisten toimintahäiriön riski saattaa olla suurentunut annettaessa tsoledronihappoa yhdessä talidomidin kanssa.
Varovaisuuteen on syytä käytettäessä tsoledronihappoa samanaikaisesti antiangiogeenisten lääkevalmisteiden kanssa, sillä leuan osteonekroosin esiintyvyyden on havaittu lisääntyneen potilailla, jotka ovat saaneet samanaikaista hoitoa em. lääkkeillä.
Raskaus ja imetys
Raskaus
Ei ole olemassa tarkkoja tietoja tsoledronihapon käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmiselle ei tunneta. Tsoledronihappoa ei pidä käyttää raskauden aikana. Hedelmällisessä iässä olevia naisia on neuvottava välttämään raskaaksi tulemista.
Imetys
Ei tiedetä, erittyykö tsoledronihappo ihmisen rintamaitoon. Tsoledronihappo on vasta-aiheista rintaruokinnan aikana (ks. kohta Vasta-aiheet).
Hedelmällisyys
Tsoledronihapon mahdollisia haittavaikutuksia vanhempien ja F1-sukupolven hedelmällisyyteen arvioitiin rotilla. Tämä johti liioiteltuun farmakologiseen vaikutukseen, jonka katsotaan liittyvän valmisteen aiheuttamaan luuston kalsiumaineenvaihdunnan estoon, joka johtaa tiineen rotan hypokalsemiaan, joka on bisfosfonaattien luokkavaikutus, sekä dystokiaan että ennenaikaiseen tutkimuksen lopetukseen. Siten näiden tulosten perusteella ei voida määrittää tsoledronihapon todellista vaikutusta ihmisten hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Haittavaikutukset, kuten huimaus ja uneliaisuus, voivat vaikuttaa ajokykyyn tai koneidenkäyttökykyyn. Siksi tsoledronihappoa käytettäessä on noudatettava varovaisuutta ajamisessa ja koneiden käytössä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisesti on ilmoitettu kolmen päivän kuluessa tsoledronihapon annosta ilmaantuvasta akuutin vaiheen reaktiosta. Sen oireita ovat luukipu, kuume, väsymys, nivelkipu, lihaskipu, vilunväristykset sekä artriitti ja sen seurauksena nivelturvotus. Nämä oireet häviävät yleensä muutaman päivän kuluessa (katso valikoitujen haittavaikutusten kuvaus).
Tärkeitä tunnistettuja riskejä, jotka liittyvät tsoledronihapon käyttöön hyväksytyissä käyttöaiheissa, ovat:
munuaisten vajaatoiminta, leuan osteonekroosi, akuutin vaiheen reaktio, hypokalsemia, eteisvärinä, anafylaksi, interstitiaalinen keuhkosairaus. Näiden tunnistettujen riskien yleisyydet on esitetty taulukossa 2.
Haittavaikutukset taulukkomuodossa
Seuraavat taulukossa 2 esitetyt haittavaikutukset on koottu kliinisistä tutkimuksista ja markkinoilletulon jälkeen saaduista raporteista, jotka koskevat pääasiassa pitkäaikaista hoitoa 4 mg:lla tsoledronihappoa:
Taulukko 2
Haittatapahtumat on luokiteltu yleisyyden mukaan aloittaen yleisimmistä seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
| Veri ja imukudos | |
| Yleinen | Anemia |
| Melko harvinainen | Trombosytopenia, leukopenia |
| Harvinainen | Pansytopenia |
| Immuunijärjestelmä | |
| Melko harvinainen | Yliherkkyysreaktio |
| Harvinainen | Angioneuroottinen edeema |
| Psyykkiset häiriöt | |
| Melko harvinainen | Ahdistuneisuus, unihäiriöt |
| Harvinainen | Sekavuus |
| Hermosto | |
| Yleinen | Päänsärky |
| Melko harvinainen | Heitehuimaus, tuntoharha, makuhäiriö, heikentynyt tunto, lisääntynyt tuntoherkkyys, vapina, uneliaisuus |
| Hyvin harvinainen | Kouristuskohtaus, heikentynyt tuntoherkkyys ja tetania (hypokalsemian seurauksena) |
| Silmät | |
| Yleinen | Sidekalvotulehdus |
| Melko harvinainen | Näön sumentuminen, silmän kovakalvon tulehdus ja orbitaalinen tulehdus |
| Harvinainen | Suonikalvoston tulehdus |
| Hyvin harvinainen | Episkleriitti |
| Sydän | |
| Melko harvinainen | Kohonnut verenpaine, matala verenpaine, eteisvärinä, pyörtymiseen tai verenkierron romahtamiseen johtava matala verenpaine |
| Harvinainen | Sydämen harvalyöntisyys, sydämen rytmihäiriö (hypokalsemian seurauksena) |
| Hengityselimet, rintakehä ja välikarsina | |
| Melko harvinainen | Hengenahdistus, yskä, bronkokonstriktio |
| Harvinainen | Interstitiaalinen keuhkosairaus |
| Ruoansulatuselimistö | |
| Yleinen | Pahoinvointi, oksentelu, heikentynyt ruokahalu |
| Melko harvinainen | Ripuli, ummetus, vatsakipu, ruuansulatushäiriöt, suutulehdus, suun kuivuminen |
| Iho ja ihonalainen kudos | |
| Melko harvinainen | Kutina, ihottuma (mukaan lukien punoittava ihottuma ja täpläinen (makulaarinen) ihottuma), lisääntynyt hikoilu |
| Luusto, lihakset ja sidekudos | |
| Yleinen | Luukipu, lihaskipu, nivelkipu, laaja-alainen kipu |
| Melko harvinainen | Lihaskouristukset, leuan osteonekroosi |
| Hyvin harvinainen | Korvakäytävän osteonekroosi (bisfosfonaattien luokkahaittavaikutus) ja muiden anatomisten osien osteonekroosi (mukaan lukien reisiluu ja lonkka) |
| Munuaiset ja virtsatiet | |
| Yleinen | Munuaisten vajaatoiminta |
| Melko harvinainen | Akuutti munuaisten vajaatoiminta, hematuria, proteinuria |
| Harvinainen | Hankinnainen Fanconin oireyhtymä |
| Tuntematon | Tubulointerstitiaalinefriitti |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Yleinen | Kuume, flunssankaltaiset oireet (kuten väsymys, vilunväreet, huonovointisuus ja punoitus) |
| Melko harvinainen | Voimattomuus, raajojen turvotus, injektiokohdan reaktiot (mukaan lukien kipu, ärtyminen, turvotus, kovettuminen), rintakipu, painon nousu, anafylaktinen reaktio/sokki, nokkosihottuma |
| Harvinainen | Akuutin vaiheen reaktion oireina artriitti ja nivelturvotus |
| Tutkimukset | |
| Hyvin yleinen | Hypofosfatemia |
| Yleinen | Veren kreatiniini- ja urea-arvojen nousu, hypokalsemia |
| Melko harvinainen | Hypomagnesemia, hypokalemia |
| Harvinainen | Hyperkalemia, hypernatremia |
Valikoitujen haittavaikutusten kuvaus
Munuaisten vajaatoiminta
Tsoledronihapon käytön yhteydessä on raportoitu munuaisten toimintahäiriöitä. Yhteen kootussa turvallisuustietojen analyysissä tsoledronihapon rekisteröintiin liittyvistä kliinisistä tutkimuksista, jotka koskivat luustotapahtumien ehkäisemistä potilailla, joilla on pitkälle edennyt luustoon liittyvä syöpä, todettiin seuraavat esiintymistiheydet munuaisten vajaatoimintahaittatapahtumille, joiden epäiltiin liittyvän tsoledronihappoon (haittavaikutukset): multippeli myelooma (3,2 %), eturauhassyöpä (3,1 %), rintasyöpä (4,3 %), keuhkokasvaimet ja muut kiinteät tuumorit (3,2 %). Tekijöihin, jotka voivat lisätä munuaisten toimintakyvyn heikkenemisen mahdollisuutta, kuuluvat dehydraatio, todettu munuaisten vajaatoiminta, useat tsoledronihappo- tai muut bisfosfonaattikäyttökerrat sekä muiden munuaistoksisten lääkkeiden käyttö tai tällä hetkellä suositeltua lyhyempi infuusioaika. Munuaisten vajaatoimintaan ja dialyysiin johtanutta munuaisten toiminnan heikkenemistä on raportoitu potilailla, jotka ovat saaneet 4 mg:n aloitusannoksen tai kerta-annoksen tsoledronihappoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Leuan osteonekroosi
Potilailla, jotka ovat olleet enimmäkseen syöpäpotilaita ja jotka ovat saaneet luun resorptiota estäviä lääkevalmisteita, kuten tsoledronihappoa, on raportoitu leuan osteonekroositapauksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Moni näistä potilaista sai myös kemoterapiaa ja kortikosteroidihoitoa ja heillä oli merkkejä paikallisesta infektiosta mukaan lukien osteomyeliitti. Pääosa näistä tapauksista raportoitiin syöpäpotilailla hampaan poiston tai muun hammasleikkauksen jälkeen.
Eteisvärinä
Yhdessä kolme vuotta kestäneessä, satunnaistetussa, kaksoissokkoutetussa kontrolloidussa tutkimuksessa, jossa verrattiin tsoledronihapon (5 mg kerran vuodessa) tehoa ja turvallisuutta lumelääkkeeseen postmenopausaalisen osteoporoosin hoidossa (PMO), eteisvärinän kokonaisilmaantuvuus oli 5 mg tsoledronihappoa saaneilla potilailla 2,5 % (96 / 3 862) ja lumelääkettä saaneilla potilailla 1,9 % (75 / 3 852). Vakavina haittatapahtumina eteisvärinää ilmeni 1,3 %:lle tsoledronihappoa saaneista potilaista (51 / 3 862) ja 0,6 %:lle lumelääkettä saaneista potilaista (22 / 3 852). Tässä tutkimuksessa havaittua epätasapainoa ei ole havaittu muissa tsoledronihappotutkimuksissa, mukaan lukien tsoledronihapolla tehdyt tutkimukset, joissa syöpäpotilaat saivat 4 mg tsoledronihappoa joka 3.–4. viikko. Tässä yhdessä kliinisessä tutkimuksessa esille tullutta eteisvärinän esiintyvyyden lisääntymisen mekanismia ei tunneta.
Akuutin vaiheen reaktio
Tähän lääkkeen haittavaikutukseen kuuluu useita oireita, kuten kuume, lihaskipu, päänsärky, raajojen kipu, pahoinvointi, oksentelu, ripuli, nivelkipu sekä artriitti ja sen seurauksena nivelturvotus. Oireet alkavat ≤ 3 päivän kuluessa tsoledronihappoinfuusion annosta. Reaktioon viitataan usein nimityksellä ”flunssankaltaiset” oireet tai ”annostuksen jälkeiset” oireet.
Epätyypilliset reisiluun murtumat
Markkinoilletulon jälkeen on ilmoitettu seuraavia haittavaikutuksia (esiintyvyys harvinainen): epätyypilliset subtrokanteeriset ja diafysiaaliset reisiluun murtumat (bisfosfonaattien luokkahaittavaikutus).
Hypokalsemiaan liittyvät haittavaikutukset
Hypokalsemia on tärkeä, tsoledronihapon käyttöaiheiden mukaisessa käytössä todettu riski. Kliinisissä tutkimuksissa ja lääkkeen markkinoille tulon jälkeen todettujen tapausten arvioinnin perusteella on riittävästi näyttöä tsoledronihappohoidon ja raportoitujen hypokalsemiatapausten sekä niiden seurauksena kehittyneiden rytmihäiriötapausten yhteydestä. Lisäksi on olemassa näyttöä hypokalsemian ja sen yhteydessä raportoitujen sekundaaristen neurologisten tapahtumien, kuten kouristuskohtausten, heikentyneen tuntoherkkyyden ja tetanian, välisestä yhteydestä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haitta vaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tsoledronihapon aiheuttamasta akuutista yliannostuksesta on vähän kliinistä kokemusta. Enintään 48 mg:n tsoledronihappoannostien antamisesta vahingossa on ilmoitettu. Potilaita, jotka ovat saaneet suositusta suuremman annoksen (ks. kohta Annostus ja antotapa), tulee seurata huolellisesti, koska heillä on havaittu munuaisten toiminnan heikentymistä (munuaisten vajaatoiminta mukaan lukien) ja seerumin elektrolyyttien (mukaan lukien kalsium, fosfori ja magnesium) poikkeamia. Hypokalsemiatapauksissa on annettava kalsiumglukonaatti-infuusioita kliinisen tarpeen mukaisesti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Luun rakenteeseen ja mineralisaatioon vaikuttavat lääkkeet, bisfosfonaatit, ATC-koodi: M05BA08
Bisfosfonaatteihin kuuluva tsoledronihappo vaikuttaa ensisijaisesti luuhun. Se on osteoklastien aiheuttaman luun resorption estäjä.
Bisfosfonaattien selektiivinen vaikutus luuhun perustuu niiden voimakkaaseen affiniteettiin mineralisoituneeseen luuhun. Tarkkaa osteoklastien toiminnan estävää mekanismia molekyylitasolla ei kuitenkaan vielä tunneta. Pitkäaikaisten eläintutkimusten mukaan tsoledronihappo estää luun resorptiota vaikuttamatta haitallisesti luun muodostukseen, mineralisaatioon tai mekaanisiin ominaisuuksiin.
Sen lisäksi, että tsoledronihappo on tehokas luun resorption estäjä, sillä on myös useita kasvainten kasvua estäviä ominaisuuksia, jotka saattavat myötävaikuttaa sen kokonaistehoon metastaattisen luusairauden hoidossa. Prekliinisissä tutkimuksissa on osoitettu seuraavat ominaisuudet:
- in vivo: luun osteoklastisen resorption esto, mikä muuttaa luuytimen mikroympäristöä tehden sen epäsuotuisammaksi kasvainsolujen kasvulle; verisuonten kasvun estäminen ja kivun esto
- in vitro: osteoblastien lisääntymisen esto, suora kasvainsoluihin kohdistuva sytostaattinen ja proapoptoottinen toiminta, synergistinen sytostaattinen vaikutus muiden syöpälääkkeiden kanssa, adheesion ja invaasion esto
Luustotapahtumien ehkäisemistä koskevien kliinisten tutkimusten tulokset potilailla, joilla on luusto on liittyvä pitkälle edennyt syöpä
Ensimmäisessä satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa tutkimuksessa verrattiin 4 mg:n tsoledronihappohoitoa lumelääkkeeseen luustoon liittyvien tapahtumien ehkäisemisessä (Skeletal Related Events, SRE) eturauhassyöpäpotilailla. Tsoledronihappo 4 mg pienensi merkitsevästi niiden potilaiden osuutta, joille ilmeni vähintään yksi luustoon liittyvä tapahtuma (SRE), pidensi mediaaniaikaa ensimmäisen luustoon liittyvän tapahtuman ilmaantumiseen yli viidellä kuukaudella ja vähensi tapahtumien vuosittaista ilmaantuvuutta potilasta kohden eli luustosairastuvuutta. Monitapahtuma-analyysi osoitti luustoon liittyvien tapahtumien kehittymisriskin pienentyneen 36 % 4 mg:n tsoledronihapporyhmässä lumelääkeryhmään verrattuna. Tsoledronihappoa 4 mg saaneet potilaat raportoivat vähemmän kivun lisääntymistä kuin potilaat, jotka saivat lumelääkettä. Erot olivat merkitseviä kuukausina 3, 9, 21 ja 24. Tsoledronihappoa (4 mg) saaneiden potilaiden ryhmässä havaittiin vähemmän patologisia murtumia. Hoidon vaikutukset olivat heikompia potilailla, joilla oli blastisia leesioita. Tehokkuustulokset on esitetty taulukossa 3.
Toisessa tutkimuksessa, johon osallistui potilaita, joilla oli jokin muu kiinteä kasvain kuin rinta- tai eturauhassyöpä, tsoledronihappo 4 mg vähensi merkitsevästi niiden potilaiden osuutta, joilla oli luustoon liittyviä tapahtumia, pidensi mediaaniaikaa ensimmäisen luustoon liittyvän tapahtuman ilmaantumiseen yli kahdella kuukaudella ja vähensi luustosairastuvuutta. Monitapahtuma-analyysi osoitti luustoon liittyvien tapahtumien kehittymisriskin pienentyneen 30,7 % 4 mg:n tsoledronihapporyhmässä lumelääkeryhmään verrattuna. Tehokkuustulokset on esitetty taulukossa 4.
Taulukko 3: Tehokkuustulokset (hormonihoitoa saaneet eturauhassyöpäpotilaat)
| SRE (+TIH) | Murtumat* | Luun sädehoito | ||||
| Tsoledroni-happo 4 mg | Lumelääke | Tsoledroni-happo 4 mg | Lumelääke | Tsoledroni-happo 4 mg | Lumelääke | |
| n | 214 | 208 | 214 | 208 | 214 | 208 |
| Niiden potilaiden osuus, joille ilmeni SRE (%) | 38 | 49 | 17 | 25 | 26 | 33 |
| p-arvo | 0,028 | 0,052 | 0,119 | |||
Mediaaniaika SRE:n ilmaantumiseen (päivinä) | 488 | 321 | NR | NR | NR | 640 |
| p-arvo | 0,009 | 0,020 | 0,055 | |||
| Luustosairastuvuus | 0,77 | 1,47 | 0,20 | 0,45 | 0,42 | 0,89 |
| p-arvo | 0,005 | 0,023 | 0,060 | |||
| Useiden tapahtumien riskin pieneneminen ** (%) | 36 | - | NA | NA | NA | NA |
| p-arvo | 0,002 | NA | NA | |||
* Sisältää nikamamurtumat ja muut kuin nikamamurtumat
** Sisältää kaikki luustotapahtumat; niiden kokonaismäärän sekä ajan jokaisen tapahtuman ilmaantumiseen tutkimuksen aikana
NR Ei saavutettu
NA Ei käytettävissä
Taulukko 4: Tehokkuustulokset (muut kiinteät kasvaimet kuin rinta- tai eturauhassyöpä)
| SRE (+TIH) | Murtumat* | Luun sädehoito | ||||
| Tsoledroni-happo 4 mg | Lumelääke | Tsoledroni-happo 4 mg | Lumelääke | Tsoledroni-happo 4 mg | Lumelääke | |
| n | 257 | 250 | 257 | 250 | 257 | 250 |
| Niiden potilaiden osuus, joille ilmeni SRE (%) | 39 | 48 | 16 | 22 | 29 | 34 |
| p-arvo | 0,039 | 0,064 | 0,173 | |||
Mediaaniaika SRE:n ilmaantumiseen (päivinä) | 236 | 155 | NR | NR | 424 | 307 |
| p-arvo | 0,009 | 0,020 | 0,079 | |||
| Luustosairastuvuus | 1,74 | 2,71 | 0,39 | 0,63 | 1,24 | 1,89 |
| p-arvo | 0,012 | 0,066 | 0,099 | |||
| Useiden tapahtumien riskin pieneneminen ** (%) | 30,7 | - | NA | NA | NA | NA |
| p-arvo | 0,003 | NA | NA | |||
* Sisältää nikamamurtumat ja muut kuin nikamamurtumat
** Sisältää kaikki luustotapahtumat; niiden kokonaismäärän sekä ajan jokaisen tapahtuman ilmaantumiseen tutkimuksen aikana
NR Ei saavutettu
NA Ei käytettävissä
Kolmannessa vaiheen 3 satunnaistetussa kaksoissokkotutkimuksessa verrattiin tsoledronihappo 4 mg:aa pamidronaatti 90 mg:aan annosteltuina joka 3.–4. viikko. Tutkimukseen osallistui potilaita, joilla oli multippeli myelooma tai rintasyöpä, johon liittyi ainakin yksi luuleesio. Tulokset osoittivat, että tsoledronihappo 4 mg oli yhtä tehokas kuin 90 mg pamidronaattia luustotapahtumien ehkäisyssä. Monitapahtuma-analyysi paljasti merkitsevän, 16 % riskin pienentymisen potilailla, joita hoidettiin tsoledronihappo 4 mg:lla, verrattuna pamidronaattihoitoa saaneisiin potilaisiin. Tehokkuustulokset on esitetty taulukossa 5.
Taulukko 5: Tehokkuustulokset (potilaat, joilla on rintasyöpä tai multippeli myelooma)
| SRE (+TIH) | Murtumat* | Luun sädehoito | ||||
| Tsoledroni-happo 4 mg | Pamidronaatti 90 mg | Tsoledroni-happo 4 mg | Pamidronaatti 90 mg | Tsoledroni-happo 4 mg | Pamidronaatti 90 mg | |
| n | 561 | 555 | 561 | 555 | 561 | 555 |
| Niiden potilaiden osuus, joille ilmeni SRE (%) | 48 | 52 | 37 | 39 | 19 | 24 |
| p-arvo | 0,198 | 0,653 | 0,037 | |||
| Mediaaniaika SRE:n ilmaantumiseen (päivinä) | 376 | 356 | NR | 714 | NR | NR |
| p-arvo | 0,151 | 0,672 | 0,026 | |||
| Luustosairastu-vuus | 1,04 | 1,39 | 0,53 | 0,60 | 0,47 | 0,71 |
| p-arvo | 0,084 | 0,614 | 0,015 | |||
| Useiden tapahtumien riskin pieneneminen ** (%) | 16 | - | NA | NA | NA | NA |
| p-arvo | 0,030 | NA | NA | |||
* Sisältää nikamamurtumat ja muut kuin nikamamurtumat
** Sisältää kaikki luustotapahtumat; niiden kokonaismäärän sekä ajan jokaisen tapahtuman ilmaantumiseen tutkimuksen aikana
NR Ei saavutettu
NA Ei käytettävissä
Tsoledronihappoa 4 mg tutkittiin myös kaksoissokkoutetussa, satunnaistetussa, lumekontrolloidussa tutkimuksessa 228 potilaalla, joilla oli rintasyövän aiheuttamia dokumentoituja luumetastaaseja. Tutkimuksessa arvioitiin tsoledronihapon 4 mg vaikutusta luustotapahtumien (skeletal related event, SRE) esiintyvyyssuhteeseen, joka laskettiin jakamalla luustotapahtumien kokonaismäärä (hyperkalsemiaa lukuun ottamatta, aiempien luunmurtumien suhteen korjattuna) koko riskijaksolla. Potilaat saivat vuoden ajan joko 4 mg tsoledronihappoa tai lumelääkettä joka neljäs viikko. Potilaat jakautuivat tasaisesti tsoledronihappo- ja lumeryhmiin.
Luustotapahtumien suhde oli tsoledronihapolla 0,628 ja lumelääkkeellä 1,096 (tapahtumat/henkilövuosi). Tsoledronihappoa saaneessa ryhmässä 29,8 %:lla potilaista oli vähintään yksi luustotapahtuma (hyperkalsemiaa lukuun ottamatta), kun lumeryhmässä vastaava luku oli 49,6 % (p = 0,003). Tsoledronihapporyhmässä ensimmäisen luustotapahtuman kehittymiseen kuluvan ajan mediaania ei saavutettu tutkimuksen loppuun mennessä, ja se piteni huomattavasti lumehoitoon verrattuna (p = 0,007). Monitapahtuma-analyysissa tsoledronihappo 4 mg pienensi luustotapahtumien riskiä 41 %:lla (riskisuhde = 0,59; p = 0,019) lumehoitoon verrattuna.
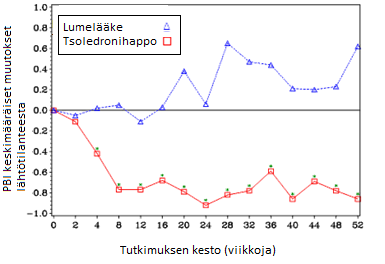
Tsoledronihappoa saaneessa ryhmässä todettiin lumelääkkeeseen verrattuna kivun (Brief Pain Inventory (BPI) -asteikolla arvioituna) tilastollisesti merkitsevä väheneminen neljän viikon kuluttua ja tutkimuksen jokaisena mittausajankohtana sen jälkeen (kuva 1). Kipu tsoledronihapporyhmässä oli johdonmukaisesti perustason alapuolella ja kivun lievittymisen myötä kivun arvioinnissa annetut pistemäärät pienenivät.
Kuva 1. Keskimääräiset muutokset lähtötilanteesta BPI-asteikolla. Tilastollisesti merkitsevät erot on merkitty (*p < 0,05) vertailuhoitojen välille (4 mg tsoledronihappoa verrattuna lumelääkkeeseen).
Kasvaimen aiheuttaman hyperkalsemian hoitoa koskevien k liinisten tutkimusten tulokset
Kasvaimen aiheuttamaa hyperkalsemiaa koskevat kliiniset tutkimukset ovat osoittaneet, että tsoledronihapon vaikutukselle ovat ominaisia seerumin kalsiumpitoisuuden pieneneminen ja kalsiumin erittyminen virtsaan. Vaiheen I annostutkimuksissa lievää tai kohtalaista kasvaimen aiheuttamaa hyperkalsemiaa sairastaneilla potilailla tutkitut vaikuttavat annokset olivat noin 1,2–2,5 mg.
Tsoledronihapon (4 mg) ja pamidronaatin (90 mg) vaikutusten vertaamiseksi kasvaimen aiheuttamaa hyperkalsemiaa sairastaville potilaille tehtyjen kahden keskeisen monikeskustutkimuksen tulokset yhdistettiin etukäteen suunnitellussa analyysissa. Korjattu seerumin kalsiumpitoisuus normalisoitui nopeammin päivänä 4 annoksella 8 mg tsoledronihappoa ja päivänä 7 annoksilla 4 mg ja 8 mg tsoledronihappoa. Todettiin seuraavat vasteprosentit:
Taulukko 6: Täydellisen vasteen saaneiden potilaiden osuus eri päivinä yhdistetyissä kasvaimen aiheuttamaa hyperkalsemiaa koskevissa tutkimuksissa
| Päivä 4 | Päivä 7 | Päivä 10 | |
| Tsoledronihappo 4 mg (n = 86) | 45,3 % (p = 0,104) | 82,6 % (p = 0,005)* | 88,4 % (p = 0,002)* |
| Tsoledronihappo 8 mg (n = 90) | 55,6 % (p = 0,021)* | 83,3 % (p = 0,010)* | 86,7 % (p = 0,015)* |
| Pamidronaatti 90 mg (n = 99) | 33,3 % | 63,6 % | 69,7 % |
*p-arvot pamidronaattiin nähden.
Veren kalsiumpitoisuuden normalisoitumiseen kulunut mediaaniaika oli 4 päivää. Mediaaniaika relapsiin (albumiinin suhteen korjatun seerumin kalsiumpitoisuuden suureneminen uudelleen arvoon 2,9 mmol/l) oli 30–40 päivää tsoledronihappoa saaneilla ja 17 päivää 90 mg pamidronaattia saaneilla potilailla (p = 0,001 tsoledronihappoa 4 mg ja p = 0,007 tsoledronihappoa 8 mg saaneiden potilaiden ryhmässä). Mainittujen kahden tsoledronihappoannoksen välillä ei ollut tilastollisesti merkitsevää eroa.
Kliinisissä tutkimuksissa hoidettiin uudelleen 8 mg:lla tsoledronihappoa 69 potilasta, joiden tila uusiutui tai jotka eivät reagoineet ensimmäiseen hoitoon (tsoledronihappo 4 mg, 8 mg tai pamidronaatti 90 mg). Näiden potilaiden vasteprosentti oli noin 52. Koska potilaita hoidettiin uudelleen vain 8 mg:n annoksella, tietoja 4 mg:n tsoledronihappoannokseen vertaamiseksi ei ole.
Kliinisissä tutkimuksissa potilailla, joilla oli kasvaimen aiheuttama hyperkalsemia, yleinen turvallisuusprofiili kaikissa kolmessa hoitoryhmässä (4 tai 8 mg tsoledronihappoa tai 90 mg pamidronaattia) oli samankaltainen haittavaikutusten tyypin ja vaikeusasteen suhteen.
Pediatriset potilaat
Kliinisten tutkimusten tulokset vaikeaa synnynnäistä luutumisvajausta (osteogenesis imperfecta) sairastavilla 1–17-vuotiailla lapsipotilailla
Laskimoon annettavan tsoledronihapon vaikutuksia vaikeaa synnynnäistä luutumisvajausta (tyypit I, III ja IV) sairastavilla 1–17-vuotiailla lapsipotilailla verrattiin laskimoon annettavaan pamidronaattihoitoon yhdessä kansainvälisessä, satunnaistetussa, avoimessa monikeskustutkimuksessa, jonka tsoledronihapporyhmään kuului 74 ja pamidronaattiryhmään 76 potilasta. Tutkimuksen hoitovaihe kesti 12 kk, ja sitä edelsi 4–9 viikon seulontavaihe, jonka aikana osallistujat käyttivät D-vitamiini- ja kalsiumlisää vähintään 2 viikon ajan. 1–< 3-vuotiaat potilaat saivat kliinisen tutkimusohjelman puitteissa 0,025 mg/kg tsoledronihappoa (maksimikerta-annos 0,35 mg) 3 kuukauden välein ja 3–17-vuotiaat potilaat taas 0,05 mg/kg tsoledronihappoa (maksimikerta-annos 0,83 mg) 3 kuukauden välein. Jatkotutkimus tehtiin, jotta voitiin arvioida kerran tai kahdesti vuodessa annosteltavan tsoledronihapon yleistä pitkäaikaisturvallisuutta ja sen pitkäaikaisturvallisuutta munuaisten kannalta 12 kuukauden pituisessa jatkotutkimuksessa lapsilla, jotka olivat suorittaneet vuoden kestäneen tsoledronihappo- tai pamidronaattihoidon loppuun varsinaisen tutkimuksen puitteissa.
Tutkimuksen ensisijaisena päätetapahtumana oli lannerangan luun mineraalitiheyden (BMD) prosentuaalinen muutos lähtötilanteeseen nähden 12 kuukauden hoidon jälkeen. Arvioidut hoidon vaikutukset luun mineraalitiheyteen olivat samankaltaisia, mutta tutkimusasetelma ei ollut riittävän häiriövapaa tsoledronihapon vähintään samanveroisen tehon vahvistamiseksi. Erityisesti ei ollut selkeää näyttöä tehosta murtumien esiintyvyyteen tai kipuun. Vaikeaa synnynnäistä luutumisvajausta sairastavista potilaista alaraajojen pitkien luiden murtumahaittavaikutuksia ilmoitettiin noin 24 %:lla (reisiluu) ja 14 %:lla (sääriluu) tsoledronihappoa saaneista potilaista ja 12 %:lla (reisiluu) ja 5 %:lla (sääriluu) pamidronaattihoitoa saaneista potilaista taudin tyypistä ja murtumien syystä riippumatta. Murtumien kokonaisilmaantuvuus oli kuitenkin verrattavissa potilailla, jotka saivat tsoledronihappoa (43 %, 32/74)ja pamidronaattia (41 %, 31/76). Murtumariskin tulkintaa vaikeuttaa se, että vaikeaa synnynnäistä luutumisvajausta sairastavilla esiintyy yleisesti murtumia itse tautiprosessin vuoksi.
Tässä populaatiossa havaitut haittavaikutukset olivat samanlaisia kuin on aiemmin havaittu pitkälle edenneitä, luustoon liittyviä syöpätauteja sairastavilla aikuisilla (ks. kohta Haittavaikutukset). Haittavaikutukset esitetään taulukossa 7 yleisyyden mukaan ryhmiteltyinä. Ryhmittelyssä käytetään seuraavaa vakiintunutta luokitusta: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 7: Vaikeaa synnynnäistä luutumisvajausta sairastavilla lapsilla havaitut haittavaikutukset1
| Hermosto | |
| Yleinen | Päänsärky |
| Sydän | |
| Yleinen | Takykardia |
| Hengityselimet, rintakehä ja välikarsina | |
| Yleinen | Nenänielutulehdus |
| Ruoansulatuselimistö | |
| Hyvin yleinen | Oksentelu, pahoinvointi |
| Yleinen | Vatsakipu |
| Luusto, lihakset ja sidekudos | |
| Yleinen | Raajojen kipu, nivelkipu, luusto- ja lihaskipu |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | Kuume, väsymys |
| Yleinen | Akuutin vaiheen reaktiot, kipu |
| Tutkimukset | |
| Hyvin yleinen | Hypokalsemia |
| Yleinen | Hypofosfatemia |
1Haittavaikutuksia, joiden esiintyvyystiheys oli < 5 %, arvioitiin lääketieteellisesti ja kävi ilmi, että nämä tapaukset ovat johdonmukaisia tsoledronihapon hyvin vakiintuneen turvallisuusprofiilin kanssa (ks. kohta Haittavaikutukset).
Vaikeaa synnynnäistä luutumisvajausta sairastavilla lapsilla tsoledronihappohoitoon näyttää liittyvän pamidronaattihoitoa suurempi akuutin vaiheen reaktioiden, hypokalsemian ja selittämättömän takykardian riski, mutta tämä eroavaisuus pieneni seuraavien infuusioiden jälkeen.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset tsoledronihapon käytöstä kaikkien pediatristen potilasryhmien hoidossa sekä kasvaimen aiheuttamassa hyperkalsemiassa että luustotapahtumien ehkäisemisessä potilailla, joilla on pitkälle edennyt luustoon liittyvä syöpä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Annettaessa 5 ja 15 minuutin kertainfuusiona ja toistoannostelussa 2, 4, 8 ja 16 mg tsoledronihappoa 64 potilaalle, joilla oli luustometastaaseja, saatiin seuraavat farmakokineettiset tiedot, joiden todettiin olevan annoksesta riippumattomia.
Tsoledronihappoinfuusion aloittamisen jälkeen lääkepitoisuudet plasmassa suurenevat nopeasti saavuttaen korkeimman pitoisuuden infuusion lopussa. Tämän jälkeen pitoisuudet nopeasti pienenevät ollen alle 10 % korkeimmasta arvosta neljän tunnin kuluttua ja alle 1 % 24 tunnin kuluttua, jonka jälkeen seuraa pidempi jakso, jolloin pitoisuus on hyvin pieni, alle 0,1 % korkeimmasta arvosta, ennen seuraavaa tsoledronihappoinfuusiota päivänä 28.
Laskimoon annettu tsoledronihappo eliminoituu kolmivaiheisesti: se poistuu systeemisestä verenkierrosta nopeasti kahdessa vaiheessa; puoliintumisajat ovat t½α 0,24 ja t½β 1,87 tuntia. Tätä seuraa pitkä eliminaatiovaihe ja terminaalinen puoliintumisaika t½γ on 146 tuntia. Tsoledronihappoa ei kertynyt plasmaan 28 päivän välein tapahtuvan toistuvan annon jälkeen. Tsoledronihappo ei metaboloidu, vaan se erittyy muuttumattomana munuaisten kautta. Ensimmäisten 24 tunnin aikana 39 ± 16 % annoksesta erittyy virtsaan ja loppu sitoutuu lähinnä luukudokseen. Luukudoksesta se vapautuu hyvin hitaasti takaisin systeemiseen verenkiertoon ja eliminoituu munuaisten kautta. Elimistön kokonaispuhdistuma on annoksesta riippumatta 5,04 ± 2,5 l/h, eikä sukupuoli, ikä, rotu tai paino vaikuta siihen. Infuusioajan pidentäminen 5 minuutista 15 minuuttiin pienensi tsoledronihappopitoisuutta 30 % infuusion lopussa, mutta ei vaikuttanut plasman AUC-arvoihin.
Tsoledronihapon farmakokineettisten parametrien vaihtelu potilaiden kesken oli suuri, kuten muillakin bisfosfonaateilla.
Tsoledronihapon farmakokinetiikasta ei ole tietoa hyperkalsemiaa eikä maksan vajaatoimintaa sairastavien potilaiden osalta. Tsoledronihappo ei estä ihmisen P450-entsyymejä in vitro, eikä se muunnu elimistössä. Eläinkokeissa < 3 % annoksesta erittyi ulosteeseen, mikä viittaa siihen, ettei maksan toiminnalla ole oleellista merkitystä tsoledronihapon farmakokinetiikassa.
Tsoledronihapon munuaispuhdistuma korreloi kreatiniinipuhdistuman kanssa munuaispuhdistuman ollessa 75 ± 33 % kreatiniinipuhdistumasta. Keskiarvo 64 tutkitulla syöpäpotilaalla oli 84 ± 29 ml/min (vaihteluväli 22–143 ml/min). Populaatioanalyysin perusteella potilaalla, jonka kreatiniinipuhdistuma on 20 ml/min (vaikea munuaisten vajaatoiminta), vastaava ennustettu tsoledronihapon puhdistuma on 37 %; ja potilaalla, jonka kreatiniinipuhdistuma on 50 ml/min (kohtalainen vajaatoiminta), vastaava ennustettu tsoledronihapon puhdistuma on 72 % arvosta, joka on potilaalla, jolla kreatiniinipuhdistuma on 84 ml/min. Farmakokinetiikasta vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (kreatiniinipuhdistuma < 30 ml/min) on vain vähän tietoa.
In vitro- tutkimuksessa tsoledronihapolla oli alhainen affiniteetti ihmisen veren sellulaarisiin komponentteihin (keskimääräinen veren ja plasman konsentraatioiden suhde oli 0,59 konsentraatioiden vaihteluvälin ollessa 30–5 000 ng/ml). Plasman proteiineihin sitoutuminen on vähäistä sitoutumattoman tsoledronihappo-osuuden vaihdellessa 60 %:sta 77 %:iin tsoledronihappopitoisuuksilla 2 ng/ml ja 2 000 ng/ml.
Erityisryhmät
Pediatriset potilaat
Rajalliset farmakokinetiikan tiedot vaikeaa synnynnäistä luutumisvajausta sairastavista lapsista viittaavat siihen, että tsoledronihapon farmakokinetiikka on 3–17-vuotiailla lapsilla samankaltainen kuin samanlaisia annoksia (mg/kg) käyttävillä aikuisilla. Ikä, paino, sukupuoli ja kreatiniinipuhdistuma eivät nähtävästi vaikuta systeemiseen tsoledronihappoaltistukseen.
Prekliiniset tiedot turvallisuudesta
Akuutti toksisuus
Suurin ei-letaali kerta-annos laskimoon oli hiirellä 10 mg/kg ja rotalla 0,6 mg/kg.
Subkrooninen ja krooninen toksisuus
Tsoledronihappo oli hyvin siedetty, kun sitä annettiin rotalle ihon alle ja koiralle laskimoon enimmillään 0,02 mg/kg vuorokaudessa neljän viikon ajan. Rotilla 0,001 mg/kg vuorokaudessa ihon alle ja koirilla 0,005 mg/kg kerran 2–3 vuorokauden välein laskimoon enimmillään 52 viikon ajan olivat myös hyvin siedettyjä.
Yleisin havainto tutkimuksissa toistuvilla annoksilla oli primaarin hohkaluun lisääntyminen kasvavien eläinten pitkien luiden metafyysialueilla lähes kaikilla annostasoilla osoituksena yhdisteen farmakologisesta luun resorptiota estävästä aktiivisuudesta.
Munuaisvaikutuksia koskevat turvallisuusmarginaalit olivat kapeat pitkäaikaisissa toistuvaa parenteraalista annostusta käyttäen tehdyissä eläintutkimuksissa, mutta kumulatiivinen haittavaikutukseton taso (NOAEL) kerta-antoa (1,6 mg/kg) ja toistuvaa antoa (0,06–0,6 mg/kg/vrk) käyttäen enimmillään kuukauden kestäneissä tutkimuksissa ei viitannut munuaisvaikutuksiin, kun käytettiin annoksia, jotka olivat vähintään ihmiselle tarkoitetun maksimaalisen hoitoannoksen suuruisia. Pitkäaikainen toistuva anto annoksina, jotka vastaavat ihmiselle tarkoitettua tsoledronihapon suurinta hoitoannosta, aiheutti toksisia vaikutuksia muissa elimissä, kuten maha-suolikanavassa, maksassa, pernassa ja keuhkoissa sekä laskimon punktiokohdassa.
Lisääntymistoksisuus
Tsoledronihappo oli teratogeeninen rotalla ≥ 0,2 mg/kg:n ihonalaisina annoksina. Vaikka teratogeenisuutta tai sikiötoksisuutta ei kaniinilla havaittu, emoon kohdistuvaa toksisuutta havaittiin. Rotilla havaittiin synnytyksen vaikeutumista pienimmällä annostasolla (0,01 mg/kg).
Mutageenisuus ja karsinogeenisuus
Mutageenisuuskokeiden perusteella tsoledronihappo ei ollut mutageeninen eivätkä karsinogeenisuustutkimukset viitanneet siihen, että tsoledronihappo olisi karsinogeeninen.
Farmaseuttiset tiedot
Apuaineet
- mannitoli (E421)
- natriumsitraatti (E331)
- injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa kalsiumia sisältävien liuosten kanssa eikä sitä saa sekoittaa tai antaa laskimoon muiden lääkevalmisteiden kanssa samalla infuusiolinjalla.
Kestoaika
Avaamaton injektiopullo: 2 vuotta.
Avaamaton pussi: 3 vuotta.
Avaamisen jälkeen:
Kemiallinen ja fysikaalinen säilyvyys on osoitettu 24 tunnin ajan 2–8 °C:ssa ja 25 °C:ssa.
Mikrobiologisista syistä infuusioneste on käytettävä heti. Jos liuosta ei käytetä heti, käytön aikainen säilytysaika ja olosuhteet ennen käyttöä ovat käyttäjän vastuulla eivätkä normaalisti saa ylittää 24 tuntia 2‑8 °C:ssa. Jääkaappikylmän liuoksen on annettava lämmetä huoneenlämpötilaan ennen antoa.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Lääkevalmisteen säilytysolosuhteet avaamisen jälkeen, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ZOLEDRONIC ACID STADA infuusioneste, liuos
4 mg/100 ml (L:ei) 100 ml (125,76 €)
PF-selosteen tieto
100 ml infuusionestettä on pakattu:
- kirkkaaseen tyypin I lasiseen injektiopulloon, jonka sisäpuoli on päällystetty piidioksidilla tai
- kirkkaaseen tyypin I borosilikaattilasiseen injektiopulloon tai
- muoviseen injektiopulloon, joka on valmistettu kirkkaasta, värittömästä syklo-olefiinikopolymeeristä (COC), ja suljettu tyypin I bromibutyylikumitulpalla ja alumiini-polypropeenirepäisykannella tai
- polypropeeni (PP) -pusseihin, joissa on polypropeenikorkit.
Zoledronic acid Stada 4 mg/100 ml infuusioneste, liuos toimitetaan pakkauksissa, jotka sisältävät:
1 injektiopullo
4 injektiopulloa
1 pussi
4 pussia
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas ja väritön liuos.
pH: 5,5–7,0
Osmolaalisuus: 0,27–0,33 Osmol/kg
Käyttö- ja käsittelyohjeet
Lisätietoja tsoledronihapon käsittelystä, mukaan lukien ohjeet pienennettyjen tsoledronihappoannosten valmistamisesta käyttövalmiin injektiopullon avulla, on kerrottu kohdassa Annostus ja antotapa.
Infuusio täytyy valmistaa aseptisen menetelmin. Vain kertakäyttöön.
Vain kirkas hiukkasia sisältämätön liuos, jonka väri ei ole muuttunut, on käyttökelpoinen.
Terveydenhoidon ammattilaiset eivät saa hävittää käyttämätöntä tsoledronihappoa tavalliseen viemäriin.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ZOLEDRONIC ACID STADA infuusioneste, liuos
4 mg/100 ml 100 ml
- Ei korvausta.
ATC-koodi
M05BA08
Valmisteyhteenvedon muuttamispäivämäärä
09.01.2025
Yhteystiedot
PL 1310, Puolikkotie 8, 02230 Espoo (käyntiosoite)
00101 Helsinki
0207 416 888