
ENTRESTO tabletti, kalvopäällysteinen 24/26 mg, 49/51 mg, 97/103 mg
Vaikuttavat aineet ja niiden määrät
Entresto 24 mg/26 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 24,3 mg sakubitriilia ja 25,7 mg valsartaania (sakubitriilin ja valsartaanin natriumsuolakompleksina).
Entresto 49 mg/51 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 48,6 mg sakubitriilia ja 51,4 mg valsartaania (sakubitriilin ja valsartaanin natriumsuolakompleksina).
Entresto 97 mg/103 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 97,2 mg sakubitriilia ja 102,8 mg valsartaania (sakubitriilin ja valsartaanin natriumsuolakompleksina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti)
Kliiniset tiedot
Käyttöaiheet
Aikuisten sydämen vajaatoiminta
Entresto on tarkoitettu aikuispotilaiden oireisen kroonisen sydämen vajaatoiminnan hoitoon, kun sairauteen liittyy alentunut ejektiofraktio (ks. kohta Farmakodynamiikka).
Pediatrinen sydämen vajaatoiminta
Entresto on tarkoitettu vähintään yhden vuoden ikäisten lasten ja nuorten oireisen kroonisen sydämen vajaatoiminnan hoitoon, kun sairauteen liittyy vasemman kammion systolinen toimintahäiriö (ks. kohta Farmakodynamiikka).
Annostus ja antotapa
Annostus
Yleisiä huomioitavia seikkoja
Entresto‑valmistetta ei pidä käyttää yhdessä angiotensiinikonvertaasin (ACE:n) estäjän tai angiotensiini II‑reseptorin salpaajan (ATR:n salpaajan) kanssa. Entresto‑valmisteen käyttö samanaikaisesti ACE:n estäjän kanssa suurentaa mahdollista angioedeeman riskiä, joten Entresto-hoito tulee aloittaa aikaisintaan 36 tuntia ACE:n estäjän käytön lopettamisen jälkeen (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Entresto‑valmisteen sisältämän valsartaanin hyötyosuus on suurempi kuin valsartaanin hyötyosuus käytettäessä muita markkinoilla olevia tablettimuotoisia valsartaanivalmisteita (ks. kohta Farmakokinetiikka).
Jos annos unohtuu, tulee potilaan ottaa seuraava annos sen normaalina ottoajankohtana.
Aikuisten sydämen vajaatoiminta
Entresto-valmisteen suositeltu aloitusannos on yksi 49 mg/51 mg:n tabletti kaksi kertaa vuorokaudessa, paitsi alla kuvatuissa tilanteissa. Tavoiteannoksen, yksi 97 mg/103 mg:n tabletti kahdesti vuorokaudessa, saavuttamiseksi annos on kaksinkertaistettava 2–4 viikon kuluessa, huomioiden lääkkeen siedettävyys potilaalla (ks. kohta Farmakodynamiikka).
Jos potilaalla ilmenee lääkkeen siedettävyyteen liittyviä ongelmia (systolinen verenpaine [SBP] ≤ 95 mmHg, oireinen hypotensio, hyperkalemia, munuaisten toimintahäiriö), suositellaan muiden samanaikaisessa käytössä olevien lääkevalmisteiden annosten säätämistä, Entresto-valmisteen annoksen tilapäistä pienentämistä tai Entresto-hoidon keskeyttämistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
PARADIGM-HF-tutkimuksessa Entresto-valmistetta annettiin yhdessä muiden sydämen vajaatoimintaan tarkoitettujen hoitojen kanssa, ACE:n estäjän tai toisen ATR:n salpaajan tilalla (ks. kohta Farmakodynamiikka). Kokemusta on vähän potilaista, jotka eivät käytä ACE:n estäjää tai ATR:n salpaajaa tai käyttävät edellä mainittuja lääkkeitä pienellä annostuksella, ja siksi näille potilaille suositellaan kaksi kertaa vuorokaudessa otettavaa 24 mg/26 mg:n aloitusannosta ja hidasta annoksen suurentamista (kaksinkertaistaminen 3–4 viikon välein) (ks. ”TITRATION” kohdassa Farmakodynamiikka).
Hoitoa ei pidä aloittaa potilaille, joiden seerumin kaliumpitoisuus on > 5,4 mmol/l tai systolinen verenpaine on < 100 mmHg (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Aloitusannosta 24 mg/26 mg kahdesti vuorokaudessa on harkittava potilaille, joiden systolinen verenpaine on välillä 100–110 mmHg.
Pediatrinen sydämen vajaatoiminta
Taulukossa 1 on esitetty pediatrisille potilaille suositellut annokset. Suositeltu annos otetaan suun kautta kahdesti vuorokaudessa. Annosta suurennetaan potilaan sietokyvyn mukaan 2–4 viikon välein tavoiteannokseen asti.
Entresto kalvopäällysteiset tabletit eivät sovellu lapsille, jotka painavat alle 40 kg. Näille potilaille on saatavilla Entresto rakeet.
Taulukko 1 Annostitraussuositukset
Potilaan paino | Annetaan kahdesti vuorokaudessa | |||
Puolitettu aloitusannos* | Aloitusannos | Keskitason annos | Tavoiteannos | |
< 40 kg painavat pediatriset potilaat | 0,8 mg/kg# | 1,6 mg/kg# | 2,3 mg/kg# | 3,1 mg/kg# |
40 – < 50 kg painavat pediatriset potilaat | 0,8 mg/kg# | 24 mg/26 mg | 49 mg/51 mg | 72 mg/78 mg |
≥ 50 kg painavat pediatriset potilaat | 24 mg/26 mg | 49 mg/51 mg | 72 mg/78 mg | 97 mg/103 mg |
*Aloitusannoksen puolittamista suositellaan potilaille, jotka eivät ole käyttäneet ACE:n estäjää tai ATR:n salpaajaa tai ovat käyttäneet edellä mainittuja lääkkeitä pienellä annostuksella, potilaille, joilla on munuaisten vajaatoiminta (glomerulusten laskennallinen suodatusnopeus [eGFR] < 60 ml/min/1,73 m2), ja potilaille, joilla on keskivaikea maksan vajaatoiminta (ks. kohta Erityisryhmät).
#Määrät 0,8 mg/kg, 1,6 mg/kg, 2,3 mg/kg ja 3,1 mg/kg viittaavat sakubitriilin ja valsartaanin yhteenlaskettuun määrään. Määrä annetaan rakeina.
Potilaille, jotka eivät käytä ACE:n estäjää tai ATR:n salpaajaa tai käyttävät edellä mainittuja lääkkeitä pienellä annostuksella, suositellaan aloitusannoksen puolittamista. 40 – < 50 kg painaville pediatrisille potilaille suositellaan aloitusannosta 0,8 mg/kg kahdesti vuorokaudessa (rakeina). Hoidon aloittamisen jälkeen annosta suurennetaan normaaliin aloitusannokseen taulukossa 1 esitettyjen annostitraussuositusten mukaisesti 3–4 viikon välein.
Esimerkiksi 25 kg painavan pediatrisen potilaan, joka ei ole aiemmin käyttänyt ACE:n estäjää, on aloitettava puolitetulla aloitusannoksella 20 mg (25 kg ×0,8 mg/kg) kahdesti vuorokaudessa, rakeina annettuna. Pyöristettynä lähimpään kokonaisten kapseleiden määrään, tämä vastaa kahta 6 mg/6 mg sakubitriili/valsartaani-kapselia kahdesti vuorokaudessa.
Hoitoa ei pidä aloittaa potilaille, joiden seerumin kaliumpitoisuus on > 5,3 mmol/l tai systolinen verenpaine on alle iänmukaisen 5. persentiilin. Jos potilaalla ilmenee lääkkeen siedettävyyteen liittyviä ongelmia (systolinen verenpaine alle iänmukaisen 5. persentiilin, oireinen hypotensio, hyperkalemia, munuaisten toimintahäiriö), suositellaan muiden samanaikaisessa käytössä olevien lääkevalmisteiden annosten säätämistä, Entresto‑valmisteen annoksen tilapäistä pienentämistä tai Entresto‑hoidon keskeyttämistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Erityisryhmät
Iäkkäät
Annos on säädettävä iäkkäiden potilaiden kohdalla munuaistoiminnan mukaan.
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilaille, joilla on lievä munuaisten vajaatoiminta (eGFR 60–90 ml/min/1,73 m2).
Keskivaikeaa munuaisten vajaatoimintaa (eGFR 30–60 ml/min/1,73 m2) sairastavien potilaiden hoidossa on harkittava aloitusannoksen puolittamista. Koska vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min/1,73 m2) sairastavista potilaista on vain hyvin vähän kliinistä kokemusta (ks. kohta Farmakodynamiikka), Entresto-valmisteen käytössä tällaisten potilaiden hoidossa on noudatettava varovaisuutta ja aloitusannoksen puolittamista suositellaan. 40 – < 50 kg painaville pediatrisille potilaille suositellaan aloitusannosta 0,8 mg/kg kahdesti vuorokaudessa (rakeina). Hoidon aloittamisen jälkeen annosta suurennetaan annostitraussuositusten mukaisesti 2–4 viikon välein.
Loppuvaiheen munuaissairaudesta kärsivien potilaiden hoidosta ei ole kokemusta, eikä Entresto-valmisteen käyttöä näin ollen suositella.
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen annettaessa Entresto-valmistetta potilaille, joilla on lievä maksan vajaatoiminta (Child–Pughin luokitus A).
Kliinistä kokemusta keskivaikeaa maksan vajaatoimintaa (Child–Pughin luokitus B) sairastavien potilaiden hoidosta ja sellaisten potilaiden hoidosta, joiden aspartaattiaminotransferaasiarvot (ASAT) / alaniiniaminotransferaasiarvot (ALAT) ovat yli kaksinkertaiset suhteessa viitearvojen ylärajaan, on vain rajallisesti. Entresto-valmisteen käytössä tällaisten potilaiden hoidossa on noudatettava varovaisuutta ja aloitusannoksen puolittamista suositellaan (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). 40 – < 50 kg painaville pediatrisille potilaille suositellaan aloitusannosta 0,8 mg/kg kahdesti vuorokaudessa (rakeina). Hoidon aloittamisen jälkeen annosta suurennetaan annostitraussuositusten mukaisesti 2–4 viikon välein.
Entresto-valmisteen käyttö vaikeaa maksan vajaatoimintaa, biliaarista kirroosia tai kolestaasia (Child–Pughin luokitus C) sairastaville potilaille on vasta-aiheista (ks. kohta Vasta-aiheet).
Pediatriset potilaat
Entresto-valmisteen turvallisuutta ja tehoa alle 1 vuoden ikäisten lasten hoidossa ei ole varmistettu. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdassa Farmakodynamiikka, ei voida antaa suosituksia annostuksesta.
Antotapa
Suun kautta.
Entresto-valmiste voidaan ottaa joko aterian yhteydessä tai tyhjään mahaan (ks. kohta Farmakokinetiikka). Tablettien kanssa on juotava lasillinen vettä. Tabletin jakamista tai murskaamista ei suositella.
Vasta-aiheet
- Yliherkkyys vaikuttaville aineille tai kohdassa Apuaineet mainituille apuaineille.
- Samanaikainen ACE:n estäjän käyttö (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset). Entresto-valmiste tulee antaa aikaisintaan 36 tuntia ACE:n estäjän käytön lopettamisen jälkeen.
- Tiedossa oleva aiempaan ACE:n estäjän tai ATR:n salpaajan käyttöön liittynyt angioedeema (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Perinnöllinen tai idiopaattinen angioedeema (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Samanaikainen käyttö aliskireeniä sisältävien lääkevalmisteiden kanssa potilaille, joilla on diabetes mellitus tai munuaisten vajaatoiminta (eGFR < 60 ml/min/1,73 m2) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
- Vaikea maksan vajaatoiminta, biliaarinen kirroosi tai kolestaasi (ks. kohta Annostus ja antotapa).
- Raskauden toinen ja kolmas kolmannes (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Reniini-angiotensiini-aldosteronijärjestelmän (RAA-järjestelmä) kaksoissalpaus
- Angioedeemariskin suurentumisen vuoksi sakubitriili/valsartaani-valmisteen anto yhdistelmänä ACE:n estäjän kanssa on vasta-aiheista (ks. kohta Vasta-aiheet). Sakubitriili/valsartaani-hoito tulee aloittaa aikaisintaan 36 tuntia ACE:n estäjän viimeisen annoksen ottamisen jälkeen. Jos sakubitriili/valsartaani-hoito lopetetaan, ACE:n estäjän käyttö tulee aloittaa aikaisintaan 36 tuntia viimeisen sakubitriili/valsartaani-annoksen ottamisen jälkeen. (ks. kohdat Annostus ja antotapa, Vasta-aiheet ja Yhteisvaikutukset).
- Sakubitriili/valsartaani-valmisteen antamista yhdistelmänä suorien reniinin estäjien, kuten aliskireenin, kanssa ei suositella (ks. kohta Yhteisvaikutukset). Sakubitriili/valsartaani-valmisteen anto yhdistelmänä aliskireeniä sisältävien lääkevalmisteiden kanssa potilaille, joilla on diabetes mellitus tai munuaisten vajaatoiminta (eGFR < 60 ml/min/1,73 m2), on vasta-aiheista (ks. kohdat Vasta-aiheet ja Yhteisvaikutukset).
- Entresto-valmiste sisältää valsartaania ja siksi sitä ei pidä antaa samanaikaisesti toisen ATR:n salpaajaa sisältävän lääkevalmisteen kanssa (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Hypotensio
Hoitoa ei pidä aloittaa, jos potilaan systolinen verenpaine ei ole aikuispotilailla ≥ 100 mmHg tai pediatrisilla potilailla ≥ iänmukainen 5. persentiili. Tutkimuksiin ei ole osallistunut potilaita, joiden systolinen verenpaine on ollut alle näiden arvojen (ks. kohta Farmakodynamiikka). Kliinisissä tutkimuksissa symptomaattista hypotensiota on ilmoitettu sakubitriili/valsartaani-valmistetta käyttäneillä aikuispotilailla (ks. kohta Haittavaikutukset) ja erityisesti vähintään 65-vuotiailla potilailla, joilla on munuaissairaus ja potilailla, joilla on matala systolinen verenpaine (< 112 mmHg). Sakubitriili/valsartaani-hoitoa aloitettaessa tai annostitrauksen yhteydessä on seurattava jatkuvasti potilaan verenpainetta. Hypotension ilmetessä suositellaan tilapäistä sakubitriili/valsartaani-annoksen pienentämistä tai valmisteen käytön keskeyttämistä (ks. kohta Annostus ja antotapa). Diureettien ja samanaikaisesti käytettävien verenpainelääkkeiden annoksen muuttamista ja hypotension muiden syiden (esim. hypovolemian) hoitoa on harkittava. Symptomaattinen hypotensio kehittyy todennäköisemmin potilaalle, jonka nestetilavuus on vähentynyt esimerkiksi diureettilääkityksen, ruokavalion suolarajoituksen, ripulin tai oksentelun vuoksi. Natriumvaje ja/tai vähentynyt nestetilavuus on korjattava ennen sakubitriili/valsartaani-hoidon aloittamista, mutta korjaustoimet on harkittava huolellisesti ottaen huomioon tilavuusylikuormituksen riski.
Heikentynyt munuaisten toiminta
Tutkittaessa sydämen vajaatoimintaa sairastavia potilaita, tutkimukseen tulee aina sisällyttää munuaisten toiminnan arviointi. Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla on suurempi hypotensioriski (ks. kohta Annostus ja antotapa). Kliinistä kokemusta on erittäin vähän potilaista, joilla on vaikea munuaisten vajaatoiminta (arvioitu GFR < 30 ml/min/1,73 m2), ja näillä potilailla hypotensioriski voi olla suurin (ks. kohta Annostus ja antotapa). Kokemusta loppuvaiheen munuaissairaudesta kärsivien potilaiden hoidosta ei ole lainkaan, joten sakubitriili/valsartaani-valmisteen käyttöä ei suositella.
Munuaisten vajaatoiminta
Sakubitriili/valsartaani-valmisteen käytön yhteydessä munuaisten toiminta saattaa heikentyä. Kuivuminen tai samanaikainen tulehduskipulääkkeiden (NSAID-lääkkeet) käyttö saattaa suurentaa riskiä edelleen (ks. kohta Yhteisvaikutukset). Annoksen pienentämistä on harkittava potilailla, joiden munuaisten toiminta heikkenee kliinisesti merkittävästi.
Hyperkalemia
Hoitoa ei pidä aloittaa aikuispotilaille, joiden seerumin kaliumpitoisuus on > 5,4 mmol/l, tai pediatrisille potilaille, joiden seerumin kaliumpitoisuus on > 5,3 mmol/l. Sakubitriili/valsartaani-valmisteen käytön yhteydessä hyperkalemian riski saattaa suurentua, mutta myös hypokalemiaa voi ilmetä (ks. kohta Haittavaikutukset). Seerumin kaliumarvojen seuranta on suositeltavaa, varsinkin jos potilaalla on riskitekijöitä, kuten munuaisten vajaatoiminta, diabetes mellitus tai aldosteronin vajaaeritystä, tai jos potilas saa ravinnosta runsaasti kaliumia tai käyttää mineralokortikoidiantagonisteja (ks. kohta Annostus ja antotapa). Jos potilaalla ilmenee kliinisesti merkittävää hyperkalemiaa, samanaikaisesti käytettävien lääkevalmisteiden annoksen muuttamista tai tilapäistä annoksen pienentämistä tai hoidon keskeyttämistä suositellaan. Jos potilaan seerumin kaliumarvo on > 5,4 mmol/l, hoidon lopettamista on harkittava.
Angioedeema
Sakubitriili/valsartaani-hoitoa saaneilla potilailla on ilmoitettu angioedeemaa. Jos potilaalle kehittyy angioedeema, sakubitriili/valsartaani-hoito on keskeytettävä välittömästi ja potilasta on hoidettava ja seurattava asianmukaisesti, kunnes oireet ja merkit ovat kadonneet täysin ja pysyvästi. Hoitoa ei saa aloittaa uudelleen. Kun potilaalla on ollut vahvistettu kasvoihin ja huuliin rajoittunut angioedeema, se on yleensä hävinnyt ilman hoitoa. Antihistamiineista on kuitenkin ollut apua oireiden lievittämisessä.
Angioedeema, johon liittyy kurkunpään turpoaminen, saattaa johtaa kuolemaan. Jos angioedeema vaikuttaa kieleen, äänihuulten alueeseen tai kurkunpäähän, se todennäköisesti ahtauttaa hengitysteitä. Tällöin potilaalle on annettava nopeasti asianmukaista hoitoa, kuten adrenaliiniliuosta 1 mg/1 ml (0,3–0,5 ml) ja/tai on ryhdyttävä toimenpiteisiin, jotka ovat tarpeen varmistamaan, että potilaan hengitystiet pysyvät auki.
Potilaita, joilla oli aiemmin ilmennyt angioedeemaa, ei tutkittu. Näiden potilaiden hoidossa sakubitriili/valsartaani-valmisteella suositellaan varovaisuutta, koska heillä saattaa olla suurentunut angioedeemariski. Sakubitriili/valsartaani-valmisteen anto on vasta-aiheista potilaille, joilla on aiemmin ilmennyt angioedeemaa ACE:n estäjän tai ATR:n salpaajan käytön yhteydessä, tai joilla on perinnöllinen tai idiopaattinen angioedeema (ks. kohta Vasta-aiheet).
Tummaihoisilla potilailla on lisääntynyt alttius angioedeeman kehittymiselle (ks. kohta Haittavaikutukset).
Suoliston angioedeemasta on saatu ilmoituksia potilailla, joita on hoidettu angiotensiini II -reseptorin antagonisteilla mukaan lukien valsartaani (ks. kohta Haittavaikutukset). Näillä potilailla ilmeni vatsakipua, pahoinvointia, oksentelua ja ripulia. Oireet hävisivät angiotensiini II -reseptorin antagonistien käytön lopettamisen jälkeen. Jos potilaalla diagnosoidaan suoliston angioedeema, sakubitriili/valsartaanin käyttö on lopetettava ja aloitettava asianmukainen seuranta, kunnes oireet ovat täysin hävinneet.
Munuaisvaltimon ahtauma
Sakubitriili/valsartaani saattaa suurentaa veren urea- ja seerumin kreatiniinipitoisuuksia potilailla, joilla on molemminpuolinen tai toispuolinen munuaisvaltimon ahtauma. Varovaisuutta on noudatettava hoidettaessa potilaita, joilla on munuaisvaltimon ahtauma, ja munuaisten toiminnan seuraaminen on suositeltavaa.
Potilaat, joiden New York Heart Association (NYHA) -toimintakykyluokka on IV
Varovaisuutta on noudatettava aloitettaessa sakubitriili/valsartaani-hoito potilaille, joiden NYHA-toimintakykyluokka on IV, koska kliinistä kokemusta tästä potilasryhmästä on vähän.
B-tyypin natriureettinen peptidi (BNP)
BNP ei sovellu sydämen vajaatoiminnan biomerkkiaineeksi sakubitriili/valsartaani-hoitoa saaville potilaille, koska se on neprilysiinin substraatti (ks. kohta Farmakodynamiikka).
Potilaat, joilla on maksan vajaatoiminta
Kliinistä kokemusta on rajallisesti keskivaikeaa maksan vajaatoimintaa (Child–Pughin luokitus B) sairastavien potilaiden tai sellaisten potilaiden hoidosta, joiden ASAT/ALAT-arvot ovat yli kaksinkertaiset suhteessa viitearvojen ylärajaan. Näiden potilaiden altistuminen lääkkeelle voi olla tavallista suurempi ja käytön turvallisuutta ei ole osoitettu. Tällaisten potilaiden hoidossa suositellaan noudattamaan varovaisuutta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka). Sakubitriili/valsartaani-hoito on vasta-aiheinen potilaille, joilla on vaikea maksan vajaatoiminta, biliaarinen kirroosi tai kolestaasi (Child–Pughin luokitus C) (ks. kohdat Vasta-aiheet).
Psyykkiset häiriöt
Sakubitriili/valsartaanin käyttöön on yhdistetty psyykkisiä oireita kuten hallusinaatioita, vainoharhaisuutta ja unihäiriöitä, jotka ovat yhteydessä psykoottisiin tiloihin. Mikäli potilas kokee tällaisia oireita, tulee harkita sakubitriili/valsartaani-hoidon lopettamista.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 97 mg/103 mg:n annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Yhteisvaikutukset, joiden perusteella käyttö on vasta-aiheista
ACE:n estäjät
Sakubitriili/valsartaani-valmisteen käyttö samanaikaisesti ACE:n estäjien kanssa on vasta-aiheista, sillä neprilysiinin ja ACE:n samanaikainen esto saattaa lisätä angioedeeman riskiä. Sakubitriili/valsartaani-hoito tulee aloittaa aikaisintaan 36 tuntia ACE:n estäjän viimeisen annoksen ottamisen jälkeen. ACE:n estäjän käyttö tulee aloittaa aikaisintaan 36 tuntia viimeisen sakubitriili/valsartaani-annoksen ottamisen jälkeen (ks. kohdat Annostus ja antotapa ja Vasta-aiheet).
Aliskireeni
Sakubitriili/valsartaani-valmisteen käyttö samanaikaisesti aliskireeniä sisältävien lääkevalmisteiden kanssa on vasta-aiheista potilaille, joilla on diabetes mellitus tai munuaisten vajaatoiminta (eGFR < 60 ml/min/1,73 m2) (ks. kohta Vasta-aiheet). Sakubitriili/valsartaani-valmisteen antoa yhdistelmänä suorien reniinin estäjien, kuten aliskireenin, kanssa ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Sakubitriili/valsartaani-lääkkeen ja aliskireenin yhdistelmän käyttöön voi mahdollisesti liittyä useammin haittavaikutuksia, kuten hypotensiota, hyperkalemiaa ja heikentynyttä munuaisten toimintaa (akuutti munuaisten vajaatoiminta mukaan lukien) (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Yhteisvaikutukset, joiden perusteella samanaikaista käyttöä ei suositella
Sakubitriili/valsartaani-valmiste sisältää valsartaania ja siksi sitä ei pidä antaa samanaikaisesti muuta ATR:n salpaajaa sisältävän lääkevalmisteen kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varovaisuutta edellyttävät yhteisvaikutukset
OATP1B1:n ja OATP1B3:n substraatit, esim. statiinit
In vitro -tulokset osoittavat, että sakubitriili estää OATP1B1- ja OATP1B3-kuljettajaproteiineja. Sen vuoksi Entresto saattaa lisätä OATP1B1:n ja OATP1B3:n substraattien, kuten statiinien, systeemistä altistusta. Sakubitriili/valsartaani-valmisteen ja atorvastatiinin samanaikainen antaminen suurensi atorvastatiinin ja sen metaboliittien Cmax-arvon enimmillään kaksinkertaiseksi ja AUC-arvon enimmillään1,3-kertaiseksi. Sakubitriili/valsartaani-valmisteen ja statiinien samanaikaisessa käytössä on noudatettava varovaisuutta. Entresto-valmisteen ja simvastatiinin samanaikaisen antamisen yhteydessä ei havaittu kliinisesti merkityksellisiä yhteisvaikutuksia.
PDE5:n estäjät, kuten sildenafiili
Kun vakaassa tilassa oleville hypertensiopotilaille annettiin sakubitriili/valsartaani-valmisteen lisäksi kerta-annos sildenafiilia, verenpaine laski huomattavasti enemmän kuin pelkästään sakubitriili/valsartaani-valmisteella. Siksi on noudatettava varovaisuutta, kun sakubitriili/valsartaani-hoitoa saaville potilaille aloitetaan sildenafiilihoito tai hoito jollakin muulla PDE5:n estäjällä.
Kalium
Kaliumia säästävien diureettien (triamtereenin tai amiloridin), mineralokortikoidiantagonistien (kuten spironolaktonin tai eplerenonin), kaliumlisien, kaliumia sisältävien suolan korvikkeiden tai muiden lääkkeiden (kuten hepariinin) samanaikainen käyttö saattaa suurentaa seerumin kalium- ja kreatiniinipitoisuuksia. Seerumin kaliumarvojen seuranta on suositeltavaa, jos sakubitriili/valsartaani-valmistetta annetaan samanaikaisesti näiden lääkeaineiden kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Steroideihin kuulumattomat tulehduskipulääkkeet (NSAID-lääkkeet), mukaan lukien selektiiviset syklo-oksigenaasi 2:n (COX-2) estäjät
Sakubitriili/valsartaani-valmisteen ja NSAID-lääkkeiden samanaikainen käyttö saattaa lisätä munuaisten toiminnan heikkenemisen riskiä iäkkäillä potilailla, potilailla, joiden nestetilavuus on vähentynyt (kuten diureetteja käyttävillä potilailla), tai potilailla, joiden munuaisten toiminta on heikentynyt. Sen vuoksi suositellaan munuaisten toiminnan seurantaa, kun NSAID-lääkkeitä samanaikaisesti käyttäville potilaille aloitetaan sakubitriili/valsartaani-hoito tai sitä muutetaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Litium
Litiumin ja ACE:n estäjien tai ATR:n salpaajien, mukaan lukien sakubitriili/valsartaanin, samanaikaisen käytön aikana on ilmoitettu ohimenevää seerumin litiumpitoisuuksien suurenemista ja toksisuutta. Siksi tämän yhdistelmän käyttöä ei suositella. Seerumin litiumpitoisuuden huolellista seurantaa suositellaan, jos yhdistelmän käyttö on tarpeen. Jos käytetään myös diureettia, litiumtoksisuuden riski saattaa lisääntyä entisestään.
Furosemidi
Sakubitriili/valsartaani-valmisteen ja furosemidin samanaikainen anto ei vaikuttanut sakubitriili/valsartaani-valmisteen farmakokinetiikkaan, mutta pienensi furosemidin Cmax-arvoa 50 % ja AUC-arvoa 28 %. Vaikka virtsan volyymissä ei todettu merkityksellistä muutosta, natriumin erittyminen virtsaan väheni 4 ja 24 tunnin kuluessa samanaikaisesta annosta. Furosemidin keskimääräinen vuorokausiannos pysyi samana lähtötilanteesta PARADIGM-HF-tutkimuksen loppuun sakubitriili/valsartaani-valmistetta saaneilla potilailla.
Nitraatit, esim. nitroglyseriini
Sakubitriili/valsartaani-valmisteen ja laskimoon annetun nitroglyseriinin välillä ei todettu yhteisvaikutuksia verenpaineen alenemisen suhteen. Nitroglyseriini ja sakubitriili/valsartaani-valmisteen samanaikaiseen antoon liittyi 5 sydämen lyönnin ero sykkeessä (per minuutti) pelkkään nitroglyseriiniin verrattuna. Sakubitriili/valsartaani-valmisteen samanaikainen käyttö kielen alle, suun kautta tai ihon kautta annosteltavien nitraattien kanssa voi aiheuttaa samanlaisen vaikutuksen sydämen sykkeeseen. Yleensä annosmuutoksiin ei ole tarvetta.
OATP- ja MRP2-kuljettajaproteiinit
Sakubitriilin aktiivinen metaboliitti (LBQ657) ja valsartaani ovat OATP1B1:n, OATP1B3:n, OAT1:n ja OAT3:n substraatteja; valsartaani on myös MRP2:n substraatti. Sakubitriili/valsartaani-valmisteen samanaikainen käyttö OATP1B1:n, OATP1B3:n tai OAT3:n estäjien (kuten rifampisiinin, siklosporiinin), OAT1:n estäjien (kuten tenofoviirin, sidofoviirin) tai MRP2:n estäjien (kuten ritonaviirin) kanssa saattaa sen vuoksi lisätä systeemistä altistusta LBQ657:lle tai valsartaanille. Asianmukaista huolellisuutta on noudatettava, kun aloitetaan tai lopetetaan tällaisten lääkevalmisteiden samanaikainen käyttö.
Metformiini
Sakubitriili/valsartaani-valmisteen ja metformiinin samanaikainen anto pienensi sekä metformiinin Cmax- että AUC-arvoa 23 %. Näiden löydösten kliinistä merkitystä ei tunneta. Potilaan kliininen tila on sen vuoksi arvioitava aloitettaessa sakubitriili/valsartaani-hoitoa metformiinia saaville potilaille.
Ei merkittäviä yhteisvaikutuksia
Sakubitriili/valsartaani-valmisteen käytön aikana ei havaittu kliinisesti merkityksellisiä yhteisvaikutuksia, kun sitä annettiin samanaikaisesti digoksiinin, varfariinin, hydroklooritiatsidin, amlodipiinin, omepratsolin, karvedilolin tai levonorgestreelin ja etinyyliestradiolin yhdistelmän kanssa.
Raskaus ja imetys
Raskaus
Sakubitriili/valsartaani-valmisteen käyttöä ei suositella ensimmäisen raskauskolmanneksen aikana, ja valmisteen käyttö on vasta-aiheista toisen ja kolmannen raskauskolmanneksen aikana (ks. kohta Vasta-aiheet).
Valsartaani
Epidemiologisten tutkimusten tulokset viittaavat siihen, että altistuminen ACE:n estäjille ensimmäisen raskauskolmanneksen aikana lisää sikiön epämuodostumien riskiä. Tulokset eivät kuitenkaan ole vakuuttavia, mutta pientä riskin suurenemista ei voida sulkea pois. ATR:n salpaajien käyttöön liittyvästä riskistä ei ole vertailevien epidemiologisten tutkimusten tuloksia, mutta näiden lääkkeiden käyttöön voi liittyä sama riski kuin ACE:n estäjiin. Jos ATR:n salpaajia käyttävä nainen aikoo tulla raskaaksi, hänelle on vaihdettava muu, raskauden aikana turvalliseksi todettu verenpainelääkitys, ellei ATR:n salpaajien käyttöä pidetä välttämättömänä. Kun raskaus todetaan, ATR:n salpaajien käyttö on lopetettava heti, ja tarvittaessa on aloitettava muu lääkitys. Tiedetään, että altistuminen ATR:n salpaajille toisen ja kolmannen raskauskolmanneksen aikana on haitallista sikiön kehitykselle (munuaisten toiminnan heikkeneminen, lapsiveden määrän pieneneminen, kallon luutumisen hidastuminen) ja vastasyntyneen kehitykselle (munuaisten toiminnan pettäminen, hypotensio, hyperkalemia).
Jos sikiö on toisen ja kolmannen raskauskolmanneksen aikana altistunut ATR:n salpaajille, suositellaan sikiölle tehtäväksi munuaisten toiminnan ja kallon ultraäänitutkimus. Imeväisikäisiä, joiden äiti on käyttänyt ATR:n salpaajia, on seurattava huolellisesti hypotension varalta (ks. kohta Vasta-aiheet).
Sakubitriili
Sakubitriilin käytöstä raskaana olevien naisten hoidossa ei ole kokemusta. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Sakubitriili/valsartaani
Sakubitriili/valsartaani-valmisteen käytöstä raskaana olevien naisten hoidossa ei ole kokemusta. Sakubitriili/valsartaani-valmisteella suoritetuissa eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imetys
Rajalliset tiedot osoittavat, että sakubitriili ja sen aktiivinen metaboliitti LBQ657 erittyvät ihmisillä äidinmaitoon hyvin pieninä määrinä, ja imeväisen suhteellinen annos on arviolta 0,01 % sakubitriilia ja 0,46 % aktiivista metaboliittia LBQ657, kun sakubitriili/valsartaani-valmistetta annetaan imettäville naisille annoksella 24 mg/26 mg kahdesti vuorokaudessa. Samoissa tiedoissa valsartaani oli havaitsemisrajan alapuolella. Sakubitriilin/valsartaanin vaikutuksista imetettävään vauvaan ei ole riittävästi tietoja. Entresto-valmisteen käyttöä ei suositella imettäville naisille imetettävään vauvaan kohdistuvien haittavaikutusten mahdollisen riskin vuoksi.
Hedelmällisyys
Sakubitriili/valsartaani-valmisteen vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Koiras- ja naarasrotilla tehdyissä tutkimuksissa ei osoitettu hedelmällisyyden heikkenemistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Sakubitriili/valsartaani-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Ajettaessa tai koneita käytettäessä on otettava huomioon, että valmiste voi joskus aiheuttaa heitehuimausta tai väsymystä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kaikista yleisimmin raportoidut haittavaikutukset aikuisilla sakubitriili/valsartaani-hoidon aikana ovat olleet hypotensio (17,6 %), hyperkalemia (11,6 %) ja munuaisten vajaatoiminta (10,1 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Angioedeemaa on raportoitu sakubitriili/valsartaani-valmistetta saaneilla potilailla (0,5 %) (ks. kohta Valikoitujen haittavaikutusten kuvaus).
Haittavaikutustaulukko
Haittavaikutukset on järjestetty elinjärjestelmäluokituksen mukaisesti ja esiintymistiheyden mukaan yleisimmistä alkaen seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2 Luettelo haittavaikutuksista
| Elinjärjestelmä | Suositeltu termi | Yleisyysluokka |
| Veri ja imukudos | Anemia | Yleinen |
| Immuunijärjestelmä | Yliherkkyys | Melko harvinainen |
| Aineenvaihdunta ja ravitsemus | Hyperkalemia* | Hyvin yleinen |
| Hypokalemia | Yleinen | |
| Hypoglykemia | Yleinen | |
| Hyponatremia | Melko harvinainen | |
| Psyykkiset häiriöt | Hallusinaatiot** | Harvinainen |
| Unihäiriöt | Harvinainen | |
| Vainoharhaisuus | Hyvin harvinainen | |
| Hermosto | Heitehuimaus | Yleinen |
| Päänsärky | Yleinen | |
| Pyörtyminen | Yleinen | |
| Asentohuimaus | Melko harvinainen | |
| Myoklonus | Tuntematon | |
| Kuulo ja tasapainoelin | Kiertohuimaus | Yleinen |
| Verisuonisto | Hypotensio* | Hyvin yleinen |
| Ortostaattinen hypotensio | Yleinen | |
| Hengityselimet, rintakehä ja välikarsina | Yskä | Yleinen |
| Ruoansulatuselimistö | Ripuli | Yleinen |
| Pahoinvointi | Yleinen | |
| Gastriitti | Yleinen | |
| Suoliston angioedeema | Hyvin harvinainen | |
| Iho ja ihonalainen kudos | Kutina | Melko harvinainen |
| Ihottuma | Melko harvinainen | |
| Angioedeema* | Melko harvinainen | |
| Munuaiset ja virtsatiet | Munuaistoiminnan huononeminen* | Hyvin yleinen |
| Munuaisten vajaatoiminta (munuaisten vajaatoiminta, munuaisten äkillinen vajaatoiminta) | Yleinen | |
| Yleisoireet ja antopaikassa todettavat haitat | Väsymys | Yleinen |
| Voimattomuus | Yleinen |
* Ks. kohta Valikoitujen haittavaikutusten kuvaus.
** Mukaan lukien auditiiviset ja visuaaliset hallusinaatiot
Valikoitujen haittavaikutusten kuvaus
Angioedeema
Angioedeemaa on ilmoitettu sakubitriili/valsartaani-valmistetta saaneilla potilailla. PARADIGM-HF-tutkimuksessa angioedeemaa raportoitiin 0,5 %:lla sakubitriili/valsartaani-valmistetta saaneista potilaista ja 0,2 %:lla enalapriilia saaneista potilaista. Angioedeeman ilmaantuvuus oli suurempi mustaihoisilla sakubitriili/valsartaani-valmistetta saaneilla (2,4 %) ja enalapriilia saaneilla (0,5 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hyperkalemia ja kaliumpitoisuus seerumissa
PARADIGM‑HF-tutkimuksessa sakubitriili/valsartaani-valmistetta saaneista potilaista 11,6 %:lla ilmoitettiin hyperkalemiaa ja 19,7 %:lla potilaista oli yli 5,4 mmol/l:n seerumin kaliumpitoisuus, kun vastaavat luvut enalapriilihoitoa saaneilla potilailla olivat 14,0 % ja 21,1 %.
Verenpaine
PARADIGM-HF-tutkimuksessa sakubitriili/valsartaani-valmistetta saaneista potilaista 17,6 %:lla raportoitiin hypotensiota ja 4,76 %:lla kliinisesti merkitsevää alhaista systolista verenpainetta (< 90 mmHg ja > 20 mmHg:n alenema lähtötasosta). Vastaavat luvut enalapriilihoitoa saaneilla potilailla olivat 11,9 % ja 2,67 %.
Munuaisten vajaatoiminta
PARADIGM-HF-tutkimuksessa sakubitriili/valsartaani-valmistetta saaneista potilaista 10,1 %:lla raportoitiin munuaisten vajaatoimintaa, kun vastaava luku enalapriilihoitoa saaneilla potilailla oli 11,5 %.
Pediatriset potilaat
Sakubitriili/valsartaani‑hoidon turvallisuutta verrattuna enalapriiliin arvioitiin 52 viikkoa kestäneessä, satunnaistetussa, aktiivikontrolloidussa PANORAMA‑HF‑tutkimuksessa, johon osallistui 375 sydämen vajaatoimintaa sairastavaa pediatrista potilasta (ikä 1 kk – < 18 v). Pitkäkestoiseen, avoimeen jatkotutkimukseen (PANORAMA-HF OLE) osallistuneiden 215 potilaan hoidon keston mediaani oli 2,5 vuotta ja potilaita hoidettiin korkeintaan 4,5 vuoden ajan. Turvallisuusprofiili oli molemmissa tutkimuksissa samankaltainen kuin aikuispotilailla. Turvallisuustietoa saatiin vain vähän sellaisilta potilailta, joiden ikä oli 1 kk – < 1 v.
Saatavilla on vain vähän turvallisuustietoa sellaisilta pediatrisilta potilailta, jotka sairastavat keskivaikeaa maksan vajaatoimintaa tai keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostukseen liittyvää tietoa ihmisistä on vähän. Terveillä vapaaehtoisilla aikuisilla tutkittiin kerta-annoksena annettua 583 mg sakubitriilia ja 617 mg valsartaania sisältävää annosta sekä toistuvia 437 mg sakubitriilia ja 463 mg valsartaania sisältäviä annoksia (14 päivää) ja ne siedettiin hyvin.
Yliannostuksen todennäköisin oire on hypotensio, joka johtuu sakubitriili/valsartaani-valmisteen verenpainetta alentavista vaikutuksista. Potilaalle on annettava oireenmukaista hoitoa.
Lääkevalmiste ei todennäköisesti eliminoidu hemodialyysissä, koska se sitoutuu voimakkaasti proteiineihin (ks. kohta Farmakokinetiikka).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: reniini-angiotensiinijärjestelmään vaikuttavat lääkeaineet; angiotensiini II -reseptorin salpaajat (ATR-salpaajat), muut yhdistelmävalmisteet, ATC-koodi: C09DX04
Vaikutusmekanismi
Sakubitriili/valsartaani-valmisteen vaikutusmekanismi perustuu sen toimintaan angiotensiinireseptorin ja neprilysiinin estäjänä, sillä sen sisältämän sakubitriiliaihiolääkkeen aktiivinen metaboliitti LBQ657 estää neprilysiiniä (neutraaliendopeptidaasi; NEP) ja samanaikaisesti valsartaani salpaa tyypin 1 angiotensiini II (AT1) -reseptoria. Sakubitriili/valsartaani-valmisteen toisiaan täydentävät kardiovaskulaariset hyödyt sydämen vajaatoimintaa sairastavilla potilailla johtuvat siitä, että LBQ657 lisää neprilysiinin pilkkomia peptidejä, kuten natriureettisia peptidejä (NP) ja samanaikaisesti valsartaani estää angiotensiini II:n vaikutuksia. Natriureettiset peptidit vaikuttavat aktivoimalla membraaniin sitoutuneita guanylyylisyklaasiin kytkettyjä reseptoreja, minkä seurauksena toisiolähetin, syklisen guanosiinimonofosfaatin (cGMP), pitoisuudet suurenevat, mikä saattaa johtaa vasodilataatioon, natriureesiin, diureesiin, antihypertrofisiin ja antifibroottisiin vaikutuksiin. Lisäksi glomerulusten suodatusnopeus suurenee, munuaisten verenkierto lisääntyy, reniinin ja aldosteronin vapautuminen estyy ja sympaattinen aktiivisuus vähenee.
Valsartaani estää angiotensiini II:n haitallisia kardiovaskulaarisia ja munuaisiin kohdistuvia vaikutuksia salpaamalla selektiivisesti AT1-reseptoria. Lisäksi se estää angiotensiini II:sta riippuvaista aldosteronin vapautumista. Tämä estää RAA-järjestelmän jatkuvaa aktivaatiota, mikä johtaisi vasokonstriktioon, munuaisperäiseen natriumin ja nesteen kertymiseen, solujen kasvun ja lisääntymisen aktivoitumiseen ja sen seurauksena maladaptiivisiin kardiovaskulaarisiin rakennemuutoksiin.
Farmakodynaamiset vaikutukset
Sakubitriili/valsartaani-valmisteen farmakodynaamisia vaikutuksia arvioitiin antamalla kerta-annoksia ja toistuvia annoksia terveille tutkittaville ja sydämen vajaatoimintaa sairastaville potilaille ja vaikutukset ovat yhdenmukaiset samanaikaisen neprilysiinin eston ja reniini-angiotensiini-aldosteronijärjestelmän salpauksen kanssa. Seitsemän päivää kestäneessä valsartaanikontrolloidussa tutkimuksessa sakubitriili/valsartaani-valmisteen antaminen potilaille, joilla oli alentunut ejektiofraktio (HFrEF), johti alussa lisääntyneeseen natriureesiin, lisäsi virtsan cGMP:tä ja pienensi keskialueen A-tyypin natriureettisen propeptidin (MR-proANP) ja N-terminaalisen B-tyypin natriureettisen propeptidin (NT-proBNP) pitoisuuksia plasmassa valsartaaniin verrattuna. 21 päivää kestäneessä HFrEF-potilailla tehdyssä tutkimuksessa hoito sakubitriili/valsartaani-valmisteella vähensi lähtötilanteeseen verrattuna huomattavasti virtsan A-tyypin natriureettista peptidiä (ANP:tä) ja cGMP:tä ja plasman cGMP:tä sekä vähensi plasman NT-proBNP:tä, aldosteronia ja endoteliini 1:tä. Samalla myös AT1-reseptori salpautui, minkä osoittivat plasman reniiniaktiivisuuden lisääntyminen ja plasman reniinipitoisuuksien suureneminen. PARADIGM-HF-tutkimuksessa hoito sakubitriili/valsartaani-valmisteella vähensi plasman NT-proBNP:tä ja lisäsi plasman B-tyypin natriureettista peptidiä (BNP) ja virtsan cGMP:tä enalapriiliin verrattuna. PANORAMA‑HF‑tutkimuksessa todettiin, että NT‑proBNP‑pitoisuudet olivat viikkojen 4 ja 12 kohdalla pienentyneet lähtötilanteesta sakubitriili/valsartaani‑valmisteella (vk 4: 40,2 %; vk 12: 49,8 %) ja enalapriililla (vk 4: 18,0 %; vk 12: 44,9 %). NT‑proBNP‑pitoisuuksien pieneneminen jatkui tutkimuksen aikana, ja viikolla 52 pienenemä lähtötilanteesta oli sakubitriili/valsartaani‑valmisteella 65,1 % ja enalapriililla 61,6 %. BNP ei ole sopiva sydämen vajaatoiminnan biomerkkiaine sakubitriili/valsartaani-valmistetta saavilla potilailla, koska BNP on neprilysiinin substraatti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). NT-proBNP ei puolestaan ole neprilysiinin substraatti ja siksi se on sopivampi biomerkkiaine.
Terveillä miespuolisilla tutkittavilla tehdyssä kattavassa kliinisessä QTc-tutkimuksessa kerta-annoksina annettujen sakubitriili/valsartaani-valmisteen 194 mg sakubitriilia ja 206 mg valsartaania ja 583 mg sakubitriilia ja 617 mg valsartaania sisältävillä annoksilla ei ollut lainkaan vaikutusta sydämen repolarisaatioon.
Neprilysiini on yksi monista entsyymeistä, jotka osallistuvat beeta-amyloidin (Aβ) poistamiseen aivoista ja selkäydinnesteestä (CSF). Kun terveille tutkittaville annettiin sakubitriili/valsartaani-valmistetta 194 mg sakubitriilia ja 206 mg valsartaania sisältävä annos kaksi kertaa vuorokaudessa kahden viikon ajan, selkäydinnesteen Aβ1-38 lisääntyi lumevalmisteeseen verrattuna; selkäydinnesteen Aβ1-40:n ja Aβ1‑42:n pitoisuuksissa ei tapahtunut muutoksia. Tämän löydöksen kliinistä merkitystä ei tiedetä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Kliininen teho ja turvallisuus
Vahvuuksiin 24 mg/26 mg, 49 mg/51 mg ja 97 mg/103 mg viitataan joissakin julkaisuissa termein 50, 100 tai 200 mg.
PARADIGM-HF
PARADIGM‑HF, keskeinen, vaiheen III tutkimus oli monikansallinen, satunnaistettu, kaksoissokkoutettu tutkimus, johon osallistui 8 442 potilasta ja jossa verrattiin sakubitriili/valsartaani-valmistetta ja enalapriilia. Molempia valmisteita annettiin sydämen vajaatoiminnan muun hoidon lisäksi aikuisille potilaille, joilla oli NYHA-luokan II–IV krooninen sydämen vajaatoiminta ja alentunut ejektiofraktio (vasemman kammion ejektiofraktio [LVEF] ≤ 40 %, muutettu myöhemmin ≤ 35 %:ksi). Ensisijainen yhdistelmäpäätemuuttuja oli kardiovaskulaarikuolema tai sydämen vajaatoiminnasta johtuva sairaalahoito. Potilaat, joiden systolinen verenpaine oli < 100 mmHg, joilla oli vaikea munuaisten vajaatoiminta (eGFR < 30 ml/min/1,73 m2) ja vaikea maksan vajaatoiminta, suljettiin pois tutkimuksen seulontavaiheessa ja siksi tällaisia potilaita ei prospektiivisesti tutkittu.
Ennen osallistumistaan tutkimukseen potilaita oli hoidettu hyvin ja he olivat saaneet tavanomaista hoitoa ACE:n estäjillä tai ATR:n salpaajilla (> 99 %), beetasalpaajilla (94 %), mineralokortikoidiantagonisteilla (58 %) ja diureeteilla (82 %). Seurannan keston mediaani oli 27 kuukautta ja potilaita hoidettiin korkeintaan 4,3 vuoden ajan.
Potilaiden täytyi keskeyttää hoitonsa ACE:n estäjällä tai ATR:n salpaajalla ja siirtyä sekventiaaliseen yksöissokkoutettuun alkuseurantajaksoon, jonka aikana he saivat 10 mg enalapriilia kaksi kertaa vuorokaudessa, minkä jälkeen he siirtyivät yksöissokkoutettuun hoitoon, jonka aikana he saivat 100 mg sakubitriili/valsartaani-valmistetta kaksi kertaa vuorokaudessa, josta annos suurennettiin 200 mg:aan kaksi kertaa vuorokaudessa (tänä aikana tapahtuneet hoidon keskeytykset, ks. kohta Haittavaikutukset). Tämän jälkeen tutkittavat satunnaistettiin tutkimuksen kaksoissokkoutettuun vaiheeseen, jonka aikana he saivat joko 200 mg sakubitriili/valsartaani-valmistetta tai 10 mg enalapriilia kaksi kertaa vuorokaudessa [sakubitriili/valsartaani (n = 4 209); enalapriili (n = 4 233)].
Tutkimuspopulaation keski-ikä oli 64 vuotta ja 19 % tutkittavista oli vähintään 75-vuotiaita. Satunnaistamishetkellä 70 % potilaista luokiteltiin NYHA-luokkaan II, 24 % luokkaan III ja 0,7 % luokkaan IV. Keskimääräinen LVEF oli 29 % ja 963 (11,4 %) potilaalla lähtötilanteen LVEF oli > 35 % ja ≤ 40%.
Tutkimuksen päättyessä 76 % sakubitriili/valsartaani-ryhmän potilaista käytti edelleen 200 mg:n tavoiteannosta kaksi kertaa vuorokaudessa (keskimääräinen vuorokausiannos oli 375 mg). Tutkimuksen päättyessä 75 % enalapriiliryhmän potilaista käytti edelleen 10 mg:n tavoiteannosta kaksi kertaa vuorokaudessa (keskimääräinen vuorokausiannos oli 18,9 mg).
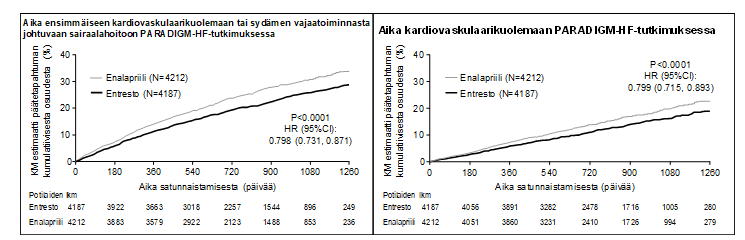
Sakubitriili/valsartaani-hoito oli tehokkaampaa kuin enalapriilihoito, sillä se pienensi kardiovaskulaarikuoleman tai sydämen vajaatoiminnasta johtuvien sairaalahoitojen riskin 21,8 %:iin verrattuna 26,5 %:iin enalapriilihoitoa saaneilla potilailla. Absoluuttinen riski pieneni 4,7 % kardiovaskulaarikuoleman ja sydämen vajaatoiminnasta johtuvan yhdistelmämuuttujan osalta, 3,1 % pelkän kardiovaskulaarikuoleman suhteen ja 2,8 % pelkän ensimmäisen sydämen vajaatoiminnasta johtuvan sairaalahoidon suhteen. Ensisijaisen yhdistelmäpäätemuuttujan suhteellinen riski pieneni 20 % enalapriiliin nähden (ks. taulukko 3). Tämä vaikutus havaittiin varhain ja se säilyi koko tutkimuksen ajan (ks. kuva 1). Molemmat ensisijaisen yhdistelmäpäätemuuttujan komponentit vaikuttivat riskin pienenemiseen. Kardiovaskulaarikuolemantapauksista 45 % oli äkillisiä kuolemantapauksia, ja niiden määrä pieneni 20 %:lla sakubitriili/valsartaani-hoitoa saaneilla potilailla verrattuna enalapriililla hoidettuihin potilaisiin (riskisuhde 0,80, p = 0,0082). Kardiovaskulaarikuolemantapauksista 26 % johtui pumppausvajavuudesta, ja niiden määrä pieneni 21 %:lla sakubitriili/valsartaani-hoitoa saaneilla potilailla verrattuna enalapriililla hoidettuihin potilaisiin (riskisuhde 0,79, p = 0,0338).
Tämä riskin pieneneminen havaittiin kaikissa alaryhmissä, jotka perustuivat sukupuoleen, ikään, rotuun, maantieteelliseen sijaintiin, NYHA-luokitukseen (II/III), ejektiofraktioon, munuaisten toimintaan, diabetes- tai hypertensiotaustaan, aiempaan sydämen vajaatoiminnan vuoksi saatuun hoitoon ja eteisvärinään.
Sakubitriili/valsartaani-hoito pidensi elossaoloaikaa ja vähensi merkittävästi kaikki kuolinsyyt kattavaa kuolleisuutta 2,8 % (sakubitriili/valsartaani 17 %, enalapriili 19,8 %). Suhteellinen riski pieneni 16 %:lla enalapriiliin verrattuna (ks. taulukko 3).
Taulukko 3 Hoidon vaikutus ensisijaiseen yhdistelmäpäätemuuttujaan, sen osatekijöihin ja kaikki kuolinsyyt kattavaan kuolleisuuteen, seurannan mediaani 27 kuukautta
Sakubitriili/valsartaani N = 4 187♯ n (%) | Enalapriili N = 4 212♯ n (%) | Riskisuhde (95 %:n luottamusväli) | Suhteellisen riskin vähenemä | p-arvo *** | |
Kardiovaskulaarikuoleman ja sydämen vajaatoiminnasta johtuvan sairaalahoidon ensisijainen yhdistelmäpäätemuuttuja* | 914 (21,83) | 1 117 (26,52) | 0,80 (0,73; 0,87) | 20 % | 0,0000002 |
Ensisijaisen yhdistelmäpäätemuuttujan yksittäiset osatekijät | |||||
Kardiovaskulaarikuolema** | 558 (13,33) | 693 (16,45) | 0,80 (0,71; 0,89) | 20 % | 0,00004 |
Ensimmäinen sairaalahoito sydämen vajaatoiminnan vuoksi | 537 (12,83) | 658 (15,62) | 0,79 (0,71; 0,89) | 21 % | 0,00004 |
Toissijainen päätetapahtuma | |||||
Kaikki kuolinsyyt kattava kuolleisuus | 711 (16,98) | 835 (19,82) | 0,84 (0,76; 0,93) | 16 % | 0,0005 |
*Ensisijainen päätemuuttuja määriteltiin aikana ensimmäiseen tapahtumaan, joita olivat kardiovaskulaarikuolema tai sairaalahoito sydämen vajaatoiminnan vuoksi.
**Kardiovaskulaarikuolema kattaa potilaat, jotka kuolivat viimeiseen tietojenkeräyspäivään mennessä aiemmasta sairaalahoidosta riippumatta.
***Yksisuuntainen p-arvo
♯ Koko analyysisarja
Kuva 1 Kaplan–Meier-käyrät ensisijaiselle yhdistelmäpäätemuuttujalle ja kardiovaskulaarikuoleman osatekijälle
TITRATION
TITRATION oli 12 viikon pituinen turvallisuus- ja siedettävyystutkimus, johon osallistui 538 potilasta, joilla oli krooninen sydämen vajaatoiminta (NYHA-luokka II–IV) ja systolinen toimintahäiriö (vasemman kammion ejektiofraktio ≤ 35 %) ja jotka eivät olleet aiemmin saaneet ACE:n estäjiä tai ATR:n salpaajia tai jotka ennen tutkimukseen osallistumistaan olivat saaneet vaihtelevia annoksia ACE:n estäjiä tai ATR:n salpaajia. Potilaille annettu sakubitriili/valsartaani-valmisteen aloitusannos oli 50 mg kaksi kertaa vuorokaudessa ja annos suurennettiin 100 mg:aan kaksi kertaa vuorokaudessa ja sitten tavoiteannokseen 200 mg kaksi kertaa vuorokaudessa joko 3 viikon tai 6 viikon kuluessa.
Suurempi osa potilaista, jotka eivät olleet aiemmin saaneet ACE:n estäjiä tai ATR:n salpaajia tai jotka saivat pieniannoksista hoitoa (joka vastasi < 10 mg enalapriilia vuorokaudessa), pystyi saavuttamaan ja jatkamaan 200 mg:n sakubitriili/valsartaani-annoksen ottamista, kun annos suurennettiin 6 viikon aikana (84,8 %) verrattuna 3 viikon aikana (73,6 %) tapahtuneeseen annoksen suurentamiseen. Yhteensä 76 % potilaista saavutti sakubitriili/valsartaani-valmisteen tavoiteannoksen 200 mg kaksi kertaa vuorokaudessa ja jatkoi sitä keskeyttämättä hoitoa tai pienentämättä annosta 12 viikon ajan.
Pediatriset potilaat
PANORAMA‑HF
PANORAMA‑HF oli monikansallinen, satunnaistettu, kaksoissokkoutettu vaiheen 3 tutkimus, jossa verrattiin sakubitriili/valsartaani‑valmistetta enalapriiliin. Tutkimukseen osallistui 375 pediatrista potilasta (ikä 1 kk – < 18 v), joiden sydämen vajaatoiminta johtui systeemisestä vasemman kammion systolisesta toimintahäiriöstä (LVEF ≤ 45 % tai ejektiofraktion lyhenemä ≤ 22,5 %). Tutkimuksen ensisijaisena tavoitteena oli selvittää, oliko sakubitriili/valsartaani‑valmiste enalapriilia parempi 52 viikkoa kestäneessä hoidossa sydämen vajaatoimintaa sairastavilla pediatrisilla potilailla, kun paremmuusvertailun perustana oli global rank ‑päätetapahtuma. Tätä ensisijaista global rank ‑päätetapahtumaa varten potilaat asetettiin (hoitotuloksiin perustuvaan nousevaan) paremmuusjärjestykseen seuraavien kliinisten tapahtumien perusteella: kuolema, elintoimintoja tukevan mekaanisen hoidon aloitus, kiireellisen sydänsiirron odotuslistalle siirtyminen, sydämen vajaatoiminnan paheneminen, toimintakykyluokka (NYHA‑/ROSS‑pisteet) ja potilaan ilmoittamat sydämen vajaatoiminnan oireet (potilaan yleisarvio Patient Global Impression Scale [PGIS] ‑asteikolla). Tutkimuksesta suljettiin pois potilaat, joilla oli systeeminen oikea kammio, vain yksi kammio tai restriktiivinen tai hypertrofinen kardiomyopatia. Sakubitriili/valsartaani‑valmisteen tavoiteltu ylläpitoannos oli 2,3 mg/kg kahdesti vuorokaudessa pediatrisilla potilailla, joiden ikä oli 1 kk – < 1 v, ja 3,1 mg/kg kahdesti vuorokaudessa potilailla, joiden ikä oli 1 v – < 18 v. Enimmäisannos oli 200 mg kahdesti vuorokaudessa. Enalapriilin tavoiteltu ylläpitoannos oli 0,15 mg/kg kahdesti vuorokaudessa pediatrisilla potilailla, joiden ikä oli 1 kk – < 1 v, ja 0,2 mg/kg kahdesti vuorokaudessa potilailla, joiden ikä oli 1 v – < 18 v. Enimmäisannos oli 10 mg kahdesti vuorokaudessa.
Tutkimuksen potilaista 9 oli iältään 1 kk – < 1 v, 61 oli iältään 1 v – < 2 v, 85 oli iältään 2 – < 6 v ja 220 oli iältään 6 – < 18 v. Lähtötilanteessa 15,7 % potilaista edusti NYHA‑/ROSS‑luokkaa I, 69,3 % luokkaa II, 14,4 % luokkaa III ja 0,5 % luokkaa IV. Keskimääräinen LVEF oli 32 %. Sydämen vajaatoiminnan syy liittyi yleisimmin kardiomyopatiaan (63,5 %). Ennen tutkimukseen osallistumista potilaita oli hoidettu yleisimmin ACE:n estäjillä / ATR:n salpaajilla (93 %), beetasalpaajilla (70 %), aldosteroniantagonisteilla (70 %) ja diureeteilla (84 %).
Ensisijaisen global rank ‑päätetapahtuman Mann–Whitney‑kerroin oli 0,907 (95 %:n luottamusväli 0,72–1,14), sakubitriili/valsartaani‑hoidon kannalta suotuisa (ks. taulukko 4). Sakubitriili/valsartaani‑valmiste ja enalapriili tuottivat vertailukelpoiset, kliinisesti merkitykselliset kohenemat lähtötilanteesta toissijaisissa, NYHA‑/ROSS‑luokkaa ja PGIS‑pistemäärää koskeneissa päätetapahtumissa. Viikon 52 kohdalla NYHA‑/ROSS‑toimintakykyluokka oli muuttunut lähtötilanteesta seuraavasti: sakubitriili/valsartaani: kohentunut 37,7 %:lla, ei muutosta 50,6 %:lla, huonontunut 11,7 %:lla potilaista; enalapriili: kohentunut 34,0 %:lla, ei muutosta 56,6 %:lla, huonontunut 9,4 %:lla potilaista. PGIS‑pistemäärä taas oli muuttunut lähtötilanteesta seuraavasti: sakubitriili/valsartaani: kohentunut 35,5 %:lla, ei muutosta 48,0 %:lla, huonontunut 16,5 %:lla potilaista; enalapriili: kohentunut 34,8 %:lla, ei muutosta 47,5 %:lla, huonontunut 17,7 %:lla potilaista. NT‑proBNP‑pitoisuus pieneni huomattavasti lähtötilanteesta molemmissa hoitoryhmissä. NT‑proBNP‑pitoisuuden pienenemä Entrestolla oli suuruudeltaan samaa luokkaa kuin aikuisilla sydämen vajaatoimintaa sairastavilla potilailla PARADIGM‑HF‑tutkimuksessa. Koska sakubitriili/valsartaani‑hoito paransi hoitotuloksia ja pienensi NT‑proBNP‑pitoisuuksia PARADIGM‑HF‑tutkimuksessa, katsottiin, että NT‑proBNP‑pitoisuuksien pieneneminen sekä PANORAMA‑HF‑tutkimuksessa havaittu oireiden lievittyminen ja toimintakyvyn koheneminen lähtötilanteeseen verrattuna olivat kohtuullisia perusteita päätellä, että hoitoon liittyy kliinisiä hyötyjä sydämen vajaatoimintaa sairastaville pediatrisille potilaille. Alle 1‑vuotiaita potilaita oli niin vähän, ettei sakubitriili/valsartaani‑hoidon tehoa voitu arvioida tässä ikäryhmässä.
Taulukko 4 Hoitovaikutus ensisijaisen global rank ‑päätetapahtuman kohdalla PANORAMA‑HF‑tutkimuksessa
Sakubitriili/valsartaani N = 187 | Enalapriili N = 188 | Hoitovaikutus | |
Ensisijainen global rank ‑päätetapahtuma | Suotuisan lopputuloksen todennäköisyys (%)* | Suotuisan lopputuloksen todennäköisyys (%)* | Kerroin** (95 %:n luottamusväli) |
52,4 | 47,6 | 0,907 (0,72–1,14) |
*Suotuisan lopputuloksen todennäköisyys tai annetun hoidon Mann–Whitney‑todennäköisyys arvioitiin voittojen prosenttiosuuden perusteella global rank ‑pistemäärän parivertailussa sakubitriili/valsartaani‑hoitoa saaneiden ja enalapriilihoitoa saaneiden potilaiden välillä (kukin suurempi pistemäärä laskettiin yhdeksi voitoksi ja kukin tasapistemäärä puolikkaaksi voitoksi).
**Mann–Whitney‑kerroin laskettiin jakamalla enalapriilin arvioitu Mann–Whitney‑todennäköisyys sakubitriili/valsartaani‑valmisteen arvioidulla Mann–Whitney‑todennäköisyydellä, jolloin < 1:n kerroin on sakubitriili/valsartaani‑valmisteelle suotuisa ja > 1:n kerroin enalapriilille suotuisa.
Farmakokinetiikka
Sakubitriili/valsartaani-valmisteen sisältämän valsartaanin biologinen hyötyosuus on suurempi kuin muilla markkinoilla olevien tablettimuotoisilla valmisteilla: sakubitriili/valsartaani-valmisteen sisältämä 26 mg, 51 mg ja 103 mg valsartaania on vastaava kuin muilla tablettimuotoisilla markkinoilla olevilla valmisteilla, jotka sisältävät 40 mg, 80 mg ja 160 mg valsartaania.
Aikuiset
Imeytyminen
Suun kautta antamisen jälkeen sakubitriili/valsartaani hajoaa valsartaaniksi ja sen aihiolääkkeeksi sakubitriiliksi. Sakubitriili metaboloituu edelleen aktiiviseksi metaboliitiksi LBQ657:ksi. Valsartaani saavuttaa huippupitoisuutensa plasmassa 2 tunnin kuluttua, sakubitriili 1 tunnin kuluttua ja LBQ657 2 tunnin kuluttua. Sakubitriilin absoluuttisen oraalisen hyötyosuuden arvioidaan olevan yli 60 % ja valsartaanin 23 %.
Kun sakubitriili/valsartaani-valmistetta annettiin kaksi kertaa vuorokaudessa, sakubitriilin, LBQ657:n ja valsartaanin vakaan tilan pitoisuudet saavutettiin kolmessa vuorokaudessa. Sakubitriili ja valsartaani eivät vakaassa tilassa akkumuloidu merkittävästi, mutta LBQ657 akkumuloituu 1,6-kertaisesti. Valmisteen antaminen aterian yhteydessä ei vaikuta kliinisesti merkityksellisellä tavalla sakubitriilin, LBQ657:n ja valsartaanin systeemisiin altistuksiin. Sakubitriili/valsartaani-valmiste voidaan ottaa joko aterian yhteydessä tai tyhjään mahaan.
Jakautuminen
Sakubitriili, LBQ657 ja valsartaani sitoutuvat voimakkaasti plasman proteiineihin (94–97 %). Kun verrattiin altistuksia plasmassa ja selkäydinnesteessä, todettiin, että LBQ657:n kyky läpäistä veri-aivoeste on vähäinen (0,28 %). Keskimääräinen valsartaanin näennäinen jakautumistilavuus oli 75 litraa ja sakubitriilin 103 litraa.
Biotransformaatio
Sakubitriili muuttuu karboksyyliesteraasien 1b:n ja 1c:n vaikutuksesta helposti LBQ657:ksi. LBQ657 ei metaboloidu edelleen merkittävästi. Valsartaanin metaboloituminen on hyvin vähäistä; vain noin 20 % annoksesta saadaan talteen metaboliitteina. Plasmasta on todettu valsartaanin hydroksyylimetaboliittia pieninä pitoisuuksina (< 10 %).
Sakubitriilin ja valsartaanin CYP450-entsyymivälitteinen metabolia on hyvin vähäistä, joten CYP450-entsyymeihin vaikuttavilla lääkevalmisteilla ei odoteta olevan vaikutusta farmakokinetiikkaan.
In vitro -metaboliatutkimukset osoittavat, että mahdollisuus CYP450-entsyymivälitteisille yhteisvaikutuksille on vähäinen, koska sakubitriili/valsartaani-valmiste metaboloituu rajoitetusti CYP450-entsyymien välityksellä. Sakubitriili/valsartaani-valmiste ei indusoi eikä inhiboi CYP450-entsyymejä.
Eliminaatio
Suun kautta antamisen jälkeen 52–68 % sakubitriilista (pääasiassa LBQ657:nä) ja noin 13 % valsartaanista ja sen metaboliiteista erittyy virtsaan; 37–48 % sakubitriilista (pääasiassa LBQ657:nä) ja 86 % valsartaanista ja sen metaboliiteista erittyy ulosteeseen.
Sakubitriili, LBQ657 ja valsartaani poistuvat plasmasta niin, että sakubitriilin keskimääräinen eliminaatiopuoliintumisaika (T½) on noin 1,43 tuntia, LBQ657:n 11,48 tuntia ja valsartaanin 9,90 tuntia.
Lineaarisuus/ei-lineaarisuus
Sakubitriilin, LBQ657:n ja valsartaanin farmakokinetiikka oli suunnilleen lineaarinen sakubitriili/valsartaani-valmisteen annosalueella, joka vaihteli annoksesta 24 mg sakubitriilia/26 mg valsartaania annokseen 97 mg sakubitriilia/103 mg valsartaania.
Erityisryhmät
Iäkkäät
Yli 65-vuotiailla tutkittavilla LBQ657-altistus on 42 % suurempi ja valsartaanialtistus 30 % suurempi kuin nuoremmilla tutkittavilla.
Munuaisten vajaatoiminta
Lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla munuaisten toiminnan ja systeemisen altistuksen välillä todettiin korrelaatio LBQ657:llä. Altistuminen LBQ657:lle keskivaikeaa munuaisten vajaatoimintaa (30 ml/min/1,73 m2 ≤ eGFR <60 ml/min/1,73 m2) sairastavilla potilailla oli 1,4 kertaa suurempi ja altistuminen vaikeaa munuaisten vajaatoimintaa (15 ml/min/1,73 m2 ≤ eGFR <30 ml/min/1,73 m2) sairastavilla potilailla 2,2 kertaa suurempi kuin potilailla, joilla oli lievä munuaisten vajaatoiminta (60 ml/min/1,73 m2 ≤ eGFR <90 ml/min/1,73 m2), suurin PARADIGM-HF-tutkimukseen osallistunut potilasryhmä. Altistuminen valsartaanille oli samankaltainen potilailla, joilla oli keskivaikea tai vaikea munuaisten vajaatoiminta kuin potilailla, joilla oli lievä munuaisten vajaatoiminta. Dialyysihoitoa saavilla potilailla ei ole tehty tutkimuksia. LBQ657 ja valsartaani sitoutuvat kuitenkin voimakkaasti plasman proteiineihin eivätkä siten todennäköisesti poistu tehokkaasti dialyysissä.
Maksan vajaatoiminta
Samankaltaisiin terveisiin tutkittaviin verrattuna lievää maksan vajaatoimintaa sairastavilla potilailla sakubitriilialtistus suureni 1,5-kertaiseksi ja keskivaikeaa maksan vajaatoimintaa sairastavilla 3,4-kertaiseksi. Vastaavat LBQ657-altistukset suurenivat 1,5- ja 1,9-kertaisiksi ja valsartaanialtistukset 1,2- ja 2,1-kertaisiksi. Terveisiin kaltaistettuihin tutkittaviin verrattuna LBQ657:n vapaiden pitoisuuksien aiheuttama altistus suureni kuitenkin 1,47‑kertaisesti potilailla, joilla oli lievä maksan vajaatoiminta, ja 3,08‑kertaisesti potilailla, joilla oli keskivaikea maksan vajaatoiminta. Valsartaanin vapaiden pitoisuuksien aiheuttamat altistukset suurenivat vastaavasti 1,09‑kertaisesti ja 2,20‑kertaisesti. Sakubitriili/valsartaani-valmistetta ei ole tutkittu vaikeaa maksan vajaatoimintaa, sappikirroosia tai kolestaasia sairastavilla potilailla (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Sukupuolen vaikutus
Sakubitriili/valsartaani-valmisteen (sakubitriilin, LBQ657:n ja valsartaanin) farmakokinetiikka on samanlainen mies- ja naispuolisilla tutkittavilla.
Pediatriset potilaat
Sakubitriili/valsartaani‑valmisteen farmakokinetiikkaa arvioitiin sydämen vajaatoimintaa sairastavilla pediatrisilla potilailla ikäryhmissä 1 kk – < 1 v ja 1 v – < 18 v. Tutkimuksessa todettiin, että sakubitriili/valsartaani‑valmisteen farmakokineettinen profiili on samankaltainen pediatrisilla ja aikuispotilailla.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, karsinogeenisuutta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset (sakubitriili/valsartaani-valmistetta ja/tai sen komponentteja sakubitriilia tai valsartaania sisältävät tutkimukset mukaan lukien) eivät viittaa erityiseen vaaraan ihmisille.
Hedelmällisyys, lisääntyminen ja kehitys
Sakubitriili/valsartaani-hoito organogeneesin aikana lisäsi rottien alkio/sikiökuolleisuutta vähintään 49 mg sakubitriilia/51 mg valsartaania/kg sisältävällä vuorokausiannoksella (≤ 0,72-kertainen ihmisille suositeltuun enimmäisannokseen [MRHD] nähden AUC-arvosta laskettuna) ja kaneilla vähintään 4,9 mg sakubitriilia/5,1 mg valsartaania/kg sisältävällä vuorokausiannoksella (2-kertainen MRHD-annokseen nähden valsartaanin AUC-arvosta laskettuna ja 0,03-kertainen LBQ657:n AUC-arvosta laskettuna). Sikiökauden hydrokefalian pienen ilmaantuvuuden perusteella valmiste on teratogeeninen. Teratogeenisuus liittyy emolle toksisiin annoksiin, mikä todettiin kaneilla vähintään 4,9 mg sakubitriilia/5,1 mg valsartaania/kg sisältävällä sakubitriili/valsartaani-annoksella vuorokaudessa. Kardiovaskulaarisia poikkeavuuksia (pääosin kardiomegaliaa) havaittiin kanisikiöillä annostasoilla, jotka eivät olleet emolle toksisia (1,46 mg sakubitriili/1,54 mg valsartaani/kg/vrk). Kaneilla todettiin myös kahden luustoon liittyvän muutoksen lievää lisääntymistä sikiöillä (sikiöaikainen rintalastan kehityshäiriö, rintalastan kaksiosainen luutuminen), kun emoille annettiin sakubitriili/valsartaani-valmistetta annoksella 4,9 mg sakubitriilia/5,1 mg valsartaania/kg/vrk. Sakubitriili/valsartaani-valmisteen haitalliset vaikutukset alkioon ja sikiöön johtuvat ATR:n salpaajavaikutuksista (ks. kohta Raskaus ja imetys).
Organogeneesin aikana sakubitriilihoito aiheutti alkio- ja sikiökuolemia ja alkioon ja sikiöön kohdistuvaa toksisuutta (alentuneita sikiöiden painoja ja luuston epämuodostumia) kaneilla annoksilla, joihin liittyi emotoksisuutta (500 mg/kg/vrk; LBQ657:n AUC-arvon perusteella 5,7-kertainen MRHD-annokseen nähden). Lievää yleistä luutumisen hidastumista todettiin annoksilla > 50 mg/kg/vrk. Tätä löydöstä ei pidetä haitallisena. Näyttöä alkio- tai sikiötoksisuudesta tai teratogeenisuudesta ei todettu sakubitriilia saaneilla rotilla. Sakubitriilin annos, joka ei aiheuta haittavaikutuksia (NOAEL) alkiolle tai sikiölle, oli vähintään 750 mg/kg/vrk rotilla ja 200 mg/kg/vrk kaneilla (LBQ657:n AUC-arvon perusteella 2,2-kertainen MRHD-annokseen nähden).
Rottien pre- ja postnataalista kehitystä selvittäneet tutkimukset, jotka tehtiin sakubitriilillä suurina annoksina, jotka olivat korkeintaan 750 mg/kg vuorokaudessa (2,2-kertaiset MRHD-annokseen nähden AUC-arvosta laskettuna), ja valsartaanilla annoksina, jotka olivat korkeintaan 600 mg/kg vuorokaudessa (0,86-kertaiset MRHD-annokseen nähden AUC-arvosta laskettuna), osoittavat että sakubitriili/valsartaani-hoito organogeneesin, tiineyden ja imetyksen aikana saattaa vaikuttaa poikasten kehitykseen ja selviytymiseen.
Muut prekliiniset löydökset
Sakubitriili/valsartaani
Sakubitriili/valsartaani-valmisteen vaikutuksia selkäydinnesteen ja aivokudoksen beeta-amyloidipitoisuuksiin arvioitiin nuorilla (2–4-vuotiailla) cynomolgus-apinoilla, jotka saivat sakubitriili/valsartaani-valmistetta (24 mg sakubitriilia/26 mg valsartaania/kg vuorokaudessa) kahden viikon ajan. Tässä tutkimuksessa cynomolgus-apinoiden selkäydinnesteen beeta-amyloidipuhdistuma väheni, mikä samalla suurensi selkäydinnesteen Aβ1‑40-, Aβ1‑42- ja Aβ1‑38-pitoisuuksia. Aivojen Aβ-pitoisuuksissa ei havaittu vastaavaa suurenemista. Kahden viikon pituisessa tutkimuksessa terveillä vapaaehtoisilla ihmisillä ei havaittu selkäydinnesteen Aβ1‑40- tai Aβ1‑42-pitoisuuksien suurenemista (ks. kohta Farmakodynamiikka). Toksikologisessa tutkimuksessa cynomolgus-apinoilla, jotka saivat sakubitriili/valsartaani-valmistetta 146 mg sakubitriilia/154 mg valsartaania/kg vuorokaudessa 39 viikon ajan, näyttöä ei saatu amyloidiplakeista aivoissa. Tässä tutkimuksessa amyloidin määrää ei kuitenkaan mitattu kvantitatiivisesti.
Sakubitriili
Sakubitriililla hoidetuilla nuorilla rotilla (7–70 päivää syntymän jälkeen) todettiin iänmukaisen luumassan kehittymisen ja luiden pidentymisen vähentymistä, kun AUC‑altistus sakubitriilin aktiiviselle metaboliitille LBQ657:lle oli noin kaksinkertainen verrattuna sakubitriili/valsartaani‑valmisteen pediatriseen kliiniseen annokseen 3,1 mg/kg kahdesti vuorokaudessa. Näiden löydösten syntymekanismia nuorissa rotissa ei tunneta, minkä vuoksi myöskään niiden merkitys pediatrisille ihmispotilaille ei ole tiedossa. Aikuisilla rotilla tehdyssä tutkimuksessa havaittiin vain vähäinen ohimenevä luun mineraalitiheyttä vähentävä vaikutus, mutta ei vaikutusta muihin luun kasvuun vaikuttaviin parametreihin, mikä viittaa siihen, että sakubitriililla ei normaalitilanteessa ole merkityksellistä vaikutusta aikuisten potilaiden luustoon. Sakubitriilin lievää ohimenevää häiritsevää vaikutusta aikuisten murtumien parantumisen varhaisvaiheessa ei kuitenkaan voida sulkea pois. Pediatrisia potilaita koskevat kliiniset tiedot (PANORAMA‑HF‑tutkimuksesta) eivät sisältäneet näyttöä siitä, että sakubitriili/valsartaani‑valmisteella olisi vaikutusta painoon, pituuteen, päänympärykseen tai luunmurtumien yleisyyteen. Tutkimuksessa ei mitattu luuntiheyttä. Pediatrisia potilaita koskevat pitkäaikaistiedot (PANORAMA-HF OLE) eivät sisältäneet näyttöä sakubitriili/valsartaani-valmisteen haittavaikutuksista (luun) kasvuun tai murtumien lukumäärään.
Valsartaani
Valsartaanilla hoidetuilla nuorilla rotilla (7–70 päivää syntymän jälkeen), jopa 1 mg/kg/vrk annokset aiheuttivat pysyviä korjautumattomia muutoksia munuaisissa, kuten munuaistiehyeen sairautta (johon joskus liittyi munuaistiehyeen epiteelin kuoliota) ja munuaisaltaan laajentumaa. Nämä munuaismuutokset ovat odotettavissa olevia angiotensiinikonvertaasin ja angiotensiini II -reseptorin tyypin 1 salpaajien liioiteltuja farmakologisia vaikutuksia rotilla, joita on hoidettu ensimmäisten 13 elinpäivän aikana. Tämä ajanjakso vastaa ihmisen raskauden 36. viikkoa, joka voi joskus pidentyä jopa 44 viikkoon hedelmöitymisestä. Munuaistoiminnan kypsymistä tapahtuu ihmisellä koko ensimmäisen elinvuoden ajan. Löydösten kliinistä merkitystä alle 1 vuoden ikäisille pediatrisille potilaille ei siten voida poissulkea. Prekliiniset tiedot eivät kuitenkaan viittaa turvallisuusriskiin yli 1‑vuotiailla pediatrisilla potilailla.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa
Matalasubstituutioasteinen hydroksipropyyliselluloosa
Krospovidoni, tyyppi A
Magnesiumstearaatti
Talkki
Kolloidinen, vedetön piidioksidi
Kalvopäällyste
Entresto 24 mg/26 mg kalvopäällysteiset tabletit
Hypromelloosi, substituutiotyyppi 2910 (3 mPa s)
Titaanidioksidi (E171)
Makrogoli 4000
Talkki
Punainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Entresto 49 mg/51 mg kalvopäällysteiset tabletit
Hypromelloosi, substituutiotyyppi 2910 (3 mPa s)
Titaanidioksidi (E171)
Makrogoli 4000
Talkki
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Entresto 97 mg/103 mg kalvopäällysteiset tabletit
Hypromelloosi, substituutiotyyppi 2910 (3 mPa s)
Titaanidioksidi (E171)
Makrogoli 4000
Talkki
Punainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ENTRESTO tabletti, kalvopäällysteinen
24/26 mg (L:ei) 28 fol (80,29 €)
49/51 mg (L:ei) 28 fol (80,29 €), 56 fol (154,06 €), 168 fol (432,91 €)
97/103 mg (L:ei) 56 fol (154,06 €), 168 fol (432,91 €)
PF-selosteen tieto
PVC/PVDC-läpipainopakkaus.
Entresto 24 mg/26 mg kalvopäällysteiset tabletit
Pakkauskoot: 14, 20, 28, 56 tai 196 kalvopäällysteistä tablettia ja monipakkaus, joka sisältää 196 (7 pakkausta à 28 kpl) kalvopäällysteistä tablettia.
Entresto 49 mg/51 mg kalvopäällysteiset tabletit
Pakkauskoot: 14, 20, 28, 56, 168 tai 196 kalvopäällysteistä tablettia ja monipakkaus, joka sisältää 168 (3 pakkausta à 56 kpl) tai 196 (7 pakkausta à 28 kpl) kalvopäällysteistä tablettia.
Entresto 97 mg/103 mg kalvopäällysteiset tabletit
Pakkauskoot: 14, 20, 28, 56, 168 tai 196 kalvopäällysteistä tablettia ja monipakkaus, joka sisältää 168 (3 pakkausta à 56 kpl) tai 196 (7 pakkausta à 28 kpl) kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Entresto 24 mg/26 mg kalvopäällysteiset tabletit
Violetinvalkoinen, soikea, kaksoiskupera kalvopäällysteinen tabletti, jossa viistetyt reunat, ei jakouurretta, toiselle puolelle on kaiverrettu "NVR" ja toiselle puolelle "LZ". Tabletti on kooltaan noin 13,1 mm x 5,2 mm.
Entresto 49 mg/51 mg kalvopäällysteiset tabletit
Vaaleankeltainen, soikea, kaksoiskupera kalvopäällysteinen tabletti, jossa viistetyt reunat, ei jakouurretta, toiselle puolelle on kaiverrettu "NVR" ja toiselle puolelle "L1". Tabletti on kooltaan noin 13,1 mm x 5,2 mm.
Entresto 97 mg/103 mg kalvopäällysteiset tabletit
Vaaleanpunainen, soikea, kaksoiskupera kalvopäällysteinen tabletti, jossa viistetyt reunat, ei jakouurretta, toiselle puolelle on kaiverrettu "NVR" ja toiselle puolelle "L11". Tabletti on kooltaan noin 15,1 mm x 6,0 mm.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ENTRESTO tabletti, kalvopäällysteinen
24/26 mg 28 fol
49/51 mg 28 fol, 56 fol, 168 fol
97/103 mg 56 fol, 168 fol
- Alempi erityiskorvaus (65 %). Sakubitriilin ja valsartaanin yhdistelmävalmiste: Kroonisen sydämen vajaatoiminnan hoito erityisin edellytyksin (289).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Sakubitriilin ja valsartaanin yhdistelmävalmiste: Kroonisen sydämen vajaatoiminnan hoito erityisin edellytyksin (381).
ATC-koodi
C09DX04
Valmisteyhteenvedon muuttamispäivämäärä
12.01.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com