
ADYNOVI injektiokuiva-aine ja liuotin, liuosta varten 1000 IU
Huomioitavaa

Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
ADYNOVI 250 IU / 2 ml injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 250 IU:ta ihmisen hyytymistekijä VIII:aa (rDNA), rurioktokogi alfa pegolia, joka vastaa pitoisuutta 125 IU/ml sen jälkeen, kun se on sekoitettu 2 ml:aan liuotinta.
ADYNOVI 500 IU / 2 ml injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 500 IU:ta ihmisen hyytymistekijä VIII:aa (rDNA), rurioktokogi alfa pegolia, joka vastaa pitoisuutta 250 IU/ml sen jälkeen, kun se on sekoitettu 2 ml:aan liuotinta.
ADYNOVI 1 000 IU / 2 ml injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää nimellisesti 1 000 IU:ta ihmisen hyytymistekijä VIII:aa (rDNA), rurioktokogi alfa pegolia, joka vastaa pitoisuutta 500 IU/ml sen jälkeen, kun se on sekoitettu 2 ml:aan liuotinta.
Vahvuus (kansainvälisinä yksikköinä, IU) määritetään käyttämällä Euroopan farmakopean kromogeenistä hyytymistestiä. ADYNOVI-valmisteen spesifinen aktiivisuus on noin 3 800–6 000 IU/mg proteiinia.
Rurioktokogi alfa pegoli (PEGyloitu ihmisen hyytymistekijä VIII [rDNA]) on proteiini, jossa on 2 332 aminohappoa, jonka molekyylipaino on noin 280 kDa:ta, ja joka on konjugoitu molekyylipainoltaan 20 kDa polyetyleeniglykolin (PEG) avulla. Se on tuotettu yhdistelmä-DNA-tekniikalla kiinankääpiöhamsterin munasarjan (CHO) solulinjassa.
Apuaineet, joiden vaikutus tunnetaan
Yksi kuiva-aineinjektiopullo sisältää 0,45 mmol (10 mg) natriumia ja 0,5 mg polysorbaattia 80 (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten (injektiokuiva-aine, liuosta varten).
Kuiva-aine: Valkoinen tai vaalea irtonainen jauhe.
Liuotin: Kirkas ja väritön liuos.
Kliiniset tiedot
Käyttöaiheet
Vähintään 12-vuotiaiden hemofilia A (synnynnäinen hyytymistekijä VIII:n puutos) -potilaiden verenvuodon hoito ja estohoito.
Ehto
Hoito pitää toteuttaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito pitää toteuttaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Hoidon seuranta
Suositellaan, että potilaalle annettava annos ja toistuvien infuusioiden antotiheys hoidon aikana selvitetään määrittämällä ensin tekijä VIII:n pitoisuus asianmukaisesti. Yksittäisten potilaiden vaste tekijä VIII:lle voi vaihdella, ja eri potilaiden puoliintumisajat ja saanto voivat olla erilaiset. Ali- tai ylipainoisten potilaiden kehonpainoon perustuvaa annosta voi olla tarpeen muuttaa. Erityisesti suurten kirurgisten toimenpiteiden yhteydessä korvaushoidon tarkka seuranta koagulaatioanalyysin (plasman tekijä VIII:n aktiivisuuden) avulla on välttämätöntä.
Kenttätutkimus osoitti, että plasman tekijä VIII -pitoisuutta voi seurata joko kromogeenisen substraattitestin tai yksivaiheisen hyytymistestin avulla, joita käytetään tavanomaisesti kliinisissä laboratorioissa.
Annostus
Korvaushoidon annos ja kesto riippuvat tekijä VIII:n puutoksen vaikeudesta, verenvuodon sijainnista ja laajuudesta ja potilaan kliinisestä tilasta.
Annetun hyytymistekijä VIII:n yksikkömäärä ilmoitetaan kansainvälisinä yksikköinä (IU), jotka liittyvät hyytymistekijä VIII ‑valmisteiden nykyiseen WHO-konsentraattistandardiin (World Health Organization, Maailman terveysjärjestö). Tekijä VIII:n aktiivisuus plasmassa ilmaistaan joko prosenttilukuna (suhteessa normaaliin ihmisen plasmaan) tai mieluummin kansainvälisinä yksikköinä (suhteessa kansainväliseen standardiin, joka koskee tekijä VIII:aa plasmassa).
Yksi kansainvälinen yksikkö (IU) hyytymistekijä VIII:aa vastaa hyytymistekijä VIII:n määrää yhdessä millilitrassa normaalia ihmisen plasmaa.
Tarvittaessa toteutettava hoito
Tarvittavan tekijä VIII -annoksen laskeminen perustuu empiiriseen havaintoon, jonka mukaan 1 IU tekijä VIII:aa painokiloa kohden lisää plasman tekijä VIII:n aktiivisuutta 2 IU:lla/dl. Tarvittava annos määritetään seuraavan kaavan avulla:
Tarvittava kansainvälinen yksikkömäärä (IU) = kehonpaino (kg) x tekijä VIII:n pitoisuuden haluttu lisäys (%) x 0,5
Annettavalla annoksella ja antotiheydellä on aina pyrittävä yksilölliseen kliiniseen tehokkuuteen.
Jos potilaalla on jokin seuraavista verenvuototapahtumista, FVIII-aktiivisuus ei saa laskea taulukossa mainitun pitoisuuden alapuolelle (% normaalista tai IU/dl) vastaavan hoitojakson aikana.
Taulukkoa 1 voidaan käyttää annostusohjeena verenvuotojen ja kirurgisten toimenpiteiden yhteydessä.
Taulukko 1: Annostusohje verenvuototapahtumia ja leikkauksia varten
Verenvuodon laajuus / kirurgisen toimenpiteen tyyppi | Tekijä VIII:n tarvittava pitoisuus (% tai IU/dl) | Annosten antotiheys (tunteina) / hoidon kesto (päivinä) |
Verenvuoto | ||
Alkava hemartroosi, lihasverenvuoto tai suun verenvuoto | 20–40 | Injektio toistetaan 12–24 tunnin välein vähintään 1 päivän ajan, kunnes kipuna ilmenevä vuotoepisodi on mennyt ohi tai paraneminen on tapahtunut. |
Laajempi hemartroosi, lihasverenvuoto tai hematooma | 30–60 | Injektio toistetaan 12–24 tunnin välein 3–4 tai useamman päivän ajan, kunnes kipu lakkaa ja akuutti toimintakyvyn vajaus korjautuu. |
Hengenvaaralliset verenvuodot | 60–100 | Injektio toistetaan 8–24 tunnin välein, kunnes vaara on ohi. |
Leikkaus | ||
Pieni Hampaanpoisto mukaan lukien. | 30–60 | Injektio toistetaan 24 tunnin välein vähintään 1 päivän ajan, kunnes paraneminen on tapahtunut. |
Suuri | 80–100 (leikkausta ennen ja sen jälkeen) | Injektio toistetaan 8–24 tunnin välein, kunnes haava on parantunut riittävästi. Hoitoa jatketaan sen jälkeen vielä vähintään 7 vuorokauden ajan, jotta tekijä VIII -aktiivisuus pysyy 30–60 %:n (IU/dl) tasolla. |
Estohoito
Pitkäaikaisessa estohoidossa suositeltu annos on 40–50 IU ADYNOVI-valmistetta painokiloa kohden kaksi kertaa viikossa, 3–4 päivän välein annettuna. Annosten ja antovälien säätämistä voidaan harkita saavutettujen tekijä VIII -pitoisuuksien ja potilaskohtaisen vuototaipumuksen perusteella (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Pediatriset potilaat
Tarvittaessa toteutettavan hoidon annostus pediatrisille potilaille (12–18-vuotiaille) on sama kuin aikuispotilaiden annostus. 12 – < 18-vuotiaiden potilaiden estohoito ei eroa aikuispotilaiden estohoidosta. Saatavissa oleva tieto ADYNOVI-valmisteen käytöstä alle 12 vuoden ikäisten lasten hoidossa on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka. Annosten ja antovälien säätämistä voidaan harkita saavutettujen tekijä VIII -pitoisuuksien ja potilaskohtaisen vuototaipumuksen perusteella (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Antotapa
ADYNOVI-valmiste annetaan laskimoon.
Antonopeus on määritettävä potilaan sietotaso huomioiden, ja se voi olla enintään 10 ml/min.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle, oktokogi alfa -isäntämolekyylille tai kohdassa Apuaineet mainituille apuaineille.
Tunnettu hiiri- tai hamsteriproteiinin aiheuttama allerginen reaktio.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyys
ADYNOVI-hoidon yhteydessä on raportoitu allergisia yliherkkyysreaktioita, mukaan lukien anafylaksia. Lääkevalmiste sisältää jäämiä hiiri- ja hamsteriproteiineista. Jos potilaalla ilmenee yliherkkyysoireita, häntä on kehotettava keskeyttämään lääkkeen käyttö välittömästi ja ottamaan yhteyttä lääkäriin. Potilaille on kerrottava yliherkkyysreaktioiden varhaisista merkeistä, kuten nokkosihottumasta, yleistyneestä urtikariasta, puristavasta tunteesta rinnassa, vinkuvasta hengityksestä, alentuneesta verenpaineesta ja anafylaksiasta.
Sokin saanutta potilasta on hoidettava yleisten sokinhoito-ohjeiden mukaisesti.
Inhibiittorit
Tekijä VIII:aa neutraloivien vasta aineiden (inhibiittoreiden) muodostuminen on tunnettu komplikaatio hemofilia A ‑potilaiden hoidossa. Inhibiittorit ovat yleensä IgG-immunoglobuliineja, jotka estävät tekijä VIII ‑hyytymistoiminnan aktivoitumisen ja joiden määrä ilmaistaan Bethesda-yksikköinä (Bethesda Units, BU) millilitrassa plasmaa. Määrityksessä käytetään modifioitua menetelmää. Inhibiittoreiden muodostumisen riski riippuu taudin vaikeusasteesta ja altistumisesta tekijä VIII:lle. Riski on suurin 50 ensimmäisen altistumispäivän aikana, ja jatkuu koko loppuelämän ajan, mutta inhibiittorien muodostuminen on melko harvinaista.
Inhibiittorien muodostumisen kliininen merkitys riippuu inhibiittorititteristä. Riittämättömän kliinisen vasteen riski on pienempi, jos potilaan titteri on alhainen verrattuna tilanteeseen, jossa potilaalla on korkean titterin inhibiittoreita.
Hyytymistekijä VIII ‑valmisteilla hoidettavien potilaiden inhibiittoreiden esiintyvyyttä on seurattava tarkkaan asianmukaisin kliinisin havainnoin ja laboratoriokokein. Jos odotettua tekijä VIII:n aktiivisuuden plasmapitoisuutta ei saavuteta tai jos verenvuotoa ei saada hallintaan asianmukaisella annoksella, on potilaalta testattava tekijä VIII:n inhibiittorin esiintyminen. Jos potilaalla on korkea inhibiittoripitoisuus, tekijä VIII ‑hoito ei ehkä ole tehokasta ja on harkittava muita terapeuttisia vaihtoehtoja. Näiden potilaiden hoidon on tapahduttava sellaisten lääkäreiden valvonnassa, joilla on kokemusta hyytymistekijä VIII:lle inhibiittoreita kehittäneiden hemofiliapotilaiden hoitamisesta.
Sydän- ja verisuonitapahtumat
Jos potilaalla on sydämeen ja verisuoniin liittyviä riskitekijöitä, korvaushoito tekijä VIII:lla voi lisätä sydän- ja verisuonitapahtumien riskiä.
Katetriin liittyvät komplikaatiot hoidon aikana
Jos keskuslaskimokatetria (CVAD) tarvitaan, siihen liittyvien komplikaatioiden, kuten paikallisten infektioiden, bakteremian ja katetrointikohdan tromboosin riski on arvioitava.
Apuaineisiin liittyvät huomioon otettavat asiat
Natrium
Tämä lääkevalmiste sisältää 10 mg natriumia per injektiopullo, mikä vastaa 0,5 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille. Ruumiinpainosta ja annostuksesta riippuen potilaalle voidaan antaa enemmän kuin yksi injektiopullo. Tämä täytyy ottaa huomioon, jos natriumin määrää potilaan ruokavaliossa säännöstellään.
Polysorbaatti 80
Tämä lääkevalmiste sisältää 0,5 mg polysorbaattia 80 per injektiopullo, mikä vastaa 0,25 mg:aa/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Valmisteen nimen ja eränumeron kirjaaminen aina ADYNOVI-valmistetta potilaalle annettaessa on erittäin suositeltavaa, jotta potilas ja lääkevalmisteen erä pystytään yhdistämään toisiinsa.
Pediatriset potilaat
Luetellut varoitukset ja varotoimet pätevät sekä aikuisiin että lapsiin.
Yhteisvaikutukset
Ihmisen hyytymistekijä VIII (rDNA) ‑valmisteilla ei ole raportoitu olevan yhteisvaikutuksia muiden lääkevalmisteiden kanssa.
Raskaus ja imetys
Tekijä VIII:sta ei ole tehty lisääntymistutkimuksia eläimillä. Kokemusta tekijä VIII:n käytöstä raskauden ja imetyksen aikana ei ole, koska hemofilia A ‑tapaukset ovat naisilla harvinaisia. Tämän vuoksi tekijä VIII:aa saa käyttää raskauden tai imetyksen aikana vain silloin, jos käyttö on selvästi aiheellista.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
ADYNOVI-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Yliherkkyys- tai allergiareaktioita (joita ovat esimerkiksi angioödeema, pistoskohdan poltto ja kirvely, vilunväreet, punoitus, levinnyt urtikaria, päänsärky, nokkosihottuma, alhainen verenpaine, uneliaisuus, pahoinvointi, levottomuus, takykardia, puristava tunne rinnassa, pistely, oksentelu, vinkuva hengitys) on havaittu harvoin, ja ne voivat joissakin tapauksissa edetä vaikeaksi anafylaksiaksi (mukaan lukien sokki).
Neutraloivia vasta-aineita (inhibiittoreita) voi kehittyä hemofilia A ‑potilaille, jotka saavat tekijä VIII ‑hoitoa, kuten ADYNOVI-hoitoa. Mikäli tällaisia inhibiittoreita ilmaantuu, se näkyy riittämättömänä kliinisenä vasteena hoidolle. Tällaisissa tapauksissa on suositeltavaa ottaa yhteyttä erikoistuneeseen hemofiliakeskukseen (ks. kohta Farmakodynamiikka).
Haittavaikutustaulukko
ADYNOVI-valmisteen turvallisuutta arvioitiin 478 yksittäisen potilaan perusteella. Nämä potilaat sairastivat vaikeaa hemofilia A:ta (tekijä VIII alle 1 % normaalista). Heistä 360 oli saanut aiemmin hoitoa ja 118 oli aiemmin hoitamattomia, ja he saivat vähintään yhden annoksen ADYNOVI-valmistetta 7 valmistuneessa prospektiivisessa avoimessa kliinisessä monikeskustutkimuksessa.
Alla esitetty taulukko noudattaa MedDRA-järjestelmän elinjärjestelmäluokitusta (elinjärjestelmiä ja suositettavia termejä).
Yleisyys on arvioitu seuraavan tavan mukaisesti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2: ADYNOVI-valmisteen raportoidut haittavaikutukset
| MedDRAnelinjärjestelmäluokitus | Haittavaikutukset | Esiintyvyys |
| Veri ja imukudos | Hyytymistekijä VIII:n inhibition | Melko harvinainen (PTP)1 Hyvin yleinen (PUP)1 |
| Immuunijärjestelmä | Yliherkkyys | Melko harvinainen |
| Anafylaktinen reaktio2 | Tuntematon | |
| Hermosto | Päänsärky | Yleinen |
| Huimaus | Yleinen | |
| Silmät | Silmien hyperemia | Melko harvinainen |
| Verisuonisto | Punastuminen | Melko harvinainen |
| Ruoansulatuselimistö | Ripuli | Yleinen |
| Oksentelu | Yleinen | |
| Pahoinvointi | Yleinen | |
| Hengityselimet, rintakehä ja välikarsina | Yskä | Hyvin yleinen |
| Iho ja ihonalainen kudos | Ihottuma | Yleinen |
| Kutiseva ihottuma | Melko harvinainen | |
| Urtikaria | Yleinen | |
| Silmäluomen ihottuma | Melko harvinainen | |
| Tutkimukset | Suurentunut eosinofiilimäärä | Melko harvinainen |
| Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioon liittyvä reaktio | Melko harvinainen |
1 Yleisyys perustuu kaikilla tekijä VIII -valmisteilla tehtyihin tutkimuksiin, joihin osallistui vaikeaa hemofilia A:ta sairastavia potilaita. PTP = aiemmin hoidetut potilaat, PUP = aiemmin hoitamattomat potilaat Esitetyt yleisyydet on laskettu käyttämällä kaikkia valmisteeseen liittyviä ja muita haittavaikutuksia. 2 Haittavaikutus havaittu myyntiluvan myöntämisen jälkeisessä valvonnassa. | ||
Kuvaus valikoiduista haittavaikutuksista
Yliherkkyys
Todettuja yliherkkyystapahtumia oli kaksi. Yksi tapahtuma oli lievä, tilapäinen, ei-vakava ihottuma, joka ilmeni 2-vuotiaalla potilaalla, jolle oli kehittynyt ihottuma ensimmäisellä ADYNOVI-valmisteen käyttökerralla kahdesta hoitokerrasta. Toinen tapahtuma oli keskivaikea, ohimenevä, ei-vakava tapahtuma, johon liittyi eryteema, takykardia ja yskä ja joka ilmeni 9,6 kuukauden ikäisellä potilaalla.
Immunogeenisuus
Yhdellekään tutkittavalle, jotka osallistuivat yhteen tai useampaan kuudesta valmistuneesta, aiemmin hoitoa saaneilla potilailla tehdystä kliinisestä tutkimuksesta, ei kehittynyt hyytymistekijä VIII:n persistenttejä neutraloivia (estäviä) vasta-aineita tasolla ≥ 0,6 BU/ml (Nijmegenin muunnetun Bethesda-määrityksen mukaan). Yhdelle potilaalle kehittyi ohimenevä tekijä VIII:n inhibiittori, joka oli positiivisuuden alarajalla (0,6 BU) yksilöllisen estolääkityksen aikana, kun tekijä VIII:n tavoitetasona oli 8–12 %.
Pediatriset potilaat
ADYNOVI-valmistetta ei ole tarkoitettu alle 12 vuoden ikäisten lasten hoitoon (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Lasten haittavaikutusten esiintyvyys, tyyppi ja vaikeusaste ovat odotettavasti samoja kuin aikuisilla. ADYNOVI-valmisteen turvallisuutta arvioitiin 38:lla <6-vuotiaalta tutkittavalla, joille oli kertynyt yhteensä 2 880 altistumispäivää, ja 34:llä 6 – < 12-vuotiaalla aiemmin hoitamattomalla potilaalla , joille oli kertynyt yhteensä 2 975 altistumispäivää. Iän keskiarvo (SD) oli ensimmäisessä tutkimuksessa 3,3 (1,55) ja toisessa tutkimuksessa 8,1 (1,92) vuotta.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa lääkevalmisteen myyntiluvan myöntämisen jälkeisistä epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin.
Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostusoireista ADYNOVI-valmisteen tai rekombinantin hyytymistekijä VIII:n yhteydessä ei ole raportoitu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: hemostaatit, veren hyytymistekijä VIII, ATC-koodi: B02BD02.
Tekijä VIII:n ja von Willebrand ‑tekijän muodostama kompleksi koostuu kahdesta molekyylistä (tekijä VIII:sta ja von Willebrand ‑tekijästä), joiden fysiologiset tehtävät ovat erilaiset. Kun tekijä VIII infusoidaan hemofiliaa sairastavaan potilaaseen, se sitoutuu von Willebrand -tekijään potilaan verenkierrossa. Aktivoitu tekijä VIII toimii aktiivisen tekijä IX:n kofaktorina, ja ne kiihdyttävät yhdessä tekijä X:n muuttumista aktiiviseksi tekijä X:ksi. Aktivoitu tekijä X muuntaa protrombiinin trombiiniksi. Trombiini muuntuu sitten fibrinogeeniksi, jonka ansiosta veri voi hyytyä. Hemofilia A on sukupuoleen yhteydessä oleva perinnöllinen verenhyytymishäiriö, joka johtuu tekijä VIII:n alentuneesta tasosta ja johtaa runsaaseen verenvuotoon niveliin, lihaksiin tai sisäelimiin, joko spontaanisti tai onnettomuudessa tai leikkauksessa saadun vamman seurauksena. Korvaushoidolla lisätään tekijä VIII:n pitoisuuksia plasmassa, jolloin tekijän puutos korjaantuu tilapäisesti ja vuototaipumus korjaantuu.
Rurioktokogi alfa pegoli on pegyloitu rekombinantti ihmisen hyytymistekijä VIII, jolla on pidentynyt puoliintumisaika. Rurioktokogi alfa pegoli on oktokogi alfan kovalenttinen konjugaatti, joka koostuu 2 332 aminohaposta ja joka on valmistettu polyetyleeniglykolireagenssin (PEG) (molekyylipaino 20 kDa) avulla. Rurioktokogi alfa pegolin terapeuttinen aktiivisuus on johdettu oktokogi alfasta, joka valmistetaan rekombinantilla DNA-tekniikalla kiinankääpiöhamsterin munasarjan solulinjasta. Oktokogi alfa konjugoidaan sitten kovalentisti PEG-reagenssilla. PEG-osa konjugoidaan oktokogi alfaan plasman puoliintumisajan pidentämiseksi.
Kliininen teho ja turvallisuus
ADYNOVI-valmisteen turvallisuutta, tehoa ja farmakokinetiikkaa arvioitiin keskeisessä avoimessa, prospektiivisessa, kliinisessä monikeskustutkimuksessa, jossa kaksi kertaa viikossa annettavan estohoidon tehoa verrattiin tarvittaessa toteutettavaan hoitoon ja jossa määritettiin hemostaattinen teho verenvuototapahtumien hoidossa. Yhteensä 137 miespuolista (12–65-vuotiasta) aiemmin hoidettua potilasta, joilla oli vaikea hemofilia A, sai vähintään yhden ADYNOVI-infuusion. Kaksikymmentäviisi näistä 137 tutkittavasta oli nuoria (12 – < 18-vuotiaita).
Estohoito aikuisilla ja nuorilla
Keskeisessä tutkimuksessa tutkittavat saivat joko estohoitoa (n = 120) ADYNOVI-valmisteella annoksella 40–50 IU/kg kaksi kertaa viikossa tai tarvittaessa toteutettavaa hoitoa (n = 17) ADYNOVI-valmisteella annoksella 10–60 IU/kg 6 kuukauden ajan. Mediaaniannosväli oli 3,6 päivää ja keskimääräinen annos (SD) oli 48,7 (4,4) IU/kg. Satakahdeksantoista tutkittavaa (98 %) 120:stä estohoitoa saaneesta tutkittavasta jatkoi suositeltua aloitushoitoa ilman annosmuutosta, ja 2 tutkittavan annos nostettiin tasolle 60 IU/kg estohoidon aikana kohdenivelien verenvuodon vuoksi.
Protokollan mukaisessa (per protocol) potilasjoukossa (eli populaatiossa, joka sai tutkimussuunnitelmassa määritettyjen annosvaatimusten mukaiset annokset) yhteensä 101 tutkittavaa sai estohoitoryhmässä hoitoa kaksi kertaa viikossa ja 17:ää tutkittavaa hoidettiin tarvittaessa toteutettavan hoidon ryhmässä tapahtumien mukaisesti. Vuotuisen verenvuototapahtumien määrän (annualised bleed rate, ABR) mediaani oli tarvittaessa annettavan hoidon ryhmässä 41,5 ja kaksi kertaa viikossa estohoitoa saaneiden ryhmässä 1,9. Vuotuisen nivelvuotutapahtumien määrän mediaani (Q1; Q3) oli tarvittaessa toteutettavan hoidon ryhmässä 38,1 (24,5; 44,6) verrattuna estohoitoryhmän määrään 0,0 (0,0; 2,0). Vuotuinen spontaanien verenvuototapahtumien mediaani oli 21,6 (11,2; 33,2) tarvittaessa annettavan hoidon ryhmässä verrattuna estohoitoryhmän määrään 0,0 (0,0; 2,2). Täysanalysoidun populaation tulokset olivat samanlaiset kuin protokollan mukaisessa potilasjoukossa. Huomattavaa on, että vuotuinen verenvuototapahtumien määrä ei ole vertailukelpoinen eri tekijäpitoisuuksien ja eri kliinisten tutkimusten välillä.
Neljälläkymmenellä tutkittavalla 101:stä (40 %) ei ilmennyt verenvuototapahtumia, 58 tutkittavalla 101:stä (57 %) ei ilmennyt nivelverenvuototapahtumia, ja 58 tutkittavalla 101:stä (57%) ei ilmennyt spontaaneja verenvuototapahtumia estohoitoryhmässä. Kaikilla tutkittavilla, jotka kuuluivat tarvittaessa toteutettavan hoidon ryhmään, ilmeni verenvuototapahtuma, mukaan lukien nivelverenvuototapahtuma tai spontaani verenvuototapahtuma.
Verenvuototapahtumien hoito aikuisilla ja nuorilla
Keskeisen tutkimuksen protokollan mukaisessa potilajoukossa ADYNOVI-valmisteella hoidettiin yhteensä 518 verenvuototapahtumaa. Näistä 361 verenvuototapahtumaa (n = 17 tutkittavaa) ilmeni tarvittaessa toteutettavan hoidon ryhmässä ja 157 tapahtumaa (n = 61 tutkittavaa) estohoitoryhmässä. Infuusiokohtainen mediaaniannos kaikkien verenvuototapahtumien hoidossa protokollan mukaisessa potilasjoukossa oli 32,0 IU/kg (kvartiiliväli: 21,5). Yhteensä 95,9 % verenvuototapahtumista saatiin hallintaan 1–2 infuusiolla, ja 85,5 % saatiin hallintaan vain 1 infuusiolla. 518 verenvuototapahtumasta 96,1 %:ssa ADYNOVI-hoidon vaste arvioitiin erinomaiseksi (täydellinen kivun helpotus ja verenvuodon objektiivisten merkkien häviäminen yhden infuusion jälkeen) tai hyväksi (selvä kivun helpotus ja/tai verenvuodon merkkien lievittyminen yhden infuusion jälkeen).
Perioperatiivinen hoito (kirurginen estohoito) aikuisilla ja nuorilla
Kirurgisessa tutkimuksessa 21 eri tutkittavalle tehtiin yhteensä 21 suurta leikkaustoimenpidettä ja 5 pientä lisäleikkausta, ja ne kaikki arvioitiin. Suurissa leikkauksissa preoperatiivinen latausannos oli 36–109 IU/kg (mediaani: 63 IU/kg) ja postoperatiivinen kokonaisannos oli 186–1320 IU/kg (mediaani: 490 IU/kg). Suurissa leikkauksissa kokonaisannoksen mediaani oli 553 IU/kg (vaihteluväli: 248–1 394 IU/kg), ja pienissä leikkauksissa kokonaisannoksen mediaani oli 106 IU/kg (vaihteluväli: 76–1 32 IU/kg).
Kaikissa 26 (21 suuressa, 5 pienessä) toimenpiteessä perioperatiivinen hemostaattinen teho arvioitiin erinomaiseksi (verta menetettiin enintään saman verran kuin on odotettavissa ei-hemofiiliselle potilaalle tehdyssä samantyyppisessä toimenpiteessä, ja verensiirroissa tarvittiin veren komponentteja enintään saman verran kuin on odotettavissa ei-hemofiilisen populaation verensiirroissa). Havaitun leikkauksen aikana menetetyn verimäärän mediaani (kvartiiliväli) oli 10,0 (20,0) ml (n = 14) verrattuna suurten ortopedisten leikkausten ennustettuun keskimääräiseen menetettyyn verimäärään 150,0 (140,0) ml (n = 14).
Pitkäkestoinen estohoito aikuisilla, nuorilla ja pediatrisilla potilailla
Pitkäaikaista turvallisuus- ja tehoa selvittäneessä tutkimuksessa ADYNOVI-valmisteen käyttöä verenvuotokohtausten estolääkityksenä ja hoitona arvioitiin 216 pediatrisella ja aikuisella aiemmin hoitoa saaneella, vaikeaa hemofilia A:ta sairastavalla potilaalla, jotka olivat aiemmin osallistuneet muihin ADYNOVI-tutkimuksiin tai jotka eivät olleet saaneet aiemmin ADYNOVI-hoitoa. Hoitoryhmän potilaat saivat kaksi kertaa viikossa kiinteän annoksen 40–50 IU/kg, jos potilaan ikä oli ≥ 12 vuotta, tai 40–60 IU/kg, jos potilaan ikä oli < 12 vuotta. Annosta säädettiin enimmillään 80 IU:n/kg annokseen kaksi kertaa viikossa, jos tämä oli tarpeen tekijä VIII ‑minimipitoisuuden pitämiseksi tasolla > 1 %. Tutkimushenkilöt, jotka valitsivat yksilöllisen (farmakokineettisesti mukautetun) estohoidon, saivat enimmillään 80 IU:n/kg infuusioannoksen vähintään kaksi kertaa viikossa tekijä VIII -minimipitoisuuden pitämiseksi tasolla ≥ 3 %. Vuotuiset verenvuototapahtumien määrät estohoito-ohjelman, verenvuotokohdan ja etiologian mukaan on esitetty taulukossa 3.
Taulukko 3: Vuotuinen verenvuototapahtumien määrä estohoidon mukaan (ITT-potilasjoukko)
Verenvuotokohtien etiologia | Kaksi kertaa viikossa (N = 186) | 5 päivän välein (N = 56) | 7 päivän välein (N = 15) | Farmakokineettisesti mukautettua (N = 25) |
Keskiarvo (Piste-estimaatti 95 %:n luottamusvälillä) | ||||
Yleinen | 2,2 (1,85; 2,69) | 2,1 (1,54; 2,86) | 2,7 (1,44; 5,20) | 2,6 (1,70; 4,08) |
Nivel | 1,2 (0,96; 1,58) | 1,1 (0,81; 1,55) | 2,0 (0,90; 4,62) | 1,4 (0,91; 2,17) |
Spontaani | 1,2 (0,92; 1,56) | 1,3 (0,87; 2,01) | 1,8 (0,78; 4,06) | 1,0 (0,54; 1,71) |
Piste-estimaatit ja 95 %:n luottamusvälit saatu yleistetystä lineaarisesta mallista, jossa negatiivinen binomijakauma sovitettiin logaritmiseen linkkifunktioon. Potilaat, jotka saivat useiden hoito-ohjelmien mukaisia annoksia, on sisällytetty useiden hoito-ohjelmien yhteenvetoihin. Sisältää kaikki tutkimuksessa mukana olleet tutkittavat (Kaksi kertaa viikossa annettavassa annostuksessa ja farmakokineettisesti mukautetussa annostuksessa mukana aikuiset ja pediatriset potilaat (< 18 vuotta). 5 päivän ja 7 päivän välein annettavissa annostuksissa ei ollut mukana yhtään tutkittavaa iältään < 12 vuotta). ITT = hoitoaiepopulaatio, N = analyysiin sisällytettyjen tutkimushenkilöiden lukumäärä a Tekijä VIII -tavoiteaktiivisuuden minimitaso ≥ 3 % normaalista | ||||
Huomattavaa on, että vuotuinen verenvuototapahtumien määrä ei ole vertailukelpoinen eri tekijäpitoisuuksien ja eri kliinisten tutkimusten välillä.
Pitkän aikavälin hemostaattista tehoa arvioitiin 910:ssä ADYNOVI-valmisteella hoidetussa verenvuotokohtauksessa, ja tulos oli erinomainen tai hyvä 88,5 %:ssa verenvuotokohtauksista. Kaikissa ikäryhmissä ja sekä kiinteäannoksisessa että farmakokineettisesti mukautetussa annosohjelmassa > 85 % verenvuotohoidoista arvioitiin erinomaisiksi tai hyviksi. Suurin osa verenvuotokohtauksista hoidettiin yhdellä (74,0 %) tai kahdella (15,4 %) infuusiolla.
Yksilöllistä estohoitoa koskeva kliininen PROPEL-tutkimus aikuisilla ja nuorilla
ADYNOVI-valmisteen turvallisuutta ja tehoa arvioitiin prospektiivisessa, satunnaistetussa, avoimessa monikeskustutkimuksessa 121:llä (satunnaistettuja 115) nuorella (ikä 12–18 vuotta) ja aikuisella aiemmin hoitoa saaneella potilaalla, joilla oli vaikea hemofilia A ja jossa hoitojakson kesto oli 12 kuukautta. Tutkimuksessa verrattiin kahta farmakokineettisesti ohjattua ADYNOVI-estohoito-ohjelmaa, joissa hyytymistekijä VIII:n tavoiteminimitaso oli 1–3 %, kun potilaat saivat annoksen kaksi kertaa viikossa (N = 57), tai 8–12 %, kun potilaat saivat annoksen joka toinen päivä (N = 58). Vertailu tehtiin arvioimalla niiden potilaiden osuus, joilla vuotuinen verenvuototapahtumien kokonaismäärä oli 0 toisen 6 kuukauden tutkimusjakson aikana.
Keskimääräiset estolääkitysannokset 1–3 %:n ja 8–12 %:n minimitason ryhmissä olivat 3 866,1 IU/kg vuodessa (keskimääräinen [keskihajonta] infuusiomäärä/viikko = 2,3 [0,58]) ja 7 532,8 IU/kg vuodessa (keskimääräinen [keskihajonta] infuusiomäärä/viikko = 3,6 [1,18]). Kun annosta oli säädetty ensimmäisen 6 kuukauden estohoitojakson aikana, mediaaniminimitasot toisella 6 kuukauden jaksolla (yksivaiheisen hyytymismäärityksen perusteella ja laskettuna suunnitellun infuusiovälin loppuun) olivat 1–3 %:n minimitason ryhmässä 2,10 IU/dl – 3,00 IU/dl ja 8–12 %:n minimitason ryhmässä 10,70 IU/dl – 11,70 IU/dl. Tämä osoitti, että annostus kahdessa estohoito-ohjelmassa oli yleisesti ottaen riittävä haluttujen tekijä VIII -minimitasojen saavuttamiseksi ja ylläpitämiseksi.
Tutkimuksen ensisijaista päätetapahtumaa, eli potilaiden osuutta, joilla vuotuinen verenvuototapahtumien kokonaismäärä oli 0 toisen 6 kuukauden tutkimusjakson aikana; ei saavutettu ITT-potilasjoukossa (p = 0,0545), mutta saavutettiin protokollan mukaisessa potilasjoukossa (p = 0,0154). Niiden protokollan mukaiseen potilasjoukkoon satunnaistettujen tutkimushenkilöiden osuudet, joilla vuotuinen verenvuototapahtumien kokonaismäärä, vuotuinen spontaanien verenvuototapahtumien määrä ja vuotuinen spontaanien nivelvuototapahtumien (annualized joint bleeding rate, AJBR) määrä oli 0 toisella 6 kuukauden tutkimusjaksolla, on esitetty taulukossa 4.
Taulukko 4: Vuotuinen verenvuototapahtumien määrä = 0, toinen 6 kuukauden tutkimusjakso
Niiden tutkimushenkilöiden osuus, joilla ei ollut verenvuotoja 6 kuukauteen (Piste-estimaatti 95 %:n luottamusvälillä) | ||
ITT-potilasjoukko | ||
1–3 %:n minimitaso (N = 57) | 8–12 %:n minimitaso (N = 58) | |
Vuotuinen verenvuototapahtumien kokonaismäärä = 0 | 0,421 (0,292; 0,549) | 0,621 (0,491; 0,750) |
Vuotuinen spontaanien verenvuototapahtumien määrä = 0 | 0,596 (0,469; 0,724) | 0,760 (0,645; 0,875) |
Vuotuinen spontaanien nivelvuototapahtumien määrä = 0 | 0,649 (0,525; 0,773) | 0,850 (0,753; 0,947) |
Vuotuinen verenvuototapahtumien määrä määritellään jakamalla verenvuototapahtumien määrä havainnointijakson pituudella vuosina. | ||
Niiden tutkimushenkilöiden osuus, joilla ei ollut verenvuototapahtumia 6 kuukauteen (Piste-estimaatti – 95 %:n luottamusväli) | ||
Protokollan mukainen potilasjoukko | ||
1–3 %:n minimitaso (N = 52) | 8–12 %:n minimitaso (N = 43) | |
Vuotuinen verenvuototapahtumien kokonaismäärä = 0 | 0,404 (0,270; 0,549) | 0,674 (0,515; 0,809) |
Vuotuinen spontaanien verenvuototapahtumien määrä = 0 | 0,596 (0,451; 0,730) | 0,814 (0,666; 0,916) |
Vuotuinen spontaanien nivelvuototapahtumien määrä = 0 | 0,654 (0,509; 0,780) | 0,907 (0,779; 0,974) |
Protokollan mukainen potilasjoukko = kaikki tutkimushenkilöt, jotka suorittivat toisen 6 kuukauden estohoitojakson ilman merkittäviä tutkimustuloksiin vaikuttavia poikkeamia protokollasta. Vuotuinen verenvuototapahtumien määrä määritellään jakamalla verenvuototapahtumien määrä havainnointijakson pituudella vuosina. | ||
Huomattavaa on, että vuotuinen verenvuototapahtumien määrä ei ole vertailukelpoinen eri tekijäpitoisuuksien ja eri kliinisten tutkimusten välillä.
Vuotuinen verenvuototapahtumien kokonaismäärä, vuotuinen spontaanien vernvuototapahtumien määrä ja vuotuinen spontaanien nivelvuototapahtumien määrä toisella 6 kuukauden tutkimusjaksolla on esitetty taulukossa 5.
Taulukko 5: Vuotuinen verenvuototapahtumien määrä toisella 6 kuukauden tutkimusjaksolla
(ITT-potilasjoukko) | ||||
1–3 %:n minimitaso (N = 57) | 8–12 %:n minimitaso (N = 53) | |||
Mediaani | Keskiarvo (keskihajonta) | Mediaani | Keskiarvo (keskihajonta) | |
Vuotuinen verenvuototapahtumien kokonaismäärä | 2,0 | 3,6 (7,5) | 0,0 | 1,6 (3,4) |
Vuotuinen spontaanien verenvuototapahtumien määrä | 0,0 | 2,5 (6,6) | 0,0 | 0,7 (1,7) |
Vuotuinen spontaanien nivelvuototapahtumien määrä | 0,0 | 2,0 (6,4) | 0,0 | 0,5 (1,7) |
ABR = vuotuinen verenvuototapahtumien määrä, ABJR = vuotuinen nivelvuototapahtumien määrä Vuotuinen verenvuototapahtumien määrä määritellään jakamalla verenvuototapahtumien määrä havainnointijakson pituudella vuosina. | ||||
Protokollan mukainen potilasjoukko | ||||
1–3 %:n minimitaso (N = 52) | 8–12 %:n minimitaso (N = 43) | |||
Mediaani | Keskiarvo (keskihajonta) | Mediaani | Keskiarvo (keskihajonta) | |
Vuotuinen verenvuototapahtumien kokonaismäärä | 2,0 | 2,4 (3,2) | 0,0 | 2,1 (4,2) |
Vuotuinen spontaanien verenvuototapahtumien määrä | 0,0 | 1,6 (2,6) | 0,0 | 0,8 (2,4) |
Vuotuinen spontaanien nivelvuototapahtumien määrä | 0,0 | 1,0 (1,8) | 0,0 | 0,7 (2,2) |
Protokollan mukainen potilasjoukko = kaikki tutkimushenkilöt, jotka suorittivat toisen 6 kuukauden estohoitojakson ilman merkittäviä tutkimustuloksiin vaikuttavia poikkeamia protokollasta. Vuotuinen verenvuototapahtumien määrä määritellään jakamalla verenvuototapahtumien määrä havainnointijakson pituudella vuosina. | ||||
ADYNOVI-valmisteella hoidettiin yhteensä 242 verenvuotokohtausta 66 tutkimushenkilöllä; näistä 155 verenvuotoa ilmeni 40 tutkimushenkilöllä 1–3 %:n minimitason ryhmässä ja 87 verenvuotoa 26 tutkimushenkilöllä 8–12 %:n minimitason ryhmässä. Suurin osa verenvuodoista (86,0 %, 208/242) hoidettiin 1 tai 2 infuusiolla, ja verenvuodon hoito arvioitiin verenvuototapahtuman päätyttyä erinomaiseksi tai hyväksi 84,7 %:ssa (205/242) tapahtumista.
Pediatriset potilaat
Pediatrinen tutkimus aiemmin hoidetuilla potilailla, joiden ikä oli < 12 vuotta
Pediatrisessa tutkimuksessa yhteensä 66 vaikeaa hemofilia A:ta sairastavaa, aiemmin hoidettua potilasta sai hoitoa (32 tutkittavaa, joiden ikä oli < 6 vuotta ja 34 tutkittavaa, joiden ikä oli 6 – < 12 vuotta). Estohoito oli 40–60 IU/kg ADYNOVI-valmistetta kaksi kertaa viikossa. Keskimääräinen annos (SD) oli 54,3 (6,3) IU/kg, ja infuusioiden mediaaniantotiheys viikossa oli 1,87. 65 protokollan mukaiseen potilasjoukkoon kuuluvan tutkittavan vuotuisen verenvuototapahtumien määrän mediaani oli 2,0 (kvartiiliväli: 3,9), ja sekä vuotuisen spontaanien verenvuototapahtumien että vuotuisen nivelverenvuototapahtumien määrän mediaani oli 0 (kvartiiliväli: 1,9). Kahdellakymmenelläneljällä tutkittavalla 65:stä (37 %) ei ilmennyt verenvuototapahtumia, 47 tutkittavalla 65:stä (72 %) ei ilmennyt nivelverenvuototapahtumia, ja 43 tutkittavalla 65:stä (66 %) ei ilmennyt spontaaneja verenvuototapahtumia estohoidossa.
Pediatrisen tutkimuksen aikana havaituista 70 verenvuototapahtumasta 82,9 % saatiin hallintaan 1 infuusiolla ja 91,4 % saatiin hallintaan 1 tai 2 infuusiolla. 63 verenvuototapauksessa 70:stä (90,0 %) verenvuodon hallinta arvioitiin erinomaiseksi (täydellinen kivun helpotus ja verenvuodon objektiivisten merkkien häviäminen yhden infuusion jälkeen) tai hyväksi (selvä kivun helpotus ja/tai verenvuodon merkkien lievittyminen yhden infuusion jälkeen).
Pediatrinen tutkimus aiemmin hoitamattomilla potilailla, joiden ikä oli < 6 vuotta
ADYNOVI-valmistetta ei ole tarkoitettu alle 12 vuoden ikäisten lasten hoitoon (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
ADYNOVI-valmisteen turvallisuutta ja tehoa arvioitiin kaikkiaan 120:lla hoitoa saaneella, aiemmin hoitamattomalla pediatrisella potilaalla, joilla oli vaikea hemofilia A. Estohoito oli 20–50 IU/kg ADYNOVI-valmistetta vähintään kerran viikossa tai tarvittaessa kaksi tai useamman kerran viikossa, ja keskimääräinen annos (SD) oli 45,5 (7,83) IU/kg infuusiota kohti.
Negatiiviseen binomiseen yleistettyyn lineaariseen malliin (generalized linear model, GLM) perustuva vuotuinen verenvuototapahtumien kokonaismäärän keskimääräinen piste-estimaatti (95 %:n luottamusväli) oli 1,70 (1,20; 2,42) kerran viikossa annettavan estohoidon yhteydessä; 0,96 (0,70; 1,31) kaksi tai useamman kerran viikossa annettavan estohoidon yhteydessä ja 2,32 (1,67; 3,22) tarvittaessa toteutettavan hoidon yhteydessä.
Huomattavaa on, että vuotuinen verenvuototapahtumien määrä ei ole vertailukelpoinen eri tekijäpitoisuuksien ja eri kliinisten tutkimusten välillä.
Aiemmin hoitamattomilla potilailla tehdyn pediatrisen tutkimuksen aikana hoidettiin yhteensä 775 verenvuototapahtumaa. Niistä 614 (79,2 %) saatiin hallintaan 1 infuusiolla ja 87 (11,2 %) saatiin hallintaan 2 infuusiolla. 517 tehon osalta arvioidusta verenvuototapahtumasta 492:ssa (95,1 %) verenvuodon hallinta arvioitiin erinomaiseksi tai hyväksi.
Farmakokinetiikka
ADYNOVI-valmisteen farmakokinetiikkaa (FK) arvioitiin vaihtovuorotutkimuksessa oktokogi alfan valmisteen kanssa 26 tutkittavan (18 aikuisen ja 8 nuoren) osalta ja 22 tutkittavan (16 aikuisen ja 6 nuoren) osalta 6 kuukauden ADYNOVI-hoidon jälkeen. Plasman tekijä VIII:n aktiivisuus mitattiin yksivaiheisella hyytymistestillä ja kromogeenisellä testillä.
ADYNOVI-valmisteella on pidennetty, 1,4–1,5-kertainen puoliintumisaika rekombinanttiin ihmisen hyytymistekijä VIII:aan (oktokogi alfa) verrattuna nuorten ja aikuisten populaatiossa yksivaiheisen hyytymistestin ja kromogeenisen testin perusteella määritettynä. Myös AUC-arvon kasvua ja puhdistuman pienenemistä oktokogi alfa -isäntämolekyyliin verrattuna havaittiin. Asteittainen palautuminen oli vastaavaa molemmilla valmisteilla. Farmakokineettisten parametrien muutos oli samanlainen aikuisten ja nuorten populaatioissa ja yksivaiheisen hyytymistestin ja kromogeenisen substraattitestin välillä.
Pediatristen potilaiden farmakokinetiikka
39:ltä alle 18-vuotiaalta tutkittavalta lasketut farmakokineettiset parametrit (intent-to-treat-analyysi) ovat saatavilla 14 lapselta (2–5-vuotiaalta), 17 vanhemmalta lapselta (6 – < 12-vuotiaalta) ja 8 nuorelta tutkittavalta (12 – < 18-vuotiaalta). Puoliintumisajan pidennys oli pediatrisessa populaatiossa 1,3–1,5-kertainen sekä yksivaiheista hyytymistestiä että kromogeenistä testiä käytettäessä. ADYNOVI-valmisteen keskimääräinen puhdistuma (kehonpainoon perustuva) oli suurempi ja keskimääräinen puoliintumisaika pienempi alle 12-vuotiailla lapsilla kuin aikuisilla. Alle 12-vuotiaat lapset saattavat tarvita suuremman annoksen, ks. kohta Annostus ja antotapa.
Taulukko 6: Farmakokineettiset parametrit kromogeenistä hyytymistestiä käytettäessä
(Aritmeettinen keskiarvo ± SD)
Farmakokineettiset parametrit | ADYNOVI Aikuiset (vähintään 18-vuotiaat)N = 18 Annos: 45 ± 5 IU/kg | ADYNOVI Nuoret (12 – < 18-vuotiaat)N = 8 Annos: 45 ± 5 IU/kg | ADYNOVI Pediatriset potilaat (6 – < 12-vuotiaat)N = 17 Annos: 50 ± 10 IU/kg | ADYNOVI Pediatriset potilaat (< 6-vuotiaat)N = 14 Annos: 50 ± 10 IU/kg |
Terminaalinen puoliintumisaika (h) | Yksilöllinen FK ja täydellinen näytteenottoa | Populaation FK ja harva näytteenottob | ||
MRT (h) | 15,01 ± 3,89 | 13,80 ± 4,01 | 11,93 ± 2,58 | 12,99 ± 8,75 |
CL (ml/[kg·h])d | 19,70 ± 5,05 | 17,73 ± 5,44 | 17,24 ± 3,73 | 18,74 ± 12,60 |
Asteittainen palautuminen ([IU/dl]/ [IU/kg]) | 2,16 ± 0,75 | 2,58 ± 0,84 | 2,80 ± 0,67 | 3,49 ± 1,21 |
AUC0‑Inf (IU·h/dl) | 2,87 ± 0,61 | 2,34 ± 0,62 | nac (2,19 ± 0,40) | nac (1,90 ± 0,27) |
Vss (dl/kg) | 2 589 ± 848 | 1 900 ± 841 | 2 259 ± 514 | 2 190 ± 1 593 |
Cmax (IU/dl) | 0,40 ± 0,09 | 0,54 ± 0,22 | 0,46 ± 0,04 | 0,54 ± 0,03 |
Terminaalinen puoliintumisaika (h) | 145 ± 29 | 117 ± 28 | nac (130 ± 24) | nac (117 ± 16) |
Lyhenteet: Cmax = suurin havaittu aktiivisuus, AUC = käyrän alle jäävä pinta-ala, MRT = keskimääräinen viipymäaika, CL = puhdistuma, Vss = kehonpainon mukaan mukautettu jakautumistilavuus vakaassa tilassa
a Yksilöllinen FK ja 12 infuusionjälkeistä näytettä.
b Populaatiofarmakokineettinen malli ja 3 infuusionjälkeistä näytettä satunnaistetun näytteenottoaikataulun perusteella
c NA, ei sovellu, sillä asteittainen palautuminen ja lasten Cmax määritettiin yksilöllisen FK:n perusteella. Asteittaisen palautumisen ja Cmax:n tulokset, jotka määritettiin yksilöllisen FK:n mukaan, ovat suluissa.
d Tutkittavan 122001 puhdistuma-arvoa 12,18 ml/(kg·h) ikäryhmässä 12 – < 18-vuotiaat ei laskettu mukaan puhdistuma-analyysiin.
Asteittaista palautumista tutkittiin lisäksi 99:llä aiemmin hoitamattomalla < 6-vuotiaalla potilaalla, ja se pysyi suhteellisen vakaana hoidon keston ajan. Keskimääräiset (SD) IR-arvot olivat 1,670 (0,594) (IU/dl)/(IU/kg) lähtötilanteessa (n = 99), 1,617 (0,640) (IU/dl)/(IU/kg) käynnillä 3 (altistumispäiviä 15 ± 1) (n = 47), 1,805 (0,593) (IU/dl)/(IU/kg) käynnillä 6 (altistumispäiviä 40 ± 3) (n = 87) ja 1,873 (0,440) (IU/dl)/(IU/kg) tutkimuksen päättyessä/keskeytyessä (altistumispäiviä 100–110) (n = 89).
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevassa, jaavanmakakeille (cynomolgus-apinoille) tehdyssä tutkimuksessa kahdella keskikokoisen annoksen (350 IU/kg) ryhmään kuuluvalla eläimellä ilmeni vakuolisaatiota munuaisissa. Vakuolisaatio ei parantunut kahden viikon jälkeen. Prekliinisessä tutkimuksessa havaitun munuaisten vakuolisaation merkitystä ihmiselle ei tunneta.
Ei-kliiniset tiedot rajoittuvat 1 kuukauden altistukseen, eikä ADYNOVI-valmistetta ole tutkittu nuorille eläimille tehdyissä tutkimuksissa. Siksi ei ollut mahdollista määrittää PDG:n kudoksiin/elimiin kertymisen mahdollisia riskejä, jotka koskevat ADYNOVI-valmisteen pitkäaikaista käyttöä pediatristen potilaiden hoitoon. ADYNOVI-valmistetta ei ole tutkittu geenitoksisuutta, karsinogeenisuutta tai lisääntymistoksisuutta koskevissa tutkimuksissa.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine
Mannitoli (E421)
Trehaloosidihydraatti
Histidiini
Glutationi
Natriumkloridi
Kalsiumklorididihydraatti (E509)
Tris(hydroksimetyyli)aminometaani
Polysorbaatti 80 (E433)
Liuotin
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo
2 vuotta.
Ennen avaamista lääkevalmistetta voidaan säilyttää huoneenlämmössä (enintään 30 °C:ssa) enintään 3 kuukauden ajan. 3 kuukauden huoneenlämmössä säilyttämisen päättymisaika on kirjattava valmisteen pakkaukseen. Tämä päivämäärä ei saa milloinkaan olla myöhäisempi kuin alun perin ulkopakkauksessa mainittu päivämäärä. Tämän ajanjakson lopussa valmistetta ei saa laittaa takaisin jääkaappiin, vaan se on käytettävä tai hävitettävä.
Käyttökuntoon saattamisen jälkeen
Valmisteen on osoitettu säilyvän kemiallisesti ja fysikaalisesti stabiilina 3 tuntia enintään 30 °C:ssa. Mikrobiologisista syistä valmiste olisi käytettävä heti, ellei sekoitusmenetelmä sulje pois mikrobikontaminaation riskiä. Jos valmistetta ei käytetä välittömästi, käytönaikainen säilytysaika ja -olosuhteet ovat käyttäjän vastuulla. Älä säilytä kylmässä.
Säilytys
Säilytä jääkaapissa (2 °C - 8 °C).
Ei saa jäätyä.
ADYNOVI ja BAXJECT II Hi‑Flow -laite: Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
ADYNOVI BAXJECT III -järjestelmässä: Pidä suljettu läpipainopakkaus ulkopakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ADYNOVI injektiokuiva-aine ja liuotin, liuosta varten
1000 IU (L:ei) 1 kpl (1000 IU+2 ml (BaxJect III)) (747,11 €)
PF-selosteen tieto
Tyypin I lasinen injektiopullo, joka on suljettu klorobutyylikumitulpalla ja joka sisältää 250 IU, 500 IU tai 1 000 IU kuiva-ainetta.
Tyypin I lasinen injektiopullo, joka on suljettu klorobutyyli- tai bromobutyylikumitulpalla ja joka sisältää 2 ml injektionesteisiin käytettävää vettä.
Tämä lääkevalmiste toimitetaan jommassakummassa seuraavista pakkaustyypeistä:
- ADYNOVI ja BAXJECT II Hi‑Flow -laite: Yksi pakkaus sisältää kuiva-aineinjektiopullon ja liuotininjektiopullon sekä laitteen käyttökuntoon saattamista varten (BAXJECT II Hi‑Flow).
- ADYNOVI BAXJECT III -järjestelmässä: Yksi pakkaus sisältää käyttövalmiin BAXJECT III -järjestelmän suljetussa läpipainopakkauksessa, jossa on kuiva-aineinjektiopullo ja liuotininjektiopullo, jotka on jo koottu yhteen käyttökuntoon saattamista varten.
Valmisteen kuvaus:
Kuiva-aine: Valkoinen tai vaalea irtonainen jauhe.
Liuotin: Kirkas ja väritön liuos.
Käyttö- ja käsittelyohjeet
Käyttövalmiiksi saatettu lääkevalmiste on tarkistettava silmämääräisesti. Siinä ei saa näkyä ennen antamista hiukkasia tai väriä. Liuoksen tulee olla kirkasta tai lievästi opalisoivaa. Älä käytä sameita liuoksia tai liuoksia, joissa on saostumia.
Käyttövalmiiksi saatetun liuoksen pH on 6,7–7,3. Osmolaliteetti on ≥380 mOsm/kg.
Valmistelu ja käyttökuntoon saattaminen BAXJECT II Hi‑Flow -laitteen avulla:
Käytä käyttövalmiiksi saattamisessa vain pakkauksen mukana toimitettua liuotininjektiopulloa ja sekoituslaitetta.
1. Käytä käyttökuntoon saattamisen aikana antiseptista (puhdasta ja vähäbakteerista) tekniikkaa ja tasaista työskentelypintaa.
2. Anna kuiva-aine- ja liuotininjektiopullojen lämmetä huoneenlämpöisiksi (15 °C – 25 °C) ennen käyttöä.
3. Poista muovikorkit kuiva-aine- ja liuotininjektiopulloista.
4. Puhdista kumitulpat desinfiointipyyhkeellä ja anna kuivua ennen käyttöä.
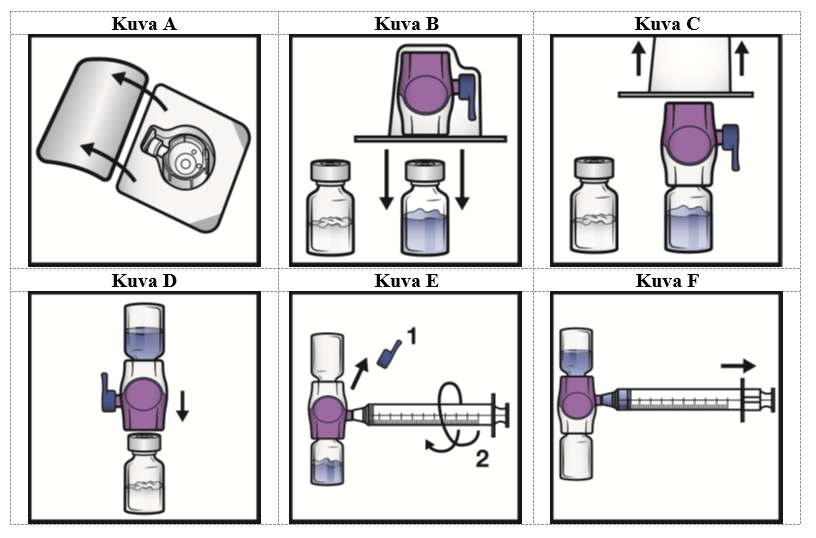
5. Avaa BAXJECT II Hi‑Flow -laitteen pakkaus vetämällä kansi pois ilman, että kosketat sisäpuolta (kuva A). Älä poista laitetta pakkauksesta.
6. Käännä pakkaus ylösalaisin. Työnnä läpinäkyvä muovipiikki kokonaan suoraan alas liuotininjektiopullon tulpan läpi (kuva B).
7. Tartu BAXJECT II Hi‑Flow -pakkaukseen sen reunasta ja vedä pakkaus irti laitteesta (kuva C). Älä poista sinistä korkkia BAXJECT II Hi‑Flow -laitteesta. Älä kosketa esillä olevaa purppuranpunaista muovipiikkiä.
8. Käännä järjestelmä ylösalaisin siten, että liuotininjektiopullo on yläpuolella. Työnnä purppuranpunainen muovipiikki nopeasti kokonaan suoraan alas kuiva-aineinjektiopullon tulpan läpi (kuva D). Tyhjiö vetää liuottimen kuiva-aineinjektiopulloon.
9. Pyöritä varovasti, kunnes kuiva-aine on liuennut kokonaan. Älä säilytä valmistetta jääkaapissa käyttökuntoon saattamisen jälkeen.
Antaminen
- Tarkista ennen antamista silmämääräisesti, ettei käyttökuntoon saatetussa liuoksessa näy hiukkasia tai värinmuutoksia.
Käyttökuntoon saatettu liuos näyttää kirkkaalta ja värittömältä.
- Älä käytä sitä, jos siinä näkyy hiukkasia tai värinmuutoksia.
Anna liuos mahdollisimman pian, mutta ei yli 3 tuntia käyttökuntoon saattamisen jälkeen.
Antamisen vaiheet
1. Poista sininen korkki BAXJECT II Hi‑Flow -laitteesta (kuva E). Älä vedä ilmaa ruiskuun. Liitä ruisku BAXJECT II Hi‑Flow -laitteeseen. Luer-lock-ruiskun käyttöä suositellaan.
2. Käännä järjestelmä ylösalaisin (kuiva-aineinjektiopullo on nyt yläpuolella). Vedä käyttökuntoon saatettu liuos ruiskuun vetämällä mäntää taakse hitaasti (kuva F).
3. Irrota ruisku. Liitä sopiva neula ja injektoi laskimoon. Jos potilaan on tarkoitus saada enemmän kuin yksi injektiopullo ADYNOVI-valmistetta, samaan ruiskuun voi vetää usean injektiopullon sisällön.
Kunkin ADYNOVI-injektiopullon sekoittamiseen liuottimen kanssa tarvitaan erillinen BAXJECT II Hi‑Flow -laite.
4. Anna valmiste enintään 5 minuutin kuluessa (enimmäisinfuusionopeus on 10 ml/min).
Valmisteen nimen ja eränumeron kirjaaminen aina ADYNOVI-valmistetta annettaessa on erittäin suositeltavaa. Kuiva-aineinjektiopullossa on repäisyetiketit.
Käyttökuntoon saattaminen BAXJECT III ‑järjestelmän avulla
Ei saa käyttää, jos läpipainopakkauksen kansi ei ole tiiviisti kiinni.
1. Jos valmistetta säilytetään vielä jääkaapissa, ota suljettu läpipainopakkaus (sisältää kuiva-aine- ja liuotininjektiopullot esikoottuina käyttökuntoon saattamiseen tarkoitetun järjestelmän kanssa) jääkaapista ja anna sen lämmetä huoneenlämpöiseksi (15 °C – 25 °C).
2. Pese kätesi huolellisesti saippualla ja lämpimällä vedellä.
3. Avaa ADYNOVI-läpipainopakkaus poistamalla kansi. Poista BAXJECT III ‑järjestelmä läpipainopakkauksesta.
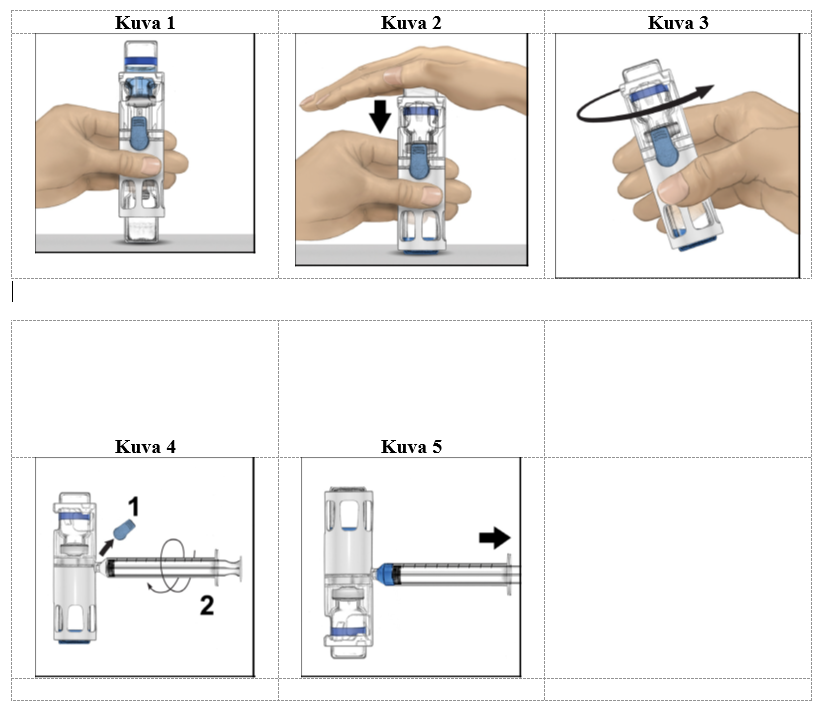
4. Aseta kuiva-aineinjektiopullo tasaiselle pinnalle niin, että liuotininjektiopullo on ylimpänä (kuva 1). Liuotininjektiopullossa on sininen raita. Älä poista sinistä korkkia, ennen kuin myöhemmässä vaiheessa niin neuvotaan.
5. Pitele kuiva-aineinjektiopulloa toisella kädellä BAXJECT III ‑järjestelmässä ja paina liuotininjektiopulloa tasaisesti alaspäin toisella kädellä, kunnes järjestelmä on painunut kokonaan kasaan ja liuotin virtaa kuiva-aineinjektiopulloon (kuva 2). Älä kallista järjestelmää, ennen kuin siirto on valmis.
6. Tarkista, että liuottimen siirto on valmis. Pyöritä varovasti, kunnes kaikki materiaali on liuennut (kuva 3). Varmista, että kuiva-aine on liuennut kokonaan. Muuten kaikki käyttökuntoon saatettu liuos ei kulkeudu laitteen suodattimen läpi. Valmiste liukenee nopeasti (tavallisesti alle minuutissa). Käyttökuntoon saattamisen jälkeen liuoksen pitäisi olla kirkasta ja väritöntä eikä siinä saa näkyä hiukkasia.
Antaminen
- Tarkista ennen antamista silmämääräisesti, ettei käyttökuntoon saatetussa liuoksessa näy hiukkasia tai värinmuutoksia.
Käyttökuntoon saatettu liuos näyttää kirkkaalta ja värittömältä.
- Älä käytä sitä, jos siinä näkyy hiukkasia tai värinmuutoksia.
Anna liuos mahdollisimman pian, mutta ei yli 3 tuntia käyttökuntoon saattamisen jälkeen.
Antamisen vaiheet
1. Poista sininen korkki BAXJECT III -laitteesta (kuva 4). Älä vedä ilmaa ruiskuun. Liitä ruisku BAXJECT III -laitteeseen. Luer-lock-ruiskun käyttöä suositellaan.
2. Käännä järjestelmä ylösalaisin (kuiva-aineinjektiopullo on nyt yläpuolella). Vedä käyttökuntoon saatettu liuos ruiskuun vetämällä mäntää taakse hitaasti (kuva 5).
3. Irrota ruisku. Liitä sopiva neula ja injektoi laskimoon. Jos potilaan on tarkoitus saada enemmän kuin yksi injektiopullo ADYNOVI-valmistetta, samaan ruiskuun voi vetää usean injektiopullon sisällön.
4. Anna valmiste enintään 5 minuutin kuluessa (enimmäisinfuusionopeus on 10 ml/min).
Valmisteen nimen ja eränumeron kirjaaminen aina ADYNOVI-valmistetta annettaessa on erittäin suositeltavaa. Läpipainopakkauksessa on repäisyetiketit.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ADYNOVI injektiokuiva-aine ja liuotin, liuosta varten
1000 IU 1 kpl
- Ylempi erityiskorvaus (100 %). Krooniset hyytymishäiriöt (126).
- Peruskorvaus (40 %).
ATC-koodi
B02BD02
Valmisteyhteenvedon muuttamispäivämäärä
07.04.2026
Yhteystiedot
 TAKEDA OY
TAKEDA OY PL 1406, Ilmalankuja 3
00101 Helsinki
0800 774 051
www.takeda.fi
etunimi.sukunimi@takeda.com