
VELTASSA jauhe oraalisuspensiota varten 1 g, 8,4 g, 16,8 g
Vaikuttavat aineet ja niiden määrät
Veltassa 1 g jauhe oraalisuspensiota varten
Yksi annospussi sisältää 1 g patiromeeria (patiromeerisorbiteksikalsiumina).
Veltassa 8,4 g jauhe oraalisuspensiota varten
Yksi annospussi sisältää 8,4 g patiromeeria (patiromeerisorbiteksikalsiumina).
Veltassa 16,8 g jauhe oraalisuspensiota varten
Yksi annospussi sisältää 16,8 g patiromeeria (patiromeerisorbiteksikalsiumina).
Veltassa 25,2 g jauhe oraalisuspensiota varten
Yksi annospussi sisältää 25,2 g patiromeeria (patiromeerisorbiteksikalsiumina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Jauhe oraalisuspensiota varten.
Kliiniset tiedot
Käyttöaiheet
Veltassa on tarkoitettu aikuisten ja 12–17‑vuotiaiden nuorten hyperkalemian hoitoon.
Annostus ja antotapa
Veltassa alkaa vaikuttaa 4 – 7 tuntia annon jälkeen. Sitä ei saa käyttää korvaamaan hengenvaarallisen hyperkalemian ensihoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Veltassa-valmistetta otetaan kerran vuorokaudessa. Suositeltu Veltassa-aloitusannos vaihtelee iän mukaan. Halutun annoksen saavuttamiseksi voidaan käyttää useita annospusseja.
Vuorokausiannosta voidaan sovittaa yhden viikon välein tai harvemmin seerumin kaliumpitoisuuden ja halutun tavoitealueen perusteella. Seerumin kaliumpitoisuutta on seurattava kliinisen tarpeen mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Hoitavan lääkärin on määritettävä hoidon kesto yksilöllisesti seerumin kaliumpitoisuuden hallintatarpeen perusteella. Jos seerumin kaliumpitoisuus laskee halutun alueen alapuolelle, annosta pitää pienentää tai valmisteen käyttö lopettaa.
Veltassa-valmisteen ja muiden suun kautta otettavien lääkevalmisteiden annon välissä on oltava 3 tuntia (ks. kohta Yhteisvaikutukset).
Aikuiset
Suositeltu aloitusannos on 8,4 g patiromeeria kerran vuorokaudessa. Vuorokausiannosta voidaan suurentaa tai pienentää 8,4 g:lla tarvittaessa halutun tavoiteannoksen saavuttamiseksi aina 25,2 g:n enimmäisvuorokausiannokseen asti.
12–17‑vuotiaat nuoret
Suositeltu aloitusannos on 4 g patiromeeria kerran vuorokaudessa. Patiromeerin vuorokausiannosta tulee säätää seerumin kaliumpitoisuuden ja halutun tavoiteannoksen perusteella aina 25,2 g:n enimmäisvuorokausiannokseen asti. Jos potilas tarvitsee yli 7 g:n annoksia, suositellaan siirtymistä 8,4 g:n patiromeeriannospusseihin.
Annoksen unohtuminen
Jos annos unohdetaan ottaa, unohdettu annos on otettava mahdollisimman pian samana päivänä. Unohdettua annosta ei pidä ottaa seuraavan annoksen kanssa.
Erityisryhmät
Iäkkäät
Tälle ryhmälle ei ole suositeltu erityisiä annos- ja anto-ohjeita.
Dialyysihoitoa saavat potilaat
Patiromeerin käytöstä dialyysipotilaille on rajoitetusti tietoja. Näihin potilaisiin ei sovellettu mitään erityisiä annos- tai anto-ohjeita kliinisissä tutkimuksissa. Dialyysihoitoa saavia pediatrisia potilaita ei ole hoidettu patiromeerilla.
Loppuvaiheen munuaissairautta (ESRD) sairastavat potilaat
Patiromeeria on tutkittu vain rajallisella määrällä potilaita, joilla glomerulusten arvioitu suodatusnopeus (eGFR) on < 15 ml/min/1,73 m².
Pediatriset potilaat
Patiromeerin turvallisuutta ja tehoa alle 12 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. 12–17-vuotiaista nuorista saadut tiedot rajoittuvat 6 kuukautta kestäneeseen altistukseen. Varovaisuutta on siis noudatettava, jos 12–17‑vuotiaille nuorille annetaan hoitoa yli 6 kuukauden ajan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Antotapa
Suun kautta.
Veltassa on sekoitettava veteen ja sekoitettava koostumukseltaan tasaiseksi suspensioksi. Suspension valmistamiseen suositellut kokonaisvesimäärät riippuvat annoksesta:
- 1 g patiromeeria: 10 ml
- 2 g patiromeeria: 20 ml
- 3 g patiromeeria: 30 ml
- 4 g patiromeeria: 40 ml
- > 4 g patiromeeria: 80 ml.
Suspensio on valmistettava seuraavien ohjeiden mukaan:
- Puolet tarvittavaan annokseen suositellusta vesimäärästä kaadetaan lasiin, minkä jälkeen siihen lisätään koko patiromeeriannos ja sekoitetaan.
- Sitten lisätään loput suositellusta vesimäärästä, ja suspensiota sekoitetaan jälleen huolellisesti.
Jauhe ei liukene. Seokseen voi lisätä tarvittaessa lisää vettä halutun koostumuksen saavuttamiseksi. Suuremmat vesimäärät voivat kuitenkin nopeuttaa jauheen sakkautumista.
Seos pitää ottaa 1 tunnin sisällä suspension valmistamisesta. Jos jauhetta jää lasiin juomisen jälkeen, siihen on lisättävä enemmän vettä, suspensiota on sekoitettava ja se on otettava välittömästi. Tämä voidaan toistaa tarvittaessa, jotta koko annos on varmasti annettu.
Seoksen valmistamiseen voi veden sijasta käyttää omien mieltymysten mukaan seuraavia nesteitä tai pehmeitä ruokia noudattamalla samoja vaiheita kuin edellä on kuvattu: omenamehua, karpalomehua, ananasmehua, appelsiinimehua, rypälemehua, päärynämehua, aprikoosinektaria, persikkanektaria, jogurttia, maitoa, sakeuttamisainetta (esimerkiksi maissitärkkelystä), omenasosetta, vanilja- ja suklaavanukasta.
Seoksen valmistukseen käytettyjen nesteiden ja pehmeiden ruokien kaliumpitoisuus jokaisen yksittäisen potilaan kaliumin saantia koskevat ruokavaliosuositukset huomioon ottaen.
Ylipäätään karpalomehun nauttiminen pitää rajoittaa kohtuullisiin määriin (esimerkiksi alle 400 ml vuorokaudessa), koska sillä voi olla yhteisvaikutuksia muiden lääkevalmisteiden kanssa.
Tämän lääkkeen voi ottaa aterian yhteydessä tai aterioista erillään. Sitä ei pidä kuumentaa (esim. mikroaaltouunissa) tai lisätä kuumiin ruokiin tai nesteisiin. Sitä ei pidä ottaa kuivassa muodossa.
Anto nenämahaletkun tai ravinnonantoletkun (PEG-letkun) kautta:
Jos patiromeeriannos on enintään 8,4 g, suspensio valmistetaan edellä suun kautta annon osiossa kuvatulla tavalla. Yli 8,4 g:n mutta korkeintaan 16,8 g:n patiromeeriannoksen kohdalla kokonaismäärän tulee olla 160 ml, ja yli 16,8 g:n mutta korkeintaan 25,2 g:n patiromeeriannoksen kohdalla kokonaismäärän tulee olla 240 ml. Näillä määrillä varmistetaan, että suspensio kulkee letkustossa helposti. Yhteensopivuus polyuretaanista, silikonista ja polyvinyylikloridista valmistettujen letkujen kanssa on osoitettu. Letkujen suositeltu halkaisija on vähintään 2,17 mm (6,5 Fr). Valmisteen annon jälkeen letku on huuhdeltava vedellä. Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet hävittämisestä.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Matala magnesiumpitoisuus
Kliinisissä tutkimuksissa 7,1 %:lla patiromeerilla hoidetuista aikuispotilaista seerumin magnesiumarvot olivat < 1,4 mg/dl ( 0,58 mmol/l), ja 0,3 %:lla seerumin magnesiumpitoisuus laski < 1,0 mg/dl (0,4 mmol/l). Keskimääräiset seerumin magnesiumpitoisuuden laskut olivat 0,137 mg/dl (0,0564 mmol/l) tai vähemmän. Pediatrisilla potilailla tehdyssä kliinisessä tutkimuksessa seerumin magnesiumpitoisuuden keskimääräinen lasku viikolla 26 oli 0,35 mg/dl (0,1440 mmol/l). Pediatrisen kliinisen tutkimuksen aikana yhdenkään potilaan seerumin magnesiumpitoisuus ei laskenut tasolle < 1,4 mg/dl (0,58 mmol/l).
Seerumin magnesiumpitoisuutta on seurattava vähintään 1 kuukauden ajan hoidon aloittamisesta sekä hoidon aikana kliinisen tarpeen mukaan, ja magnesiumlisää on harkittava potilaille, joiden seerumin magnesiumpitoisuus pienenee (ks. kohta Haittavaikutukset).
Ruoansulatuselimistö
Kliinisiin tutkimuksiin ei osallistunut potilaita, joilla oli aiemmin ollut suolitukos, vaikeita ruoansulatuselimistön häiriöitä tai nielemishäiriöitä tai joille oli tehty suuri ruoansulatuselimistön leikkaus. Muiden kaliumin sitojien kanssa on raportoitu ruoansulatuskanavan iskemiaa, nekroosia ja/tai suolten puhkeamista. Patiromeerin annon hyötyjä ja riskejä on harkittava tarkoin ennen hoitoa ja hoidon aikana aikuisilla ja pediatrisilla potilailla, joilla on tai on ollut vakavia ruoansulatuselimistön häiriöitä.
Patiromeerihoidon lopettaminen
Kun patiromeerihoito lopetetaan, seerumin kaliumpitoisuus voi nousta johtaen toistuvaan hyperkalemiaan, erityisesti, jos reniini-angiotensiini-aldosteronijärjestelmää (RAA) estävää hoitoa jatketaan. Potilaita on neuvottava olemaan lopettamatta hoitoa keskustelematta ensin lääkärin kanssa. Seerumin kaliumpitoisuus voi nousta jo 2 vuorokauden kuluttua viimeisen patiromeeriannoksen jälkeen. Pediatristen potilaiden seerumin kaliumpitoisuuksista patiromeerihoidon lopettamisen yhteydessä on vain vähän tietoa.
Seerumin kaliumpitoisuus
Seerumin kaliumpitoisuutta on seurattava tavanomaisten käytäntöjen mukaan, kun se on kliinisesti aiheellista, muun muassa sen jälkeen, kun on tehty muutoksia seerumin kaliumpitoisuuteen vaikuttaviin lääkevalmisteisiin (esim. RAAS‑estäjät tai diureetit) ja sen jälkeen, kun patiromeeriannosta on sovitettu tai hoito on lopetettu.
Kliinisten tietojen rajoitukset
Vakava hyperkalemia
Kokemukset ovat rajallisia potilaista, joiden seerumin kaliumpitoisuus on suurempi kuin 6,5 mmol/l. Pediatrisessa populaatiossa kokemukset rajoittuvat potilaisiin, joiden seerumin kaliumpitoisuus on enintään 6,2 mmol/l. Veltassa-valmistetta ei saa käyttää hengenvaarallisen hyperkalemian ensihoitona, koska sen vaikutus alkaa viiveellä (ks. kohta Annostus ja antotapa).
Pitkäaikainen altistuminen
Kliinisissä tutkimuksissa vähintään yhden vuoden ajan altistuneista aikuisista on vain vähän tietoja. Pediatrisilla potilailla tehtyihin kliinisiin tutkimuksiin ei ole liittynyt yli 6 kuukautta pidempää altistumista. Varovaisuutta on siis noudatettava, jos 12–17‑vuotiaille nuorille annetaan hoitoa yli 6 kuukauden ajan.
Tietoa sorbitolista
Veltassa sisältää sorbitolia osana vastaionikompleksia. Sorbitolipitoisuus on noin 4 g (10,4 kcal) per 8,4 g patiromeeria ja noin 0,5 g (1,2 kcal) per 1 g patiromeeria. Potilaiden, joilla on perinnöllinen fruktoosi-intoleranssi, ei pidä käyttää tätä lääkettä.
Tietoa kalsiumista
Veltassa sisältää kalsiumia osana vastaionikompleksia. Kalsium vapautuu osittain ja osa siitä voi imeytyä (ks. kohta Farmakodynamiikka). Tämän lääkevalmisteen annon hyötyjä ja riskejä on harkittava tarkoin aikuisilla ja pediatrisilla potilailla, joilla on hyperkalsemian riski. Seerumin kalsiumpitoisuutta on seurattava vähintään 1 kuukauden ajan hoidon aloittamisesta sekä hoidon aikana kliinisen tarpeen mukaan.
Yhteisvaikutukset
Patiromeerin vaikutus muihin lääkevalmisteisiin
Patiromeeri voi sitoa joitakin suun kautta samanaikaisesti otettavia lääkevalmisteita, mikä voi heikentää niiden imeytymistä ruoansulatuskanavasta. Samanaikaisesti annettujen lääkevalmisteiden hyötyosuuden suurenemista ei todettu lääkkeiden yhteisvaikutuksia selvittäneissä tutkimuksissa. Patiromeeri ei imeydy eikä metaboloidu elimistössä, joten sen vaikutukset muiden lääkevalmisteiden toimintaan ovat vähäisiä.
Varotoimenpiteenä ja alla olevien tietojen perusteella patiromeerin ja muiden suun kautta otettavien lääkevalmisteiden annon välillä on siksi oltava vähintään 3 tuntia.
In vivo -tutkimukset
Patiromeerin samanaikainen anto ei vaikuttanut amlodipiinin, sinakalseetin, klopidogreelin, furosemidin, litiumin, metoprololin, trimetopriimin, verapamiilin ja varfariinin hyötyosuuteen mitattuna käyrän alapuolisella alueella (AUC). Näiden lääkevalmisteiden ja patiromeerin ottamisen välillä ei tarvitse pitää taukoa.
Patiromeerin samanaikainen anto johti siprofloksasiinin, levotyroksiinin ja metformiinin heikentyneeseen hyötyosuuteen. Yhteisvaikutuksia ei kuitenkaan ilmennyt, kun patiromeerin ja näiden lääkevalmisteiden ottamisen välinen aika oli 3 tuntia.
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
In vitro -tutkimukset
In vitro -tutkimukset eivät ole osoittaneet, että patiromeerilla olisi mahdollisia yhteisvaikutuksia seuraavien vaikuttavien aineiden kanssa: allopurinoli, amoksisilliini, apiksabaani, asetyylisalisyylihappo, atorvastatiini, atsilsartaani, benatsepriili, bumetanidi, kanagliflotsiini, kandesartaani, kaptopriili, kefaleksiini, dapagliflotsiini, digoksiini, empagliflotsiini, enalapriili, eplerenoni, finerenoni, fosinopriili, glipitsidi, irbesartaani, lisinopriili, losartaani, olmesartaani, perindopriili, fenytoiini, kinapriili, ramipriili, riboflaviini, rivaroksabaani, sakubitriili, sevelameeri, spironolaktoni, takrolimuusi, torasemidi, trandolapriili ja valsartaani.
In vitro -tutkimukset ovat osoittaneet, että patiromeerilla saattaa olla yhteisvaikutuksia bisoprololin, karvedilolin, mykofenolaattimofetiilin, nebivololin, kinidiinin ja telmisartaanin kanssa.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja patiromeerin käytöstä raskaana oleville naisille.
Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Varmuuden vuoksi patiromeerin käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei ole odotettavissa vaikutuksia vastasyntyneisiin tai imeväisiin, sillä patiromeerin systeeminen altistus imettävälle naiselle on merkityksetön. On päätettävä, lopetetaanko imetys vai lopetetaanko/vältetäänkö patiromeerihoito ottaen huomioon imetyksestä aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Patiromeerin vaikutuksesta ihmisten hedelmällisyyteen ei ole tietoa. Eläinkokeissa ei ole havaittu vaikutuksia lisääntymiskykyyn tai hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Patiromeerilla ei ole haitallista vaikutusta tai on vain vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Suurin osa tutkimuksissa aikuispotilailla raportoiduista haittavaikutuksista (AR) oli hypomagnesemia (1,8 %) ja ruoansulatuselimistön häiriöitä, ja useimmin raportoidut haittavaikutukset olivat ummetus (3,7 %), ripuli (3 %), vatsakipu (1,4 %), pahoinvointi (1,3 %) ja ilmavaivat (1 %). Ruoansulatuselimistön häiriöt olivat yleensä lieviä tai kohtalaisen vaikeita, ne eivät vaikuttaneet olevan suhteessa annokseen, ne paranivat yleensä spontaanisti tai hoidon avulla, eikä yhdenkään raportoitu olleen vakava. Hypomagnesemia oli lievää tai kohtalaisen vaikeaa, ja 0,3 %:lla potilaista seerumin magnesiumpitoisuus aleni tasolle < 0,4 mmol/l (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa raportoidut haittavaikutukset luokitellaan seuraavassa elinjärjestelmän ja yleisyyden mukaan. Yleisyydet määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
MedDRA-elinjärjestelmä | Yleinen
| Melko harvinainen
| Tuntematon | ||
|---|---|---|---|---|---|
Immuunijärjestelmä |
|
| Yliherkkyys(1,2) | ||
Aineenvaihdunta ja ravitsemus | Hypomagnesemia |
|
| ||
| Ruoansulatuselimistö | Ummetus(3)* Ripuli(4)* Vatsakipu(5) Pahoinvointi Ilmavaivat* | Oksentelu |
| ||
* Haittavaikutuksia raportoitiin myös pediatrisissa kliinisissä tutkimuksissa
1 Markkinoille tulon jälkeen raportoidut haittavaikutukset.
2 Yliherkkyysreaktioihin sisältyivät ihottuma, nokkosihottuma, suuontelon ja huulten turvotus, ja ne olivat lieviä tai keskivaikeita.
3 Ummetus on yhdistelmätermi, johon sisältyvät ensisijaiset termit ummetus ja kova uloste.
4 Ripuli on yhdistelmätermi, johon sisältyvät ripuli ja tiheä suolen toimintaa.
5 Vatsakipu on yhdistelmätermi, johon sisältyvät suositeltavat termit vatsavaivat, vatsakipu, ylävatsakipu ja alavatsakipu.
Pediatriset potilaat
Patiromeerin turvallisuutta hyperkalemian hoidossa on tutkittu yhdessä tutkimuksessa, johon osallistui 23 pediatrista iältään 6–17‑vuotiasta potilasta. Pediatrisilla potilailla todettu haittavaikutusprofiili vastasi suurilta osin aikuisilla todettua turvallisuusprofiilia. Patiromeerin turvallisuutta ei ole tutkittu alle 6‑vuotiailla potilailla.
Haittavaikutuksia raportoitiin yhteensä 4 tutkittavalla, joista kolme kuului 12 – < 18‑vuotiaiden ikäryhmään ja yksi 6 – < 12‑vuotiaiden ikäryhmään. Kahdella näistä tutkittavista haittavaikutukset olivat ruoansulatuselimistön haittavaikutusten ryhmään kuuluvia eli ripulia, ummetusta, tiheää ulostamista ja ilmavaivoja. Muita haittavaikutuksia olivat veren kalsiumpitoisuuden suureneminen ja hypokalemia. Haittavaikutukset eivät olleet vakavia, ja ne olivat vaikeusasteeltaan lieviä tai keskivaikeita.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Koska patiromeerin yliannostus voi aiheuttaa hypokalemiaa, seerumin kaliumpitoisuutta on seurattava. Patiromeeri poistuu elimistöstä noin 24 – 48 tunnissa ruoansulatuskanavan keskimääräisen läpikulkuajan perusteella. Jos lääketieteellisten toimenpiteiden katsotaan olevan tarpeen, seerumin kaliumpitoisuuden ennalleen palauttamiseen tarvittavia asianmukaisia toimenpiteitä voidaan harkita.
Pediatriset potilaat
Patiromeeria ei ole tutkittu 12–17-vuotiailla nuorilla yli 25,2 g:n vuorokausiannoksina eikä 6 – < 12‑vuotiailla lapsilla yli 12 g:n vuorokausiannoksina. Koska liian suuret patiromeeriannokset voivat aiheuttaa hypokalemiaa, seerumin kaliumpitoisuutta on seurattava. Jos lääketieteellisten toimenpiteiden katsotaan olevan tarpeen, seerumin kaliumpitoisuuden ennalleen palauttamiseen tarvittavia asianmukaisia toimenpiteitä voidaan harkita.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: hyperkalemia- ja hyperfosfatemialääkkeet. ATC-koodi: V03AE09
Vaikutusmekanismi
Patiromeeri on imeytymätön kationinvaihtajapolymeeri, joka sisältää kalsiumsorbitoliyhdistelmää vastaionina.
Patiromeeri lisää kaliumin erittymistä ulosteeseen sitomalla kaliumia maha-suolikanavan luumenissa. Kaliumin sitoutuminen vähentää vapaan kaliumin pitoisuutta ruoansulatuskanavan luumenissa, mistä seuraa seerumin kaliumpitoisuuden pieneneminen.
Farmakodynaamiset vaikutukset
Terveillä aikuisilla tutkittavilla patiromeeri lisäsi suhteessa annokseen kaliumin erittymistä ulosteeseen ja vastaavasti vähensi kaliumin erittymistä virtsaan ilman, että seerumin kaliumpitoisuudessa tapahtui muutosta. 25,2 g patiromeeria annettuna kerran vuorokaudessa 6 vuorokauden ajan lisäsi kaliumin erittymistä ulosteeseen keskimäärin 1 283 mg/vrk ja vähensi kaliumin erittymistä virtsaan keskimäärin 1 438 mg/vrk. Kalsiumin erittyminen vuorokausivirtsaan lisääntyi lähtötilanteesta 53 mg/vrk.
Avoimessa tutkimuksessa, jossa arvioitiin vaikutuksen alkamiseen kuluvaa aikaa, seerumin kaliumpitoisuuden tilastollisesti merkittävää pienenemistä havaittiin hyperkaleemisilla potilailla 7 tuntia ensimmäisen annoksen jälkeen. Patiromeerihoidon lopettamisen jälkeen kaliumpitoisuus pysyi vakaana 24 tuntia viimeisen annoksen jälkeen, minkä jälkeen se kohosi jälleen 4 vuorokauden seurantajakson aikana.
Kliininen teho ja turvallisuus
Vaiheen 2 ja 3 kliinisissä tutkimuksissa kaikkiaan 99,5 % potilaista sai lähtötilanteessa hoitona RAA-järjestelmän estäjää, 87,0 %:lla potilaista oli krooninen munuaissairaus, jossa eGFR oli < 60 ml/min/1,73 m2, 65,6 %:lla diabetes mellitus ja 47,5 %:lla sydämen vajaatoiminta.
Tutkimus 1 (OPAL-HK)
Patiromeerin turvallisuus ja teho osoitettiin kaksiosaisessa, yksöissokkoutetussa, satunnaistetussa hoidon lopettamista koskeneessa tutkimuksessa, jossa arvioitiin tätä hoitoa hyperkaleemisten, kroonista munuaissairautta sairastavien aikuispotilaiden hoidossa vähintään yhden RAA-järjestelmän estäjän (eli angiotensiiniä konvertoivan entsyymin estäjän [ACEI], angiotensiini II:n reseptorin salpaajan [ARB] tai mineralokortikosteroidireseptorin antagonistin [MRA]) sisältävillä vakailla annoksilla.
Osassa A 243 potilasta sai patiromeerihoitoa 4 viikon ajan. Potilaat, joiden seerumin kaliumpitoisuus oli lähtötilanteessa 5,1 – < 5,5 mmol/l (mEql/l), saivat aloitusannoksena 8,4 g patiromeeria vuorokaudessa (jaettuna annoksena), ja potilaat, joiden seerumin kaliumpitoisuus oli lähtötilanteessa 5,5 – < 6,5 mmol/l, saivat aloitusannoksena 16,8 g patiromeeria vuorokaudessa (jaettuna annoksena). Annos sovitettiin tarvittaessa seerumin kaliumpitoisuuden perusteella aloittaen arvioinnit päivänä 3 ja sitten viikoittaisilla käynneillä 4 viikon hoitojakson loppuun asti. Tavoitteena oli säilyttää seerumin kaliumpitoisuus tavoitealueella (3,8 – < 5,1 mmol/l). Patiromeerin keskimääräiset vuorokausiannokset olivat 13 g potilaille, joiden seerumin kaliumpitoisuus oli 5,1 – < 5,5 mmol/l, ja 21 g potilaille, joiden seerumin kaliumpitoisuus oli 5,5 – < 6,5 mmol/l.
Potilaiden keski-ikä oli 64 vuotta (54 % oli vähintään 65‑vuotiaita, 17 % vähintään 75‑vuotiaita), 58 % potilaista oli miehiä ja 98 % valkoihoisia. Noin 97 %:lla potilaista oli korkea verenpaine, 57 %:lla oli tyypin 2 diabetes ja 42 %:lla oli sydämen vajaatoiminta.
Taulukossa 1 esitetään seerumin keskimääräiset kaliumpitoisuudet ja seerumin kaliumpitoisuuksien muutokset osan A lähtötilanteesta osan A viikkoon 4. Osan A toissijaisena tuloksena 76 %:lla (95 %:n CI: 70 %, 81 %) potilaista seerumin kaliumpitoisuus oli tavoitealueella 3,8 – < 5,1 mmol/l osan A viikolla 4.
Taulukko 1: Patiromeerihoitovaihe (osa A): ensisijainen päätetapahtuma
| Lähtötilanteen kalium | Koko ryhmä (n = 237) | ||
5,1 – < 5,5 mmol/l (n = 90) | 5,5 – < 6,5 mmol/l (n = 147) | ||
| Seerumin kaliumpitoisuus (mmol/l) | |||
| Lähtötilanne, keskiarvo (SD) | 5,31 (0,57) | 5,74 (0,40) | 5,58 (0,51) |
| Viikon 4 muutos lähtötilanteesta, keskiarvo ± SE (95 %:n CI) | −0,65 ± 0,05 (−0,74, −0,55) | −1,23 ± 0,04 (−1,31, −1,16) | −1,01 ± 0,03 (−1,07, −0,95) |
| p‑arvo | < 0,001 | ||
Osassa B 107 potilasta, joiden seerumin kaliumpitoisuus oli osan A lähtötilanteessa 5,5 – < 6,5 mmol/l, joiden seerumin kaliumpitoisuus oli tavoitealueella (3,8 – < 5,1 mmol/l) osan A viikolla 4 ja jotka saivat yhä RAA-järjestelmää estävää hoitoa, satunnaistettiin jatkamaan patiromeerihoitoa tai saamaan lumelääkettä 8 viikon ajan, minkä tarkoituksena oli arvioida patiromeerihoidon lopettamisen vaikutusta seerumin kaliumpitoisuuteen. Patiromeerihoitoon satunnaistettujen potilaiden vuorokausiannos oli 21 g osan B alussa ja osan B aikana.
Osan B ensisijainen päätetapahtuma oli seerumin kaliumpitoisuuden muutos osan B lähtötilanteesta ensimmäiseen käyntiin, jolla potilaan seerumin kaliumpitoisuus oli alueen 3,8 – < 5,5 mmol/l ulkopuolella, tai osan B viikkoon 4, jos potilaan seerumin kaliumpitoisuus pysyi alueen sisällä. Osassa B lumelääkettä saavien potilaiden seerumin kaliumpitoisuus nousi merkitsevästi suhteessa patiromeerihoitoa jatkaneisiin potilaisiin (p < 0,001).
Seerumin kaliumpitoisuus oli lumelääkettä saaneilla potilailla useammin ≥ 5,1 mmol/l milloin tahansa osan B aikana (91 % [95 %:n CI: 83 %, 99 %]) kuin patiromeeripotilailla (43 % [95 %:n CI: 30 %, 56 %]), p < 0,001. Seerumin kaliumpitoisuus oli lumelääkettä saaneilla potilailla useammin ≥ 5,5 mmol/l milloin tahansa osan B aikana (60 % [95 %:n CI: 47 %, 74 %]) kuin patiromeerihoitoa saaneilla potilailla (15 % [95 %:n CI: 6 %, 24 %]), p < 0,001.
Osassa B arvioitiin myös, voiko patiromeeri mahdollistaa RAA-järjestelmää estävän hoidon samanaikaisen käytön: Viisikymmentäkaksi prosenttia (52 %) lumelääkettä saavista tutkittavista lopetti RAA-järjestelmää estävän hoidon toistuvan hyperkalemian vuoksi, kun patiromeerihoitoa saaneista tutkittavista sen lopetti 5 %.
Tutkimus 2 (AMETHYST-DN)
Patiromeerihoidon vaikutusta 52 viikkoon asti arvioitiin avoimessa tutkimuksessa, johon osallistui 304 hyperkaleemista potilasta, joilla oli krooninen munuaissairaus ja tyypin 2 diabetes mellitus ja jotka saivat tasaisia annoksia RAA-järjestelmää estäviä lääkkeitä. Potilaiden keski-ikä oli 66 vuotta (59,9 % oli vähintään 65‑vuotiaita, 19,7 % vähintään 75‑vuotiaita), 63 % potilaista oli miehiä, ja kaikki olivat valkoihoisia. Seerumin pienentynyt kaliumpitoisuus pysyi patiromeerihoidolla yli 1 vuoden kestäneen hoidon ajan, kuten kuvasta 1 näkee. Hypokalemian ilmaantuminen oli vähäistä (2,3 %) ja suurin osa tutkittavista (97,7 %) saavutti seerumin tavoitekaliumpitoisuuden ja pysyi siinä (ylläpitojaksolla seerumin kaliumpitoisuus oli kaiken kaikkiaan tavoitealueella noin 80 % ajasta). Potilaiden, joiden seerumin kaliumpitoisuus oli lähtötilanteessa > 5,0 – 5,5 mmol/l ja jotka saivat aloitusannoksena 8,4 g patiromeeria vuorokaudessa, keskimääräinen vuorokausiannos oli 14 g, ja potilaiden, joiden seerumin kaliumpitoisuus oli lähtötilanteessa > 5,5 – < 6,0 mmol/l ja jotka saivat aloitusannoksena 16,8 g patiromeeria vuorokaudessa, keskimääräinen vuorokausiannos oli 20 g koko tutkimuksen ajan.
Kuva 1: Keskimääräinen (95 %:n CI) seerumin kaliumpitoisuus ajan mukaan
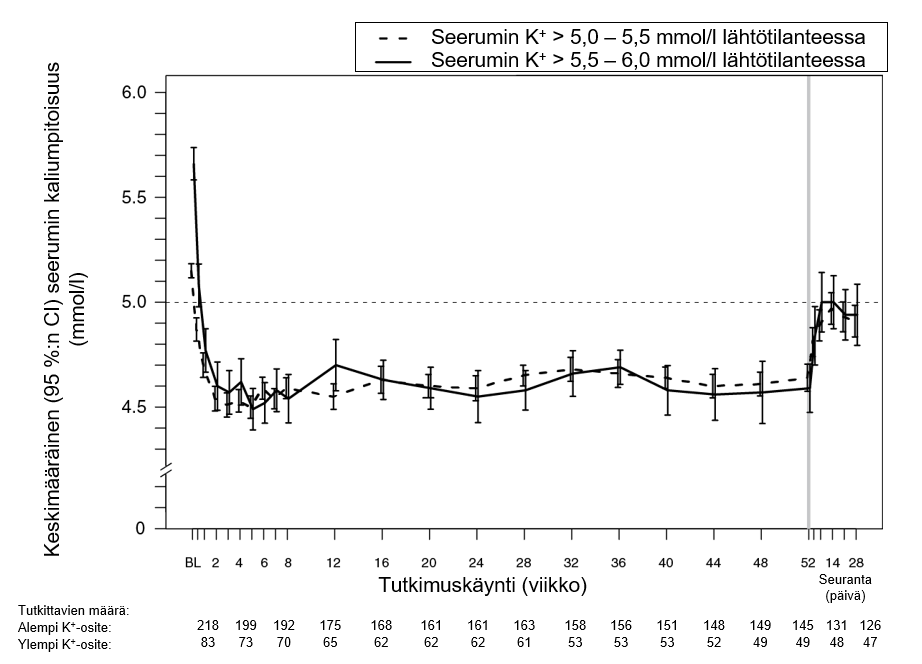
Tutkimus 3 (PEARL-HF)
Mahdollisuutta käyttää patiromeeria samanaikaisesti spironolaktonihoidon kanssa tutkittiin satunnaistetussa, kaksoissokkoutetussa lumelääkekontrolloidussa tutkimuksessa sydämen vajaatoimintaa sairastavilla potilailla, joilla MRA:n käyttö oli kliinisesti aiheellista. Potilaat aloittivat spironolaktonihoidon 25 mg:n vuorokausiannoksella samaan aikaan kuin satunnaistetun hoidon (patiromeeri 12,6 g kahdesti päivässä tai lumelääke), ja annos suurennettiin 50 mg:aan vuorokaudessa päivän 14 jälkeen, jos seerumin kaliumpitoisuus oli > 3,5 ja ≤ 5,1 mmol/l. Satunnaistettujen ja tutkimushoitoa (patiromeeri 56; lumelääke 49) saaneiden 105 potilaan keski-ikä oli 68,3 vuotta, 60,6 % oli miehiä, 97,1 % oli valkoihoisia, ja glomerulusten keskimääräinen arvioitu suodatusnopeus oli 81,3 ml/min. Seerumin kaliumpitoisuus oli lähtötilanteessa 4,71 mmol/l potilaissa ja vastaavasti 4,68 mmol/l lumelääkettä saaneissa potilaissa.
Ensisijainen tehoa mittaava päätetapahtuma, muutos seerumin kaliumpitoisuuden lähtötasosta 28 päivän hoitojakson loppuun saakka, oli merkitsevästi pienempi (p < 0,001) patiromeeriryhmässä (neliösumman keskiarvo [SEM]: −0,21 [0,07] mmol/l) lumelääkeryhmään verrattuna (neliösumman keskiarvo [SEM]: +0,23 [0,07] mmol/l). Lisäksi patiromeeriryhmässä oli myös vähemmän potilaita, joiden seerumin kaliumarvot olivat > 5,5 mmol/l (7,3 % vs. 24,5 %; p = 0,027), ja enemmän potilaita, jotka saivat spironolaktonia 50 mg vuorokaudessa (90,9 % vs. 73,5 %, p = 0,022).
Tutkimus 4 (AMBER)
Patiromeerin kykyä mahdollistaa samanaikainen spironolaktonihoito resistenttiä korkeaa verenpainetta ja kroonista munuaissairautta sairastavilla potilailla tutkittiin edelleen satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa 12 viikkoa kestävässä tutkimuksessa. Normokaleemiset potilaat aloittivat spironolaktonin annoksella 25 mg kerran vuorokaudessa samanaikaisesti satunnaistetun hoitonsa (patiromeeri 8,4 g kerran vuorokaudessa tai lumelääke) kanssa. Patiromeeri/lumelääke titrattiin viikoittain (korkeintaan annokseen 25,2 g kerran vuorokaudessa), jotta seerumin kaliumtasot pysyisivät arvoissa ≥ 4,0 mmol/l ja ≤ 5,1 mmol/l. Viikolla 3 tai sen jälkeen spironolaktoniannosta nostettiin annokseen 50 mg kerran vuorokaudessa tutkittavilla, joiden systolinen verenpaine oli ≥ 120 mmHg ja seerumin kaliumtaso ≤ 5,1 mmol/l.
Tutkimushoitoa (patiromeeri 147; lumelääke 148) saavien 295 satunnaistetun potilaan keski-ikä oli 68,1 vuotta, 51,9 % potilaista oli miehiä, 98,3 % valkoihoisia ja glomerulusten keskimääräinen arvioitu suodatusnopeus oli 35,73 ml/min/1,73 m2. Satunnaistamisen aikaisen lähtötason seerumin kaliumarvo oli keskiarvoltaan 4,74 mmol/l patiromeerihoitoa saaneilla potilailla ja 4,69 mmol/l lumelääkettä saaneilla potilailla. Ensisijainen tehoa mittaava päätetapahtuma, spironolaktonihoitoa jatkavien tutkittavien määrä viikolla 12, oli huomattavasti korkeampi (p < 0,0001) patiromeerihoitoa saavassa ryhmässä (85,7 %) kuin lumelääkettä saavassa ryhmässä (66,2 %). Huomattavasti useammat saivat spironolaktonia 50 mg/vrk (69,4 % vs. 51,4 %).
Yleisesti patiromeeriryhmän potilaat jatkoivat spironolaktonin käyttöä 7,1 vuorokautta pidempään (95 %:n CI 2,2–12,0; p = 0,0045) kuin lumelääkeryhmän potilaat, ja he saivat myös huomattavasti korkeammat kumulatiiviset annokset spironolaktonia (2 942,3 (SE 80,1) mg vs. 2 580,7 (SE 95,8) mg, p = 0,0021).
Lisäksi huomattavasti harvemman patiromeeriryhmän potilaan seerumin kaliumarvo oli ≥ 5,5 mmol/l (35,4 % vs. 64,2 %, p < 0,001).
Viikolla 12 systolisen verenpaineen keskiarvo oli laskenut 11,0 mmHg (SD 15,34) spironolaktoni + lumelääke -ryhmässä ja vastaavasti 11,3 mmHg (SD 14,11) spironolaktoni + patiromeeri -ryhmässä. Lähtötasoon verrattavat arvojen laskut olivat tilastollisesti merkitseviä kummassakin hoitoryhmässä (p < 0,0001), mutta ryhmien välillä ei ollut tilastollisesti merkitsevää eroa.
Ruoan vaikutus
Avoimessa tutkimuksessa 114 hyperkalemiaa sairastavaa potilasta satunnaistettiin saamaan patiromeeria kerran vuorokaudessa joko ruoan kanssa tai ilman ruokaa. Seerumin kaliumarvo hoidon päättyessä, seerumin kaliumarvon muutos lähtötilanteesta ja keskimääräinen patiromeeriannos olivat samaa luokkaa molemmissa ryhmissä.
Pediatriset potilaat
Avoimessa moniannostutkimuksessa arvioitiin patiromeerioraalisuspension tehoa, turvallisuutta ja siedettävyyttä 6 – < 18‑vuotiailla lapsilla ja nuorilla, joilla oli dialyysihoitoa edellyttämätön krooninen munuaissairaus ja hyperkalemia. Tutkimuksesta suljettiin pois potilaat, joilla oli jokin vaikea ruoansulatuskanavan diagnoosi tai joille oli tehty jokin ruoansulatuskanavan leikkaus. Tutkimuksessa oli kaksi hoitovaihetta: ensin 14 vuorokauden pituinen annoksenhakuvaihe ja sen jälkeen enintään 24 viikon pituinen pitkäkestoinen hoitovaihe. Hoitoviikkoja oli yhteensä enintään 26. Tutkimuksessa oli kaksi ikäryhmää, 12 – < 18-vuotiaat ja 6 – < 12-vuotiaat, ja kummassakin ryhmässä patiromeerin aloitusannokset valittiin mediaanipainon perusteella. Patiromeeria annettiin kerran vuorokaudessa jauheena oraalisuspensiota varten.
Kaikkiaan 23 tutkittavaa (joista 14 tutkittavaa oli 12 – < 18‑vuotiaita ja 9 tutkittavaa oli 6 – < 12‑vuotiaita) suoritti annoksenhakuvaiheen loppuun asti, ja 21 tutkittavaa (joista 12 tutkittavaa oli 12 – < 18‑vuotiaita ja 9 tutkittavaa oli 6 – < 12‑vuotiaita) suoritti pitkäkestoisen hoitovaiheen loppuun asti. Yksikään tutkittava ei keskeyttänyt tutkimusta turvallisuusongelmien takia. Tämän tutkimuksen ensisijainen tehon päätetapahtuma oli seerumin kaliumpitoisuuden muutos lähtötilanteesta päivänä 14. Molemmissa ikäryhmissä kaliumpitoisuuksien todettiin pienentyneen päivään 14 mennessä: kaliumpitoisuuden keskimääräinen (keskihajonta) muutos lähtötilanteesta oli ‑0,50 (0,542) mEq/l 12 – < 18‑vuotiaiden ryhmässä ja -0,14 (0,553) mEq/l 6 – < 12‑vuotiaiden ryhmässä, ja se säilyi koko tutkimuksen ajan niin kauan kuin hoitoa jatkettiin (kuva 2).
Toissijaisia tehon päätetapahtumia olivat niiden tutkittavien osuudet, joiden seerumin kaliumpitoisuus oli tavoitealueella (3,8–5,0 mEq/l) päivänä 14 (annoksenhakuvaihe) ja käyntikohtaisesti milloin tahansa kuukauden 6 loppuun asti (pitkäkestoinen hoitovaihe). 12 – < 18-vuotiaiden ryhmässä tavoitealueella olevan seerumin kaliumpitoisuuden saavutti 50,0 % tutkittavista päivänä 14 ja 81,8 % tutkittavista viikolla 26. Patiromeeriannos 4,2 g/vrk vaikuttaisi olevan sopiva aloitusannos tälle ryhmälle. 6 – < 12-vuotiaiden ryhmässä tavoitealueella olevan seerumin kaliumpitoisuuden saavutti vain 12,5 % tutkittavista päivänä 14 ja 22,2 % tutkittavista viikolla 26.
Kuva 2.Keskimääräinen (±95 %:n CI) seerumin kaliumpitoisuus (turvallisuuspopulaatio, N = 23)
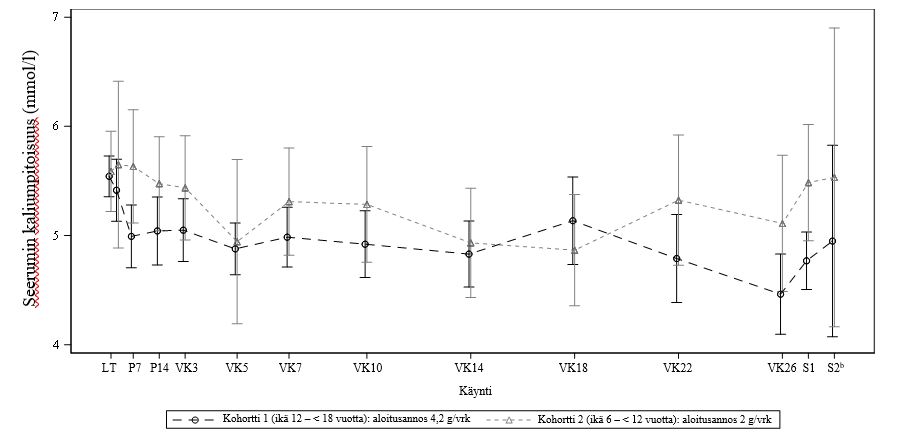
Huom: a Lähtötilanteen arvo oli viimeinen ei-puuttuva keskuslaboratorion arvo, joka kerättiin ennen ensimmäisen patiromeeriannoksen antopäivää ja -kellonaikaa.
b Seuranta 2 oli valinnainen tutkimuspaikkakäynti ja voitiin järjestää puhelimitse.
Tietoja seerumin kaliumpitoisuuksista dialyysin aloituspäivänä tai sen jälkeen ei otettu mukaan.
LT = lähtötilanne; CI = luottamusväli; P = päivä; S = seuranta; VK = viikko.
12 – < 18-vuotiaiden ryhmässä tutkittaville määrätyn patiromeeriannoksen mediaani oli 4,2 g/vrk päivänä 14 ja 8,4 g/vrk hoidon lopussa, ja seerumin kaliumarvon keskimääräinen muutos lähtötilanteesta oli -0,50 mEq/l päivänä 14 ja -1,08 mEq/l hoidon lopussa. 6 – < 12-vuotiaiden ryhmässä tutkittaville määrätyn patiromeeriannoksen mediaani oli 6,0 g/vrk päivänä 14 ja 8,0 g/vrk hoidon lopussa, ja seerumin kaliumarvon keskimääräinen muutos lähtötilanteesta oli -0,14 mEq/l päivänä 14 ja -0,50 mEq/l hoidon lopussa. 12–17-vuotiaiden tutkittavien annos-vastetulokset vaikuttivat osoittavan kvalitatiivisesti, että suurempi patiromeeriannos pienensi seerumin kaliumpitoisuutta enemmän hoitojen välisenä aikana. Kuitenkin 6 – < 12-vuotiaiden ryhmässä annoksenhakutulokset eivät olleet täysin vakuuttavia. Näin ollen patinomeerin lisäarviointi 6 – < 12-vuotiailla tutkittavilla vaaditaan hyöty-riskiarvion määrittämiseen.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Veltassa-valmisteen käytöstä hyperkalemian hoidossa alle 6‑vuotiailla lapsilla (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Patiromeeri toimii sitoutumalla kaliumiin maha-suolikanavassa, ja siten seerumipitoisuus ei vaikuta sen tehoon. Tämän lääkevalmisteen liukenemattomuuden ja imeytymättömyyden vuoksi monia klassisia farmakokineettisiä tutkimuksia ei voida suorittaa.
Patiromeeri poistuu elimistöstä noin 24–48 tunnin kuluttua nauttimisesta ruoansulatuskanavan keskimääräisen läpikulkuajan perusteella.
Prekliiniset tiedot turvallisuudesta
Rotille ja koirille tehdyissä radiomerkityissä tutkimuksissa patiromeeri ei imeytynyt elimistöön ja poistui ulosteen mukana. Rotille tehdyssä koko kehon kvantitatiivisessa autoradiografia-analyysissä osoitettiin, että radioaktiivisuus rajoittui maha-suolikanavaan niin, että muissa kudoksissa tai elimissä ei ollut havaittua radioaktiivisuustasoa.
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Patiromeeri ei ollut geenitoksinen takaisinmutaatiokokeissa (Amesin testi), kromosomipoikkeavuustesteissä tai rotan mikrotumatesteissä.
Karsinogeenisuutta koskevia tutkimuksia ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Ksantaanikumi (lisätietoa sorbitolista, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Säilytä ja kuljeta kylmässä (2°C – 8°C).
Jos Veltassa-valmistetta säilytetään huoneenlämmössä (alle 25°C), se pitää käyttää 6 kuukauden sisällä jääkaapista ottamisen jälkeen.
Veltassa-valmistetta ei saa käyttää annospussiin painetun viimeisen käyttöpäivän jälkeen kummassakaan säilytysolosuhteessa.
Seos pitää ottaa 1 tunnin sisällä suspension valmistamisesta.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VELTASSA jauhe oraalisuspensiota varten
1 g (L:ei) 60 x 1 g (485,97 €)
8,4 g (L:ei) 30 x 8,4 g (251,01 €)
16,8 g (L:ei) 30 x 16,8 g (251,01 €)
PF-selosteen tieto
Veltassa 1 g jauhe oraalisuspensiota varten
1 g patiromeeria jauheena annospusseissa, jotka koostuvat viidestä kerroksesta: polyeteeni, alumiini, polyeteeni, polyesteri ja paperi.
Pakkauskoot: 60 annospussia sisältävä rasia.
Veltassa 8,4 g jauhe oraalisuspensiota varten
8,4 g patiromeeria jauheena annospusseissa, jotka koostuvat viidestä kerroksesta: polyeteeni, alumiini, polyeteeni, polyesteri ja paperi.
Pakkauskoot: 30, 60 tai 90 annospussia sisältävät rasiat ja moniannospakkaukset, joissa on kolme 30 annospussin pakkausta.
Veltassa 16,8 g jauhe oraalisuspensiota varten
16,8 g patiromeeria jauheena annospusseissa, jotka koostuvat viidestä kerroksesta: polyeteeni, alumiini, polyeteeni, polyesteri ja paperi.
Pakkauskoot: 30, 60 tai 90 annospussia sisältävät rasiat.
Veltassa 25,2 g jauhe oraalisuspensiota varten
25,2 g patiromeeria jauheena annospusseissa, jotka koostuvat viidestä kerroksesta: polyeteeni, alumiini, polyeteeni, polyesteri ja paperi.
Pakkauskoot: 30, 60 tai 90 annospussia sisältävät rasiat.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Vaalea tai vaaleanruskea jauhe, jossa valkoisia hiukkasia.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VELTASSA jauhe oraalisuspensiota varten
8,4 g 30 x 8,4 g
16,8 g 30 x 16,8 g
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Natriumsirkoniumsyklosilikaatti ja patiromeeri: Kroonista munuaissairautta sairastavien aikuispotilaiden hyperkalemian hoito erityisin edellytyksin / Natriumsirkoniumsyklosilikaatti: Sydämen vajaatoimintaa sairastavien aikuispotilaiden hyperkalemian hoito erityisin edellytyksin (3020).
VELTASSA jauhe oraalisuspensiota varten
1 g 60 x 1 g
- Ei korvausta.
ATC-koodi
V03AE09
Valmisteyhteenvedon muuttamispäivämäärä
06.06.2024
Yhteystiedot
Gustav III:s Boulevard 46
SE-169 73 Solna
Sverige
+46 8 558 066 00
www.viforpharma.se
Info.nordic@viforpharma.com