
REPATHA injektioneste, liuos, esitäytetty kynä 140 mg
Vaikuttavat aineet ja niiden määrät
Yksi esitäytetty kynä sisältää 140 mg evolokumabia 1 millilitrassa liuosta.
Repatha on ihmisen monoklonaalinen IgG2-vasta-aine, joka tuotetaan kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-menetelmällä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste), esitäytetty kynä (SureClick).
Kliiniset tiedot
Käyttöaiheet
Hyperkolesterolemia ja sekamuotoinen dyslipidemia
Repatha on tarkoitettu aikuisille primaarisen (heterotsygoottisen familiaalisen ja ei‑familiaalisen) hyperkolesterolemian tai sekamuotoisen dyslipidemian hoitoon ja 10 vuotta täyttäneille lapsipotilaille heterotsygoottisen familiaalisen hyperkolesterolemian hoitoon, ruokavalion ohella:
- yhdessä statiinin kanssa tai statiinin ja muiden veren rasva-arvoja alentavien hoitojen kanssa potilaille, joiden LDL‑kolesterolipitoisuus ei laske tavoitetasolle suurimmalla siedetyllä statiiniannoksella, tai
- yksinään tai yhdessä muiden veren rasva-arvoja alentavien hoitojen kanssa, kun potilas ei siedä statiinia tai statiini on vasta-aiheinen.
Homotsygoottinen familiaalinen hyperkolesterolemia
Repatha on tarkoitettu aikuisille ja 10 vuotta täyttäneille lapsipotilaille, joilla on homotsygoottinen familiaalinen hyperkolesterolemia, yhdessä muiden rasva-arvoja alentavien hoitojen kanssa.
Varmistettu ateroskleroottinen sydän- tai verisuonisairaus
Repatha on tarkoitettu aikuisille, joilla on todettu ateroskleroottinen sydän- tai verisuonisairaus (sydäninfarkti, aivohalvaus tai perifeerinen valtimosairaus), vähentämään sydän- ja verisuonisairauksien riskiä LDL‑kolesterolipitoisuutta pienentämällä, yhdistettynä muiden riskitekijöiden korjaamiseen:
- yhdessä suurimman siedetyn statiiniannoksen ja mahdollisesti muiden veren rasva-arvoja alentavien hoitojen kanssa tai
- yksinään tai yhdessä muiden veren rasva-arvoja alentavien hoitojen kanssa, kun potilas ei siedä statiinia tai statiini on vasta-aiheinen.
Tutkimustuloksia Repathan vaikutuksista LDL‑kolesteroliin ja sydän- ja verisuonitautitapahtumiin sekä tutkittuihin potilasryhmiin kuvataan kohdassa Farmakodynamiikka.
Annostus ja antotapa
Ennen evolokumabin aloittamista on suljettava pois hyperlipidemian tai sekamuotoisen dyslipidemian sekundaariset syyt (esim. nefroottinen oireyhtymä, hypotyreoosi).
Annostus
Primaarinen hyperkolesterolemia ja sekamuotoinen dyslipidemia (mukaan lukien heterotsygoottinen familiaalinen hyperkolesterolemia)
Aikuiset ja 10 vuotta täyttäneet lapset
Suositeltu evolokumabiannostus on joko 140 mg kahden viikon välein tai 420 mg kerran kuukaudessa. Nämä annostukset ovat kliinisesti samanarvoisia.
Aikuisten ja 10 vuotta täyttäneiden lasten homotsygoottinen familiaalinen hyperkolesterolemia
Suositeltu aloitusannostus on 420 mg kerran kuukaudessa. Kun hoito on kestänyt 12 viikkoa, annostelua voidaan tihentää siten, että 420 mg:n annos annetaan kahden viikon välein, ellei kliinisesti merkittävää vastetta ole saavutettu. Afereesihoitoa saaville potilaille voidaan antaa aloitusannostuksena 420 mg kahden viikon välein heidän afereesiaikataulunsa mukaisesti.
Aikuisten varmistettu ateroskleroottinen sydän- tai verisuonisairaus
Suositeltu evolokumabiannostus on joko 140 mg kahden viikon välein tai 420 mg kerran kuukaudessa. Nämä annostukset ovat kliinisesti samanarvoisia.
Erityisryhmät
Iäkkäät (≥ 65‑vuotiaat) potilaat
Annosta ei tarvitse muuttaa iäkkäitä potilaita hoidettaessa.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa munuaisten vajaatoiminnan vuoksi (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa lievän maksan vajaatoiminnan vuoksi. Katso kohdasta Varoitukset ja käyttöön liittyvät varotoimet ohjeet kohtalaista ja vaikeaa maksan vajaatoimintaa sairastavien potilaiden hoidosta.
Pediatriset potilaat
Repatha-valmisteen turvallisuutta ja tehoa ei ole varmistettu alle 10‑vuotiaiden lasten heterotsygoottisen familiaalisen hyperkolesterolemian eikä homotsygoottisen familiaalisen hyperkolesterolemian hoidossa eikä lapsipotilaiden muuntyyppisen hyperlipidemian hoidossa.
Antotapa
Ihon alle.
Evolokumabi annetaan injektiona ihon alle vatsan, reiden tai olkavarren alueelle. Pistoskohtaa on vaihdettava joka pistokerralla, eikä pistosta pidä antaa ihoalueelle, joka aristaa, punoittaa tai tuntuu kovalta tai jossa on mustelma.
Evolokumabia ei saa antaa laskimoon eikä lihakseen.
140 mg:n annos annetaan yhdellä esitäytetyllä kynällä.
420 mg:n annos annetaan kolmella esitäytetyllä kynällä peräkkäisinä pistoksina 30 minuutin aikana.
Repatha on tarkoitettu potilaiden itse pistettäväksi asianmukaisen opetuksen jälkeen. Evolokumabiannokset voi pistää myös joku muu, joka on saanut koulutuksen tämän lääkkeen pistämiseen.
Vain yhtä käyttökertaa varten.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Maksan vajaatoiminta
Evolokumabin kokonaisaltistuksen on havaittu olevan pienempi kohtalaista maksan vajaatoimintaa sairastavilla potilailla, mikä voi heikentää LDL‑kolesterolipitoisuutta pienentävää tehoa. Siksi saattaa olla perusteltua seurata tarkoin näiden potilaiden tilaa.
Vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavia potilaita ei ole tutkittu (ks. kohta Farmakokinetiikka). Evolokumabin käytössä on noudatettava varovaisuutta, jos potilaalla on vaikea maksan vajaatoiminta.
Natriumsisältö
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan "natriumiton".
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Kliinisissä tutkimuksissa arvioitiin statiinien ja evolokumabin farmakokineettistä yhteisvaikutusta. Evolokumabin puhdistuma oli noin 20 % suurempi potilailla, jotka saivat samanaikaisesti statiineja. Puhdistuman suureneminen johtuu osittain siitä, että statiinit suurentavat PCSK9:n (Proprotein Convertase Subtilisin/Kexin Type 9) pitoisuutta. Tämä ei heikentänyt evolokumabin farmakodynaamista vaikutusta lipideihin. Statiinien annostusta ei tarvitse muuttaa, kun niitä annetaan yhtaikaa evolokumabin kanssa.
Evolokumabin farmakokineettisiä ja farmakodynaamisia yhteisvaikutuksia ei ole tutkittu muiden rasva-arvoja alentavien lääkevalmisteiden kuin statiinien ja etsetimibin kanssa.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja Repathan käytöstä raskaana oleville naisille.
Eläinkokeissa ei ole havaittu suoria eikä epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Repathaa ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä evolokumabihoitoa.
Imetys
Ei tiedetä, erittyykö evolokumabi ihmisen rintamaitoon.
Rintamaitoa saavaan vastasyntyneeseen/imeväiseen kohdistuvaa riskiä ei voida sulkea pois.
On päätettävä lopetetaanko rintaruokinta vai lopetetaanko Repatha-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Evolokumabin vaikutuksesta ihmisen hedelmällisyyteen ei ole tutkimustietoa. Eläinkokeissa ei havaittu hedelmällisyyttä mittaaviin tulostapahtumiin kohdistuvia vaikutuksia altistustasoilla, jotka olivat huomattavasti suurempia pitoisuus-aikakäyrän alle jäävän pinta-alan (AUC) perusteella kuin potilaiden evolokumabialtistus annostuksen ollessa 420 mg kerran kuukaudessa (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Repatha-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Tiivistelmä turvallisuustiedoista
Yleisimmin raportoidut haittavaikutukset suositeltuja annoksia käytettäessä ovat nenänielun tulehdus (7,4 %), ylähengitystieinfektio (4,6 %), selkäkipu (4,4 %), nivelsärky (3,9 %), influenssa (3,2 %) ja injektiokohdan reaktiot (2,2 %). Turvallisuusprofiili oli homotsygoottista familiaalista hyperkolesterolemiaa sairastavilla potilailla samanlainen kuin primaarista hyperkolesterolemiaa ja sekamuotoista dyslipidemiaa sairastavilla potilailla.
Haittavaikutustaulukko
Keskeisissä kliinisissä vertailututkimuksissa esiintyneet sekä spontaanisti raportoidut haittavaikutukset on lueteltu alla taulukossa 1 elinjärjestelmän ja esiintymistiheyden mukaan seuraavaa käytäntöä noudattaen: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1000, < 1/100), harvinainen (≥ 1/10 000, < 1/1000) ja hyvin harvinainen (< 1/10 000).
Taulukko 1. Haittavaikutukset
Elinjärjestelmä (MedDRA) | Haittavaikutukset | Yleisyys |
Infektiot | Influenssa | Yleinen |
Nenänielun tulehdus | Yleinen | |
Ylähengitystieinfektio | Yleinen | |
Immuunijärjestelmä | Yliherkkyys | Yleinen |
Ihottuma | Yleinen | |
Nokkosihottuma | Melko harvinainen | |
| Hermosto | Päänsärky | Yleinen |
Ruoansulatuselimistö | Pahoinvointi | Yleinen |
Iho ja ihonalainen kudos | Angioedeema | Harvinainen |
Luusto, lihakset ja sidekudos | Selkäkipu | Yleinen |
Nivelsärky | Yleinen | |
| Myalgia | Yleinen | |
Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan reaktiot1 | Yleinen |
Influenssan kaltainen sairaus | Melko harvinainen | |
| 1 Ks. kohta Tärkeimpien haittavaikutusten kuvaus. | ||
Turvallisuusprofiili oli yhdenmukainen tutkittavilla, joilla lähtötason jälkeinen LDL‑kolesteroliarvo oli joko < 0,65 mmol/l (25 mg/dl) tai < 1,03 mmol/l (40 mg/dl), verrattuna tutkittaviin, joilla lähtötason jälkeinen LDL‑kolesteroliarvo oli korkeampi (≥ 1,03 mmol/l [40 mg/dl]). Repatha-altistuksen keston mediaani (Q1, Q3) oli 84,2 (78,1; 89,8) kuukautta niillä tutkittavilla, joilla Repatha-hoitoa jatkettiin, ja 59,8 (52,8; 60,3) kuukautta niillä lumevalmistetta saaneilla tutkittavilla, joiden hoito vaihdettiin Repathaan avoimessa jatkotutkimuksessa.
Tärkeimpien haittavaikutusten kuvaus
Injektiokohdan reaktiot
Yleisimmin esiintyneet injektiokohdan reaktiot olivat injektiokohdan mustelmat, punoitus, verenvuoto, kipu ja turvotus.
Pediatriset potilaat
Repatha‑valmisteen turvallisuus ja teho on varmistettu lapsipotilaiden heterotsygoottisen ja homotsygoottisen familiaalisen hyperkolesterolemian hoidossa. Repathan vaikutuksia on tutkittu kliinisessä tutkimuksessa, jossa oli mukana 158 heterotsygoottista familiaalista hyperkolesterolemiaa sairastavaa 10 vuotta täyttänyttä mutta alle 18‑vuotiasta potilasta. Uusia turvallisuutta koskevia huolenaiheita ei havaittu, ja turvallisuustiedot tässä lapsipotilaiden joukossa olivat yhdenmukaiset tuotteen tunnetun turvallisuusprofiilin kanssa heterotsygoottista familiaalista hyperkolesterolemiaa sairastavilla aikuisilla. Kliinisissä tutkimuksissa oli mukana 26 homotsygoottista familiaalista hyperkolesterolemiaa sairastavaa 10 vuotta täyttänyttä mutta alle 18‑vuotiasta lapsipotilasta, joita hoidettiin Repathalla. Turvallisuudessa ei havaittu eroa lasten ja aikuisten homotsygoottista familiaalista hyperkolesterolemiaa sairastavien potilaiden välillä.
Iäkkäät potilaat
Kliinisissä kaksoissokkotutkimuksissa evolokumabia saaneista 18 546 potilaasta 7656 (41,3 %) oli vähintään 65‑vuotiaita ja 1500 (8,1 %) oli vähintään 75‑vuotiaita. Hoidon turvallisuudessa ja tehossa ei havaittu yleisiä eroja näiden ikäryhmien ja nuorempien potilaiden välillä.
Immunogeenisuus
Kliinisissä tutkimuksissa sitoutuvia vasta-aineita todettiin 0,3 prosentilla potilaista (48 potilaalla 17 992:sta), jotka olivat saaneet vähintään yhden evolokumabiannoksen. Potilailta, joiden seerumissa todettiin sitoutuvia vasta-aineita, tutkittiin vielä neutraloivat vasta-aineet. Yhdelläkään näistä potilaista ei esiintynyt neutraloivia vasta-aineita. Sitoutuvien evolokumabin vasta-aineiden esiintyminen ei vaikuttanut evolokumabin farmakokineettiseen profiiliin, kliiniseen vasteeseen eikä turvallisuuteen.
Evolokumabin vasta-aineiden kehittymistä ei havaittu Repatha‑hoitoa saaneiden lapsipotilaiden kliinisissä tutkimuksissa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta: www.fimea.fi.
Yliannostus
Eläinkokeissa ei havaittu haittavaikutuksia, kun altistus oli jopa 300 kertaa suurempi kuin potilaiden altistus evolokumabiannostuksen ollessa 420 mg kerran kuukaudessa.
Evolokumabin yliannokseen ei ole spesifistä hoitoa. Yliannostapauksessa potilasta on hoidettava oireidenmukaisesti, ja tukihoitotoimenpiteet on aloitettava tarvittaessa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Lipidejä muuntavat lääkeaineet, muut lipidejä muuntavat lääkeaineet. ATC-koodi: C10AX13
Vaikutusmekanismi
Evolokumabi sitoutuu selektiivisesti PCSK9:ään ja estää veressä olevan PCSK9:n sitoutumisen maksasolujen pinnan LDL-reseptoreihin (LDLR), mikä puolestaan estää PCSK9:n säätelemää LDL-reseptorien tuhoutumista. Maksan LDL-reseptorien määrän suureneminen pienentää vastaavasti seerumin LDL-kolesterolipitoisuutta (LDL‑Kol).
Farmakodynaamiset vaikutukset
Kliinisissä tutkimuksissa evolokumabi pienensi sitoutumattoman PCSK9:n, LDL‑kolesterolin, kokonaiskolesterolin, apolipoproteiini B:n (ApoB:n) ja ei‑HDL‑kolesterolin pitoisuutta, kokonaiskolesterolin ja HDL‑kolesterolin suhdetta, ApoB/ApoA1‑suhdetta ja VLDL‑kolesterolin, triglyseridien ja lipoproteiini (a):n (Lp(a):n) pitoisuutta ja suurensi HDL‑kolesterolin ja ApoA1:n pitoisuutta primaarista hyperkolesterolemiaa ja sekamuotoista dyslipidemiaa sairastavilla potilailla.
Kun evolokumabia annettiin kerta-annoksena 140 mg tai 420 mg ihon alle, veressä olevan sitoutumattoman PCSK9:n alhaisin pitoisuus saavutettiin 4 tunnissa. Sitä seurasi LDL‑kolesterolitason lasku, joka saavutti alhaisimman tason keskimäärin 14 vuorokauden kuluttua 140 mg:n annoksen jälkeen ja 21 vuorokauden kuluttua 420 mg:n annoksen jälkeen. Sitoutumattoman PCSK9:n ja seerumin lipoproteiinien muutokset palautuivat ennalleen evolokumabihoidon lopettamisen jälkeen. Sitoutumattoman PCSK9:n tai LDL‑kolesterolin pitoisuus ei noussut lähtötason yläpuolelle, kun evolokumabi poistui elimistöstä. Tämä viittaa siihen, ettei hoidon aikana esiinny kompensoivia mekanismeja, jotka lisäävät PCSK9:n ja LDL‑kolesterolin tuotantoa.
Annostukset 140 mg ihon alle kahden viikon välein ja 420 mg ihon alle kerran kuukaudessa olivat samanarvoisia keskimääräisen LDL‑kolesterolitason laskun (viikkojen 10 ja 12 mittausten keskiarvon) perusteella, ja lasku lähtötasosta oli 57–72 % lumeryhmään verrattuna. Evolokumabi laski LDL‑kolesteroliarvoa yhtä paljon yksinään käytettynä kuin yhteiskäytössä muiden rasva-arvoja pienentävien lääkkeiden kanssa.
Kliininen teho primaarisen hyperkolesterolemian ja sekamuotoisen dyslipidemian hoidossa
Evolokumabi laski LDL‑kolesterolipitoisuutta noin 55–75 % jo ensimmäisellä viikolla, ja vaikutus säilyi pitkäaikaishoidossa. Maksimaalinen vaste saavutettiin yleensä 1–2 viikon kuluessa, kun annostus oli 140 mg kahden viikon välein tai 420 mg kerran kuukaudessa. Evolokumabi oli tehokas kaikissa alaryhmissä lumevalmisteeseen ja etsetimibiin verrattuna, eikä seuraaviin kriteereihin perustuvien alaryhmien välillä ollut havaittavia eroja: ikä, etninen tausta, sukupuoli, alue, painoindeksi, NCEP-kriteerien (National Cholesterol Education Program) mukainen riski, nykyinen tupakointi, sepelvaltimotaudin riskitekijät lähtötilanteessa, perinnöllinen varhaisen sepelvaltimotaudin riski, sokeriaineenvaihdunnan tila (tyypin 2 diabetes, metabolinen oireyhtymä tai ei kumpaakaan), hypertensio, statiinin annos ja intensiteetti, sitoutumattoman PCSK9:n pitoisuus lähtötilanteessa, LDL‑kolesterolin lähtöarvo ja triglyseridipitoisuus lähtötilanteessa.
Evolokumabi laski LDL‑kolesteroliarvoa ≥ 50 % viikkojen 10 ja 12 mittausten keskiarvon perusteella 80−85 prosentilla kaikista jompaakumpaa annostusta saaneista primaarista hyperlipidemiaa sairastaneista potilaista. Jompaakumpaa evolokumabiannostusta saaneista potilaista jopa 99 prosentilla LDL‑kolesteroli laski tasolle < 2,6 mmol/l, ja jopa 95 % saavutti LDL-kolesterolitason < 1,8 mmol/l viikkojen 10 ja 12 mittausten keskiarvon perusteella.
Yhdistelmähoitona statiinin tai statiinin ja muiden veren rasva-arvoja alentavien hoitojen kanssa
LAPLACE‑2 oli kansainvälinen kaksoissokkoutettu satunnaistettu 12 viikon monikeskustutkimus, jossa oli mukana 1896 primaarista hyperkolesterolemiaa tai sekamuotoista dyslipidemiaa sairastavaa potilasta. Potilaat saivat satunnaistetusti evolokumabia yhdessä eri statiinien (rosuvastatiinin, simvastatiinin tai atorvastatiinin) kanssa. Evolokumabia verrattiin lumevalmisteeseen rosuvastatiini- ja simvastatiiniryhmissä ja lumevalmisteeseen ja etsetimibiin atorvastatiiniryhmässä.
Repatha laski LDL‑kolesteroliarvoa merkitsevästi lähtötasosta viikkojen 10 ja 12 mittausten keskiarvon perusteella lumevalmisteeseen verrattuna rosuvastatiini- ja simvastatiiniryhmissä ja lumevalmisteeseen ja etsetimibiin verrattuna atorvastatiiniryhmässä (p < 0,001). Repatha pienensi merkitsevästi kokonaiskolesteroli-, ApoB- ja ei‑HDL‑kolesterolipitoisuutta, kokonaiskolesterolin ja HDL‑kolesterolin suhdetta, ApoB/ApoA1‑suhdetta, VLDL‑kolesteroli-, triglyseridi- ja Lp(a)‑pitoisuutta ja suurensi HDL‑kolesterolipitoisuutta lähtötasosta viikkojen 10 ja 12 mittausten keskiarvon perusteella lumevalmisteeseen verrattuna rosuvastatiini- ja simvastatiiniryhmissä (p < 0,05) ja pienensi merkitsevästi kokonaiskolesteroli-, ApoB- ja ei-HDL-kolesterolipitoisuutta, kokonaiskolesterolin ja HDL‑kolesterolin suhdetta, ApoB/ApoA1‑suhdetta ja Lp(a)‑arvoa lumevalmisteeseen ja etsetimibiin verrattuna atorvastatiiniryhmässä (p < 0,001) (ks. taulukot 2 ja 3).
RUTHERFORD‑2 oli kansainvälinen kaksoissokkoutettu satunnaistettu 12 viikon monikeskus-lumevertailututkimus, jossa oli mukana 329 heterotsygoottista familiaalista hyperkolesterolemiaa sairastavaa potilasta, jotka käyttivät rasva-arvoja alentavia lääkkeitä. Repatha pienensi merkitsevästi LDL‑kolesterolipitoisuutta lähtötasosta viikkojen 10 ja 12 mittausten keskiarvon perusteella lumevalmisteeseen verrattuna (p < 0,001). Repatha pienensi merkitsevästi kokonaiskolesteroli-, ApoB- ja ei‑HDL‑kolesterolipitoisuutta, kokonaiskolesterolin ja HDL‑kolesterolin suhdetta, ApoB/ApoA1‑suhdetta, VLDL‑kolesteroli-, triglyseridi- ja Lp(a)‑pitoisuutta ja suurensi HDL‑kolesteroli- ja ApoA1‑pitoisuutta lähtötasosta viikkojen 10 ja 12 mittausten keskiarvon perusteella lumevalmisteeseen verrattuna (p < 0,05) (ks. taulukko 2).
Taulukko 2. Evolokumabin hoitovaikutukset lumevalmisteeseen verrattuna primaarista hyperkolesterolemiaa ja sekamuotoista dyslipidemiaa sairastavilla potilailla – prosentuaalinen muutos (keskiarvo) lähtötasosta viikkojen 10 ja 12 mittaustulosten keskiarvon perusteella (%, 95 % CI)
Tutkimus | Annostusohjelma | LDL-Kol (%) | Ei-HDL-Kol (%) | ApoB (%) | Kol (%) | Lp(a) (%) | VLDL‑Kol (%) | HDL-Kol (%) | Trigly (%) | ApoA1 (%) | Kol/ HDL-Kol-suhde % | ApoB/ ApoA1-suhde % |
LAPLACE-2 (HMD) (rosuvastatiini-, simvastatiini- & atorvastatiini-ryhmät yhdistettyinä) | 140 mg Q2W (N = 555) | −72b (‑75,‑69) | −60b (‑63,‑58) | −56b (‑58,‑53) | −41b (‑43,‑39) | −30b (‑35,‑25) | −18b (‑23,‑14) | 6b (4,8) | −17b (‑22,‑13) | 3b (1,5) | −45b (‑47,‑42) | −56b (‑59,‑53) |
420 mg QM (N = 562) | −69b (‑73,‑65) | −60b (‑63,‑57) | −56b (‑58,‑53) | −40b (‑42,‑37) | −27b (‑31,‑24) | −22b (‑28,‑17) | 8b (6,10) | −23b (‑28,‑17) | 5b (3,7) | −46b (‑48,‑43) | −58b (‑60,‑55) | |
RUTHERFORD-2 (HeFH) | 140 mg Q2W (N = 110) | −61b (‑67,‑55) | −56b (‑61,‑51) | −49b (‑54,‑44) | −42b (‑46,‑38) | −31b (‑38,‑24) | −22b (‑29,‑16) | 8b (4,12) | −22b (‑29,‑15) | 7a (3,12) | −47b (‑51,‑42) | −53 (‑58,‑48) |
420 mg QM (N = 110) | −66b (‑72,‑61) | −60b (‑65,‑55) | −55b (‑60,‑50) | −44b (‑48,‑40) | −31b (‑38,‑24) | −16b (‑23,‑8) | 9b (5,14) | −17b (‑24,‑9) | 5a (1,9) | −49b (‑54,‑44) | −56b (‑61,‑50) | |
| Lyhenteet: Q2W = kahden viikon välein, QM = kerran kuukaudessa, HMD = primaarinen hyperkolesterolemia ja sekamuotoinen dyslipidemia, HeFH = heterotsygoottinen familiaalinen hyperkolesterolemia, a p-arvo < 0,05 lumevalmisteeseen verrattuna, b p-arvo < 0,001 lumevalmisteeseen verrattuna. | ||||||||||||
Potilaat, jotka eivät siedä statiineja
GAUSS‑2 oli kansainvälinen kaksoissokkoutettu satunnaistettu 12 viikon monikeskus-vertailututkimus, jossa vertailuaineena oli etsetimibi. Tutkimuksessa oli mukana 307 potilasta, jotka eivät sietäneet statiineja tai statiinin tehokasta hoitoannosta. Repatha pienensi LDL‑kolesterolipitoisuutta merkitsevästi etsetimibiin verrattuna (p < 0,001). Repatha pienensi merkitsevästi kokonaiskolesteroli-, ApoB- ja ei‑HDL‑kolesterolipitoisuutta, kokonaiskolesterolin ja HDL‑kolesterolin suhdetta, ApoB/ApoA1‑suhdetta ja Lp(a)‑arvoa lähtötasosta viikkojen 10 ja 12 mittausten keskiarvon perusteella etsetimibiin verrattuna (p < 0,001) (ks. taulukko 3).
Hoito ilman statiinia
MENDEL‑2 oli kansainvälinen kaksoissokkoutettu satunnaistettu 12 viikon monikeskus-vertailututkimus, jossa Repathan vertailuaineina olivat lumevalmiste ja etsetimibi. Tutkimuksessa oli mukana 614 primaarista hyperkolesterolemiaa ja sekamuotoista dyslipidemiaa sairastavaa potilasta. Repatha pienensi merkitsevästi LDL‑kolesterolipitoisuutta lähtötasosta viikkojen 10 ja 12 mittausten keskiarvon perusteella sekä lumevalmisteeseen että etsetimibiin verrattuna (p < 0,001). Repatha pienensi merkitsevästi kokonaiskolesteroli-, ApoB- ja ei‑HDL‑kolesterolipitoisuutta, kokonaiskolesterolin ja HDL‑kolesterolin suhdetta, ApoB/ApoA1‑suhdetta ja Lp(a)‑arvoa lähtötasosta viikkojen 10 ja 12 mittausten keskiarvon perusteella sekä lumevalmisteeseen että etsetimibiin verrattuna (p < 0,001) (ks. taulukko 3).
Taulukko 3. Evolokumabin hoitovaikutukset etsetimibiin verrattuna primaarista hyperkolesterolemiaa ja sekamuotoista dyslipidemiaa sairastavilla potilailla – prosentuaalinen muutos (keskiarvo) lähtötasosta viikkojen 10 ja 12 mittaustulosten keskiarvon perusteella (%, 95 % CI)
Tutkimus | Annostusohjelma | LDL-Kol (%) | Ei-HDL-Kol (%) | ApoB (%) | Kol (%) | Lp(a) (%) | VLDL-Kol (%) | HDL-Kol (%) | Trigly (%) | ApoA1 (%) | Kol/ HDL-Kol-suhde % | ApoB/ ApoA1-suhde % |
LAPLACE-2 (HMD) (atorvastatiiniryhmät yhdistettyinä) | 140 mg Q2W (N = 219) | −43c (‑50, ‑37) | −34c (‑39, ‑30) | −34c (‑38, ‑30) | −23c (‑26, ‑19) | −30c (‑35, ‑25) | −1 (‑7, 5) | 7c (4, 10) | −2 (‑9, 5) | 7c (4, 9) | −27c (‑30, ‑23) | −38c (‑42, ‑34) |
420 mg QM (N = 220) | −46c (‑51, ‑40) | −39c (‑43, ‑34) | −40c (‑44, ‑36) | −25c (‑29, ‑22) | −33c (‑41, ‑26) | −7 (‑20, 6) | 8c (5, 12) | −8 (‑21, 5) | 7c (2, 11) | −30c (‑34, ‑26) | −42c (‑47, ‑38) | |
GAUSS-2 (potilaat, jotka eivät siedä statiineja) | 140 mg Q2W (N = 103) | −38b (‑44, ‑33) | −32b (‑36, ‑27) | −32b (‑37, ‑27) | −24b (‑28, ‑20) | −24b (‑31, ‑17) | −2 (‑10, 7) | 5 (1, 10) | −3 (‑11, 6) | 5a (2, 9) | −27b (‑32, ‑23) | −35b (‑40, ‑30) |
420 mg QM (N = 102) | −39b (‑44, ‑35) | −35b (‑39, ‑31) | −35b (‑40, ‑30) | −26b (‑30, ‑23) | −25b (‑34, ‑17) | −4 (‑13, 6) | 6 (1, 10) | −6 (‑17, 4) | 3 (‑1, 7) | −30b (‑35, ‑25) | −36b (‑42, ‑31) | |
MENDEL-2 (hoito ilman statiinia) | 140 mg Q2W (N = 153) | −40b (‑44, ‑37) | −36b (‑39, ‑32) | −34b (‑37, ‑30) | −25b (‑28, ‑22) | −22b (‑29, ‑16) | −7 (‑14, 1) | 6a (3, 9) | −9 (‑16, ‑1) | 3 (0, 6) | −29b (‑32, ‑26) | −35b (‑39, ‑31) |
420 mg QM (N = 153) | −41b (‑44, ‑37) | −35b (‑38, ‑33) | −35b (‑38, ‑31) | −25b (‑28, ‑23) | −20b (‑27, ‑13) | −10 (‑19, ‑1) | 4 (1, 7) | −9 (‑18, 0) | 4a (1, 7) | −28b (‑31, ‑24) | −37b (‑41, ‑32) | |
| Lyhenteet: Q2W = kahden viikon välein, QM = kerran kuukaudessa, HMD = primaarinen hyperkolesterolemia ja sekamuotoinen dyslipidemia, a p‑arvo < 0,05 etsetimibiin verrattuna, b p-arvo < 0,001 etsetimibiin verrattuna, c nimellinen p‑arvo < 0,001 etsetimibiin verrattuna. | ||||||||||||
Pitkäaikainen teho primaarisen hyperkolesterolemian ja sekamuotoisen dyslipidemian hoidossa
DESCARTES oli kansainvälinen kaksoissokkoutettu satunnaistettu 52 viikon monikeskus-lumevertailututkimus. Tutkimuksessa oli mukana 901 hyperlipidemiapotilasta, jotka saivat pelkkää ruokavaliohoitoa, atorvastatiinia tai atorvastatiinin ja etsetimibin yhdistelmähoitoa. Repatha 420 mg kerran kuukaudessa pienensi LDL‑kolesterolipitoisuutta merkitsevästi lähtötasosta viikon 52 mittauksen perusteella lumevalmisteeseen verrattuna (p < 0,001). Hoitovaikutukset säilyivät vuoden ajan, mistä oli osoituksena viikolla 12 todetun LDL‑kolesteroliarvon laskun säilyminen viikolle 52 asti. Viikolla 52 todettu LDL‑kolesteroliarvon lasku lähtötasosta lumevalmisteeseen verrattuna oli yhdenmukainen kaikissa hoitoryhmissä, joissa rasva-arvoja alentava perushoito oli optimoitu LDL‑kolesterolipitoisuuden ja sydän- ja verisuonitautiriskin suhteen.
Repatha pienensi merkitsevästi kokonaiskolesteroli-, ApoB- ja ei‑HDL‑kolesterolipitoisuutta, kokonaiskolesterolin ja HDL‑kolesterolin suhdetta, ApoB/ApoA1‑suhdetta, VLDL‑kolesteroli-, triglyseridi- ja Lp(a)‑pitoisuutta ja suurensi HDL‑kolesteroli- ja ApoA1‑pitoisuutta viikon 52 mittauksen perusteella lumevalmisteeseen verrattuna (p < 0,001) (ks. taulukko 4).
Taulukko 4. Evolokumabin hoitovaikutukset lumevalmisteeseen verrattuna primaarista hyperkolesterolemiaa ja sekamuotoista dyslipidemiaa sairastavilla potilailla – prosentuaalinen muutos (keskiarvo) lähtötasosta viikolla 52 (%, 95 % CI)
Tutkimus | Annostusohjelma | LDL-Kol (%) | Ei-HDL-Kol (%) | ApoB (%) | Kol (%) | Lp(a) (%) | VLDL-Kol (%) | HDL-Kol (%) | Trigly (%) | ApoA1 (%) | Kol/ HDL-Kol-suhde % | ApoB/ ApoA1-suhde % |
DESCARTES | 420 mg QM (N = 599) | −59b (‑64, ‑55) | −50b (‑54, ‑46) | −44b (‑48, ‑41) | −33b (‑36, ‑31) | −22b (‑26, ‑19) | −29b (‑40, ‑18) | 5b (3, 8) | −12b (‑17, ‑6) | 3a (1, 5) | −37b (‑40, ‑34) | −46b (‑50, ‑43) |
| Lyhenteet: QM = kerran kuukaudessa, a nimellinen p-arvo < 0,001 lumevalmisteeseen verrattuna, b p-arvo < 0,001 lumevalmisteeseen verrattuna. | ||||||||||||
OSLER ja OSLER‑2 olivat kaksi satunnaistettua avointa vertailevaa jatkotutkimusta, joissa arvioitiin Repathan pitkäaikaista turvallisuutta ja tehoa potilailla, jotka olivat mukana päätutkimuksessa hoidon loppuun asti. Kummassakin jatkotutkimuksessa potilaat jaettiin satunnaistetusti 2:1 Repathaa ja tavanomaista hoitoa saavaan ryhmään (evolokumabiryhmä) tai pelkkää tavanomaista hoitoa saavaan ryhmään (vertailuryhmä) ensimmäisen tutkimusvuoden ajaksi. Ensimmäisen vuoden lopussa (OSLER-tutkimuksessa viikolla 52 ja OSLER‑2-tutkimuksessa viikolla 48) potilaat siirtyivät Repatha-jaksoon, jossa kaikki potilaat saivat avointa Repatha-hoitoa vielä 4 vuoden (OSLER) tai 2 vuoden (OSLER‑2) ajan.
OSLER-tutkimukseen otettiin mukaan yhteensä 1324 potilasta. Repatha 420 mg kerran kuukaudessa laski LDL‑kolesteroliarvoa merkitsevästi lähtötasosta viikon 12 ja viikon 52 mittausten perusteella vertailuryhmään verrattuna (nimellinen p-arvo < 0,001). Hoitovaikutukset säilyivät 272 viikon ajan, minkä osoittaa LDL‑kolesteroliarvon laskun säilyminen päätutkimuksen viikolta 12 avoimen jatkotutkimuksen viikolle 260. OSLER‑2-tutkimukseen otettiin mukaan yhteensä 3681 potilasta. Repatha laski LDL‑kolesteroliarvoa merkitsevästi lähtötasosta viikon 12 ja viikon 48 mittausten perusteella vertailuryhmään verrattuna (nimellinen p-arvo < 0,001). Hoitovaikutukset säilyivät, mistä on osoituksena viikolla 12 todetun LDL‑kolesteroliarvon laskun säilyminen avoimen jatkotutkimuksen viikolle 104. Repatha pienensi merkitsevästi kokonaiskolesteroli-, ApoB- ja ei‑HDL‑kolesterolipitoisuutta, kokonaiskolesterolin ja HDL‑kolesterolin suhdetta, ApoB/ApoA1‑suhdetta sekä VLDL‑kolesteroli-, triglyseridi- ja Lp(a)‑pitoisuutta ja suurensi HDL‑kolesteroli- ja ApoA1‑pitoisuutta lähtötasosta OSLER-tutkimuksessa viikon 52 ja OSLER‑2-tutkimuksessa viikon 48 mittausten perusteella vertailuryhmään verrattuna (nimellinen p‑arvo < 0,001). LDL‑kolesteroli ja muut lipidiparametrit palautuivat lähtötasolle 12 viikon kuluessa Repatha-hoidon lopettamisen jälkeen OSLER- tai OSLER‑2-tutkimuksen alussa. Viitteitä arvojen tilapäisestä huononemisesta (rebound) ei havaittu.
TAUSSIG oli 5 vuoden avoin monikeskus-jatkotutkimus, jossa arvioitiin Repathan pitkäaikaista turvallisuutta ja tehoa muihin rasva-arvoja alentaviin hoitoihin yhdistettynä vaikean familiaalisen hyperkolesterolemian (FH), myös homotsygoottisen familiaalisen hyperkolesterolemian, hoidossa. TAUSSIG-tutkimukseen otettiin mukaan yhteensä 194 vaikeaa familiaalista hyperkolesterolemiaa (ei‑HoFH) ja 106 homotsygoottista familiaalista hyperkolesterolemiaa sairastavaa potilasta. Kaikki tutkimukseen otetut potilaat saivat aluksi Repathaa 420 mg kerran kuukaudessa, lukuun ottamatta potilaita, jotka saivat lipidiafereesihoitoa tutkimuksen sisäänottovaiheessa. Näille potilaille Repatha-hoito aloitettiin annostuksella 420 mg kahden viikon välein. Potilaille, jotka eivät saaneet afereesihoitoa, annostiheys voitiin nostaa tasolle 420 mg kahden viikon välein LDL‑kolesterolivasteen ja PCSK9‑pitoisuuden perusteella. Repathan pitkäaikainen käyttö osoitti, että hoitovaikutus on pitkäkestoinen, mistä oli osoituksena vaikeaa familiaalista hyperkolesterolemiaa (ei‑HoFH) sairastavien potilaiden LDL‑kolesteroliarvon lasku (ks. taulukko 5).
Myös muiden lipidiparametrien (kokonaiskolesterolin, ApoB:n, ei‑HDL‑kolesterolin, kokonaiskolesterolin ja HDL‑kolesterolin suhteen ja ApoB/ApoA1‑suhteen) muutokset osoittivat, että pitkäaikaisen Repatha-hoidon teho säilyy pitkään vaikeaa familiaalista hyperkolesterolemiaa (ei‑HoFH) sairastavilla potilailla.
Taulukko 5. Evolokumabin vaikutus LDL-kolesteroliin vaikean familiaalisen hyperkolesterolemian (ei‑HoFH) hoidossa – prosentuaalinen muutos lähtötasosta (keskiarvo) avoimen jatkotutkimuksen (OLE) viikkoon 216 mennessä (ja vastaava 95 % CI)
Potilasjoukko (N) | OLE viikko 12 (n = 191) | OLE viikko 24 (n = 191) | OLE viikko 36 (n = 187) | OLE viikko 48 (n = 187) | OLE viikko 96 (n = 180) | OLE viikko 144 (n = 180) | OLE viikko 192 (n = 147) | OLE viikko 216 (n = 96) |
Vaikea FH (ei‑HoFH) (N = 194) | −54,9 (−57,4, −52,4) | −54,1 (−57,0, −51,3) | −54,7 (−57,4, −52,0) | −56,9 (−59,7, −54,1) | −53,3 (−56,9, −49,7) | −53,5 (−56,7, −50,2) | −48,3 (−52,9, −43,7) | −47,2 (−52,8, −41,5) |
| Lyhenteet: OLE = avoin jatkotutkimus (open-label extension), N (n) = Arvioitavissa olevien potilaiden lukumäärä (N) ja niiden potilaiden lukumäärä, joiden LDL-kolesteroliarvo oli määritetty tietyllä aikataulun mukaisella käynnillä (n) vaikean familiaalisen hyperkolesterolemian (ei‑HoFH) loppuanalyysissä. | ||||||||
Heterotsygoottisen familiaalisen hyperkolesterolemian hoito lapsipotilailla
HAUSER-RCT oli kaksoissokkoutettu satunnaistettu lumekontrolloitu ja rinnakkaisryhmissä toteutettu 24 viikon monikeskustutkimus, jossa oli mukana 158 heterotsygoottista familiaalista hyperkolesterolemiaa (HeFH) sairastavaa, 10 vuotta täyttänyttä mutta alle 18‑vuotiasta lapsipotilasta. Potilaiden edellytettiin noudattavan vähärasvaista ruokavaliota ja saavan perushoitona optimaalista rasva-arvoja alentavaa lääkehoitoa (statiinihoitoa optimaalisella annoksella ilman annoksen nostamisen tarvetta). Tutkimukseen otetut potilaat satunnaistettiin suhteessa 2:1 saamaan 24 viikkoa Repathaa 420 mg ihon alle kerran kuukaudessa tai lumevalmistetta.
Tutkimuksessa tehon ensisijainen päätetapahtuma oli LDL‑kolesterolipitoisuuden prosentuaalinen muutos lähtötasosta viikolla 24. Repatha- ja lumeryhmän välinen ero LDL‑kolesterolipitoisuuden keskimääräisessä prosentuaalisessa muutoksessa lähtötasosta viikolla 24 oli 38 % (95 % CI: 45 %, 31 %; p < 0,0001). LDL‑kolesterolipitoisuus oli pienentynyt (p < 0,0001) lähtötasosta viikolla 24 Repatha‑ryhmässä 44 % (keskivirhe, SE 2 %) ja lumevalmistetta saaneiden ryhmässä 6 % (SE 3 %). Tulos on laskettu pienimpien neliösummien keskiarvona. Absoluuttisten LDL‑kolesterolipitoisuuksien keskiarvo viikolla 24 oli Repatha-ryhmässä 2,69 mmol/l (104 mg/dl) ja lumevalmistetta saaneessa ryhmässä 4,45 mmol/l (172 mg/dl). LDL‑kolesterolipitoisuuden pieneneminen havaittiin ensimmäisessä lähtötason jälkeisessä arvioinnissa viikolla 12 ja säilyi koko tutkimuksen ajan.
Tutkimuksen toissijainen päätetapahtuma oli LDL‑kolesterolipitoisuuden keskimääräinen prosentuaalinen muutos lähtötasosta viikoilla 22 ja 24, missä viikko 22 edustaa ihon alle kerran kuukaudessa annosteluvälin huippupitoisuutta ja viikko 24 annosteluvälin jäännöspitoisuutta, antaen tietoa Repatha‑hoidon ajan suhteen keskiarvoistetusta vaikutuksesta koko annosteluvälin ajalta. Repatha- ja lumevalmistehoidon ero LDL‑kolesterolipitoisuuden keskimääräisessä prosentuaalisessa muutoksessa lähtötasosta viikkojen 22 ja 24 keskiarvoon oli 42 % (laskettu pienimpien neliösummien keskiarvoista, 95 % CI: 48 %, 36 %; p < 0,0001). Tulokset on esitelty laajemmin taulukossa 6.
Taulukko 6. Repathan hoitovaikutukset lumevalmisteeseen verrattuna heterotsygoottista familiaalista hyperkolesterolemiaa sairastavilla lapsipotilailla – prosentuaalinen muutos (keskiarvo) lähtötasosta viikolla 24 (%, 95 % CI)
Tutkimus | Annostusohjelma | LDL-Kol (%) | Ei-HDL-Kol (%) | ApoB (%) | Kol/ (%) | ApoB/ (%) |
HAUSER-RCT (HeFH-lapsipotilaat) | 420 mg QM (N = 104) | −38,3 (−45,5, −31,1) | −35,0 (−41,8, −28,3) | −32,5 (−38,8, −26,1) | −30,3 (−36,4, −24,2) | −36,4 (−43,0, −29,8) |
QM = kerran kuukaudessa (ihon alle), CI (Confidence Interval) = luottamusväli, LDL-Kol = LDL (Low Density Lipoprotein) -kolesteroli; HDL-Kol = HDL (High Density Lipoprotein) -kolesteroli, ApoB = apolipoproteiini B, ApoA1 = apolipoproteiini A1, Kol = kokonaiskolesteroli Kaikki mukautetut p-arvot < 0,0001 N = satunnaistettujen ja annoksen saaneiden potilaiden määrä koko analyysijoukossa | ||||||
HAUSER‑OLE oli 80 viikon avoin, yhden haaran monikeskustutkimus, jossa arvioitiin Repathaa. Tutkimukseen osallistui 150 10–17‑vuotiasta HeFH-lapsipotilasta, jotka siirtyivät HAUSER‑RCT-tutkimuksesta, ja 13 de novo HoFH-lapsipotilasta. Potilaiden edellytettiin noudattavan vähärasvaista ruokavaliota ja saavan perushoitona rasva-arvoja alentavaa lääkehoitoa. Kaikki tähän tutkimukseen osallistuneet HeFH-potilaat saivat Repathaa 420 mg ihon alle kerran kuukaudessa (altistuksen mediaanikesto: 18,4 kuukautta). Laskennallisen LDL-kolesterolin keskimääräiset (SE) prosentuaaliset muutokset lähtötasosta olivat −44,4 % (1,7 %) viikolla 12, −41,0 % (2,1 %) viikolla 48 ja −35,2 % (2,5 %) viikolla 80.
Lipidejä koskevien muiden päätetapahtumien keskimääräiset (SE) prosentuaaliset muutokset lähtötasosta viikolle 80 olivat −32,1 % (2,3 %) ei‑HDL‑Kol, −25,1 % (2,3 %) ApoB, −28,5 % (2,0 %) Kol/HDL‑Kol-suhde, −30,3 % (2,2 %) ApoB/ApoA1-suhde ja −24,9 % (1,9 %) Kol.
Homotsygoottisen familiaalisen hyperkolesterolemian hoito
TESLA oli 12 viikon kansainvälinen kaksoissokkoutettu, satunnaistettu monikeskus-lumevertailututkimus, jossa oli mukana 49 homotsygoottista familiaalista hyperkolesterolemiaa sairastavaa 12–65‑vuotiasta potilasta. Repatha 420 mg kerran kuukaudessa yhdistettynä muihin rasva-arvoja alentaviin lääkkeisiin (esim. statiineihin, sappihappoja sitoviin aineisiin) laski merkitsevästi LDL‑kolesteroli- ja ApoB‑arvoja viikon 12 mittausten perusteella lumevalmisteeseen verrattuna (p < 0,001) (ks. taulukko 7). Myös muiden lipidiparametrien (kokonaiskolesterolin, ei‑HDL‑kolesterolin, kokonaiskolesterolin ja HDL‑kolesterolin suhteen ja ApoB/ApoA1‑suhteen) muutokset osoittivat Repathan hoitotehon homotsygoottista familiaalista hyperkolesterolemiaa sairastavilla potilailla.
Taulukko 7. Evolokumabin hoitovaikutukset lumevalmisteeseen verrattuna homotsygoottista familiaalista hyperkolesterolemiaa sairastavilla potilailla – prosentuaalinen muutos (keskiarvo) lähtötasosta viikolla 12 (%, 95 % CI)
Tutkimus | Annostusohjelma | LDL-Kol (%) | Ei-HDL-Kol (%) | ApoB (%) | Kol (%) | Lp(a) (%) | VLDL-Kol (%) | HDL-Kol (%) | Trigly (%) | Kol/ HDL-Kol-suhde % | ApoB/ ApoA1-suhde % |
TESLA (HoFH) | 420 mg QM (N = 33) | −32b (−45, −19) | −30a (−42, −18) | −23b (−35, −11) | −27a (−38, −16) | −12 (−25, 2) | −44 (−128, 40) | −0,1 (−9, 9) | 0,3 (−15, 16) | −26a (−38, −14) | −28a (−39, −17) |
| Lyhenteet: HoFH = homotsygoottinen familiaalinen hyperkolesterolemia, QM = kerran kuukaudessa, a nimellinen p‑arvo < 0,001 lumevalmisteeseen verrattuna, b p‑arvo < 0,001 lumevalmisteeseen verrattuna. | |||||||||||
Pitkäaikainen teho homotsygoottisen familiaalisen hyperkolesterolemian hoidossa
TAUSSIG-tutkimuksessa pitkäaikaishoitona annetun Repathan hoitoteho oli pitkäkestoinen. Tästä oli osoituksena noin 20–30 prosentin lasku LDL‑kolesteroliarvossa homotsygoottista familiaalista hyperkolesterolemiaa sairastavilla potilailla, jotka eivät saaneet afereesihoitoa, ja noin 10–30 prosentin lasku afereesihoitoa saavilla homotsygoottista familiaalista hyperkolesterolemiaa sairastavilla potilailla (ks. taulukko 8). Myös muiden lipidiparametrien (kokonaiskolesterolin, ApoB:n, ei‑HDL‑kolesterolin, kokonaiskolesterolin ja HDL‑kolesterolin suhteen ja ApoB/ApoA1‑suhteen) muutokset osoittivat, että pitkäaikaisen Repatha-hoidon teho on pitkäkestoinen homotsygoottista familiaalista hyperkolesterolemiaa sairastavilla potilailla. LDL‑kolesterolin lasku ja muiden lipidiparametrien muutokset ovat 14 nuorella (ikäjakauma ≥ 12 ‑ < 18 vuotta) homotsygoottista familiaalista hyperkolesterolemiaa sairastavalla potilaalla vertailukelpoiset koko homotsygoottista familiaalista hyperkolesterolemiaa sairastavassa potilasjoukossa havaittujen muutosten kanssa.
Taulukko 8. Evolokumabin vaikutus LDL-kolesteroliin homotsygoottista familiaalista hyperkolesterolemiaa sairastavilla potilailla – prosentuaalinen muutos lähtötasosta (keskiarvo) avoimen jatkotutkimuksen (OLE) viikkoon 216 mennessä (ja vastaava 95 % CI)
Potilas- joukko (N) | OLE viikko 12 | OLE viikko 24 | OLE viikko 36 | OLE viikko 48 | OLE viikko 96 | OLE viikko 144 | OLE viikko 192 | OLE viikko 216 |
HoFH (N = 106) | −21,2 (−26,0, −16,3) (n = 104) | −21,4 (−27,8, −15,0) (n = 99) | −27,0 (−32,1, −21,9) (n = 94) | −24,8 (−31,4, −18,3) (n = 93) | −25,0 (−31,2, −18,8) (n = 82) | −27,7 (−34,9, −20,5) (n = 79) | −27,4 (−36,9, −17,8) (n = 74) | −24,0 (−34,0, −14,0) (n = 68) |
Ei afereesihoitoa (N = 72) | −22,7 (−28,1, −17,2) (n = 70) | −25,8 (−33,1, −18,5) (n = 69) | −30,5 (−36,4, −24,7) (n = 65) | −27,6 (−35,8, −19,4) (n = 64) | −23,5 (−31,0, −16,0) (n = 62) | −27,1 (−35,9, −18,3) (n = 60) | −30,1 (−37,9, −22,2) (n = 55) | −23,4 (−32,5, −14,2) (n = 50) |
Afereesihoito (N = 34) | −18,1 (−28,1, −8,1) (n = 34) | −11,2 (−24,0, 1,7) (n = 30) | −19,1 (−28,9, −9,3) (n = 29) | −18,7 (−29,5, −7,9) (n = 29) | −29,7 (−40,6, −18,8) (n = 20) | −29,6 (−42,1, −17,1) (n = 19) | −19,6 (−51,2, 12,1) (n = 19) | −25,9 (−56,4, 4,6) (n = 18) |
| Lyhenteet: OLE = avoin jatkotutkimus (open-label extension). N (n) = Arvioitavissa olevien potilaiden lukumäärä (N) ja niiden potilaiden lukumäärä, joiden LDL-arvo oli määritetty tietyllä aikataulun mukaisella käynnillä (n) homotsygoottisen familiaalisen hyperkolesterolemian (HoFH) loppuanalyysissä. | ||||||||
HAUSER-OLE oli 80 viikon avoin, yhden haaran monikeskustutkimus, jossa arvioitiin Repathan turvallisuutta, siedettävyyttä ja tehoa LDL‑kolesterolin alentamisessa 10 vuotta täyttäneillä mutta alle 18‑vuotiailla HoFH-lapsipotilailla. Tutkimukseen osallistui 12 potilasta. Potilaiden edellytettiin noudattavan vähärasvaista ruokavaliota ja saavan perushoitona rasva-arvoja alentavaa lääkehoitoa. Kaikki tutkimukseen osallistuneet potilaat saivat Repathaa 420 mg ihon alle kerran kuukaudessa. LDL‑kolesterolipitoisuuden mediaani (Q1, Q3) lähtötilanteessa oli 10,29 mmol/l (8,87; 12,28 mmol/l) [398 mg/dl (343, 475 mg/dl)]. LDL‑kolesterolipitoisuuden prosentuaalisen muutoksen mediaani (Q1, Q3) lähtötasosta viikolla 80 oli −14 % (−41, 4). LDL‑kolesterolipitoisuuden pieneneminen havaittiin ensimmäisessä arvioinnissa viikolla 12 ja säilyi koko tutkimuksen ajan. Pienentyminen vaihteli välillä 12 % (−3, 32) ja 15 % (−4, 39) (mediaani (Q1, Q3)). Tulokset on esitelty laajemmin taulukossa 9.
Taulukko 9. Evolokumabin hoitovaikutukset lumevalmisteeseen verrattuna homotsygoottista familiaalista hyperkolesterolemiaa sairastavilla potilailla – prosentuaalinen muutos lähtötasosta viikolla 80 (mediaani (Q1, Q3))
Tutkimus | Annostusohjelma | LDL-Kol (%) | Ei-HDL-Kol (%) | ApoB (%) | Kol/ (%) | ApoB/ (%) |
HAUSER-OLE (HoFH-lapsipotilaat) | 420 mg QM (N = 12) | −14,3 (−40,6, 3,5) | −13 (−40,7, 2,7) | −19,1 (−33,3, 11,6) | −3,7 (−41,6, 7,6) | −3 (−35,7, 9,3) |
QM = kerran kuukaudessa (ihon alle), LDL-Kol = LDL (Low Density Lipoprotein) -kolesteroli; HDL-Kol = HDL (High Density Lipoprotein) -kolesteroli, ApoB = apolipoproteiini B, ApoA1 = apolipoproteiini A1, Kol = kokonaiskolesteroli N = satunnaistettujen ja annoksen saaneiden potilaiden määrä välianalyysijoukossa | ||||||
Vaikutus ateroskleroottisen sairauden taakkaan
Kaksoissokkoutetussa satunnaistetussa 78 viikon lumevertailututkimuksessa arvioitiin 420 mg:n annoksina kerran kuukaudessa annetun Repathan vaikutuksia ateroskleroottisen sairauden taakkaan suonensisäisellä kaikukuvauksella (IVUS) mitattuna. Tutkimuksessa oli mukana 968 sepelvaltimotautia sairastavaa potilasta, jotka saivat vakaana perushoitona optimaalista statiinihoitoa. Repatha vähensi sekä aterooman prosentuaalista osuutta suonen seinämässä (PAV; 1,01 % [95 prosentin luottamusväli (95 % CI) 0,64–1,38], p < 0,0001) että ateroomamassan kokonaistilavuutta (TAV; 4,89 mm3 [95 % CI 2,53–7,25], p < 0,0001) lumevalmisteeseen verrattuna. Aterooman prosentuaalisen osuuden perusteella mitattuna ateroskleroosin vähenemistä havaittiin 64,3 prosentilla (95 % CI 59,6–68,7) Repathaa saaneista ja 47,3 prosentilla (95 % CI 42,6–52,0) lumevalmistetta saaneista potilaista. Kokonaistilavuuden perusteella mitattuna ateroskleroosin vähenemistä havaittiin 61,5 prosentilla (95 % CI 56,7–66,0) Repathaa saaneista ja 48,9 prosentilla (95 % CI 44,2–53,7) lumevalmistetta saaneista potilaista. Tutkimuksessa ei selvitetty korrelaatiota ateroskleroottisen sairauden lievittymisen ja sydän- ja verisuonitautitapahtumien välillä.
Vaikutus sepelvaltimoiden ateroskleroottisten plakkien morfologiaan
Repathan (420 mg kerran kuukaudessa) vaikutuksia sepelvaltimoiden ateroskleroottisiin plakkeihin arvioitiin valokerroskuvauksella (OCT) kaksoissokkoutetussa satunnaistetussa lumekontrolloidussa 52 viikon mittaisessa tutkimuksessa. Tutkimukseen osallistuneiden aikuispotilaiden hoito aloitettiin 7 vuorokauden kuluessa ST‑nousuttoman äkillisen sepelvaltimo‑oireyhtymän (NSTEACS) ilmenemisestä. Potilailla oli käytössä suurin siedetty statiiniannos. Ensisijaisena päätetapahtumana oli sidekudoskaton pienimmän paksuuden absoluuttinen muutos lähtötilanteeseen verrattuna vastaavassa valtimosegmentissä. Muutos laskettiin pienimpien neliösummien keskiarvona (95 % CI). Paksuus lisääntyi lähtötilanteeseen verrattuna Repatha‑ryhmässä 42,7 μm (32,4–53,1) ja lumeryhmässä 21,5 μm (10,9–32,1), eli ero lumevalmisteeseen oli 21,2 μm (4,7–37,7) (p = 0,015; 38 %:n ero [p = 0,041]). Toissijaisina löydöksinä raportoitiin hoitoryhmien välisiä eroja, kuten sidekudoskaton pienimmän paksuuden keskiarvon muutos (kasvua 32,5 µm [12,7–52,4]; p = 0,016) ja maksimaalisen lipidikaaren absoluuttinen muutos (–26° [–49,6...–2,4]; p = 0,041).
Sydän- ja verisuonisairauksien riskin pienentäminen aikuisilla, joilla on todettu ateroskleroottinen sydän- tai verisuonisairaus
Repathan hoitotulostutkimus (FOURIER) oli satunnaistettu, tapahtumaohjattu kaksoissokkotutkimus, jossa oli mukana 27 564 tutkittavaa, joiden ikäjakauma oli 40–86 vuotta (keski-ikä 62,5 vuotta) ja joilla oli todettu ateroskleroottinen sydän- tai verisuonisairaus: 81 prosentilla oli ollut sydäninfarktitapahtuma ja 19 prosentilla aivohalvaustapahtuma ja 13 prosentilla oli perifeerinen valtimosairaus. Yli 99 % potilaista sai kohtalaisen tai suuren intensiteetin statiinia ja vähintään yhtä muuta sydän- ja verisuonisairauksien lääkettä, kuten verihiutaleiden estäjiä, beetasalpaajia, angiotensiinikonvertaasin (ACE) estäjiä tai angiotensiinireseptorin salpaajia. LDL‑kolesterolipitoisuuden mediaani (Q1, Q3) lähtötilanteessa oli 2,4 mmol/l (2,1; 2,8). Absoluuttinen sydän- ja verisuonisairauksien riski oli tasapainossa hoitoryhmien välillä, ja indeksitapahtuman lisäksi kaikilla potilailla oli vähintään yksi merkittävä tai kaksi vähäisempää sydän- ja verisuonisairauksien riskitekijää: 80 prosentilla oli hypertensio, 36 prosentilla oli diabetes ja 28 % tupakoi päivittäin. Potilaat jaettiin satunnaistetusti suhteessa 1:1 joko Repathaa (140 mg kahden viikon välein tai 420 mg kerran kuukaudessa) tai vastaavaa lumevalmistetta saavaan ryhmään. Potilaiden seuranta-ajan keskiarvo oli 26 kuukautta.
LDL‑kolesteroliarvon huomattava alenema säilyi koko tutkimuksen ajan. Saavutettu LDL‑kolesterolipitoisuuden mediaani oli 0,8–0,9 mmol/l jokaisessa mittauksessa, ja 25 prosentilla potilaista LDL‑kolesterolipitoisuus oli alle 0,5 mmol/l. Saavutetuista hyvin alhaisista LDL‑kolesteroliarvoista huolimatta uusia turvallisuusongelmia ei havaittu (ks. kohta Haittavaikutukset). Uusien diabetestapausten ja kognitiivisten tapahtumien esiintymistiheys oli samalla tasolla potilailla, joiden LDL‑kolesterolipitoisuus oli < 0,65 mmol/l, kuin niillä, joiden LDL‑kolesteroliarvo oli korkeampi.
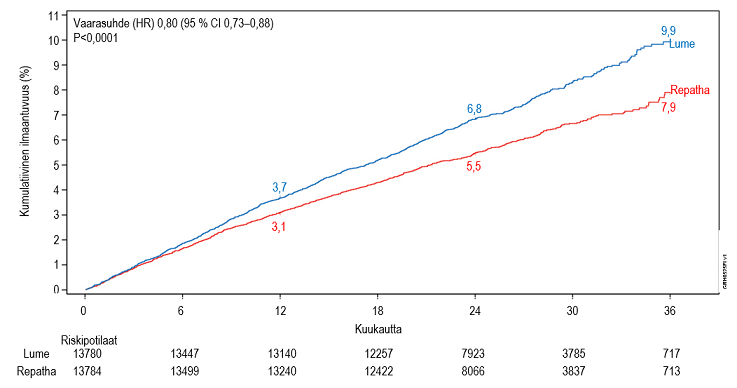
Repatha pienensi merkittävästi sydän- ja verisuonitautitapahtumien riskiä, jonka kriteerinä oli aika ensimmäiseen sydän- ja verisuonitautikuolemaan, sydäninfarktiin, aivohalvaukseen, sepelvaltimoiden revaskularisaatioon tai epästabiilista angina pectoriksesta johtuvaan sairaalahoitoon (ks. taulukko 10). Ensisijaisen päätetapahtuman ja tärkeimpien toissijaisten päätetapahtumien yhdistelmän Kaplan–Meier-kuvaajat erosivat toisistaan noin 5 kuukauden kohdalla (ks. kuva 1, kolmen vuoden Kaplan–Meier-kuvaaja MACE-tapahtumista). Yhdistettyjen MACE-tapahtumien (sydän- ja verisuonitautikuolema, sydäninfarkti tai aivohalvaus) suhteellinen riski pieneni merkitsevästi, 20 %. Hoitovaikutus oli yhdenmukainen kaikissa alaryhmissä (joihin kuuluivat ikä, sairaustyyppi, LDL‑kolesterolin lähtöarvo, statiinin intensiteetti lähtötilanteessa, etsetimibin käyttö ja diabetes), ja se perustui sydäninfarktin, aivohalvauksen ja sepelvaltimoiden revaskularisaation riskin pienenemiseen. Sydän- ja verisuonitautikuolleisuudessa tai kokonaiskuolleisuudessa ei havaittu merkitsevää eroa, mutta tällaisen eron osoittaminen ei ollut tutkimusasetelman tavoitteena.
Taulukko 10. Evolokumabin vaikutus merkittäviin sydän- ja verisuonitautitapahtumiin
Lume (N = 13 780) n (%) | Evolokumabi (N = 13 784) n (%) | Vaarasuhde (HR)a (95 % CI) | p-arvob | |
MACE+ (MACE, sepelvaltimoiden revaskularisaatio tai epästabiilista angina pectoriksesta johtuva sairaalahoito) | 1563 (11,34) | 1344 (9,75) | 0,85 (0,79–0,92) | < 0,0001 |
MACE (sydän- ja verisuonitautikuolema, sydäninfarkti tai aivohalvaus) | 1013 (7,35) | 816 (5,92) | 0,80 (0,73–0,88) | < 0,0001 |
Sydän- ja verisuonitautikuolema | 240 (1,74) | 251 (1,82) | 1,05 (0,88–1,25) | 0,62 |
Kuolleisuus, kaikki kuolinsyyt | 426 (3,09) | 444 (3,22) | 1,04 (0,91–1,19) | 0,54 |
Sydäninfarkti (kuolemaan johtanut tai muu) | 639 (4,64) | 468 (3,40) | 0,73 (0,65–0,82) | < 0,0001c |
Aivohalvaus (kuolemaan johtanut tai muu)d | 262 (1,90) | 207 (1,50) | 0,79 (0,66–0,95) | 0,0101c |
Sepelvaltimoiden revaskularisaatio | 965 (7,00) | 759 (5,51) | 0,78 (0,71–0,86) | < 0,0001c |
Epästabiilista angina pectoriksesta johtuva sairaalahoitoe | 239 (1,7) | 236 (1,7) | 0,99 (0,82–1,18) | 0,89 |
a Perustuu Coxin malliin ositettuna vuorovaikutteisen äänipalvelujärjestelmän (Interactive Voice Response System, IVRS) kautta kerättyjen, satunnaistamisen osituksessa käytettyjen tekijöiden mukaan. b Kaksisuuntainen logrank-testi ositettuna IVRS-järjestelmän kautta kerättyjen, satunnaistamisen osituksessa käytettyjen tekijöiden mukaan. c Nimellinen merkitsevyys. d Hoidon vaikutus aivohalvauksiin perustui iskeemisen aivohalvauksen riskin pienenemiseen; sillä ei ollut vaikutusta verenvuodon aiheuttamaan eikä määrittämättömään aivohalvaukseen. e Aika epästabiilista angina pectoriksesta johtuvaan sairaalahoitoon arvioitiin ad hoc. | ||||
Kuva 1. Aika MACE‑tapahtumaan (sydän- ja verisuonitautikuolema, sydäninfarkti tai aivohalvaus), 3 vuoden Kaplan–Meier
FOURIER OLE (FOURIER avoin jatkotutkimus) koostui kahdesta avoimesta yhden haaran monikeskustutkimuksesta (tutkimus 1 ja tutkimus 2), joissa arvioitiin Repathan pitkäaikaista turvallisuutta, siedettävyyttä ja tehoa potilailla, joilla oli todettu sydän- ja verisuonisairaus ja jotka olivat loppuun asti mukana FOURIER-tutkimuksessa. Mukaan otetut potilaat saivat Repathaa joko 140 mg kahden viikon välein tai 420 mg kerran kuukaudessa noin 5 vuoden ajan. Samalla jatkettiin kohtalaisen (22,2 %:lla potilaista) tai suuren intensiteetin (74,8 %:lla potilaista) perushoitoa statiinilla. Tutkimuksessa 1 vähintään yhden Repatha-annoksen saaneista 5 031 potilaasta 2 499 oli saanut FOURIER-tutkimuksessa Repathaa ja 2 532 potilasta lumevalmistetta. Tutkimuksessa 2 vähintään yhden Repatha-annoksen saaneista 1 599 potilaasta 854 oli saanut FOURIER-tutkimuksessa Repathaa ja 745 potilasta lumevalmistetta. Kun tutkimukset 1 ja 2 päättyivät, Repatha-kokonaisaltistus oli FOURIER-tutkimuksessa Repatha-hoitoon satunnaistetuilla tutkimuksen 1 potilailla enimmillään 8,4 vuotta (mediaani 85,4 kuukautta) ja tutkimuksen 2 potilailla enimmillään 8,0 vuotta (mediaani 80,2 kuukautta) ja vastaavasti FOURIER-tutkimuksessa lumeryhmään satunnaistetuilla tutkimuksen 1 potilailla enimmillään 5,25 vuotta (mediaani 60,0 kuukautta) ja tutkimuksen 2 potilailla enimmillään 4,9 vuotta (mediaani 55,1 kuukautta).
Tutkimusten 1 ja 2 yhdistettyjen tulosten mukaan 72,4 %:lla (n = 4 802) potilaista saavutettu alhaisin lähtötason jälkeinen LDL‑kolesteroliarvo oli < 0,65 mmol/l (25 mg/dl), 87,0 % (n = 5 765) potilaista saavutti LDL‑kolesteroliarvon < 1,03 mmol/l (40 mg/dl), ja 11,9 %:lla (n = 792) potilaista kaikkien lähtötason jälkeisten LDL‑kolesterolimittausten arvo oli ≥ 1,03 mmol/l (40 mg/dl). Kun tarkastellaan potilaita, jotka saavuttivat lähtötilanteen jälkeen alhaisen LDL‑kolesteroliarvon (< 0,65 mmol/l [25 mg/dl] tai < 1,03 mmol/l [40 mg/dl]), hoidon aikaisia haittatapahtumia ilmeni kaiken kaikkiaan 80,0 %:lla potilaista, jotka saavuttivat LDL‑kolesteroliarvon < 0,65 mmol/l (25 mg/dl), ja 82,7 %:lla potilaista, jotka saavuttivat LDL‑kolesteroliarvon < 1,03 mmol/l (40 mg/dl). Vastaava ilmaantuvuusluku oli 85,0 % potilailla, joiden LDL‑kolesteroliarvo oli ≥ 1,03 mmol/l (40 mg/dl). Hoidon aikana ilmeni vakavia haittatapahtumia kaiken kaikkiaan 37,7 %:lla niistä potilaista, jotka saavuttivat LDL‑kolesteroliarvon < 0,65 mmol/l (25 mg/dl), ja 40,0 %:lla niistä potilaista, jotka saavuttivat LDL‑kolesteroliarvon < 1,03 mmol/l (40 mg/dl). Vastaava ilmaantuvuusluku oli 41,5 % potilailla, joiden LDL‑kolesteroliarvo oli ≥ 1,03 mmol/l (40 mg/dl).
LDL‑kolesteroliarvon prosentuaalinen pieneneminen (keskiarvo) lähtötasosta pysyi vakaana avoimessa jatkotutkimusvaiheessa: tutkimuksessa 1 vaihteluväli oli 53,4–59,1 %, ja tutkimuksessa 2 se oli 62,5–67,2 % riippumatta siitä, mihin hoitoryhmään potilas oli FOURIER-tutkimuksessa alun perin satunnaistettu. Tämä vaikuttaisi olevan syynä siihen, että todettujen eksploratiivisten yhdistettyjen sydän- ja verisuonitautien päätetapahtumien (sydän- ja verisuonitautikuolema, sydäninfarkti ja aivohalvaus) ilmaantuvuus oli numeerisesti pienempi potilailla, jotka saivat Repathaa sekä FOURIER- että FOURIER OLE -tutkimuksessa, kuin potilailla, jotka saivat FOURIER-tutkimuksessa lumevalmistetta ja FOURIER OLE -tutkimuksessa Repathaa.
Kaiken kaikkiaan näissä tutkimuksissa ei tunnistettu uusia turvallisuuslöydöksiä.
Vaikutus LDL-kolesteroliin äkillisen sepelvaltimo-oireyhtymän (ACS) akuutissa vaiheessa
EVOPACS oli yhdessä maassa tehty, 8 viikkoa kestänyt kaksoissokkoutettu, satunnaistettu monikeskus-lumevertailututkimus, jossa oli mukana 308 äkillistä sepelvaltimo-oireyhtymää sairastavaa potilasta, joilla evolokumabihoito aloitettiin sairaalassa 24–72 tunnin sisällä oireiden ilmenemisestä.
Jos potilaat eivät ennen seulontaa saaneet statiinihoitoa tai saivat muuta statiinihoitoa kuin 40 mg atorvastatiinia, hoito lopetettiin ja aloitettiin hoito 40 mg:n atorvastatiiniannoksella kerran vuorokaudessa. Satunnaistaminen ositettiin tutkimuskeskuksen mukaan ja sen mukaan, oliko potilaalla vakaa statiinihoito ≥ 4 viikkoa ennen potilaan ottamista tutkimukseen. Suurimmalla osalla tutkittavista (241 [78 %]) ei ollut vakaata statiinihoitoa ≥ 4 viikon ajan ennen seulontaa, eikä suurimmalla osalla (235 [76 %]) ollut statiinihoitoa lähtötilanteessa. Viikkoon 4 mennessä 281 (97 %) tutkittavaa sai suuren intensiteetin statiinia. Kerran kuukaudessa annettu 420 mg:n annos evolokumabia pienensi LDL‑kolesterolipitoisuutta merkitsevästi lähtötasosta viikkoon 8 mennessä lumevalmisteeseen verrattuna (p < 0,001). Laskennallinen LDL-kolesteroliarvo oli pienentynyt lähtötasosta viikolla 8 evolokumabiryhmässä 77,1 % (15,8 %) (keskiarvo (SD)) ja lumevalmistetta saaneessa ryhmässä 35,4 % (26,6 %). Pienimpien neliösummien (LS) keskiarvojen ero (95 % CI) oli 40,7 % (36,2–45,2 %). Lähtötilanteen LDL-kolesteroliarvot olivat 3,61 mmol/l (139,5 mg/dl) evolokumabiryhmässä ja 3,42 mmol/l (132,2 mg/dl) lumevalmistetta saaneessa ryhmässä. LDL-kolesteroliarvojen pieneneminen tässä tutkimuksessa oli yhdenmukaista aiempien tutkimusten kanssa, joissa evolokumabia annettiin vakaan lipidilääkityksen lisäksi. Sen osoittivat tämän tutkimuksen viikolla 8 mitatut hoitoa saaneiden LDL-kolesterolitasot (kuvastaen suuren intensiteetin statiinihoidon vaikutuksen vakiintumista molemmissa hoitohaaroissa). LDL-kolesterolitasot olivat 0,79 mmol/l (30,5 mg/dl) evolokumabia ja atorvastatiinia saaneessa ryhmässä ja 2,06 mmol/l (79,7 mg/dl) lumevalmistetta ja atorvastatiinia saaneessa ryhmässä.
Evolokumabin vaikutukset tässä potilasjoukossa olivat yhdenmukaisia aiemmissa evolokumabin kliinisen kehitysohjelman tutkimuksissa havaittujen vaikutusten kanssa eikä uusia turvallisuutta koskevia huolenaiheita havaittu.
Farmakokinetiikka
Imeytyminen ja jakautuminen
Kun terveille aikuisille annettiin evolokumabia 140 mg tai 420 mg kerta-annoksena ihon alle, seerumin huippupitoisuuksien mediaani saavutettiin 3–4 vuorokaudessa. Ihon alle annetun 140 mg:n kerta-annoksen jälkeen Cmax-arvojen keskiarvo (SD) oli 13,0 (10,4) μg/ml ja AUClast-arvojen keskiarvo (SD) 96,5 (78,7) vrk•μg/ml. Ihon alle annetun 420 mg:n kerta-annoksen jälkeen Cmax-keskiarvo (SD) oli 46,0 (17,2) µg/ml ja AUClast-keskiarvo (SD) 842 (333) vrk•µg/ml. Kolme ihon alle annettua 140 mg:n annosta olivat biologisesti samanarvoiset yhden ihon alle annetun 420 mg:n annoksen kanssa. Farmakokineettisistä malleista määritetty absoluuttinen hyötyosuus oli 72 % ihon alle annetun annoksen jälkeen.
Kun evolokumabia annettiin 420 mg kerta-annoksena laskimoon, vakaan tilan aikaisen jakautumistilavuuden keskiarvon (SD) arvioitiin olevan 3,3 (0,5) litraa, mikä viittaa siihen, että evolokumabin jakautuminen kudoksiin on vähäistä.
Biotransformaatio
Evolokumabi muodostuu yksinomaan aminohapoista ja hiilihydraateista luontaisen immunoglobuliinin tavoin, joten se ei todennäköisesti eliminoidu metaboloitumalla maksassa. Se metaboloituu ja eliminoituu todennäköisesti immunoglobuliinipuhdistuman reittejä pitkin ja pilkkoutuu pieniksi peptideiksi ja yksittäisiksi aminohapoiksi.
Eliminaatio
Evolokumabin efektiivisen puoliintumisajan arvioitiin olevan 11–17 vuorokautta.
Systeeminen evolokumabialtistus oli suuria statiiniannoksia saavilla primaarista hyperkolesterolemiaa tai sekamuotoista dyslipidemiaa sairastavilla potilailla hieman pienempi kuin pieniä tai kohtalaisia statiiniannoksia saavilla (AUClast-arvojen suhde 0,74 [90 % CI 0,29–1,9]). Puhdistuman suureneminen noin 20 % johtuu osittain siitä, että statiinit suurentavat PCSK9-pitoisuutta. Tämä ei heikentänyt evolokumabin farmakodynaamista vaikutusta lipideihin. Populaatiofarmakokineettisessä analyysissä ei havaittu huomattavia eroja seerumin evolokumabipitoisuuksissa (ei-familiaalista tai familiaalista) hyperkolesterolemiaa sairastavilla potilailla, jotka käyttivät samanaikaisesti statiineja.
Lineaarisuus/ei-lineaarisuus
Laskimoon annetun 420 mg:n kerta-annoksen jälkeen systeemisen puhdistuman keskiarvon (SD) arvioitiin olevan 12 (2) ml/h. Kliinisissä tutkimuksissa, joissa annettiin toistuvia annoksia ihon alle 12 viikon ajan, lääkeainealtistus suureni suoraan suhteessa annokseen, kun annos oli 140 mg tai suurempi. Seerumin jäännöspitoisuuksissa havaittiin noin 2–3-kertainen kumuloituminen annettaessa 140 mg:n annoksia kahden viikon välein (Cmin (SD) 7,21 (6,6)) tai 420 mg:n annoksia kerran kuukaudessa (Cmin (SD) 11,2 (10,8)), ja jäännöspitoisuudet seerumissa lähenivät vakaata tilaa, kun hoito oli kestänyt 12 viikkoa.
Seerumin lääkeainepitoisuuksissa ei havaittu ajasta riippuvia muutoksia 124 viikon jakson aikana.
Munuaisten vajaatoiminta
Munuaisten vajaatoiminta ei vaadi annoksen muuttamista. Evolokumabin kliinisten tutkimusten tulosten perusteella evolokumabin farmakokinetiikassa ei havaittu eroa lievää tai kohtalaista munuaisten vajaatoimintaa sairastavien potilaiden ja niiden potilaiden välillä, joiden munuaiset toimivat normaalisti.
Kliinisessä tutkimuksessa, johon osallistuneilla 18 potilaalla oli joko normaali munuaisten toiminta (glomerulusten laskennallinen suodatusnopeus [eGFR] ≥ 90 ml/min/1,73 m2, n = 6), vaikea munuaisten vajaatoiminta (eGFR 15–29 ml/min/1,73 m2, n = 6) tai hemodialyysilla hoidettava loppuvaiheen munuaissairaus (ESRD) (n = 6), arvioitu altistus sitoutumattomalle evolokumabille ihon alle annetun 140 mg:n kerta-annoksen jälkeen oli Cmax-arvon perusteella 30 % pienempi vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla ja 45 % pienempi hemodialyysilla hoidettavaa loppuvaiheen munuaissairautta sairastavilla potilailla. AUClast-arvon perusteella arvioitu altistus oli noin 24 % pienempi vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla ja noin 45 % pienempi hemodialyysilla hoidettavaa loppuvaiheen munuaissairautta sairastavilla potilailla. Farmakokineettisten erojen tarkat syyt eivät ole tiedossa, mutta nämä erot eivät selity potilaiden painoeroilla. Tulosten tulkinnassa on syytä huomioida eräitä tekijöitä, mukaan lukien pieni otoskoko sekä tutkittavien välinen suuri vaihtelu. Evolokumabin farmakodynamiikka ja turvallisuus olivat vaikeaa munuaisten vajaatoimintaa ja loppuvaiheen munuaissairautta sairastavilla potilailla samanlaiset kuin potilailla, joiden munuaisten toiminta oli normaali, eikä LDL-kolesteroliarvon laskussa havaittu kliinisesti merkittäviä eroja. Näin ollen annosta ei tarvitse muuttaa vaikean munuaisten vajaatoiminnan tai hemodialyysilla hoidettavan loppuvaiheen munuaissairauden vuoksi.
Maksan vajaatoiminta
Lievä maksan vajaatoiminta (Child-Pugh-luokka A) ei vaadi annoksen muuttamista. Evolokumabia annettiin 140 mg:n kerta-annoksina ihon alle 8 potilaalle, joilla oli lievä maksan vajaatoiminta, 8 potilaalle, joilla oli kohtalainen maksan vajaatoiminta, ja 8 terveelle tutkittavalle. Evolokumabialtistuksen havaittiin olevan noin 40–50 % pienempi kuin terveillä tutkittavilla. PCSK9:n lähtötason ja PCSK9:n neutraloitumisasteen ja neutraloitumiseen kuluvan ajan havaittiin kuitenkin olevan lievää tai kohtalaista maksan vajaatoimintaa sairastavilla potilailla samanlaiset kuin terveillä tutkittavilla. Tämän seurauksena myös LDL‑kolesterolipitoisuuden absoluuttiseen laskuun kuluva aika ja laskun laajuus olivat samanlaiset. Evolokumabia ei ole tutkittu vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavien potilaiden hoidossa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Paino
Paino oli merkittävä kovariaatti populaatiofarmakokineettisessä analyysissä. Se vaikutti evolokumabin jäännöspitoisuuksiin, mutta LDL‑kolesteroliarvon laskuun sillä ei ollut vaikutusta. Kun evolokumabia annettiin toistuvina annoksina 140 mg ihon alle 2 viikon välein, viikon 12 jäännöspitoisuus oli 69 kg painavien potilaiden ryhmässä 147 % suurempi ja 93 kg painavien ryhmässä 70 % pienempi kuin tyypillisellä 81 kg painavalla tutkittavalla todettu jäännöspitoisuus. Painon vaikutus oli vähäisempi, kun evolokumabia annettiin toistuvina annoksina 420 mg ihon alle kerran kuukaudessa.
Muut erityisryhmät
Populaatiofarmakokineettiset analyysit viittaavat siihen, ettei annostusta tarvitse muuttaa iän, etnisen taustan eikä sukupuolen perusteella. Potilaan paino vaikutti evolokumabin farmakokinetiikkaan, mutta sillä ei ollut havaittavaa vaikutusta LDL‑kolesteroliarvon laskuun. Siksi annosta ei tarvitse muuttaa potilaan painon perusteella.
Repathan farmakokinetiikkaa arvioitiin 103:lla heterotsygoottista familiaalista hyperkolesterolemiaa sairastavalla 10 vuotta täyttäneellä mutta alle 18-vuotiaalla lapsipotilaalla (HAUSER-RCT). Kun Repathaa oli annettu kerran kuukaudessa 420 mg ihon alle, seerumin keskimääräinen (SD) jäännöspitoisuus viikolla 12 oli 22,4 (14,7) mikrog/ml, viikolla 22 64,9 (34,4) mikrog/ml ja viikolla 24 25,8 (19,2) mikrog/ml. Repathan farmakokinetiikkaa arvioitiin 12:lla homotsygoottista familiaalista hyperkolesterolemiaa sairastavalla 10 vuotta täyttäneellä mutta alle 18-vuotiaalla lapsipotilaalla (HAUSER-OLE). Kun Repathaa oli annettu kerran kuukaudessa 420 mg ihon alle, seerumin keskimääräinen (SD) jäännöspitoisuus viikolla 12 oli 20,3 (14,6) mikrog/ml ja viikolla 80 17,6 (28,6) mikrog/ml.
Prekliiniset tiedot turvallisuudesta
Evolokumabi ei ollut karsinogeeninen hamstereille altistustasoilla, jotka olivat huomattavasti suurempia kuin evolokumabia 420 mg kerran kuukaudessa saavien potilaiden altistus. Evolokumabin mutageenisuutta ei ole arvioitu.
Hamstereilla ja jaavanmakakeilla ei havaittu urosten eikä naaraiden hedelmällisyyteen kohdistuvia vaikutuksia altistustasoilla, jotka olivat huomattavasti suurempia kuin evolokumabia 420 mg kerran kuukaudessa saavien potilaiden altistus.
Jaavanmakakeilla ei havaittu alkion- tai sikiönkehitykseen eikä postnataaliseen kehitykseen (6 kuukauden ikään asti) kohdistuvia vaikutuksia altistustasoilla, jotka olivat huomattavasti suurempia kuin evolokumabia 420 mg kerran kuukaudessa saavien potilaiden altistus.
Lukuun ottamatta T‑soluista riippuvaisen vasta-ainevasteen heikkenemistä kolmen kuukauden ajan evolokumabia saaneilla KLH:lla (keyhole limpet haemocyanin) immunisoiduilla jaavanmakakeilla, haittavaikutuksia ei havaittu hamstereilla (enintään 3 kuukautta) eikä jaavanmakakeilla (enintään 6 kuukautta), kun altistustasot olivat huomattavasti suurempia kuin evolokumabia 420 mg kerran kuukaudessa saavien potilaiden altistus. Tavoiteltu farmakologinen vaikutus eli seerumin LDL‑kolesteroli- ja kokonaiskolesterolipitoisuuden pieneneminen tuli esiin näissä tutkimuksissa, ja arvot palautuivat ennalleen hoidon lopettamisen jälkeen.
Jaavanmakakeilla, jotka saivat evolokumabia yhdessä rosuvastatiinin kanssa 3 kuukauden ajan, ei havaittu haittavaikutuksia altistustasoilla, jotka olivat huomattavasti suurempia kuin evolokumabia 420 mg kerran kuukaudessa saavien potilaiden altistus. Seerumin LDL‑kolesteroli- ja kokonaiskolesterolipitoisuuden lasku oli selvempi kuin aikaisemmin, kun evolokumabia annettiin yksinään, ja arvot palautuivat ennalleen hoidon lopettamisen jälkeen.
Farmaseuttiset tiedot
Apuaineet
Proliini
Väkevä etikkahappo
Polysorbaatti 80
Natriumhydroksidi (pH:n säätämiseen)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta.
Kun Repatha on otettu pois jääkaapista, sitä voidaan säilyttää huoneenlämmössä (enintään 25 °C) alkuperäispakkauksessa, ja se on käytettävä 1 kuukauden kuluessa.
Säilytys
Säilytä jääkaapissa (2°C ‑ 8°C). Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
REPATHA injektioneste, liuos, esitäytetty kynä
140 mg (L:ei) 6 x 1 ml (3x2x1 ml) (1339,95 €)
PF-selosteen tieto
1 ml liuosta kertakäyttöisessä esitäytetyssä kynässä, joka on valmistettu tyypin I lasista ja jossa on ruostumattomasta teräksestä valmistettu 27 G:n neula.
Pakkauskoot: kerrannaispakkaus, jossa on 6 (3 pakkausta, kussakin 2) esitäytettyä kynää.
Valmisteen kuvaus:
Liuos on kirkas tai opalisoiva, väritön tai kellertävä eikä siinä ole käytännöllisesti katsoen lainkaan hiukkasia.
Käyttö- ja käsittelyohjeet
Liuos on tarkistettava ennen käyttöä. Liuosta ei saa injisoida, jos siinä on hiukkasia tai se on sameaa tai sen väri on muuttunut. Jotta pistos olisi miellyttävämpi, lääkevalmisteen annetaan lämmetä huoneenlämpöiseksi (enintään 25 °C) ennen pistämistä. Koko sisältö injisoidaan.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
REPATHA injektioneste, liuos, esitäytetty kynä
140 mg 6 x 1 ml
- Alempi erityiskorvaus (65 %). Alirokumabi ja evolokumabi familiaalisen hyperkolesterolemian hoidossa (lapset ja nuoret): Familiaalisen hyperkolesterolemian hoito lapsille ja nuorille erityisin edellytyksin (254), Alirokumabi ja evolokumabi familiaalisen hyperkolesterolemian hoidossa (aikuiset): Familiaalisen hyperkolesterolemian hoito erityisin edellytyksin (292), Alirokumabi ja evolokumabi: Krooniseen sepelvaltimotautiin liittyvän hyperkolesterolemian ja sekamuotoisen dyslipidemian hoito erityisin edellytyksin (294).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Alirokumabi, evolokumabi ja inklisiraani familiaalisen hyperkolesterolemian hoidossa (aikuiset): Familiaalisen hyperkolesterolemian hoito erityisin edellytyksin (388), Alirokumabi, evolokumabi ja inklisiraani: Hyperkolesterolemian ja sekamuotoisen dyslipidemian hoito erityisin edellytyksin (3015), Alirokumabi ja evolokumabi familiaalisen hyperkolesterolemian hoidossa (lapset ja nuoret): Familiaalisen hyperkolesterolemian hoito lapsille ja nuorille erityisin edellytyksin (3074).
ATC-koodi
C10AX13
Valmisteyhteenvedon muuttamispäivämäärä
12.12.2025
Yhteystiedot
Keilaranta 10, PL 86
02101 Espoo
09 5490 0500