
TRANSLARNA rakeet oraalisuspensiota varten 125 mg, 250 mg, 1000 mg
Huomioitavaa
▼Tähän lääkkeeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti uutta turvallisuutta koskevaa tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Translarna 125 mg rakeet oraalisuspensiota varten
Yksi pussi sisältää 125 mg atalureenia.
Translarna 250 mg rakeet oraalisuspensiota varten
Yksi pussi sisältää 250 mg atalureenia.
Translarna 1 000 mg rakeet oraalisuspensiota varten
Yksi pussi sisältää 1 000 mg atalureenia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Rakeet oraalisuspensiota varten.
Kliiniset tiedot
Käyttöaiheet
Translarna on tarkoitettu dystrofiinigeenin nonsense-mutaatiosta johtuvan Duchennen lihasdystrofian hoitoon vähintään kaksivuotiaille liikuntakykyisille potilaille (ks. kohta Farmakodynamiikka).
Dystrofiinigeenin nonsense-mutaatio on varmistettava geenitesteillä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ehto
Hoidon saa aloittaa vain erikoislääkäri, jolla on kokemusta Duchennen tai Beckerin lihasdystrofian hoidosta.
Annostus ja antotapa
Translarna-hoidon saa aloittaa vain erikoislääkäri, jolla on kokemusta Duchennen tai Beckerin lihasdystrofian hoidosta.
Annostus
Atalureenia annetaan suun kautta kolme annosta joka päivä.
Ensimmäinen annos otetaan aamulla, toinen päivällä ja kolmas illalla. Suositellut annosvälit ovat kuusi tuntia aamu- ja päiväannoksen välillä, kuusi tuntia päivä- ja ilta-annoksen välillä ja 12 tuntia ilta- ja aamuannoksen välillä.
Suositeltu annos on 10 mg painokiloa kohden aamulla, 10 mg painokiloa kohden päivällä ja 20 mg painokiloa kohden illalla (vuorokausiannosyhteensä 40 mg painokiloa kohden).
Translarnaa on saatavana 125 mg:n, 250 mg:n ja 1 000 mg:n pusseissa. Seuraavassa taulukossa on esitetty painoluokittain, mikä vahvuus sopii mitäkin suositeltua annosta varten.
Painoluokka (kg) | Pussien määrä | |||||||||
Aamu | Päivä | Ilta | ||||||||
125 mg:n pusseja | 250 mg:n pusseja | 1 000 mg:n pusseja | 125 mg:n pusseja | 250 mg:n pusseja | 1 000 mg:n pusseja | 125 mg:n pusseja | 250 mg:n pusseja | 1 000 mg:n pusseja | ||
12 | 14 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
15 | 16 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 |
17 | 20 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
21 | 23 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 0 |
24 | 26 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 2 | 0 |
27 | 31 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 2 | 0 |
32 | 35 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 2 | 0 |
36 | 39 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 3 | 0 |
40 | 44 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 3 | 0 |
45 | 46 | 0 | 2 | 0 | 0 | 2 | 0 | 1 | 3 | 0 |
47 | 55 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 1 |
56 | 62 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 1 | 1 |
63 | 69 | 0 | 3 | 0 | 0 | 3 | 0 | 0 | 1 | 1 |
70 | 78 | 0 | 3 | 0 | 0 | 3 | 0 | 0 | 2 | 1 |
79 | 86 | 0 | 3 | 0 | 0 | 3 | 0 | 0 | 3 | 1 |
87 | 93 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 3 | 1 |
94 | 105 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 2 |
106 | 111 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 2 |
112 | 118 | 0 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 2 |
119 | 125 | 0 | 1 | 1 | 0 | 1 | 1 | 0 | 2 | 2 |
Viivästynyt tai unohtunut annos
Jos atalureenin aamu- tai päiväannoksen ottamisessa on alle kolmen tunnin viive tai ilta-annoksen ottamisessa on alle kuuden tunnin viive, annos otetaan ilman, että seuraavien annosten aikatauluun tehdään muutoksia. Jos aamu- tai päiväannos viivästyy yli kolme tuntia tai jos ilta-annos viivästyy yli kuusi tuntia, annosta ei oteta, ja sen jälkeen jatketaan tavanomaisen annostusaikataulun mukaisesti. Potilas ei saa ottaa kaksinkertaista tai ylimääräistä annosta, jos annos unohtuu. On tärkeää, että potilaalle annetaan oikea annos. Teho saattaa laskea, jos annosta nostetaan suositeltua annosta suuremmaksi.
Erityisryhmät
Iäkkäät potilaat
Atalureenin turvallisuutta ja tehoa vähintään 65-vuotiaiden potilaiden hoidossa ei ole vielä varmistettu. (ks. kohta Farmakokinetiikka.)
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min) tai loppuvaiheen munuaissairautta sairastavien potilaiden hoitamista tällä valmisteella ei suositella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa potilailla, joilla on lievä, keskitason tai vakava maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Pediatrisia potilaita, joiden paino on ≥ 12 kg, hoidetaan painoluokkasuositusten mukaisesti (katso taulukko yllä). Suositellut annokset ovat samat kaikille ikäluokille, eli 10 mg/kg aamuisin, 10 mg/kg päivällä ja 20 mg/kg illalla (jolloin kokonaisannos päivän aikana on 40 mg/kg painon mukaan).
Translarnan turvallisuutta ja tehoa alle 12 kg:n ja vähintään kuuden kuukauden ja enintään kahden vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Translarna sekoitetaan nesteeseen tai puolikiinteään ruokaan ja annetaan suun kautta. Pussin saa avata vasta juuri ennen annoksen valmistamista. Yhden pussin koko sisältö sekoitetaan vähintään 30 millilitraan nestettä (vettä, maitoa tai hedelmämehua), tai kolmeen ruokalusikalliseen puolikiinteätä ruokaa (jugurttia tai omenasosetta). Annos on sekoitettava hyvin ennen ottamista. Nesteen tai puolikiinteän ruoan määrää voidaan lisätä potilaan niin halutessa. Potilaan on otettava koko annos.
Ohjeet lääkevalmisteen saattamisesta käyttövalmiiksi ennen lääkkeen antoa, ks. kohta Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aminoglykosidien samanaikainen laskimonsisäinen käyttö (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset muiden lääkevalmisteiden kanssa sekä muut yhteisvaikutukset.
Varoitukset ja käyttöön liittyvät varotoimet
Potilaat, joilla ei ole nonsense-mutaatiota
Potilaan dystrofiinigeenissä on oltava perussairauteen liittyvä ja geenitestillä varmistettu nonsense-mutaatio. Atalureenia ei saa antaa potilaille, joilla ei ole nonsense-mutaatiota.
Munuaisten vajaatoiminta
Lisääntynyttä altistumista atalureenille ja sen metaboliitille on ilmoitettu vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min) sairastavilla potilailla. Metaboliitin toksisuus on tuntematon. Suurempaan atalureenialtistumiseen liittyi mahdollista tehon heikkenemistä. Siksi vaikeaa munuaisten vajaatoimintaa tai loppuvaiheen munuaissairautta sairastaville potilaille saa antaa atalureenihoitoa vain, jos odotettavissa oleva kliininen hyöty on mahdollista riskiä suurempi, ja potilaita on seurattava tiiviisti metaboliitin mahdollisen toksisuuden ja tehon heikkenemisen varalta. Pienemmän atalureeniannoksen käyttämistä tulee harkita.
Hoitoa ei saa aloittaa aiemmin hoitamattomille potilaille, joiden eGFR-arvo on < 30 ml/min (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Lipidiprofiilin muutokset
Koska kliinisissä tutkimuksissa ilmoitettiin joillakin potilailla lipidiprofiilin muutoksia (triglyseridien ja kolesterolin pitoisuuksien nousua), atalureenia saavien nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa (nmDMD) sairastavien potilaiden kokonaiskolesterolia, LDL- ja HDL-kolesterolia sekä triglyseridejä on suositeltavaa seurata vuosittain tai useammin, jos potilaan kliininen tila niin vaatii.
Hypertensio systeemisten kortikosteroidien samanaikaisen käytön yhteydessä
Koska kliinisissä tutkimuksissa ilmoitettiin joillakin potilailla hypertensiota systeemisten kortikosteroidien samanaikaisen käytön yhteydessä, atalureenia saavien nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa sairastavien potilaiden, jotka saavat samanaikaisesti kortikosteroideja, systolista ja diastolista lepopainetta on suositeltavaa seurata kuuden kuukauden välein tai useammin, jos potilaan kliininen tila niin vaatii.
Munuaisten toiminnan seuranta
Koska nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa koskevissa kontrolloiduissa tutkimuksissa havaittiin seerumin keskimääräisen kreatiniinin, veren ureatypen (BUN) ja kystatiini C:n pientä nousua, atalureenia saavien nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa sairastavien potilaiden seerumin kreatiniinia, veren ureatyppeä ja kystatiini C:tä ja on suositeltavaa seurata 6–12 kuukauden välein tai useammin, jos potilaan kliininen tila niin vaatii.
Mahdolliset yhteisvaikutukset muiden lääkevalmisteiden kanssa
Varovaisuutta on noudatettava, jos atalureenin kanssa annetaan lääkevalmisteita, jotka ovat UGT1A9:n induktoreja, tai OAT1:n tai OAT3:n substraatteja (ks. kohta Yhteisvaikutukset muiden lääkevalmisteiden kanssa sekä muut yhteisvaikutukset).
Aminoglykosidit
Aminoglykosidien on osoitettu vähentävän atalureenin vaikutusta luentaan in vitro. Lisäksi atalureenin todettiin lisäävän laskimonsisäisesti annettavien aminoglykosidien munuaistoksisuutta. Näiden lääkevalmisteiden samanaikaista käyttöä atalureenin kanssa on vältettävä (ks. kohta Vasta-aiheet). Koska ei tiedetä, miten atalureeni lisää laskimonsisäisesti annettavien aminoglykosidien munuaistoksisuutta, muiden munuaistoksisten lääkevalmisteiden käyttöä atalureenin kanssa ei suositella. Jos tätä ei voida välttää (esim. MRSA:n hoito vankomysiinillä), munuaisten toimintaa on suositeltavaa seurata huolellisesti (ks. kohta Yhteisvaikutukset muiden lääkevalmisteiden kanssa sekä muut yhteisvaikutukset).
Yhteisvaikutukset
Aminoglykosidit
Atalureenia ei saa käyttää samanaikaisesti laskimonsisäisesti annettavien aminoglykosidien kanssa, koska kliinisessä tutkimuksessa on havaittu nonsense-mutaatiosta johtuvaa kystistä fibroosia sairastavien potilaiden munuaisten toiminnan heikkenemistä (ks. kohta Vasta-aiheet).
Seerumin kreatiniini nousi useilla nonsense-mutaatiosta johtuvaa kystistä fibroosia sairastavilla potilailla, jotka saivat atalureenia ja laskimonsisäisesti annettavia aminoglykosideja yhdessä muiden kystisen fibroosin pahenemisvaiheiden hoitoon tarkoitettujen antibioottien kanssa. Seerumin kreatiniini palautui normaaliksi kaikissa tapauksissa, kun aminoglykosidin laskimonsisäinen käyttö lopetettiin ja Translarnan käyttöä joko jatkettiin tai se keskeytettiin. Nämä löydökset viittaavat siihen, että Translarnan ja laskimonsisäisesti annettavien aminoglykosidien samanaikainen käyttö voi vahvistaa aminoglykosidien munuaistoksista vaikutusta. Jos laskimonsisäisesti annettavien aminoglykosidien käyttö on välttämätöntä, Translarna-hoito on lopetettava. Hoitoa voidaan jatkaa kaksi vuorokautta sen jälkeen, kun aminoglykosidin anto on lopetettu. Atalureenin ja muiden munuaistoksisten lääkevalmisteiden samanaikaisen käytön vaikutuksia ei tunneta.
Kuivuminen saattaa vaikuttaa asiaan joissakin tapauksissa. Potilaiden nesteytyksen on oltava riittävä atalureenin käytön aikana. Ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Muiden lääkevalmisteiden vaikutukset atalureenin farmakokinetiikkaan
In vitro -tutkimusten mukaan atalureeni on UGT1A9:n substraatti. Samanaikainen rifampisiinin anto, joka on vahva metabolisten entsyymien induktori (mukaan lukien UGT1A9), vähensi atalureenialtistusta 29 %:lla. Näiden löydösten merkitystä ihmisille ei tiedetä. Varovaisuutta on noudatettava, kun atalureenia annetaan yhdessä sellaisten lääkevalmisteiden kanssa, jotka ovat UGT1A9:n induktoreja (esim. rifampisiini).
Atalureenin vaikutus muiden lääkevalmisteiden farmakokinetiikkaan
In vitro -tutkimusten mukaan atalureenilla on potentiaalia estää UGT1A9:n, orgaanisen anionin kuljettaja 1:n (OAT1), orgaanisen anionin kuljettaja 3:n (OAT3) ja orgaanisen anioninkuljetuspolypeptidin 1B3:n (OATP1B3) toimintaa. Atalureenin ja mykofenolaattimofetiilin samanaikainen anto terveille tutkittaville ei vaikuttanut atalureenin aktiivisen metaboliitin, mykofenolihapon (UGT1A9:n substraatti) altistukseen. Annoksen muuttaminen ei ole tarpeen annettaessa atalureenia samanaikaisesti lääkevalmisteiden kanssa, jotka ovat UGT1A9:n substraatteja.
Kliinisessä tutkimuksessa, jossa tutkittiin atalureenin potentiaalia estää OATP1B3-kuljetusjärjestelmää käytettäessä 80 mg:n kerta-annosta telmisartaania, joka on in-vitro valikoiva OATP1B3:n substraatti, atalureeni lisäsi telmisartaanialtistuksen määrää 28 %. Tätä vaikutusta ei pidetä kliinisesti merkittävänä. 40 mg:n telmisartaaniannoksella vaikutus voi kuitenkin olla korostuneempi. Tästä johtuen on noudatettava varovaisuutta, kun atalureenia annetaan yhdessä sellaisten lääkevalmisteiden kanssa, jotka ovat OAT1:n tai OATP1B3:n substraatteja, sillä näiden lääkevalmisteiden (esim. oseltamiviiri, asikloviiri, kaptopriili, furosemidi, bumetanidi, valsartaani, pravastatiini, rosuvastatiini, atorvastatiini ja pitavastatiini) pitoisuus saattaa nousta.
On myös noudatettava varovaisuutta, kun atalureenia annetaan yhdessä OAT3:n substraattien kanssa (esim. siprofloksasiini), erityisesti sellaisten OAT3-substraattien kanssa, joiden terapeuttinen ikkuna on kapea. Kliinisessä tutkimuksessa siprofloksasiinialtistuksen määrä oli 32 % korkeampi atalureenin läsnä ollessa.
Erillisessä kliinisessä tutkimuksessa adefoviirialtistuksen määrä oli 60 % korkeampi atalureenin läsnä ollessa. Varovaisuutta on noudatettava, kun atalureenia annetaan yhdessä adefoviirin kanssa.
In vitro -tutkimusten mukaan atalureenin ei odoteta estävän p-gp-välitteistä kuljetusta tai sytokromi P450 -välitteistä metaboliaa. Atalureenin ei myöskään odoteta indusoivan sytokromi P450 -isoentsyymejä in vivo.
Kortikosteroidien (deflatsakortin, prednisonin tai prednisolonin) ja atalureenin samanaikainen käyttö ei vaikuttanut atalureenin pitoisuuteen plasmassa. Kortikosteroidien pitoisuuksissa plasmassa ei havaittu kliinisesti merkittäviä muutoksia, kun niitä käytettiin samanaikaisesti atalureenin kanssa. Nämä tiedot viittaavat siihen, että kortikosteroidien ja atalureenin välillä ei ole ilmeistä lääkkeiden välistä yhteisvaikutusta, eikä annosten muuttaminen ole tarpeen.
Lääkevalmisteet, jotka vaikuttavat p-glykoproteiinin kuljettajaan
Atalureeni ei ole in vitro p-glykoproteiinin kuljettajan substraatti. P-glykoproteiinin kuljettajaa estävät lääkevalmisteet eivät todennäköisesti vaikuta atalureenin farmakokinetiikkaan.
Raskaus ja imetys
Raskaus
Atalureenin käytöstä raskaana oleville naisille ei ole riittävästi tietoa. Eläinkokeissa on havaittu lisääntymistoksisuutta ainoastaan käytettäessä annoksia, jotka aiheuttivat toksisuutta emälle (ks. kohta Prekliiniset tiedot turvallisuudesta).
Varmuuden vuoksi atalureenin käyttöä on suositeltavaa välttää raskauden aikana
Imetys
Ei tiedetä, erittyvätkö atalureeni tai sen metaboliitit ihmisen rintamaitoon. Olemassa olevat farmakokineettiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet atalureenin/metaboliittien erittyvän rintamaitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea.
Rintaruokinta on lopetettava atalureenihoidon ajaksi.
Hedelmällisyys
Ei-kliiniset tiedot eivät viittaa rotilla tehdyn urosten ja naaraiden hedelmällisyystutkimuksen perusteella vaaraan ihmisille (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Atalureenin vaikutusta ajokykyyn, pyöräilyyn tai koneiden käyttökykyyn ei ole testattu. Jos huimausta ilmenee, on noudatettava varovaisuutta autoa ajettaessa, pyöräiltäessä tai koneita käytettäessä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Atalureenin turvallisuusprofiili perustuu yhdistettyihin tietoihin kahdesta satunnaistetusta, kaksoissokkoutetusta, 48 viikkoa kestäneestä lumekontrolloidusta tutkimuksesta, jotka tehtiin 232 miespuoliselle nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa sairastavalle potilaalle, joita hoidettiin suositellulla 40 mg/kg/vrk annoksella (10, 10, 20 mg painokiloa kohden; n=172) tai 80 mg/kg/vrk annoksella (20, 20, 40 mg painokiloa kohden; n=60), lumelääkkeellä hoidettuihin potilaisiin (n=172) verrattuna.
Yleisimmät haittavaikutukset kahdessa lumekontrolloidussa tutkimuksessa olivat oksentelu, ripuli, pahoinvointi, päänsärky, ylävatsakipu ja ilmavaivat, joita kaikkia ilmeni vähintään 5 prosentilla kaikista atalureenilla hoidetuista potilaista. Molemmissa tutkimuksissa 1/232 (0,43 %) atalureenilla hoidettua potilasta keskeytti hoidon haittavaikutuksena olleen ummetuksen takia ja 1/172 (0,58 %) lumelääkkeellä hoidetuista potilaista keskeytti hoidon taudin etenemisen (liikuntakyvyn menettämisen) haittavaikutuksen takia.
Atalureenin farmakokinetiikkaa ja turvallisuutta tutkittiin avoimessa tutkimuksessa, johon osallistui 2–5‑vuotiaita potilaita (n = 14). Iältään 2–5-vuotiaat potilaat raportoivat enemmän pahoinvointia (7,1 %), kuumetta (42,9 %), korvatulehduksia (28,6 %) ja ihottumaa (21,4 %) kuin vähintään 5-vuotiaat potilaat. Näitä tiloja raportoidaan kuitenkin nuoremmilla lapsilla yleensä useammin. Turvallisuustiedot 28 viikon hoidosta osoitti atalureenilla samanlaisen turvallisuusprofiilin 2–5-vuotiailla potilailla kuin vähintään 5-vuotiailla potilailla.
Haittavaikutukset olivat yleensä vaikeusasteeltaan lieviä tai kohtalaisia, ja hoitoon liittyvistä vakavista haittatapahtumista ei ilmoitettu atalureenilla hoidetuilla potilailla näissä kahdessa tutkimuksessa.
Taulukko haittavaikutuksista
Ilmoitetut haittavaikutukset nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa sairastavilla potilailla, jotka saivat suositellun annoksen 40 mg/kg/vrk atalureenia kahdessa lumekontrolloidussa tutkimuksessa on esitetty taulukossa 1. Haittavaikutukset, joista on ilmoitettu useammalla kuin yhdellä potilaalla 40 mg/kg/vrk –ryhmässä ja suuremmalla esiintymistiheydellä kuin lumeryhmässä, on esitetty MedDRAn elinjärjestelmäluokan, suositellun termin ja yleisyyden mukaan. Yleisyysryhmät määritellään seuraavan menetelmän mukaisesti: hyvin yleinen (≥ 1/10) ja yleinen (≥ 1/100, < 1/10).
Taulukko 1. Haittavaikutukset, joista on ilmoitettu useammalla kuin yhdellä nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa sairastavalla potilaalla esiintymistiheydellä, joka on suurempi kuin lumelääkkeellä kahdessa lumekontrolloidussa tutkimuksessa (yhdistetty analyysi)
Elinluokka | Hyvin yleinen | Yleinen | Tuntematon |
Aineenvaihdunta ja ravitsemus |
| Heikentynyt ruokahalu, hypertriglyseridemia | Lipidiprofiilin muutos (triglyseridien ja kolesterolin pitoisuuden nousu) |
Hermosto |
| Päänsärky |
|
Verisuonisto |
| Hypertensio |
|
Hengityselimet, rintakehä ja välikarsina |
| Yskä, nenäverenvuoto |
|
Ruoansulatuselimistö | Oksentelu | Pahoinvointi, ylävatsakipu, ilmavaivat, vatsavaivat, ummetus |
|
Iho ja ihonalainen kudos |
| Punoittava ihottuma |
|
Luusto, lihakset ja sidekudos |
| Raajakipu, muskuloskeletaalinen rintakipu |
|
Munuaiset ja virtsatiet |
| Hematuria, kastelu | Munuaisten toimintakokeiden muutokset (kreatiniinin, veren ureatypen ja kystatiini C:n nousu) |
Yleisoireet ja antopaikassa todettavat haitat |
| Kuume, painonlasku |
|
Duchennen lihasdystrofiaa sairastavilla potilailla tehdyssä 48 viikkoa kestävässä avoimessa jatkotutkimuksessa saatu turvallisuusprofiili oli samanlainen sekä liikuntakykyisillä että liikuntakyvyttömillä potilailla. Pitkän aikavälin turvallisuustietoja ei ole saatavilla.
Tiettyjen haittavaikutusten kuvaus (laboratorioarvojen poikkeavuudet)
Seerumin lipidit
Seerumin lipidiarvoissa eli kolesterolissa ja triglyserideissä havaittiin nousua. Joissakin raportoiduissa tapauksissa tämä nousu epänormaaleihin korkeisiin arvoihin havaittiin jo neljän viikon jälkeen.
Munuaisten toimintakokeet
Nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa koskevissa satunnaistetuissa, lumekontrolloiduissa tutkimuksissa havaittiin seerumin kreatiniinin, veren ureatypen ja kystatiini C:n vähäistä nousua. Arvot vakiintuivat yleensä tutkimuksen alkuvaiheessa eivätkä nousseet enempää, kun hoitoa jatkettiin.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle.
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Terveillä vapaaehtoisilla koehenkilöillä, jotka saivat atalureenia suun kautta kerta-annoksena 200 mg painokiloa kohden, ilmeni ohimenevää ja lievää päänsärkyä, pahoinvointia, oksentelua ja ripulia. Näillä potilailla ei havaittu vakavia haittavaikutuksia. Jos on aihetta epäillä yliannostusta, potilaalle on annettava oireenmukaista hoitoa. Terveydenhuollon ammattilaista on konsultoitava ja potilaan kliinistä tilaa on tarkkailtava huolellisesti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä:Muut tuki- ja liikuntaelinten sairauksien lääkkeet, ATC-koodi: M09AX03
Vaikutusmekanismi
DNA:n nonsense-mutaatio aiheuttaa lähetti-RNA:n ennenaikaisen lopetuskodonin. Tämä lähetti-RNA:n ennenaikainen lopetuskodoni aiheuttaa sairauden lopettamalla translaation ennen kuin täysimittainen proteiini on tuotettu. Atalureenin ansiosta ribosomit voivat lukea lähetti-RNA:ta, joka sisältää tällaisen lopetuskodonin, jolloin voidaan tuottaa täysimittainen proteiini.
Farmakodynaamiset vaikutukset
Soluille, joissa on nonsense-mutaatio, ja atalureeniliuoksessa viljellyille kalanpoikasille tehdyt ei-kliiniset in vitro -kokeet ovat osoittaneet, että atalureeni mahdollisti luennan ribosomeissa ilmentäen käänteistä U:n muotoista pitoisuus-vastesuhdetta. In vivo -annos-vastesuhteen epäillään myös olevan käänteisen U:n muotoinen, mutta in vivo -tietoja ei ollut tarpeeksi, jotta tämä hypoteesi olisi voitu vahvistaa nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa koskevassa hiirimallissa ja ihmisillä.
Ei-kliiniset in vitro -tutkimukset viittaavat siihen, että jatkuva altistus atalureenille voi olla tärkeää vaikutuksen maksimoinnin kannalta ja että vaikuttavan aineen vaikutukset ribosomien ennenaikaisten lopetuskodonien luentaan loppuvat pian atalureenin annon lopettamisen jälkeen.
Kliininen teho ja turvallisuus
Translarnan tehoa ja turvallisuutta arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa, jotka koskivat nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa. Ensisijaisena tehon päätetapahtumana molemmissa tutkimuksissa oli muutos 6 minuutin kävelyetäisyydessä (6MWD) viikolla 48. Muita päätetapahtumia molemmissa tutkimuksissa olivat 6 minuutin kävelyetäisyydessä pysyvään 10 prosentin heikkenemiseen kuluva aika, muutos 10 metrin juoksemiseen/kävelemiseen kuluvassa ajassa viikolla 48, muutos 4 portaan nousemiseen kuluvassa ajassa viikolla 48 ja muutos 4 portaan laskeutumiseen kuluvassa ajassa viikolla 48. Potilailla oli oltava dokumentoitu vahvistus siitä, että dystrofiinigeenissä oli määritetty nonsense-mutaatio geenisekvensoinnin avulla.
Tutkimuksessa 1 arvioitiin 174 5–20-vuotiasta miespuolista potilasta. Kaikkien potilaiden oli pystyttävä kävelemään ≥ 75 metriä ilman apuvälineitä seulonnassa käytetyssä kuuden minuutin kävelytestissä. Potilaiden enemmistö (90 %) oli valkoihoisia kaikissa hoitoryhmissä. Potilaat satunnaistettiin suhteessa 1:1:1 saamaan atalureenia tai lumelääkettä kolmesti päivässä (aamulla, päivällä ja illalla). 57 potilasta sai atalureenia 40 mg/kg/vrk (10 mg, 10 mg ja 20 mg painokiloa kohden), 60 potilasta sai atalureenia 80 mg/kg/vrk (20 mg, 20 mg ja 40 mg painokiloa kohden) ja 57 potilasta sai lumelääkettä.
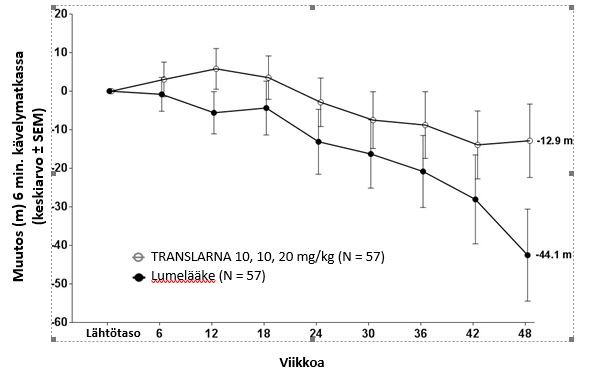
Tutkimuksessa 1 ensisijaisen päätetapahtuman post hoc -analyysi osoitti, että potilaiden, jotka saivat atalureenia 40 mg/kg/vrk, kuuden minuutin kävelytestissä kulkema matka lyheni lähtötasosta viikkoon 48 mennessä keskimäärin 12,9 metriä ja lumelääkettä saaneiden keskimäärin 44,1 metriä (kuva 1). Kuuden minuutin kävelytestissä lähtötasosta viikkoon 48 mennessä havaittu keskimääräinen muutos oli atalureenia 40 mg/kg/vrk saaneessa ryhmässä 31,3 metriä parempi kuin lumeryhmässä (p = 0,056). Tilastollisessa mallianalyysissä keskimääräinen ero oli 31,7 metriä (korjattu p=0.0367). Atalureeniannoksen 80 mg/kg/vrk ja lumeryhmän välillä ei ollut eroa.
Nämä tulokset viittaavat siihen, että atalureeniannos 40 mg/kg/vrk hidastaa kävelykyvyn menetystä potilailla, jotka sairastavat nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa.
Kuva 1. Keskimääräinen muutos kuuden minuutin kävelyetäisyydessä (tutkimus 1)
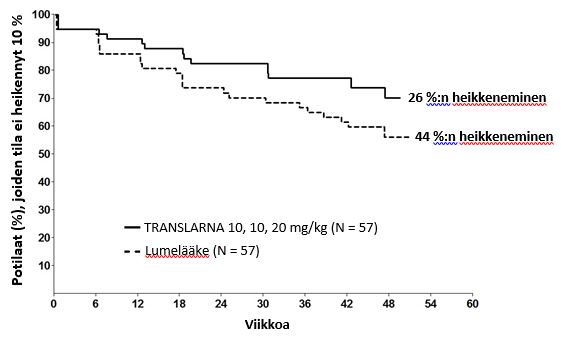
Kuuden minuutin kävelytestissä kuljetun matkan 10 prosentin pysyvään heikkenemiseen kuluneen ajan post hoc -analyysi osoitti, että tila oli viikolla 48 edennyt 26 prosentilla potilaista, jotka saivat atalureenia 40 mg/kg/vrk. Vastaava osuus lumeryhmässä oli 44 prosenttia (p = 0,0652) (kuva 2). Atalureeniannoksen 80 mg/kg/vrk ja lumeryhmän välillä ei ollut eroa. Nämä tulokset viittaavat siihen, että kuuden minuutin kävelytestin tulos heikkeni 48 viikon aikana pienemmällä osuudella potilaista, jotka saivat atalureenia 40 mg/kg/vrk.
Kuva 2. Kaplan-Meierin käyrä, joka kuvaa kuuden minuutin kävelytestin tuloksen 10 prosentin pysyvään heikkenemiseen kuluvaa aikaa (tutkimus 1)
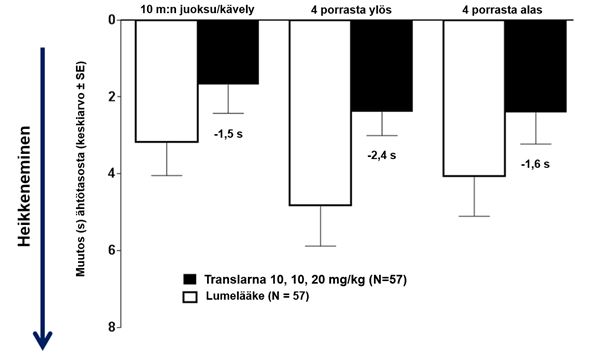
Toimintakykytesteissä (TFT) mitattiin aikaa, joka kului 10 metrin juoksemiseen tai kävelemiseen, neljän portaan nousemiseen ja neljän portaan laskeutumiseen. Atalureenihoitoa saaneilla 10 metrin juoksemiseen tai kävelemiseen, neljän portaan nousemiseen ja neljän portaan laskeutumiseen kulunut aika piteni vähemmän, mikä osoittaa, että nonsense-mutaatiosta johtuva Duchennen lihasdystrofia eteni hitaammin kuin lumeryhmässä.
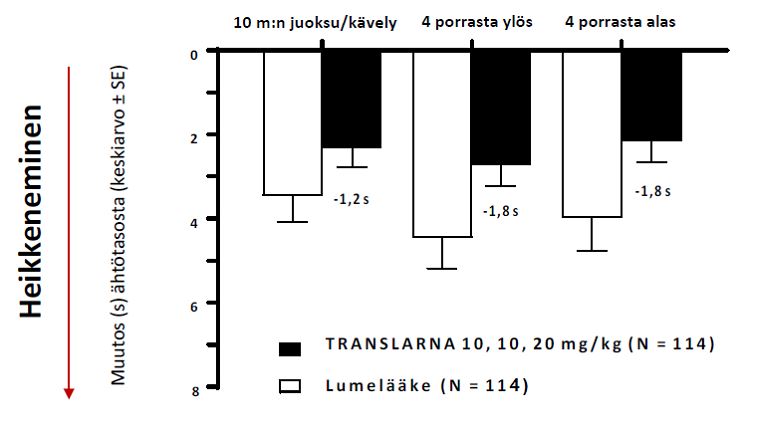
Keskimääräinen muutos toimintakykytesteissä lähtötasosta viikkoon 48 oli parempi potilailla, jotka saivat atalureenia 40 mg/kg/vrk: 10 metrin juoksuun tai kävelyyn kului 1,5 sekuntia vähemmän, neljän portaan kiipeämiseen 2,4 sekuntia vähemmän ja neljän portaan laskeutumiseen 1,6 sekuntia vähemmän (kuva 3).
Kuva 3: Keskimääräinen muutos toimintakykytesteissä (tutkimus 1)
Kuuden minuutin kävelytestin tulokset potilailla, joilla lähtötason tulos oli alle 350 metriä
Potilailla, joiden lähtötason tulos kuuden minuutin kävelytestissä oli alle 350 metriä, lähtötasosta viikkoon 48 mennessä havaittu keskimääräinen muutos oli atalureenia 40 mg/kg/vrk saaneessa ryhmässä 68 metriä parempi kuin lumeryhmässä (p = 0,0053).
Näillä potilailla keskimääräinen muutos toimintakykytesteissä lähtötasosta viikkoon 48 oli parempi potilailla, jotka saivat atalureenia 40 mg/kg/vrk: 10 metrin juoksuun tai kävelyyn kului 3,5 sekuntia vähemmän, neljän portaan kiipeämiseen 6,4 sekuntia vähemmän ja neljän portaan laskeutumiseen 5,0 sekuntia vähemmän.
Tutkimuksessa 2 arvioitiin 230 7–14-vuotiasta miespotilasta. Kaikkien potilaiden oli pystyttävä kävelemään ≥ 150 metriä ja vähemmän kuin 80 % ennakoidusta matkasta ilman apuvälineitä seulonnassa käytetyssä kuuden minuutin kävelytestissä. Potilaiden enemmistö (76 %) oli valkoihoisia molemmissa hoitoryhmissä. Potilaat satunnaistettiin suhteessa 1:1 saamaan atalureenia tai lumelääkettä kolmesti päivässä (aamulla, päivällä ja illalla). Potilaat saivat atalureenia 40 mg/kg/vrk (n=115) tai lumelääkettä (n=115) kolmesti päivässä (aamulla, päivällä ja illalla).
Atalureenilla hoidetut potilaat saivat kliinistä hyötyä numeerisesti suotuisina muutoksina mitattuna ensisijaisissa ja toissijaisissa tehon päätetapahtumissa. Koska ensisijainen päätetapahtuma (muutos 6 minuutin kävelytestissä lähtötasolta viikolle 48) ei saavuttanut tilastollista merkitsevyyttä (p≤0.05), kaikkia muita p-arvoja on pidettävä nimellisinä.
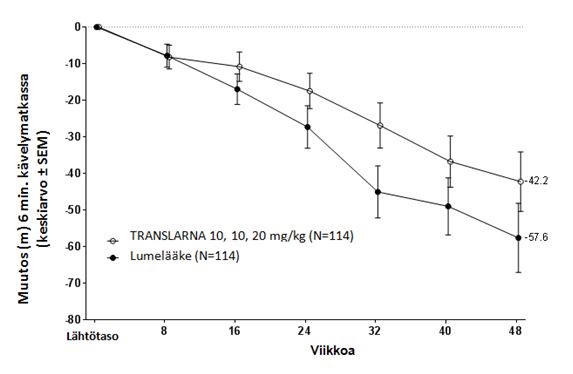
ITT-populaatiossa ero atalureeni- ja lumeryhmän välillä keskimääräisessä havaitussa 6 minuutin kävelytestin tuloksessa lähtötasolta viikolle 48 oli 15,4 metriä parempi atalureeni 40 mg/kg/vrk –ryhmässä kuin lumeryhmässä. Tilastollisessa mallissa estimoitu keskimääräinen ero oli 13,0 metriä (p=0,213), ks. kuva 4. Ero atalureenin ja lumelääkkeen välillä pysyi voimassa viikolta 16 alkaen tutkimuksen loppuun saakka.
Kuva 4. Keskimääräinen muutos 6 minuutin kävelyetäisyydessä (tutkimus 2)
48 viikon aikana atalureenilla hoidetuilla potilailla oli vähemmän lihasten toiminnan heikentymistä, mikä näkyi pienempänä lisääntymisenä 10 metrin juoksemiseen/kävelemiseen, 4 portaan nousemiseen ja 4 portaan laskeutumiseen kuluvassa ajassa atalureenilla hoidetussa ryhmässä lumelääkkeeseen verrattuna. Atalureenille suotuisat erot lumelääkkeeseen verrattuna ajastettujen toimintakykytestien keskimääräisissä muutoksissa ITT-populaatiossa viikolla 48 saavuttivat kliinisesti merkittävän eron kynnyksen (muutokset ~1–1,5 sekuntia).
Keskimääräinen muutos ajastetuissa toimintakykytesteissä lähtötasolta viikolle 48 oli parempi atalureeni 40 mg/kg/vrk –ryhmässä kuin lumelääkettä käytettäessä havaitussa 10 metrin juoksemiseen/kävelemiseen kuluvassa ajassa (1,2 sekuntia parempi, p=0,117), 4 portaan nousemiseen kuluvassa ajassa (1,8 sekuntia parempi, p=0,058) ja 4 portaan laskeutumiseen kuluvassa ajassa (1,8 sekuntia parempi, p=0,012), ks. kuva 5.
Kuva 5. Keskimääräinen muutos ajastetuissa toimintakykytesteissä (tutkimus 2)
6 minuutin kävelytestissä 10 prosentin heikkenemiseen kuluva aika määriteltiin viimeisenä aikana, jolloin kuuden minuutin kävelytestin tulos ei ollut 10 prosenttia heikompi kuin lähtötasolla. ITT-populaatiossa atalureennin riskisuhde lumelääkkeeseen verrattuna oli 0,75 (p=0,160), joka vastaa 6 minuutin kävelytestin tuloksen 10 prosentin heikentymisen riskin vähenemistä 25 prosentilla.
Pediatriset potilaat
Translarnan turvallisuus, farmakokinetiikka ja eksploratiivinen teho arvioitiin avoimessa tutkimuksessa 2–5-vuotiailla lapsilla, jotka sairastavat nonsense-mutaatioista johtuvaa Duchennen lihasdystrofiaa. Translarnan tehokkuus 2–5-vuotiailla lapsilla on johdettu vähintään 5-vuotiaiden potilaiden vastaavasta.
Kliinisessä tutkimuksessa, jossa tutkittiin monoterapiana toteutetun atalureenihoidon tehokkuutta ja turvallisuutta potilailla, joilla on nonsense-mutaatioita sisältävä kystinen fibroosi, ei havaittu tilastollisesti merkittävää vaikutusta ensisijaisessa kliinisessä tulosmittarissa ja tärkeimmissä toissijaisissa kliinisissä tulosmittareissa (ppFEV1 ja keuhko-oireiden pahenemistahti) aikuisilla ja yli 6-vuotiailla lapsilla.
Avoin eksploratiivinen tutkimus (tutkimus 045) tehtiin 20:llä 2–7-vuotiaalla tutkittavalla, joilla oli nonsense-mutaatioista johtuva Duchennen lihasdystrofia (nmDMD), kvantitatiivisten dystrofiinitasojen tutkimiseksi lihaskudoksessa ennen 40 viikon atalureenihoitoa ja sen jälkeen. Dystrofiini mitattiin käyttämällä elektrokemiluminesenssi (ECL)- ja immunohistokemia (IHC) -määrityksiä. Kultakin tutkittavalta otettiin kolme neulabiopsiaa kaksoiskantalihaksesta ja etummaisesta säärilihaksesta lähtötilanteessa ja hoidon lopussa. Tutkimukseen 045 sisältyi myös toiminnallisten tulosten arviointi (ts. tarkistettu North Star Ambulatory Assessment [rNSAA] ja ajastettuja toimintakykytestejä (Timed Function Tests [TFTs])).
Lähtötason dystrofiinipitoisuuden mediaani ECL-määrityksellä mitattuna oli 0,42 % normaalista (vaihteluväli 0,00–41,85 %). Tutkimuksen lopussa dystrofiinipitoisuuden mediaani oli 0,33 % normaalista (vaihteluväli 0,04–48,55 %).
IHC-määrityksissä dystrofiinipositiivisten kuitujen mediaaniprosentti lähtötilanteessa oli 73 % (vaihteluväli 0,42–99,6 %). Tutkimuksen lopussa dystrofiinipositiivisten kuitujen mediaaniprosentti oli 66 % (vaihteluväli 0,51–99,77 %).
Tutkimuksen lopussa keskimääräinen (mediaani) paheneminen lähtötasosta rNSAA:n osalta oli 0,1 (1,0) pistettä kokonaispistemäärässä ja keskimääräinen (mediaani) muutos lähtötasosta koskien aikaa pystyyn nousemiseen, juoksemiseen tai kävelemiseen 10 metriä, 4 porrasaskelman kiipeämiseen ja 4 porrasaskelman laskeutumiseen oli vastaavasti -1,56 (-0,6), -0,41 (-0,35), -1,09 (-0,5), ja -2,43 (-0,7) sekuntia.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset atalureenin käytöstä kahdessa pediatrisessa potilasryhmässä (nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa sairastavat alle 28 vuorokauden ikäiset vastasyntyneet ja vauvat, joiden ikä on vähintään 28 vuorokautta ja alle 6 kuukautta) pediatrisen tutkimussuunnitelman päätöksen mukaan myönnetyssä käyttöaiheessa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset atalureenin käytöstä yhdessä pediatrisessa potilasryhmässä (nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa sairastavat lapset, joiden ikä on vähintään kuusi kuukautta ja alle kaksi vuotta) pediatrisen tutkimussuunnitelman päätöksen mukaan myönnetyssä käyttöaiheessa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Tämä lääkevalmiste on saanut ns. ehdollisen myyntiluvan. Se tarkoittaa, että lääkevalmisteesta odotetaan uutta tietoa.
Euroopan lääkevirasto arvioi vähintään joka vuosi tätä lääkevalmistetta koskevat uudet tiedot ja tarvittaessa päivittää valmisteyhteenvedon.
Farmakokinetiikka
Kun atalureenia annettiin painon perusteella (mg/kg), nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa sairastavien lasten ja nuorten vakaan tilan altistus (AUC) oli samankaltainen hyvin laajalla painoalueella. Vaikka atalureeni ei käytännössä liukene veteen, atalureeni imeytyy helposti, kun se annetaan suspensiona suun kautta.
Atalureenin yleiset ominaisuudet lääkkeen antamisen jälkeen
Imeytyminen
Atalureenin huippupitoisuus plasmassa saavutetaan noin 1,5 tunnin kuluttua annostelusta, kun potilas saa lääkevalmistetta 30 minuutin kuluessa ateriasta. Radioaktiivisesti merkittyä atalureenia koskevassa kerta-annostutkimuksessa virtsassa mitatun radioaktiivisuuden perusteella atalureenin biologisen hyötyosuuden arvioidaan olevan ≥55 prosenttia. Atalureenin pitoisuudet plasmassa vakaassa tilassa kasvavat suhteessa annoksen suurenemiseen. Vakaan tilan pitoisuudet plasmassa ovat suhteessa annokseen, kun atalureenin annos on 10–50 mg painokiloa kohden. Kertymistä ei ilmene toistuvan annostuksen jälkeen.
Jakautuminen
Atalureeni sitoutuu in vitro 99,6-prosenttisesti ihmisen plasmaproteiineihin eikä pitoisuus plasmassa vaikuta sitoutumiseen. Atalureeni ei jakaudu punasoluihin.
Biotransformaatio
Atalureeni metabolisoituu konjugoitumalla uridiinidifosfaattiglukuronyylitransferaasin (UGT) kautta, lähinnä maksan, suoliston ja munuaisten UGT1A9-entsyymien kautta.
Ainoa metaboliitti, joka havaittiin plasmassa in vivo, kun radioaktiivisesti merkittyä atalureenia oli annettu suun kautta, oli atalureeni-O-1β-asyyliglukuronidi. Ihmisen altistus tälle metaboliitille oli noin 8 prosenttia atalureenin AUC-arvosta plasmassa.
Eliminaatio
Atalureenin puoliintumisaika plasmassa on 2–6 tuntia. Annos tai toistuva annostus eivät vaikuta puoliintumisaikaan. Atalureenin eliminaatio riippuu todennäköisesti atalureenin glukuronidaatiosta maksassa ja munuaisissa, minkä jälkeen glukuronidimetaboliitti erittyy munuaisten ja maksan kautta.
Kun radioaktiivisesti merkittyä atalureenia annettiin kerta-annos suun kautta, noin puolet annetusta radioaktiivisesta annoksesta erittyi ulosteeseen ja loput virtsaan. Virtsassa muuttumattoman atalureenin määrä on < 1 prosentti annetusta annoksesta ja asyyliglukuronidimetaboliitin määrä 49 prosenttia annetusta annoksesta.
Lineaarisuus/ei-lineaarisuus
Vakaan tilan pitoisuudet plasmassa ovat suhteessa annokseen, kun atalureenin annos on 10–50 mg painokiloa kohden. Kertymistä ei ilmene toistuvan annostuksen jälkeen. Terveistä koehenkilöistä saatujen tietojen perusteella atalureenin suhteellinen biologinen hyötyosuus on vakaassa tilassa noin 40 prosenttia pienempi kuin ensimmäisen annoksen jälkeen. Suhteellisen biologisen hyötyosuuden pienenemisen arvioidaan alkavan noin 60 tuntia ensimmäisen annoksen jälkeen. Vakaa tila saavutetaan noin kahden viikon kuluttua, kun atalureenia annetaan kolmesti päivässä.
Ominaisuudet tietyissä potilasryhmissä
Ikä
2–57 vuoden ikäisiä potilaita koskevien tietojen perustella iällä ei ole ilmeistä vaikutusta atalureenin altistukselle plasmassa. Annosta ei tarvitse säätää iän mukaan.
Atalureenin farmakokinetiikkaa on arvioitu neljän viikon aikana tutkimuksessa PTC124-GD-030. Atalureenin plasmakertymät 2–5-vuotiailla potilailla vastasivat niitä, jotka nähtiin yli 5-vuotiailla potilailla, jotka saivat 10/10/20 mg/kg annostusta.
Sukupuoli
Naisia ei tutkittu kliinisissä tutkimuksissa, jotka koskivat nonsense-mutaatiosta johtuvaa Duchennen lihasdystrofiaa. Muissa potilasryhmissä sukupuolella ei kuitenkaan ollut ilmeistä vaikutusta atalureenin altistukselle plasmassa.
Rotu
On epätodennäköistä, että UGT1A9-polymorfismi vaikuttaa merkittävästi atalureenin farmakokinetiikkaan valkoihoisessa väestössä. Koska kliinisiin tutkimuksiin osallistui vain pieni määrä muiden rotujen edustajia, UGT1A9:n vaikutuksesta muissa etnisissä ryhmissä ei voida tehdä johtopäätöksiä.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta.
Farmakokineettisessä tutkimuksessa, johon osallistui eriasteista munuaisten vajaatoimintaa sairastavia tutkittavia, kerta-annoksen jälkeen plasman atalureenialtistuminen muuttui -13 % lievää munuaisten vajaatoimintaa sairastavilla, 27 % keskivaikea munuaisten vajaatoimintaa sairastavilla ja 61 % vaikeaa munuaisten vajaatoimintaa sairastavilla tutkittavilla ja 46 % loppuvaiheen munuaissairautta sairastavilla tutkittavilla verrattuna tutkittaviin, joiden munuaiset toimivat normaalisti. Lisäksi atalureenin metaboliitin pitoisuuden on ilmoitettu suurentuneen 3–8-kertaiseksi vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min) sairastavilla potilailla. Toistuvassa annostelussa atalureenin ja sen metaboliitin pitoisuuksien nousun odotetaan olevan vakaassa tilassa suurempi vaikeaa munuaisten vajaatoimintaa ja loppuvaiheen munuaissairautta sairastavilla potilailla kuin potilailla, joiden munuaiset toimivat normaalisti. Vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min) tai loppuvaiheen munuaissairautta sairastaville potilaille saa antaa atalureenihoitoa vain, jos odotettavissa oleva kliininen hyöty on mahdollista riskiä suurempi (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoiminta
Mitään annoksen muutosta ei tarvita potilailla, joilla on minkään tasoinen maksan vajaatoiminta, perustuen joko lievästä, keskitason tai vakavasta maksan vajaatoiminnasta kärsivien ryhmille tehtyyn farmakokineettiseen arvioon verrattuna terveiden potilaiden vertailuryhmään. Vertailussa ei havaittu mitään eroa atalureenin kokonaisaltistumisessa vertailuryhmän ja lievästä, keskitason tai vakavasta maksan vajaatoiminnasta kärsivien ryhmien välillä. Noin 40 %:n alentuminen atalureenin keskimääräisessä kokonaisaltistumisessa havaittiin keskitason maksan vajaatoiminnasta kärsivässä ryhmässä verrattuna vertailuryhmään, luultavasti johtuen pienestä kohorttikoosta ja vaihtelusta.
Liikuntakyvyttömät potilaat
Vakaan tilan suhteellisessa biologisessa hyötyosuudessa tai näennäisessä puhdistumassa ei ollut ilmeisiä liikuntakyvyn menetyksestä johtuvia eroja. Annosta ei tarvitse muuttaa, jos potilas on menettämässä liikuntakykynsä.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta ja geenitoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Käytettävissä olivat lisääntymistoksisuuden vakiotutkimukset. Vaikutusta urosten ja naaraiden hedelmällisyyteen ei havaittu, mutta varhaisnuoruudessa annetun hoidon vaikutusta aikuisiän hedelmällisyyteen ei tutkittu. Rotilla ja kaneilla havaittiin alkio-sikiötoksisuutta (esim. aikaisten sikiön resorptioiden määrän nousua, alkion menetystä kohtuun kiinnittymisen jälkeen, elinkykyisten sikiöiden määrän laskua) ja merkkejä viivästyneestä kehityksestä (luustomuutosten määrän kasvua) maternaalisen toksisuuden annostasoilla. Altistus tasolla, joka ei aiheuta havaittavaa haittavaikutusta (NOAEL), vastasi kaneilla ihmisten systeemistä altistusta (40 mg/kg/vrk) ja oli rotilla nelinkertainen. Radioaktiivisesti merkityn atalureenin osoitettiin siirtyvään istukkaan rotilla. Emälle annetun suhteellisen pienen annoksen 30 mg/kg kertatestissä radioaktiivisuuden pitoisuus sikiössä oli ≤ 27 prosenttia maternaalisesta pitoisuudesta. Rottien prenataalisessa/postnataalisessa kehitystoksisuustutkimuksessa, jossa altistus oli viisi kertaa suurempi kuin ihmisten altistus, havaittiin merkittävää toksisuutta emälle sekä vaikutuksia jälkeläisen painoon ja ambulatoriseen kehitykseen. Maternaalinen systeeminen altistus tasolla, joka ei aiheuta havaittavaa vaikutusta (NOEL) neonataalitoksisuuden osalta, oli kolme kertaa suurempi kuin ihmisen altistus. Kun emälle annettiin suhteellisen pieni 30 mg/kg:n kerta-annos radioaktiivisesti merkittyä atalureenia, suurin mitattu radioaktiivisuuspitoisuus rotan maidossa oli 37 prosenttia pitoisuudesta emän plasmassa. Radioaktiivisuus poikasen plasmassa vahvisti imeytymisen maidosta poikasiin.
Munuaistoksisuutta (distaalisen nefronin nefroosia) ilmeni suun kautta tapahtuvaa toistuvaa altistusta koskevissa hiirillä tehdyissä tutkimuksissa, kun systeeminen altistus oli 0,3 kertaa suurempi kuin vakaan tilan AUC potilailla, jotka saavat Translarnaa 10 mg painokiloa kohden aamulla, 10 mg painokiloa kohden päivällä ja 20 mg painokiloa kohden illalla tai enemmän.
Transgeenisessa hiirimallissa, joka kesti 26 viikkoa ja jossa tutkittiin karsinogeenisuutta, ei saatu näyttöä karsinogeenisuudesta. Rotilla tehdyssä kaksivuotisessa karsinogeenisuustutkimuksessa todettiin yksi hibernoomatapaus. Kun altistus oli paljon suurempi kuin potilailla, todettiin lisäksi (harvinaisten) virtsarakon kasvaimien määrän kasvua. Virtsarakon kasvaimilla ei todennäköisesti ole merkitystä ihmisille.
Toinen rotilla tehdyistä kahdesta 26 viikkoa kestäneestä toistuvaa altistusta koskevasta tutkimuksesta, joka aloitettiin 4–5 viikon ikäisillä rotilla, osoitti rotilla harvinaisten pahanlaatuisten hibernoomien annossidonnaista ilmaantuvuuden nousua. Lisäksi yksi pahanlaatuinen hibernooma todettiin kaksivuotisessa rottien karsinogeenisuustutkimuksessa, kun käytössä oli suurin annos. Tämän kasvaintyypin taustailmaantuvuus on hyvin pieni niin rotilla kuin ihmisillä, eikä tiedetä, mikä niitä aiheuttaa rottatutkimuksissa (ja miten se liittyy atalureenihoitoon). Tämän merkitystä ihmisille ei tiedetä.
Yksivuotisessa tutkimuksessa 10–12 viikon ikäisillä koirilla todettiin lisämunuaisen löydöksiä (pesäkkeinen tulehdus ja lisämunuaiskuoren glukokortikoideja tuottavien alueiden degeneraatio) sekä kortisolintuotannon lievää heikentymistä eksogeenisen kortikotropiinistimulaation jälkeen. Näitä löydöksiä todettiin koirilla, kun systeeminen altistus oli 0,8 kertaa suurempi kuin vakaan tilan AUC potilailla, jotka saavat Translarnaa 40 mg/kg/vrk tai enemmän. Rotilla tehdyssä jakautumistutkimuksessa havaittiin atalureenin suuria pitoisuuksia lisämunuaisissa.
Edellä mainittujen vaikutusten lisäksi toistuvaa altistusta koskevissa tutkimuksissa havaittiin useita muita vähemmän vakavia vaikutuksia ja erityisesti painonnousun ja syömisen vähenemistä sekä maksan painonnousua ilman histologista yhteyttä. Tämän kliininen merkitys on epäselvä. Rotilla ja koirilla tehdyissä tutkimuksissa ilmeni myös muutoksia plasman lipideissä (kolesterolissa ja triglyserideissä), mikä viittaa rasva-aineenvaihdunnan muutoksiin.
Kolme kuukautta kestäneessä vastasyntyneillä (1 viikon ikäisillä) koirilla tehdyssä tutkimuksessa, jota seurasi 3 kuukautta kestänyt toipumisjakso, ei todettu haitallisia löydöksiä myöskään lisämunuaisessa. Koirien systeemiset altistukset vakaassa tilassa vastasivat korkeimmillaan potilailla todettuja vakaan tilan AUC-arvoja. Vastasyntyneillä (1 viikon ikäisillä) koirilla tehdyissä alustavissa tutkimuksissa osa eläimistä ei sietänyt systeemistä altistusta, joka oli 5–10-kertainen verrattuna potilailla todettuihin vakaan tilan AUC-arvoihin.
Farmaseuttiset tiedot
Apuaineet
Polydekstroosi (E1200), makrogoli, poloksameeri, mannitoli (E421), krospovidoni, hydroksietyyliselluloosa, keinotekoinen vanilja-aromi (maltodekstriini, keinotekoiset aromit ja propyleeniglykoli), kolloidinen vedetön piidioksidi (E551), magnesiumstearaatti.
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta
Jokainen annos on suositeltavaa antaa heti valmistamisen jälkeen. Valmistettu annos säilyy enintään 24 tuntia jääkaapissa (2–8 °C) ja kolme tuntia huoneenlämmössä (15–30 °C). Sen jälkeen käyttämättä jäänyt annos on hävitettävä.
Säilytys
Tämä lääke ei vaadi erityisiä säilytysolosuhteita.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TRANSLARNA rakeet oraalisuspensiota varten
125 mg (L:ei) 30 x 125 mg (3581,51 €)
250 mg (L:ei) 30 x 250 mg (6960,71 €)
1000 mg (L:ei) 30 x 1000 mg (27235,91 €)
PF-selosteen tieto
Kuumasaumattu laminoitu alumiinifoliopussi: polyeteenitereftalaatti (lapsisuojaus), polyeteeni (värjäys ja polyesterin ja folion saumaus), alumiinifolio (kosteussuoja), liima-aine (polyuretaani), etyleenikopolymeeri ja metakryylihappo (pakkauksen tiiviys).
Pakkauskoko: 30 pussia.
Valmisteen kuvaus:
Valkoiset tai luonnonvalkoiset rakeet.
Käyttö- ja käsittelyohjeet
Pussin saa avata vasta juuri ennen annoksen valmistamista. Yhden pussin koko sisältö sekoitetaan vähintään 30 millilitraan nestettä (vettä, maitoa tai hedelmämehua) tai kolmeen ruokalusikalliseen puolikiinteätä ruokaa (jugurttia tai omenasosetta). Annos on sekoitettava hyvin ennen ottamista. Nesteen tai puolikiinteän ruoan määrää voidaan lisätä potilaan niin halutessa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TRANSLARNA rakeet oraalisuspensiota varten
125 mg 30 x 125 mg
250 mg 30 x 250 mg
1000 mg 30 x 1000 mg
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Atalureeni: Duchennen lihasdystrofian hoito vähintään 2-vuotiaille potilaille erityisin edellytyksin (3056).
ATC-koodi
M09AX03
Valmisteyhteenvedon muuttamispäivämäärä
28.10.2024
Yhteystiedot
5th Floor, 3 Grand Canal Plaza, Grand Canal Street Upper
4 Dublin
Ireland
+358 (0) 974790388