
ALUNBRIG tabletti, kalvopäällysteinen 90 mg + 180 mg, 30 mg, 90 mg, 180 mg
Vaikuttavat aineet ja niiden määrät
Alunbrig 30 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 30 mg brigatinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 56 mg laktoosimonohydraattia.
Alunbrig 90 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 90 mg brigatinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 168 mg laktoosimonohydraattia.
Alunbrig 180 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 180 mg brigatinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 336 mg laktoosimonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Alunbrig on tarkoitettu monoterapiana edennyttä anaplastista lymfoomakinaasi (ALK) ‑positiivista ei‑pienisoluista keuhkosyöpää (NSCLC) sairastavien aikuispotilaiden hoitoon, joita ei ole aiemmin hoidettu ALK‑estäjällä.
Alunbrig on tarkoitettu monoterapiana edennyttä ALK ‑positiivista NSCLC sairastavien aikuispotilaiden hoitoon, joita on aiemmin hoidettu kritsotinibillä.
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Alunbrig‑hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
ALK‑positiivista ei‑pienisoluista keuhkosyöpää sairastavien potilaiden soveltuvuus hoitoon on varmistettava validoidulla ALK‑määrityksellä (ks. kohta Farmakodynamiikka). ALK‑positiivisen ei‑pienisoluisen keuhkosyövän määritys on suoritettava laboratoriossa, jonka osaaminen käytettävässä erityistekniikassa on osoitettu.
Annostus
Suositeltu Alunbrig‑aloitusannos on 90 mg kerran vuorokaudessa ensimmäisten 7 vrk ajan ja sen jälkeen 180 mg kerran vuorokaudessa.
Jos Alunbrig‑hoito keskeytetään vähintään 14 vuorokaudeksi jostakin muusta syystä kuin haittavaikutusten vuoksi, hoito on aloitettava uudestaan annostuksella 90 mg kerran vuorokaudessa 7 vrk ajaksi, minkä jälkeen annos nostetaan aiemmin siedetylle tasolle.
Jos annos jää väliin tai potilas oksentaa annoksen ottamisen jälkeen, lisäannosta ei pidä ottaa, vaan seuraava annos on otettava hoitoaikataulun mukaisena ajankohtana.
Hoitoa jatketaan niin pitkään kuin siitä havaitaan olevan kliinistä hyötyä.
Annosmuutokset
Annostelun keskeytys ja/tai annoksen pienentäminen saattaa olla tarpeen yksilöllisen turvallisuuden ja siedettävyyden perusteella.
Alunbrig‑annoksen pienentäminen esitetään yhteenvetona taulukossa 1.
Taulukko 1: Suositeltava Alunbrig‑annoksen pienentäminen
Annos | Annoksen pienennystasot | ||
Ensimmäinen | Toinen | Kolmas | |
90 mg x 1 (ensimmäiset 7 vrk) | pienennetään tasolle 60 mg kerran vuorokaudessa | lopetetaan pysyvästi | ei oleellinen |
180 mg x 1 | pienennetään tasolle 120 mg kerran vuorokaudessa | pienennetään tasolle 90 mg kerran vuorokaudessa | pienennetään tasolle 60 mg kerran vuorokaudessa |
Alunbrig‑hoito on lopettava pysyvästi, jos potilas ei siedä 60 mg vuorokausiannosta.
Suositukset Alunbrig‑annosmuutoksista haittavaikutusten hallinnassa esitetään yhteenvetona taulukossa 2.
Taulukko 2: Suositeltavat Alunbrig‑annosmuutokset haittavaikutusten yhteydessä
Haittavaikutus | Vaikeusaste* | Annosmuutokset |
Interstitiaalinen keuhkosairaus (ILD) / pneumoniitti | Aste 1 |
|
Aste 2 |
| |
Aste 3 tai 4 |
| |
Hypertensio | Asteen 3 hypertensio (systolinen verenpaine ≥ 160 mmHg tai diastolinen ≥ 100 mmHg, lääketieteellinen interventio aiheellinen, enemmän kuin yksi verenpainelääke tai aiempaa intensiivisempi hoito aiheellista) |
|
Asteen 4 hypertensio (henkeä uhkaavat seuraukset, kiireellinen hoito tarpeen) |
| |
Bradykardia (syke alle 60/min) | Oireinen bradykardia |
|
Bradykardia, henkeä uhkaavat seuraukset, kiireellinen toimenpide aiheellinen |
| |
Kreatiinikinaasiarvon kohoaminen | Asteen 3 tai 4 kreatiinikinaasiarvon kohoaminen (> 5,0 × ULN) ja asteen ≥ 2 lihaskipu tai ‑heikkous |
|
Lipaasi‑ tai amylaasiarvon kohoaminen | Asteen 3 lipaasi‑ tai amylaasiarvon kohoaminen (> 2,0 × ULN) |
|
Asteen 4 lipaasi‑ tai amylaasiarvon kohoaminen (> 5,0 × ULN) |
| |
Maksatoksisuus | Asteen ≥ 3 kohoaminen (> 5,0 × ULN), joko alaniiniaminotransferaasi (ALAT) tai aspartaattiaminotransferaasi (ASAT) ja bilirubiini ≤ 2 × ULN |
|
Asteen ≥ 2 kohoaminen ALAT‑ tai ASAT‑arvo (> 3 x ULN) ja samanaikainen kokonaisbilirubiiniarvon kohoaminen > 2 x ULN (ei kolestaasia eikä hemolyysiä) |
| |
Hyperglykemia | Asteen 3 (yli 250 mg/dl tai 13,9 mmol/l) tai suurempi |
|
Näköhäiriöt | Aste 2 tai 3 |
|
Aste 4 |
| |
Muut haittavaikutukset | Aste 3 |
|
Aste 4 |
| |
ULN = viitealueen yläraja | ||
*Asteet perustuvat National Cancer Institute ‑organisaation Common Terminology Criteria for Adverse Events ‑kriteereihin, versio 4.0 (NCI CTCAE v4).
Erityiset potilasryhmät
Iäkkäät
Rajalliset tiedot Alunbrig‑valmisteen turvallisuudesta ja tehosta 65 vuotta täyttäneillä potilailla viittaavat siihen, ettei annoksen muuttaminen ole tarpeen iäkkäillä potilailla (ks. kohta Haittavaikutukset). Yli 85‑vuotiaista potilaista ei ole tietoa saatavilla.
Maksan vajaatoiminta
Alunbrig‑annosta ei tarvitse muuttaa, jos potilaalla on lievä maksan vajaatoiminta (Child–Pugh‑luokka A) tai keskivaikea maksan vajaatoiminta (Child–Pugh‑luokka B). Potilaille, joilla on vaikea maksan vajaatoiminta (Child–Pugh‑luokka C), suositellaan pienempää aloitusannosta, 60 mg kerran vuorokaudessa ensimmäisten 7 vrk ajan ja sen jälkeen 120 mg kerran vuorokaudessa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Alunbrig‑annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta (glomerulusten laskennallinen suodatusnopeus (eGFR) ≥ 30 ml/min). Potilaille, joilla on vaikea munuaisten vajaatoiminta (eGFR < 30 ml/min), suositellaan pienempää aloitusannosta, 60 mg kerran vuorokaudessa ensimmäisten 7 vrk ajan ja sen jälkeen 90 mg kerran vuorokaudessa (ks. kohta Farmakokinetiikka). Vaikeaa munuaisten vajaatoimintaa sairastavia potilaita on seurattava tarkoin uusien tai pahenevien, mahdollisesti interstitiaaliseen keuhkosairauteen / pneumoniittiin viittaavien hengitystieoireiden (esim. hengenahdistus, yskä) varalta etenkin ensimmäisenä viikkona (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Alunbrig‑valmisteen turvallisuutta ja tehoa alle 18‑vuotiaiden potilaiden hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Alunbrig otetaan suun kautta. Tabletit nielaistaan kokonaisena veden kera. Alunbrig voidaan ottaa ruoan kanssa tai ilman ruokaa.
Greippi ja greippimehu voivat suurentaa brigatinibin pitoisuuksia plasmassa, joten niitä on vältettävä (ks. kohta Yhteisvaikutukset).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Keuhkohaittavaikutukset
Alunbrig‑hoitoa saaneilla potilailla voi esiintyä vakavia, henkeä uhkaavia ja kuolemaan johtavia keuhkohaittavaikutuksia, mukaan lukien haittoja, jotka viittaavat interstitiaaliseen keuhkosairauteen / pneumoniittiin (ks. kohta Haittavaikutukset).
Useimmat keuhkohaittavaikutukset havaittiin ensimmäisten 7 hoitovuorokauden aikana. Asteen 1–2 keuhkohaittavaikutukset korjaantuivat keskeyttämällä lääkehoito tai muuttamalla annosta. Korkeampi ikä ja lyhyempi aikaväli (alle 7 vrk) viimeisen kritsotinibiannoksen ja ensimmäisen Alunbrig‑annoksen välillä yhdistettiin toisistaan riippumatta näiden keuhkohaittavaikutusten suurentuneeseen esiintyvyyteen. Nämä tekijät on otettava huomioon Alunbrig‑hoitoa aloitettaessa. Pivotaalitutkimuksista suljettiin pois potilaat, joilla oli ollut interstitiaalinen keuhkosairaus tai lääkehoidosta johtuva pneumoniitti.
Joillakin potilailla esiintyi pneumoniittia myöhemmin Alunbrig‑hoidon aikana.
Potilaita on seurattava uusien tai pahenevien hengitystieoireiden (esim. hengenahdistus, yskä) varalta etenkin ensimmäisen hoitoviikon aikana. Jos potilaalla on pahenevia hengitystieoireita, pneumoniitin mahdollisuus on tutkittava viipymättä. Jos pneumoniittia epäillään, Alunbrig‑hoito on keskeytettävä ja potilas on tutkittava muiden mahdollisten oireiden aiheuttajien varalta (esim. keuhkoembolia, kasvaimen eteneminen ja infektioperäinen keuhkokuume). Annosta on muutettava vastaavasti (ks. kohta Annostus ja antotapa).
Hypertensio
Alunbrig‑hoitoa saaneilla potilailla on esiintynyt hypertensiota (ks. kohta Haittavaikutukset).
Verenpainetta on seurattava säännöllisesti Alunbrig‑hoidon aikana. Hypertensiota on hoidettava verenpaineen hoitosuositusten mukaisesti. Syketiheyttä on seurattava tavallista tiheämmin, jos bradykardiaa tunnetusti aiheuttavan lääkevalmisteen samanaikaista käyttöä ei voida välttää. Jos hypertensio on vaikea (≥ aste 3), Alunbrig‑hoito on keskeytettävä, kunnes hypertensio on korjaantunut asteeseen 1 tai lähtötasolle. Annosta on muutettava vastaavasti (ks. kohta Annostus ja antotapa).
Bradykardia
Alunbrig‑hoitoa saaneilla potilailla on esiintynyt bradykardiaa (ks. kohta Haittavaikutukset). Varovaisuutta on noudatettava käytettäessä Alunbrig‑valmistetta samanaikaisesti tunnetusti bradykardiaa aiheuttavien aineiden kanssa. Syketiheyttä ja verenpainetta on seurattava säännöllisesti.
Jos potilaalla on oireista bradykardiaa, Alunbrig‑hoito on keskeytettävä ja samanaikaisesti käytettävät, tunnetusti bradykardiaa aiheuttavat lääkevalmisteet on arvioitava. Tilanteen korjaannuttua annosta on muutettava vastaavasti (ks. kohta Annostus ja antotapa). Jos bradykardia on henkeä uhkaavaa eikä samanaikaista siihen vaikuttavaa lääkitystä ole käytössä tai bradykardia uusiutuu, Alunbrig‑hoito on lopetettava (ks. kohta Annostus ja antotapa).
Näköhäiriöt
Alunbrig‑hoitoa saaneilla potilailla on esiintynyt näköhäiriöitä (ks. kohta Haittavaikutukset). Potilaita on ohjeistettava ilmoittamaan kaikista näköoireista. Uusien tai pahenevien vaikeiden näköoireiden kohdalla on harkittava silmälääkärin suorittamaa arviointia ja annoksen pienentämistä (ks. kohta Annostus ja antotapa).
Kreatiinikinaasiarvon kohoaminen
Alunbrig‑hoitoa saaneilla potilailla on esiintynyt kreatiinikinaasiarvon kohoamista (ks. kohta Haittavaikutukset). Potilaita on ohjeistettava ilmoittamaan selittämättömästä lihaskivusta, ‑arkuudesta tai ‑heikkoudesta. Kreatiinikinaasiarvoja on seurattava säännöllisesti Alunbrig‑hoidon aikana. Kreatiinikinaasiarvon kohoamisen vaikeusasteesta riippuen, ja jos kohoamiseen liittyy lihaskipua tai ‑heikkoutta, Alunbrig‑hoito on keskeytettävä ja annosta muutettava vastaavasti (ks. kohta Annostus ja antotapa).
Haimaentsyymiarvojen kohoaminen
Alunbrig‑hoitoa saaneilla potilailla on esiintynyt kohonneita amylaasi‑ ja lipaasiarvoja (ks. kohta Haittavaikutukset). Lipaasi‑ ja amylaasiarvoja on seurattava säännöllisesti Alunbrig‑hoidon aikana. Laboratorioarvojen poikkeavuuksien vaikeusasteesta riippuen Alunbrig‑hoito on keskeytettävä ja annosta muutettava vastaavasti (ks. kohta Annostus ja antotapa).
Maksatoksisuus
Alunbrig‑hoitoa saaneilla potilailla on esiintynyt maksaentsyymiarvojen (ASAT, ALAT) ja bilirubiinin kohoamista (ks. kohta Haittavaikutukset.) Maksan toiminta, mukaan lukien ASAT, ALAT ja kokonaisbilirubiini, on määritettävä ennen Alunbrig‑hoidon aloittamista ja sen jälkeen 2 viikon välein ensimmäisten 3 hoitokuukauden aikana. Sen jälkeen arvoja on seurattava säännöllisesti. Laboratorioarvojen poikkeavuuksien vaikeusasteesta riippuen hoito on keskeytettävä ja annosta muutettava vastaavasti (ks. kohta Annostus ja antotapa).
Hyperglykemia
Alunbrig‑hoitoa saaneilla potilailla on esiintynyt seerumin glukoosipitoisuuden nousua. Seerumin paastoglukoosi on määritettävä ennen Alunbrig‑hoidon aloittamista, ja arvoja on seurattava säännöllisesti tämän jälkeen. Diabeteksen hoito on aloitettava tai optimoitava tarpeen mukaana. Jos riittävää glukoositasapainoa ei saavuteta optimaalisella lääkehoidolla, Alunbrig‑hoito on keskeytettävä, kunnes veren glukoositasapaino on riittävä. Arvojen korjaannuttua annoksen pienentämistä taulukon 1 mukaisesti voidaan harkita tai Alunbrig‑hoito voidaan lopettaa pysyvästi.
Lääkeaineinteraktiot
Vahvojen CYP3A:n estäjien samanaikaista käyttöä Alunbrig‑valmisteen kanssa on vältettävä. Jos vahvojen CYP3A:n estäjien samanaikaista käyttöä ei voida välttää, Alunbrig‑annosta on pienennettävä tasolta 180 mg tasolle 90 mg tai tasolta 90 mg tasolle 60 mg. Vahvan CYP3A:n estäjähoidon lopettamisen jälkeen Alunbrig‑hoito on aloitettava uudestaan annoksella, jota potilas sieti ennen vahvan CYP3A:n estäjähoidon aloittamista.
Vahvojen ja kohtalaisten CYP3A:n induktorien samanaikaista käyttöä Alunbrig‑valmisteen kanssa on vältettävä (ks. kohta Yhteisvaikutukset). Jos kohtalaisten CYP3A:n induktorien samanaikaista käyttöä ei voida välttää, Alunbrig-annosta voidaan suurentaa 30 mg kerrallaan sen jälkeen, kun hoitoa on annettu nykyisellä Alunbrig-annoksella 7 vuorokauden ajan ja se on ollut siedettyä. Annosta saa suurentaa enintään sen verran, että se on kaksi kertaa sen Alunbrig-annoksen suuruinen, joka oli siedetty ennen kohtalaisen CYP3A:n induktorin käytön aloittamista. Kohtalaisen CYP3A:n induktorin käytön lopettamisen jälkeen Alunbrig-hoitoa jatketaan käyttämällä sitä annosta, joka oli siedetty ennen kohtalaisen CYP3A:n induktorin käytön aloittamista.
Valoherkkyys ja valoihottuma
Alunbrig-valmisteella hoidetuilla potilailla on ilmennyt herkistymistä auringonvalolle (ks. kohta Haittavaikutukset). Potilaita tulee neuvoa välttämään pitkäkestoista altistumista auringolle Alunbrig-valmisteen käytön aikana ja vähintään 5 päivän ajan hoidon lopettamisen jälkeen. Potilaita tulee neuvoa käyttämään ulkona ollessaan hattua ja suojaavaa vaatetusta sekä käyttämään laajakirjoista UVA/UVB‑aurinkosuojavoidetta (ultavioletti A- ja B-säteet) ja huulirasvaa (suojakerroin ≥ 30) mahdolliselta auringonpolttamalta suojaamiseksi. Vaikeiden valoherkkyysreaktioiden yhteydessä (aste ≥ 3) Alunbrig-hoito tulee keskeyttää siihen asti, että iho on toipunut lähtötilannetta vastaavaksi. Annosta tulee muuttaa tilanteen mukaan (ks. kohta Annostus ja antotapa).
Hedelmällisyys
Naisia, jotka voivat tulla raskaaksi, on neuvottava käyttämään tehokasta, ei‑hormonaalista ehkäisyä Alunbrig‑hoidon aikana ja vähintään 4 kuukauden ajan viimeisen annoksen jälkeen. Miehiä, joiden kumppani voi tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä hoidon aikana ja vähintään 3 kuukauden ajan viimeisen Alunbrig‑annoksen jälkeen (ks. kohta Raskaus ja imetys).
Laktoosi
Alunbrig sisältää laktoosimonohydraattia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi‑intoleranssi, täydellinen laktaasinpuutos tai glukoosi‑galaktoosi‑imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) tablettia kohden eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Aineet, jotka saattavat suurentaa plasman brigatinibipitoisuuksia
CYP3A:n estäjät
In vitro ‑tutkimuksissa todettiin, että brigatinibi on CYP3A4/5:n substraatti. Kun terveille henkilöille annettiin samanaikaisesti toistuvia 200 mg itrakonatsoliannoksia kahdesti vuorokaudessa (itrakonatsoli on vahva CYP3A:n estäjä) ja 90 mg kerta‑annos brigatinibia, brigatinibin Cmax suureni 21 %, AUC0‑INF suureni 101 % (2‑kertaiseksi) ja AUC0‑120 suureni 82 % (< 2‑kertaiseksi), verrattuna pelkkään 90 mg brigatinibikerta‑annokseen. Vahvojen CYP3A:n estäjien, mm. tietyt viruslääkkeet (esim. indinaviiri, nelfinaviiri, ritonaviiri, sakinaviiri), makrolidiantibiootit (esim. klaritromysiini, telitromysiini, troleandomysiini), sienilääkkeet (esim. ketokonatsoli, vorikonatsoli) ja nefatsodoni, samanaikaista käyttöä Alunbrig‑valmisteen kanssa on vältettävä. Jos vahvojen CYP3A:n estäjien samanaikaista käyttöä ei voida välttää, Alunbrig‑annosta on pienennettävä noin 50 % (tasolta 180 mg tasolle 90 mg tai tasolta 90 mg tasolle 60 mg). Vahvan CYP3A:n estäjähoidon lopettamisen jälkeen Alunbrig‑hoito on aloitettava uudestaan annoksella, jota potilas sieti ennen vahvan CYP3A:n estäjähoidon aloittamista.
Keskivahvat CYP3A:n estäjät (esim. diltiatseemi ja verapamiili) voivat suurentaa brigatinibin AUC‑arvoa noin 40 % fysiologisella farmakokinetiikkamallilla tehtyjen simulaatioiden perusteella. Keskivahvan CYP3A:n estäjän käyttö ei edellytä Alunbrig‑annoksen muutoksia. Potilasta on seurattava tarkoin, jos Alunbrig‑hoitoa käytetään samanaikaisesti keskivahvojen CYP3A:n estäjien kanssa.
Myös greippi ja greippimehu voivat suurentaa brigatinibin pitoisuuksia plasmassa, joten niitä on vältettävä (ks. kohta Annostus ja antotapa).
CYP2C8:n estäjät
In vitro ‑tutkimuksissa todettiin, että brigatinibi on CYP2C8:n substraatti. Kun terveille henkilöille annettiin samanaikaisesti toistuvia 600 mg gemfibrotsiiliannoksia (vahva CYP2C8:n estäjä) kahdesti vuorokaudessa ja 90 mg kerta‑annos brigatinibia, brigatinibin Cmax pieneni 41 %, AUC0‑INF pieneni 12 % ja AUC0‑120 pieneni 15 % verrattuna pelkkään 90 mg brigatinibikerta‑annokseen. Gemfibrotsiilin vaikutus brigatinibin farmakokinetiikkaan ei ole kliinisesti merkityksellinen, ja pienentyneen brigatinibialtistuksen taustamekanismi on tuntematon. Annosta ei tarvitse muuttaa samanaikaisessa käytössä vahvojen CYP2C8:n estäjien kanssa.
P‑gp:n ja BCRP:n estäjät
Brigatinibi on in vitro P‑glykoproteiinin (P‑gp) ja rintasyöpäresistenssiproteiinin (BCRP) substraatti. Koska brigatinibin liukoisuus ja läpäisevyys ovat suuria, P‑gp:n ja BCRP:n eston ei odoteta johtavan kliinisesti merkittävään systeemisen brigatinibialtistuksen muutokseen. P‑gp:n ja BCRP:n estäjien käyttö ei edellytä Alunbrig‑annoksen muutoksia.
Aineet, jotka saattavat pienentää plasman brigatinibipitoisuuksia
CYP3A:n induktorit
Kun terveille henkilöille annettiin samanaikaisesti toistuvia 600 mg/vrk rifampisiiniannoksia (rifampisiini on vahva CYP3A:n induktori) ja 180 mg kerta‑annos brigatinibia, brigatinibin Cmax pieneni 60 %, AUC0‑INF pieneni 80 % (5‑kertaisesti) ja AUC0‑120 pieneni 80 % (5‑kertaisesti) verrattuna pelkkään 180 mg brigatinibikerta‑annokseen. Alunbrig‑hoidon kanssa on vältettävä samanaikaista vahvojen CYP3A:n induktorien, mm. rifampisiinin, karbamatsepiinin, fenytoiinin, rifabutiinin, fenobarbitaalin ja mäkikuisman käyttöä.
Keskivahvat CYP3A:n induktorit voivat pienentää brigatinibin AUC‑arvoa noin 50 % fysiologisella farmakokinetiikkamallilla tehtyjen simulaatioiden perusteella. Alunbrig‑hoidon kanssa on vältettävä samanaikaista keskivahvojen CYP3A:n induktorien, mm. efavirentsin, modafiniilin, bosentaanin, etraviriinin ja nafsilliinin käyttöä. Jos kohtalaisten CYP3A:n induktorien samanaikaista käyttöä ei voida välttää, Alunbrig-annosta voidaan suurentaa 30 mg kerrallaan sen jälkeen, kun hoitoa on annettu nykyisellä Alunbrig-annoksella 7 vuorokauden ajan ja se on ollut siedettyä. Annosta saa suurentaa enintään sen verran, että se on kaksi kertaa sen Alunbrig-annoksen suuruinen, joka oli siedetty ennen kohtalaisen CYP3A:n induktorin käytön aloittamista. Kohtalaisen CYP3A:n induktorin käytön lopettamisen jälkeen Alunbrig-hoitoa jatketaan käyttämällä sitä annosta, joka oli siedetty ennen kohtalaisen CYP3A:n induktorin käytön aloittamista.
Aineet, joiden pitoisuuksiin plasmassa brigatinibi voi vaikuttaa
CYP3A:n substraatit
In vitro ‑tutkimukset hepatosyyteissä ovat osoittaneet, että brigatinibi on CYP3A4:n induktori. Kun syöpää sairastaville potilaille annettiin samanaikaisesti useita 180 mg:n vuorokausiannoksia Alunbrig‑valmistetta ja suun kautta 3 mg:n kerta-annos midatsolaamia, joka on herkkä CYP3A:n substraatti, midatsolaamin Cmax-arvo pieneni 16 %:lla, AUC0-INF-arvo 26 %:lla ja AUC0-last‑arvo 30 %:lla verrattuna suun kautta yksinään annettuun 3 mg:n midatsolaamiannokseen. Brigatinibi pienentää sellaisten samanaikaisesti käytettävien lääkevalmisteiden pitoisuutta plasmassa, jotka metaboloituvat pääasiassa CYP3A‑välitteisesti. Tästä syystä Alunbrig‑valmisteen samanaikainen käyttö sellaisten CYP3A:n substraattien kanssa, joiden terapeuttinen leveys on kapea (esim. alfentaniili, fentanyyli, kinidiini, siklosporiini, sirolimuusi, takrolimuusi), on vältettävä, sillä niiden teho voi heikentyä.
Alunbrig voi indusoida myös muita entsyymejä ja kuljettajaproteiineja (esim. CYP2C, P‑gp) samalla mekanismilla, joka vastaa CYP3A‑induktiosta (esim. pregnaani X ‑reseptorin aktivaatio).
Kuljettajaproteiinien substraatit
Brigatinibin samanaikainen käyttö P‑gp‑substraattien (esim. digoksiini, dabigatraani, kolkisiini, pravastatiini), BCRP:n substraattien (esim. metotreksaatti, rosuvastatiini, sulfasalatsiini), orgaanisten kationien kuljettajaproteiini 1:n (OCT1) substraattien sekä monilääke‑ ja toksiinipoistajaproteiini 1:n (MATE1) ja 2K:n (MATE2K) substraattien kanssa voi suurentaa niiden pitoisuuksia plasmassa. Potilaita on seurattava tiiviisti, kun Alunbrig‑valmistetta käytetään samanaikaisesti näiden kapean terapeuttisen leveyden kuljettajaproteiinien substraattien (esim. digoksiini, dabigatraani, metotreksaatti) kanssa.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset / Ehkäisy miehille ja naisille
Hedelmällisessä iässä olevia, Alunbrig‑hoitoa saavia naisia, tulee kehottaa välttämään raskaaksi tulemista, ja Alunbrig‑hoitoa saavia miehiä on kehotettava välttämään lapsen siittämistä hoidon aikana. Naisia, jotka voivat tulla raskaaksi, on neuvottava käyttämään tehokasta, ei‑hormonaalista ehkäisyä Alunbrig‑hoidon aikana ja vähintään 4 kuukauden ajan viimeisen annoksen jälkeen. Miehiä, joiden kumppani voi tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä hoidon aikana ja vähintään 3 kuukauden ajan viimeisen Alunbrig‑annoksen jälkeen.
Raskaus
Alunbrig‑valmisteen anto raskaana olevalle naiselle voi aiheuttaa haittaa sikiölle. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Ei ole olemassa kliinisiä tietoja Alunbrig‑valmisteen käytöstä raskaana olevilla naisilla. Alunbrigia ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa. Jos Alunbrig‑valmistetta käytetään raskauden aikana tai raskaus alkaa Alunbrig‑hoidon aikana, potilaalle on kerrottava sikiöön mahdollisesti kohdistuvista riskeistä.
Imetys
Ei tiedetä, erittyykö Alunbrig ihmisen rintamaitoon. Saatavana olevien tietojen perusteella ei voida sulkea pois mahdollisuutta, että lääkeaine erittyy rintamaitoon. Imetys on lopetettava Alunbrig‑hoidon ajaksi.
Hedelmällisyys
Alunbrigin vaikutuksesta ihmisen hedelmällisyyteen ei ole saatavana tietoa. Urospuolisille eläimille annetuilla toistuvaisannoksilla tehtyjen toksisuustutkimusten perusteella Alunbrig saattaa heikentää miesten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta). Löydösten kliinistä merkitystä ihmisen hedelmällisyydelle ei tunneta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Alunbrig‑valmisteella on vähäinen vaikutus ajokykyyn ja koneiden käyttökykyyn. Potilaiden on kuitenkin noudatettava varovaisuutta ajaessaan tai käyttäessään koneita, sillä Alunbrig‑hoidon aikana voi esiintyä näköhäiriöitä, huimausta tai väsymystä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät ilmoitetut haittavaikutukset (≥ 25 %) suositeltua Alunbrig‑hoitoannosta saaneilla potilailla olivat ASAT‑arvon kohoaminen, kreatiinikinaasiarvon kohoaminen, hyperglykemia, lipaasiarvon kohoaminen, hyperinsulinemia, ripuli, ALAT‑arvon kohoaminen, amylaasiarvon kohoaminen, anemia, pahoinvointi, väsymys, hypofosfatemia, lymfosyyttien lasku, yskä, AFOS‑arvon kohoaminen, ihottuma, APTT‑arvon kohoaminen, lihaskipu, päänsärky, hypertensio, valkosolumäärän pieneneminen, hengenahdistus ja oksentelu.
Yleisimmät ilmoitetut vakavat haittavaikutukset (≥ 2 %) suositeltua Alunbrig‑hoitoannosta saaneilla potilailla (pois lukien kasvaimen etenemiseen liittyvät tapahtumat), olivat keuhkokuume, pneumoniitti, hengenahdistus ja kuume.
Haittavaikutustaulukko
Jäljempänä esitetyt tiedot kuvastavat Alunbrig‑altistusta suositellulla hoitoannoksella kolmessa kliinisessä lääketutkimuksessa: vaiheen III tutkimus (ALTA 1L) ALK‑positiivista ei‑pienisoluista keuhkosyöpää sairastavilla potilailla, joita ei ollut aiemmin hoidettu ALK‑estäjällä (N = 136), vaiheen II tutkimus (ALTA) ALK‑positiivista ei‑pienisoluista keuhkosyöpää sairastavilla potilailla, joilla tauti oli edennyt kritsotinibi‑hoidon aikana (N = 110) ja vaiheen I/II annoshaku tutkimus potilailla joilla oli edennyt pahanlaatuinen kasvain (N = 28). Näiden tutkimusten mediaani altistusaika Alunbrig‑valmisteelle oli 21,8 kuukautta niillä potilailla, jotka saivat suositeltua hoitoannosta.
Ilmoitetut haittavaikutukset esitetään taulukossa 3 järjestettynä elinjärjestelmäluokan, MedDRA‑termien ja esiintymistiheyden mukaan. Yleisyysluokat ovat hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10) ja melko harvinainen (≥ 1/1 000, < 1/100). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen yleisyyden mukaisessa järjestyksessä.
Taulukko 3: Haittavaikutukset, joita on ilmoitettu Alunbrig‑hoitoa saaneilla potilailla (Common Terminology Criteria for Adverse Events ‑haittatapahtumaluokituksen (CTCAE) version 4.03 mukaan) käytettäessä 180 mg:n annosta (N = 274)
Elinjärjestelmä | Yleisyysluokka | Haittavaikutukset† kaikki asteet | Haittavaikutukset asteet 3–4 |
Infektiot | Hyvin yleinen | Keuhkokuumea,b Ylähengitystieinfektio | |
Yleinen | Keuhkokuumea | ||
Veri ja imukudos | Hyvin yleinen | Anemia Lymfosyyttimäärän pieneneminen APTT‑arvon kohoaminen Valkosolumäärän pieneneminen Neutrofiilimäärän pieneneminen | Lymfosyyttimäärän pieneneminen |
Yleinen | Trombosyyttimäärän pieneneminen | APTT‑arvon kohoaminen Anemia | |
Melko harvinainen | Neutrofiilimäärän pieneneminen | ||
Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Hyperglykemia Hyperinsulinemiac Hypofosfatemia Hypomagnesemia Hyperkalsemia Hyponatremia Hypokalemia Ruokahalun heikentyminen | |
Yleinen | Hypofosfatemia, Hyperglykemia, Hyponatremia, Hypokalemia Ruokahalun heikentyminen | ||
Psyykkiset häiriöt | Yleinen | Unettomuus | |
Hermosto | Hyvin yleinen | Päänsärkyd Perifeerinen neuropatiae Huimaus | |
Yleinen | Muistin heikkeneminen Makuaistin häiriöt | Päänsärkyd Perifeerinen neuropatiad | |
Melko harvinainen | Huimaus | ||
Silmät | Hyvin yleinen | Näköhäiriöf | |
Yleinen | Näköhäiriöf | ||
Sydän | Yleinen | Bradykardiag QT‑ajan piteneminen EKG‑tutkimuksessa Takykardiah Sydämentykytys | QT‑ajan piteneminen EKG‑tutkimuksessa |
Melko harvinainen | Bradykardiag | ||
Verisuonisto | Hyvin yleinen | Hypertensioi | Hypertensioi |
Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Yskä Hengenahdistusj | |
Yleinen | Pneumoniittik | Pneumoniittik Hengenahdistusj | |
Ruoansulatuselimistö | Hyvin yleinen | Lipaasiarvon kohoaminen Ripuli Amylaasiarvon kohoaminen Pahoinvointi Oksentelu Vatsakipul Ummetus Suutulehdusm | Lipaasiarvon kohoaminen |
Yleinen | Suun kuivuus Dyspepsia Ilmavaivat | Amylaasiarvon kohoaminen Pahoinvointi Vatsakipul Ripuli | |
Melko harvinainen | Haimatulehdus | Oksentelu Suutulehdusm Dyspepsia Haimatulehdus | |
Maksa ja sappi | Hyvin yleinen | ASAT‑arvojen kohoaminen ALAT‑arvojen kohoaminen AFOS‑arvon kohoaminen | |
Yleinen | Veren laktaattidehydrogenaasiarvon kohoaminen Hyperbilirubinemia | ALAT‑arvojen kohoaminen ASAT‑arvojen kohoaminen AFOS‑arvon kohoaminen | |
Melko harvinainen | Hyperbilirubinemia | ||
Iho ja ihonalainen kudos | Hyvin yleinen | Ihottuman Kutinao | |
Yleinen | Ihon kuivuminen Valoherkkyysreaktiotp | Ihottuman Valoherkkyysreaktiotp | |
Melko harvinainen | Ihon kuivuminen Kutinao | ||
Luusto, lihakset ja sidekudos | Hyvin yleinen | Veren kreatiinikinaasiarvon kohoaminen Lihaskipuq Nivelkipu | Veren kreatiinikinaasiarvon kohoaminen |
Yleinen | Tuki‑ ja liikuntaelinperäinen rintakipu Kipu raajoissa Tuki‑ ja liikuntaelimistön jäykkyys | ||
Melko harvinainen | Kipu raajoissa Tuki‑ ja liikuntaelinperäinen rintakipu Lihaskipuq | ||
Munuaiset ja virtsatiet | Hyvin yleinen | Veren kreatiniiniarvon kohoaminen | |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Uupumusr Turvotuss Kuume | |
Yleinen | Ei‑sydänperäinen rintakipu Epämukava tunne rinnassa Kipu | Uupumusr | |
Melko harvinainen | Kuume Turvotuss Ei‑sydänperäinen rintakipu | ||
Tutkimukset | Yleinen | Veren kolesteroliarvon kohoaminent Painon lasku | |
Melko harvinainen | Painon lasku | ||
† Kemiallisiinja hematologisiin laboratorioarvojen muutoksiin liittyvien haittavaikutustermien yleisyys on määritelty perustuen lähtötilanteeseen nähden poikkeavien laboratorioarvojen muutosten esiintyvyyteen. a Sisältää: epätyypillinen keuhkokuume, keuhkokuume, aspiraatiokeuhkokuume, kryptokokin aiheuttama keuhkokuume, alahengitystieinfektio, virusperäinen alahengitystieinfektio, keuhkoinfektio b Sisältää asteen 5 tapahtumat c Astetta ei sovelleta d Sisältää: päänsärky, poskiontelopäänsärky, epämukava tunne päässä, migreeni, jännityspäänsärky e Sisältää: parestesiat, perifeerinen sensorinen neuropatia, dysestesia, hyperestesia, hypoestesia, neuralgia, perifeerinen neuropatia, neurotoksisuus, perifeerinen motorinen neuropatia, polyneuropatia, polttava tunne, vyöruusun jälkeinen hermosärky f Sisältää: syvyysnäön muutokset, kaihi, hankinnainen värisokeus, kaksoiskuvat, glaukooma, silmänpaineen nousu, makulaturvotus, valoherkkyys, fotopsia, verkkokalvoturvotus, näön heikkeneminen, näöntarkkuuden heikkeneminen, näkökenttäpuutos, näköhäiriö, lasiaisirtauma, lasiaiskellujat, amaurosis fugax g Sisältää: bradykardia, sinusbradykardia h Sisältää: sinustakykardia, takykardia, eteistakykardia, sydämensykkeen kiihtyminen i Sisältää: verenpaineen kohoaminen, diastolinen hypertensio, hypertensio, systolinen hypertensio j Sisältää: hengenahdistus, hengenahdistus rasituksessa k Sisältää: interstitiaalinen keuhkosairaus, pneumoniitti l Sisältää: epämukava tunne vatsassa, vatsan pullotus, vatsakipu, alavatsakipu, ylävatsakipu, epämukava tunne epigastriumissa m Sisältää: haavainen suutulehdus, stomatiitti, aftoosinen haavauma, suun haavaumat, rakkulamuodostus suun limakalvoilla n Sisältää: aknetyyppinen dermatiitti, punoitus, eksfoliatiivinen dermatiitti, ihottuma, punoittava ihottuma, makulaarinen ihottuma, makulopapulaarinen ihottuma, papulaarinen ihottuma, kutiava ihottuma, märkärakkulainen ihottuma, dermatiitti, allerginen dermatiitti, kosketusihottuma, yleistynyt punoitus, follikulaarinen ihottuma, nokkosihottuma, lääkeaineihottuma, toksinen ihottuma o Sisältää: kutina, allerginen kutina, yleistynyt kutina, kutina sukupuolielimissä, kutina ulkosynnyttimissä ja emättimessä p Sisältää: valoherkkyysreaktio, monimuotoinen valoihottuma, aurinkoihottuma q Sisältää: tuki‑ ja liikuntaelimistön kipu, lihaskipu, lihasspasmit, lihaskireys, lihasten nykiminen, epämukava tunne tuki‑ ja liikuntaelimistössä r Sisältää: astenia, väsymys s Sisältää: silmäluomien turvotus, kasvojen turvotus, ääreisosien turvotus, turvotus silmäkuopan ympärillä, turvotus kasvoissa, yleistynyt turvotus, perifeerinen turvotus, angioedeema, huulten turvotus, turvotus silmäkuopan ympärillä, ihoturvotus, silmäluomien turvotus t Sisältää: veren kolesteroliarvon kohoaminen, hyperkolesterolemia | |||
Valikoitujen haittavaikutusten kuvaus
Keuhkohaittavaikutukset
ALTA 1L ‑tutkimuksessa 2,9 %:lla potilaista oli interstitiaalinen keuhkosairaus / pneumoniitti (kaikki asteet) hoidon varhaisvaiheessa (8 vrk kuluessa) ja 2,2 %:lla potilaista oli asteen 3–4 interstitiaalinen keuhkosairaus / pneumoniitti. Mikään interstitiaalisen keuhkosairauden / pneumoniitin tapauksista ei johtanut kuolemaan. Lisäksi 3,7 %:lla potilaista oli pneumoniitti hoidon myöhemmässä vaiheessa.
ALTA‑tutkimuksessa 6,4 %:lla potilaista oli keuhkohaittavaikutuksia (kaikki asteet), mukaan lukien interstitiaalinen keuhkosairaus / pneumoniitti, keuhkokuume ja hengenahdistus, hoidon varhaisvaiheissa (9 vrk kuluessa, mediaaniaika alkamiseen: 2 vrk); 2,7 %:lla potilaista oli asteen 3‑4 keuhkohaittavaikutuksia, ja 1 potilaalla (0,5 %) oli kuolemaan johtanut keuhkokuume. Asteen 1–2 keuhkohaittavaikutusten vuoksi Alunbrig‑hoito joko keskeytettiin ja aloitettiin myöhemmin uudestaan, tai annosta pienennettiin. Varhaisia keuhkohaittavaikutuksia esiintyi myös annosnostotutkimuksessa potilailla (N = 137) (tutkimus 101), ja kolme tapausta johti kuolemaan (hypoksia, äkillinen hengitysvajausoireyhtymä ja keuhkokuume).
Lisäksi 2,3 %:lla ALTA‑tutkimuksen potilaista oli pneumoniittia myöhemmin hoidon aikana, joista 2 potilaalla oli asteen 3 pneumoniitti (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Iäkkäät
Varhaisia keuhkohaittavaikutuksia ilmoitettiin 10,1%:lla ≥ 65‑vuotiaista potilaista, kun taas < 65‑vuotiailla potilailla luku oli 3,1 %.
Hypertensio
Hypertensiota ilmoitettiin 30 %:lla Alunbrig‑hoitoa 180 mg:n annosta saaneilla potilailla. 11 %:lla oli asteen 3 hypertensio. 180 mg annosta saaneilla annosta pienennettiin hypertension vuoksi 1,5 %:lla potilaista. Kaikilla potilailla systolisen ja diastolisen verenpaineen keskiarvot nousivat ajan myötä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Bradykardia
Bradykardiaa ilmoitettiin 8,4 %:lla Alunbrig‑hoitoa 180 mg:n annosta saaneista potilaista.
Alle 50/min syketiheyttä ilmoitettiin 8,4 %:lla potilaista 180 mg:n hoitoryhmässä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Näköhäiriöt
Näköhäiriöitä ilmoitettiin haittavaikutuksina 14 %:lla Alunbrig‑hoitoa 180 mg annosta saaneista potilaista. Näistä kolme oli asteen 3 haittavaikutuksia (1,1 %), mukaan lukien makulaturvotus ja kaihi.
Annosta pienennettiin näköhäiriöiden vuoksi kahdella potilaalla (0,7 %) 180 mg:n hoitoryhmässä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Perifeerinen neuropatia
Perifeeristä neuropatiaa ilmoitettiin haittavaikutuksena 20 %:lla 180 mg:n hoitoryhmän potilaista. 33 %:lla potilaista kaikki perifeeriseen neuropatiaan liittyvät haittavaikutukset korjaantuivat. Perifeerisen neuropatian keston mediaani oli 6,6 kk ja pisin kesto 28,9 kk.
Kreatiinikinaasiarvon kohoaminen
ALTA 1L‑ ja ALTA‑tutkimuksissa kreatiinikinaasiarvon kohoamista ilmoitettiin 64 %:lla Alunbrig‑hoitoa 180 mg:n annosta saaneista potilaista. Asteen 3–4 kreatiinikinaasiarvon kohoamista esiintyi 18 %:lla. Mediaaniaika kreatiinikinaasiarvon kohoamiseen oli 28 vrk.
Annosta pienennettiin kreatiinikinaasiarvon suurenemisen vuoksi 10 %:lla potilaista 180 mg:n hoitoryhmässä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Haimaentsyymiarvojen kohoaminen
Amylaasiarvon kohoamista ilmoitettiin 47 %:lla ja lipaasiarvon kohoamista 54 %:lla Alunbrig‑hoitoa 180 mg:n annosta saaneista potilaista. Amylaasiarvojen kohoamista asteeseen 3 ja 4 esiintyi 7,7 %:lla potilaista ja lipaasiarvojen kohoamista 15 %:lla potilaista. Mediaaniaika amylaasiarvojen kohoamiseen oli 16 vrk ja lipaasiarvojen kohoamiseen 29 vrk.
Annosta pienennettiin lipaasiarvon kohoamisen vuoksi 4,7 %:lla potilaista ja amylaasiarvon kohoamisen vuoksi 2,9 %:lla potilaista 180 mg:n hoitoryhmässä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Maksaentsyymiarvojen nousu
ALAT‑arvon kohoamista ilmoitettiin 49 %:lla ja ASAT‑arvon kohoamista 68 %:lla Alunbrig‑hoitoa 180 mg:n hoito‑ohjelmalla saaneista potilaista. ALAT‑arvon kohoamista asteeseen 3 ja 4 esiintyi 4,7 %:lla potilaista ja ASAT‑arvon vastaavaa kohoamista 3,6 %:lla potilaista.
Annosta pienennettiin ALAT‑arvojen kohoamisen vuoksi 0,7 %:lla potilaista ja ASAT‑arvojen kohoamisen vuoksi 1,1 %:lla potilaista 180 mg:n hoitoryhmässä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Hyperglykemia
Potilaista 61 %:lle kehittyi hyperglykemiaa. Asteen 3 hyperglykemiaa esiintyi 6,6 %:lla potilaista.
Yhdelläkään potilaalla annosta ei pienennetty hyperglykemian vuoksi.
Valoherkkyys ja valoihottuma
Seitsemästä kliinisestä tutkimuksesta saadut yhdistetyt tiedot, jotka koskevat 804 Alunbrig-valmisteen eri annostuksilla hoidettua potilasta, osoittivat, että valoherkkyyttä ja valoihottumaa raportoitiin 5,8 %:lla potilaista ja asteen 3–4 reaktioita ilmeni 0,7 %:lla potilaista. Potilaista 0,4 %:lla pienennettiin annosta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta‑tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle: www-sivusto: www.fimea.fi, Lääkealan turvallisuus- ja kehittämiskeskus Fimea, Lääkkeiden haittavaikutusrekisteri, PL 55, 00034 FIMEA
Yliannostus
Spesifistä vastalääkettä Alunbrig‑valmisteen yliannokselle ei tunneta. Yliannostustapauksessa potilasta on seurattava haittavaikutusten varalta (ks. kohta Haittavaikutukset) ja hänelle on järjestettävä asianmukaista elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: syöpälääkkeet ja immuunivasteen muuntajat, proteiinikinaasin estäjät, ATC‑koodi: L01ED04
Vaikutusmekanismi
Brigatinibi on tyrosiinikinaasin estäjä, jonka vaikutus kohdistuu ALK:iin, c‑ros‑onkogeeniin 1 (ROS1) ja insuliininkaltaisen kasvutekijä 1:n reseptoriin (IGF‑1R). Brigatinibi esti in vivo‑ ja in vitro ‑tutkimuksissa ALK:n autofosforylaatiota ja ALK‑välitteistä alavirran signalointiproteiini STAT3:n fosforylaatiota.
Brigatinibi esti in vitro sellaisten solulinjojen proliferaatiota, jotka ilmensivät EML4‑ALK‑ ja NPM‑ALK‑fuusioproteiineja. Lääkeaineen osoitettiin estävän annosriippuvaisesti EML4‑ALK‑positiivisten NSCLC‑ksenograftien kasvua hiirellä. Brigatinibi esti EML4‑ALK:n mutatoituneita muotoja ilmentävien solujen elinkykyisyyttä in vitro ja in vivo. Nämä mutantoituneet muodot, mukaan lukien G1202R ja L1196M, on yhdistetty resistenssiin ALK ‑inhibiitoreille.
Sydämen elektrofysiologia
101‑tutkimuksessa tutkittiin Alunbrigin vaikutuksia QT‑ajan pitenemiseen 123 potilaalla, joilla oli pitkälle edennyt maligniteetti ja jotka saivat 30–240 mg brigatinibiannoksia kerran vuorokaudessa. Keskimääräisen QTcF‑ajan (Fridericia‑korjaus) suurin muutos lähtötilanteesta oli alle 10 msek. Altistus‑QT‑analyysi ei viitannut pitoisuusriippuvaiseen QTc‑ajan pitenemiseen.
Kliininen teho ja turvallisuus
ALTA 1L
Alunbrig‑valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa (1:1), avoimessa monikeskustutkimuksessa (ALTA 1L), johon osallistui 275 edennyttä ALK‑positiivista ei‑pienisoluista keuhkosyöpää (NSCLC) sairastavaa aikuispotilasta, jotka eivät olleet aiemmin saaneet ALK‑kohdennettua hoitoa. Soveltuvuuskriteerien mukaan tutkimukseen voitiin ottaa potilaita, joilla oli paikallisen hoitokäytännön mukaisen testauksen perusteella dokumentoitu ALK‑uudelleenjärjestymä ja ECOG‑toimintakykyluokka 0–2. Potilailla oli saanut olla korkeintaan yksi aiempi paikallisesti edenneen tai etäpesäkkeisen taudin solunsalpaajahoitojakso. Neurologiselta tilaltaan vakaat potilaat, joilla oli hoidettuja tai hoitamattomia keskushermostoetäpesäkkeitä, mukaan lukien leptomeningeaalisia etäpesäkkeitä, soveltuivat tutkimukseen. Potilaat, joilla oli ollut interstitiaalinen keuhkosairaus, lääkehoitoon liittyvä pneumoniitti tai sädehoitoon liittyvä pneumoniitti, suljettiin pois.
Potilaat satunnaistettiin suhteessa 1:1 saamaan joko 180 mg Alunbrig‑valmistetta kerran vuorokaudessa (jota edelsi 7 vuorokauden aloitusvaihe annostuksella 90 mg kerran vuorokaudessa; N = 137), tai 250 mg kritsotinibiä suun kautta kahdesti vuorokaudessa (N = 138). Satunnaistaminen stratifioitiin aivometastaasien (kyllä, ei) ja aiemman, paikallisesti edenneen tai etäpesäkkeisen taudin solunsalpaajahoidon (kyllä, ei) perusteella.
Niille kritsotinibihaaran potilaille, joiden tauti eteni, tarjottiin vaihtoa Alunbrig‑hoitoon. Niistä kaikista 121 potilaasta, jotka oli satunnaistettu kritsotinibihaaraan ja jotka keskeyttivät tutkimushoidon loppuanalyysiin mennessä, 99 (82 %) potilasta sai sen jälkeen ALK‑tyrosiinikinaasin estäjää (TKI). 80 (66 %) kritsotinibihaaraan satunnaistettua potilasta sai myöhempää Alunbrig‑hoitoa, mukaan lukien 65 (54 %) potilasta, jotka vaihtoivat tutkimuksen eri haaraan.
Tärkein tulosmuuttuja oli etenemisvapaa aika (PFS) RECIST‑kriteerien (Response Evaluation Criteria in Solid Tumours) version 1.1 mukaan sokkoutetun riippumattoman arviointitoimikunnan (Blinded Independent Review Committee, BIRC) arvioimana. Muita BIRC:n arvioimia tulosmuuttujia olivat varmistettu objektiivinen vaste (ORR), vasteen kesto (DOR), vasteen saavuttamiseen kulunut aika, taudin hallinta (DCR), intrakraniaalinen objektiivinen vaste, intrakraniaalinen etenemisvapaa aika sekä intrakraniaalisen vasteen kesto. Tutkijan arvioimia tulosmuuttujia olivat etenemisvapaa aika ja kokonaiselossaolo.
ALTA 1 L tutkimuksessa saavutettiin sen ensisijainen päätetapahtuma primaarianalyysissä (11 kuukauden seuranta‑aika Alunbrig‑haarassa), jossa osoitettiin BIRC:n arvion perusteella tilastollisesti merkitsevä paraneminen taudin etenemisvapaassa ajassa. Tutkimussuunnitelmassa määritellyssä välianalyysissä, jonka katkaisupäivä oli 28.6.2019, Alunbrig‑haarassa mediaani seuranta‑aika oli 24,9 kuukautta. BIRC:n arvion perusteella määritelty etenemisvapaan elinajan (PFS) mediaani ITT‑populaatiossa oli 24 kuukautta Alunbrig‑haarassa ja 11 kuukautta kritsotinibihaarassa (suhteellinen riskitiheys, HR = 0,49 [95 %:n lv (0,35, 0,68)], p < 0,0001).
Tutkimussuunnitelmassa määritellyn loppuanalyysin tulokset esitetään alla. Viimeiseen potilaaseen oltiin viimeisen kerran yhteydessä 29.1.2021 ja seurannan keston mediaani oli 40,4 kuukautta Alunbrig‑haarassa.
Taulukko 4: ALTA 1L ‑tutkimuksen tehotulokset (ITT‑populaatio)
Tehoparametrit | Alunbrig N = 137 | kritsotinibi N = 138 | ||
Seuranta‑ajan mediaani (kk)a | 40,4 (vaihteluväli: 0,0‑52,4) | 15,2 (vaihteluväli: 0,1–51,7) | ||
Ensisijaiset tehon parametrit |
|
| ||
Etenemisvapaa aika (BIRC) | ||||
Potilaiden lukumäärä, joilla tapahtumia, n (%) | 73 (53,3 %) | 93 (67,4 %) | ||
Etenevä tauti, n (%) | 66 (48,2 %)b | 88 (63,8 %)c | ||
Kuolema, n (%) | 7 (5,1 %) | 5 (3,6 %) | ||
Mediaani (kk) (95 % lv) | 24,0 (18,5, 43,2) | 11,1 (9,1, 13,0) | ||
Riskisuhde (95 % lv) | 0,48 (0,35, 0,66) | |||
Log‑rank p‑arvod | < 0,0001 | |||
Toissijaiset tehon parametrit |
| |||
Varmistettu objektiivinen vaste (BIRC) | ||||
Vasteen saaneet, n (%) (95 % lv) | 102 (74,5 %) (66,3, 81,5) | 86 (62,3 %) (53,7, 70,4) | ||
p‑arvod,e | 0,0330 | |||
Täydellinen vaste, % | 24,1 % | 13,0 % | ||
Osittainen vaste, % | 50,4 % | 49,3 % | ||
Varmistetun vasteen kesto (BIRC) | ||||
Mediaani (kk) (95 % lv) | 33,2 (22,1,EA) | 13,8 (10,4, 22,1) | ||
Kokonaiselossaolof | ||||
Tapahtumien lukumäärä, n (%) | 41 (29,9 %) | 51 (37,0 %) | ||
Mediaani (kk) (95 % lv) | EA (EA, EA) | EA (EA, EA) | ||
Riskisuhde (95 % lv) | 0,81 (0,53, 1,22) | |||
Log‑rank p‑arvod | 0,3311 | |||
Kokonaiselossaolo 36 kuukauden kohdalla | 70,7 % | 67,5 % | ||
BIRC = Blinded Independent Review Committee; EA = ei arvioitavissa; lv = luottamusväli
Tämän taulukon tulokset perustuvat tehon loppuanalyysiin, jossa viimeiseen potilaaseen oltiin viimeisen kerran yhteydessä 29.1.2021.
a koko tutkimuksen seurannan kesto
b sisältää 3 potilasta, joille annettu palliatiivista sädehoitoa aivoihin
c sisältää 9 potilasta, joille annettu palliatiivista sädehoitoa aivoihin
dositettu log‑rank testiin ja Cochran Mantel‑Haenszel testiin lähtötilanteen keskushermostoetäpesäkkeiden esiintymisen ja aiemman paikallisesti edenneeseen tai etäpesäkkeiseen tautiin saadun solunsalpaajahoidon perusteella
e Cochran Mantel‑Haenszel –testistä
f Niille kritsotinibihaaran potilaille, joiden tauti eteni, tarjottiin vaihtoa Alunbrig-hoitoon
Kuva 1: Kaplan‑Meierin‑kuvaaja riippumattoman arviointitoimikunnan arvioimasta etenemisvapaasta ajasta ALTA 1L ‑tutkimuksessa
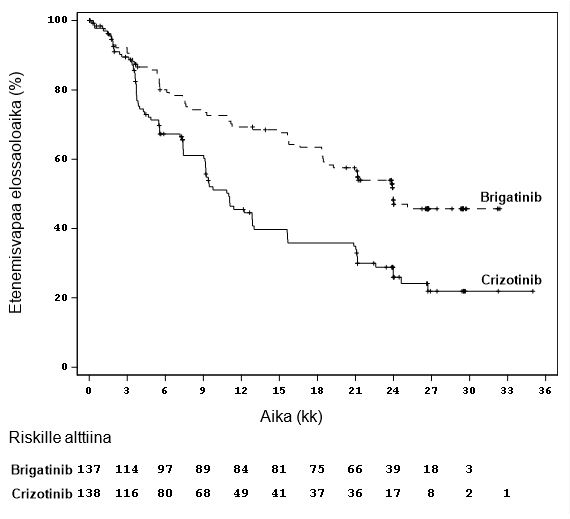
Tämän kuvan tulokset perustuvat tehon loppuanalyysiin, jossa viimeiseen potilaaseen oltiin viimeisen kerran yhteydessä 29.1.2021.
Taulukossa 5 esitetään yhteenvetona riippumattoman arviointitoimikunnan RECIST‑kriteerein (versio 1.1) arvioima intrakraniaalinen teho potilailla, joilla oli lähtötilanteessa aivometaaseja. Intrakraniaalinen teho esitetään myös erikseen potilailla, joilla on mitattavissa olevia aivometastaaseja (pisimmältä läpimitaltaan ≥ 10 mm).
Taulukko 5: BIRC:n arvioima intrakraniaalinen teho ALTA 1L ‑tutkimuksen potilailla
Tehoparametrit | Potilaat, joilla oli lähtötilanteessa mitattavissa olevia aivometastaaseja | |
Alunbrig N = 18 | kritsotinibi N = 23 | |
Varmistettu intrakraniaalinen objektiivinen vaste | ||
Vasteen saaneet, n (%) (95 % lv) | 14 (77,8 %) (52,4, 93,6) | 6 (26,1 %) (10,2, 48,4) |
p‑arvoa,b | 0,0014 | |
Täydellinen vaste % | 27,8 % | 0,0 % |
Osittainen vaste % | 50,0 % | 26,1 % |
Varmistetun intrakraniaalisen vasteen kestoc | ||
Mediaani (kk) (95 % lv) | 27,9 (5,7, EA) | 9,2 (3,9, EA) |
| Kaikki potilaat, joilla oli lähtötilanteessa aivometastaaseja | |
Alunbrig N = 47 | kritsotinibi N = 49 | |
Varmistettu intrakraniaalinen objektiivinen vaste | ||
Vasteen saaneet, n (%) (95 % lv) | 31 (66,0 %) (50,7, 79,1) | 7 (14,3 %) (5,9, 27,2) |
p‑arvoa,b | < 0,0001 | |
Täydellinen vaste (%) | 44,7 % | 2,0 % |
Osittainen vaste (%) | 21,3 % | 12,2 % |
Varmistetun intrakraniaalisen vasteen kestoc | ||
|
|
|
Mediaani (kk) (95 % lv) | 27,1 (16,9, 42,8) | 9,2 (3,9, EA) |
Intrakraniaalinen etenemisvapaa aikad |
|
|
Potilaiden lukumäärä, joilla todettiin tapahtumia, n (%) | 27 (57,4 %) | 35 (71,4 %) |
Taudin eteneminen, n (%) | 27 (57,4 %)e | 32 (65,3 %)f |
Kuolema, n (%) | 0 (0,0 %) | 3 (6,1 %) |
Mediaani (kk) (95 % lv) | 24,0 (12,9, 30,8) | 5,5 (3,7, 7,5) |
Riskisuhde (95 % lv) | 0,29 (0,17, 0,51) | |
Log‑rank p‑arvoa | < 0,0001 | |
lv = luottamusväli; EA = ei arvioitavissa
Tämän taulukon tulokset perustuvat tehon loppuanalyysiin, jossa viimeiseen potilaaseen oltiin viimeisen kerran yhteydessä 29.1.2021.
aOsitettu log‑rank testiin ja Cochran Mantel Heanszel testiin aiemman paikallisesti edenneeseen tai etäpesäkkeiseen tautiin saadun solunsalpaajahoidon perusteella
b Cochran Mantel‑Haenszel ‑testistä
c mitattu ensimmäisen varmistetun intrakraniaalisen vasteen ajankohdasta intrakraniaalisen taudin etenemisen (uusi intrakraniaalinen leesio, intrakraniaalisen kohdeleesion läpimitan kasvu > 20 % nadiirista tai intrakraniaalisen ei‑kohdeleesion yksiselitteinen eteneminen) ajankohtaan tai kuolemaan tai sensurointiin.
d mitattu satunnaistamisajankohdasta intrakraniaalisen taudin etenemisen (uusi intrakraniaalinen leesio, intrakraniaalisen kohdeleesion läpimitan kasvu > 20 % nadiirista tai intrakraniaalisen ei‑kohdeleesion yksisielitteinen eteneminen) ajankohtaan tai kuolemaan tai sensurointiin.
e sisältää 1 potilaan, jolle annettu palliatiivista sädehoitoa aivoihin
f sisältää 3 potilasta, joille annettu palliatiivista sädehoitoa aivoihin
ALTA
Alunbrig‑valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa (1:1), avoimessa monikeskustutkimuksessa (ALTA) 222 aikuispotilaalla, joiden paikallisesti levinnyt tai etäpesäkkeinen ALK‑positiivinen ei‑pienisoluinen keuhkosyöpä oli edennyt kritsotinibihoidon aikana. Soveltuvuuskriteerien mukaan tutkimukseen voitiin ottaa potilaita, joilla oli validoidun testin perusteella dokumentoitu ALK‑uudelleenjärjestymä, ECOG‑toimintakykyluokka 0–2 ja jotka olivat saaneet solunsalpaajahoitoa. Lisäksi mukaan otettiin potilaita, joilla oli keskushermostometastaaseja, kunhan potilaan neurologinen tila oli vakaa eikä vaatinut kortikosteroidiannoksen suurentamista. Potilaat, joilla oli ollut interstitiaalinen keuhkosairaus tai lääkehoitoon liittyvä pneumoniitti, suljettiin pois.
Potilaat satunnaistettiin suhteessa 1:1 saamaan Alunbrig‑hoitoa joko 90 mg kerran vuorokaudessa (90 mg hoito‑ohjelma, N = 112) tai 180 mg kerran vuorokaudessa, jolloin hoitoa edelsi 7 vrk aloitusvaihe annostuksella 90 mg kerran vuorokaudessa (180 mg hoito‑ohjelma, N = 110). Seurannan mediaanikesto oli 22,9 kk. Satunnaistaminen stratifioitiin aivometastaasien (kyllä, ei) ja parhaan aiemman kritsotinibihoitovasteen (täydellinen tai osittainen vaste, muu vaste/tuntematon) perusteella.
Tärkein tulosmuuttuja oli varmistettu objektiivinen vaste (ORR) RECIST v1.1‑kriteerien (Response Evaluation Criteria in Solid Tumors) mukaan tutkijan arvioimana. Muita tulosmuuttujia olivat varmistettu ORR riippumattoman arviointitoimikunnan (IRC) arvioimana; vasteen saavuttamiseen kulunut aika; etenemisvapaa aika (PFS); vasteen kesto (DOR); kokonaiselossaolo sekä intrakraniaalinen ORR ja intrakraniaalinen DOR riippumattoman arviointitoimikunnan arvioimana.
ALTA‑tutkimuksessa lähtötilanteen demografiset ja sairautta koskevat ominaisuudet olivat: iän mediaani 54 v (vaihteluväli 18–82 v; 23 % vähintään 65 v), 67 % valkoihoisia ja 31 % aasialaisia, 57 % naisia, 36 %:lla ECOG‑luokka 0 ja 57 %:lla ECOG‑luokka 1, 7 %:lla ECOG‑luokka 2, 60 % ei koskaan tupakoineita, 35 % aiemmin tupakoineita, 5 % tupakoitsijoita, 98 %:lla levinneisyysaste IV, 97 %:lla adenokarsinooma ja 74 %:lla anamneesissa solunsalpaajahoito. Yleisimmät rintakehän ulkopuoliset etäpesäkkeiden sijaintipaikat olivat: 69 %:lla aivot (näistä 62 % oli saanut sädehoitoa aivoihin), 39 %:lla luusto ja 26 %:lla maksa.
ALTA‑analyysin tehotulokset esitetään yhteenvetona taulukossa 6 ja tutkijan arvioiman etenemisvapaan ajan Kaplan–Meier‑käyrä esitetään kuvassa 2.
Taulukko 6: ALTA‑tutkimuksen tehotulokset (ITT populaatio)
Tehoparametri | Tutkijan arvio | Riippumattoman arviointitoimikunnan arvio | ||
90 mg hoito‑ohjelma* N = 112 | 180 mg hoito‑ohjelma† N = 110 | 90 mg hoito‑ohjelma* N = 112 | 180 mg hoito‑ohjelma† N = 110 | |
Objektiivinen vaste | ||||
(%) | 46 % | 56 % | 51 % | 56 % |
lv‡ | (35, 57) | (45, 67) | (41, 61) | (47, 66) |
Vasteen saavuttamiseen kulunut aika | ||||
Mediaani (kk) | 1,8 | 1,9 | 1,8 | 1,9 |
Vasteen kesto | ||||
Mediaani (kk) | 12,0 | 13,8 | 16,4 | 15,7 |
95 % lv | (9,2, 17,7) | (10,2, 19,3) | (7,4, 24,9) | (12,8, 21,8) |
Etenemisvapaa aika | ||||
Mediaani (kk) | 9,2 | 15,6 | 9,2 | 16,7 |
95 % lv | (7,4, 11,1) | (11,1, 21) | (7,4, 12,8) | (11,6, 21,4) |
Kokonaiselossaolo | ||||
Mediaani (kk) | 29,5 | 34,1 | NA | NA |
95 % lv | (18,2, Ei arv.) | (27,7, Ei arv.) | NA | NA |
12 kk elossaolon todennäköisyys (%) | 70,3 % | 80,1 % | NA | NA |
lv = luottamusväli; Ei arv. = ei arvioitavissa, NA = ei oleellinen
*90 mg kerran vuorokaudessa
†180 mg kerran vuorokaudessa; 7 vrk aloitusvaihe annostuksella 90 mg kerran vuorokaudessa
‡Tutkijan arvioiman ORR:n luottamusväli on 97,5 % ja riippumattoman arviointitoimikunnan arvioima ORR on 95 %
Kuva 2:Tutkijan arvioima systeeminen etenemisvapaa aika: ITT‑populaatio hoitoryhmän mukaan (ALTA)
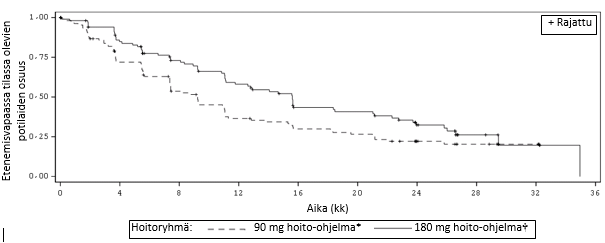
Lyhenteet: ITT = hoitoaikeen mukainen
Huom. Etenemisvapaan ajan määritelmänä oli aika hoidon aloittamisesta päivään, jolloin taudin eteneminen havaittiin ensi kertaa tai jolloin potilas kuoli (ensin tapahtunut valittiin).
*90 mg kerran vuorokaudessa
†180 mg kerran vuorokaudessa; 7 vrk aloitusvaihe annostuksella 90 mg kerran vuorokaudessa
Riippumattoman arviointitoimikunnan arvio intrakraniaalisesta ORR:sta ja intrakraniaalisen vasteen kestosta ALTA‑tutkimuksen potilailla, joilla oli lähtötilanteessa mitattavissa olevia aivometastaaseja (pisimmältä läpimitaltaan ≥ 10 mm) esitetään yhteenvetona taulukossa 7.
Taulukko 7: Intrakraniaalinen teho potilailla, joilla oli lähtötilanteessa mitattavissa olevia aivometastaaseja (ALTA)
Teho IRC:n arvioimana | Potilaat, joilla oli lähtötilanteessa mitattavissa olevia aivometastaaseja | |
90 mg hoito‑ohjelma* (N = 26) | 180 mg hoito‑ohjelma† (N = 18) | |
Intrakraniaalinen objektiivinen vaste | ||
(%) | 50 % | 67 % |
95 % lv | (30, 70) | (41, 87) |
Intrakraniaalisen taudin hallinta | ||
(%) | 85 % | 83 % |
95 % lv | (65, 96) | (59, 96) |
Intrakraniaalisen vasteen kesto‡, | ||
Mediaani (kk) | 9,4 | 16,6 |
95 % lv | (3,7, 24,9) | (3,7, Ei arv.) |
Lv = luottamusväli; Ei arv. = Ei arvioitavissa
*90 mg kerran vuorokaudessa
†180 mg kerran vuorokaudessa; 7 vrk aloitusvaihe annostuksella 90 mg kerran vuorokaudessa
‡Tapahtumiin sisältyi intrakraniaalinen taudin eteneminen (uusia leesioita, intrakraniaalisen kohdeleesion halkaisijan kasvu ≥ 20 % pienimpään halkaisijaan verrattuna tai intrakraniaalisen ei‑kohdeleesion todennettu eteneminen) tai kuolema.
Potilailla, joilla oli lähtötilanteessa aivometastaaseja, intrakraniaalisen taudin hallintaprosentti oli 90 mg:n ryhmässä 77,8 % (95 % lv 67,2–86,3) (N = 81) ja 180 mg:n ryhmässä 85,1 % (95 % lv 75–92,3) (N = 74).
Tutkimus 101
Erillisessä annoshakututkimuksessa 25 potilasta, joilla oli kritsotinibihoidon aikana edennyt ALK‑positiivinen ei‑pienisoluinen keuhkosyöpä, saivat Alunbrig‑valmistetta 180 mg kerran vuorokaudessa; 7 vrk pituisen aloitusvaiheen aikana annostus oli 90 mg kerran vuorokaudessa. Näistä 19 potilasta sai tutkijan arvioiman varmistetun objektiivisen vasteen (76 %; 95 % lv: 55–91), ja Kaplan Maier (KM)‑estimoitu vasteen keston mediaani näillä 19:lla vasteen saavuttaneella oli 26,1 kk (95 % lv: 7,9–26,1). KM‑estimoitu etenemisvapaan ajan mediaani oli 16,3 kuukautta (95 % lv: 9,2, Ei arv.), ja 12 kk kokonaiselossaolon todennäköisyys oli 84,0 % (95 % lv: 62,8–93,7).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Alunbrig‑valmisteen käytöstä kaikkien pediatristen potilasryhmien hoidossa keuhkosyövässä (pienisoluinen ja ei‑pienisoluinen keuhkosyöpä) (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Tutkimuksessa 101 suun kautta otettavan brigatinibin kerta‑annoksen (30–240 mg) jälkeen huippupitoisuuden saavuttamiseen kulunut mediaaniaika (Tmax) oli 1–4 h annoksesta. Kerta‑annoksen jälkeen ja vakaassa tilassa systeeminen altistus oli suhteessa annokseen annosalueella 60–240 mg kerran vuorokaudessa. Vähäistä kumulaatiota havaittiin toistuvassa annostelussa (kumulaatiokertoimen geometrinen keskiarvo: 1,9–2,4). Brigatinibin vakaan tilan Cmax‑arvon geometrinen keskiarvo oli 90 mg:n annoksella kerran vuorokaudessa 552 ng/ml ja 180 mg:n annoksella kerran vuorokaudessa 1 452 ng/ml, kun taas 90 mg:n annoksella AUC0–τ oli 8 165 h∙ng/ml ja 180 mg annoksella 20 276 h∙ng/ml. Brigatinibi on P‑gp‑ ja BCRP‑kuljettajaproteiinien substraatti.
Terveillä henkilöillä, yön yli paastoon verrattuna, runsasrasvainen ateria pienensi brigatinibin Cmax‑arvoa 13 %, muttei vaikuttanut AUC‑arvoon. Brigatinibi voidaan ottaa ruoan kanssa tai ilman ruokaa.
Jakautuminen
Brigatinibi sitoutuu kohtalaisesti (91 %) ihmisen plasman proteiineihin. Sitoutumisaste ei riipu pitoisuudesta. Veressä ja plasmassa olevien pitoisuuksien suhde on 0,69. Kun potilaille annettiin 180 mg brigatinibia kerran vuorokaudessa, näennäisen jakautumistilavuuden geometrinen keskiarvo (Vz/F) oli vakaassa tilassa 307 l, mikä viittaa kohtalaiseen jakautumiseen kudoksiin.
Biotransformaatio
In vitro ‑tutkimusten perusteella brigatinibi metaboloituu pääasiassa CYP2C8‑ ja CYP3A4‑välitteisesti ja paljon pienemmässä määrin CYP3A5‑välitteisesti.
Kun 180 mg kerta‑annos [14C]‑brigatinibia annettiin suun kautta terveille henkilöille, kaksi tärkeintä metabolista puhdistumisreittiä olivat N‑demetylaatio ja kysteiinikonjugaatio. Tarkkailtaessa yhdistettynä virtsaan ja ulosteeseen erittyviä määriä 48 % radioaktiivisesta annoksesta erittyi muuttumattomana brigatinibina, 27 % N‑desmetyylibrigatinibina (AP26123) ja 9,1 % brigatinibikysteiinikonjugaattina. Pääasiallinen verenkierron radioaktiivinen komponentti oli muuttumaton brigatinibi (92 %) ja AP26123 (3,5 %), jonka on in vitro havaittu olevan pääasiallinen metaboliitti. Vakaassa tilassa potilailla plasman AP26123‑pitoisuuden AUC oli < 10 % brigatinibialtistuksesta. In vitro kinaasi‑ ja solumäärityksissä metaboliitti AP26123 esti ALK‑kinaasia noin 3 kertaa heikommin kuin brigatinibi.
Eliminaatio
Potilailla, joille annettiin 180 mg brigatinibia kerran vuorokaudessa, näennäisen oraalisen puhdistuman (CL/F) geometrinen keskiarvo vakaassa tilassa oli 8,9 l/h ja eliminaation puoliintumisaika plasmasta (mediaani) 24 h.
Brigatinibi erittyy pääasiallisesti ulosteeseen. Kun kuudelle terveelle miespuoliselle henkilölle annettiin suun kautta 180 mg kerta‑annos [14C]‑brigatinibia, 65 % annoksesta havaittiin ulosteessa ja 25 % virtsassa. Muuttumaton brigatinibi vastasi 41 % kokonaisradioaktiivisuudesta ulosteessa ja 86 % kokonaisradioaktiivisuudesta virtsassa. Loput olivat metaboliitteja.
Erityisryhmät
Maksan vajaatoiminta
Brigatinibin farmakokinetiikkaa arvioitiin terveillä henkilöillä, joilla maksan toiminta oli normaali (N = 9), potilailla, joilla oli lievä maksan vajaatoiminta (Child–Pugh‑luokka A, N = 6), keskivaikea maksan vajaatoiminta (Child–Pugh‑luokka B, N = 6) tai vaikea maksan vajaatoiminta (Child–Pugh‑luokka C, N = 6). Brigatinibin farmakokinetiikka oli terveillä, maksan toiminnaltaan normaaleilla henkilöillä samankaltainen kuin potilailla, joilla oli lievä (Child–Pugh‑luokka A) tai keskivaikea (Child–Pugh‑luokka B) maksan vajaatoiminta. Sitoutumaton AUC0–INF oli vaikeaa maksan vajaatoimintaa (Child–Pugh‑luokka C) sairastavilla 37 % suurempi kuin terveillä henkilöillä, joiden maksan toiminta oli normaali (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Populaatiofarmakokinetiikan analyysien mukaan brigatinibin farmakokinetiikka on samankaltainen munuaistoiminnaltaan normaaleilla potilailla ja potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta (eGFR ≥ 30 ml/min). Farmakokinetiikan tutkimuksessa sitoutumaton AUC0–INF oli vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min, N = 6) sairastavilla 94 % suurempi kuin potilailla, joiden munuaistoiminta on normaali (eGFR ≥ 90 ml/min, N = 8) (ks. kohta Annostus ja antotapa).
Etninen tausta ja sukupuoli
Populaatiofarmakokinetiikan analyysien mukaan etninen tausta ja sukupuoli eivät vaikuttaneet brigatinibin farmakokinetiikkaan.
Ikä, paino ja albumiinipitoisuudet
Populaatiofarmakokinetiikan analyysien mukaan paino, ikä ja albumiinipitoisuudet eivät vaikuttaneet brigatinibin farmakokinetiikkaan kliinisesti merkittävästi.
Prekliiniset tiedot turvallisuudesta
Brigatinibin farmakologista turvallisuutta arvioineissa tutkimuksissa tunnistettiin potentiaalisia keuhkovaikutuksia (hengitystaajuuden muutokset tasolla 1–2 kertaa ihmisen Cmax), kardiovaskulaarivaikutuksia (syketiheyden ja verenpaineen muutokset tasolla 0,5 kertaa ihmisen Cmax) ja munuaisvaikutuksia (heikentynyt munuaistoiminta tasolla 1–2,5 kertaa ihmisen Cmax). Tutkimustulokset eivät sen sijaan viitanneet potentiaaliseen QT‑ajan pidentymiseen eivätkä hermoston toimintaan kohdistuviin vaikutuksiin.
Kliiniselle käytölle mahdollisesti relevantit haittavaikutukset, joita havaittiin eläimillä kliinistä altistusta vastaavilla altistustasoilla, liittyivät ruoansulatuskanavaan, luuytimeen, silmiin, kiveksiin, maksaan, munuaisiin, luustoon ja sydämeen. Nämä vaikutukset korjautuivat yleensä lääkkeettömän toipumisjakson aikana, huomattavina poikkeuksina silmiin ja kiveksiin kohdistuvat vaikutukset, jotka eivät korjautuneet.
Toistuvaisannoksilla tehdyissä toksisuustutkimuksissa apinoilla havaittiin keuhkomuutoksia (alveolien vaahtosoluja), kun altistustaso oli ≥ 0,2‑kertainen verrattuna ihmisen AUC‑arvoon. Nämä muutokset olivat kuitenkin minimaalisia ja samankaltaisia kuin hoitamattomilla apinoilla taustalöydöksinä ilmoitetut muutokset, eikä näillä apinoilla havaittu kliinistä näyttöä hengitysvaikeuksista.
Brigatinibin karsinogeenisuutta ei ole tutkittu.
Brigatinibi ei ollut mutageeninen in vitro bakteerien käänteismutaatiokokeessa (Amesin testi) eikä nisäkässolujen kromosomipoikkeavuustestissä, mutta se lisäsi hieman mikrotumien määrää rotan luuytimen mikrotumatestissä. Mikrotumainduktion mekanismi oli poikkeava kromosomin segregaatio (aneugeenisuus) eikä klastogeeninen vaikutus kromosomeihin. Vaikutus havaittiin, kun altistus oli noin viisinkertainen ihmiselle 180 mg kerran vuorokaudessa ‑annostuksella aiheutuvaan altistukseen nähden.
Brigatinibi saattaa heikentää miesten hedelmällisyyttä. Toistuvaisannoksilla tehdyissä eläintutkimuksissa havaittiin kivestoksisuutta. Rotalla löydöksiä olivat kivesten, rakkularauhasten ja eturauhasen alentunut paino sekä kivesten siementiehyeiden degeneroituminen. Nämä vaikutukset eivät korjautuneet toipumisjakson aikana. Apinoilla löydöksiä olivat kivesten koon pieneneminen sekä mikroskooppinen näyttö hypospermatogeneesistä. Nämä vaikutukset korjautuivat toipumisjakson aikana. Yleisesti ottaen nämä urosrottien ja ‑apinoiden lisääntymiselimiin kohdistuneet vaikutukset ilmenivät ≥ 0,2‑kertaisilla altistustasoilla verrattuna AUC‑arvoihin, joita havaittiin 180 mg kerran vuorokaudessa ‑hoitoa saaneilla potilailla. Naaraiden lisääntymiselimistöön kohdistuvia vaikutuksia ei havaittu yleisen toksikologian tutkimuksissa rotilla ja apinoilla.
Alkion‑ ja sikiönkehitystä koskeneessa tutkimuksessa, jossa kantaville rotille annettiin päivittäin brigatinibiannoksia organogeneesin aikana, havaittiin annosriippuvaisia luustopoikkeavuuksia jo noin 0,7‑kertaisella altistustasolla verrattuna AUC‑arvoihin, joita havaittiin 180 mg kerran vuorokaudessa ‑hoitoa saaneilla potilailla. Löydöksiä olivat alkiokuolemat, sikiöiden kasvun hidastuminen ja luustomuutokset.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Laktoosimonohydraatti
Mikrokiteinen selluloosa
Natriumtärkkelysglykolaatti (tyyppi A)
Piidioksidi, hydrofobinen, kolloidinen
Magnesiumstearaatti
Tabletin päällyste
Talkki
Makrogoli
Polyvinyylialkoholi
Titaanidioksidi
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ALUNBRIG tabletti, kalvopäällysteinen
90 mg + 180 mg (L:kyllä) 7+21 fol (aloituspakkaus, 7x90 mg+21x180 mg) (4652,81 €)
30 mg (L:kyllä) 28 fol (1072,05 €)
90 mg (L:kyllä) 28 fol (3035,21 €)
180 mg (L:kyllä) 28 fol (4652,81 €)
PF-selosteen tieto
Alunbrig 30 mg kalvopäällysteiset tabletit
Pyöreä, leveäsuinen HDPE‑purkki, jossa on kaksiosainen, polypropeeninen lapsiturvallinen kierresuljin ja folioinduktiotiiviste, sisältää joko 60 tai 120 kalvopäällysteistä tablettia ja yhden HDPE‑säiliön, jossa on molekyylisuodatinperiaatteella toimivaa kuivausainetta.
Läpipainopakkaus kirkasta, lämpömuovautuvaa PCTFE:tä, jossa on lämpösinetöitävä paperilaminoitu taustakalvo, pakattu pahvipakkaukseen, sisältää joko 28, 56 tai 112 kalvopäällysteistä tablettia.
Alunbrig 90 mg kalvopäällysteiset tabletit
Pyöreä, leveäsuinen HDPE‑purkki, jossa on kaksiosainen, polypropeeninen lapsiturvallinen kierrekorkki ja folioinduktiotiiviste, sisältää joko 7 tai 30 kalvopäällysteistä tablettia ja yhden HDPE‑säiliön, jossa on molekyylisuodatinperiaatteella toimivaa kuivausainetta.
Läpipainopakkaus kirkasta, lämpömuovautuvaa PCTFE:tä, jossa on lämpösinetöitävä paperilaminoitu taustakalvo, pakattu pahvipakkaukseen, sisältää joko 7 tai 28 kalvopäällysteistä tablettia.
Alunbrig 180 mg kalvopäällysteiset tabletit
Pyöreä, leveäsuinen HDPE‑purkki, jossa on kaksiosainen, polypropeeninen lapsiturvallinen kierrekorkki ja folioinduktiotiiviste, sisältää 30 kalvopäällysteistä tablettia ja yhden HDPE‑säiliön, jossa on molekyylisuodatinperiaatteella toimivaa kuivausainetta.
Läpipainopakkaus kirkasta, lämpömuovautuvaa PCTFE:tä, jossa on lämpösinetöitävä paperilaminoitu taustakalvo, pakattu pahvipakkaukseen, sisältää 28 kalvopäällysteistä tablettia.
Hoidon aloituspakkaus Alunbrig 90 mg ja 180 mg kalvopäällysteiset tabletit
Yksi pakkaus koostuu ulkopakkauksesta ja kahdesta sisäpakkauksesta, jotka sisältävät:
-
Alunbrig 90 mg kalvopäällysteiset tabletit
1 läpipainopakkaus kirkasta, lämpömuovautuvaa PCTFE:tä, jossa on lämpösinetöitävä paperilaminoitu taustakalvo, pakattu pahvipakkaukseen, sisältää 7 kalvopäällysteistä tablettia -
Alunbrig 180 mg kalvopäällysteiset tabletit
3 läpipainopakkausta kirkasta, lämpömuovautuvaa PCTFE:tä, jossa on lämpösinetöitävä paperilaminoitu taustakalvo, pakattu pahvipakkaukseen, sisältää 21 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Alunbrig 30 mg kalvopäällysteiset tabletit
Pyöreä, valkoinen tai luonnonvalkoinen kalvopäällysteinen tabletti, halkaisija noin 7 mm, toisella puolella kaiverrus ”U3”, toisella puolella ei merkintöjä.
Alunbrig 90 mg kalvopäällysteiset tabletit
Soikea, valkoinen tai luonnonvalkoinen kalvopäällysteinen tabletti, pituus noin 15 mm, toisella puolella kaiverrus ”U7”, toisella puolella ei merkintöjä.
Alunbrig 180 mg kalvopäällysteiset tabletit
Soikea, valkoinen tai luonnonvalkoinen kalvopäällysteinen tabletti, pituus noin 19 mm, toisella puolella kaiverrus ”U13”, toisella puolella ei merkintöjä.
Käyttö- ja käsittelyohjeet
Potilasta on neuvottava säilyttämään kuivausainesäiliö purkissa. Sitä ei saa niellä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ALUNBRIG tabletti, kalvopäällysteinen
90 mg + 180 mg 7+21 fol
30 mg 28 fol
90 mg 28 fol
180 mg 28 fol
- Ylempi erityiskorvaus (100 %). Brigatinibi: Keuhkosyövän hoito erityisin edellytyksin (1521).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Brigatinibi: Keuhkosyövän hoito erityisin edellytyksin (3017).
ATC-koodi
L01ED04
Valmisteyhteenvedon muuttamispäivämäärä
24.07.2023
Yhteystiedot
 TAKEDA OY
TAKEDA OY PL 1406, Ilmalankuja 3
00101 Helsinki
0800 774 051
www.takeda.fi
etunimi.sukunimi@takeda.com