
SOOLANTRA emulsiovoide 10 mg/g
Vaikuttavat aineet ja niiden määrät
Yksi gramma emulsiovoidetta sisältää 10 mg ivermektiiniä.
Apuaineet, joiden vaikutus tunnetaan:
Yksi gramma emulsiovoidetta sisältää 35 mg setyylialkoholia, 25 mg stearyylialkoholia, 2 mg metyyliparahydroksibentsoaattia (E218), 1 mg propyyliparahydroksibentsoaattia (E216) ja 20 mg propyleeniglykolia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Emulsiovoide.
Kliiniset tiedot
Käyttöaiheet
Soolantra on tarkoitettu ruusufinnin tulehduksellisten (papulopustulaaristen) leesioiden paikallishoitoon aikuisille potilaille.
Annostus ja antotapa
Annostus
Levitetään kerran päivässä enintään 4 kuukauden ajan. Soolantraa pitäisi käyttää päivittäin hoitojakson ajan. Hoitojakso voidaan uusia. Soolantraa voidaan käyttää monoterapiana tai yhdistelmähoidon osana (ks. kohta Farmakodynamiikka).
Jos paranemista ei havaita 3 kuukauden hoidon jälkeen, hoito pitää lopettaa.
Erityisryhmät
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen.
Maksan vajaatoiminta
Käytetään varoen potilaille, joilla on vaikea maksan vajaatoiminta.
Iäkkäät potilaat
Annoksen muuttaminen iäkkäille ei ole tarpeen (ks. myös kohta Haittavaikutukset).
Pediatriset potilaat
Soolantran turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu.
Tietoja ei ole saatavilla.
Antotapa
Käytetään vain iholle.
Herneen kokoinen määrä lääkevalmistetta annostellaan kullekin kasvojen viidelle alueelle: otsa, leuka, nenä ja kumpikin poski. Lääke levitetään ohuelti koko kasvoille välttäen sen joutumista silmiin, huulille ja limakalvoille.
Soolantraa käytetään vain kasvoille.
Kädet pitää pestä lääkkeen levittämisen jälkeen.
Kosmeettisia tuotteita voi levittää sen jälkeen, kun lääke on kuivunut.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Ruusufinni voi tilapäisesti pahentua. Tilanne tavallisesti lievittyy viikon kuluessa, kun hoitoa jatketaan, kuten voi olettaa, kun kyseessä on kuolevien Demodex-punkkien aiheuttama reaktio.
Jos tauti pahenee huomattavasti tai tulee voimakas ihoreaktio, hoito pitää lopettaa.
Soolantraa ei ole tutkittu potilailla, joilla on munuaisten tai maksan vajaatoiminta.
Tämä lääkevalmiste sisältää:
- setyylialkoholia ja stearyylialkoholia, jotka voivat aiheuttaa paikallisia ihoreaktioita (esim. kosketusihottumaa),
- metyyliparahydroksibentsoaattia (E218) ja propyyliparahydroksibentsoaattia (E216), jotka voivat aiheuttaa allergisia reaktioita (mahdollisesti viivästyneitä),
- ja propyleeniglykolia, joka voi aiheuttaa ihoärsytystä.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty (ks. kohta Farmakokinetiikka Biotransformaatio).
In vitro -tutkimusten perusteella ivermektiini metaboloituu pääasiassa CYP3A4-entsyymijärjestelmän kautta. Sen vuoksi varovaisuutta on syytä noudattaa, kun ivermektiiniä annetaan samanaikaisesti voimakkaiden CYP3A4-estäjien kanssa, koska pitoisuus plasmassa voi suurentua merkitsevästi.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja ivermektiinin paikallisesta käytöstä raskaana oleville naisille. Lisääntymistoksisuutta selvittävät tutkimukset ovat osoittaneet, että suun kautta annettu ivermektiini on teratogeeninen rotilla ja kaneilla (ks. kohta Prekliiniset tiedot turvallisuudesta), mutta koska systeemialtistus on vähäinen käytettäessä valmistetta paikallisesti suositelluilla annoksilla, sikiöön kohdistuvien haittojen todennäköisyys ihmisellä on pieni. Soolantraa ei suositella käytettäväksi raskauden aikana.
Imetys
Suun kautta otettu ivermektiini erittyy ihmisen rintamaitoon pieninä pitoisuuksina. Paikallisen käytön jälkeen tapahtuvaa erittymistä ihmisen rintamaitoon ei ole tutkittu. Olemassa olevat farmakokineettiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet ivermektiinin erittyvän rintamaitoon. Imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Soolantra-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Tietoa ivermektiinin vaikutuksesta hedelmällisyyteen ihmisellä ei ole saatavilla. Rotilla ivermektiinihoidolla ei ollut vaikutusta paritteluun tai hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Soolantralla ei ole haitallista vaikutusta tai on vähäinen vaikutus ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Yleisimmin ilmoitettuja haittavaikutuksia ovat polttava tunne iholla, ihoärsytys, kutina ja kuiva iho, mitä kaikkia esiintyi korkeintaan 1 %:lla potilaista, joita hoidettiin lääkevalmisteella kliinisissä tutkimuksissa.
Haittavaikutukset ovat yleensä lieviä tai kohtalaisia, ja tavallisesti ne lievittyvät, kun hoitoa jatketaan.
Turvallisuuden suhteen ei ollut merkittäviä eroja 18-65 -vuotiaiden ja ≥ 65-vuotiaiden henkilöiden välillä.
Haittavaikutustaulukko
Haittavaikutukset on luokiteltu elinjärjestelmän ja esiintymistiheyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1000, < 1/100), harvinainen (≥ 1/10000, < 1/1000), hyvin harvinainen (< 1/10000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin) (ks. Taulukko 1).
Taulukko 1 – Haittavaikutukset
Elinjärjestelmä | Yleisyys | Haittavaikutus |
Iho ja ihonalainen kudos | Yleinen | Polttava tunne iholla |
Melko harvinainen | Ihoärsytys, kutina, kuiva iho | |
| Tuntematon | Eryteema, kosketusihottuma (allerginen tai ärsytysihottuma), kasvojen turvotus | |
| Tutkimukset | Tuntematon | Kohonneet transaminaasit* |
* Raportoitu myyntiluvan jälkeisessä haittavaikutusseurannassa
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‐haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Soolantran yliannostuksesta ei ole ilmoituksia.
Kun eläimille tarkoitettua ivermektiiniä on annettu ihmiselle vahingossa tai määrinä, jotka aiheuttavat huomattavan altistuksen, joko suun kautta, hengitettynä, injektiona tai ihoaltistuksen kautta, seuraavia haittavaikutuksia on ilmoitettu useimmin: ihottuma, edeema, päänsärky, huimaus, heikotus, pahoinvointi, oksentelu ja ripuli. Muita ilmoitettuja haittavaikutuksia ovat: kouristukset, ataksia, hengenahdistus, vatsakipu, tuntohäiriöt, urtikaria ja kosketusihottuma.
Vahingossa tapahtuneen nielemisen jälkeen tukihoitoon, jos tarpeen, pitää kuulua parenteraalinen nesteytys ja elektrolyytit, hengityksen tuki (happi ja mekaaninen ventilaatio, jos tarpeen) ja verenpainetta kohottavia aineita, jos on huomattava hypotensio. Oksennuttaminen ja/tai mahanhuuhtelu mahdollisimman pian, ja sen jälkeen puhdistava ja muu tavanomainen myrkytyksen hoito voi olla aiheellista, jos on tarpeen estää niellyn aineen imeytyminen.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut ihotautien lääkkeet, muut ihotautilääkkeet, ATC-koodi: D11AX22.
Vaikutusmekanismi
Ivermektiini kuuluu avermektiineihin. Avermektiini vaikuttaa anti-inflammatorisesti estämällä lipopolysakkaridien indusoimaa tulehduksellisten sytokiinien tuotantoa. Iholle käytetyn ivermektiinin anti-inflammatoriset vaikutukset on havaittu ihotulehduksen eläinmalleissa. Ivermektiini myös tappaa loisia, pääasiassa sitoutumalla selektiivisesti ja suurella affiniteetilla glutamaatti-välitteisiin kloridikanaviin, joita on selkärangattomien hermo- ja lihassoluissa. Soolantran vaikutusmekanismia ruusufinnin tulehduksellisten leesioiden hoidossa ei tiedetä, mutta se voi liittyä ivermektiinin tulehdusta vähentäviin vaikutuksiin sekä siihen, että se tappaa Demodex-punkkeja, joiden on raportoitu olevan yksi ihon tulehdukseen liittyvä tekijä.
Kliininen teho ja turvallisuus
Kerran päivässä ennen nukkumaanmenoa levitetyn Soolantran tehoa ruusufinnin aiheuttamien tulehduksellisten leesioiden hoidossa arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, vehikkeli-kontrolloidussa kliinisessä tutkimuksessa, joiden asetelmat olivat identtisiä. Tutkimuksissa oli 1371 vähintään 18 vuoden ikäistä henkilöä, joita hoidettiin kerran päivässä 12 viikon ajan Soolantralla tai vehikkelillä.
Tutkimushenkilöistä 96 % oli valkoihoisia ja 67 % naisia. Arvioituna 5-pisteisellä tutkijan arviointiasteikolla (Investigator Global Assessment, IGA), 79 %:lla henkilöistä tauti oli lähtötilanteessa keskivaikea (IGA = 3) ja 21 %:lla vaikea (IGA = 4).
Yhdistetyt ensisijaiset päätetapahtumat molemmissa kliinisissä tutkimuksissa olivat onnistuneiden hoitotulosten määrä IGA-asteikolla arvioituna (niiden potilaiden prosentuaalinen osuus, jotka arvioitiin oireettomiksi tai lähes oireettomiksi tutkimuksen viikolla 12) ja absoluuttinen tulehduksellisten leesioiden määrän muutos lähtötilanteesta. IGA-asteikko on määritelty seuraavasti:
Taulukko 2: Tutkijan arviointiasteikko, Investigator Global Assessment (IGA)
Aste | Pisteytys | Kliininen kuvaus |
Oireeton | 0 | Ei tulehduksellisia leesioita, ei punoitusta |
Lähes oireeton | 1 | Hyvin vähän pieniä papuloita/pustuloita, hyvin lievä punoitus |
Lievä | 2 | Vähän papuloita/pustuloita, lievä punoitus |
Keskivaikea | 3 | Useita pieniä tai suuria papuloita/pustuloita, keskivaikea punoitus |
Vaikea | 4 | Paljon pieniä tai suuria papuloita/pustuloita, vaikea punoitus |
Kummankin kliinisen tutkimuksen tulokset osoittivat, että kerran päivässä 12 viikon ajan käytetty Soolantra oli tilastollisesti merkitsevästi parempi kuin vehikkeli sekä IGA-arvioinnin perusteella että tulehduksellisten leesioiden määrällä laskettuna (p < 0,001, ks. taulukko 3 ja kuva 1, kuva 2, kuva 3 ja kuva 4).
Seuraavassa taulukossa ja kuvissa on kummankin tutkimuksen tehoa osoittavat päätetapahtumat.
Taulukko 3: Tehoa osoittavat tulokset
Tutkimus 1 | Tutkimus 2 | |||
Soolantra (N=451) | Vehikkeli (N=232) | Soolantra (N=459) | Vehikkeli (N=229) | |
Tutkijan arvio (IGA) | ||||
Henkilöiden määrä (%), jotka arvioitiin oireettomiksi tai lähes oireettomiksi viikolla 12 | 173 (38,4) | 27 (11,6) | 184 (40,1) | 43 (18,8) |
Tulehdukselliset leesiot | ||||
Tulehduksellisten leesioiden lukumäärä lähtötilanteessa (keskiarvo) | 31,0 | 30,5 | 33,3 | 32,2 |
Tulehduksellisten leesioiden lukumäärä viikolla 12 (keskiarvo) | 10,6 | 18,5 | 11,0 | 18,8 |
Tulehduksellisten leesioiden lukumäärän absoluuttisen muutoksen keskiarvo (muutos %) viikolla 12 | -20,5 (-64,9) | -12,0 (-41,6) | -22,2 (-65,7) | -13,4 (-43,4) |
Kuvat 1 ja 2: Hoidon onnistuminen (IGA-arvio) hoitoviikkojen aikana
Tutkimus 1 Tutkimus 2
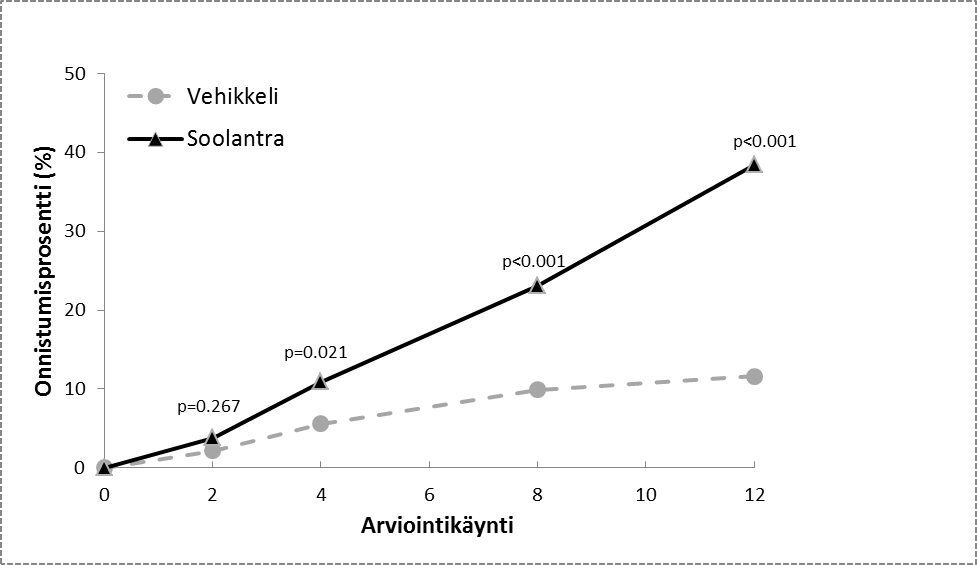
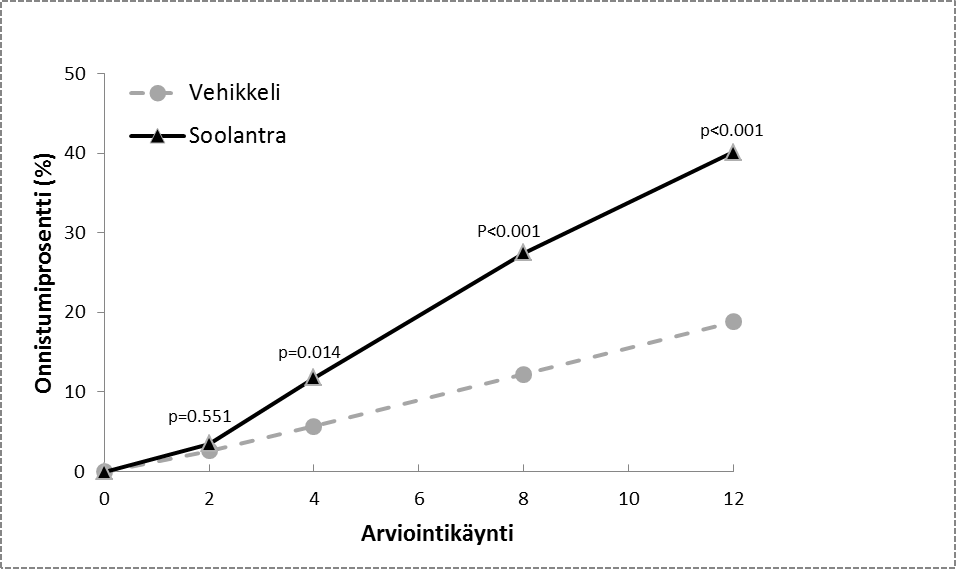
Kuvat 3 ja 4: Tulehduksellisten leesioiden lukumäärän absoluuttisen muutoksen keskiarvo hoitoviikkojen aikana
Tutkimus 1 Tutkimus 2
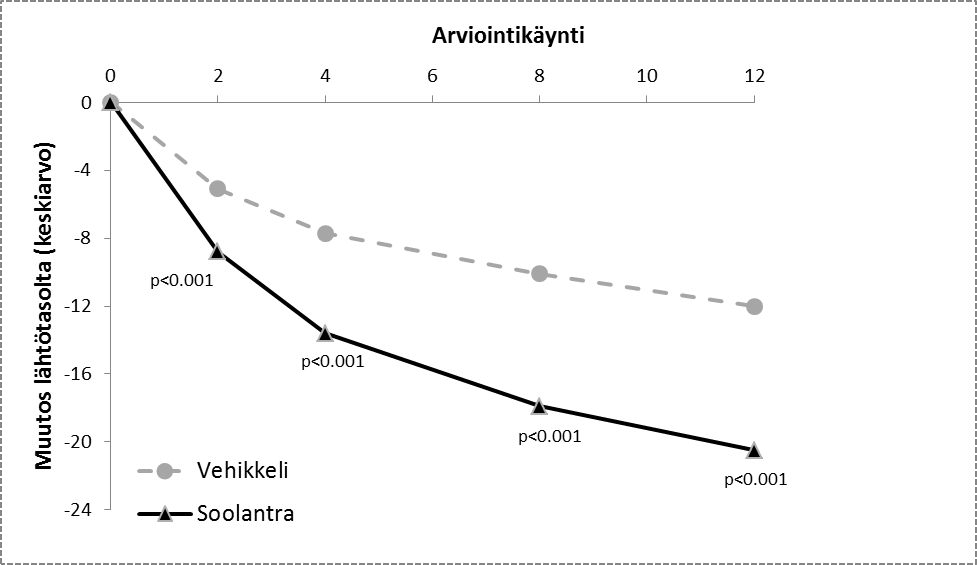
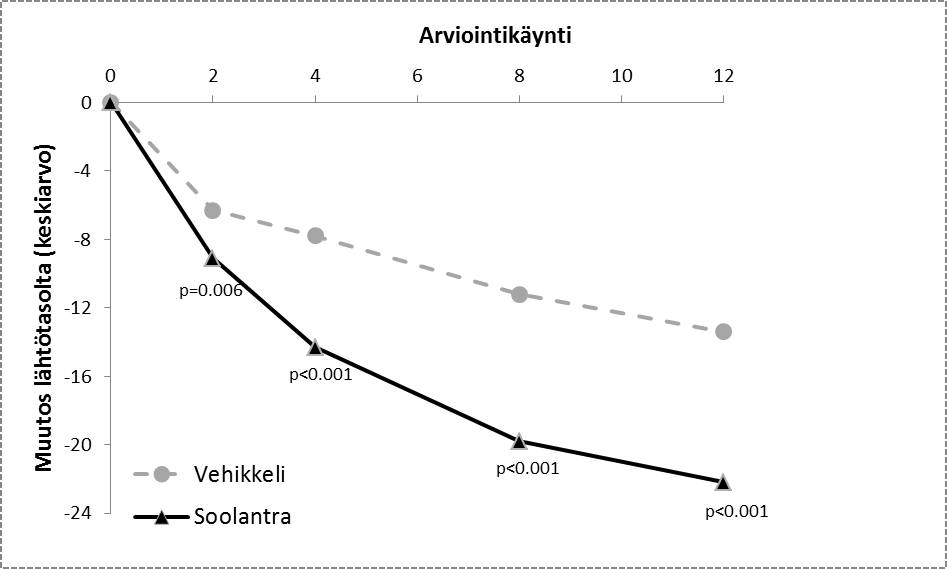
Soolantra oli tilastollisesti merkitsevästi parempi kuin vehikkelivoide yhdistetyissä tehoa mittaavissa päätetapahtumissa, ja tehon alkaminen nähtiin viikon 4 kohdalla (p < 0,05).
IGA arvioitiin kahden kliinisen tutkimuksen 40 viikon jatkotutkimuksissa ja niiden potilaiden prosentuaalinen osuus, joilla saavutettiin Soolantra-hoidon aikana IGA-pisteytys 0 tai 1, lisääntyi viikolle 52 asti. Onnistumisprosentti (IGA 0 tai 1) viikolla 52 oli 71 % tutkimuksessa 1 ja 76 % tutkimuksessa 2.
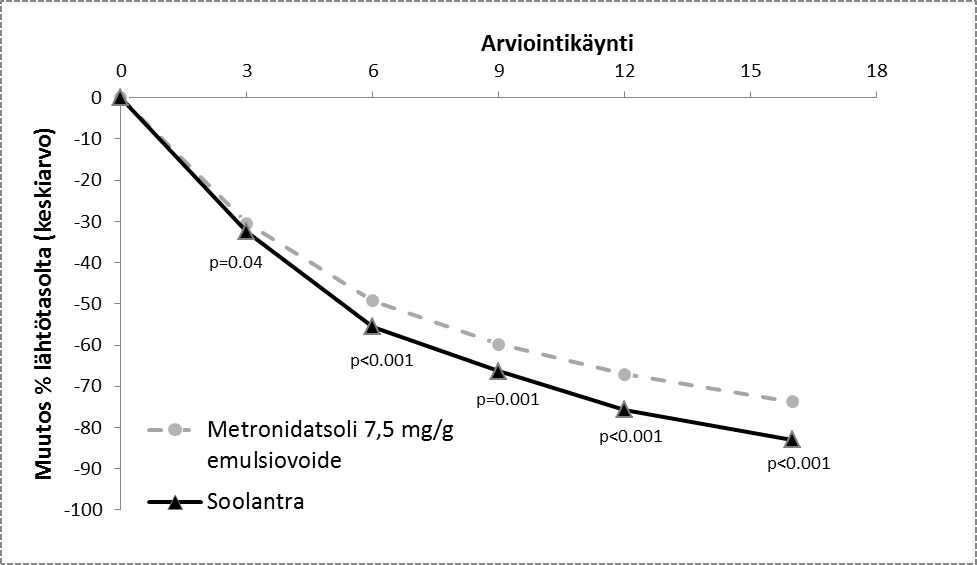
Lääkevalmisteen teho ja turvallisuus ruusufinnin tulehduksellisten leesioiden hoidossa arvioitiin myös satunnaistetussa, tutkijoiden suhteen sokeutetussa, aktiivi-kontrolloidussa kliinisessä tutkimuksessa. Tutkimuksessa oli 962 vähintään 18 vuoden ikäistä henkilöä, joita hoidettiin 16 viikon ajan joko Soolantralla kerran päivässä tai 7,5 mg/g metronidatsoli-emulsiovoiteella kaksi kertaa päivässä. Tässä tutkimuksessa 99,7 % henkilöistä oli valkoihoisia ja 65,2 % naisia; IGA-asteikolla 83,3 %:lla henkilöistä tauti arvioitiin keskivaikeaksi (IGA = 3) ja 16,7 %:lla vaikeaksi (IGA = 4) lähtötilanteessa (ks. kuva 5).
Tutkimuksen tulokset osoittivat, että Soolantra oli tilastollisesti merkitsevästi parempi kuin 7,5 mg/g metronidatsoli-emulsiovoide ensisijaisen päätetapahtuman suhteen (tulehduksellisten leesioiden lukumäärän prosentuaalisen muutoksen keskiarvo): 16 viikon hoito ivermektiinillä vähensi leesioita lähtötilanteesta 83,0 % ja metronidatsoli 73,7 % (p < 0,001). Soolantran parempaa tehoa viikolla 16 osoittivat myös IGA-asteikkoon perustuva onnistumisprosentti ja tulehduksellisten leesioiden absoluuttinen lukumäärän muutos (toissijaiset päätetapahtumat (p < 0,001)).
Kuva 5: Prosentuaalinen muutos viikkojen aikana (keskiarvo)
Noin 300 vähintään 65 vuoden ikäistä henkilöä on hoidettu lääkevalmisteella kliinisissä tutkimuksissa. Tehossa ja turvallisuudessa ei havaittu merkittäviä eroja iäkkäiden henkilöiden ja 18-65 -vuotiaiden välillä.
Turvallisuusprofiili, joka on kuvattu kohdassa Haittavaikutukset, pysyi muuttumattomana pisimmillään vuoden jatkuneessa pitkäaikaiskäytössä.
Hoito ivermektiinin ja 40 mg doksisykliiniä säädellysti vapauttavien kapselien yhdistelmällä.
ANSWER-tutkimuksessa vertailtiin Soolantran ja 40 mg doksisykliiniä säädellysti vapauttavien kapselien yhdistelmän ja ivermektiinin ja doksisykliiniä säädellysti vapauttavien kapselien sijaan annetun lumevalmisteen yhdistelmän suhteellista tehoa vaikea-asteisen ruusufinnin hoidossa. Kyseessä oli 12 viikkoa kestänyt satunnaistettu, tutkijasokkoutettu, kontrolloitu, rinnakkaisryhmillä toteutettu tutkimus. Tutkimuksessa oli mukana 273 mies- ja naistutkittavaa, jotka olivat iältään ≥ 18-vuotiaita, joiden kasvoissa oli 20–70 tulehdusvauriota (näppylöitä ja märkärakkuloita) ja joiden tutkijan arvioon perustuva IGA-pisteytys lähtötilanteessa (Baseline Investigator’s Global Assessment score) oli 4.
Ensisijainen tehon päätetapahtuma oli tulehdusvaurioiden lukumäärän prosentuaalinen muutos lähtötilanteesta viikolla 12. Ivermektiinin ja doksisykliiniä säädellysti vapauttavien kapselien yhdistelmää saaneilla havaittiin tulehdusvaurioiden merkittävästi suurempi keskimääräinen prosentuaalinen vähenemä verrattuna ivermektiinin ja lumevalmisteen yhdistelmää saaneisiin (keskiarvo ± keskihajonta: ‑80,29 ± 21,65 % vs -73,56 ± 30,52 %; p = 0,032).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Soolantra-valmisteen käytöstä kaikkien pediatristen potilasryhmien papulopustulaarisen ruusufinnin hoidossa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Ivermektiinin imeytymistä Soolantrasta arvioitiin kliinisessä tutkimuksessa, jossa hoidettiin suurimmilla mahdollisilla annoksilla vaikeaa papulopustulaarista ruusufinniä sairastavia aikuisia henkilöitä. Vakaassa tilassa (2 viikon hoidon jälkeen), suurimmat ivermektiinin pitoisuudet plasmassa (± keskihajonta) mitattiin 10 ± 8 tuntia annostelun jälkeen (Cmax: 2,1 ± 1,0 ng/ml, vaihtelu: 0,7-4,0 ng/ml) ja korkein keskiarvo (± keskihajonta) AUC0-24t oli 36 ± 16 ng.t/ml (vaihtelu: 14-75 ng.t/ml). Ivermektiinin systeemialtistuksen pitoisuudet saavuttivat tasannevaiheen kahden viikon hoidon jälkeen (vakaan tilan pitoisuus). Pitkäaikaisessa hoidossa faasi 3:n tutkimuksissa, ivermektiinin systeemialtistus oli samalla tasolla kuin kahden viikon hoidon jälkeen. Vakaassa tilassa ivermektiinin systeemialtistus (AUC0-24t: 36 ± 16 ng.t/ml) oli pienempi kuin terveillä vapaaehtoisilla todettu altistus suun kautta otetun 6 mg oraalisen kerta-annoksen jälkeen (AUC0-24t: 134 ± 66 ng.t/ml).
Jakautuminen
In vitro -tutkimus osoitti, että ivermektiinistä yli 99 % sitoutuu plasman proteiineihin, ensisijaisesti ihmisen seerumin albumiiniin. Ivermektiinin merkitsevää sitoutumista punasoluihin ei havaittu.
Biotransformaatio
Ivermektiini metaboloituu ensisijaisesti CYP3A4:n kautta, mikä on osoitettu ihmisen maksan mikrosomeilla ja rekombinantti CYP450-entsyymeillä tehdyissä in vitro -tutkimuksissa.
In vitro -tutkimukset ovat osoittaneet, että ivermektiini ei estä CYP450-isoentsyymejä 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4, 4A11 tai 2E1. Ivermektiini ei indusoi CYP450-entsyymin ilmentymistä (1A2, 2B6, 2C9 tai 3A4) viljellyissä ihmisen maksasoluissa.
Ivermektiinin kaksi päämetaboliittia identifioitiin kliinisessä farmakokineettisessä tutkimuksessa maksimaalisella altistuksella ja arvioitiin faasi 2:n kliinisissä tutkimuksissa (3’’-O-demetyyli-ivermektiini ja 4a-hydroksi-ivermektiini). Kanta-aineen tavoin metaboliittien vakaa tila saavutettiin 2 viikon hoidon aikana, eikä kumulaatiota tapahtunut 12 viikon aikana. Edelleen, metaboliittien systeeminen altistus (arvioituna Cmax- ja AUC-arvojen perusteella) vakaassa tilassa oli huomattavasti pienempi kuin mitä nähdään suun kautta otetun ivermektiinin jälkeen.
Eliminaatio
Terminaalinen puoliintumisaika oli keskimäärin 6 päivää (keskiarvo: 145 tuntia, vaihtelu 92-238 tuntia) potilailla, joita hoidettiin kerran päivässä iholle levitetyllä lääkevalmisteella 28 päivän ajan, maksimaalista altistusta mittaavassa kliinisessä farmakokineettisessä tutkimuksessa. Eliminaatio riippuu imeytymisestä Soolantran paikallisen käytön jälkeen. Ivermektiinin farmakokinetiikkaa ei ole tutkittu potilailla, joilla on munuaisten tai maksan vajaatoiminta.
Prekliiniset tiedot turvallisuudesta
Enintään 9 kuukautta kestäneissä toistuvan annon tutkimuksissa, joissa 10 mg/g ivermektiini-emulsiovoidetta annosteltiin minisikojen iholle, toksisia vaikutuksia tai paikallisia toksisia vaikutuksia ei havaittu systeemisen altistuksen pitoisuuksilla, jotka vastasivat kliinistä altistusta.
Ivermektiini ei ole genotoksinen useissa in vitro- ja in vivo -testeissä. Kahden vuoden karsinogeenisuustutkimuksissa, joissa hiiren iholle annosteltiin ivermektiini 10 mg/g emulsiovoidetta, ei havaittu kasvainten lisääntymistä.
Lisääntymistoksisuutta selvittävissä tutkimuksissa suun kautta annetulla ivermektiinillä ei havaittu teratogeenisiä vaikutuksia rotilla (kitalakihalkio) ja kaneilla (jalkojen fleksio) suurilla annoksilla (altistuksen marginaali NOAEL-tasoon vähintään 70-kertainen verrattuna kliiniseen altistukseen).
Suun kautta annetun ivermektiinin neonataalitoksisuus rotilla ei ollut suhteessa in utero -altistukseen vaan postnataaliseen, äidinmaidon kautta saatuun altistukseen, joka tuotti korkeita ivermektiinin pitoisuuksia jälkeläisten aivoihin ja plasmaan.
Ivermektiini-emulsiovoide 10 mg/g on aiheuttanut marsuilla ihoärsytystä, herkistymistä ja valoherkistymistä, mutta se ei ole valotoksinen.
Ympäristöön kohdistuvien riskien arviointi
Ivermektiini on hyvin toksinen selkärangattomille, ja vedessä, sedimentissä ja maalla oleviin eliöihin kohdistuva riski on tunnistettu. On pyrittävä estämään ympäristön, erityisesti vesistön, kontaminaatio.
Farmaseuttiset tiedot
Apuaineet
Glyseroli, isopropyylipalmitaatti, karbomeeri, dimetikoni, dinatriumedetaatti, sitruunahappomonohydraatti, setyylialkoholi, stearyylialkoholi, makrogolisetostearyylieetteri, sorbitaanistearaatti, metyyliparahydroksibentsoaatti (E218), propyyliparahydroksibentsoaatti (E216), fenoksietanoli, propyleeniglykoli, oleyylialkoholi, natriumhydroksidi ja puhdistettu vesi.
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
2 vuotta
Ensimmäisen avaamisen jälkeen: käytä 6 kuukauden kuluessa.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SOOLANTRA emulsiovoide
10 mg/g (L:ei) 30 g (34,20 €), 60 g (56,84 €)
PF-selosteen tieto
Polyeteeni (PE)/aluminiini (Al)/Polyetyleeni (PE) laminoitu muovinen valkoinen tuubi, jossa on:
- valkoinen high density-polyetyleeni (HDPE) kärki ja lapsiturvallinen polypropeeni (PP) korkki 15 g, 30 g, 45 g ja 60 g tuubissa
- Polypropeeni (PP) valkoinen kärki 2 g tuubissa (ei lapsiturvallista korkkia)
Pakkauskoot: 2 g, 15 g, 30 g, 45 g tai 60 g tuubi.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai kellertävä hydrofiilinen emulsiovoide.
Käyttö- ja käsittelyohjeet
Kontaminaatiota täytyy pyrkiä estämään tai vähentämään, erityisesti vesistöihin joutuvan kontaminaation välttämiseksi.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SOOLANTRA emulsiovoide
10 mg/g 30 g, 60 g
- Ei korvausta.
ATC-koodi
D11AX22
Valmisteyhteenvedon muuttamispäivämäärä
24.06.2024
Yhteystiedot
Seminariegatan 21
SE-752 28 Uppsala
Sverige
+46 18 444 0330
www.galdermanordic.com
nordic@galderma.com