
ORKAMBI tabletti, kalvopäällysteinen 100/125 mg, 200/125 mg
Vaikuttavat aineet ja niiden määrät
Orkambi 100 mg/125 mg tabletti, kalvopäällysteinen
Yksi kalvopäällysteinen tabletti sisältää 100 mg lumakaftoria ja 125 mg ivakaftoria.
Orkambi 200 mg/125 mg tabletti, kalvopäällysteinen
Yksi kalvopäällysteinen tabletti sisältää 200 mg lumakaftoria ja 125 mg ivakaftoria.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti)
Kliiniset tiedot
Käyttöaiheet
Orkambi-tabletit on tarkoitettu kystisen fibroosin (CF) hoitoon vähintään 6-vuotiaille potilaille, jotka ovat homotsygoottisia CFTR (cystic fibrosis transmembrane conductance regulator) -geenin F508del-mutaation suhteen (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Ehto
Valmistetta saavat määrätä vain kystisen fibroosin hoitoon perehtyneet lääkärit.
Annostus ja antotapa
Orkambi-valmistetta saavat määrätä vain kystisen fibroosin hoitoon perehtyneet lääkärit. Jos potilaan genotyyppi ei ole tiedossa, F508del-mutaation olemassaolo CFTR-geenin molemmissa alleeleissa on vahvistettava käyttämällä täsmällistä, validoitua genotyypitysmenetelmää.
Annostus
Taulukko 1: Annossuositukset vähintään 6-vuotiaille potilaille
Ikä | Vahvuus | Annos (12 tunnin välein) | |
Aamu | Ilta | ||
6–<12 vuotta | lumakaftori 100 mg / ivakaftori 125 mg | 2 tablettia | 2 tablettia |
Vähintään 12 vuotta | lumakaftori 200 mg / ivakaftori 125 mg | 2 tablettia | 2 tablettia |
Potilaat voivat aloittaa hoidon minä tahansa viikonpäivänä.
Tämä lääkevalmiste tulee ottaa rasvaa sisältävän ruoan kanssa. Rasvaa sisältävä ateria tai välipala tulee nauttia juuri ennen annoksen ottamista tai heti sen jälkeen (ks. kohta Farmakokinetiikka).
Annoksen unohtuminen
Jos annoksen unohtumisesta on kulunut alle 6 tuntia, annos tulee ottaa rasvaa sisältävän ruoan kanssa. Jos unohtumisesta on kulunut yli 6 tuntia, potilasta tulee neuvoa odottamaan seuraavaan aikataulun mukaiseen annokseen. Unohtunutta annosta ei pidä korvata ottamalla kaksinkertaista annosta.
CYP3A:n estäjien samanaikainen käyttö
Annosta ei tarvitse muuttaa, kun hoito CYP3A:n estäjillä aloitetaan potilaille, jotka parhaillaan ottavat Orkambi-valmistetta. Kun hoito aloitetaan potilaille, jotka parhaillaan ottavat voimakkaita CYP3A:n estäjiä, lumakaftorin induktiovaikutus vakaassa tilassa on kuitenkin huomioitava, ja annosta on pienennettävä yhteen tablettiin vuorokaudessa ensimmäisen hoitoviikon ajaksi. Tämän jälkeen hoitoa jatketaan suositellulla vuorokausiannoksella (ks. taulukko 2).
Taulukko 2: Hoidon aloitus potilaille, jotka ottavat voimakkaita CYP3A:n estäjiä
Ikä | Vahvuus | Hoitoviikko 1 | Hoitoviikko 2 ja myöhemmät hoitoviikot |
6–<12 vuotta | lumakaftori 100 mg / ivakaftori 125 mg | 1 tabletti vuorokaudessa | Päivästä 8 alkaen annoksen on oltava suositeltu vuorokausiannos |
Vähintään 12 vuotta | lumakaftori 200 mg / ivakaftori 125 mg |
Jos hoito keskeytetään yli viikon ajaksi voimakkaiden CYP3A:n estäjien ottamisen aikana, annosta on pienennettävä yhteen tablettiin vuorokaudessa ensimmäisen viikon ajaksi, kun hoito aloitetaan uudelleen (ks. taulukko 2). Tämän jälkeen hoitoa jatketaan suositellulla vuorokausiannoksella (ks. kohta Yhteisvaikutukset).
Erityisryhmät
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastaville potilaille. Vaikeaa munuaisten vajaatoimintaa (kreatiniinin puhdistuma 30 ml/min tai vähemmän) tai loppuvaiheen munuaissairautta sairastavien potilaiden hoidossa tulee noudattaa varovaisuutta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa lievää maksan vajaatoimintaa sairastaville potilaille (Child-Pugh-luokka A). Keskivaikeaa maksan vajaatoimintaa sairastaville potilaille (Child-Pugh-luokka B) suositellaan annoksen pienentämistä.
Tämän lääkevalmisteen käytöstä vaikeaa maksan vajaatoimintaa sairastavien potilaiden (Child-Pugh-luokka C) hoidossa ei ole kokemusta, mutta altistuksen odotetaan olevan suurempi kuin keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla. Vaikeaa maksan vajaatoimintaa sairastaville potilaille Orkambi-valmistetta on näin ollen käytettävä varoen, pienennetyllä annoksella, hoidon riskien ja hyötyjen punnitsemisen jälkeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset ja Farmakokinetiikka).
Annosmuutokset keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastaville potilaille, ks. taulukko 3.
Taulukko 3: Suositellut annosmuutokset keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastaville potilaille
Ikä | Vahvuus | Kokonaisannos vuorokaudessa | |||
Keskivaikea (Child-Pugh-luokka B) | Vaikea (Child-Pugh-luokka C) | ||||
Aamu | Ilta | Aamu | Ilta | ||
6–<12 vuotta | lumakaftori 100 mg / ivakaftori 125 mg | 2 tablettia | 1 tabletti | 1 tabletti tai harvemmin* | 1 tabletti tai harvemmin* |
Vähintään 12 vuotta | lumakaftori 200 mg / ivakaftori 125 mg | ||||
* Antoväliä on muutettava kliinisen vasteen ja siedettävyyden mukaan; antoa voidaan harventaa sekä aamu- että ilta-annokselle.
Pediatriset potilaat
Orkambi-valmisteen turvallisuutta ja tehoa alle 1 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Suun kautta.
Potilaita tulee neuvoa nielemään tabletit kokonaisina. Tabletteja ei saa pureskella, halkaista tai liuottaa.
Vasta-aiheet
Yliherkkyys vaikuttaville aineille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Kystistä fibroosia sairastavat potilaat, jotka ovat heterotsygoottisia CFTR-geeninF508del-mutaation suhteen
Lumakaftori/ivakaftori ei tehoa kystistä fibroosia sairastavilla potilailla, joilla on F508del-mutaatio yhdessä alleelissa ja toisessa alleelissa mutaatio, jonka ennustetaan johtavan CFTR:n tuotannon puutteeseen tai joka ei reagoi ivakaftoriin in vitro (ks. kohta Farmakodynamiikka).
Kystistä fibroosia sairastavat potilaat, joilla on CFTR-geenissä(luokan III) gating-mutaatio
Lumakaftoria/ivakaftoria ei ole tutkittu kystistä fibroosia sairastavilla potilailla, joilla on (luokan III) gating-mutaatio CFTR-geenin yhdessä alleelissa, riippumatta siitä, onko toisessa alleelissa F508del-mutaatiota vai ei. Koska altistus ivakaftorille vähenee huomattavasti yhteisannossa lumakaftorin kanssa, lumakaftoria/ivakaftoria ei pidä käyttää näille potilaille.
Hengityselimiin kohdistuvat haittavaikutukset
Hengityselinhaittavaikutukset (esim. epämiellyttävät tuntemukset rinnassa, hengenahdistus, bronkospasmi ja poikkeava hengitys) olivat tavallisempia lumakaftori/ivakaftori-hoidon alussa. Vakavia hengityselintapahtumia havaittiin useammin potilailla, joiden uloshengityksen sekuntitilavuus oli <40 % ennustetusta (ppFEV1-arvo <40), ja ne saattavat johtaa hoidon lopettamiseen. Kliininen kokemus potilailla, joiden ppFEV1-arvo <40, on niukkaa. Näille potilaille suositellaan lisäseurantaa hoidon alussa (ks. kohta Haittavaikutukset). Joillakin potilailla on myös havaittu ohimenevää FEV1-arvon laskua lumakaftorin/ivakaftorin käytön aloituksen jälkeen. Lumakaftorin/ivakaftorin käytön aloituksesta potilaille, joiden keuhko-oireet ovat pahentuneet, ei ole kokemusta, eikä käytön aloitusta suositella potilaille, joiden keuhko-oireet ovat pahentuneet.
Vaikutus verenpaineeseen
Verenpaineen kohoamista on havaittu joillakin lumakaftori-/ivakaftorihoitoa saaneilla potilailla. Kaikkien potilaiden verenpainetta tulee seurata määräajoin hoidon aikana (ks. kohta Haittavaikutukset).
Transaminaasiarvojen nousu ja maksavaurio
Kystistä fibroosia sairastavilla potilailla saattaa esiintyä maksan toiminnan poikkeavuuksia mukaan lukien pitkälle edennyt maksasairaus. Potilailla, joilla on pitkälle edennyt maksasairaus, on raportoitu maksan toiminnan heikkenemistä. Maksan toiminnan dekompensaatiota, mukaan lukien kuolemaan johtanutta maksan vajaatoimintaa, on raportoitu lumakaftoria/ivakaftoria saavilla kystistä fibroosia sairastavilla potilailla, joilla on ennestään maksakirroosi ja siihen liittyvä koholla oleva porttilaskimopaine. Elinsiirtoon johtavan maksan vajaatoiminnan tapauksia on raportoitu ensimmäisen 6 hoitokuukauden aikana potilailla, joista osalla on ollut olemassa oleva muilla CFTR-modulaattoreilla hoidettava pitkälle edennyt maksasairaus ja osalla ei ole ollut sitä.
Lumakaftoria/ivakaftoria on käytettävä varoen potilaille, joilla on pitkälle edennyt maksasairaus, ja vain silloin, kun odotetut hyödyt ovat riskejä suurempia. Jos lumakaftoria/ivakaftoria käytetään tällaisille potilaille, potilaita on tarkkailtava huolellisesti hoidon alussa ja annosta on pienennettävä (ks. kohdat Annostus ja antotapa, Haittavaikutukset ja Farmakokinetiikka).
Lumakaftoria/ivakaftoria saavilla kystistä fibroosia sairastavilla potilailla on raportoitu yleisesti transaminaasiarvojen kohoamista. Joissakin tapauksissa kohoaminen on yhdistetty samanaikaiseen seerumin kokonaisbilirubiiniarvon kohoamiseen. Transaminaasiarvojen kohoamista on todettu useammin pediatrisilla kuin aikuisilla potilailla (ks. kohta Haittavaikutukset).
Koska yhteyttä maksavaurioon ei voida poissulkea, on suositeltavaa mitata maksan toimintakokeiden (ALAT, ASAT ja bilirubiini) arvot ennen lumakaftori-/ivakaftorihoidon aloittamista, 3 kuukauden välein ensimmäisen vuoden aikana sekä kerran vuodessa sen jälkeen. Potilaille, joiden ALAT-, ASAT- tai bilirubiiniarvot ovat aiemmin olleet koholla, tulee harkita tiheämpää seurantaa.
Hoito on keskeytettävä ja seerumin transaminaasit ja kokonaisbilirubiini on mitattava välittömästi, jos potilaalle ilmaantuu maksavaurion kliinisiä merkkejä tai oireita. Jos ALAT tai ASAT on > 5 × viitealueen yläraja tai ALAT tai ASAT on > 3 × viitealueen yläraja sekä kokonaisbilirubiini > 2 × viitealueen yläraja ja/tai kliinisen keltaisuuden yhteydessä, hoito on keskeytettävä ja laboratorioarvoja on seurattava tarkasti, kunnes ne palautuvat normaaleiksi. Potilas on tutkittava huolellisesti mahdollisten syiden selvittämiseksi ja potilasta on seurattava tiiviisti kliinisen etenemisen varalta. Kun arvot ovat palautuneet normaalille tasolle, hoidon jatkamisen hyödyt ja riskit on arvioitava (ks. kohdat Annostus ja antotapa, Haittavaikutukset ja Farmakokinetiikka). Potilaita, joiden hoitoa jatketaan sen keskeyttämisen jälkeen, on seurattava huolellisesti.
Masennus
Lumakaftori-ivakaftorihoitoa saaneilla potilailla on raportoitu masennusta (mukaan lukien itsemurha-ajatuksia ja itsemurhayrityksiä), joka on yleensä ilmaantunut kolmen kuukauden sisällä hoidon aloittamisesta ja potilailla, joilla on aiemmin esiintynyt psyykkisiä häiriöitä (ks. kohdat Haittavaikutukset). Joissakin tapauksissa oireiden on raportoitu lievittyneen annoksen pienentämisen tai hoidon lopettamisen jälkeen. Potilaita (ja hoitajia) on neuvottava kiinnittämään huomiota masentuneisuuteen, itsemurha-ajatuksiin, epätavallisiin muutoksiin käyttäytymisessä, ahdistuneisuuteen tai unettomuuteen sekä kääntymään lääkärin puoleen välittömästi, jos näitä oireita esiintyy.
Yhteisvaikutukset lääkevalmisteiden kanssa
CYP3A:n substraatit
Lumakaftori on voimakas CYP3A:n indusoija. Samanaikaista antoa herkkien tai terapeuttiselta indeksiltään kapeiden CYP3A:n substraattien kanssa ei suositella (ks. kohta Yhteisvaikutukset).
Hormonaaliset suun kautta annettavat, pistettävät, ihon läpi annettavat ja implantoitavat ehkäisyvalmisteet eivät ole luotettavia ehkäisymenetelmiä silloin, kun niitä käytetään samanaikaisesti Orkambi-valmisteen kanssa (ks. kohta Yhteisvaikutukset).
Voimakkaat CYP3A:n indusoijat
Ivakaftori on CYP3A4:n ja CYP3A5:n substraatti. Samanaikaista antoa voimakkaiden CYP3A:n indusoijien (esim. rifampisiini, mäkikuisma [Hypericum perforatum]) kanssa ei siksi suositella (ks. kohta Yhteisvaikutukset).
Munuaisten vajaatoiminta
Lumakaftoria/ivakaftoria tulee käyttää varoen vaikeaa munuaisten vajaatoimintaa tai loppuvaiheen munuaissairautta sairastaville potilaille (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Kaihit
Lumakaftoria/ivakaftoria ja ivakaftorimonoterapiaa saaneilla pediatrisilla potilailla on raportoitu ei-synnynnäistä silmän mykiön samentumista ilman näköön kohdistuvia vaikutuksia. Vaikka osassa tapauksista oli läsnä muita riskitekijöitä (kuten kortikosteroidien käyttö ja säteilylle altistuminen), ivakaftoriin liittyvää mahdollista riskiä ei voida poissulkea (ks. kohta Prekliiniset tiedot turvallisuudesta). Pediatrisille potilaille, joille aloitetaan lumakaftori-/ivakaftorihoito, on suositeltavaa tehdä silmätutkimus ennen hoidon aloittamista ja sen aikana.
Potilaat, joille on tehty elinsiirto
Lumakaftoria/ivakaftoria ei ole tutkittu kystistä fibroosia sairastavilla potilailla, joille on tehty elinsiirto. Käyttöä elinsiirtopotilaille ei siksi suositella. Yhteisvaikutukset immunosuppressanttien kanssa, ks. kohta Yhteisvaikutukset.
Natriumsisältö
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Altistuksen ja käyttöaiheen mukaisten annosten perusteella yhteisvaikutusprofiilin katsotaan olevan sama kaikille vahvuuksille ja lääkemuodoille.
Monoterapiana annettuna lumakaftori on voimakas CYP3A:n indusoija ja ivakaftori heikko CYP3A:n estäjä. Samanaikaisesti annetut muut lääkevalmisteet saattavat vaikuttaa lumakaftoriin/ivakaftoriin, ja lumakaftori/ivakaftori saattaa niin ikään vaikuttaa muihin lääkevalmisteisiin.
Muiden lääkevalmisteiden mahdolliset vaikutukset lumakaftoriin/ivakaftoriin
CYP3A:n estäjät
Lumakaftorin/ivakaftorin samanaikainen anto voimakkaan CYP3A:n estäjän itrakonatsolin kanssa ei vaikuttanut altistukseen lumakaftorille mutta lisäsi altistusta ivakaftorille 4,3-kertaisesti. Lumakaftorin CYP3A:ta (vakaassa tilassa) indusoivan vaikutuksen vuoksi nettoaltistuksen ivakaftorille ei samanaikaisessa annossa CYP3A:n estäjän kanssa odoteta ylittävän altistusta, joka saadaan, kun ivakaftoria annetaan ilman lumakaftoria ivakaftorimonoterapialle hyväksyttynä annoksena, 150 mg 12 tunnin välein.
Annosta ei tarvitse muuttaa, kun hoito CYP3A:n estäjillä aloitetaan potilaille, jotka parhaillaan ottavat lumakaftoria/ivakaftoria. Kun lumakaftori-/ivakaftorihoito aloitetaan potilailla, jotka parhaillaan ottavat voimakkaita CYP3A:n estäjiä, annosta on kuitenkin säädettävä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Annoksen muuttamista ei suositella samanaikaisessa käytössä kohtalaisten tai heikkojen CYP3A:n estäjien kanssa.
CYP3A:n indusoijat
Lumakaftorin/ivakaftorin samanaikainen anto voimakkaan CYP3A:n indusoijan rifampisiinin kanssa vaikutti minimaalisesti altistukseen lumakaftorille mutta vähensi altistusta ivakaftorille (AUC) 57 %. Lumakaftorin/ivakaftorin samanaikaista antoa voimakkaiden CYP3A:n indusoijien kanssa ei siksi suositella (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Annoksen muuttamista ei suositella samanaikaisessa käytössä kohtalaisten tai heikkojen CYP3A:n indusoijien kanssa.
Lumakaftorin/ivakaftorin mahdolliset vaikutukset muihin lääkevalmisteisiin
CYP3A:n substraatit
Lumakaftori on voimakas CYP3A:n indusoija. Ivakaftori on monoterapiana annettuna heikko CYP3A:n estäjä. Lumakaftori-/ivakaftorihoidon nettovaikutuksen odotetaan olevan voimakas CYP3A:n induktio. Lumakaftorin/ivakaftorin ja CYP3A:n substraattien samanaikainen käyttö saattaa siksi vähentää näille substraateille altistusta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
P‑gp:n substraatit
In vitro -tutkimusten mukaan lumakaftorilla on kyky sekä estää että indusoida P-gp:tä. Tämän lisäksi ivakaftorimonoterapiaa koskeva kliininen tutkimus osoitti ivakaftorin olevan heikko P-gp:n estäjä. Näin ollen lumakaftorin/ivakaftorin ja P-gp:n substraattien (esim. digoksiinin) samanaikainen käyttö saattaa muuttaa näille substraateille altistusta.
CYP2B6:n ja CYP2C:n substraatit
Yhteisvaikutusta CYP2B6:n ja CYP2C:n substraattien kanssa ei ole tutkittu in vivo. In vitro ‑tutkimusten mukaan lumakaftori saattaa olla CYP2B6:n, CYP2C8:n, CYP2C9:n ja CYP2C19:n indusoija. Myös CYP2C8:n ja CYP2C9:n estoa on kuitenkin havaittu in vitro. Lisäksi in vitro -tutkimukset viittaavat siihen, että ivakaftori voi olla CYP2C9:n estäjä. Näin ollen lumakaftorin/ivakaftorin samanaikainen käyttö saattaa muuttaa (eli joko lisätä tai vähentää) CYP2C8:n ja CYP2C9:n substraateille altistusta, vähentää CYP2C19:n substraateille altistusta ja vähentää huomattavasti CYP2B6:n substraateille altistusta.
Lumakaftorin/ivakaftorin mahdolliset yhteisvaikutukset kuljettajaproteiinien kanssa
In vitro -kokeet osoittavat lumakaftorin olevan rintasyöpäresistenssiproteiinin (BCRP) substraatti. Orkambi-valmisteen samanaikainen anto BCRP:tä estävien lääkevalmisteiden kanssa saattaa suurentaa lumakaftorin pitoisuutta plasmassa. Lumakaftori estää orgaanisten anionien kuljettajaproteiineja 1 ja 3 (OAT1, OAT3). Lumakaftori ja ivakaftori ovat BCRP:n estäjiä. Orkambi-valmisteen samanaikainen anto sellaisten lääkevalmisteiden kanssa, jotka ovat OAT1:n/OAT3:n tai BCRP:n substraatteja, saattaa suurentaa näiden lääkevalmisteiden pitoisuutta plasmassa. Lumakaftori ja ivakaftori eivät estä OATP1B1:tä, OATP1B3:a eivätkä orgaanisten kationien kuljettajaproteiineja 1 ja 2 (OCT1, OCT2). Ivakaftori ei ole OAT1:n eikä OAT3:n estäjä.
Varmistetut ja muut mahdollisesti merkittävät yhteisvaikutukset
Taulukossa 4 on esitetty lumakaftorin/ivakaftorin varmistetut tai ennustetut vaikutukset muihin lääkevalmisteisiin tai muiden lääkevalmisteiden vaikutukset lumakaftoriin/ivakaftoriin.
Taulukon 4 tiedot ovat pääasiassa peräisin in vitro -tutkimuksista. Taulukon 4 ”Kliininen suositus” ‑sarakkeen suositukset perustuvat yhteisvaikutustutkimuksiin, kliiniseen merkitykseen tai ennustettaviin yhteisvaikutuksiin eliminaatioreittien vuoksi. Kliinisesti merkittävimmät yhteisvaikutukset on mainittu ensin.
Taulukko 4:Varmistetut ja muut mahdollisesti merkittävät yhteisvaikutukset sekä annossuositukset lumakaftorin/ivakaftorin käytölle muiden lääkevalmisteiden kanssa
Samanaikaisesti käytettävän lääkevalmisteen luokka: vaikuttavan aineen nimi | Vaikutus | Kliininen suositus |
| Kliinisesti merkittävimmät samanaikaisesti käytettävät lääkevalmisteet | ||
| Allergialääkkeet: | ||
| montelukasti | ↔ LUM, IVA | |
↓ montelukasti LUM:in aikaansaaman CYP3A:n/2C8:n/2C9:n induktion vuoksi | Montelukastiannoksen muuttamista ei suositella. Asianmukainen kliininen tarkkailu on suositeltavaa samanaikaisessa annossa lumakaftorin/ivakaftorin kanssa. Lumakaftori/ivakaftori saattaa vähentää montelukastille altistusta, jolloin montelukastin teho saattaa heikentyä. | |
| feksofenadiini | ↔ LUM, IVA | |
↑ tai ↓ feksofenadiini P-gp:n mahdollisen induktion tai eston vuoksi | Feksofenadiiniannoksen muuttaminen voi olla tarpeen halutun kliinisen vaikutuksen saavuttamiseksi. Lumakaftori/ivakaftori saattaa muuttaa feksofenadiinille altistusta. | |
| Antibiootit: | ||
| klaritromysiini, telitromysiini | ↔ LUM ↑ IVA Klaritromysiinin ja telitromysiinin aikaansaaman CYP3A:n eston vuoksi | Lumakaftori-/ivakaftoriannoksen muuttamista ei suositella, kun klaritromysiini- tai telitromysiinihoito aloitetaan potilaille, jotka parhaillaan ottavat lumakaftoria/ivakaftoria. |
↓ klaritromysiini, telitromysiini LUM:in aikaansaaman CYP3A:n induktion vuoksi | Lumakaftori-/ivakaftoriannos tulee pienentää yhteen tablettiin vuorokaudessa ensimmäisen hoitoviikon ajaksi, kun lumakaftori-/ivakaftorihoito aloitetaan potilailla, jotka parhaillaan ottavat klaritromysiiniä tai telitromysiiniä. | |
| Vaihtoehtoa näille antibiooteille, kuten atsitromysiiniä, tulee harkita. Lumakaftori/ivakaftori saattaa vähentää klaritromysiinille ja telitromysiinille altistusta, jolloin klaritromysiinin ja telitromysiinin teho saattaa heikentyä. | ||
| erytromysiini | ↔ LUM ↑ IVA Erytromysiinin aikaansaaman CYP3A:n eston vuoksi | Lumakaftori-/ivakaftoriannoksen muuttamista ei suositella samanaikaisessa annossa erytromysiinin kanssa. |
↓ erytromysiini LUM:in aikaansaaman CYP3A:n induktion vuoksi | Vaihtoehtoa erytromysiinille, kuten atsitromysiiniä, tulee harkita. Lumakaftori/ivakaftori saattaa vähentää erytromysiinille altistusta, jolloin erytromysiinin teho saattaa heikentyä. | |
| Antikonvulsantit: | ||
| karbamatsepiini, fenobarbitaali, fenytoiini | ↔ LUM ↓ IVA Näiden antikonvulsanttien aikaansaaman CYP3A:n induktion vuoksi | |
↓ karbamatsepiini, fenobarbitaali, fenytoiini LUM:in aikaansaaman CYP3A:n induktion vuoksi | Lumakaftorin/ivakaftorin samanaikaista käyttöä näiden antikonvulsanttien kanssa ei suositella. Altistus ivakaftorille ja antikonvulsantille saattaa vähentyä huomattavasti, jolloin kummankin vaikuttavan aineen teho saattaa heikentyä. | |
| Sienilääkkeet: | ||
| itrakonatsoli*, ketokonatsoli, posakonatsoli, vorikonatsoli | ↔ LUM ↑ IVA Näiden sienilääkkeiden aikaansaaman CYP3A:n eston vuoksi | Lumakaftori-/ivakaftoriannoksen muuttamista ei suositella, kun hoito näillä sienilääkkeillä aloitetaan potilaille, jotka parhaillaan ottavat lumakaftoria/ivakaftoria. |
↓ itrakonatsoli, ketokonatsoli, vorikonatsoli LUM:in aikaansaaman CYP3A:n induktion vuoksi | Lumakaftori-/ivakaftoriannos tulee pienentää yhteen tablettiin vuorokaudessa ensimmäisen hoitoviikon ajaksi, kun lumakaftori-/ivakaftorihoito aloitetaan potilaille, jotka parhaillaan ottavat näitä sienilääkkeitä. | |
↓ posakonatsoli LUM:in aikaansaaman UGT:n induktion vuoksi | Lumakaftorin/ivakaftorin samanaikaista käyttöä näiden sienilääkkeiden kanssa ei suositella. Jos samanaikainen käyttö on välttämätöntä, potilaita tulee tarkkailla lääkityksen läpäisevien sieni-infektioiden varalta. Lumakaftori/ivakaftori saattaa vähentää näille sienilääkkeille altistusta, jolloin sienilääkkeiden teho saattaa heikentyä. | |
| flukonatsoli | ↔ LUM ↑ IVA Flukonatsolin aikaansaaman CYP3A:n eston vuoksi | Lumakaftori-/ivakaftoriannoksen muuttamista ei suositella samanaikaisessa annossa flukonatsolin kanssa. |
↓ flukonatsoli LUM:in aikaansaaman induktion vuoksi. Flukonatsoli poistuu pääasiassa erittymällä munuaisten kautta muuttumattomana lääkeaineena. Flukonatsolille altistuksen on kuitenkin havaittu lievästi vähenevän voimakkaiden indusoijien käytön yhteydessä. | Flukonatsoliannoksen suurentaminen saattaa olla tarpeen, jotta haluttu kliininen vaikutus saavutetaan. Lumakaftori/ivakaftori saattaa vähentää flukonatsolille altistusta, jolloin flukonatsolin teho saattaa heikentyä. | |
| Tulehduskipulääkkeet: | ||
| ibuprofeeni | ↔ LUM, IVA | |
↓ ibuprofeeni LUM:in aikaansaaman CYP3A:n/2C8:n/2C9:n induktion vuoksi | Ibuprofeeniannoksen suurentaminen saattaa olla tarpeen, jotta haluttu kliininen vaikutus saavutetaan. Lumakaftori/ivakaftori saattaa vähentää ibuprofeenille altistusta, jolloin ibuprofeenin teho saattaa heikentyä. | |
| Mykobakteerilääkkeet: | ||
| rifabutiini, rifampisiini*, rifapentiini | ↔ LUM ↓ IVA Mykobakteerilääkkeiden aikaansaaman CYP3A:n induktion vuoksi | |
↓ rifabutiini LUM:in aikaansaaman CYP3A:n induktion vuoksi | Lumakaftorin/ivakaftorin samanaikaista käyttöä näiden mykobakteerilääkkeiden kanssa ei suositella. Samanaikaisessa käytössä altistus ivakaftorille vähenee, jolloin lumakaftorin/ivakaftorin teho saattaa heikentyä. Rifabutiiniannoksen suurentaminen saattaa olla tarpeen, jotta haluttu kliininen vaikutus saavutetaan. Lumakaftori/ivakaftori saattaa vähentää rifabutiinille altistusta, jolloin rifabutiinin teho saattaa heikentyä. | |
| ↔ rifampisiini, rifapentiini | ||
| Bentsodiatsepiinit: | ||
| midatsolaami, triatsolaami | ↔ LUM, IVA | |
↓ midatsolaami, triatsolaami LUM:in aikaansaaman CYP3A:n induktion vuoksi | Lumakaftorin/ivakaftorin samanaikaista käyttöä näiden bentsodiatsepiinien kanssa ei suositella. Lumakaftori/ivakaftori vähentää midatsolaamille ja triatsolaamille altistusta, mikä heikentää midatsolaamin ja triatsolaamin tehoa. | |
| Hormonaaliset ehkäisyvalmisteet: | ||
| etinyyliestradioli, noretisteroni ja muut progestiinit | ↓ etinyyliestradioli, noretisteroni ja muut progestiinit LUM:in aikaansaaman CYP3A:n/UGT:n induktion vuoksi | Hormonaaliset suun kautta annettavat, pistettävät, ihon läpi annettavat ja implantoitavat ehkäisyvalmisteet eivät ole luotettavia ehkäisymenetelmiä silloin, kun niitä käytetään samanaikaisesti lumakaftorin/ivakaftorin kanssa. Lumakaftori/ivakaftori saattaa vähentää näille hormonaalisille ehkäisyvalmisteille altistusta, jolloin ehkäisyvalmisteiden teho saattaa heikentyä. |
| Immunosuppressantit: | ||
| siklosporiini, everolimuusi, sirolimuusi, takrolimuusi (käytetään elinsiirron jälkeen) | ↔ LUM, IVA | |
↓ siklosporiini, everolimuusi, sirolimuusi, takrolimuusi LUM:in aikaansaaman CYP3A:n induktion vuoksi | Lumakaftorin/ivakaftorin samanaikaista käyttöä näiden immunosuppressanttien kanssa ei suositella. Lumakaftori/ivakaftori vähentää näille immunosuppressanteille altistusta, jolloin immunosuppressanttien teho saattaa heikentyä. Lumakaftorin/ivakaftorin käyttöä ei ole tutkittu elinsiirtopotilailla. | |
| Protonipumpun estäjät: | ||
| esomepratsoli, lansopratsoli, omepratsoli | ↔ LUM, IVA | |
↓ esomepratsoli, lansopratsoli, omepratsoli LUM:in aikaansaaman CYP3A:n/2C19:n induktion vuoksi | Protonipumpun estäjien annoksen suurentaminen saattaa olla tarpeen, jotta haluttu kliininen vaikutus saavutetaan. Lumakaftori/ivakaftori saattaa vähentää näille protonipumpun estäjille altistusta, jolloin protonipumpun estäjien teho saattaa heikentyä. | |
| Rohdosvalmisteet: | ||
| mäkikuisma (Hypericum perforatum) | ↔ LUM ↓ IVA Mäkikuisman aikaansaaman CYP3A:n induktion vuoksi | Lumakaftorin/ivakaftorin samanaikaista käyttöä mäkikuisman kanssa ei suositella. Samanaikaisessa käytössä altistus ivakaftorille vähenee, jolloin lumakaftorin/ivakaftorin teho saattaa heikentyä. |
| Muut kliinisesti merkittävät samanaikaisesti käytettävät lääkevalmisteet | ||
| Rytmihäiriölääkkeet: | ||
| digoksiini | ↔ LUM, IVA | |
↑ tai ↓ digoksiini P-gp:n mahdollisen induktion tai eston vuoksi | Digoksiinin pitoisuutta seerumissa tulee tarkkailla, ja annosta tulee titrata, kunnes haluttu kliininen vaikutus saavutetaan. Lumakaftori/ivakaftori saattaa muuttaa digoksiinille altistusta. | |
| Antikoagulantit: | ||
| dabigatraani | ↔ LUM, IVA | |
↑ tai ↓ dabigatraani P-gp:n mahdollisen induktion tai eston vuoksi | Asianmukainen kliininen tarkkailu on tarpeen, kun dabigatraania annetaan samanaikaisesti lumakaftorin/ivakaftorin kanssa. Dabigatraaniannoksen muuttaminen voi olla tarpeen halutun kliinisen vaikutuksen saavuttamiseksi. Lumakaftori/ivakaftori saattaa muuttaa dabigatraanille altistusta. | |
| varfariini | ↔ LUM, IVA | |
↑ tai ↓ varfariini LUM:in aikaansaaman CYP2C9:n mahdollisen induktion tai eston vuoksi | INR-arvoa tulee tarkkailla, kun varfariinin ja lumakaftorin/ivakaftorin samanaikainen käyttö on välttämätöntä. Lumakaftori/ivakaftori saattaa muuttaa varfariinille altistusta. | |
| Masennuslääkkeet: | ||
| sitalopraami, essitalopraami, sertraliini | ↔ LUM, IVA | |
↓ sitalopraami, essitalopraami, sertraliini LUM:in aikaansaaman CYP3A:n/2C19:n induktion vuoksi | Näiden masennuslääkkeiden annoksen suurentaminen saattaa olla tarpeen, jotta haluttu kliininen vaikutus saavutetaan. Lumakaftori/ivakaftori saattaa vähentää näille masennuslääkkeille altistusta, jolloin masennuslääkkeiden teho saattaa heikentyä. | |
| bupropioni | ↔ LUM, IVA | |
↓ bupropioni LUM:in aikaansaaman CYP2B6:n induktion vuoksi | Bupropioniannoksen suurentaminen saattaa olla tarpeen, jotta haluttu kliininen vaikutus saavutetaan. Lumakaftori/ivakaftori saattaa vähentää bupropionille altistusta, jolloin bupropionin teho saattaa heikentyä. | |
| Systeemiset kortikosteroidit: | ||
| metyyliprednisoloni, prednisoni | ↔ LUM, IVA | |
↓ metyyliprednisoloni, prednisoni LUM:in aikaansaaman CYP3A:n induktion vuoksi | Näiden systeemisten kortikosteroidien annoksen suurentaminen saattaa olla tarpeen, jotta haluttu kliininen vaikutus saavutetaan. Lumakaftori/ivakaftori saattaa vähentää metyyliprednisolonille ja prednisonille altistusta, jolloin metyyliprednisolonin ja prednisonin teho saattaa heikentyä. | |
| H2-salpaajat: | ||
| ranitidiini | ↔ LUM, IVA | |
↑ tai ↓ ranitidiini P-gp:n mahdollisen induktion tai eston vuoksi | Ranitidiiniannoksen muuttaminen saattaa olla tarpeen, jotta haluttu kliininen vaikutus saavutetaan. Lumakaftori/ivakaftori saattaa muuttaa ranitidiinille altistusta. | |
| Oraaliset veren glukoosipitoisuutta pienentävät lääkkeet: | ||
| repaglinidi | ↔ LUM, IVA | |
↓ repaglinidi LUM:in aikaansaaman CYP3A:n/2C8:n induktion vuoksi | Repaglinidiannoksen suurentaminen saattaa olla tarpeen, jotta haluttu kliininen vaikutus saavutetaan. Lumakaftori/ivakaftori saattaa vähentää repaglinidille altistusta, jolloin repaglinidin teho saattaa heikentyä. | |
Huom: ↑ = kohottaa, ↓ = alentaa, ↔ = ei muutosta; LUM = lumakaftori; IVA = ivakaftori.
* Perustuu kliinisiin yhteisvaikutustutkimuksiin. Kaikki muut esitetyt yhteisvaikutukset ovat ennustettuja.
Väärät positiiviset tulokset THC-virtsakokeissa
Orkambia saaneilla potilailla on ilmoitettu virtsanseulontakokeissa vääriä positiivisia tuloksia tetrahydrokannabinolia (THC) testattaessa. Vaihtoehtoisen varmistusmenetelmän käyttöä on harkittava, jotta tulokset saadaan vahvistettua.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja (alle 300 raskaudesta) lumakaftorin/ivakaftorin käytöstä raskaana oleville naisille. Lumakaftorilla ja ivakaftorilla tehdyissä eläinkokeissa ei ole havaittu suoria tai epäsuoria kehitys- tai lisääntymistoksisia vaikutuksia; ivakaftorin aiheuttamia vaikutuksia havaittiin vain emolle myrkyllisillä annoksilla (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi lumakaftorin/ivakaftorin käyttöä on suositeltavaa välttää raskauden aikana, ellei äidin kliininen tila edellytä hoitoa lumakaftorilla/ivakaftorilla.
Imetys
Vähäiset tiedot osoittavat, että ivakaftori ja lumakaftori erittyvät äidinmaitoon ihmisillä. Lumakaftorin/ivakaftorin vaikutuksista imetettävään vauvaan ei ole riittävästi tietoja. On päätettävä, lopetetaanko imetys vai pidättäydytäänkö hoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Tietoja lumakaftorin ja/tai ivakaftorin vaikutuksesta ihmisen hedelmällisyyteen ei ole saatavilla. Lumakaftori ei vaikuttanut hedelmällisyyttä ja lisääntymiskykyä mittaaviin indekseihin uros- ja naarasrotilla. Ivakaftori huononsi hedelmällisyyttä ja lisääntymiskykyä mittaavia indeksejä uros- ja naarasrotilla (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ivakaftorilla, joka on yksi Orkambi-valmisteen vaikuttavista aineista, on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Ivakaftori saattaa aiheuttaa huimausta (ks. kohta Haittavaikutukset).
Jos potilasta huimaa Orkambi-valmisteen ottamisen aikana, häntä tulee neuvoa olemaan ajamatta ja käyttämättä koneita, kunnes oireet lakkaavat.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät haittavaikutukset ovat hengenahdistus (14,0 %), ripuli (11,0 %) ja pahoinvointi (10,2 %).
Vakavia haittavaikutuksia olivat maksa- ja sappitapahtumat, kuten transaminaasiarvojen kohoaminen (0,5 %), kolestaattinen maksatulehdus (0,3 %) ja maksaperäinen enkefalopatia (0,1 %).
Haittavaikutusten luettelo
Taulukko 5 kuvastaa haittavaikutuksia, joita raportoitiin lumakaftori-/ivakaftorihoitoa ja pelkkää ivakaftorihoitoa saaneilla potilailla kliinisissä tutkimuksissa, myyntiluvan jälkeen suoritetuissa turvallisuustutkimuksissa ja spontaaneissa raporteissa. Haittavaikutukset on lueteltu MedDRA-elinjärjestelmän ja seuraavien yleisyysluokitusten mukaan: Hyvin yleinen (≥1/10), yleinen (≥1/100, <1/10), melko harvinainen (≥1/1 000, <1/100), harvinainen (≥1/10 000, <1/1 000), hyvin harvinainen (<1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 5:Haittavaikutukset lumakaftori-/ivakaftorihoitoa saaneilla potilailla ja pelkkää ivakaftorihoitoa saaneilla potilailla
| Elinjärjestelmä | Yleisyys | Haittavaikutukset |
| Infektiot | hyvin yleinen | Nenänielutulehdus* |
| yleinen | Ylähengitystieinfektio, nuha | |
| Psyykkiset häiriöt | tuntematon | Masennus |
| Verisuonisto | melko harvinainen | Hypertensio |
| Hermosto | hyvin yleinen | Päänsärky, huimaus* |
| melko harvinainen | Maksaperäinen enkefalopatia† | |
| Kuulo ja tasapainoelin | yleinen | Korvakipu*, epämukava tunne korvassa*, tinnitus*, tärykalvon hyperemia*, korvaperäinen tasapainohäiriö* |
| melko harvinainen | Korvan tukkoisuus* | |
| Hengityselimet, rintakehä ja välikarsina | hyvin yleinen | Nenän tukkoisuus, hengenahdistus, produktiivinen yskä, lisääntynyt yskös |
| yleinen | Poikkeava hengitys, suunielun kipu, nenän sivuontelon kongestio*, rinorrea, nielun punoitus*, bronkospasmi | |
| Ruoansulatuselimistö | hyvin yleinen | Vatsakipu*, ylävatsakipu, ripuli, pahoinvointi |
| yleinen | Ilmavaivat, oksentelu | |
| Maksa ja sappi | yleinen | Transaminaasiarvojen kohoaminen |
| melko harvinainen | Kolestaattinen maksatulehdus‡ | |
| Iho ja ihonalainen kudos | yleinen | Ihottuma |
| Sukupuolielimet ja rinnat | yleinen | Epäsäännölliset kuukautiset, dysmenorrea, metrorragia, rinnan kyhmy* |
| melko harvinainen | Menorragia, amenorrea, polymenorrea, rintatulehdus*, gynekomastia*, nännisairaus*, nännikipu*, oligomenorrea | |
| Tutkimukset | hyvin yleinen | Bakteereja ysköksissä* |
| yleinen | Suurentunut veren kreatiinikinaasipitoisuus | |
| melko harvinainen | Kohonnut verenpaine |
*Kliinisissä tutkimuksissa pelkkää ivakaftoria saaneilla potilailla havaitut haittavaikutukset ja niiden yleisyys.
† Yhdellä potilaalla 738:sta
‡ Kahdella potilaalla 738:sta
Turvallisuutta koskevat tiedot 96 viikon jatkotutkimuksesta (809‑105) olivat yhdenmukaisia faasin 3 tutkimuksista saatujen turvallisuutta koskevien tietojen kanssa (tutkimukset 809‑103 ja 809‑104).
Valikoitujen haittavaikutusten kuvaus
Maksaan ja sappeen kohdistuvat haittavaikutukset
Tutkimusten 809‑103 ja 809‑104 aikana yli kahdeksan, viisi, ja kolme kertaa normaalialueen ylärajaa suurempien transaminaasiarvojen (ALAT tai ASAT) ilmaantuvuudet olivat 0,8 %, 2,0 % ja 5,2 % lumakaftori-/ivakaftorihoitoa saaneilla ja 0,5 %, 1,9 % ja 5,1 % lumelääkettä saaneilla potilailla. Transaminaasiarvoihin liittyvien haittavaikutusten ilmaantuvuus oli 5,1 % lumakaftori-/ivakaftorihoitoa saaneilla ja 4,6 % lumelääkettä saaneilla potilailla. Seitsemällä lumakaftoria/ivakaftoria saaneella potilaalla esiintyi vakavia kohonneisiin transaminaasiarvoihin liittyviä maksahaittavaikutuksia. Näistä potilaista kolmella myös kokonaisbilirubiiniarvo oli samanaikaisesti koholla. Lumakaftori-/ivakaftorihoidon keskeyttämisen jälkeen maksan toimintakokeiden tulokset palautuivat lähtötasolle tai paranivat huomattavasti kaikilla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Niistä seitsemästä lumakaftoria/ivakaftoria lumekontrolloiduissa faasin 3 tutkimuksissa saaneesta potilaasta, joilla oli ennestään maksakirroosi ja/tai koholla oleva porttilaskimopaine, yhdellä havaittiin maksan toiminnan heikkenemistä, johon liittyi kohonneet ALAT-, ASAT- ja bilirubiiniarvot sekä enkefalopatia. Haittavaikutus esiintyi viiden vuorokauden sisällä lumakaftori-/ivakaftorihoidon aloittamisesta, ja sen oireet paranivat hoidon keskeyttämisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksan toiminnan dekompensaatiota, mukaan lukien elinsiirtoon ja/tai kuolemaan johtanutta maksan vajaatoimintaa, on raportoitu markkinoille tulon jälkeen lumakaftoria/ivakaftoria saavilla kystistä fibroosia sairastavilla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hengityselimiin kohdistuvat haittavaikutukset
Tutkimuksissa 809‑103 ja 809‑104 hengityselinhaittavaikutusten (esim. epämiellyttävät tuntemukset rinnassa, hengenahdistus, bronkospasmi ja poikkeava hengitys) ilmaantuvuus oli 26,3 % lumakaftori-/ivakaftorihoitoa saaneilla ja 17,0 % lumelääkettä saaneilla potilailla. Näiden haittavaikutusten ilmaantuvuus oli suurempi potilailla, joiden hoitoa edeltävä FEV1-arvo oli pienempi. Noin kolme neljännestä näistä haittavaikutuksista ilmaantui ensimmäisen hoitoviikon aikana, ja useimmilla potilailla oireet hävisivät ilman hoidon keskeyttämistä. Suurin osa haittavaikutuksista oli vaikeusasteeltaan lieviä tai keskivaikeita (ei vakavia) eivätkä ne johtaneet hoidon keskeyttämiseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Avoimessa 24 viikon pituisessa faasin 3b kliinisessä tutkimuksessa (tutkimus 809‑106), johon osallistui 46 vähintään 12-vuotiasta potilasta, joilla oli pitkälle edennyt keuhkosairaus (ppFEV1-arvo <40) [keskimääräinen lähtötilanteen ppFEV1-arvo 29,1 (vaihteluväli: 18,3–42,0)], hengityselinhaittavaikutusten ilmaantuvuus oli 65,2 %. Alaryhmässä, jossa 28 potilasta sai hoitoa heti täydellä lumakaftori-/ivakaftoriannoksella (2 tablettia 12 tunnin välein), ilmaantuvuus oli 71,4 %, ja alaryhmässä, jossa 18 potilasta sai aluksi hoitoa pienennetyllä lumakaftori-/ivakaftoriannoksella (1 tabletti 12 tunnin välein enintään 2 viikon ajan, minkä jälkeen hoitoa jatkettiin täydellä annoksella), ilmaantuvuus oli 55,6 %. Potilaista, joiden hoito aloitettiin täydellä lumakaftori-/ivakaftoriannoksella, yhdellä potilaalla esiintyi vakava hengityselinhaittavaikutus, kolmella potilaalla annosta pienennettiin hoidon kuluessa, ja kolme potilasta lopetti hoidon. Vakavia hengityselinhaittavaikutuksia, annoksen pienennyksiä ja hoidon lopetuksia ei esiintynyt potilailla, joiden hoito aloitettiin puolikkaalla annoksella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kuukautishäiriöt
Tutkimuksissa 809‑103 ja 809‑104 kuukautishäiriöiden (kuukautisten puuttuminen, kivuliaat kuukautiset, runsas kuukautisvuoto, epäsäännölliset kuukautiset, metrorragia, harvat kuukautiset ja tiheät kuukautiset) yhteisilmaantuvuus oli 9,9 % lumakaftori-/ivakaftorihoitoa saavilla ja 1,7 % lumelääkettä saavilla naispotilailla. Näitä kuukautisiin liittyviä haittavaikutuksia esiintyi enemmän naispotilailla, jotka käyttivät hormonaalisia ehkäisyvalmisteita (25,0 %) verrattuna potilaisiin, jotka eivät käyttäneet hormonaalisia ehkäisyvalmisteita (3,5 %) (ks. kohta Yhteisvaikutukset). Suurin osa näistä haittavaikutuksista oli vaikeusasteeltaan lieviä tai kohtalaisia (ei vakavia). Lumakaftori-/ivakaftorihoitoa saavilla potilailla noin kaksi kolmannesta haittavaikutuksista parani, ja niiden keston mediaani oli 10 vuorokautta.
Verenpaineen kohoaminen
Tutkimuksissa 809‑103 ja 809‑104 verenpaineen kohoamiseen liittyviä haittavaikutuksia (esim. hypertensio, kohonnut verenpaine) raportoitiin 0,9 %:lla (7/738) lumakaftori-/ivakaftorihoitoa saaneista potilaista eikä kenelläkään lumelääkettä saaneista potilaista.
Lumakaftori-/ivakaftorihoitoa saaneilla potilailla (keskimääräinen lähtötilanteen systolinen verenpaine 114 mmHg ja diastolinen verenpaine 69 mmHg) suurin nousu lähtötilanteen keskimääräisestä systolisesta verenpaineesta oli 3,1 mmHg ja suurin nousu lähtötilanteen keskimääräisestä diastolisesta verenpaineesta 1,8 mmHg. Lumelääkettä saaneilla potilailla (keskimääräinen lähtötilanteen systolinen verenpaine 114 mmHg ja diastolinen verenpaine 69 mmHg) vastaava systolisen verenpaineen suurin nousu oli 0,9 mmHg ja vastaava diastolisen verenpaineen suurin nousu 0,9 mmHg.
Niiden potilaiden osuus, joilla systolinen verenpaine nousi arvoon >140 mmHg tai diastolinen verenpaine arvoon >90 mmHg vähintään kaksi kertaa, oli lumakaftori-/ivakaftorihoitoa saaneilla potilailla 3,4 % ja 1,5 % ja lumelääkettä saaneilla potilailla 1,6 % ja 0,5 % (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Lumakaftorin/ivakaftorin turvallisuutta koskevat tiedot arvioitiin 46:lla 1–<2-vuotiaalla (tutkimus 809-122), 60:llä 2–5-vuotiaalla (tutkimus 809‑115), 161:llä 6–<12-vuotiaalla (tutkimukset 809‑011 ja 809‑109) ja 194:llä 12–17-vuotiaalla kystistä fibroosia sairastavalla potilaalla, jotka olivat homotsygoottisia F508del-mutaation suhteen ja jotka saivat lumakaftoria/ivakaftoria kliinisissä tutkimuksissa. 12–17-vuotiaat potilaat osallistuivat tutkimuksiin 809‑103 ja 809‑104.
Yleinen turvallisuusprofiili näillä pediatrisilla potilailla oli yleisesti ottaen yhdenmukainen aikuisten potilaiden kanssa. Valikoiduista haittavaikutuksista vain harvoja on raportoitu yksinomaan pediatrisilla potilailla.
Pitkäaikaista turvallisuutta koskevat tiedot kolmesta 96 viikon pituisesta jatkotutkimuksesta, joihin osallistui 52 vähintään 1‑vuotiasta (tutkimus 809‑124), 57 vähintään 2‑vuotiasta (tutkimus 809‑116) ja 239 vähintään 6‑vuotiasta (tutkimus 809‑110) CFTR-geenin F508del-mutaation suhteen homotsygoottista potilasta, olivat yleisesti ottaen yhdenmukaisia 24‑viikkoisten päätutkimusten kanssa. Päätutkimukset tehtiin 1–<2‑vuotiailla (tutkimus 809‑122, tutkimuksen 809‑124 päätutkimus), 2–5‑vuotiailla (tutkimus 809‑115, tutkimuksen 809‑116 päätutkimus) ja 6–<12‑vuotiailla (tutkimukset 809‑011 ja 809‑109, tutkimuksen 809‑110 päätutkimukset) potilailla.
Valikoitujen, 6–<12-vuotiailla potilailla esiintyneiden haittavaikutusten kuvaus
Maksaan ja sappeen kohdistuvat haittavaikutukset
Avoimessa, 24 viikon pituisessa kliinisessä faasin 3 tutkimuksessa, johon osallistui 58 iältään 6–<12-vuotiasta potilasta (tutkimus 809‑011), yli kahdeksan, viisi, ja kolme kertaa normaalialueen ylärajaa suurempien transaminaasiarvojen (ALAT tai ASAT) ilmaantuvuudet olivat 5,3 %, 8,8 % ja 19,3 %. Kenenkään potilaan kokonaisbilirubiini ei kohonnut yli kaksi kertaa normaalialueen ylärajaa suuremmaksi. Lumakaftorin/ivakaftorin antoa jatkettiin tai se aloitettiin onnistuneesti uudelleen keskeytyksen jälkeen kaikilla potilailla, joiden transaminaasiarvot kohosivat, lukuun ottamatta yhtä potilasta, jonka hoito lopetettiin.
Lumekontrolloidussa, 24 viikon pituisessa kliinisessä faasin 3 tutkimuksessa, johon osallistui 204 iältään 6–<12-vuotiasta potilasta (tutkimus 809‑109), yli kahdeksan, viisi, ja kolme kertaa normaalialueen ylärajaa suurempien transaminaasiarvojen (ALAT tai ASAT) ilmaantuvuudet olivat 1,0 %, 4,9 % ja 12,6 % lumakaftori-/ivakaftoriryhmän potilailla ja 2,0 %, 3,0 % ja 7,9 % lumelääkeryhmän potilailla. Kenenkään potilaan kokonaisbilirubiini ei kohonnut yli kaksi kertaa normaalialueen ylärajaa suuremmaksi. Hoito lopetettiin kohonneiden transaminaasiarvojen takia kahdella potilaalla lumakaftori-/ivakaftoriryhmässä ja kahdella potilaalla lumelääkeryhmässä.
Hengityselimiin kohdistuvat haittavaikutukset
Avoimessa, 24 viikon pituisessa faasin 3 kliinisessä tutkimuksessa (tutkimus 809‑011), johon osallistui 58 iältään 6–<12-vuotiasta potilasta (keskimääräinen lähtötilanteen ppFEV1-arvo 91,4), hengityselintapahtumien ilmaantuvuus oli 6,9 % (4/58).
Lumekontrolloidussa, 24 viikon pituisessa faasin 3 kliinisessä tutkimuksessa (tutkimus 809‑109), johon osallistui 6–<12-vuotiaita potilaita (keskimääräinen lähtötilanteen ppFEV1-arvo oli 89,8), hengityselintapahtumien ilmaantuvuus oli 18,4 % lumakaftori-/ivakaftoriryhmän potilailla ja 12,9 % lumelääkeryhmän potilailla. Annosten jälkeen tehdyt spirometriatutkimukset osoittivat ppFEV1-arvon laskeneen hoidon alussa. Absoluuttinen muutos annosta edeltävästä ajankohdasta 4–6 tuntiin annoksen jälkeen oli lumakaftori-/ivakaftoriryhmän potilailla -7,7 päivänä 1 ja -1,3 päivänä 15. Viikkoon 16 mennessä annoksen jälkeistä laskua ei enää havaittu.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Lumakaftorin/ivakaftorin yliannostukseen ei ole saatavilla erityistä vastalääkettä. Yliannostusta hoidetaan tukemalla ja tarkkailemalla elintoimintoja sekä seuraamalla potilaan kliinistä tilaa.
Haittavaikutukset, joiden ilmaantuvuus oli supraterapeuttisena annosjaksona ≥5 % suurempi kuin terapeuttisena annosjaksona, olivat päänsärky, yleistynyt ihottuma ja transaminaasiarvojen kohoaminen.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut hengityselinten sairauksien lääkkeet; ATC-koodi: R07AX30
Vaikutusmekanismi
CFTR-proteiini on kloridikanava, jota esiintyy useiden elinten epiteelisolujen pinnalla. F508del-mutaatio vaikuttaa CFTR-proteiiniin usealla tavalla, pääasiassa aiheuttamalla virheen sellulaarisessa prosessoinnissa ja liikennöinnissä, mikä vähentää CFTR:n määrää solujen pinnalla. Solun pinnan saavuttavalla pienellä määrällä F508del‑CFTR:ää avoimen kanavan todennäköisyys on pieni (kanavaportin poikkeavuus). Lumakaftori vaikuttaa F508del‑CFTR:ään suoraan korjaavasti parantamalla sen sellulaarista prosessointia ja liikennöintiä, jolloin toimivan CFTR:n määrä solun pinnalla lisääntyy. Ivakaftorilla on CFTR:ää potentoiva vaikutus, joka edistää kloridikuljetusta parantamalla CFTR:n avoimen kanavan todennäköisyyttä (kanavaportin toimintaa) solun pinnalla. Yhdessä lumakaftori ja ivakaftori lisäävät F508del‑CFTR:n määrää ja parantavat sen toimintaa solun pinnalla, jolloin kloridi-ionien kuljetus lisääntyy. Tarkkoja mekanismeja, joilla lumakaftori parantaa F508del‑CFTR:n sellulaarista prosessointia ja liikennöintiä ja ivakaftori potentoi F508del‑CFTR:ää, ei tunneta.
Farmakodynaamiset vaikutukset
Vaikutukset hien kloridiin
Hien kloridin muutoksia pelkän lumakaftorihoidon tai lumakaftorin ja ivakaftorin yhdistelmän seurauksena arvioitiin kaksoissokkoutetussa faasin 2 kliinisessä tutkimuksessa kystistä fibroosia sairastavilla vähintään 18-vuotiailla potilailla. Tutkimuksessa 10 potilasta (jotka olivat homotsygoottisia F508del‑CFTR-mutaation suhteen) sai ensin pelkkää lumakaftoria 400 mg 12 tunnin välein 28 vuorokauden ajan ja sen jälkeen lisäksi ivakaftoria 250 mg 12 tunnin välein 28 vuorokauden ajan. Lumelääkettä sai 25 potilasta (jotka olivat homotsygoottisia tai heterotsygoottisia F508del-CFTR-mutaation suhteen). Hoitoero pelkän lumakaftorin (400 mg 12 tunnin välein) ja lumelääkkeen välillä hien kloridipitoisuuden keskimääräisenä muutoksena lähtötilanteesta vuorokauteen 28 mitattuna oli tilastollisesti merkitsevä: ‑8,2 mmol/l (95 %:n luottamusväli: ‑14, ‑2). Hoitoero lumakaftorin ja ivakaftorin yhdistelmän (400 mg ja 250 mg 12 tunnin välein) ja lumelääkkeen välillä hien kloridipitoisuuden keskimääräisenä muutoksena lähtötilanteesta vuorokauteen 56 mitattuna oli tilastollisesti merkitsevä: ‑11 mmol/l (95 %:n luottamusväli: ‑18, ‑4).
Tutkimuksessa 809‑109 (ks. Kliininen teho ja turvallisuus), johon osallistui 6–<12-vuotiaita F508del-CFTR-mutaation suhteen homotsygoottisia potilaita, hoitoero lumelääkkeeseen verrattuna (pienimmän neliösumman keskiarvo) hien kloridin absoluuttisena muutoksena viikolla 24 oli ‑24,9 mmol/l (nimellinen P < 0,0001). Hoitoero lumelääkkeeseen verrattuna (pienimmän neliösumman keskiarvo) hien kloridin keskimääräisenä absoluuttisena muutoksena päivänä 15 ja viikolla 4 oli -20,8 mmol/l (95 %:n luottamusväli: -23,4, -18,2; nimellinen P < 0,0001).
Muutokset FEV1-arvossa
ppFEV1-muutoksia pelkän lumakaftorihoidon tai lumakaftorin ja ivakaftorin yhdistelmän seurauksena arvioitiin myös kaksoissokkoutetussa, lumekontrolloidussa faasin 2 tutkimuksessa, johon osallistui vähintään 18-vuotiaita, kystistä fibroosia sairastavia potilaita. Hoitoero pelkän lumakaftorin (400 mg 12 tunnin välein) ja lumelääkkeen välillä ppFEV1:n keskimääräisenä absoluuttisena muutoksena arvioituna oli -4,6 prosenttiyksikköä (95 %:n luottamusväli: -9,6, 0,4) lähtötilanteesta vuorokauteen 28, 4,2 prosenttiyksikköä (95 %:n luottamusväli: -1,3, 9,7) lähtötilanteesta vuorokauteen 56, ja 7,7 prosenttiyksikköä (95 %:n luottamusväli: 2,6, 12,8; tilastollisesti merkitsevä) vuorokaudesta 28 vuorokauteen 56 (sen jälkeen kun pelkkään lumakaftorihoitoon lisättiin ivakaftori).
Sydämen sykkeen hidastuminen
Lumekontrolloiduissa 24 viikon pituisissa faasin 3 tutkimuksissa keskimääräisen sykkeen havaittiin hidastuneen lähtötilanteesta enimmillään 6 lyönnillä minuutissa päivinä 1 ja 15 noin 4–6 tuntia annon jälkeen mitattuna. Päivän 15 jälkeen sykettä ei mitattu annon jälkeen näissä tutkimuksissa. Viikosta 4 alkaen keskimääräinen sykkeen muutos ennen antoa mitattuna oli 1–2 lyöntiä minuutissa alle lähtötilanteen arvon lumakaftori-/ivakaftorihoitoa saavilla potilailla. Niiden potilaiden osuus, joilla mitattiin sykearvo <50 lyöntiä minuutissa hoidon aikana, oli lumakaftori-/ivakaftorihoitoa saaneilla potilailla 11 % ja lumelääkettä saaneilla potilailla 4,9 %.
Sydämen elektrofysiologia
Perusteellisessa QT-aikaa koskevassa kliinisessä tutkimuksessa, jossa arvioitiin lumakaftoria 600 mg kerran vuorokaudessa / ivakaftoria 250 mg 12 tunnin välein ja lumakaftoria 1 000 mg kerran vuorokaudessa / ivakaftoria 450 mg 12 tunnin välein, ei havaittu merkittäviä muutoksia QTc-ajassa tai verenpaineessa.
Kliininen teho ja turvallisuus
Tutkimukset vähintään 12-vuotiailla, kystistä fibroosia sairastavilla potilailla, jotka ovat homotsygoottisia CFTR-geenin F508del-mutaation suhteen
Lumakaftorin/ivakaftorin tehoa kystistä fibroosia sairastavilla potilailla, jotka ovat homotsygoottisia CFTR-geenin F508del-mutaation suhteen, arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa kliinisessä tutkimuksessa, joihin osallistui yhteensä 1 108 kliinisesti vakaata kystistä fibroosia sairastavaa potilasta. Potilaista 737 satunnaistettiin saamaan ja sai lumakaftoria/ivakaftoria. Kummankin tutkimuksen potilaat satunnaistettiin suhteessa 1:1:1 saamaan 600 mg lumakaftoria kerran vuorokaudessa ja 250 mg ivakaftoria 12 tunnin välein, 400 mg lumakaftoria 12 tunnin välein ja 250 mg ivakaftoria 12 tunnin välein tai lumelääkettä. Potilaat ottivat tutkimuslääkettä rasvaa sisältävän ruoan kanssa 24 viikon ajan omien kystiseen fibroosiin määrättyjen hoitojensa (esim. bronkodilaattorien, inhaloitavien antibioottien, dornaasi alfan ja hypertonisen keittosuolaliuoksen) lisäksi. Tutkimusten potilailla oli mahdollisuus jatkaa sokkoutetussa jatkotutkimuksessa.
Tutkimuksessa 809‑103 arvioitiin 549:ää kystistä fibroosia sairastavaa, vähintään 12-vuotiasta potilasta (keski-ikä 25,1 vuotta), joiden FEV1-arvo seulonnassa oli 40–90 % odotetusta (ppFEV1-arvo 40–90) (ppFEV1-keskiarvo lähtötilanteessa: 60,7 [vaihteluväli: 31,1–94,0]). Tutkimuksessa 809‑104 arvioitiin 559:ää vähintään 12-vuotiasta potilasta (keski-ikä 25,0 vuotta), joiden ppFEV1-arvo seulonnassa oli 40–90 (ppFEV1-keskiarvo lähtötilanteessa: 60,5 [vaihteluväli: 31,3–99,8]). Potilaita, joilla oli aiemmin havaittu kolonisaatiota organismeilla kuten Burkholderia cenocepacia, Burkholderia dolosa tai Mycobacterium abscessus tai joilla oli vähintään 3 poikkeavaa maksan toimintakokeiden tulosta (ALAT, ASAT, AFOS, GGT ≥3 kertaa normaalialueen ylärajaa suurempi tai kokonaisbilirubiini ≥2 kertaa normaalialueen ylärajaa suurempi), ei otettu mukaan tutkimuksiin.
Ensisijainen tehon päätetapahtuma kummassakin tutkimuksessa oli ppFEV1-arvon absoluuttinen muutos lähtötilanteesta viikolla 24. Muita tehon muuttujia olivat ppFEV1-arvon suhteellinen muutos lähtötilanteesta, painoindeksin (BMI) absoluuttinen muutos lähtötilanteesta, CFQ‑R-kyselyn hengitystä koskevan osan pistemäärän absoluuttinen muutos lähtötilanteesta, niiden potilaiden osuus, joiden ppFEV1-arvon suhteellinen muutos lähtötilanteesta oli ≥5 % viikolla 24, sekä keuhko-oireiden pahentumisten lukumäärä (mukaan lukien sairaalahoitoa tai laskimonsisäistä antibioottihoitoa vaativat tapaukset) viikon 24 loppuun mennessä.
Kummassakin tutkimuksessa lumakaftori-/ivakaftorihoito paransi ppFEV1-arvoa tilastollisesti merkitsevästi (ks. taulukko 6). ppFEV1-arvon parantuminen alkoi keskimäärin nopeasti (vuorokautena 15), ja parannus säilyi 24 viikon pituisen hoitojakson loppuun asti. Vuorokautena 15 hoitoero ppFEV1-arvon keskimääräiselle absoluuttiselle muutokselle lähtötilanteesta (95 %:n luottamusväli) lumakaftorin ja ivakaftorin yhdistelmän (400 mg ja 250 mg 12 tunnin välein) ja lumelääkkeen välillä oli tutkimusten 809‑103 ja 809‑104 yhteistuloksissa 2,51 prosenttiyksikköä (P < 0,0001). Parannuksia ppFEV1-arvossa havaittiin iästä, sairauden vaikeusasteesta, sukupuolesta ja maantieteellisestä alueesta riippumatta. Lumakaftorilla/ivakaftorilla tehtyihin faasin 3 tutkimuksiin osallistui 81 potilasta, joiden ppFEV1-arvo oli <40 lähtötilanteessa. Hoitoero tässä alaryhmässä oli samankaltainen kuin potilailla, joiden ppFEV1-arvo oli ≥40. Viikolla 24 hoitoero ppFEV1-arvon keskimääräiselle absoluuttiselle muutokselle lähtötilanteesta (95 %:n luottamusväli) lumakaftorin ja ivakaftorin yhdistelmän (400 mg ja 250 mg 12 tunnin välein) ja lumelääkkeen välillä oli tutkimusten 809‑103 ja 809‑104 yhteistuloksissa 3,39 prosenttiyksikköä (P = 0,0382) potilaille, joiden ppFEV1-arvo oli <40, ja 2,47 prosenttiyksikköä (P < 0,0001) potilaille, joiden ppFEV1-arvo oli ≥40.
Taulukko 6:Yhteenveto tutkimusten 809‑103 ja 809‑104 ensisijaisesta ja tärkeimmistä toissijaisista päätetapahtumista*
| Tutkimus 809‑103 | Tutkimus 809‑104 | Yhdistetty (tutkimukset 809‑103 ja 809‑104) | |||||
Lumelääke (n = 184) | LUM 400 mg 12 h:n välein / IVA 250 mg 12 h:n välein (n = 182) | Lumelääke (n = 187) | LUM 400 mg 12 h:n välein / IVA 250 mg 12 h:n välein (n = 187) | Lumelääke (n = 371) | LUM 400 mg 12 h:n välein / IVA 250 mg 12 h:n välein (n = 369) | ||
| ppFEV1-arvon absoluuttinen muutos viikolla 24 (prosentti-yksikköä) | Hoitoero | – | 2,41 (P = 0,0003)† | – | 2,65 (P = 0,0011)† | – | 2,55 (P < 0,0001) |
| Ryhmänsisäinen muutos | -0,73 (P = 0,2168) | 1,68 (P = 0,0051) | -0,02 (P = 0,9730) | 2,63 (P < 0,0001) | -0,39 (P < 0,3494) | 2,16 (P < 0,0001) | |
| ppFEV1-arvon suhteellinen muutos viikolla 24 (%) | Hoitoero | – | 4,15 (P = 0,0028)† | – | 4,69 (P = 0,0009)† | – | 4,4 (P < 0,0001) |
| Ryhmänsisäinen muutos | -0,85 (P = 0,3934) | 3,3 (P = 0,0011) | 0,16 (P = 0,8793) | 4,85 (P < 0,0001) | -0,34 (P = 0,6375) | 4,1 (P < 0,0001) | |
| Painoindeksin absoluuttinen muutos viikolla 24 (kg/m2) | Hoitoero | – | 0,13 (P = 0,1938) | – | 0,36 (P < 0,0001) † | – | 0,24 (P = 0,0004) |
| Ryhmänsisäinen muutos | 0,19 (P = 0,0065) | 0,32 (P < 0,0001) | 0,07 (P = 0,2892) | 0,43 (P < 0,0001) | 0,13 (P = 0,0066) | 0,37 (P < 0,0001) | |
| CFQ‑R-kyselyn hengitystä koskevan osan pistemäärän absoluuttinen muutos viikolla 24 (pistettä) | Hoitoero | – | 1,5 (P = 0,3569) | – | 2,9 (P = 0,0736) | – | 2,2 (P = 0,0512) |
| Ryhmänsisäinen muutos | 1,1 (P = 0,3423) | 2,6 (P = 0,0295) | 2,8 (P = 0,0152) | 5,7 (P < 0,0001) | 1,9 (P = 0,0213) | 4,1 (P < 0,0001) | |
| Niiden potilaiden osuus, joiden ppFEV1-arvon suhteellinen muutos oli ≥5 % viikolla 24 | % | 25 % | 32 % | 26 % | 41 % | 26 % | 37 % |
| Vetosuhde | – | 1,43 (P = 0,1208) | – | 1,90 (P = 0,0032) | – | 1,66 (P = 0,0013) | |
| Keuhko-oireiden pahentumisten lukumäärä viikon 24 loppuun mennessä | Tapahtumia (esiintyvyys / 48 viikkoa) | 112 (1,07) | 73 (0,71) | 139 (1,18) | 79 (0,67) | 251 (1,14) | 152 (0,70) |
| Esiintyvyyssuhde | – | 0,66 (P = 0,0169) | – | 0,57 (P = 0,0002) | – | 0,61 (P < 0,0001) | |
*Kummassakin tutkimuksessa käytettiin hierarkkista testausmenettelyä jokaisessa aktiivisessa hoitohaarassa ensisijaiselle ja toissijaisille päätetapahtumille vs. lumelääke. Kullakin tasolla tilastollisen merkitsevyyden edellytyksenä oli P ≤ 0,0250 ja että kaikki aiemmat testit myös täyttivät tämän merkitsevyysvaatimuksen.
† Osoittaa tilastollista merkitsevyyttä, joka vahvistettiin hierarkkisella testausmenettelyllä.
Viikolla 24 niiden potilaiden osuus, joilla ei esiintynyt keuhko-oireiden pahentumista, oli huomattavasti suurempi lumakaftori-/ivakaftorihoitoa saaneilla kuin lumelääkettä saaneilla potilailla. Tutkimusten yhteisanalyysissa pahentumisten esiintyvyyssuhde viikon 24 loppuun mennessä oli lumakaftorilla/ivakaftorilla (400 mg lumakaftoria 12 tunnin välein / 250 mg ivakaftoria 12 tunnin välein; n = 369) 0,61 (P < 0,0001), mikä oli 39 % vähemmän lumelääkkeeseen verrattuna. Tapahtumien esiintyvyys vuotta kohden (vuotuistettu 48 viikoksi) oli 0,70 lumakaftori-/ivakaftoriryhmässä ja 1,14 lumelääkeryhmässä. Lumakaftori-/ivakaftorihoito pienensi sairaalahoitoa vaativien pahentumisten riskiä merkitsevästi 61 %:lla lumelääkkeeseen nähden (esiintyvyyssuhde = 0,39, P < 0,0001; tapahtumien esiintyvyys 48:aa viikkoa kohden 0,17 lumakaftorille/ivakaftorille ja 0,45 lumelääkkeelle) ja vähensi laskimonsisäistä antibioottihoitoa vaativia pahentumisia 56 %:lla (esiintyvyyssuhde = 0,44, P < 0,0001; tapahtumien esiintyvyys 48:aa viikkoa kohden 0,25 lumakaftorille/ivakaftorille ja 0,58 lumelääkkeelle). Näitä tuloksia ei pidetty tilastollisesti merkitsevinä yksittäisten tutkimusten testaushierarkian puitteissa.
Pitkäaikaista turvallisuutta ja tehoa koskeva jatkotutkimus
Tutkimus 809‑105 oli kystistä fibroosia sairastavilla potilailla tehtävä, rinnakkaisryhmillä toteutettava, monikeskuksinen, faasin 3 jatkotutkimus, johon osallistui 12-vuotiaita ja sitä vanhempia potilaita tutkimuksista 809‑103 ja 809‑104. Tässä jatkotutkimuksessa arvioitiin pitkäaikaisen lumakaftori-/ivakaftorihoidon turvallisuutta ja tehoa. Tutkimuksessa 809‑103 tai 809‑104 hoitoa saaneista 1 108 potilaasta 1 029 (93 %) sai lääkettä ja aktiivista hoitoa (600 mg lumakaftoria kerran vuorokaudessa / 250 mg ivakaftoria 12 tunnin välein tai 400 mg lumakaftoria 12 tunnin välein / 250 mg ivakaftoria 12 tunnin välein) tutkimuksessa 809‑105 ylimääräisten enintään 96 viikon ajan (eli yhteensä enintään 120 viikon ajan). Tämän jatkotutkimuksen ensisijainen tehon analyysi käsitti tiedot tutkimuksesta 809‑105 enintään viikkoon 72 asti sekä herkkyysanalyysin, joka käsitti tiedot tutkimuksesta 809‑105 enintään viikkoon 96 asti.
Lumakaftori-/ivakaftorihoitoa tutkimuksessa 809‑103 tai 809‑104 saaneilla potilailla vaikutus oli säilynyt suhteessa lähtötilanteeseen tutkimuksen 809‑105 ylimääräisten 96 viikon jälkeen. Potilailla, jotka siirtyivät lumelääkkeestä aktiiviseen hoitoon, todettiin samankaltaisia muutoksia kuin lumakaftori-/ivakaftorihoitoa tutkimuksessa 809‑103 tai 809‑104 saaneilla potilailla (ks. taulukko 6). Tutkimuksen 809‑105 tulokset on esitetty kuvassa 1 ja taulukossa 7.
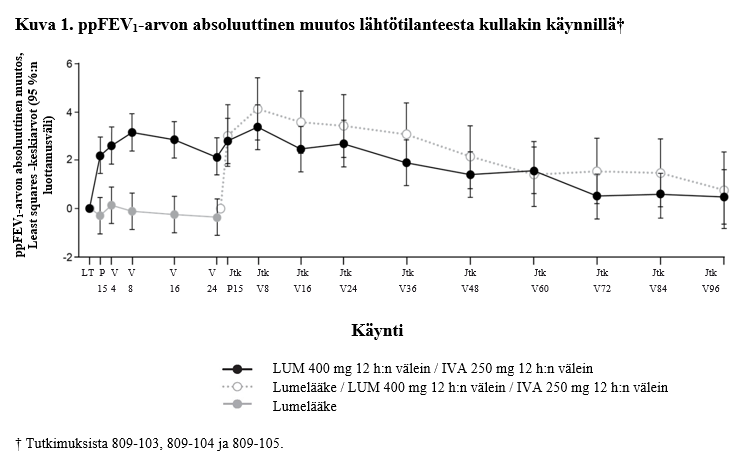
Taulukko 7: Lumakaftorin/ivakaftorin pitkäaikainen vaikutus tutkimuksessa 809‑105*
| Lähtötilanne ja päätetapahtuma | Vaihto lumelääkkeestä lumakaftoriin, 400 mg 12 tunnin välein / ivakaftoriin, 250 mg 12 tunnin välein (n = 176)** | lumakaftori, 400 mg 12 tunnin välein / ivakaftori, 250 mg 12 tunnin välein (n = 369)† | ||||
| Keskiarvo (keski-hajonta) | Least squares ‑keskiarvot (95 %:n luottamus-väli) | P-arvo | Keskiarvo (keski-hajonta) | Least squares ‑keskiarvot (95 %:n luottamus-väli) | P-arvo | |
Lähtötilanteen ppFEV1-arvo ‡ | 60,2 (14,7) | 60,5 (14,1) | ||||
| ppFEV1-arvon absoluuttinen muutos lähtötilanteesta (prosenttipistettä) | ||||||
| Jatkotutkimuksen viikko 72 | (n = 134) 1,5 (0,2, 2,9) | 0,0254 | (n = 273) 0,5 (‑0,4, 1,5) | 0,2806 | ||
| Jatkotutkimuksen viikko 96 | (n = 75) 0,8 (‑0,8, 2,3) | 0,3495 | (n = 147) 0,5 (‑0,7, 1,6) | 0,4231 | ||
| ppFEV1-arvon suhteellinen muutos lähtötilanteesta (%) | ||||||
| Jatkotutkimuksen viikko 72 | (n = 134) 2,6 (0,2, 5,0) | 0,0332 | (n = 273) 1,4 (‑0,3, 3,2) | 0,1074 | ||
| Jatkotutkimuksen viikko 96 | (n = 75) 1,1 (‑1,7, 3,9) | 0,4415 | (n = 147) 1,2 (‑0,8, 3,3) | 0,2372 | ||
| Lähtötilanteen painoindeksi (kg/m2)‡ | 20,9 (2,8) | 21,5 (3,0) | ||||
| Painoindeksin absoluuttinen muutos lähtötilanteesta (kg/m2) | ||||||
| Jatkotutkimuksen viikko 72 | (n = 145) 0,62 (0,45, 0,79) | <0,0001 | (n = 289) 0,69 (0,56, 0,81) | <0,0001 | ||
| Jatkotutkimuksen viikko 96 | (n = 80) 0,76 (0,56, 0,97) | <0,0001 | (n = 155) 0,96 (0,81, 1,11) | <0,0001 | ||
| CFQ‑R-kyselyn hengitystä koskevan osan pistemäärä (pistettä)‡ | 70,4 (18,5) | 68,3 (18,0) | ||||
| CFQ‑R-kyselyn hengitystä koskevan osan pistemäärän absoluuttinen muutos (pistettä) | ||||||
| Jatkotutkimuksen viikko 72 | (n = 135) 3,3 (0,7, 5,9) | 0,0124 | (n = 269) 5,7 (3,8, 7,5) | <0,0001 | ||
| Jatkotutkimuksen viikko 96 | (n = 81) 0,5 (‑2,7, 3,6) | 0,7665 | (n = 165) 3,5 (1,3, 5,8) | 0,0018 | ||
| Keuhko-oireiden pahentumisten lukumäärä (tapahtumaa)**† *** | ||||||
| Tapahtumien lukumäärä potilasvuotta kohden (95 %:n luottamusväli) (esiintyvyys 48 viikkoa kohden) | 0,69 (0,56, 0,85) | 0,65 (0,56, 0,75) | ||||
| Sairaalahoitoa vaativien tapahtumien lukumäärä potilasvuotta kohden (95 %:n luottamusväli) (esiintyvyys 48 viikkoa kohden) | 0,30 (0,22, 0,40) | 0,24 (0,19, 0,29) | ||||
| Laskimonsisäistä antibioottihoitoa vaativien tapahtumien lukumäärä potilasvuotta kohden (95 %:n luottamusväli) (esiintyvyys 48 viikkoa kohden) | 0,37 (0,29, 0,49) | 0,32 (0,26, 0,38) | ||||
* Yhteensä 82 % (421 tutkimukseen soveltuvasta 516:sta potilaasta) osallistui tutkimukseen 72 viikon ajan; 42 % osallistui 96 viikon ajan. Suurin osa potilaista lopetti osallistumisen muista kuin turvallisuuteen liittyvistä syistä.
** Potilailla, jotka siirtyivät tutkimuksista 809‑103 ja 809‑104 (ryhmä, joka siirtyi lumelääkehoidosta lumakaftori-/ivakaftorihoitoon), kokonaisaltistus oli enintään 96 viikkoa. Annostus lumakaftoria 400 mg 12 tunnin välein / ivakaftoria 250 mg 12 tunnin välein saavassa ryhmässä vastaa suositeltua annostusta.
*** Tapahtumien esiintyvyys potilasvuotta kohden vuotuistettiin 48 viikoksi.
† Potilailla, jotka siirtyivät tutkimuksista 809‑103 ja 809‑104 (ryhmä, joka siirtyi lumakaftori-/ivakaftorihoidosta lumakaftori-/ivakaftorihoitoon), kokonaisaltistus oli enintään 120 viikkoa. Annostus lumakaftoria 400 mg 12 tunnin välein / ivakaftoria 250 mg 12 tunnin välein saavassa ryhmässä vastaa suositeltua annostusta.
‡ Ryhmässä, joka siirtyi lumelääkehoidosta lumakaftori-/ivakaftorihoitoon (lumakaftoria 400 mg 12 tunnin välein / ivakaftoria 250 mg 12 tunnin välein), lähtötilanne oli tutkimuksen 809‑105 lähtötilanne. Ryhmässä, joka sai lumakaftoria 400 mg 12 tunnin välein / ivakaftoria 250 mg 12 tunnin välein, lähtötilanne oli tutkimusten 809‑103 ja 809‑104 lähtötilanne.
Tutkimus kystistä fibroosia sairastavilla potilailla, jotka olivat heterotsygoottisia CFTR-geenin F508del-mutaation suhteen
Tutkimus 809‑102 oli monikeskuksinen, kaksoissokkoutettu, satunnaistettu, lumekontrolloitu faasin 2 tutkimus, johon osallistui 125 vähintään 18-vuotiasta kystistä fibroosia sairastavaa potilasta, joiden ppFEV1-arvo oli 40–90 ja joilla oli F508del-mutaatio yhdessä alleelissa ja toisessa alleelissa mutaatio, jonka ennustetaan johtavan CFTR:n tuotannon puutteeseen tai CFTR-proteiiniin, joka ei reagoi ivakaftoriin in vitro.
Potilaat saivat joko lumakaftoria/ivakaftoria (n = 62) tai lumelääkettä (n = 63) omien kystiseen fibroosiin määrättyjen hoitojensa lisäksi. Ensisijainen päätetapahtuma oli keuhkojen toiminnan parantuminen, joka mitattiin ppFEV1-arvon keskimääräisenä absoluuttisena muutoksena lähtötilanteesta vuorokautena 56. Lumakaftori-/ivakaftorihoito ei parantanut ppFEV1-arvoa merkitsevästi suhteessa lumelääkkeeseen kystistä fibroosia sairastavilla potilailla, jotka olivat heterotsygoottisia CFTR-geenin F508del-mutaation suhteen (hoitoero 0,60 [P = 0,5978]), eikä merkittävää parannusta tapahtunut myöskään painoindeksissä tai painossa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Tutkimukset 6–<12-vuotiailla, kystistä fibroosia sairastavilla potilailla, jotka ovat homotsygoottisia CFTR-geenin F508del-mutaation suhteen
Tutkimus 809‑109 oli lumekontrolloitu, 24 viikon pituinen faasin 3 kliininen tutkimus, johon osallistui 204 kystistä fibroosia sairastavaa 6–<12-vuotiasta potilasta (keski-ikä 8,8 vuotta). Tutkimuksessa 809‑109 arvioitiin potilaita, joiden keuhkojen tuuletusta mittaava indeksi (LCI2.5) ensimmäisellä seulontakäynnillä oli ≥7,5 (keskimääräinen lähtötilanteen LCI2.5-arvo 10,28 [vaihteluväli: 6,55–16,38]) ja ppFEV1-arvo seulonnassa ≥70 (keskimääräinen lähtötilanteen ppFEV1-arvo 89,8 [vaihteluväli: 48,6–119,6]). Potilaat saivat joko lumakaftoria 200 mg / ivakaftoria 250 mg 12 tunnin välein (n = 103) tai lumelääkettä (n = 101) omien kystiseen fibroosiin määrättyjen hoitojensa lisäksi. Tutkimukseen ei otettu potilaita, joilla oli vähintään 2 poikkeavaa maksan toimintakokeiden tulosta (ALAT, ASAT, AFOS, GGT ≥3 kertaa normaalialueen ylärajaa suurempi, ALAT tai ASAT >5 kertaa normaalialueen ylärajaa suurempi tai kokonaisbilirubiini >2 kertaa normaalialueen ylärajaa suurempi).
Ensisijainen tehon päätetapahtuma oli LCI2.5-arvon absoluuttinen muutos lähtötilanteesta viikolla 24. Tärkeimmät toissijaiset päätetapahtumat olivat hien kloridin keskimääräinen absoluuttinen muutos lähtötilanteesta päivänä 15 sekä viikoilla 4 ja 24 (ks. Farmakodynaamiset vaikutukset), painoindeksin absoluuttinen muutos lähtötilanteesta viikolla 24 ja CFQ‑R-kyselyn hengitystä koskevan osan pistemäärän absoluuttinen muutos lähtötilanteesta viikon 24 loppuun mennessä. Nämä tulokset on esitetty taulukossa 8:
Taulukko 8: Yhteenveto tutkimuksen 809‑109 ensisijaisesta päätetapahtumasta ja tärkeimmistä toissijaisista päätetapahtumista
Lumelääke (n = 101) | LUM 200 mg / IVA 250 mg 12 h:n välein (n = 103) | ||
| Ensisijainen päätetapahtuma | |||
| Keuhkojen tuuletusta mittaavan indeksin (LCI2.5) absoluuttinen muutos lähtötilanteesta viikolla 24 | Hoitoero | – | -1,09 (P < 0,0001) |
| Ryhmänsisäinen muutos | 0,08 (P = 0,5390) | -1,01 (P < 0,0001) | |
| Tärkeimmät toissijaiset päätetapahtumat* | |||
| Painoindeksin absoluuttinen muutos viikolla 24 (kg/m2) | Hoitoero | – | 0,11 (P = 0,2522) |
| Ryhmänsisäinen muutos | 0,27 (P = 0,0002) | 0,38 (P < 0,0001) | |
| CFQ‑R-kyselyn hengitystä koskevan osan pistemäärän absoluuttinen muutos viikon 24 loppuun mennessä (pistettä) | Hoitoero | – | 2,5 (P = 0,0628) |
| Ryhmänsisäinen muutos | 3,0 (P = 0,0035) | 5,5 (P < 0,0001) | |
* Tutkimuksessa oli keskeisiä toissijaisia päätetapahtumia sekä muita toissijaisia päätetapahtumia.
ppFEV1-arvo arvioitiin kliinisesti merkityksellisenä muuna toissijaisena päätetapahtumana. Lumakaftoria/ivakaftoria saaneilla potilailla hoitoero ppFEV1-arvon absoluuttisena muutoksena lähtötilanteesta viikolla 24 oli 2,4 (P = 0,0182).
Kystistä fibroosia sairastavat, iältään vähintään 6-vuotiaat potilaat tutkimuksista 809‑011 ja 809‑109 otettiin mukaan faasin 3 jatko- ja monikeskustutkimukseen (tutkimus 809‑110). Tämän jatkotutkimuksen tarkoituksena oli arvioida pitkäaikaisen lumakaftori-/ivakaftorihoidon turvallisuutta ja tehoa. Niistä 262:sta potilaasta, jotka saivat mitä tahansa hoitoa tutkimuksessa 809‑011 tai 809‑109, 239:lle potilaalle (91 %) annettiin annos ja he saivat aktiivista hoitoa (6 – <12-vuotiaat potilaat saivat lumakaftoria 200 mg 12 tunnin välein / ivakaftoria 250 mg 12 tunnin välein; ≥12-vuotiaat potilaat saivat lumakaftoria 400 mg 12 tunnin välein / ivakaftoria 250 mg 12 tunnin välein) jatkotutkimuksessa lisäksi enintään 96 viikon ajan (eli yhteensä enintään 120 viikon ajan) (ks. kohta Haittavaikutukset). Toissijaiset tehon tulokset ja keuhko-oireiden pahentumistapahtumien lukumäärä potilasvuotta kohden on esitetty taulukossa 9.
Taulukko 9: Lumakaftorin/ivakaftorin pitkäaikainen vaikutus tutkimuksessa 809‑110
| Lähtötilanne ja päätetapahtuma | Vaihto lumelääkkeestä lumakaftoriin/ivakaftoriin (n = 96)* | lumakaftori/ivakaftori – lumakaftori/ivakaftori (n = 143)* | ||
| Keskiarvo (keskihajonta) | Least squares ‑keskiarvot (95 %:n luottamusväli) | Keskiarvo (keskihajonta) | Least squares ‑keskiarvot (95 %:n luottamusväli) | |
| n = 101 | n = 128 | |||
| Lähtötilanteen LCI2.5‡** | 10,26 (2,24) | 10,24 (2,42) | ||
| LCI2.5-arvon absoluuttinen muutos lähtötilanteesta | ||||
| Jatkotutkimuksen viikko 96 | (n = 69) -0,86 (-1,33, -0,38) | (n = 88) -0,85 (-1,25, -0,45) | ||
| n = 101 | n = 161 | |||
| Lähtötilanteen painoindeksi (kg/m2)‡ | 16,55 (1,96) | 16,56 (1,77) | ||
| Painoindeksin absoluuttinen muutos lähtötilanteesta (kg/m2) | ||||
| Jatkotutkimuksen viikko 96 | (n = 83) 2,04 (1,77, 2,31) | (n =130) 1,78 (1,56, 1,99) | ||
| n = 78 | n = 135 | |||
| CFQ‑R-kyselyn lähtötilanteen hengitystä koskevan osan pistemäärä (pistettä)‡ | 77,1 (15,5) | 78,5 (14,3) | ||
| CFQ-R-kyselyn hengitystä koskevan osan pistemäärän absoluuttinen muutos (pistettä) | ||||
| Jatkotutkimuksen viikko 96 | (n = 65) 6,6 (3,1, 10,0) | (n = 108) 7,4 (4,8, 10,0) | ||
| Keuhko-oireiden pahentumisten lukumäärä (tapahtumaa) (tutkimukset 809‑109 FAS ja ROS)† | ||||
| Tapahtuminen lukumäärä potilasvuotta kohden (95 %:n luottamusväli) | n = 96 0,30 (0,21, 0,43) | n = 103 0,45 (0,33, 0,61) | ||
*Lumelääkettä tutkimuksessa 809‑109 saaneet potilaat (n = 96), jotka siirrettiin saamaan aktiivista lumakaftori-/ivakaftorihoitoa jatkotutkimuksessa (lumelääke-lumakaftori/ivakaftori). Potilaat, jotka saivat lumakaftori-/ivakaftorihoitoa joko kantatutkimuksessa [tutkimus 809‑011 (n = 49) tai tutkimuksessa 809‑109 (n = 94)] ja jatkoivat aktiivista lumakaftori-/ivakaftorihoitoa jatkovaiheessa (lumakaftori/ivakaftori-lumakaftori/ivakaftori).
‡Lähtötilanne molemmille ryhmille (lumelääke-lumakaftori/ivakaftori ja lumakaftori/ivakaftori-lumakaftori/ivakaftori) oli tutkimuksen 809‑011 ja tutkimuksen 809‑109 (kantatutkimus) lähtötilanne, ja vastaava ”n” tarkoittaa kantatutkimuksessa määritettyä analyysia.
**Keuhkojen tuuletusta mittaavan indeksin (LCI) osatutkimukseen osallistui 117 potilasta ryhmässä lumakaftori/ivakaftori-lumakaftori/ivakaftori ja 96 potilasta ryhmässä lumelääke-lumakaftori/ivakaftori.
†FAS = koko analyysiryhmä (n = 103) kattaa potilaat, jotka saivat lumakaftoria/ivakaftoria tutkimuksissa 809‑109 ja 809‑110, ja joita arvioitiin lumakaftorin/ivakaftorin osalta koko kumulatiivisen tutkimusajanjakson ajan; ROS = jatkotutkimusryhmä (n = 96) kattaa potilaat, jotka saivat lumelääkettä tutkimuksessa 809‑109 ja lumakaftoria/ivakaftoria tutkimuksessa 809‑110, ja joita arvioitiin meneillään olevan tutkimusajanjakson kuluessa tutkimuksessa 809‑110.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Orkambi-valmisteen käytöstä kystisen fibroosin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Altistus lumakaftorille (AUC) on noin kaksi kertaa suurempi terveillä aikuisilla koehenkilöillä verrattuna altistukseen kystistä fibroosia sairastavilla potilailla. Altistus ivakaftorille on samankaltainen terveillä aikuisilla koehenkilöillä ja kystistä fibroosia sairastavilla potilailla. Kahdesti vuorokaudessa annettaessa lumakaftorin ja ivakaftorin vakaan tilan pitoisuudet plasmassa saavutettiin terveillä koehenkilöillä noin 7 vuorokauden hoidon jälkeen. Lumakaftorin kumulaatiokerroin oli noin 1,9. Altistus ivakaftorille vakaassa tilassa on pienempi kuin vuorokautena 1, mikä johtuu lumakaftorin CYP3A:ta indusoivasta vaikutuksesta (ks. kohta Yhteisvaikutukset).
Kun 400 mg lumakaftoria / 250 mg ivakaftoria annettiin aterian yhteydessä suun kautta 12 tunnin välein, keskimääräinen (±SD) vakaan tilan AUC0-12h- ja Cmax-arvo olivat lumakaftorilla 198 (64,8) μg∙h/ml ja 25,0 (7,96) μg/ml ja ivakaftorilla 3,66 (2,25) μg∙h/ml ja 0,602 (0,304) μg/ml. Kun pelkkää ivakaftoria annettiin 150 mg aterian yhteydessä suun kautta 12 tunnin välein, keskimääräinen (±SD) vakaan tilan AUC0-12h- ja Cmax-arvo olivat 9,08 (3,20) μg∙h/ml ja 1,12 (0,319) μg/ml.
Imeytyminen
Useita lumakaftoriannoksia suun kautta annettaessa altistus lumakaftorille lisääntyi yleensä suhteessa annokseen annosvälillä 50–1 000 mg 24 tunnin välein. Rasvaa sisältävän ruoan kanssa annettaessa altistus lumakaftorille lisääntyi noin 2,0-kertaisesti paastotilaan verrattuna. Lumakaftorin Tmax-arvon mediaani (vaihteluväli) on noin 4,0 tuntia (2,0, 9,0) ravitussa tilassa.
Kun ivakaftoria annettiin suun kautta useina annoksina yhdessä lumakaftorin kanssa, altistus ivakaftorille lisääntyi yleensä suhteessa annokseen annosvälillä 150–250 mg 12 tunnin välein. Altistus ivakaftorille yhdessä lumakaftorin kanssa annettuna lisääntyi noin kolminkertaisesti rasvaa sisältävän ruoan kanssa annettaessa terveillä vapaaehtoisilla. Näin ollen lumakaftori/ivakaftori tulee antaa rasvaa sisältävän ruoan kanssa. Ivakaftorin Tmax-arvon mediaani (vaihteluväli) on noin 4,0 tuntia (2,0, 6,0) ravitussa tilassa.
Jakautuminen
Lumakaftorista noin 99 % sitoutuu plasman proteiineihin, etupäässä albumiiniin. Kun kystistä fibroosia sairastaville potilaille annettiin aterian yhteydessä suun kautta 400 mg 12 tunnin välein, tyypillisten näennäisten jakautumistilavuuksien keskustilassa ja perifeerisessä tilassa (variaatiokerroin prosenttiosuutena [CV]) arvioitiin olevan 23,5 l (48,7 %) ja 33,3 l (30,5 %).
Ivakaftorista noin 99 % sitoutuu plasman proteiineihin, etupäässä happamaan alfa-1-glykoproteiiniin ja albumiiniin. Kun ivakaftoria annettiin 250 mg suun kautta 12 tunnin välein yhdessä lumakaftorin kanssa, tyypillisten näennäisten jakautumistilavuuksien keskustilassa ja perifeerisessä tilassa (CV) arvioitiin olevan 95,0 l (53,9 %) ja 201 l (26,6 %).
In vitro -tutkimukset osoittavat lumakaftorin olevan rintasyöpäresistenssiproteiinin (BCRP) substraatti.
Biotransformaatio
Lumakaftori ei metaboloidu voimakkaasti ihmisillä; suurin osa lumakaftorista erittyy muuttumattomana ulosteeseen. In vitro- ja in vivo -tiedot viittaavat siihen, että lumakaftori metaboloituu pääasiassa hapettumalla ja glukuronidoitumalla.
Ivakaftori metaboloituu voimakkaasti ihmisillä. In vitro- ja in vivo -tiedot viittaavat siihen, että ivakaftori metaboloituu pääasiassa CYP3A-välitteisesti. M1 ja M6 ovat ivakaftorin kaksi päämetaboliittia ihmisillä. M1-metaboliitti vastaa teholtaan noin yhtä kuudesosaa ivakaftorista, ja sitä pidetään farmakologisesti aktiivisena. M6-metaboliitti vastaa teholtaan alle 1/50 ivakaftorista, ja sitä ei pidetä farmakologisesti aktiivisena.
Eliminaatio
Suun kautta annettaessa suurin osa lumakaftorista (51 %) erittyy muuttumattomana ulosteeseen. Lumakaftorin erittyminen virtsan kautta muuttumattomana lääkeaineena oli vähäistä. Näennäinen terminaalinen puoliintumisaika on noin 26 tuntia. Lumakaftorin tyypillisen näennäisen puhdistuman (CL/F) (CV) arvioitiin olevan 2,38 l/h (29,4 %) kystistä fibroosia sairastavilla potilailla.
Pelkkää ivakaftoria suun kautta annettaessa suurin osa ivakaftorista (87,8 %) erittyy ulosteeseen metabolisen muuntumisen jälkeen. Ivakaftorin erittyminen virtsan kautta muuttumattomana lääkeaineena oli vähäistä. Terveillä koehenkilöillä ivakaftorin puoliintumisaika lumakaftorin kanssa annettuna on noin 9 tuntia. Ivakaftorin tyypillisen näennäisen puhdistuman (CL/F) (CV) lumakaftorin kanssa annettuna arvioitiin olevan 25,1 l/h (40,5 %) kystistä fibroosia sairastavilla potilailla.
Erityisryhmät
Maksan vajaatoiminta
Useiden lumakaftori-/ivakaftoriannosten antaminen 10 vuorokauden ajan keskivaikeaa maksan vajaatoimintaa sairastaville koehenkilöille (Child‑Pugh-luokka B, pistemäärä 7–9) lisäsi altistusta terveisiin, demografisesti kaltaistettuihin koehenkilöihin verrattuna (AUC0‑12h-arvo oli noin 50 % suurempi ja Cmax-arvo noin 30 % suurempi). Lievän maksan vajaatoiminnan (Child‑Pugh-luokka A, pistemäärä 5–6) vaikutusta lumakaftorin farmakokinetiikkaan samanaikaisessa annossa ivakaftorin kanssa ei ole tutkittu, mutta altistuksen odotetaan lisääntyvän alle 50 %:lla.
Vaikeaa maksan vajaatoimintaa sairastavia potilaita (Child-Pugh-luokka C, pistemäärä 10–15) koskevia tutkimuksia ei ole tehty, mutta altistuksen odotetaan olevan suurempi kuin keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Munuaisten vajaatoiminta
Lumakaftoria/ivakaftoria koskevia farmakokineettisiä tutkimuksia ei ole tehty munuaisten vajaatoimintaa sairastavilla potilailla. Pelkästään lumakaftoria koskevassa ihmisillä tehdyssä farmakokineettisessä tutkimuksessa lumakaftorin ja sen metaboliittien eliminaatio virtsaan oli minimaalista (vain 8,6 % kokonaisradioaktiivisuudesta poistui virtsan kautta, ja siitä 0,18 % muuttumattomana kanta-aineena). Pelkästään ivakaftoria koskevassa ihmisillä tehdyssä farmakokineettisessä tutkimuksessa ivakaftorin ja sen metaboliittien eliminaatio virtsaan oli minimaalista (vain 6,6 % kokonaisradioaktiivisuudesta poistui virtsan kautta). Puhdistumaa ja kreatiniinin puhdistumaa koskeva populaatiofarmakokineettinen analyysi ei anna viitteitä trendistä lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla tutkittavilla (ks. kohta Annostus ja antotapa).
Iäkkäät potilaat
Lumakaftorin/ivakaftorin turvallisuutta ja tehoa 65 vuoden ikäisten tai sitä vanhempien potilaiden hoidossa ei ole arvioitu.
Sukupuoli
Sukupuolen vaikutusta lumakaftorin farmakokinetiikkaan arvioitiin populaatiofarmakokineettisellä analyysillä lumakaftorin ja ivakaftorin samanaikaista antoa koskevien kliinisten tutkimusten tiedoista. Tulokset eivät viitanneet kliinisesti merkittävään eroon lumakaftorin ja ivakaftorin farmakokineettisissä parametreissa miesten ja naisten välillä. Annosta ei tarvitse muuttaa sukupuolen perusteella.
Pediatriset potilaat
Populaatiofarmakokineettisten analyysien perusteella altistukset ovat samankaltaisia aikuisilla ja pediatrisilla potilailla. Altistukset on esitetty taulukossa 10.
Taulukko 10: Keskimääräinen (SD) lumakaftori- ja ivakaftorialtistus ikäryhmän mukaan
Ikäryhmä | Annos | Keskimääräinen lumakaftorin (SD) AUCss (μg∙h/ml) | Keskimääräinen ivakaftorin (SD) AUCss (μg∙h/ml) |
6–<12-vuotiaat potilaat | lumakaftori 200 mg / ivakaftori 250 mg 12 tunnin välein | 203 (57,4) | 5,26 (3,08) |
12–<18-vuotiaat potilaat | lumakaftori 400 mg / ivakaftori 250 mg 12 tunnin välein | 241 (61,4) | 3,90 (1,56) |
≥18-vuotiaat potilaat | lumakaftori 400 mg / ivakaftori 250 mg 12 tunnin välein | 198 (64,8) | 3,66 (2,25) |
Prekliiniset tiedot turvallisuudesta
Lumakaftori
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, karsinogeenisuutta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Erillisiä lumakaftorin valotoksisuutta arvioivia tutkimuksia ei ole tehty; saatavilla olevat ei-kliiniset ja kliiniset tiedot eivät kuitenkaan viittaa valotoksisuuteen.
Ivakaftori
Haittoja on koe-eläimillä todettu vain silloin, kun on käytetty altistusta, joka ylittää suurimman ihmisille käytettävän ivakaftoriannostuksen Orkambi-valmisteena annettaessa niin huomattavasti (>25‑kertaisesti hiirillä, >45‑kertaisesti rotilla ja >35‑kertaisesti koirilla), että asialla on kliinisen käytön kannalta vain vähäinen merkitys. Genotoksisuutta ja karsinogeenisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Farmakologinen turvallisuus
Ivakaftorilla oli pitoisuudesta riippuvainen estävä vaikutus hERG (human ether‑à‑go‑go related gene) -geenin häntävirtoihin; IC15-arvo oli 5,5 µM. Ivakaftorin Cmax-arvo lumakaftorin/ivakaftorin terapeuttisella annoksella on 1,5 µM. Ivakaftorin ei kuitenkaan havaittu aiheuttavan QT-ajan pidentymistä telemetriatutkimuksessa, jossa koirille annettiin enintään 60 mg/kg:n kerta-annoksia, eikä EKG-mittauksissa toistuvaa altistusta koskevissa tutkimuksissa, joissa koirille annettiin 60 mg/kg/vrk:n annoksia enintään 1 vuoden ajan (Cmax 365 vuorokauden jälkeen = 36,2–47,6 μM). Ivakaftori kohotti koirien verenpaineparametrejä tilapäisesti suhteessa annokseen suun kautta annettuina enintään 60 mg/kg:n kerta-annoksina (ks. kohta Farmakodynamiikka).
Raskaus ja hedelmällisyys
Ivakaftori ei ollut teratogeeninen, kun sitä annettiin suun kautta tiineille rotille ja kaniineille sikiön organogeneesin aikana annoksina, jotka vastasivat 7-kertaista (altistus ivakaftorille ja metaboliiteille) ja 46-kertaista ihmisen altistusta ivakaftorille terapeuttisella lumakaftori-/ivakaftoriannoksella. Emolle toksisina annoksina rotilla ivakaftori aiheutti sikiöiden painon alentumista, kaulakylkiluiden variaatioiden, hypoplastisten kylkiluiden ja aaltoilevien kylkiluiden esiintyvyyden lisääntymistä sekä rintalastan poikkeavuuksia mukaan lukien fuusioitumista. Näiden löydösten merkitystä ihmiselle ei tunneta.
Ivakaftori huononsi hedelmällisyyttä ja lisääntymiskykyä mittaavia indeksejä uros- ja naarasrotilla 200 mg/kg/vrk:n annoksilla (altistukset vastaavat uroksilla noin 11-kertaista ja naarailla noin 7-kertaista altistusta ihmiselle suositellulla Orkambi-valmisteen ivakaftorikomponentin enimmäisannoksella, ja ne perustuvat ivakaftorin ja sen metaboliittien yhteenlaskettuihin AUC-arvoihin rotilla tehdyn 6 kuukautta kestäneen toistuvan annoksen toksisuutta koskevan tutkimuksen vuorokautena 90 altistuksella 150 mg/kg/vrk sekä rottien alkioiden ja sikiöiden kehitystä koskevan pilottitutkimuksen tiineysvuorokauden 17 altistuksella). Naaraat saivat ivakaftoria ennen tiineyttä ja tiineyden alkuvaiheessa. Vaikutuksia urosten ja naaraiden hedelmällisyyttä ja lisääntymiskykyä mittaaviin indekseihin ei havaittu ≤100 mg/kg/vrk:n annoksilla (altistukset vastaavat uroksilla noin 8-kertaista ja naarailla noin 5-kertaista altistusta ihmiselle suositellulla Orkambi-valmisteen ivakaftorikomponentin enimmäisannoksella, ja ne perustuvat ivakaftorin ja sen metaboliittien yhteenlaskettuihin AUC-arvoihin rotilla tehdyn 6 kuukautta kestäneen toistuvan annoksen toksisuutta koskevan tutkimuksen vuorokautena 90 altistuksella 100 mg/kg/vrk sekä rottien alkioiden ja sikiöiden kehitystä koskevan tutkimuksen tiineysvuorokauden 17 altistuksella). Ivakaftorin havaittiin läpäisevän tiineiden rottien ja kaniinien istukan.
Peri- ja postnataalinen kehitys
Ivakaftori ei aiheuttanut kehityshäiriöitä, kun tiineille rotille annettiin suun kautta 100 mg/kg/vrk tiineydestä poikasten vieroitukseen asti (altistus vastaa noin 4‑kertaista altistusta ihmiselle suositellulla Orkambi-valmisteen ivakaftorikomponentin enimmäisannoksella, ja se perustuu ivakaftorin ja sen metaboliittien yhteenlaskettuihin AUC-arvoihin). Yli 100 mg/kg/vrk:n annoksilla eloonjääntiä ja imetystä mittaavat indeksit olivat 92 % ja 98 % vertailuarvoista, ja nämä annokset johtivat poikasten painon alentumiseen.
Nuoret eläimet
Nuorilla rotilla havaittiin kaihia, kun niille oli annettu ivakaftoria annoksina, jotka vastasivat 0,32-kertaista ihmiselle suositeltua enimmäisannosta mitattuna systeemisenä altistuksena ivakaftorille ja sen metaboliiteille lumakaftorin kanssa, Orkambi-valmisteena annettaessa. Kaihia ei havaittu sellaisten rottaemojen sikiöillä, joita oli hoidettu sikiön organogeneesin aikana, rotanpoikasilla, jotka olivat ennen vieroitusta altistuneet jossain määrin rintamaidon kautta, tai ivakaftorilla tehdyissä toistuvan annoksen toksisuutta koskevissa tutkimuksissa. Näiden löydösten mahdollista merkitystä ihmiselle ei tunneta.
Lumakaftori ja ivakaftori
Toistuvan annoksen toksisuutta koskevat tutkimukset, joissa lumakaftoria ja ivakaftoria annettiin yhdessä, eivät viitanneet erityiseen additiivisen ja/tai synergisen toksisuuden vaaraan ihmiselle.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa
Kroskarmelloosinatrium
Hypromelloosiasetaattisuksinaatti
Povidoni (K30)
Natriumlauryylisulfaatti
Magnesiumstearaatti
Päällyste
Polyvinyylialkoholi
Titaanidioksidi (E171)
Makrogoli 3350
Talkki
Karmiini (E120)
Briljanttisininen FCF -alumiinilakka (E133)
Indigokarmiini-alumiinilakka (E132)
Painomuste
Sellakka
Musta rautaoksidi (E172)
Propyleeniglykoli
Väkevä ammoniakki
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Orkambi 100 mg/125 mg tabletti, kalvopäällysteinen
3 vuotta
Orkambi 200 mg/125 mg tabletti, kalvopäällysteinen
4 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ORKAMBI tabletti, kalvopäällysteinen
100/125 mg (L:ei) 112 fol (4x28) (11139,34 €)
200/125 mg (L:ei) 112 fol (4x28) (11139,34 €)
PF-selosteen tieto
Polyklorotrifluoroetyleenistä (PCTFE) / polyvinyylikloridista (PVC) valmistettu läpipainopakkaus, jossa on paperilla päällystetty alumiinifoliokansi.
Orkambi 100 mg/125 mg tabletti, kalvopäällysteinen
Pakkaus, joka sisältää 112 (4 pakkausta, joissa kussakin 28) kalvopäällysteistä tablettia.
Orkambi 200 mg/125 mg tabletti, kalvopäällysteinen
Monipakkaus, joka sisältää 112 (4 pakkausta, joissa kussakin 28) kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Orkambi 100 mg/125 mg tabletti, kalvopäällysteinen
Vaaleanpunaiset, soikeat tabletit (mitat 14 × 7,6 × 4,9 mm), joiden toiselle puolelle on painettu mustalla musteella ”1V125”.
Orkambi 200 mg/125 mg tabletti, kalvopäällysteinen
Vaaleanpunaiset, soikeat tabletit (mitat 14 × 8,4 × 6,8 mm), joiden toiselle puolelle on painettu mustalla musteella ”2V125”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ORKAMBI tabletti, kalvopäällysteinen
100/125 mg 112 fol
200/125 mg 112 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Ivakaftorin ja lumakaftorin yhdistelmävalmiste: Kystisen fibroosin hoito vähintään 1-vuotiaille erityisin edellytyksin (3043).
ATC-koodi
R07AX30
Valmisteyhteenvedon muuttamispäivämäärä
26.03.2026
Yhteystiedot
Unit 49, Block 5, Northwood Court, Northwood Crescent
Dublin 9, D09 T665
Ireland
+358 8009 4476
www.vrtx.com
vertexmedicalinfo@vrtx.com