
ONCASPAR injektio/infuusiokuiva-aine, liuosta varten 750 U/ml
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 3 750 yksikköä (U)** pegaspargaasia*.
Käyttökuntoon saattamisen jälkeen 1 ml liuosta sisältää 750 yksikköä pegaspargaasia (750 U/ml).
* Vaikuttava aine on Escherichia coli -bakteerissa tuotetun L-asparaginaasin kovalenttinen konjugaatti monometoksipolyetyleeniglykolin kanssa
** Yksi yksikkö määritellään entsyymimääränä, joka tarvitaan vapauttamaan 1 µmol ammoniakkia minuutissa, kun pH on 7,3 ja lämpötila 37 °C
Tämän lääkevalmisteen vahvuutta ei pidä verrata toisen, samaan terapeuttiseen ryhmään kuuluvan pegyloidun tai pegyloimattoman proteiinin voimakkuuteen. Lisätietoja, ks. kohta Farmakodynamiikka.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektio-/infuusiokuiva-aine liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Oncaspar on tarkoitettu 0–18 vuoden ikäisten pediatristen potilaiden ja aikuisten potilaiden antineoplastisen yhdistelmähoidon osaksi akuuttiin lymfoblastileukemiaan (ALL).
Ehto
Valmistetta saavat määrätä ja antaa vain sellaiset lääkärit ja terveydenhuollon ammattilaiset, jotka ovat perehtyneet antineoplastisten valmisteiden käyttöön. Valmistetta saa antaa vain sairaalassa, jossa on asianmukaiset elvytyslaitteet.
Annostus ja antotapa
Oncaspar-valmistetta saavat määrätä ja antaa lääkärit ja/tai terveydenhuollon ammattilaiset, joilla on kokemusta antineoplastisten valmisteiden käytöstä. Sitä saa antaa vain sairaalaympäristössä, jossa on käytettävissä soveltuvat elvytyslaitteet. Potilaita on seurattava tarkasti haittavaikutusten varalta koko antojakson ajan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Oncaspar-valmistetta annetaan tavallisesti osana yhdistelmäsolunsalpaajahoito-ohjelmia muiden antineoplastisten aineiden kanssa (ks. myös kohta Yhteisvaikutukset).
Suositeltu esilääkitys
Anna potilaille esilääkityksenä parasetamolia, jotakin H1-reseptorisalpaajaa (esim. difenhydramiinia) ja jotakin H2-reseptorisalpaajaa (esim. famotidiinia) 30–60 minuuttia ennen Oncaspar-valmisteen antamista sekä infuusio- että yliherkkyysreaktioiden riskin ja vaikeusasteen vähentämiseksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat ja ≤ 21 vuoden ikäiset aikuiset
Suositeltava annos potilaille, joiden kehon pinta-ala (BSA) on ≥ 0,6 m2 ja jotka ovat ≤ 21 vuoden ikäisiä, on 2 500 U pegaspargaasia (vastaa 3,3 ml:aa Oncaspar-valmistetta) kehon pinta-alan neliömetriä (m²) kohden 14 päivän välein.
Lapsille, joiden kehon pinta-ala on < 0,6 m², pitää antaa 82,5 U pegaspargaasia (vastaa 0,1 ml:aa Oncaspar-valmistetta) kehon painokiloa kohden 14 päivän välein.
> 21 vuoden ikäiset aikuiset
Ellei toisin määrätä, suositeltava annostus > 21 vuoden ikäisille aikuisille on 2 000 U pegaspargaasia (vastaa 2,67 ml:a Oncaspar-valmistetta)/m2 BSA 14 päivän välein.
Hoitoa voidaan seurata seerumin asparaginaasin minimiaktiivisuuden perusteella. Tämä mitataan ennen seuraavaa pegaspargaasin antokertaa. Jos asparaginaasin aktiivisuusarvot eivät nouse tavoitetasolle, vaihtamista toiseen asparaginaasivalmisteeseen voidaan harkita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Erityisryhmät
Munuaisten toimintahäiriö
Koska pegaspargaasi on proteiini, jolla on suuri molekyylipaino, se ei erity munuaisten kautta, eikä munuaisten toimintahäiriötä sairastavien potilaiden annostusta tarvitse muuttaa.
Maksan toimintahäiriö
Maksan toimintahäiriötä sairastavien potilaiden annostusta ei tarvitse muuttaa.
Vanhukset
Yli 65 vuoden ikäisistä potilaista on saatavilla vain vähän tietoja.
Antotapa
Oncaspar-valmiste voidaan antaa injektiona lihakseen (i.m.) tai infuusiona laskimoon (i.v.).
Kun käytetään pieniä määriä, valmiste on suositeltavaa antaa lihakseen. Kun Oncaspar-valmiste annetaan lihaksensisäisenä injektiona, yhteen kohtaan saa injisoida enintään 2 ml, kun kyseessä on lapsi tai nuori, ja enintään 3 ml, kun kyseessä on aikuinen. Jos valmistetta annetaan suurempi määrä, annos on jaettava ja annettava useaan injektiokohtaan.
Kun Oncaspar annetaan infuusiona laskimoon, se annetaan tavallisesti 1–2 tunnin aikana 100 ml:ssa natriumkloridi-injektionestettä, joka on vahvuudeltaan 9 mg/ml (0,9 %), tai 5 %:n vahvuista dekstroosiliuosta.
Laimennettu liuos voidaan antaa yhdessä potilaan jo menossa olevan infuusion kanssa, joka on joko natriumkloridiliuosta (9 mg/ml) tai 5 %:n vahvuista glukoosiliuosta. Älä infusoi muita lääkevalmisteita saman laskimoyhteyden kautta Oncaspar-valmisteen annon aikana.
Ks. kohdasta Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Vaikea maksan toimintahäiriö (bilirubiini > 3 kertaa normaalin yläraja [ULN], transaminaasit > 10 kertaa ULN).
Vakava tromboosi aiemman L-asparaginaasihoidon yhteydessä.
Aikaisempi haimatulehdus, mukaan lukien aiempaan L-asparaginaasihoitoon liittyvä haimatulehdus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vakavat verenvuototapahtumat aiemman L-asparaginaasihoidon yhteydessä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Asparaginaasivasta-aineet
Asparaginaasin vasta-aineiden esiintymiseen saattaa liittyä matalia asparaginaasin aktiivisuustasoja, jotka johtuvatnäiden vasta-aineiden mahdollisesta neutraloivasta vaikutuksesta. Tällaisissa tapauksissa vaihtamista toiseen asparaginaasivalmisteeseen on harkittava.
Seerumin tai plasman asparaginaasin aktiivisuustaso voidaan mitata, jotta voidaan sulkea pois asparaginaasin aktiivisuuden kiihtyvä lasku.
Yliherkkyys
Yliherkkyysreaktioita pegaspargaasille, mukaan lukien hengenvaarallista anafylaksiaa, voi esiintyä hoidon aikana, mukaan lukien potilailla, joilla on tunnettu yliherkkyys E. coli ‑peräisille asparaginaasivalmisteille. Muita yliherkkyysreaktioita voivat olla angioödeema, huulten turvotus, silmien turvotus, eryteema, alentunut verenpaine, bronkospasmi, dyspnea, kutina ja ihottuma (ks. kohdat Vasta-aiheet ja Haittavaikutukset).
Anna potilaille esilääkitys 30–60 minuuttia ennen Oncaspar-valmisteen antamista (ks. kohta Annostus ja antotapa).
Tavanomaisena varotoimenpiteenä potilasta on seurattava tunnin ajan annon jälkeen; elvytyslaitteet ja muut asianmukaiset välineet anafylaksian hoitoon (adrenaliini, happi, laskimoon annettavat steroidit, jne.) on pidettävä saatavilla. Oncaspar-valmisteen anto on lopetettava, jos potilas saa vakavia yliherkkyysreaktioita (ks. kohdat Vasta-aiheet ja Haittavaikutukset). Antihistamiinien, kortikosteroidien ja vasopressorien anto voi olla aiheellinen vastatoimenpide oireiden vaikeuden mukaan.
Vaikutukset haimaan
Haimatulehduksia, mukaan lukien kuolemaan johtaneita hemorragisen tai nekrotisoivan haimatulehduksen tapauksia, on raportoitu Oncaspar-valmistetta saaneilla potilailla (ks. kohta Haittavaikutukset).
Potilaille on kerrottava haimatulehduksen merkeistä ja oireista, jotka voivat hoitamattomina johtaa kuolemaan.
Mikäli haimatulehdusta epäillään, Oncaspar-hoito on lopetettava; mikäli haimatulehdus vahvistuu, Oncaspar-hoitoa ei saa aloittaa uudelleen.
Seerumin amylaasi- ja/tai lipaasitasoja on seurattava usein haimatulehduksen varhaisten merkkien tunnistamiseksi. Veren glukoositasoja on seurattava, koska heikentynyt glukoositoleranssi voi ilmetä käytettäessä Oncaspar-valmistetta ja prednisonia samanaikaisesti.
Koagulopatia
Pegaspargaasia saavilla potilailla voi esiintyä vakavia tromboottisia tapahtumia, mukaan lukien sagittaalinen sinustromboosi (ks. kohta Haittavaikutukset). Oncaspar-valmisteen anto on lopetettava, jos potilaalla ilmenee tromboottisia tapahtumia.
Pegaspargaasia saavilla potilailla voi esiintyä protrombiiniajan (PT) ja osittaisen tromboplastiiniajan (PTT) lisääntymistä, hypofibrinogenemiaa ja antitrombiini III:n määrän pienenemistä. Koagulaatioparametreja on seurattava lähtötilanteessa ja määräajoin hoidon aikana ja sen jälkeen erityisesti, kun muita lääkevalmisteita, joilla on antikoagulanttivaikutuksia (esimerkiksi asetyylisalisyylihappoa ja tulehduskipulääkevalmisteita) käytetään samanaikaisesti (ks. kohta Yhteisvaikutukset) tai kun annetaan samanaikaisesti kemoterapiaa, joka sisältää metotreksaattia, daunorubisiinia tai kortikosteroideja. Kun potilaan fibrinogeenitaso on selvästi laskenut tai hänellä on antitrombiini III:n (ATIII) puutos, harkitse asianmukaista korvaushoitoa.
Osteonekroosi
Osteonekroosi (avaskulaarinen nekroosi) on glukokortikoidien samanaikaisen käytön yhteydessä hyperkoagulaation mahdollinen komplikaatio, jota havaitaan lapsilla ja nuorilla, ja yleisemmin tytöillä (ks. kohdat Yhteisvaikutukset ja Haittavaikutukset). Sen vuoksi nuorten ja lapsipotilaiden huolellinen seuranta on suositeltavaa, jotta osteonekroosin kliiniset oireet tai löydökset havaitaan. Kunkin potilaan hoitosuunnitelman pitää perustua hoitavan lääkärin kliiniseen arvioon, joka puolestaan perustuu akuutin lymfoblastileukemian tavanomaisten hoito-ohjeistojen mukaiseen yksilöllisen hyöty–riskiarvioon ja tukihoitoperiaatteisiin.
Vaikutukset maksaan
Yhdistelmähoidosta Oncaspar-valmisteen ja muiden hepatotoksisten valmisteiden kanssa voi seurata vaikea maksatoksisuus.
Varovaisuutta on noudatettava, kun Oncaspar-valmistetta annetaan yhdessä hepatotoksisten valmisteiden kanssa, erityisesti, jos potilaalla on maksan toimintahäiriö. Potilasta on seurattava maksan toimintaparametrien muuttumisen varalta.
On olemassa mahdollinen hepatotoksisuuden lisääntynyt riski Philadelphia-kromosomipositiivisilla potilailla, joiden tyrosiinikinaasi-inhibiittorihoito (esim. imatinibi) yhdistetään L-asparaginaasihoitoon. Tämä on otettava huomioon harkittaessa Oncaspar-valmisteen käyttöä näissä potilasryhmissä.
Maksan veno-okklusiivista sairautta (VOD), joka voi joskus olla vakava, henkeä uhkaava ja mahdollisesti kuolemaan johtava sairaus, on havaittu potilailla, joita on hoidettu Oncaspar-valmisteella yhdessä tavanomaisen solunsalpaajahoidon kanssa, myös monivaiheisen solunsalpaajahoidon induktiovaiheen aikana (ks. kohta Haittavaikutukset).
VOD:n merkkejä ja oireita ovat nopea painonnousu, nesteen kertyminen ja askites, hepatomegalia, trombosytopenia ja bilirubiinin määrän nopea nousu. VOD:n ehkäisemiseksi on olennaista tunnistaa sen riskitekijät, kuten jo olemassa oleva maksasairaus tai aiemmin esiintynyt VOD. On ehdottoman välttämätöntä tunnistaa VOD nopeasti, ja siihen on annettava asianmukaista hoitoa. Potilaita, joilla tämä tila ilmenee, tulee hoitaa tavanomaisen hoitokäytännön mukaisesti.
Hyperbilirubinemiavaaran vuoksi on suositeltavaa tarkkailla bilirubiinitasoja lähtötilanteessa ja aina ennen kutakin annosta.
Vaikutukset keskushermostoon
Yhdistelmähoidosta Oncaspar-valmisteen kanssa voi seurata keskushermoston toksisuus. Enkefalopatiatapauksia (mukaan lukien reversiibeliä posteriosta leukoenkefalopatiaoireyhtymää) on raportoitu (ks. kohta Haittavaikutukset).
Oncaspar voi aiheuttaa keskushermosto-oireita, jotka ilmenevät uneliaisuutena, sekavuutena ja kouristuksina. Potilaita on seurattava tarkasti tällaisten oireiden varalta, erityisesti, jos Oncaspar-valmistetta käytetään neurotoksisten valmisteiden yhteydessä (kuten vinkristiinin ja metotreksaatin kanssa; ks. kohta Yhteisvaikutukset).
Myelosuppressio
Pegaspargaasi voi aiheuttaa myelosuppressiota joko suorasti tai epäsuorasti (muuttamalla muiden aineiden, kuten metotreksaatin tai 6-merkaptopuriinin, myelosuppressoivia vaikutuksia). Tämän vuoksi Oncaspar voi lisätä infektioiden riskiä.
Kiertävien lymfoblastien määrän väheneminen on usein merkittävää, ja hoidon aloittamisen jälkeisinä ensimmäisinä päivinä leukosyyttien määrä on usein normaali tai liian vähäinen. Tähän voi liittyä seerumin virtsahappopitoisuuden merkittävää suurenemista. Potilaalle voi kehittyä uraattinefropatia. Hoidon vaikutuksen seuraamiseksi perifeeristä verisolujen määrää ja potilaan luuydintä on seurattava tarkasti.
Hyperammonemia
Asparaginaasi välittää asparagiinin ja glutamiinin nopeaa muuttumista asparagiinihapoksi ja glutamiinihapoksi ammoniakin ollessa molempien reaktioiden yhteinen sivutuote (ks. kohta Farmakodynamiikka). Asparaginaasin antaminen laskimoon voi siten saada seerumin ammoniakkitasot nousemaan voimakkaasti antamisen jälkeen.
Hyperammonemian oireet ovat usein ohimeneviä ja niitä voivat olla pahoinvointi, oksentelu, päänsärky, huimaus ja ihottuma. Vaikeissa tapauksissa voi kehittyä enkefalopatia erityisesti iäkkäille. Enkefalopatiaan saattaa liittyä maksan vajaatoiminta ja se voi olla hengenvaarallista tai johtaa kuolemaan. Jos hyperammonemian oireita esiintyy, ammoniakkitasoja on seurattava tarkasti.
Ehkäisy
Oncaspar-hoidon aikana ja vähintään 6 kuukautta Oncaspar-valmisteen käytön jälkeen on käytettävä tehokasta, muuta kuin suun kautta otettavaa ehkäisyä. Koska suun kautta otettavien ehkäisyvalmisteiden ja pegaspargaasin välistä epäsuoraa yhteisvaikutusta ei voida sulkea pois, suun kautta otettavan ehkäisyvalmisteen käyttö ei ole hyväksyttävä ehkäisymenetelmä (ks. kohdat Yhteisvaikutukset ja Raskaus ja imetys).
Natriumsisältö
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Pegaspargaasin aiheuttama seerumin proteiinipitoisuuden pieneneminen voi lisätä muiden proteiiniin sitoutuvien lääkevalmisteiden toksisuutta.
Lisäksi pegaspargaasi voi proteiinisynteesiä ja solunjakautumista estämällä häiritä muiden sellaisten lääkeaineiden toimintaa, joiden teho edellyttää solunjakautumista, esimerkkinä metotreksaatti.
Metotreksaatti ja sytarabiini voivat vaikuttaa Oncaspar-valmisteeseen eri tavoin: näiden aineiden aiempi anto voi lisätä pegaspargaasin vaikutusta synergistisesti. Jos näitä aineita annetaan pegaspargaasin jälkeen, pegaspargaasin teho voi heikentyä antagonistisesti.
Pegaspargaasi voi häiritä muiden lääkevalmisteiden metaboliaa ja puhdistumaa sen proteiinisynteesiin ja maksan toimintaan kohdistuvien vaikutustensa vuoksi sekä käytettäessä sitä samanaikaisesti muiden CYP-entsyymeihin vaikuttavien kemoterapiavalmisteiden kanssa.
Oncaspar-valmisteen käyttö voi aiheuttaa hyytymistekijöiden vaihtelua. Tämä voi edistää verenvuototaipumusta ja/tai tromboosien esiintymistä. Siksi on oltava varovainen, kun antikoagulantteja, kuten kumariinia, hepariinia, dipyridamolia, asetyylisalisyylihappoa tai ei-steroidaalisia tulehduskipulääkevalmisteita annetaan samanaikaisesti tai kun käytetään samanaikaisesti kemoterapiaa, joka sisältää metotreksaattia, daunorubisiinia tai kortikosteroideja.
Kun glukokortikoideja (esim. prednisonia) ja pegaspargaasia annetaan samanaikaisesti, koagulaatioparametrien muutokset (esimerkiksi fibrinogeenipitoisuuden lasku ja antitrombiini III:n puutos, ATIII) voivat korostua.
Samanaikaisessa käytössä pegaspargaasi voi mahdollisesti suuremman deksametasonialtistuksen vuoksi lisätä glukokortikoidien indusoiman osteonekroosin riskiä lapsilla ja nuorilla, ja sen ilmaantuvuus on tytöillä suurempi (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Välittömästi edeltävä tai samanaikainen vinkristiinihoito voi lisätä pegaspargaasin toksisuutta. Oncaspar-valmisteen anto ennen vinkristiiniä saattaa lisätä vinkristiinin neurotoksisuutta. Siksi vinkristiini on annettava vähintään 12 tuntia ennen Oncaspar-valmisteen antoa, jotta toksisuus voidaan minimoida.
Pegaspargaasin ja suun kautta otettavien ehkäisyvalmisteiden epäsuoraa yhteisvaikutusta ei voida sulkea pois, sillä pegaspargaasin hepatotoksisuus voi heikentää suun kautta otettavien ehkäisyvalmisteiden poistumaa maksan kautta. Siksi Oncaspar-valmisteen käyttöä samanaikaisesti suun kautta otettavien ehkäisyvalmisteiden kanssa ei suositella. Hedelmällisessä iässä olevien naisten on käytettävä muuta kuin suun kautta otettavaa ehkäisyä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys).
Samanaikainen rokottaminen elävillä rokotteilla voi lisätä sellaisten vaikeiden infektioiden riskiä, jotka johtuvat pegaspargaasin immunosuppressiivisesta vaikutuksesta, potilaan perussairaudesta ja yhdistelmäsolunsalpaajahoidosta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Rokottaminen elävillä rokotteilla tulisi tehdä aikaisintaan 3 kuukautta koko leukemiahoidon lopettamisen jälkeen.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset / ehkäisy miehille ja naisille
Miesten ja naisten on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 6 kuukautta Oncaspar-valmisteen käytön jälkeen. Koska suun kautta otettavien ehkäisyvalmisteiden ja pegaspargaasin välistä epäsuoraa yhteisvaikutusta ei voida sulkea pois, suun kautta otettavia ehkäisyvalmisteita ei pidetä riittävän turvallisina tällaisessa kliinisessä tilanteessa. Hedelmällisessä iässä olevien naisten on käytettävä muuta kuin suun kautta otettavia ehkäisyvalmisteita (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Raskaus
On vain vähän tietoja L-asparaginaasin käytöstä ja ei lainkaan tietoja Oncaspar-valmisteen käytöstä raskaana oleville naisille. Pegaspargaasilla ei ole tehty lisääntymistä koskevia eläinkokeita, mutta L-asparaginaasilla tehdyissä eläinkokeissa on havaittu teratogeenisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta). Tämän vuoksi ja Oncaspar-valmisteen farmakologisten ominaisuuksien vuoksi Oncaspar-valmistetta ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa pegaspargaasilla.
Imetys
Ei tiedetä, erittyykö pegaspargaasi rintamaitoon. Sen farmakologisten ominaisuuksien perusteella rintaruokittuun vastasyntyneeseen/imeväiseen kohdistuvaa riskiä ei voida sulkea pois. Varotoimenpiteenä imetys on lopetettava Oncaspar-hoidon aikana, eikä sitä pidä aloittaa uudelleen ennen kuin Oncaspar-hoidon keskeyttämisen jälkeen.
Hedelmällisyys
Tutkimuksia pegaspargaasin vaikutuksesta hedelmällisyyteen ei ole tehty.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Oncaspar-valmisteella on huomattava vaikutus ajokykyyn tai koneidenkäyttökykyyn. Seuraavia haittavaikutuksia on raportoitu potilailla, jotka ovat saaneet Oncaspar-valmistetta ja muuta kemoterapiavalmistetta: uneliaisuutta, sekavuutta, huimausta, pyörtymistä ja kohtauksia.
Potilaita on neuvottava olemaan ajamatta tai käyttämättä koneita Oncaspar-hoidon aikana, jos heillä ilmenee näitä tai muita haittavaikutuksia, jotka voivat heikentää heidän ajokykyään tai koneidenkäyttökykyään (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Tässä osiossa kuvatut haittavaikutukset ovat peräisin kliinisten tutkimusten tiedoista ja Oncaspar-valmisteen markkinoille tulon jälkeisistä, akuuttia lymfoblastileukemiaa (ALL) sairastavia potilaita koskevista kokemuksista. Turvallisuusprofiili perustuu satunnaistettuihin, kontrolloituihin, prospektiivisiin, avoimiin monikeskustutkimuksiin, joissa Oncaspar-annos 2 500 U/m2 annettiin laskimoon vertailuhoitona (tutkimukset DFCI 11-001 ja AALL07P4). Lisäksi turvallisuusprofiilin määrittämisessä otettiin huomioon tietoja myös muista Oncaspar-tutkimuksista, kuten tutkimuksesta, jossa verrattiin pegaspargaasin nestemäisen valmistemuodon ja lyofilisoidun valmistemuodon farmakokinetiikkaa (CL2-95014-002), sen roll-over-tutkimuksesta (CL2-95014-003) sekä tutkimuksista, joissa valmiste annettiin lihakseen (tutkimukset CCG‑1962 ja CCG‑1991) (tiedot tutkimuksista CCG-1962 ja CCG-1991, ks. kohta Farmakodynamiikka).
Yleisimmät Oncaspar-valmisteeseen liittyvät haittavaikutukset (joita havaittiin vähintään kahdessa tutkimuksessa esiintymistiheydellä > 10 %) olivat alaniiniaminotransferaasiarvon nousu, aspartaattiaminotransferaasiarvon nousu, veren bilirubiinimäärän suureneminen,aktivoidun osittaisen tromboplastiiniajan piteneminen, hypertriglyseridemia, hyperglykemia ja kuumeinen neutropenia.
Yleisimmät vaikeat Oncaspar-valmisteeseen liittyvät haittavaikutukset (luokaltaan 3 tai 4), joita havaittiin tutkimuksissa DFCI 11‑001 ja AALL07P4 esiintymistiheydellä > 5 %, olivat alaniiniaminotransferaasiarvon nousu, aspartaattiaminotransferaasiarvon nousu, veren bilirubiinimäärän suureneminen, kuumeinen neutropenia, hyperglykemia, lipaasiarvon nousu ja haimatulehdus.
Haittavaikutustaulukko
Haittavaikutukset ja niiden esiintymistiheydet esitetään taulukossa 1. Haittavaikutusten esiintymistiheydet on määritetty seuraavalla tavalla: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Oncaspar-hoidon yhteydessä raportoidut haittavaikutukset
| MedDRAn elinjärjestelmäluokitus | Haittavaikutukset |
|---|---|
| Infektiot | Yleiset: infektiot, sepsis |
| Veri ja imukudos | Hyvin yleiset: kuumeinen neutropenia |
| Yleiset: anemia, koagulopatia | |
| Tuntematon: luuytimen vajaatoiminta | |
| Immuunijärjestelmä | Hyvin yleiset: yliherkkyys, nokkosihottuma, anafylaktinen reaktio |
| Tuntematon: anafylaktinen sokki | |
| Aineenvaihdunta ja ravitsemus | Hyvin yleiset: vähentynyt ruokahalu, hyperglykemia |
| Yleiset: hyperlipidemia, hyperkolesterolemia | |
| Tuntematon: diabeettinen ketoasidoosi, hypoglykemia | |
| Psyykkiset häiriöt | Tuntematon: sekavuustila |
| Hermosto | Yleiset: kohtaus, perifeerinen motorinen neuropatia, synkopee |
| Harvinaiset: posteriorinen reversiibeli leukoenkefalopatiaoireyhtymä | |
| Tuntematon: uneliaisuus, vapina* | |
| Verisuonisto | Hyvin yleiset: embolia** |
| Yleiset: tromboosi*** | |
| Tuntematon: aivoverisuonitapahtuma, verenvuoto, superiorisen sagittaalisen sinuksen tromboosi | |
| Hengityselimet, rintakehä ja välikarsina | Yleiset: hypoksia |
| Ruoansulatuselimistö | Hyvin yleiset: haimatulehdus, ripuli, vatsakipu, pahoinvointi |
| Yleiset: oksentelu, stomatiitti, askites | |
| Harvinaiset: nekrotisoiva haimatulehdus, hemorraginen haimatulehdus | |
| Tuntematon: haiman pseudokysta, parotiitti* | |
| Maksa ja sappi | Yleiset: hepatotoksisuus, rasvamaksa |
| Harvinaiset: maksanekroosi, keltaisuus, kolestaasi, maksan vajaatoiminta | |
| Tuntematon: veno-okklusiivinen sairaus | |
| Iho ja ihonalainen kudos | Hyvin yleiset: ihottuma |
| Tuntematon: toksinen epidermaalinen nekrolyysi* | |
| Luusto, lihakset ja sidekudos | Yleiset: raajakipu |
| Tuntematon: osteonekroosi (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset) | |
| Munaiset ja virtsatiet | Tuntematon: akuutti munuaisten vajaatoiminta* |
| Yleisoireet ja antopaikassa todettavat haitat | Tuntematon: kuume |
| Tutkimukset | Hyvin yleiset: painonlasku, hypoalbuminemia, alaniiniaminotransferaasiarvon nousu, aspartaattiaminotransferaasiarvon nousu, hypertriglyseridemia, veren fibrinogeenimäärän pieneneminen, lipaasiarvon nousu, amylaasipitoisuuden suureneminen, aktivoidun osittaisen tromboplastiiniajan piteneminen, veren bilirubiinipitoisuuden suureneminen, antitrombiini III:n määrän pieneneminen****, neutrofiilimäärän pieneneminen**** |
| Yleiset: protrombiiniajan piteneminen, INR-arvon nousu, hypokalemia, veren kolesteroliarvon nousu, hypofibrinogenemia, gammaglutamyylitransferaasiarvon nousu | |
| Tuntematon: veren ureapitoisuuden suureneminen, pegaspargaasin lääkevasta-aineet, verihiutalemäärän pieneneminen, hyperammonemia |
*Haittavaikutuksia havaittu toisten luokan asparaginaasien kanssa
**Keuhkoembolia-, laskimotromboosi-, raajan laskimotromboosi- ja pinnallisia tromboflebiittitapauksia havaittiin DFCI 11-001 ‑tutkimuksessa.
***Selitys: keskushermoston tromboosi
**** Antitrombiini III:n ja neutrofiilien määrän pienenemistä havaittiin tutkimuksissa CL2-95014-002 ja CL2 95014 003
Kuvaus valikoiduista haittavaikutuksista
Asparaginaasihoidon yhteydessä on havaittu esiintyneen seuraavia haittavaikutuksia. Vaikka niitä ei ole yhdistetty erityisesti pegaspargaasiin, niitä voi esiintyä Oncaspar-valmisteen käytön yhteydessä:
Veri ja imukudos
Oncaspar voi aiheuttaa lievää tai kohtalaista luuydinlamaa ja vaikuttaa kaikkiin kolmeen verisolulinjaan.
Noin puolet kaikista vakavista verenvuototapauksista ja trombooseista vaikuttavat aivoverisuoniin ja voivat aiheuttaa esimerkiksi aivohalvauksia, kohtauksen, päänsärkyä tai tajuttomuuskohtauksia.
Hermosto
Oncaspar voi aiheuttaa keskushermoston häiriöitä, jotka ilmenevät kouristuksina ja harvemmin sekavuutena ja uneliaisuutena (lievästi heikentyneenä tajuntana).
Harvinaisissa tapauksissa voi ilmetä reversiibeli posteriorinen leukoenkafalopatiaoireyhtymä (RPLS).
Hyvin harvinaisissa tapauksissa on ilmoitettu sormien lievästä vapinasta.
Ruoansulatuselimistö
Noin puolelle potilaista kehittyy lieviä tai kohtalaisia ruoansulatuselimistön reaktioita, kuten ruokahaluttomuutta, pahoinvointia, oksentelua, vatsakramppeja, ripulia ja painon laskua.
Akuuttia haimatulehdusta voi esiintyä yleisesti. Yksittäisiä pseudokystatapauksia on raportoitu (enintään neljä kuukautta viimeisen hoidon jälkeen).
Hemorragisia tai nekroottisia haimatulehduksia esiintyy harvoin. Yhdestä haimatulehdustapauksesta samanaikaisen akuutin parotiitin kanssa on raportoitu L-asparaginaasihoidon yhteydessä. Yksittäisiä kuolemaan johtaneita hemorragisia tai nekroottisia haimatulehdustapauksia on raportoitu.
Seerumin amylaasipitoisuus voi nousta Oncaspar-hoidon aikana ja myös sen päättymisen jälkeen.
Munuaiset ja virtsatiet
Akuutti munuaisten vajaatoiminta voi kehittyä harvoissa tapauksissa L-asparaginaasia sisältävien hoitojen aikana.
Iho ja ihonalainen kudos
Iholla voi ilmetä allergisia reaktioita. L-asparaginaasin yhteydessä on raportoitu yhdestä toksisen epidermaalisen nekrolyysin tapauksesta (Lyellin oireyhtymä).
Umpieritys
Endokriinisen haimatoiminnan muutoksia on havaittu yleisesti, ja ne ilmenevät pääasiassa epänormaalin glukoosimetabolian muodossa. Sekä diabeettistä ketoasidoosia että hyperosmolaarista hyperglykemiaa on kuvattu, ja ne reagoivat tavallisesti insuliiniin.
Aineenvaihdunta ja ravitsemus
Seerumin lipiditasojen muutos havaittiin, ja seerumin lipidiarvojen muutokset, joihin ei useimmissa tapauksissa liity kliinisiä oireita, ovat hyvin yleisiä.
Seerumin ureapitoisuuden suurenemista esiintyy säännöllisesti, se on annoksesta riippumatonta ja lähes aina munuaisten metabolisen epätasapainon esiasteen oire.
Yleisoireet ja antopaikassa todettavat haitat
Kuumetta voi esiintyä injektion jälkeen, ja se menee tavallisesti ohi itsestään.
Immuunijärjestelmä
Pegaspargaasille spesifejä vasta-aineita on havaittu. Ne liittyivät melko harvoin yliherkkyysreaktioihin. Myös kliinistä tehoa neutraloivia vasta-aineita havaittiin.
Hoidon aikana voi ilmetä yliherkkyysreaktioita Oncaspar-valmisteelle, mukaan lukien henkeä uhkaava anafylaksia, angioedeemaa, huulten turpoamista, silmien turpoamista, ihon punoitusta, verenpaineen laskua, bronkospasmi, hengenahdistusta, kutinaa ja ihottumaa (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Maksa- ja sappihäiriöt
Maksaparametrien muutokset ovat yleisiä. Seerumin transaminaasi- ja bilirubiinipitoisuuksien annoksesta riippumatonta suurenemista esiintyy yleisesti.
Nopea painonnousu, nesteen kertyminen ja askites, hepatomegalia, joihin liittyy seerumin bilirubiinin nopea nousu ja persistentti trombosytopenia, voivat viitata riskiin sairastua vaikea-asteiseen VOD:hen, joka hoitamattomana voi johtaa kuolemaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Rasvamaksaa esiintyy hyvin yleisesti. Kolestaasia, keltaisuutta, maksasolunekroosia ja kuolemaan johtavaa maksan vajaatoimintaa on raportoitu.
Heikentynyt proteiinisynteesi voi aiheuttaa seerumin proteiinipitoisuuden pienenemistä. Suurimmalla osalla potilaista esiintyy hoidon aikana annoksesta riippumatonta seerumin albumiinipitoisuuden pienenemistä.
Oncaspar-valmisteen haittavaikutustyypit ovat samankaltaisia natiivin pegyloimattoman L‑asparaginaasin (esimerkiksi natiivin E. coli -asparaginaasin) yhteydessä havaittujen haittavaikutusten kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
FI-00034 Fimea
Yliannostus
Oncaspar-valmisteen käytön yhteydessä on raportoitu tahattomia yliannostustapauksia. Yliannostuksen jälkeen on esiintynyt maksaentsyymiarvojen kohoamista, ihottumaa ja hyperbilirubinemiaa. Spesifiä farmakologista hoitoa yliannostukseen ei ole. Yliannostustapauksessa potilasta on seurattava tarkasti haittavaikutusten merkkien ja oireiden varalta ja hoidettava oireenmukaisella ja tukevalla hoidolla.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset ja immunomoduloivat aineet, muut antineoplastiset aineet, ATC-koodi: L01XX24
Vaikutusmekanismi
L-asparaginaasin vaikutusmekanismi on L-asparagiiniaminohapon entsymaattinen pilkkoutuminen asparagiinihapoksi ja ammoniakiksi. L-asparagiinin ehtyminen verestä aiheuttaa proteiinisynteesin, DNA-synteesin ja RNA-synteesin estymistä. Tämä koskee erityisesti leukemiablasteja, jotka eivät kykene syntetisoimaan L-asparagiinia ja läpikäyvät sen vuoksi apoptoosin.
Normaalit solut sen sijaan kykenevät L-asparagiinin syntetisoimiseen, ja L-asparagiinin nopea ehtyminen L-asparaginaasientsyymihoidon aikana vaikuttaa niihin vähemmän. Pegylointi ei muuta L-asparaginaasin entsymaattisia ominaisuuksia, mutta se vaikuttaa entsyymin farmakokinetiikkaan ja immunogeenisuuteen.
Farmakodynaamiset vaikutukset
L-asparaginaasin leukemiaa parantava teho liittyy L-asparagiinin pitkittyneeseen ehtymiseen verestä ja aivo-selkäydinnesteestä (CSF). Oncaspar-valmisteen farmakodynaaminen vaikutus arvioitiin lihakseen (tutkimus CCG-1962) ja laskimoon (AALL07P4) antamisen jälkeen.
Tutkimuksessa CCG-1962 Oncaspar-valmisteen farmakodynamiikkaa arvioitiin asparagiinin sarjamittauksilla seerumista (n = 57) ja aivo-selkäydinnesteeestä (n = 50) äskettäin diagnosoidulla pediatrisella potilaalla, joilla oli normaalin riskin akuutti lymfoblastileukemia ja jotka saivat kolme Oncaspar-annosta lihakseen (2 500 yksikköä/m2 BSA), yhden induktiohoidon aikana ja kaksi viivästetyn tehostuksen hoitojakson aikana. Seerumin asparagiinipitoisuuden pieneneminen oli havaittavissa 4. päivänä ensimmäisen induktioannoksen jälkeen. Pitoisuus saavutti ilmeisen pohja-arvonsa 10. päivään mennessä annoksen jälkeen. Seerumin asparagiinipitoisuudet noin 1 µM säilyivät noin 3 viikon ajan. Asparagiinipitoisuus pieneni < 3 µM:iin, kun asparaginaasin aktiivisuus oli > 0,1 U/ml. Aivo-selkäydinnesteen asparagiinitaso 2,3 µM ennen hoitoa laski 1,1 µM:iin induktion päivänä 7 ja 0,6 µM:iin päivänä 28 (ks. Kliininen teho ja turvallisuus).
Tutkimuksessa AALL07P4 Oncaspar-valmisteen farmakodynaaminen vaikutus arvioitiin 47 evaluoitavissa olevalla tutkittavalla, joilla oli korkean riskin B-esiaste-ALL ja jotka saivat laskimoinfuusiona Oncaspar-annoksia 2 500 U/m2 BSA induktio- ja konsolidaatiovaiheen aikana. Plasman L‑asparagiinipitoisuudet olivat ehtyneet kvantifioinnin määritysrajan alle 24 tuntia induktion jälkeen ja ensimmäisen Oncaspar-konsolidaatioannoksen jälkeen. Ehtyminen säilyi noin kaksi viikkoa. Aivo-selkäydinnesteen asparagiinipitoisuudet pienenivät neljänteen induktioannoksen jälkeiseen päivään mennessä ja pysyivät pääosin havaitsemattomissa 18. päivään annoksen antamisen jälkeen.
Näiden kahden tutkimuksen tulosten perusteella Oncaspar-annos 2 500 U/m2 BSA annettuna lihakseen (CCG‑1962) ja laskimoon (AALL07P4) ylläpitää L-asparagiinin ehtymistä noin kaksi viikkoa annostuksen jälkeen.
Kliininen teho ja turvallisuus
Oncaspar-valmisteen teho ja turvallisuus arvioitiin kolmen sellaisen kliinisen tutkimuksen perusteella, joissa Oncaspar-injektio-/infuusionesteliuosta käytettiin ensimmäisen linjan ALL-hoidossa. Tutkimukseen CCG-1962 osallistui normaalin riskin ALL-potilaita, tutkimukseen AALL07P4 korkean riskin ALL-potilaita ja tutkimukseen DFCI 11-001 sekä normaalin että korkean riskin ALL-potilaita.
Oncaspar-valmisteen tehon arviointi relapsoivien tai refraktoristen, aiemmin kliinisen allergisen reaktion natiivista E. coli -L-asparaginaasista saaneiden ALL-potilaiden hoidossa perustui kuudesta avoimesta tutkimuksesta [ASP-001, ASP-201A, ASP-302, ASP-304, ASP-400 ja ASP-001C/003C] poimittuun 94 potilaan ryhmään.
Ensilinja (akuuttia lymfoblastileukemiaa sairastavat potilaat, jotka eivät ole yliherkkiä natiiville E. coli -L-asparaginaasille)
Oncaspar-valmisteen turvallisuutta ja tehoa arvioitiin avoimessa, satunnaistetussa aktiivikontrolloidussa monikeskustutkimuksessa (tutkimus CCG-1962). Tässä tutkimuksessa 118 1‑9 vuoden ikäistä pediatrista potilasta, joilla oli aiemmin hoitamaton normaalin riskin akuutti lymfoblastileukemia, satunnaistettiin suhteessa 1:1 saamaan Oncaspar-valmistetta tai natiivia E. coli -L‑asparaginaasia osana yhdistelmähoitoa. Oncaspar-valmistetta annettiin lihakseen annostuksella 2 500 yksikköä/m2 BSA 4 viikon induktiovaiheen päivänä 3 ja kummankin 8 viikon viivästetyn tehostuksen (DI) hoitovaiheen päivänä 3. Natiivia E. coli -L-asparaginaasia annettiin lihakseen annostuksella 6 000 yksikköä/m2 BSA kolme kertaa viikossa yhteensä 9 annoksen ajan induktiovaiheen aikana ja yhteensä 6 annoksen ajan jokaisen viivästetyn tehostuksen hoitovaiheen aikana.
Ensisijainen tehon määritys perustui Oncaspar-valmistetta saaneen ja natiivia E. coli -L‑asparaginaasia saaneen ryhmän samankaltaiseen asparagiinin ehtymiseen. Tutkimussuunnitelmassa määritetty tavoite oli asparagiinin ehtyminen seerumin pitoisuustasolle ≤1 μM. Niiden potilaiden suhteellinen osuus, joiden ehtymisen taso oli tällainen, oli samanlainen 2 tutkimusryhmän välillä hoidon kaikkien 3 vaiheen aikana tutkimussuunnitelmassa määritetyissä aikapisteissä.
Hoidon kaikissa vaiheissa seerumin asparagiinipitoisuudet pienenivät 4 päivän sisällä ensimmäisestä asparaginaasiannoksesta hoitovaiheessa ja pysyivät pieninä noin 3 viikon ajan sekä Oncaspar-valmistetta että natiivia E. coli -L‑asparaginaasia saaneessa ryhmässä. Seerumin asparagiinipitoisuudet induktiovaiheen aikana näkyvät kuvassa 1. Seerumin asparagiinin ehtymisen mallit kahdessa viivästetyn tehostuksen vaiheessa vastaavat seerumin asparagiinin ehtymisen mallia induktiovaiheessa.
Kuva 1: Seerumin keskimääräinen (± keskivirhe) asparagiinipitoisuus tutkimuksen CCG-1962 induktiovaiheen aikana
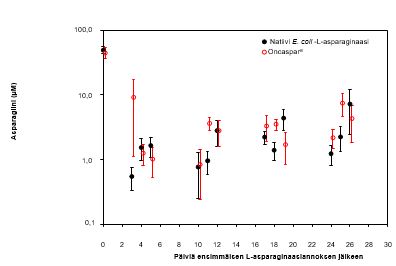
Huomautus: Oncaspar (2 500 yksikköä/m2 BSA lihakseen) annettiin 4 viikon induktiovaiheen päivänä 3. Natiivi E. coli -L‑asparaginaasi (6 000 yksikköä/m2 BSA lihakseen) annettiin 3 kertaa viikossa 9 annoksen ajan induktiovaiheen aikana.
50 potilaan aivo-selkäydinnesteen asparagiinipitoisuudet määritettiin induktiovaiheen aikana. Aivo-selkäydinnesteen asparagiinipitoisuus laski keskimääräisestä hoitoa edeltävästä 3,1 µM:n pitoisuudesta 1,7 µM:iin päivänä 4 ± 1 ja 1,5 µM:iin 25 ± 1 päivää Oncaspar-valmisteen annon jälkeen. Nämä tulokset olivat samanlaiset kuin natiivin E. coli -L‑asparaginaasin hoitohaarassa.
Oncaspar-valmistetta saaneen ja natiivia E. coli -L-asparaginaasia saaneen ryhmän päätetapahtumattomista elossaoloasteista (EFS) on yhteenveto taulukossa 2. Tutkimusta CCG-1962 ei ollut suunniteltu arvioimaan päätetapahtumattomien elossaoloasteiden eroja.
Taulukko 2: Päätetapahtumaton elossaoloaste 3, 5 ja 7 vuoden kuluttua (tutkimus CCG-1962)
| Oncaspar | Natiivi E. coli -L-asparaginaasi | |
3 vuoden EFS-aste, % (95 % CI) | 83 (73, 93) | 79 (68, 90) |
5 vuoden EFS-aste, % (95 % CI) | 78 (67, 88) | 73 (61, 85) |
7 vuoden EFS-aste, % (95 % CI) | 75 (63, 87) | 66 (52, 80) |
Tutkimuksessa CCG-1962 yleisimmät haittavaikutukset olivat infektiot, mukaan lukien kaksi hengenvaarallista infektiota (1 potilas kussakin haarassa). Yleisesti ottaen luokkien 3 ja 4 haittavaikutusten esiintyvyys ja tyyppi olivat samanlaiset kahden hoitoryhmän välillä. Kaksi Oncaspar-ryhmän potilasta sai allergisia reaktioita viivästetyn tehostuksen (DI) aikana, tyyppi DI #1 (luokan 1 allerginen reaktio ja luokan 3 nokkosihottuma).
Äskettäin diagnosoiduille, 1 – < 31 vuoden ikäisille potilaille, joilla oli korkean riskin B-esiaste-ALL, tehtiin pilottitutkimus (tutkimus AALL07P42). Tämä oli avoin, kontrolloitu, satunnaistettu tutkimus, jossa tutkittavaa pegyloitua asparaginaasivalmistetta verrattiin Oncaspar-valmisteeseen monen aineen solunsalpaajahoidon osana ALL:n ensimmäisen linjan hoidossa. Valkosolukriteerit olivat a) 1–10-vuotiailla: valkosoluja ≥ 50 000/μl; b) 10–30-vuotiailla: mikä tahansa valkosolumäärä; c) aiempaa steroidihoitoa saaneilla: mikä tahansa valkosolumäärä. Potilaat eivät olleet saaneet saada sytotoksista solunsalpaajahoitoa, paitsi steroideja ja intratekaalista sytarabiinia. Tähän tutkimukseen ilmoittautui yhteensä 166 potilasta, joista 54 potilasta satunnaistettiin saamaan Oncaspar-hoitoa annostuksella 2 500 U/m2 BSA, ja 111 potilasta satunnaistettiin saamaan tutkittavaa pegyloitua asparaginaasivalmistetta. Oncaspar-valmistetta annettiin laskimoon annostuksella 2 500 yksikköä/m2 BSA induktio-, konsolidaatio, viivästetyn tehostusvaiheen ja väliaikaisen ylläpitovaiheen aikana potilaille, joilla oli korkean riskin akuutti lymfoblastileukemia ja jotka saivat vahvennettua Berlin-Frankfurt-Münster-hoitoa. Oncaspar-hoitoryhmän niiden potilaiden prosenttiosuus, joilla oli evaluoitavissa oleva minimaalinen jäännöstauti negatiivinen (< 0,1 % leukemiasoluja luuytimessä) induktiovaiheen päivänä 29, oli 80 % (40/50). 4 vuoden kuluttua Oncaspar-hoitoryhmän päätetapahtumaton elossaoloaste oli 81,8 % [95 % CI 62,9–91,7 %] ja kokonaiselossaoloaste (OS) 90,4 % [95 % CI 78,5–95,9 %]. Kaiken kaikkiaan Oncaspar-valmistetta saavassa ryhmässä yliherkkyyden kaikkien luokkien esiintyvyys oli 5,8 %, anafylaktisia reaktioita oli 19,2 % ja haimatulehduksia 7,7 %. Luokan 3 tai suuremman luokan kuumeista neutropeniaa oli 15,4 %.
Dana‑Farber Cancer Institute (DFCI) ‑laitoksen suorittama tutkimus DFCI 11‑001 on meneillään oleva, aktiivikontrolloitu, satunnaistettu monikeskustutkimus laskimoon annettavasta tutkittavasta pegyloidusta asparaginaasivalmisteesta verrattuna Oncaspar-valmisteeseen. Tutkimukseen osallistuu yli 1-vuotiaita lapsia ja alle 22-vuotiaita nuoria, joilla on äskettäin diagnosoitu ALL, jota hoidetaan DFCI:n rutiininomaisella ALL-hoidolla. Yhteensä 239 potilasta satunnaistettiin, ja näistä 237 sai tutkimuslääkehoitoa (146 miestä ja 91 naista); näistä 119 potilasta (115 ALL-diagnosoitua) hoidettiin Oncaspar-annoksella 2 500 U/m2. Hoitoa annettiin induktiovaiheessa (päivänä 7) ja sen jälkeen 2 viikon välein yhteensä 30 viikkoa induktiovaiheen jälkeisenä hoitona. Sekä B- että T-ALL-potilaiden satunnaistus stratifioitiin riskiryhmän (normaali / korkea / erittäin korkea riski) perusteella. Niiden Oncaspar-ryhmän potilaiden prosenttiosuus, joilla oli evaluoitavissa oleva vähäinen minimaalinen jäännöstauti induktiovaiheen lopussa (< 0,001 havaittava sairaus) päivänä 32, oli 87,9 % (80/91). Tässä tutkimuksessa yhden vuoden EFS-aste oli 98,0 [95 %:n CI 92,3, 99,5]; yhden vuoden OS-arvo oli 100 [95 %:n CI 100, 100].
Akuuttia lymfoblastileukemiaa sairastavat potilaat, jotka olivat yliherkkiä natiiville E. coli -L-asparaginaasille
Kuudessa avoimessa tutkimuksessa arvioitiin Oncaspar-valmistetta relapsoivien/refraktoristen hematologisten sairauksien hoidossa. Näissä tutkimuksissa yhteensä 94 akuutin lymfoblastileukemian diagnoosin saanutta potilasta, jotka olivat saaneet aiemmin kliinisen allergisen reaktion natiivista E. coli -L-asparaginaasista, altistettiin Oncaspar-valmisteelle. Yksi potilas sai Oncaspar-valmistetta annostuksilla 250 ja 500 yksikköä/m2 BSA laskimoon. Loput potilaat saivat 2 000 tai 2 500 U/m2 BSA lihakseen tai laskimoon. Potilaat saivat Oncaspar-valmistetta yhtenä lääkeaineena tai yhdessä monen lääkeaineen solunsalpaajahoidossa. Yhteensä viidessä tutkimuksessa, jotka analysoitiin 65 Oncaspar-valmisteelle altistetun ALL-potilaan perusteella käyttäen koko tutkimuksen aikana korkeinta hoitovastetta, täydellinen remissio havaittiin 30 potilaalla (46 %), osittainen remissio 7 potilaalla (11 %) ja hematologinen parannus 1 potilaalla (2 %). Toisessa tutkimuksessa, jossa 29 yliherkkää ALL-potilasta altistettiin Oncaspar-valmisteelle, 11 potilaan vaste arvioitiin induktiovaiheen aikana. Näistä 3 potilasta (27 %) saavutti täydellisen remission, 1 potilas (9 %) osittaisen remission, 1 potilas (9 %) hematologista parantumista ja 2 potilasta (18 %) hoitotehon. Hoitoteho määriteltiin kliiniseksi paranemiseksi, joka ei täyttänyt muiden suotuisten tulosten kriteereitä. Ylläpitovaiheen aikana arvioitiin 19 potilasta, joista 17 (89 %) saavutti täydellisen remission ja 1 potilas (5 %) hoitotehon.
Farmakokinetiikka
Oncaspar-valmisteen farmakokineettiset ominaisuudet perustuivat asparaginaasiaktiivisuuteen, joka mitattiin entsyymimäärityksellä lihakseen (CCG-1962) ja laskimoon (AALL07P4, DFCI 11-001) antamisen jälkeen.
Tutkimuksessa CCG‑1962 keskimääräinen asparaginaasin aktiivisuus saavutti huippuarvon 1 U/ml päivänä 5 injektion jälkeen. Keskimääräinen puoliintumisaika injektiokohdasta absorboitumisen jälkeen oli 1,7 päivää ja eliminaation puoliintumisaika oli 5,5 päivää. Jakautumistilavuudeksi vakaassa tilassa arvioitiin 1,86 l/m2/vrk ja puhdistumaksi 0,169 l/m2/vrk.
Tutkimuksessa AALL07P4 laskettiin farmakokinetiikkaparametrit yhden 2 500 U/m2 ‑infuusioannoksen jälkeen induktion aikana tilamallittomalla analyysillä peräkkäisistä plasmanäytteistä. Tulokset on esitetty taulukossa 3 (ks. kohta Farmakodynamiikka). Oncaspar-valmisteen Cmax- ja AUC-arvot olivat matalampia miehillä, suuremman BMI:n tutkittavilla sekä yli 10-vuotiailla tutkittavilla. Induktion aikana yhden Oncaspar-infuusioannoksen 2 500 U/m2 jälkeen asparaginaasiaktiivisuus ≥ 0,1 U/ml säilyi enintään 18 päivää annostelun jälkeen 95,3 %:lla tutkittavista.
Taulukko 3: Farmakokineettiset parametrit yhden Oncaspar-infuusioannoksen 2 500 U/m2 BSA jälkeen induktion aikana (N = 47, tutkimus AALL07P4) | |
| Farmakokineettiset parametrit | Aritmeettinen keskiarvo (SD) |
Cmax (mU/ml)* | 1638 (459,1) |
Tmax (h)* | 1,25 (1,08; 5,33)† |
AUC0-t (mU•vrk/ml)* | 14810 (3555) |
AUC0–∞ (mU•vrk/ml)ǂ | 16570 (4810) |
t1/2 (vrk)ǂ | 5,33 (2,33) |
CL (L/vrk)ǂ | 0,2152 (0,1214) |
Vss (L)ǂ | 1,95 (1,13) |
* N = 47 evaluoitavaa tutkittavaa. † Mediaani (10. ja 90. persentiili). ǂ N = 46 evaluoitavaa tutkittavaa. | |
Tutkimuksessa DFCI 11‑001 asparaginaasiaktiviteetin arviot tehtiin yhden Oncaspar-infuusioannoksen 2 500 U/m2 BSA jälkeen induktion aikana ja kahden viikon välein induktion jälkeisenä aikana (ks. kohta Farmakodynamiikka). Induktion aikana plasman asparaginaasiaktiivisuus ≥ 0,1 U/ml säilyi 93,5 %:lla tutkittavista 18 päivää antamisen jälkeen. Induktion jälkeisen vaiheen aikana asparaginaasiaktiivisuuden pohja-arvo yli 0,4 U/ml säilyi 100 %:lla tutkittavista viikosta 7 viikkoon 25. Nämä tulokset osoittavat, että kun Oncaspar 2 500 U/m2 BSA annetaan yhtenä ja kahden viikon välein toistettavana annoksena, kliinisesti relevantti asparaginaasiaktiivisuus pysyy yllä koko annostusvälin (ts. kaksi viikkoa).
Äskettäin ALL-diagnoosin saaneet potilaat saivat yhden lihaksensisäisen (i.m.) pistoksen Oncaspar-valmistetta (2 500 U m2 BSA) tai E. coli -bakteerissa tuotettua natiivia asparaginaasia (25 000 U/m2 BSA) tai Erwinia-bakteerissa tuotettua natiivia asparaginaasia (25 000 U/m2 BSA). Oncaspar-valmisteen puoliintumisaika plasmassa oli tilastollisesti merkittävästi pidempi (5,7 päivää) kuin E. coli - (1,3 päivää) ja Erwinia-bakteereissa (0,65 päivää) tuotettujen natiivien asparaginaasien. Leukemiasolujen välitön solukuolema in vivo rodamiinifluoresenssin avulla mitattuna oli sama kaikkien kolmen L-asparaginaasivalmisteen kohdalla.
ALL-potilaita, joilla oli useita relapseja, hoidettiin joko Oncaspar-valmisteella tai E. coli -bakteerissa tuotetulla natiivilla asparaginaasilla osana induktiohoitoa. Oncaspar-valmistetta annettiin 2 500 U/m2 BSA lihakseen induktiohoidon päivinä 1 ja 15. Oncaspar-valmisteen keskimääräinen puoliintumisaika plasmassa oli 8 päivää ei-yliherkillä potilailla (AUC 10,35 U/ml/vrk) ja 2,7 päivää yliherkillä potilailla (AUC 3,52 U/ml/vrk).
Erityisryhmät
Kontrolloituja tutkimuksia ei suunniteltu arvioimaan Oncaspar-valmistetta virallisesti erityisryhmissä. Oncaspar-valmisteen populaatiofarmakokineettinen arvio perustui tutkimuksista AALL07P4 (i.v.), DFCI 11-001 (i.v.) ja CCG-1962 (i.m.) saatuihin tietoihin, joissa havaittiin puhdistuman (lineaarisen ja saturoituvan) suurenevan likimäärin suhteessa kehon pinta-alaan (BSA) ja jakaantumistilavuuden suurenevan hieman tarkemmin suhteessa kehon pinta-alaan. Tässä analyysissä ei tunnistettu tilastollisesti merkitseviä farmakokineettisten ominaisuuksien eroja mies- ja naispuolisten tutkittavien välillä.
Munuaisten ja maksan vajaatoiminnan vaikutusta Oncaspar-valmisteen farmakokinetiikkaan ei ole arvioitu. Koska pegaspargaasi on proteiini, jolla on suuri molekyylipaino, se ei erity munuaisten kautta, eikä Oncaspar-valmisteen farmakokinetiikan muutoksia munuaisten toimintahäiriötä sairastavilla potilailla ennakoida.
Koska Oncaspar-valmisteen metaboliasta vastaavat proteolyyttiset entsyymit jakautuvat kaikkialle kudoksiin, maksan tarkka rooli on tuntematon. Maksan toiminnan heikentymisen ei kuitenkaan odoteta aiheuttavan kliinisiä olennaisia ongelmia Oncaspar-valmisteen käyttöön.
Iäkkäistä potilaista ei ole saatavilla tietoja.
Prekliiniset tiedot turvallisuudesta
Oncaspar-valmisteen kahden lääkemuodon, injektio-/infuusionesteliuoksen ja liuosta varten tarkoitetun kuiva-aineen, välinen ei-kliininen vertailukelpoisuus farmakokinetiikan/farmakodynamiikan osalta osoitettiin koirilla laskimonsisäisen kerta-annoksen ja laskimonsisäisten toistuvien annosten (500 U/kg) jälkeen. Seuraavassa mainitut tutkimukset tehtiin injektio-/infuusionesteliuosvalmisteella.
Akuutti toksisuus
Vain hyvin suuret hiirille yhtenä annoksena intraperitoneaalisesti annetut pegaspargaasiannokset (25 000–100 000 yksikköä kehon painokiloa kohden) aiheuttivat kuoleman 14 %.lle kaikista hoidetuista hiiristä. Lievää hepatotoksisuutta havaittiin samoilla annoksilla. Haittavaikutuksia olivat painonlasku, piloerektio ja heikentynyt aktiivisuus. Pernan painon lasku voi olla potentiaalinen merkki hoidon immunosuppressiivisesta vaikutuksesta.
Pegaspargaasi oli hyvin siedetty sekä rotilla että koirilla, kun sitä annettiin laskimoon yhtenä annoksena, joka oli enintään 500 U/kg elopainoa.
Toistuvan altistuksen aiheuttama toksisuus
4 viikon tutkimus rotilla, joita hoidettiin annoksella 400 U/kg/vrk pegaspargaasia intraperitoneaalisesti annettuna aiheutti ruoan nauttimisen ja ruumiinpainon laskua kontrolliryhmään verrattuna.
3 kuukauden tutkimus, jossa hiirille annettiin enintään 500 U/kg pegaspargaasia intraperitoneaalisesti tai lihakseen, aiheutti lieviä hepatosellulaarisia muutoksia vain suurimmalla intraperitoneaalisella annoksella.
Väliaikaisesti heikentynyttä ruumiinpainon nousua ja leukosyyttien kokonaismäärän väliaikaista laskua havaittiin koirilla, joita hoidettiin pegaspargaasiannoksella 1 200 U/kg viikoittain 2 viikon ajan. Myös alaniiniaminotransferaasin aktiivisuutta esiintyi yhdellä neljästä koirasta.
Immunogeenisuus
Immunogeenista vastetta ei havaittu hiirillä tehdyssä 12 viikon tutkimuksessa, jossa pegaspargaasia annettiin viikoittain 10,5 U/hiiri lihakseen tai vatsaonteloon.
Lisääntymiseen vaikuttava toksisuus
Pegaspargaasilla ei ole tehty lisääntymiseen vaikuttavaa toksisuutta koskevia tutkimuksia.
L-asparaginaasilla tehdyt sikiötoksisuustutkimukset osoittivat teratogeenista potentiaalia rotilla, joita hoidettiin raskauspäivinä 6–15 teratogeenisten vaikutusten NOAEL-arvolla, laskimoon annetulla annoksella 300 U/kg. Kaniineilla 50 tai 100 yksikön annos painokiloa kohden laskimoon annettuna raskauspäivinä 8 ja 9 aiheutti elinkelpoisille sikiöille parantumattomia epämuodostumia. NOAEL-arvoa ei ole määritetty. Hoitoalueella olevilla annoksilla havaittiin useita epämuodostumia ja sikiöitä tappavia vaikutuksia. Hedelmällisyyteen tai peri- ja postnataalisen kehitykseen kohdistuvia vaikutuksia koskevia tutkimuksia ei ole tehty.
Karsinogeenisuus, mutageenisuus, hedelmällisyys
Pegaspargaasilla ei ole tehty pitkäaikaisia karsinogeenisuustutkimuksia tai eläinten hedelmällisyyteen kohdistuvia vaikutuksia.
Pegaspargaasi ei ollut mutageeninen Amesin testin perusteella, kun käytettiin Salmonella typhimurium -kantoja.
Farmaseuttiset tiedot
Apuaineet
Dinatriumfosfaattiheptahydraatti
Natriumdivetyfosfaattimonohydraatti
Natriumkloridi
Sakkaroosi
Natriumhydroksidi (pH:n säätämistä varten)
Suolahappo (pH:n säätämistä varten)
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo:
3 vuotta.
Käyttökuntoon saatettu liuos
On osoitettu, että liuos pysyy käytössä kemiallisesti ja fysikaalisesti stabiilina 24 tunnin ajan alle 25 °C:ssa. Mikrobiologisista syistä lääkevalmiste tulisi käyttää heti, ellei käyttökuntoon saattamisessa ole hyödynnetty menetelmää, joka estää mikrobikontaminaation. Jos lääkevalmistetta ei käytetä heti, käytönaikainen säilytysaika ja säilytysolosuhteet ovat käyttäjän vastuulla.
Laimennettu liuos
On osoitettu, että liuos pysyy käytössä kemiallisesti ja fysikaalisesti stabiilina 48 tunnin ajan 2–8 °C:ssa. Mikrobiologisista syistä lääkevalmiste tulisi käyttää heti. Jos sitä ei käytetä heti, käytönaikainen säilytysaika ja säilytysolosuhteet ennen käyttöä ovat käyttäjän vastuulla eivätkä normaalisti saa ylittää 24:ää tuntia 2 °C – 8 °C:ssa, jos valmistetta ei ole saatettu käyttökuntoon/laimennettu kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä jääkaapissa (2°C - 8°C).
Ei saa jäätyä.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ONCASPAR injektio/infuusiokuiva-aine, liuosta varten
750 U/ml (L:ei) 1 kpl (3750 U (5 ml)) (2061,30 €)
PF-selosteen tieto
Tyypin I piilasipullo, jossa on klooributyylielastomeeritulppa ja korkkina 20 mm:n alumiininen irti napsautettava flip-off-sinetti. Pullo sisältää 3 750 U pegaspargaasia.
Pakkauskoko on yksi.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen jauhe.
Käyttö- ja käsittelyohjeet
Tämä lääkevalmiste voi aiheuttaa ärsytystä kosketuksen yhteydessä. Jauhetta on tämän vuoksi käsiteltävä ja annettava erityisen varovaisesti. Höyryn hengittämistä ja kosketusta ihoon ja limakalvoihin, erityisesti silmiin, on vältettävä. Jos lääkevalmistetta pääsee kosketuksiin silmien, ihon tai limakalvojen kanssa, huuhtele heti runsaalla vedellä vähintään 15 minuutin ajan.
Oncaspar täytyy antaa laskimoon tai lihakseen sen jälkeen, kun valmiste on saatettu käyttökuntoon. Jauhe täytyy liuottaa 5,2 ml:aan injektionesteisiin käytettävää vettä ennen antoa (ks. kohta Annostus ja antotapa).
Käsittelyohjeet
- Henkilökunta täytyy kouluttaa lääkevalmisteiden käsittelyyn ja siirtämiseen (raskaana olevien työntekijöiden ei pidä käsitellä tätä lääkevalmistetta).
- Käsittelyssä täytyy noudattaa aseptista tekniikkaa.
- Käsittelyssä täytyy noudattaa antineoplastisten aineiden asianmukaiseen käsittelyyn tarkoitettuja menettelytapoja.
- Oncaspar-valmistetta käsiteltäessä on suositeltavaa käyttää kertakäyttökäsineitä ja suojavaatteita.
- Kaikki antoon tai puhdistukseen käytetyt välineet, mukaan lukien käsineet, on laitettava vaaralliselle jätteelle tarkoitettuihin säkkeihin, jotta ne voidaan polttaa korkeassa lämpötilassa.
Käyttökuntoon saattaminen
- Injektiopulloon injektoidaan ruiskulla ja 21 gaugen neulalla 5,2 ml injektionesteisiin käytettävää vettä.
- Injektiopulloa tulee pyörittää hellävaroen, kunnes jauhe on liuennut nesteeseen.
- Käyttökuntoon saattamisen jälkeen liuoksen tulee olla kirkasta ja väritöntä, eikä siinä saa olla näkyviä hiukkasia. Älä käytä, jos käyttökuntoon saatettu liuos on samea tai siihen on muodostunut hiukkasia. Ei saa ravistaa.
- Liuos on käytettävä 24 tunnin kuluessa käyttökuntoon saattamisesta, ja sitä on säilytettävä alle 25 °C:ssa.
Anto
- Parenteraaliset lääkevalmisteet on tarkastettava hiukkasten varalta ennen antoa. Vain kirkas, väritön liuos, jossa ei näy hiukkasia, voidaan käyttää.
- Lääkevalmiste tulee antaa laskimoon tai lihakseen. Liuos on annettava hitaasti. Jos lääke annetaan lihaksensisäisenä injektiona, tilavuus ei saa olla yli 2 ml hoidettaessa lapsia ja nuoria tai yli 3 ml hoidettaessa aikuisia.
Jos lääkevalmiste annetaan laskimoon, käyttökuntoon saatettu liuos on laimennettava 100 ml:aan 0,9 %:n vahvuista natriumkloridi-injektionestettä (9 mg/ml) tai 5 %:n vahvuista glukoosiliuosta.
Laimennettu liuos voidaan antaa 1–2 tunnin aikana yhdessä potilaan jo menossa olevan infuusion kanssa, joka on joko natriumkloridiliuosta 9 mg/ml (0,9 %) tai 5 %:n vahvuista glukoosiliuosta. Älä infusoi muita lääkevalmisteita saman laskimoyhteyden kautta Oncaspar-valmisteen annon aikana (ks. kohta Annostus ja antotapa).
Laimennuksen jälkeen liuos on käytettävä heti. Jos sen käyttäminen heti ei ole mahdollista, laimennettua liuosta voidaan säilyttää 2–8 °C:ssa korkeintaan 48 tunnin ajan (ks. kohta Kestoaika).
Hävittäminen
Oncaspar on tarkoitettu vain kertakäyttöön. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ONCASPAR injektio/infuusiokuiva-aine, liuosta varten
750 U/ml 1 kpl
- Ei korvausta.
ATC-koodi
L01XX24
Valmisteyhteenvedon muuttamispäivämäärä
04.05.2026
Yhteystiedot
Äyritie 12 A
01510 Vantaa
040 901 2978
www.servierfinland.fi
info.finland@servier.com