
ZYDELIG tabletti, kalvopäällysteinen 100 mg, 150 mg
Vaikuttavat aineet ja niiden määrät
Zydelig 100 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 100 mg idelalisibia.
Apuaine, jonka vaikutus tunnetaan
Yksi tabletti sisältää 0,1 mg paraoranssia (E110) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Zydelig 150 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 150 mg idelalisibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti.
Kliiniset tiedot
Käyttöaiheet
Zydelig on tarkoitettu yhdistelmänä rituksimabin kanssa sellaisten kroonista lymfaattista leukemiaa (KLL) sairastavien aikuispotilaiden hoitoon:
- jotka ovat saaneet aiemmin vähintään yhtä hoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), tai
- ensilinjan hoitona potilaille, joilla on 17p‑deleetio tai TP53-mutaatio ja joille mikään muu hoito ei tule kyseeseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Zydelig on tarkoitettu monoterapiaksi sellaisten follikulaarista lymfoomaa (FL) sairastavien aikuispotilaiden hoitoon, jotka ovat hoitoresistenttejä kahdelle aiemmalle hoitolinjalle (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Zydelig-hoitoa saa antaa vain lääkäri, jolla on kokemusta syöpähoidoista.
Annostus
Suositeltu annos on 150 mg idelalisibia kahdesti päivässä. Hoitoa on jatkettava taudin etenemiseen tai kestämättömien toksisten vaikutusten ilmaantumiseen asti.
Jos potilas unohtaa ottaa Zydelig-annoksen ja muistaa sen 6 tunnin kuluessa annoksen normaalista ottamisajankohdasta, hänen tulee ottaa unohtunut annos mahdollisimman pian, ja seuraava annos normaalin annostusaikataulun mukaisesti. Jos potilas unohtaa ottaa annoksen ja muistaa sen vasta yli 6 tunnin kuluttua, potilaan ei tule ottaa unohtunutta annosta, vaan hänen tulee ottaa seuraava annoksensa normaalin annostusaikataulun mukaisesti.
Annoksen muuttaminen
Kohonneet maksan transaminaasit
Zydelig-hoito on keskeytettävä, jos maksan transaminaasit nousevat asteelle 3 tai 4 (alaniiniaminotransferaasi [ALAT]/aspartaattiaminotransferaasi [ASAT] > 5 x normaaliarvojen yläraja [ULN]). Kun arvot ovat palanneet asteelle 1 tai alemmas (ALAT/ASAT ≤ 3 x ULN), hoitoa voidaan jatkaa annoksella 100 mg kahdesti päivässä.
Jos arvot eivät nouse uudelleen, annos voidaan hoitavan lääkärin harkinnan mukaan nostaa takaisin 150 mg:aan kahdesti päivässä.
Jos arvot nousevat uudelleen, Zydelig-hoito on keskeytettävä eikä sitä saa jatkaa, ennen kuin arvot ovat palanneet asteelle 1 tai alemmas, minkä jälkeen hoito voidaan hoitavan lääkärin harkinnan mukaan aloittaa uudelleen annoksella 100 mg kahdesti päivässä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Ripuli/koliitti
Zydelig-hoito on keskeytettävä, jos potilaalla esiintyy asteen 3 tai 4 ripulia/koliittia. Kun ripuli/koliitti on palannut asteelle 1 tai alemmas, hoitoa voidaan jatkaa annoksella 100 mg kahdesti päivässä. Jos ripuli/koliitti ei uusiudu, annos voidaan hoitavan lääkärin harkinnan mukaan nostaa takaisin 150 mg:aan kahdesti päivässä (ks. kohta Haittavaikutukset).
Pneumoniitti
Zydelig-hoito on keskeytettävä, jos potilaalla epäillään pneumoniittia. Kun pneumoniitti on parantunut ja Zydelig-hoiton uudelleen aloittaminen on tarpeen, voidaan harkita hoidon jatkamista annoksella 100 mg kahdesti päivässä. Zydelig-hoito on lopetettava pysyvästi, jos potilaalla todetaan keskivaikea tai vaikea oireinen pneumoniitti tai organisoituva keuhkokuume (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Ihottuma
Zydelig-hoito on keskeytettävä, jos potilaalla esiintyy asteen 3 tai 4 ihottumaa. Kun ihottuma on palannut asteelle 1 tai alemmas, hoitoa voidaan jatkaa annoksella 100 mg kahdesti päivässä. Jos ihottuma ei uusiudu, annos voidaan hoitavan lääkärin harkinnan mukaan nostaa takaisin 150 mg:aan kahdesti päivässä (ks. kohta Haittavaikutukset).
Neutropenia
Zydelig-hoito on keskeytettävä potilailla, joiden neutrofiilien absoluuttinen määrä (ANC) on alle 500/mm3. ANC-arvo on tutkittava vähintään viikoittain, kunnes ANC on ≥ 500/mm3, jolloin hoitoa voidaan jatkaa annoksella 100 mg kahdesti päivässä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
| ANC 1 000 - < 1 500/mm3 | ANC 500 - < 1 000/mm3 | ANC < 500/mm3 |
| Pidä Zydelig-annostus samana. | Pidä Zydelig-annostus samana.
Mittaa ANC-arvo vähintään | Keskeytä Zydelig-valmisteen
Mittaa ANC-arvo vähintään |
Erityisryhmät
Iäkkäät potilaat
Erityinen annoksen muuttaminen ei ole tarpeen iäkkäille (≥ 65-vuotiaille) potilaille (ks. kohta Farmakokinetiikka).
Heikentynyt munuaisten toiminta
Annoksen muuttaminen ei ole tarpeen lievästi (kreatiniinipuhdistuma [CrCl] = 60–80 ml/min), kohtalaisesti (CrCl = 30–59 ml/min) tai vaikeasti (CrCl = 15–29 ml/min) heikentynyttä munuaisten toimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka).
Heikentynyt maksan toiminta
Annoksen muuttaminen ei ole tarpeen aloitettaessa Zydelig-hoitoa lievästi (Child-Pugh-luokka A) tai kohtalaisesti (Child-Pugh-luokka B) heikentynyttä maksan toimintaa sairastavilla potilailla, mutta haittavaikutusten tarkempi seuranta on suositeltavaa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Vaikeasti heikentynyttä maksan toimintaa sairastavien potilaiden annossuositusten laatimiseksi ei ole riittävästi tietoa. Siksi on suositeltavaa noudattaa varovaisuutta annettaessa Zydelig-valmistetta tälle potilasryhmälle, ja haittavaikutusten tarkempi seuranta on suositeltavaa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Zydelig-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Zydelig otetaan suun kautta. Potilaita on neuvottava nielemään tabletit kokonaisina. Kalvopäällysteistä tablettia ei saa pureskella tai murskata. Kalvopäällysteiset tabletit voidaan ottaa joko ruoan kanssa tai tyhjään mahaan (ks. kohta Farmakokinetiikka).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Vakavat infektiot
Zydelig-hoitoa ei pidä aloittaa potilaille, joilla todetaan viitteitä aktiivisesta systeemisestä bakteeri-, sieni- tai virusinfektiosta.
Idelalisibia käytettäessä on esiintynyt vakavia ja kuolemaan johtaneita infektioita, mukaan lukien opportunistisia infektioita, kuten Pneumocystis jirovecii -keuhkokuumetta (PJP) ja sytomegaloviruksen (CMV) aiheuttamaa infektiota. Sen vuoksi kaikille potilaille on annettava PJP:n estohoitoa koko idelalisibihoidon ajan ja 2–6 kuukauden ajan hoidon keskeyttämisen jälkeen. Hoidonjälkeisen estohoidon kesto on määritettävä kliinisen harkinnan pohjalta, ja määrityksessä voidaan ottaa huomioon potilaan riskitekijät, kuten samanaikainen kortikosteroidihoito tai pitkittynyt neutropenia (ks. kohta Haittavaikutukset).
Potilaita on tarkkailtava hengitystieoireiden ja -löydösten varalta koko hoidon ajan. Potilaita on neuvottava ilmoittamaan viipymättä uusista hengitystieoireista.
Jos potilaan CMV-serologia idelalisibihoitoa aloitettaessa on positiivinen tai on olemassa muuta näyttöä siitä, että potilaalla on aiemmin ollut CMV-infektio, suositellaan potilaan seurantaa säännöllisin kliinisin tutkimuksin ja laboratoriokokein CMV-infektion varalta. Potilaita, joilla todetaan CMV-viremia ilman kliinisiä merkkejä CMV-infektiosta, on tarkkailtava huolellisesti. Jos potilaalla todetaan näyttöä CMV-viremiasta ja kliinisiä merkkejä CMV-infektiosta, idelalisibihoidon keskeyttämistä infektion paranemiseen saakka on harkittava. Jos idelalisibihoidon jatkamisen hyötyjä pidetään riskejä suurempina, ennakoivan CMV-hoidon antamista on harkittava.
Progressiivisen multifokaalisen leukoenkefalopatian (PML) tapauksia on raportoitu idelalisibin käytön jälkeen, kun sitä ennen tai samanaikaisesti on annettu immunosuppressiivisia hoitoja, joihin on liittynyt PML:ään. Lääkärien on otettava huomioon PML:n mahdollisuus erotusdiagnoosissa potilailla, joilla on uusia tai pahenevia neurologisia, kognitiivisia tai käyttäytymiseen liittyviä oireita tai merkkejä. Jos epäillään PML:ää, asianmukaiset diagnostiset tutkimukset on aloitettava ja hoito keskeytettävä, kunnes PML on suljettu pois. Jos on olemassa vähäinenkin epäilys PML:stä, on harkittava lähetettä neurologille sekä asianmukaisia PML:n diagnostisia toimenpiteitä, kuten magneettikuvausta mielellään varjoaineella, JC-viruksen DNA-määritystä aivo-selkäydinnesteestä ja neurologisten tutkimusten toistamista.
Neutropenia
Hoidon aikana ilmaantuvaa asteen 3 tai 4 neutropeniaa, mukaan lukien kuumeista neutropeniaa, on esiintynyt idelalisibilla hoidetuilla potilailla. Verisolujen määrä on tutkittava kaikilta potilailta vähintään 2 viikon välein idelalisibihoidon ensimmäisten 6 kuukauden ajan ja vähintään viikoittain niiltä potilailta, joiden ANC on alle 1 000/mm3 (ks. kohta Annostus ja antotapa).
Maksatoksisuus
Idelalisibilla tehdyissä kliinisissä tutkimuksissa on havaittu ALAT- ja ASAT-arvojen asteen 3 ja 4 nousuja (> 5 x ULN). Hepatosellulaarisia vaurioita, mukaan lukien maksan vajaatoiminta, on myös ilmoitettu. Maksan transaminaasien lisääntymistä havaittiin yleensä ensimmäisten 12 hoitoviikon aikana, ja transaminaasiarvot palautuivat normaaleiksi, kun annoksen antaminen keskeytettiin (ks. kohta Annostus ja antotapa). 26 %:lla potilaista, joiden idealisibihoitoa jatkettiin pienemmällä annoksella, ALAT/ASAT-arvot kohosivat uudelleen. Zydelig-hoito on keskeytettävä, jos ALAT/ASAT-arvot kohoavat asteelle 3 tai 4, ja maksan toimintaa on tarkkailtava. Hoitoa voidaan jatkaa pienemmällä annoksella, kun arvot ovat palanneet asteelle 1 tai alemmas (ALAT/ASAT ≤ 3 x ULN).
ALAT-, ASAT- ja kokonaisbilirubiiniarvoja on tarkkailtava kaikilla potilailla 2 viikon välein ensimmäisten 3 hoitokuukauden ajan ja sen jälkeen kliinisen tarpeen mukaan. Jos ALAT- ja/tai ASAT-arvoissa havaitaan asteen 2 tai suurempia nousuja, potilaiden ASAT-, ALAT- ja kokonaisbilirubiiniarvot on tutkittava viikoittain, kunnes arvot palautuvat asteelle 1 tai alemmas.
Heikentynyt maksan toiminta
Haittavaikutusten tarkempi seuranta on suositeltavaa heikentynyttä maksan toimintaa sairastavilla potilailla, sillä altistumisen odotetaan kasvavan tässä potilasryhmässä, erityisesti vaikeasti heikentynyttä maksan toimintaa sairastavien potilaiden kohdalla. Idelalisibin kliinisiin tutkimuksiin ei otettu mukaan vaikeasti heikentynyttä maksan toimintaa sairastavia potilaita. On suositeltavaa noudattaa varovaisuutta annettaessa Zydelig-valmistetta tälle potilasryhmälle.
Krooninen hepatiitti
Idelalisibia ei ole tutkittu potilailla, joilla on krooninen aktiivinen hepatiitti, mukaan lukien virushepatiitti. Varovaisuutta on noudatettava annettaessa Zydelig-valmistetta potilaille, joilla on aktiivinen hepatiitti.
Ripuli/koliitti
Vaikeita lääkkeeseen liittyneitä koliittitapauksia ilmeni suhteellisen myöhään (kuukausia) hoidon aloittamisen jälkeen, toisinaan oireet pahenivat nopeasti, mutta ne korjautuivat muutaman viikon sisällä annoksen antamisen keskeyttämisestä ja oireenmukaisella lisähoidolla (esim. tulehduslääkkeet, kuten enteerinen budesonidi) (ks. kohta Annostus ja antotapa).
Tulehduksellista suolistosairautta sairastaneiden potilaiden hoidosta on hyvin vähän kokemuksia.
Pneumoniitti ja organisoituva keuhkokuume
Idelalisibia käytettäessä on ilmoitettu pneumoniitti- ja organisoituvan keuhkokuumeen tapauksia (joista osa on johtanut kuolemaan). Jos potilaalla todetaan vakava keuhkoihin kohdistuva haittavaikutus, idelalisibihoito on keskeytettävä ja potilas tutkittava haittavaikutuksen etiologian selvittämiseksi. Jos potilaalla todetaan joko keskivaikea tai vaikea oireinen pneumoniitti tai organisoituva keuhkokuume, asianmukainen hoito on aloitettava ja idelalisibin käyttö on lopetettava pysyvästi.
Vakavat ihoreaktiot
Idelalisibin käytön yhteydessä on ilmennyt Stevens-Johnsonin oireyhtymää, toksista epidermaalista nekrolyysiä sekä yleisoireista eosinofiilista oireyhtymää (DRESS). Kuolemaan johtaneita Stevens‑Johnsonin oireyhtymän ja toksisen epidermaalisen nekrolyysin tapauksia on ilmoitettu, kun idelalisibia annettiin samanaikaisesti muiden lääkevalmisteiden kanssa, jotka on liitetty näihin oireyhtymiin. Idelalisibin antaminen on keskeytettävä, jos potilaalla epäillään Stevens‑Johnsonin oireyhtymää, toksista epidermaalista nekrolyysiä tai yleisoireista eosinofiilistä oireyhtymää, ja potilas on tutkittava ja hoidettava sen mukaisesti. Jos diagnoosiksi varmistuu Stevens‑Johnsonin oireyhtymä, toksinen epidermaalinen nekrolyysi tai yleisoireinen eosinofiilinen oireyhtymä, idelalisibihoito on lopetettava pysyvästi.
CYP3A:n induktorit
Altistus idelalisibille saattaa vähentyä, kun sitä annetaan yhdessä CYP3A:n induktorien, kuten rifampisiinin, fenytoiinin, mäkikuisman (Hypericum perforatum) tai karbamatsepiinin, kanssa. Koska idelalisibin plasmapitoisuuden pieneneminen voi johtaa tehon vähenemiseen, Zydelig-valmisteen antamista yhdessä kohtalaisten tai voimakkaiden CYP3A:n induktorien kanssa on vältettävä (ks. kohta Yhteisvaikutukset).
CYP3A:n substraatit
Idelalisibin pääasiallinen metaboliitti GS-563117 on voimakas CYP3A4:n estäjä. Sen vuoksi idelalisibi saattaa aiheuttaa yhteisvaikutuksia CYP3A:n metaboloimien lääkevalmisteiden kanssa, mikä saattaa johtaa muun lääkevalmisteen kohonneeseen pitoisuuteen seerumissa (ks. kohta Yhteisvaikutukset). Kun idelalisibia annetaan yhdessä muiden lääkevalmisteiden kanssa, ohjeet samanaikaisesta annostelusta CYP3A4:n estäjien kanssa on tarkastettava muiden lääkevalmisteiden valmisteyhteenvedoista. Idelalisibin käyttöä samanaikaisesti CYP3A:n substraattien kanssa, jotka aiheuttavat vakavia ja/tai hengenvaarallisia haittavaikutuksia (esim. alfutsosiini, amiodaroni, sisapridi, pimotsidi, kinidiini, ergotamiini, dihydroergotamiini, ketiapiini, lovastatiini, simvastatiini, sildenafiili, midatsolaami, triatsolaami), on vältettävä, ja jos mahdollista, on käytettävä muita lääkevalmisteita, jotka ovat vähemmän herkkiä CYP3A4:n estolle.
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä idelalisibihoidon aikana ja 1 kuukauden ajan hoidon päättymisen jälkeen (ks. kohta Raskaus ja imetys). Hormoniehkäisyvalmisteita käyttävien naisten tulisi käyttää mekaanista ehkäisyä lisäehkäisynä, sillä tällä hetkellä ei tiedetä, heikentääkö idelalisibi hormoniehkäisyvalmisteiden tehoa.
Apuaineet, joiden vaikutus tunnetaan
Zydelig sisältää paraoranssi-atsoväriä (E110), joka voi aiheuttaa allergisia reaktioita.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Idelalisibi metaboloituu pääasiassa aldehydioksidaasin vaikutuksesta ja pienemmässä määrin CYP3A:n ja glukuronidaation (UGT1A4) kautta. Sen pääasiallinen metaboliitti on GS-563117, joka ei ole farmakologisesti aktiivinen. Idelalisibi ja GS-563117 ovat P-pg- ja BCRP-substraatteja.
Muiden lääkevalmisteiden vaikutus idelalisibin farmakokinetiikkaan
CYP3A:n induktorit
Eräässä kliinisessä lääkkeiden yhteisvaikutustutkimuksessa todettiin, että idelalisibin 150 mg:n kerta-annoksen antaminen yhdessä rifampisiinin kanssa (voimakas CYP3A:n induktori) pienensi idelalisibin AUCinf-arvoa ~75 %. Zydelig-valmisteen antamista yhdessä kohtalaisten tai voimakkaiden CYP3A:n induktorien, kuten rifampisiinin, fenytoiinin, mäkikuisman tai karbamatsepiinin kanssa tulee välttää, koska se voi johtaa tehon vähenemiseen.
CYP3A:n/P-gp:n estäjät
Eräässä kliinisessä lääkkeiden yhteisvaikutustutkimuksessa todettiin, että annettaessa idelalisibin 400 mg:n kerta-annos yhdessä 400 mg kerran päivässä annosteltavan ketokonatsolin (voimakas CYP3A:n, P-gp:n ja BCRP:n estäjä) kanssa suurensi idelalisibin Cmax-arvoa 26 % ja AUCinf-arvoa 79 %. Idelalisibin aloitusannoksen muuttamista ei pidetä tarpeellisena, kun sitä annetaan samanaikaisesti CYP3A:n/P-gp:n estäjien kanssa, mutta haittavaikutusten tarkempi seuranta on suositeltavaa.
Idelalisibin vaikutus muiden lääkevalmisteiden farmakokinetiikkaan
CYP3A:n substraatit
Idelalisibin pääasiallinen metaboliitti GS-563117 on voimakas CYP3A:n estäjä. Eräässä kliinisessä lääkkeiden yhteisvaikutustutkimuksessa todettiin, että idelalisibin antaminen yhdessä midatsolaamin (herkkä CYP3A:n substraatti) kanssa suurensi midatsolaamin Cmax-arvoa ~140 % ja AUCinf-arvoa ~440 % johtuen siitä, että GS-563117 inhiboi CYP3A:ta. Idelalisibin antaminen yhdessä CYP3A:n substraattien kanssa voi lisätä niiden systeemisiä altistuksia sekä lisätä tai pidentää niiden terapeuttista vaikutusta ja haittavaikutuksia. In vitro CYP3A4:n esto oli pysyvä, ja normaaliin entsyymien toimintaan palautumisen arvioidaan sen vuoksi kestävän useita päiviä idelalisibin annon lopettamisen jälkeen.
Mahdolliset yhteisvaikutukset idelalisibin ja sellaisten samanaikaisesti annettujen lääkevalmisteiden kanssa, jotka ovat CYP3A:n substraatteja, on luetteloitu taulukossa 1 (lisääntyminen on merkitty “↑”:lla). Tämä luettelo ei ole täydellinen, ja se on tarkoitettu ainoastaan ohjeistukseksi. Yleisesti ohjeet samanaikaisesta annostelusta CYP3A4:n estäjien kanssa on tarkastettava muiden lääkevalmisteiden valmisteyhteenvedoista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukko 1: Yhteisvaikutukset idelalisibin ja muiden CYP3A4 substraateiksi luokiteltavien lääkevalmisteiden kanssa
| Lääkevalmiste | Idelalisibin odotettu vaikutus lääkevalmisteen tasoon | Kliininen suositus samanaikaisessa käytössä idelalisibin kanssa |
| ADRENERGISIA ALFA-1-RESEPTOREITA SALPAAVAT LÄÄKEAINEET | ||
| Alfutsosiini | ↑ seerumipitoisuudet | Idelalisibia ei saa antaa samanaikaisesti alfutsosiinin kanssa. |
| KIPULÄÄKKEET | ||
| Fentanyyli, alfentaniili, metadoni, buprenorfiini/naloksoni | ↑ seerumipitoisuudet | Haittavaikutusten (esim. hengityslama, sedaatio) tarkka seuranta on suositeltavaa. |
| RYTMIHÄIRIÖLÄÄKKEET | ||
Amiodaroni, kinidiini Bepridiili, disopyramidi, | ↑ seerumipitoisuudet ↑ seerumipitoisuudet | Idelalisibia ei saa antaa Kliininen seuranta on suositeltavaa. |
| SYÖPÄLÄÄKKEET | ||
| Tyrosiinikinaasin estäjät kuten dasatinibi ja nilotinibi, myös vinkristiini ja vinblastiini | ↑ seerumipitoisuudet | Näiden syöpälääkkeiden toleranssin tarkka seuranta on suositeltavaa. |
| ANTIKOAGULANTIT | ||
| Varfariini | ↑ seerumipitoisuudet | Kansainvälisen vakioidun suhdeluvun (INR, international normalised ratio) seuranta on suositeltavaa samanaikaisessa annossa sekä idelalisibihoidon päättymisen yhteydessä. |
| EPILEPSIALÄÄKKEET | ||
| Karbamatsepiini | ↑ seerumipitoisuudet | Epilepsialääkkeen lääketasoja on seurattava. |
| MASENNUSLÄÄKKEET | ||
| Tratsodoni | ↑ seerumipitoisuudet | Masennuslääkkeen annoksen huolellinen säätäminen ja masennuslääkkeen vasteen seuranta on suositeltavaa. |
| KIHTILÄÄKKEET | ||
| Kolkisiini | ↑ seerumipitoisuudet | Kolkisiiniannoksen pienentäminen saattaa olla tarpeen. Idelalisibia ei saa antaa samanaikaisesti kolkisiinin kanssa potilaille, joilla on heikentynyt munuaisten tai maksan toiminta. |
| VERENPAINELÄÄKKEET | ||
| Amlodipiini, diltiatseemi, felodipiini, nifedipiini, nikardipiini | ↑ seerumipitoisuudet | Terapeuttisen vaikutuksen ja haittavaikutusten kliininen seuranta on suositeltavaa. |
| INFEKTIOLÄÄKKEET | ||
| Sienilääkkeet | ||
| Ketokonatsoli, itrakonatsoli, posakonatsoli, vorikonatsoli | ↑ seerumipitoisuudet | Kliininen seuranta on suositeltavaa. |
| Mykobakteerilääkkeet | ||
| Rifabutiini | ↑ seerumipitoisuudet | Rifabutiiniin liittyvien haittavaikutusten ml. neutropenian ja uveiitin tarkempi seuranta on suositeltavaa. |
| HCV-proteaasin estäjät | ||
| Bosepreviiri, telapreviiri | ↑ seerumipitoisuudet | Kliininen seuranta on suositeltavaa. |
| Makrolidiantibiootit | ||
| Klaritromysiini, telitromysiini | ↑ seerumipitoisuudet | Klaritromysiinin annosta ei tarvitse muuttaa potilailla, joilla on normaali munuaisten toiminta tai lievästi heikentynyt munuaisten toiminta (kreatiniinipuhdistuma [CrCl] 60–90 ml/min). Kliinistä seurantaa suositellaan potilailla, joilla CrCl < 90 ml/min. Potilailla, joiden CrCl < 60 ml/min, suositellaan vaihtoehtoisten bakteerilääkkeiden harkitsemista. Telitromysiinin osalta suositellaan kliinistä seurantaa. |
| PSYKOOSILÄÄKKEET/NEUROLEPTIT | ||
| Ketiapiini, pimotsidi | ↑ seerumipitoisuudet | Idelalisibia ei saa antaa Vaihtoehtoisia lääkevalmisteita, |
| ENDOTELIINIRESEPTORIN ANTAGONISTIT | ||
| Bosentaani | ↑ seerumipitoisuudet | Varovaisuutta on noudatettava ja potilaita on seurattava bosentaaniin liittyvän toksisuuden varalta. |
| TORAJYVÄALKALOIDIT | ||
| Ergotamiini, dihydroergotamiini | ↑ seerumipitoisuudet | Idelalisibia ei saa antaa samanaikaisesti ergotamiinin tai dihydroergotamiinin kanssa. |
| RUOANSULATUSKANAVAN MOTILITEETTIIN VAIKUTTAVAT LÄÄKKEET | ||
| Sisapridi | ↑ seerumipitoisuudet | Idelalisibia ei saa antaa samanaikaisesti sisapridin kanssa. |
| GLUKOKORTIKOIDIT | ||
Inhaloitavat/nasaalit Suun kautta otettava budesonidi | ↑ seerumipitoisuudet ↑ seerumipitoisuudet |
Kliininen seuranta on suositeltavaa |
| HMG Co-A -REDUKTAASIN ESTÄJÄT | ||
Lovastatiini, simvastatiini Atorvastatiini | ↑ seerumipitoisuudet ↑ seerumipitoisuudet | Idelalisibia ei saa antaa Kliininen seuranta on suositeltavaa ja |
| IMMUNOSUPPRESSANTIT | ||
| Siklosporiini, sirolimuusi, takrolimuusi | ↑ seerumipitoisuudet | Terapeuttinen seuranta on suositeltavaa. |
| INHALOITAVAT BEETA-AGONISTIT | ||
| Salmeteroli | ↑ seerumipitoisuudet | Salmeterolin ja idelalisibin samanaikainen anto ei ole suositeltavaa. Yhdistelmä voi johtaa salmeteroliin liittyvien kardiovaskulaaristen haittavaikutusten riskin lisääntymiseen, ml. QT-ajan pidentyminen, palpitaatiot ja sinustakykardia. |
| FOSFODIESTERAASIN ESTÄJÄT | ||
Sildenafiili Tadalafiili Sildenafiili, tadalafiili | ↑ seerumipitoisuudet ↑ seerumipitoisuudet ↑ seerumipitoisuudet | Keuhkoverenpainetaudissa: Varovaisuutta on noudettava, ml. Erektiohäiriöissä: |
| SEDATIIVIT/UNILÄÄKKEET | ||
Midatsolaami (suun kautta Buspironi, kloratsepaatti, | ↑ seerumipitoisuudet ↑ seerumipitoisuudet | Idelalisibia ei saa antaa Sedatiivin/unilääkkeen pitoisuuden |
CYP2C8:n substraatit
In vitro idelalisibi sekä esti että indusoi CYP2C8:aa, mutta ei tiedetä, vastaako tämä in vivo -vaikutusta CYP2C8:n substraatteihin. Varovaisuutta on noudatettava, jos Zydelig-valmistetta käytetään yhdessä kapean terapeuttisen indeksin omaavien lääkevalmisteiden kanssa, jotka ovat CYP2C8:n substraatteja (paklitakseli).
Indusoitavien entsyymien substraatit (esim. CYP2C9, CYP2C19, CYP2B6 ja UGT)
In vitro, idelalisibi oli useiden entsyymien indusoija. Siksi pienemmän altistuksen ja siten alentuneen tehon riskiä indusoitavien entsyymien kuten CYP2C9, CYP2C19, CYP2B6 ja UGT substraattien osalta ei voida poissulkea. Varovaisuutta on noudatettava, jos Zydelig-valmistetta käytetään yhdessä kapean terapeuttisen indeksin omaavien lääkevalmisteiden kanssa, jotka ovat näiden entsyymien substraatteja (varfariini, fenytoiini, S-mefenytoiini).
BCRP, OATP1B1, OATP1B3 ja P-gp:n substraatit
Yhteiskäytössä rosuvastatiinin tai digoksiinin kanssa idelalisibi 150 mg kahdesti päivässä terveille henkilöille jatkuvasti annosteltuna ei muuttanut merkittävästi rosuvastatiinin (AUC 90 % CI: 87, 121) tai digoksiinin (AUC 90 % CI: 98, 111) altistuksia, mikä viittaa siihen, että idelalisibi ei estä BCRP:tä, OATP1B1/1B3:a tai systeemistä P-gp:tä kliinisesti merkittävällä tavalla. P-gp:n eston riskiä mahasuolikanavassa, mikä voisi johtaa suoliston P-gp:n estolle herkkien substraattien kuten dabigatraanieteksilaatin pitoisuuden kasvuun, ei voida poissulkea.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy
Eläintutkimusten tulosten perusteella idelalisibi saattaa vahingoittaa sikiötä. Naisten on vältettävä raskaaksituloa Zydelig-hoidon aikana ja 1 kuukauden ajan hoidon päättymisen jälkeen. Tämän vuoksi naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä Zydelig-hoidon aikana ja 1 kuukauden ajan hoidon päättymisen jälkeen. Tällä hetkellä ei tiedetä, heikentääkö idelalisibi hormoniehkäisyvalmisteiden tehoa, minkä vuoksi hormoniehkäisyvalmisteita käyttävien naisten tulisi käyttää mekaanista ehkäisyä lisäehkäisynä.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja idelalisibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Zydelig-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyvätkö idelalisibi ja sen metaboliitit ihmisen rintamaitoon.
Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Imetys on lopetettava Zydelig-hoidon ajaksi.
Hedelmällisyys
Idelalisibin vaikutuksesta hedelmällisyyteen ihmisellä ei ole tietoa. Eläinkokeissa on havaittu, että idelalisibilla saattaa olla haitallisia vaikutuksia hedelmällisyyteen ja sikiön kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Zydelig-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kliinisissä tutkimuksissa, joihin osallistuneilla tutkittavilla oli hematologisia maligniteetteja ja he saivat idelalisibia, yleisimmin raportoituja haittavaikutuksia olivat infektiot (70 %), neutropenia (55 %), transaminaasien nousu (53 %), ripuli (48 %), triglyseridien nousu (47 %), kuume (36 %), ihottuma (30 %) ja lymfosytoosi (21 %). Yleisimmin raportoituja vaikeita haittavaikutuksia (≥ aste 3) olivat infektiot (39 %), neutropenia (33 %), ripuli/koliitti (22 %), transaminaasien nousu (15 %) ja lymfosytoosi (13 %).
Haittavaikutustaulukko
Haittavaikutusten arviointi perustuu kahteen faasin 3 tutkimukseen (tutkimukset 312-0116 ja 312-0119) ja kuuteen faasin 1 ja 2 tutkimukseen. Tutkimus 312-0116 oli satunnaistettu, lumelääkekontrolloitu kaksoissokkotutkimus, jossa 110 aiemmin hoidettua KLL:aa sairastavaa koehenkilöä sai idelalisibia + rituksimabia. Tästä tutkimuksesta 86 koehenkilöä, jotka oli satunnaistettu saamaan lumelääkettä + rituksimabia, osallistui lisäksi jatkotutkimukseen (tutkimus 312-0117), jossa he saivat idelalisibia monoterapiana. Tutkimus 312-0119 oli satunnaistettu, kontrolloitu, avoin tutkimus, jossa 173 aiemmin hoidettua KLL:aa sairastavaa koehenkilöä sai idelalisibia + ofatumumabia. Faasin 1 ja 2 tutkimuksissa arvioitiin idelalisibin turvallisuutta yhteensä 536 koehenkilöllä, joilla oli hematologisia maligniteetteja, sisältäen 400 koehenkilöä, jotka saivat idelalisibia (mikä tahansa annos) monoterapiana ja 136 koehenkilöä, jotka saivat idelalisibia yhdessä CD20:n monoklonaalisen vasta-aineen (rituksimabin tai ofatumumabin) kanssa.
Taulukossa 2 on esitetty ne lääkkeiden haittavaikutukset, joita on ilmoitettu käytettäessä idelalisibia yksin tai yhdessä CD20:n monoklonaalisten vasta-aineiden (rituksimabin tai ofatumumabin) kanssa. Haittavaikutukset on lueteltu elinjärjestelmien ja esiintymistiheyden mukaan. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 2: Kliinisissä tutkimuksissa ilmoitetut lääkkeiden haittavaikutukset koehenkilöillä, joilla oli hematologisia maligniteetteja ja jotka saivat idelalisibia, ja markkinoille tulon jälkeen ilmoitetut haittavaikutukset
| Haittavaikutukset | Kaikki asteet | Aste ≥ 3 |
| Infektiot | ||
| Infektiot (mukaan lukien Pneumocystis jirovecii ‑keuhkokuume ja CMV‑infektio)* | Hyvin yleinen | Hyvin yleinen |
| Veri ja imukudos | ||
| Neutropenia | Hyvin yleinen | Hyvin yleinen |
| Lymfosytoosi** | Hyvin yleinen | Hyvin yleinen |
| Hengityselimet, rintakehä ja välikarsina | ||
| Pneumoniitti | Yleinen | Yleinen |
| Organisoituva keuhkokuume**** | Melko harvinainen | Melko harvinainen |
| Ruoansulatuselimistö | ||
| Ripuli/koliitti | Hyvin yleinen | Hyvin yleinen |
| Maksa ja sappi | ||
| Transaminaasin nousu | Hyvin yleinen | Hyvin yleinen |
| Hepatosellulaarinen vaurio | Yleinen | Yleinen |
| Iho ja ihonalainen kudos | ||
| Ihottuma*** | Hyvin yleinen | Yleinen |
| Stevens-Johnsonin oireyhtymä/ toksinen epidermaalinen nekrolyysi**** | Harvinainen | Harvinainen |
| Yleisoireinen eosinofiilinen oireyhtymä (DRESS)**** | Tuntematon | Tuntematon |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Kuume | Hyvin yleinen | Yleinen |
| Tutkimukset | ||
| Triglyseridien nousu | Hyvin yleinen | Yleinen |
* Sisältää sekä opportunistiset infektiot että bakteeri‑ ja virusinfektiot, kuten keuhkokuumeen, bronkiitin ja sepsiksen.
** Idelalisibin aiheuttamaa lymfosytoosia ei pidä tulkita taudin etenemiseksi, jos muita kliinisiä löydöksiä ei ole (ks. kohta Farmakodynamiikka).
*** Sisältää suositellut termit yleistynyt kesivä ihottuma, lääkeaineihottuma, ihottuma, punoittava ihottuma, yleistynyt ihottuma, täpläinen ihottuma, täpläinen ja näppyläinen ihottuma, näppyläinen ihottuma, kutiseva ihottuma, märkärakkulainen ihottuma, rakkulainen ihottuma, näppylät, koholäiskät ja hilseilevä ihottuma.
**** Havaittu markkinoille tulon jälkeen saatujen tietojen perusteella.
Valikoitujen haittavaikutusten kuvaus
Infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Idelalisibilla tehdyissä kliinisissä tutkimuksissa havaittiin, että kaikkien infektioiden, myös 3. ja 4. asteen infektioiden, esiintymistiheys oli idelalisibihoitoryhmissä suurempi kuin vertailuryhmissä. Yleisimpiä olivat hengityselinten infektiot ja septiset tapahtumat. Useissa tapauksissa patogeenia ei tunnistettu; tunnistettujen patogeenien joukossa oli kuitenkin sekä tavanomaisia että opportunistisia patogeeneja, mukaan lukien Pneumocystis jirovecii ja CMV. Lähes kaikki PJP‑infektiot, joihin sisältyi myös kuolemaan johtaneita tapauksia, ilmenivät potilailla, jotka eivät olleet saaneet PJP‑estohoitoa. PJP‑tapauksia on esiintynyt idelalisibihoidon lopettamisen jälkeen.
Ihottuma
Ihottuma oli tavallisesti lievää tai kohtalaista ja johti hoidon keskeyttämiseen 2,1 %:lla koehenkilöistä. Tutkimuksissa 312-0116/0117 ja 312-0119 ihottumaa (ilmoitettu termein yleistynyt kesivä ihottuma, lääkeaineihottuma, ihottuma, punoittava ihottuma, yleistynyt ihottuma, täpläinen ihottuma, täpläinen ja näppyläinen ihottuma, näppyläinen ihottuma, kutiseva ihottuma, märkärakkulainen ihottuma, rakkulainen ihottuma, näppylät sekä koholäiskät) esiintyi 31,1 %:lla idelalisibia + CD20:n monoklonaalista vasta-ainetta (rituksimabia tai ofatumumabia) saaneista koehenkilöistä ja 8,2 %:lla pelkkää CD20:n monoklonaalista vasta-ainetta (rituksimabia tai ofatumumabia) saaneista koehenkilöistä. Näistä 5,7 %:lla idelalisibia + CD20:n monoklonaalista vasta-ainetta (rituksimabia tai ofatumumabia) saaneista ja 1,5 %:lla pelkkää CD20:n monoklonaalista vasta-ainetta (rituksimabia tai ofatumumabia) saaneista oli asteen 3 ihottuma. Yhdelläkään potilaalla ei ollut asteen 4 haittavaikutusta. Ihottuma hävisi tavallisesti, kun sitä hoidettiin (esim. paikallisilla ja/tai suun kautta otettavilla steroideilla, difenhydramiinilla) ja kun annoksen antaminen keskeytettiin vakavissa tapauksissa (ks. kohta Prekliiniset tiedot turvallisuudesta, fototoksisuus).
Vakavat ihoreaktiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Stevens-Johnsonin oireyhtymää, toksista epidermaalista nekrolyysiä sekä yleisoireista eosinofiilistä oireyhtymää (DRESS) on ilmennyt, kun idelalisibia on annettu samanaikaisesti muiden lääkevalmisteiden kanssa, jotka on liitetty näihin oireyhtymiin (bendamustiini, rituksimabi, allopurinoli ja amoksisilliini sekä sulfametoksatsoli/trimetopriimi). Stevens-Johnsonin oireyhtymää tai toksista epidermaalista nekrolyysiä ilmeni yhden kuukauden sisällä lääkeyhdistelmän antamisesta. Kuolemaan johtaneita tapauksia on ilmennyt.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksessa potilasta on tarkkailtava myrkytysoireiden varalta (ks. kohta Haittavaikutukset). Zydelig-valmisteen yliannostusta tulee hoitaa oireenmukaisesti, mukaan lukien potilaan elintoimintojen ja kliinisen tilan tarkkailu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, proteiinikinaasin estäjät, fosfatidyyli-inositoli-3-kinaasin (Pi3K) estäjät, ATC-koodi: L01EM01
Vaikutusmekanismi
Idelalisibi inhiboi fosfatidyyli-inositoli-3-kinaasia p110δ (PI3Kδ), joka on hyperaktiivinen B-solumaligniteeteissa ja on tärkeä useille sellaisille signaalien välitysreiteille, jotka edistävät malignien solujen proliferaatiota, eloonjäämistä, kotiutumista ja retentiota imukudoksissa ja luuytimessä. Idelalisibi estää selektiiviesti adenosiini-5’-trifosfaatin (ATP) sitoutumista katalyyttiseen domeeniin PI3Kδ, minkä seurauksena keskeisen lipidirakenteisen toisiolähetin, fosfatidyyli-inositolin fosforylaatio estyy ja Akt:n (proteiinikinaasi B) fosforylaatio estyy.
Idelalisibi indusoi apoptoosia ja estää proliferaatiota maligneista B-soluista peräisin olevissa solulinjoissa ja ensisijaisissa kasvainsoluissa. CXCL12- ja CXCL13-kemokiinien indusoimaa, vastaavien kemokiinireseptorien CXCR4 ja CXCR5 signaalinvälitystä inhiboimalla idelalisibi inhiboi malignien B-solujen kotiutumista ja retentiota kasvaimen mikroympäristössä, mukaan lukien imukudokset ja luuydin.
Kliinisissä tutkimuksissa ei ole löydetty mekanismiin liittyviä selityksiä resistenssin kehittymiselle idelalisibihoitoa kohtaan. Lisätutkimuksia tästä aiheesta ei ole suunnitteilla nykyisissä B‑solumaligniteettia koskevissa tutkimuksissa.
Farmakodynaamiset vaikutukset
Elektrokardiografia
Idelalisibin (150 mg ja 400 mg) vaikutusta QT/QTc-aikaan arvioitiin lumelääke- ja aktiivikontrolloidussa (moksifloksasiini 400 mg) eräässä vaihtovuoroisessa tutkimuksessa 40 terveellä henkilöllä. Käytettäessä annosta, joka oli 2,7-kertainen suurimpaan suositeltuun annokseen nähden, idelalisibi ei pidentänyt QT/QTc-aikaa (ts. < 10 ms).
Lymfosytoosi
Idelalisibihoitoa aloitettaessa on havaittu tilapäistä lymfosyyttimäärien suurenemista (≥ 50 % enemmän kuin lähtötilanteessa ja absoluuttinen lymfosyyttimäärä yli 5 000/mm3). Tätä ilmenee noin kahdella kolmasosalla KLL-potilaista, jotka saavat idelalisibimonoterapiaa ja neljäsosalla KLL-potilaista, jotka saavat idelalisibiyhdistelmähoitoa. Yksittäisiä lymfosytoositapauksia ilmenee tavallisesti kahden ensimmäisen idelalisibihoitoviikon aikana, ja niihin liittyy usein lymfadenopatian heikkeneminen. Tämä havaittu lymfosytoosi on farmakodynaaminen vaikutus, jota ei pidä tulkita taudin etenemiseksi, jos muita kliinisiä löydöksiä ei ole.
Kliininen teho kroonisessa lymfaattisessa leukemiassa
Idelalisibi yhdistelmänä rituksimabin kanssa
Tutkimus 312-0116 oli faasin 3 satunnaistettu, lumelääkekontrolloitu kaksoissokkotutkimus, johon osallistui 220 aiemmin hoidettua KLL:aa sairastavaa, hoitoa tarvitsevaa koehenkilöä, joille sytotoksisen kemoterapian ei arvioitu sopivan. Koehenkilöt satunnaistettiin suhteessa 1:1 saamaan 8 jaksoa rituksimabia (ensimmäinen jakso annoksella 375 mg/m2 kehon pinta-alasta, seuraavat jaksot annoksella 500 mg/m2 kehon pinta-alasta) joko kahdesti päivässä suun kautta otettavan lumelääkkeen tai kahdesti päivässä otettavan 150 mg idelalisibin kanssa taudin etenemiseen tai kestämättömien toksisten vaikutusten ilmaantumiseen asti.
Mediaani-ikä oli 71 vuotta (vaihteluväli: 47-92). Koehenkilöistä 78,2 % oli yli 65-vuotiaita; 65,5 % oli miespuolisia ja 90,0 % oli valkoihoisia; 64,1 %:lla oli Rai-aste III tai IV ja 55,9 %:lla oli Binet'n aste C. Useimmilla koehenkilöillä oli sytogeneettisiä huonon ennusteen tekijöitä: 43,2 %:lla oli kromosomaalinen 17p-deleetio ja/tai kasvainproteiini 53:n (TP53) mutaatio ja 83,6 %:lla immunoglobuliinin raskaan ketjun muuttuvan alueen (IGHV) mutatoitumattomia geenejä.
Mediaaniaika KLL-diagnoosista satunnaistamiseen oli 8,5 vuotta. Koehenkilöiden CIRS (Cumulative Illness Rating Scale) -pistemäärän mediaani oli 8. Aikaisempien hoitojen mediaanilukumäärä oli 3,0. Lähes kaikki (95,9 %) koehenkilöt olivat saaneet aikaisemmin CD20:n monoklonaalisia vasta-aineita. Ensisijainen päätetapahtuma oli etenemisvapaa elossaoloaika (PFS, progression free survival). Tehoa koskevat tulokset on esitetty taulukoissa 3 ja 4. Etenemisvapaan elossaoloajan Kaplan-Meierin käyrä on esitetty kuvassa 1.
Rituksimabiin + lumelääkkeeseen verrattuna idelalisibi + rituksimabi -hoito johti tilastollisesti merkitsevään ja kliinisesti merkittävään fyysisen, sosiaalisen ja toiminnallisen hyvinvoinnin paranemiseen, Functional Assessment of Cancer Therapy: Leukaemia (FACT-LEU) - kyselylomakkeen leukemiaspesifien muuttujien paranemiseen sekä tilastollisesti merkitsevään ja kliinisesti merkittävään ahdistuneisuuden, masennuksen ja tavallisten toimintojen paranemiseen mitattuna EQ-5D (EuroQoL Five-Dimensions) -kyselylomakkeella.
Taulukko 3: Tehoa koskevat tulokset tutkimuksesta 312-0116
| Idelalisibi + R N = 110 | Lumelääke + R N = 110 | ||
| PFS | Mediaani (kk) (95 % CI) | 19,4 (12,3; NR) | 6,5 (4,0; 7,3) |
| Riskisuhde (HR) (95 % CI) | 0,15 (0,09; 0,24) | ||
| P-arvo | < 0,0001 | ||
| ORR* | n (%) (95 % CI) | 92 (83,6 %) (75,4; 90,0) | 17 (15,5 %) (9,3; 23,6) |
| Kerroinsuhde (95 % CI) | 27,76 (13,40; 57,49) | ||
| P-arvo | < 0,0001 | ||
| LNR** | n/N (%) (95 % CI) | 102/106 (96,2 %) (90,6; 99,0) | 7/104 (6,7 %) (2,7; 13,4) |
| Kerroinsuhde (95 % CI) | 225,83 (65,56; 777,94) | ||
| P-arvo | < 0,0001 | ||
| OS^ | Mediaani (kk) (95 % CI) | NR (NR; NR) | 20,8 (14,8; NR) |
| Riskisuhde (HR) (95 % CI) | 0,34 (0,19; 0,60) | ||
| P-arvo | 0,0001 | ||
CI: luottamusväli; R: rituksimabi; n: reagoivien koehenkilöiden lukumäärä; N: koehenkilöiden lukumäärä ryhmää kohti; NR: ei saavutettu (not reached). PFS:n, kokonaishoitovasteen (ORR, overall response rate) ja imusolmukkeiden hoitovasteen (LNR, lymph node response rate) analyysit perustuivat riippumattoman arviointiryhmän (IRC, independent review committee) suorittamaan arviointiin.
* ORR on määritelty niiden koehenkilöiden osuudeksi, jotka saavuttivat vuoden 2013 NCCN (National Comprehensive Cancer Network) -vastekriteerien ja Cheson (2012) mukaisen täydellisen vasteen (CR, complete response) tai osittaisen vasteen (PR, partial response).
** LNR määriteltiin niiden koehenkilöiden osuudeksi, jotka saavuttivat ≥ 50 % laskun indeksivaurioiden suurimpien kohtisuorien halkaisijoiden tulojen summassa. Tähän analyysiin sisällytettiin vain ne koehenkilöt, joilla oli sekä lähtötason arviointi että ≥ 1 arvioitavissa olevia lähtötason jälkeisiä arviointeja.
^ Kokonaiselinaika-analyysi (OS, overall survival) sisältää tiedot koehenkilöistä, jotka saivat lumelääkettä + R tutkimuksessa 312-0116 ja sen jälkeen idelalisibia jatkotutkimuksessa, perustuen hoitoaikeen mukaiseen
analyysiin (ITT).
Taulukko 4: Yhteenveto PFS:stä ja vasteista ennalta määritetyissä alaryhmissä tutkimuksesta 312-0116
| Idelalisibi + R | Lumelääke + R | |
| 17p-deleetio/TP53-mutaatio | N = 46 | N = 49 |
| PFS:n mediaani (kk) (95 % CI) | NR (12,3; NR) | 4,0 (3,7; 5,7) |
| Riskisuhde (HR) (95 % CI) | 0,13 (0,07; 0,27) | |
| ORR (95 % CI) | 84,8 % (71,1; 93,7) | 12,2 % (4,6; 24,8) |
| Mutatoitumaton IGHV | N = 91 | N = 93 |
| PFS:n mediaani (kk) (95 % CI) | 19,4 (13,9; NR) | 5,6 (4,0; 7,2) |
| Riskisuhde (HR) (95 % CI) | 0,14 (0,08; 0,23) | |
| ORR (95 % CI) | 82,4 % (73,0; 89,6) | 15,1 % (8,5; 24,0) |
| Ikä ≥ 65 vuotta | N = 89 | N = 83 |
| PFS:n mediaani (kk) (95 % CI) | 19,4 (12,3; NR) | 5,7 (4,0; 7,3) |
| Riskisuhde (HR) (95 % CI) | 0,14 (0,08; 0,25) | |
| ORR (95 % CI) | 84,3 % (75,0; 91,1) | 16,9 % (9,5; 26,7) |
CI: luottamusväli; R: rituksimabi; N: koehenkilöiden lukumäärä ryhmää kohti; NR: ei saavutettu (not reached)
Kuva 1: Etenemisvapaata elossaoloaikaa koskeva Kaplan-Meierin käyrä tutkimuksesta 312-0116 (hoitoaikeen mukainen (ITT) potilasryhmä)
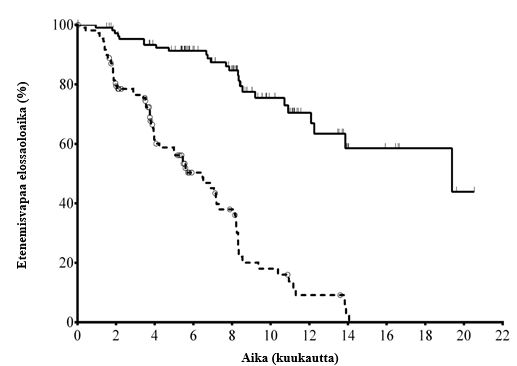
| Riskiryhmään kuuluvien N (tapahtumat) | ||||||||||||
| Idelalisibi + R | 110 (0) | 101 (3) | 93 (7) | 73 (9) | 59 (14) | 31 (19) | 20 (21) | 9 (24) | 7 (24) | 4 (24) | 1 (25) | 0 (25) |
| Lumelääke + R | 110 (0) | 84 (21) | 48 (38) | 29 (46) | 20 (53) | 9 (63) | 4 (67) | 1 (69) | 0 (70) | 0 (70) | 0 (70) | 0 (70) |
Yhtenäinen viiva: idelalisibi + R (N = 110), katkoviiva: lumelääke + R (N = 110)
R: rituksimabi; N: koehenkilöiden lukumäärä ryhmää kohti
PFS:n analyysi perustui riippumattoman arviointiryhmän (IRC) suorittamaan arviointiin. Lumelääke- + R-ryhmän koehenkilöiden osalta yhteenveto sisältää tiedot idelalisibin ensimmäiseen annosteluun asti jatkotutkimuksessa.
Tutkimukseen 101‑08/99 osallistui 64 koehenkilöä, joilla oli aiemmin hoitamaton KLL, mukaan lukien 5 koehenkilöä, joilla oli pienilymfosyyttinen lymfooma (SLL). Koehenkilöt saivat 150 mg idelalisibia kahdesti päivässä ja rituksimabia 375 mg/m2 kehon pinta-alasta viikottain 8 annoksen ajan. ORR oli 96,9 %, 12 koehenkilöä saavutti CR:n (18,8 %) ja 50 PR:n (78,1 %) mukaan lukien 3 CR:ta ja 6 PR:ta koehenkilöillä, joilla oli 17p‑deleetio ja/tai TP53‑mutaatio sekä 2 CR:ta ja 34 PR:ta koehenkilöillä, joilla oli mutatoitumaton IGHV. Vasteen mediaanikestoa ei ole saavutettu.
Idelalisibi yhdistelmänä ofatumumabin kanssa
Tutkimus 312‑0119 oli faasin 3 satunnaistettu, avoin, rinnakkaisryhmillä toteutettu monikeskustutkimus. Tutkimukseen osallistui 261 aiemmin hoidettua KLL:aa sairastavaa koehenkilöä, joilla oli mitattavissa oleva lymfadenopatia, jotka tarvitsivat hoitoa, ja joiden KLL oli edennyt < 24 kuukauden kuluttua viimeisimmän aiemman hoidon lopettamisen jälkeen. Koehenkilöt satunnaistettiin suhteessa 2:1 saamaan idelalisibia 150 mg kahdesti vuorokaudessa ja 12 ofatumumabi-infuusiota 24 viikon ajan tai saamaan pelkästään 12 ofatumumabi-infuusiota 24 viikon ajan. Ensimmäinen ofatumumabi-infuusio annettiin 300 mg:n annoksena, jonka jälkeen jatkettiin 1 000 mg:n annoksella idelalisibi + ofatumumabi -ryhmässä ja 2 000 mg:n annoksella pelkän ofatumumabin ryhmässä. Annokset annettiin ensin viikon välein 7 annoksen ajan ja sitten 4 viikon välein 4 annoksen ajan. Idelalisibia annettiin taudin etenemiseen tai kestämättömien toksisten vaikutusten ilmaantumiseen asti.
Mediaani-ikä oli 68 vuotta (vaihteluväli: 61‑74). Koehenkilöistä 64,0 % oli yli 65‑vuotiaita; 71,3 % oli miespuolisia ja 84,3 % oli valkoihoisia; 63,6 %:lla oli Rai-aste III tai IV ja 58,2 %:lla oli Binet'n aste C. Useimmilla koehenkilöillä oli sytogeneettisiä huonon ennusteen tekijöitä: 39,5 %:lla oli kromosomaalinen 17p‑deleetio ja/tai kasvainproteiini 53:n (TP53) mutaatio ja 78,5 %:lla immunoglobuliinin raskaan ketjun muuttuvan alueen (IGHV) mutatoitumattomia geenejä. Mediaaniaika diagnoosista oli 7,7 vuotta. Koehenkilöiden CIRS (Cumulative Illness Rating Scale) -pistemäärän mediaani oli 4. Aikaisempien hoitojen mediaanilukumäärä oli 3,0. Ensisijainen päätetapahtuma oli etenemisvapaa elossaoloaika (PFS, progression free survival). Tehoa koskevat tulokset on esitetty taulukoissa 5 ja 6. Etenemisvapaan elossaoloajan Kaplan‑Meierin käyrä on esitetty kuvassa 2.
Taulukko 5: Tehoa koskevat tulokset tutkimuksesta 312‑0119
| Idelalisibi + O N = 174 | Ofatumumabi N = 87 | ||
| PFS Mediaani (kk) (95 % CI) Riskisuhde (HR) (95 % CI) P-arvo | 16,3 (13,6; 17,8) | 8,0 (5,7; 8.2) | |
| 0,27 (0,19; 0,39) | |||
| < 0,0001 | |||
| ORR* n (%) (95 % CI) Kerroinsuhde /95 % CI) P-arvo | 131 (75,3 %) (68,2; 81,5) | 16 (18,4 %) (10,9; 28,1) | |
| 15,94 (7,8; 32,58) | |||
| < 0,0001 | |||
| LNR** n/N (%) (95 % CI) Kerroinsuhde (95 % CI) P‑arvo | 153/164 (93,3 %) (88,3; 96,6) | 4/81 (4,9 %) (1,4; 12,2) | |
| 486,96 (97,91; 2 424,85) | |||
| < 0,0001 | |||
| OS Mediaani (kk) (95 % CI) Riskisuhde (HR) (95 % CI) P‑arvo | 20,9 (20,9; NR) | 19,4 (16,09; NR) | |
| 0,74 (0,44; 1,25) | |||
| 0,27 | |||
CI: luottamusväli; O: ofatumumabi; n: reagoivien koehenkilöiden lukumäärä; N: koehenkilöiden lukumäärä ryhmää kohti; NR: ei saavutettu (not reached). PFS:n, kokonaishoitovasteen (ORR, overall response rate) ja imusolmukkeiden hoitovasteen (LNR, lymph node response rate) analyysit perustuivat riippumattoman arviointiryhmän (IRC, independent review committee) suorittamaan arviointiin.
* ORR on määritelty niiden koehenkilöiden osuudeksi, jotka saavuttivat täydellisen vasteen (CR, complete response) tai osittaisen vasteen (PR, partial response), joka säilyi vähintään 8 viikon ajan.
** LNR määriteltiin niiden koehenkilöiden osuudeksi, jotka saavuttivat ≥ 50 % laskun indeksivaurioiden suurimpien kohtisuorien halkaisijoiden tulojen summassa. Tähän analyysiin sisällytettiin vain ne koehenkilöt, joilla oli sekä lähtötason arviointi että ≥ 1 arvioitavissa olevia lähtötason jälkeisiä arviointeja.
Taulukko 6: Yhteenveto PFS:stä ja vasteista ennalta määritetyissä alaryhmissä tutkimuksesta 312‑0119
| Idelalisibi + O | Ofatumumabi | |
| 17p‑deleetio/TP53‑mutaatio | N = 70 | N = 33 |
| PFS:n mediaani (kk) (95 % CI) | 13,7 (11,0; 17,8) | 5,8 (4,5; 8,4) |
| Riskisuhde (HR) (95 % CI) | 0,32 (0,18; 0,57) | |
| ORR (95 % CI) | 72,9 % (60,9; 82,8) | 15,2 % (5,1; 31,9) |
| Mutatoitumaton IGHV | N = 137 | N = 68 |
| PFS:n mediaani (kk) (95 % CI) | 14,9 (12,4; 17,8) | 7,3 (5,3; 8,1) |
| Riskisuhde (HR) (95 % CI) | 0,25 (0,17; 0,38) | |
| ORR (95 % CI) | 74,5 % (66,3; 81,5) | 13,2 % (6,2; 23,6) |
| Ikä ≥ 65 vuotta | N = 107 | N = 60 |
| PFS:n mediaani (kk) (95 % CI) | 16,4 (13,4; 17,8) | 8,0 (5,6; 8,4) |
| Riskisuhde (HR) (95 % CI) | 0,30 (0,19; 0,47) | |
| ORR (95 % CI) | 72,0 % (62,5; 80,2) | 18,3 % (9,5; 30,4) |
CI: luottamusväli; O: ofatumumabi; N: koehenkilöiden lukumäärä ryhmää kohti
Kuva 2: Etenemisvapaata elossaoloaikaa koskeva Kaplan-Meierin käyrä tutkimuksesta 312‑0119 (hoitoaikeen mukainen (ITT) potilasryhmä)
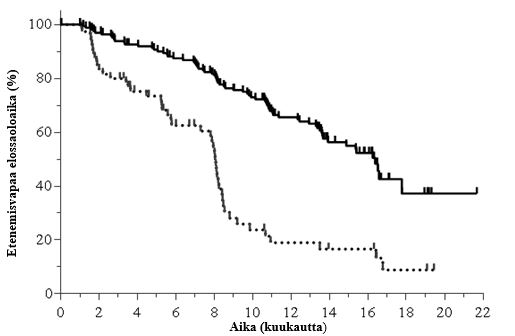
| Riskiryhmään kuuluvien N (tapahtumat) | ||||||||||||
| Idelalisibi + O | 174 (0) | 162 (6) | 151 (13) | 140 (22) | 129 (31) | 110 (45) | 82 (57) | 44 (67) | 37 (70) | 7 (76) | 1 (76) | 0 (76) |
| Ofatumumabi | 87 (0) | 60 (14) | 47 (21) | 34 (30) | 26 (34) | 11 (49) | 8 (51) | 6 (52) | 6 (52) | 2 (54) | 0 (54) | 0 (54) |
Yhtenäinen viiva: idelalisibi + O (N = 174), katkoviiva: ofatumumabi (N = 87)
O: ofatumumabi; N: koehenkilöiden lukumäärä ryhmää kohti
Kliininen teho follikulaarisessa lymfoomassa
Idelalisibin turvallisuutta ja tehoa arvioitiin yksihaaraisessa kliinisessä monikeskustutkimuksessa (tutkimus 101-09) 125 koehenkilöllä, joilla oli indolentti non-Hodgkin-B-solulymfooma (iNHL, joista: FL, n = 72; SLL, n = 28; lymfoplasmasyyttinen lymfooma/Waldenströmin makroglobulinemia [LPL/WM], n = 10; ja marginaalivyöhykkeen lymfooma [MZL], n = 15). Kaikki koehenkilöt olivat hoitoresistenttejä rituksimabille ja 124 koehenkilöä 125:stä oli hoitoresistenttejä ainakin yhdelle alkyloivalle aineelle. 112 koehenkilöä (89,6 %) oli hoitoresistenttejä tutkimuksen alkua viimeksi edeltäneelle hoidolle.
Tutkimukseen osallistuneesta 125 koehenkilöstä 80 (64 %) oli miespuolisia, mediaani-ikä oli 64 vuotta (vaihteluväli: 33-87) ja 110 (89 %) oli valkoihoisia. Koehenkilöt saivat 150 mg idelalisibin suun kautta kahdesti päivässä taudin todettuun etenemiseen tai kestämättömien toksisten vaikutusten ilmaantumiseen asti.
Ensisijainen päätetapahtuma oli ORR, joka määritettiin niiden koehenkilöiden osuudeksi, jotka saavuttivat CR:n tai PR:n (perustuen malignin lymfoomaa koskevan vasteen tarkistettuihin kriteereihin [Cheson]), ja Waldenströmin makroglobulinemiaa sairastavilla koehenkilöillä niiden koehenkilöiden osuudeksi, jotka saavuttivat vähäisen vasteen (MR, minor response) (perustuen Waldenströmin makroglobulinemian vastearviointiin [Owen]). Vasteen kesto oli toissijainen päätetapahtuma, ja se määritettiin ajaksi ensimmäisestä dokumentoidusta vasteesta (CR, PR tai MR) ensimmäiseen tietoon sairauden etenemisestä tai mistä tahansa syystä johtuneeseen kuolemaan. Tehoa koskevat tulokset on esitetty taulukossa 7.
Taulukko 7: Yhteenveto tehosta tutkimuksessa 101-09 (IRC:n arviointi)
| Ominaisuus | iNHL-kokonaiskohortti (N = 125) n (%) | FL-alaryhmä (N = 72) n (%) |
ORR* Vastekategoria*† | 72 (57,6 %)
13 (10,4 %) | 40 (55,6 %)
12 (16,7 %) |
| Vasteen kesto (kk) mediaani (95 % CI) | 12,5 (7,4; 22,4) | 11,8 (6,2; 26,9) |
| PFS (kk) mediaani (95 % CI) | 11,1 (8,3; 14,0) | 11,0 (8,0; 14,0) |
| OS (kk) mediaani (95 % CI) | 48,6 (33,9; 71,7) | 61,2 (38,1; NR) |
CI: luottamusväli; n: reagoivien koehenkilöiden lukumäärä;
NR: ei saavutettu (not reached)
* Riippumattoman arviointiryhmän (IRC, independent review committee) määrittämä vaste, jossa ORR = täydellinen vaste (CR) + osittainen vaste (PR) + vähäinen vaste (MR) WM-koehenkilöillä.
† iNHL-kokonaiskohortissa yhdellä koehenkilöllä (0,6 %), jolla oli WM, paras kokonaisvaste oli MR.
Kaikkien koehenkilöiden vasteen mediaanikesto oli 12,5 kuukautta (SLL-koehenkilöillä 12,5 kuukautta, FL-koehenkilöillä 11,8 kuukautta, LPL/WM-koehenkilöillä 20,4 kuukautta ja MZL-koehenkilöillä 18,4 kuukautta). Niiden 122 koehenkilön joukossa, joilla oli mitattavissa olevia imusolmukkeita sekä lähtötasossa että lähtötason jälkeen, 71 koehenkilöä (58,2 %) saavutti ≥ 50 % laskun lähtötasosta indeksivaurioiden halkaisijoiden tulojen summassa (SPD, sum of the products of the diameters). Hoitoon vastaamattomista 53 koehenkilöstä 41:llä (32,8 %) oli vakaa tauti, 10:llä (8,0 %) oli etenevä sairaus ja 2:n (1,6 %) sairaus ei ollut arvioitavissa. Mediaani-OS, mukaan lukien kaikkien 125 koehenkilön pitkäaikaisseuranta, oli 48,6 kuukautta. Mediaani-OS, mukaan lukien kaikkien FL-koehenkilöiden pitkäaikaisseuranta, oli 61,2 kuukautta.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset idelalisibin käytöstä kypsien B‑solujen neoplasmien hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Sen jälkeen, kun idelalisibin kerta-annos oli annettu potilaalle suun kautta ruoan nauttimisen jälkeen, huippupitoisuudet plasmassa havaittiin 2-4 tunnin kuluttua annoksen ottamisesta ja paastotilassa 0,5-1,5 tunnin kuluttua annoksen ottamisesta.
Sen jälkeen, kun idelalisibia oli annettu 150 mg kahdesti päivässä, keskimääräiset (vaihteluväli) Cmax- ja AUC-arvot vakaassa tilassa olivat 1 953 (272; 3 905) ng/ml ja 10 439 (2 349; 29 315) ng•h/ml idelalisibin osalta ja 4 039 (669; 10 897) ng/ml ja 39 744 (6 002; 119 770) ng•h/ml GS-563117:n osalta. Idelalisibin altistus plasmassa (Cmax ja AUC) on lähes suhteessa annokseen välillä 50-100 mg ja vähemmän kuin suhteessa annokseen yli 100 mg:n annoksilla.
Ruokailun vaikutus
Paastotilaan verrattuna idelalisibin aiemman kapselikoostumuksen antaminen rasvaisen aterian yhteydessä ei aiheuttanut muutosta Cmax-arvoon ja suurensi keskimääräistä AUCinf-arvoa 36 %. Idelalisibia voidaan antaa ruokailuajoista riippumatta.
Jakautuminen
Idelalisibi on sitoutunut 93-94-prosenttisesti ihmisen plasman proteiineihin kliinisesti havaituilla pitoisuuksilla. Keskimääräinen veren ja plasman pitoisuuksien suhdeluku oli noin 0,5. Idelalisibin ilmeinen jakautumistilavuus oli keskimäärin noin 96 l.
Biotransformaatio
Idelalisibi metaboloituu pääasiassa aldehydioksidaasin vaikutuksesta ja pienemmässä määrin CYP3A:n ja UGT1A4:n kautta. Pääasiallisella ja ainoalla verenkierrossa tavattavalla metaboliitilla, GS-563117:lla, ei ole vaikutusta PI3Kδ:aan.
Eliminaatio
Idelalisibin terminaalinen eliminaation puoliintumisaika oli 8,2 (vaihteluväli: 1,9; 37,2) tuntia ja idelalisibin ilmeinen puhdistuma oli 14,9 (vaihteluväli: 5,1; 63,8) l/h sen jälkeen, kun idelalisibia on annettu 150 mg kahdesti päivässä suun kautta. Yhden suun kautta otetun 150 mg:n [14C]-leimatun idelalisibiannoksen ottamisen jälkeen noin 78 % erittyi ulosteeseen ja 15 % virtsaan. 48 tunnin kuluessa virtsassa havaitusta kokonaisradioaktiivisuudesta 23 % ja ulosteessa 144 tunnin kuluessa havaitusta kokonaisradioaktiivisuudesta 12 % oli muuttumatonta idelalisibia.
In vitro yhteisvaikutustiedot
In vitro tiedot osoittavat, että idelalisibi ei inhiboi metaboloivia entsyymejä CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A tai UGT1A1 eikä kuljettajia OAT1, OAT3 tai OCT2.
GS-563117 ei inhiboi metaboloivia entsyymejä CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 tai UGT1A1 tai kuljettajia P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3 tai OCT2.
Erityisryhmät
Sukupuoli ja rotu
Väestön farmakokineettiset analyysit osoittivat, ettei sukupuolella ja rodulla ollut kliinisesti merkittävää vaikutusta idelalisibin tai GS-563117:n altistuksiin.
Iäkkäät potilaat
Väestön farmakokineettiset analyysit osoittivat, ettei iällä ollut kliinisesti merkittävää vaikutusta idelalisibin tai GS-563117:n altistuksiin, mukaan lukien iäkkäät potilaat (65-vuotiaat ja vanhemmat) verrattuna nuorempiin potilaisiin.
Heikentynyt munuaisten toiminta
Idelalisibin farmakokinetiikkaa ja turvallisuutta koskeva tutkimus tehtiin terveillä henkilöillä ja vaikeasti heikentynyttä munuaisten toimintaa sairastavilla koehenkilöillä (arvioitu CrCl 15-29 ml/min). Koehenkilöillä, joilla oli vaikeasti heikentynyt munuaisten toiminta, 150 mg:n kerta-annoksen jälkeen ei havaittu kliinisesti merkittäviä muutoksia altistuksissa idelalisibille tai GS-563117:lle verrattuna terveisiin henkilöihin.
Heikentynyt maksan toiminta
Idelalisibin farmakokinetiikkaa ja turvallisuutta koskeva tutkimus tehtiin terveillä henkilöillä ja kohtalaisesti (Child-Pugh-luokka B) tai vaikeasti (Child-Pugh-luokka C) heikentynyttä maksan toimintaa sairastavilla koehenkilöillä. 150 mg:n kerta-annoksen jälkeen idelalisibin AUC (yhteensä, ts. sitoutunut plus sitoutumaton) oli ~60 % korkeampi kohtalaisesti ja vaikeasti heikentynyttä maksan toimintaa sairastavilla koehenkilöillä verrattuna kaltaistettuihin verrokkeihin. Idelalisibin AUC (sitoutumaton) proteiineihin sitoutumiseen liittyvien erojen huomioinnin jälkeen oli ~80 % (1,8 kertaa) korkeampi kohtalaisesti heikentynyttä maksan toimintaa sairastavilla koehenkilöillä ja ~152 % (2,5 kertaa) korkeampi vaikeasti heikentynyttä maksan toimintaa sairastavilla koehenkilöillä verrattuna kaltaistettuihin verrokkeihin.
Pediatriset potilaat
Idelalisibin farmakokinetiikkaa pediatrisilla potilailla ei ole vahvistettu (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttama toksisuus
Idelalisibi aiheutti imukudoskatoa pernassa, kateenkorvassa, imusolmukkeissa ja suoliston imukudoksessa. Yleisesti ottaen B-lymfosyyteista riippuvilla alueilla vaikutus oli suurempi kuin T-lymfosyyteista riippuvilla alueilla. Rotilla idelalisibi voi estää T-riippuvaista vasta-ainemuodostusta. Idelalisibi ei kuitenkaan estänyt isännän normaalia reaktiota Staphylococcus aureus -bakteerille eikä voimistanut syklofosfamidin myelosuppressiivista vaikutusta. Idelalisibilla ei katsota olevan laaja-alaista immunosuppressiivista vaikutusta.
Idelalisibi aiheutti tulehduksellisia muutoksia sekä rotilla että koirilla. Korkeintaan 4 viikkoa kestävissä tutkimuksissa maksan nekroosia havaittiin rotilla 7-kertaisilla ja koirilla 5-kertaisilla altistuksilla verrattuna ihmisen altistukseen AUC:n perusteella. Seerumin transaminaasien nousut korreloivat maksan nekroosin kanssa koirilla, mutta niitä ei havaittu rotilla. Heikentynyttä maksan toimintaa tai kroonista transaminaasien nousua ei havaittu rotilla tai koirilla tutkimuksissa, jotka kestivät 13 viikkoa tai pidempään.
Genotoksisuus
Idelalisibi ei indusoinut mutaatioita mikrobeilla tehdyissä mutageenisuustestissä (Ames), ei ollut klastogeeninen kromosomipoikkeamatestissä in vitro, jossa käytettiin ihmisen perifeerisen veren lymfosyytteja, eikä ollut genotoksinen rotan mikrotumatestissä in vivo.
Karsinogeenisuus
Idelalisibin karsinogeenisuutta arvioitiin 26 viikkoa kestäneessä tutkimuksessa, jossa käytettiin RasH2-transgeenisiä hiiriä, sekä 2 vuotta kestäneessä tutkimuksessa, jossa käytettiin rottia. Idelalisibi ei ollut karsinogeeninen altistuksilla, jotka olivat uroshiirillä korkeintaan 1,4‑kertaisia ja naarashiirillä korkeintaan 7,9‑kertaisia verrattuna potilaisiin, joilla oli hematologisia maligniteetteja ja jotka saivat idelalisibia suositellun 150 mg:n annoksen kahdesti päivässä. Urosrottien idelalisibialtistuksilla, jotka olivat enintään 0,4‑kertaisia verrattuna suositellulla annoksella aikaansaatuun altistukseen ihmisillä, haiman saarekesolukasvainten havaittiin pienellä ilmaantuvuudella lisääntyneen annoksesta riippuvaisella tavalla. Samaa ilmiötä ei havaittu naarasrotilla, joilla altistus oli 0,62‑kertainen.
Lisääntymis- ja kehitystoksisuus
Alkion-/sikiönkehitystä arvioivassa tutkimuksessa rotilla havaittiin lisääntyneitä alkion kuolemia kiinnittymisen jälkeen, epämuodostumia (häntänikamien ja joissain tapauksissa myös ristinikamien puuttuminen), luuston muutoksia ja sikiöiden alentunutta ruumiinpainoa. Epämuodostumia havaittiin vähintään 12-kertaisilla altistuksilla verrattuna ihmisen altistukseen AUC:n perusteella. Vaikutuksia alkion-/sikiönkehitykseen ei tutkittu toisella lajilla.
Kivesten siementiehyiden rappeutumista havaittiin 2-13 viikon toistuvan annoksen tutkimuksissa koirilla ja rotilla, mutta ei 26 viikkoa tai pidempään kestävissä tutkimuksissa. Urosrottien hedelmällisyystutkimuksessa havaittiin lisäkivesten ja kivesten painojen pienenemistä mutta haittavaikutuksia parittelu- tai hedelmällisyysparametreihin, ja spermatogeneesin muuttumista tai estymistä ei havaittu. Lääkkeellä ei ollut vaikutusta naarasrottien hedelmällisyyteen.
Fototoksisuus
Fototoksisuuden potentiaalin arviointi hiiren alkion fibroblastisolulinjassa BALB/c 3T3 oli tulokseton idelalisibin osalta sytotoksisuuden vuoksi in vitro -analyysissä. Pääasiallinen metaboliitti GS-563117 saattaa lisätä fototoksisuutta, kun solut altistuvat samanaikaisesti UVA-valolle. On olemassa potentiaalinen riski, että idelalisibi saattaa aiheuttaa hoidetuilla potilailla valoherkkyyttä pääasiallisen metaboliittinsa GS-563117 kautta.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa
Hydroksipropyyliselluloosa (E463)
Kroskarmelloosinatrium
Natriumtärkkelysglykolaatti
Magnesiumstearaatti
Tabletin päällys
Poly(vinyylialkoholi) (E1203)
Makrogoli (E1521)
Titaanidioksidi (E171)
Talkki (E553B)
Paraoranssi (E110) (vain Zydelig 100 mg kalvopäällysteiset tabletit)
Punainen rautaoksidi (E172) (vain Zydelig 150 mg kalvopäällysteiset tabletit)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
5 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ZYDELIG tabletti, kalvopäällysteinen
100 mg (L:ei) 60 kpl (3802,84 €)
150 mg (L:ei) 60 kpl (3802,84 €)
PF-selosteen tieto
Suurtiheyspolyeteenistä (HDPE) valmistettu purkki, jossa on polypropeenista valmistettu lapsiturvallinen korkki ja joka sisältää 60 kalvopäällysteistä tablettia ja polyesteripehmusteen.
Yksi pakkaus sisältää 1 purkin.
Valmisteen kuvaus:
Zydelig 100 mg kalvopäällysteiset tabletit
Oranssi, soikea, kalvopäällysteinen tabletti, kooltaan 9,7 mm x 6,0 mm, yhdellä puolella merkintä ″GSI″ ja toisella puolella merkintä ″100″.
Zydelig 150 mg kalvopäällysteiset tabletit
Vaaleanpunainen, soikea, kalvopäällysteinen tabletti, kooltaan 10,0 mm x 6,8 mm, yhdellä puolella merkintä ″GSI″ ja toisella puolella merkintä ″150″.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ZYDELIG tabletti, kalvopäällysteinen
100 mg 60 kpl
150 mg 60 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Idelalisibi: Kroonisen lymfaattisen leukemian ja follikulaarisen lymfooman hoito aikuispotilailla erityisin edellytyksin (372).
ATC-koodi
L01EM01
Valmisteyhteenvedon muuttamispäivämäärä
20.06.2024
Yhteystiedot
Karhumäentie 3
01530 Vantaa
Suomi
09 42726918
www.gilead.se/utility/contact
nordics.medinfo@gilead.com