
XALKORI kapseli, kova 200 mg, 250 mg, rakeet, avattavat kapselit 20 mg, 50 mg, 150 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Yleinen
Vaikuttavat aineet ja niiden määrät
XALKORI 200 mg kovat kapselit
Yksi kova kapseli sisältää 200 mg kritsotinibia.
XALKORI 250 mg kovat kapselit
Yksi kova kapseli sisältää 250 mg kritsotinibia.
XALKORI 20 mg rakeet avattavissa kapseleissa
Yksi kapseli sisältää 20 mg kritsotinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi avattava kapseli sisältää 6 mg sakkaroosia.
XALKORI 50 mg rakeet avattavissa kapseleissa
Yksi kapseli sisältää 50 mg kritsotinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi avattava kapseli sisältää 14 mg sakkaroosia.
XALKORI 150 mg rakeet avattavissa kapseleissa
Yksi kapseli sisältää 150 mg kritsotinibia.
Apuaine, jonka vaikutus tunnetaan
Yksi avattava kapseli sisältää 43 mg sakkaroosia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, kova.
Rakeet avattavissa kapseleissa.
Kliiniset tiedot
Käyttöaiheet
XALKORI on tarkoitettu monoterapiana
- ensilinjan hoidoksi aikuisille, joilla on edennyt anaplastinen lymfoomakinaasi (ALK) ‑positiivinen ei-pienisoluinen keuhkosyöpä (non-small cell lung cancer, NSCLC)
- aikuisille aiemmin hoidetun edenneen ALK-positiivisen ei-pienisoluisen keuhkosyövän hoitoon
- aikuisille edenneen ROS1-positiivisen ei-pienisoluisen keuhkosyövän hoitoon
- pediatrisille (≥ 1 – < 18‑vuotiaille) potilaille uusiutuneen tai refraktorisen systeemisen ALK‑positiivisen anaplastisen suurisoluisen lymfooman (anaplastic large cell lymphoma, ALCL) hoitoon
- pediatrisille (≥ 1 – < 18‑vuotiaille) potilaille uusiutuneen tai refraktorisen ALK-positiivisen leikkaukseen soveltumattoman tulehduksellisen myofibroblastituumorin (inflammatory myofibroblastic tumour, IMT) hoitoon.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
XALKORI-hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
ALK- ja ROS1-määritys
Arvioitaessa potilaiden sopivuutta XALKORI-hoitoon kasvaimesta on tehtävä joko ALK- tai ROS1-määritys validoidulla menetelmällä (ks. kohdasta Farmakodynamiikka tietoa kliinisissä tutkimuksissa käytetyistä määritysmenetelmistä).
Ei-pienisoluisen keuhkosyövän ALK-positiivisuus, ei‑pienisoluisen keuhkosyövän ROS1-positiivisuus, anaplastisen suurisoluisen lymfooman ALK-positiivisuus tai tulehduksellisen myofibroblastituumorin ALK-positiivisuus tulee osoittaa ennen kritsotinibihoidon aloittamista. ALK-määritys on tehtävä laboratoriossa, joka on perehtynyt hyödynnettävän erityistekniikan käyttöön (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Aikuispotilaat, joilla on edennyt ALK‑positiivinen tai ROS1‑positiivinen ei-pienisoluinen keuhkosyöpä (NSCLC)
Kritsotinibin suositeltu annostus on 250 mg kaksi kertaa vuorokaudessa (500 mg vuorokaudessa) yhtäjaksoisesti.
Pediatriset potilaat, joilla on ALK‑positiivinen anaplastinen suurisoluinen lymfooma (ALCL) tai ALK‑positiivinen tulehduksellinen myofibroblastituumori (IMT)
Pediatrisille potilaille suositeltu kritsotinibin aloitusannos perustuu kehon pinta-alaan. Kritsotinibin suositeltu annostus ALCL:ää tai IMT:tä sairastaville pediatrisille potilaille on 280 mg/m2 suun kautta kaksi kertaa vuorokaudessa, kunnes sairaus etenee tai ilmenee toksisuutta, joka ei ole hyväksyttävissä.
Suositeltu annostus pediatrisille potilaille, joiden kehon pinta-ala on ≥ 1,34 m2, on esitetty taulukossa 1. Oikean annoksen saamiseksi voidaan tarvittaessa yhdistää erivahvuisia kritsotinibikapseleita.
Taulukko 1. Pediatriset potilaat, joiden kehon pinta-ala on ≥ 1,34 m2: kritsotinibikapseleiden*suositeltu aloitusannostus
| Kehon pinta-ala** | Annos (kaksi kertaa vuorokaudessa) | Kokonaisannos vuorokaudessa |
| 1,34–1,51 m2 | 400 mg (2 × 200 mg:n kapseli) | 800 mg |
| 1,52–1,69 m2 | 450 mg (1 × 200 mg:n kapseli + 1 × 250 mg:n kapseli) | 900 mg |
| ≥ 1,70 m2 | 500 mg (2 × 250 mg:n kapseli) | 1000 mg |
* Viittaa XALKORI 200 mg ja 250 mg koviin kapseleihin. ** Pediatriset potilaat, joiden kehon pinta-ala on < 1,34 m2, ks. taulukko 2. | ||
Pediatrisille potilaille, joiden kehon pinta-ala on < 1,34 m2, pitää käyttää XALKORI-valmistemuotoa rakeet avattavissa kapseleissa. Suositeltu annostus pediatrisille potilaille, joiden kehon pinta-ala on < 1,34 m2, on esitetty taulukossa 2.
Rakeet on kapseloitu kolmeksi vahvuudeksi: 20 mg, 50 mg ja 150 mg kritsotinibia. Oikean annoksen saamiseksi voidaan tarvittaessa yhdistää erivahvuisia kritsotinibirakeita avattavissa kapseleissa. Kerta-annokseen tarvitaan enintään 4 kapselia (ks. taulukko 2).
Taulukko 2. Pediatriset potilaat, joiden kehon pinta-ala on 0,38–1,33 m2: kritsotinibirakeiden*suositeltu aloitusannostus
| Kehon pinta-ala** | Annos (kaksi kertaa vuorokaudessa) | Kokonaisannos vuorokaudessa |
| 0,38–0,46 m2 | 120 mg (1 × 20 mg + 2 × 50 mg) | 240 mg |
| 0,47–0,51 m2 | 140 mg (2× 20 mg + 2 × 50 mg) | 280 mg |
| 0,52–0,61 m2 | 150 mg (1 × 150 mg) | 300 mg |
| 0,62–0,80 m2 | 200 mg (1 × 50 mg + 1 × 150 mg) | 400 mg |
| 0,81–0,97 m2 | 250 mg (2 × 50 mg + 1 × 150 mg) | 500 mg |
| 0,98–1,16 m2 | 300 mg (2 × 150 mg) | 600 mg |
| 1,17–1,33 m2 | 350 mg (1 × 50 mg + 2 × 150 mg) | 700 mg |
* Viittaa 20 mg:n, 50 mg:n ja 150 mg:n kritsotinibirakeisiin avattavissa kapseleissa. ** Suositeltavaa annostusta potilaille, joiden kehon pinta-ala on alle 0,38 m2, ei ole määritetty. Pediatriset potilaat, joiden kehon pinta-ala on ≥ 1,34 m2, ks. taulukko 1. | ||
Anna kritsotinibi pediatrisille potilaille aikuisen valvonnassa.
Annoksen muuttaminen
Yksilöllinen turvallisuus ja siedettävyys saattavat edellyttää annostelun keskeyttämistä ja/tai annoksen pienentämistä.
Aikuispotilaat, joilla on edennyt ALK-positiivinen tai ROS1-positiivinen NSCLC
Kliinisissä tutkimuksissa kritsotinibia saaneilla ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla aikuispotilailla (n = 1 722) yleisimpiä annostelun keskeyttämiseen liittyviä haittavaikutuksia (≥ 3 %) olivat neutropenia, transaminaasien nousu, oksentelu ja pahoinvointi. Yleisimpiä annoksen pienentämiseen liittyviä haittavaikutuksia (≥ 3 %) olivat transaminaasien nousu ja neutropenia. Jos annoksen pienentäminen on tarpeellista potilaalla, jota hoidetaan kritsotinibiannoksella 250 mg kaksi kertaa vuorokaudessa suun kautta, kritsotinibiannos tulee pienentää seuraavasti:
- Ensimmäinen annoslasku: XALKORI 200 mg kaksi kertaa vuorokaudessa suun kautta.
- Toinen annoslasku: XALKORI 250 mg kerran vuorokaudessa suun kautta.
- Hoito tulee lopettaa pysyvästi, jos potilas ei siedä XALKORI-annosta 250 mg kerran vuorokaudessa suun kautta.
Ohjeet annoksen pienentämiseen hematologisen ja muun kuin hematologisen toksisuuden vuoksi on esitetty taulukoissa 3 ja 4. Jos potilasta hoidetaan kritsotinibiannostuksella, joka on pienempi kuin 250 mg kaksi kertaa vuorokaudessa, noudata taulukoiden 3 ja 4 ohjeita annoksen pienentämisestä.
Taulukko 3. Aikuispotilaat: XALKORI-hoidon annosmuutos – hematologinen toksisuusa,b
| CTCAEc-vaikeusasteluokitus | XALKORI-hoito |
| Aste 3 | Keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 2, ja jatka hoitoa sitten samalla annostuksella |
| Aste 4 | Keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 2, ja jatka hoitoa sitten yhtä annostasoa pienemmällä annostuksellad,e |
a. Lymfopeniaa lukuun ottamatta (ellei siihen liity kliinisiä tapahtumia, esim. opportunisti-infektioita).
b. Potilaat, joille kehittyy neutropenia ja/tai leukopenia, ks. myös kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset.
c. Toksisuuden vaikeusasteluokitus: National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events.
d. Jos haittavaikutus uusiutuu, tulee lääkkeen annostelu keskeyttää, kunnes haittavaikutus lievittyy asteelle ≤ 2. Tämän jälkeen hoitoa jatketaan annostuksella 250 mg kerran vuorokaudessa. XALKORI-hoito on lopetettava pysyvästi, jos haittavaikutus vielä uusiutuu vaikeusasteella 4.
e. Jos potilasta hoidetaan annostuksella 250 mg kerran vuorokaudessa tai jos potilaan annos on pienennetty 250 mg:aan kerran vuorokaudessa, lopeta hoito arvioinnin ajaksi.
Taulukko 4. Aikuispotilaat: XALKORI-hoidon annosmuutos – muu kuin hematologinen
toksisuus
| CTCAEa-vaikeusasteluokitus | XALKORI-hoito |
| Asteen 3 tai 4 alaniiniaminotransferaasin (ALAT) tai aspartaattiaminotransferaasin (ASAT) nousu, johon liittyy asteen ≤ 1 bilirubiinipitoisuus | Keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 1 tai lähtötasolle, jatka hoitoa sitten annostuksella 250 mg kerran vuorokaudessa ja nosta annostus 200 mg:aan kaksi kertaa vuorokaudessa potilaan sietokyvyn mukaanb,c |
| Asteen 2, 3 tai 4 ALAT- tai ASAT-arvon nousu ja samanaikainen asteen 2, 3 tai 4 bilirubiinin nousu (kun potilaalla ei ole kolestaasia eikä hemolyysiä) | Lopeta hoito pysyvästi |
| Minkä tahansa asteen interstitiaalinen keuhkosairaus (ILD)/pneumoniitti | Keskeytä hoito, jos ILD:tä/pneumoniittia epäillään, ja lopeta hoito pysyvästi, jos hoitoon liittyvä ILD/pneumoniitti todetaand |
| Asteen 3 QTc-ajan piteneminen | Keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 1, määritä ja tarvittaessa korjaa elektrolyytit ja jatka hoitoa sitten yhtä annostasoa pienemmällä annostuksellab,c |
| Asteen 4 QTc-ajan piteneminen | Lopeta hoito pysyvästi |
Asteen 2 tai 3 bradykardiad,e Aiheuttaa oireita, voi olla vaikea ja lääketieteellisesti merkittävä, lääketieteellinen interventio aiheellinen | Keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 1 tai kunnes sydämen syke on ≥ 60 Selvitä samanaikaisesti käytössä olevat tunnetusti bradykardiaa aiheuttavat lääkevalmisteet, myös verenpainelääkkeet Jos bradykardiaan myötävaikuttava samanaikaisesti käytössä oleva lääkevalmiste tunnistetaan ja sen käyttö lopetetaan tai annostusta muutetaan, jatka XALKORI-hoitoa aikaisemmalla annostuksella sitten kun haittavaikutus on lievittynyt asteelle ≤ 1 tai kun sydämen syke on ≥ 60 Jos bradykardiaan myötävaikuttavaa samanaikaisesti käytössä olevaa lääkevalmistetta ei tunnisteta tai bradykardiaan myötävaikuttavien lääkevalmisteiden käyttöä ei lopeteta eikä annostusta muuteta, jatka XALKORI-hoitoa pienemmällä annostuksellac sitten kun haittavaikutus on lievittynyt asteelle ≤ 1 tai kun sydämen syke on ≥ 60 |
Asteen 4 bradykardiad,e,f Hengenvaarallisia seuraamuksia, kiireellinen interventio aiheellinen | Lopeta hoito pysyvästi, jos bradykardiaan myötävaikuttavaa samanaikaisesti käytössä olevaa lääkevalmistetta ei tunnisteta Jos bradykardiaan myötävaikuttava samanaikaisesti käytössä oleva lääkevalmiste tunnistetaan ja sen käyttö lopetetaan tai annostusta muutetaan, jatka XALKORI-hoitoa annostuksella 250 mg kerran vuorokaudessac sitten kun haittavaikutus on lievittynyt asteelle ≤ 1 tai kun sydämen syke on ≥ 60, ja seuraa potilasta tiheään |
| Asteen 4 silmiin liittyvä häiriö (näönmenetys) | Keskeytä hoito vakavan näönmenetyksen arvioinnin ajaksi |
a. Toksisuuden vaikeusasteluokitus: National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events
b. XALKORI-hoito on lopetettava pysyvästi, jos haittavaikutus vielä uusiutuu vaikeusasteella ≥ 3. Ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset.
c. Jos potilasta hoidetaan annostuksella 250 mg kerran vuorokaudessa tai jos potilaan annos on pienennetty 250 mg:aan kerran vuorokaudessa, lopeta hoito arvioinnin ajaksi.
d. Ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset.
e. Sydämen syke < 60 lyöntiä minuutissa.
f. Lopeta hoito pysyvästi, jos haittavaikutus vielä uusiutuu.
Pediatriset potilaat, joilla on ALK‑positiivinen ALCL tai ALK-positiivinen IMT
Jos suositellulla aloitusannoksella XALKORI-hoitoa saaneiden pediatristen potilaiden annosta on tarpeen pienentää, pediatristen potilaiden, joiden kehon pinta-ala on ≥ 1,34 m2, annoksen pienentäminen on tehtävä taulukon 5 mukaisesti.
Taulukko 5. Pediatriset potilaat, joiden kehon pinta-ala on ≥ 1,34 m2: XALKORI-kapseleiden*suositeltujen annosten pienentäminen
| Kehon pinta-ala** | Ensimmäinen annoslasku | Toinen annoslasku*** | ||
Annos (kaksi kertaa vuorokaudessa*) | Kokonaisannos vuorokaudessa | Annos (kaksi kertaa vuorokaudessa*) | Kokonaisannos vuorokaudessa | |
| 1,34–1,69 m2 | 250 mg | 500 mg | 200 mg | 400 mg |
| ≥ 1,70 m2 | 400 mg | 800 mg | 250 mg | 500 mg |
* Viittaa XALKORI 200 mg ja 250 mg koviin kapseleihin. ** Pediatriset potilaat, joiden kehon pinta-ala on < 1,34 m2, ks. taulukko 6. *** Hoito on lopetettava pysyvästi, jos potilas ei siedä kritsotinibia sen jälkeen, kun annosta on pienennetty kahdesti. | ||||
Jos suositellulla aloitusannoksella XALKORI-hoitoa saaneiden pediatristen potilaiden annosta on tarpeen pienentää, pediatristen potilaiden, joiden kehon pinta-ala on < 1,34 m2, annoksen pienentäminen on tehtävä taulukon 6 mukaisesti.
Taulukko 6. Pediatriset potilaat, joiden kehon pinta-ala on 0,38–1,33 m2: XALKORI-rakeiden*suositeltujen annosten pienentäminen
| Kehon pinta-ala** | Ensimmäinen annoslasku | Toinen annoslasku*** | ||
Annos (kaksi kertaa vuorokaudessa) | Kokonaisannos vuorokaudessa | Annos (kaksi kertaa vuorokaudessa) | Kokonaisannos vuorokaudessa | |
| 0,38–0,46 m2 | 90 mg (2 × 20 mg + 1 × 50 mg) | 180 mg | 70 mg (1 × 20 mg + 1 × 50 mg) | 140 mg |
| 0,47–0,51 m2 | 100 mg (2 × 50 mg) | 200 mg | 80 mg (4 × 20 mg) | 160 mg |
| 0,52–0,61 m2 | 120 mg (1 × 20 mg + 2 × 50 mg) | 240 mg | 90 mg (2 × 20 mg + 1 × 50 mg) | 180 mg |
| 0,62–0,80 m2 | 150 mg (1 × 150 mg) | 300 mg | 120 mg (1 × 20 mg + 2 × 50 mg) | 240 mg |
| 0,81–0,97 m2 | 200 mg (1 × 50 mg + 1 × 150 mg) | 400 mg | 150 mg (1 × 150 mg) | 300 mg |
| 0,98–1,16 m2 | 220 mg (1 × 20 mg + 1 × 50 mg + 1 × 150 mg) | 440 mg | 170 mg (1 × 20 mg + 1 × 150 mg) | 340 mg |
| 1,17–1,33 m2 | 250 mg (2 × 50 mg + 1 × 150 mg) | 500 mg | 200 mg (1 × 50 mg + 1 × 150 mg) | 400 mg |
* Viittaa 20 mg:n, 50 mg:n ja 150 mg:n kritsotinibirakeisiin avattavissa kapseleissa. ** Pediatriset potilaat, joiden kehon pinta-ala on ≥ 1,34 m2, ks. taulukko 5. *** Hoito on lopetettava pysyvästi, jos potilas ei siedä kritsotinibia sen jälkeen, kun annosta on pienennetty kahdesti. | ||||
Hematologisten ja muiden kuin hematologisten haittavaikutusten vuoksi suositellut annosmuutokset pediatrisille potilaille, joilla on ALK‑positiivinen ALCL tai ALK-positiivinen IMT, on esitetty taulukoissa 7 ja 8.
Taulukko 7. Pediatriset potilaat: XALKORI-hoidon annosmuutokset – hematologiset haittavaikutukset
| CTCAEa-vaikeusasteluokitus | XALKORI-hoito |
| Absoluuttinen neutrofiilien määrä (B‑Neut) | |
| Asteen 4 neutrofiilimäärän väheneminen | Ensimmäisellä ilmenemiskerralla: keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 2, ja jatka hoitoa sitten yhtä annostasoa pienemmällä annostuksella. Toisella ilmenemiskerralla:
|
| Verihiutaleiden määrä | |
| Asteen 3 verihiutalemäärän väheneminen (ja samanaikainen verenvuoto) | Keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 2, ja jatka hoitoa sitten samalla annostuksella. |
| Asteen 4 verihiutalemäärän väheneminen | Keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 2, ja jatka hoitoa sitten yhtä annostasoa pienemmällä annostuksella. Jos haittavaikutus uusiutuu, lopeta hoito pysyvästi. |
| Anemia | |
| Aste 3 | Keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 2, ja jatka hoitoa sitten samalla annostuksella. |
| Aste 4 | Keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 2, ja jatka hoitoa sitten yhtä annostasoa pienemmällä annostuksella. Jos haittavaikutus uusiutuu, lopeta hoito pysyvästi. |
a. Vaikeusasteluokitus perustuu Yhdysvaltain kansallisen syöpäinstituutin (National Cancer Institute, NCI) CTCAE (Common Terminology Criteria for Adverse Events) ‑luokituksen versioon 4.0. b. Hoito on lopetettava pysyvästi, jos potilas ei siedä XALKORI-hoitoa sen jälkeen, kun annosta on pienennetty kahdesti, ellei taulukoissa 5 ja 6 ole muuta ilmoitettu. | |
On suositeltavaa seurata täydellistä verenkuvaa, erittelylaskenta mukaan lukien, viikoittain ensimmäisen hoitokuukauden aikana ja sen jälkeen vähintään kuukausittain. Seurantaa pitää tehdä tiheämmin, jos havaitaan asteen 3 tai 4 poikkeavuuksia tai jos potilaalla ilmenee kuumetta tai infektio.
Taulukko 8. Pediatriset potilaat: XALKORI-hoidon annosmuutokset – muut kuin hematologiset haittavaikutukset
| CTCAEa-vaikeusasteluokitus | XALKORI-hoito |
| Asteen 3 tai 4 ALAT- tai ASAT-arvon nousu, johon liittyy asteen ≤ 1 bilirubiinipitoisuus | Keskeytä hoito, kunnes haittavaikutus lievittyy asteelle ≤ 1, ja jatka hoitoa sitten yhtä annostasoa pienemmällä annostuksella. |
| Asteen 2, 3 tai 4 ALAT- tai ASAT-arvon nousu ja samanaikainen asteen 2, 3 tai 4 bilirubiinipitoisuuden nousu (kun potilaalla ei ole kolestaasia eikä hemolyysiä) | Lopeta hoito pysyvästi. |
| Lääkkeeseen liittyvä minkä tahansa asteen interstitiaalinen keuhkosairaus (ILD) / pneumoniitti | Lopeta hoito pysyvästi. |
| Asteen 3 QTc-ajan piteneminen | Keskeytä hoito, kunnes haittavaikutus lievittyy lähtötasolle tai kunnes QTc-aika on alle 481 ms, ja jatka hoitoa sitten yhtä annostasoa pienemmällä annostuksella. |
| Asteen 4 QTc-ajan piteneminen | Lopeta hoito pysyvästi. |
Asteen 2 tai 3 bradykardiab Aiheuttaa oireita, voi olla vaikea ja lääketieteellisesti merkittävä, lääketieteellinen interventio aiheellinen | Keskeytä hoito, kunnes leposyke palaa potilaan ikää vastaavalle tasolle (perustuu iänmukaisten viitearvojen 2,5. persentiiliin):
|
Asteen 4 bradykardiab,c Hengenvaarallisia seuraamuksia, kiireellinen interventio aiheellinen | Jos bradykardiaan myötävaikuttavaa samanaikaisesti käytössä olevaa lääkevalmistetta ei tunnisteta, lopeta hoito pysyvästi. Jos bradykardiaan myötävaikuttava samanaikaisesti käytössä oleva lääkevalmiste tunnistetaan ja sen käyttö lopetetaan tai sen annosta muutetaan, jatka XALKORI-hoitoa taulukon 5c toisen annoslaskun mukaisesti, kunnes haittavaikutus on lievittynyt asteelle ≤ 1, tai kun sydämensyke on palautunut sydämen sykkeen hallintakriteerien mukaisesti tasolle, joka vastaa oireita aiheuttavaa tai vaikeaa, lääketieteellisesti merkittävää bradykardiaa, ja seuraa potilasta tiheään. |
Asteen 3 pahoinvointi Riittämätön lääkkeen saanti suun kautta yli 3 vuorokautta, vaatii lääketieteellistä interventiota | Aste 3 (huolimatta maksimaalisesta lääkehoidosta): keskeytä hoito, kunnes haittavaikutus korjaantuu, ja jatka hoitoa sitten yhtä annostasoa pienemmällä annostuksellad |
Asteen 3 tai 4 oksentelu Yli 6 oksentelukertaa 24 tunnin sisällä yli 3 vuorokautta, vaatii lääketieteellistä interventiota (letkuruokinta tai sairaalahoito); hengenvaarallisia seuraamuksia, kiireellinen interventio aiheellinen | Aste 3 tai 4 (huolimatta maksimaalisesta lääkehoidosta): keskeytä hoito, kunnes haittavaikutus korjaantuu, ja jatka hoitoa sitten yhtä annostasoa pienemmällä annostuksellad |
Asteen 3 tai 4 ripuli Ulostuskertojen lisääntyminen ≥ 7:llä vuorokaudessa lähtötilanteeseen nähden, pidätyskyvyttömyys, sairaalahoito aiheellista; hengenvaarallisia seuraamuksia, kiireellinen interventio aiheellinen | Aste 3 tai 4 (huolimatta maksimaalisesta lääkehoidosta): keskeytä hoito, kunnes haittavaikutus korjaantuu, ja jatka hoitoa sitten yhtä annostasoa pienemmällä annostuksellad |
| Asteen 1 (lieviä oireita) tai 2 (kohtalaisia oireita, jotka vaikuttavat kykyyn suoriutua ikätason mukaisista päivittäisistä toiminnoista) silmiin liittyvä häiriö | Aste 1 tai 2: Tarkkaile oireita ja ilmoita mistä tahansa oireesta silmälääkärille. Asteen 2 näköhäiriöiden tapauksessa harkitse annostuksen pienentämistä. |
| Asteen 3 tai 4 silmiin liittyvä häiriö (näönmenetys tai huomattava näkökyvyn heikkeneminen) | Aste 3 tai 4: Keskeytä hoito vaikean näönmenetyksen arvioinnin ajaksi. Jos arvioinnissa ei tunnisteta muuta syytä näönmenetykseen, lopeta hoito pysyvästi. |
a. Vaikeusasteluokitus perustuu Yhdysvaltain kansallisen syöpäinstituutin (National Cancer Institute, NCI) CTCAE (Common Terminology Criteria for Adverse Events) ‑luokituksen versioon 4.0. b. Leposyke on hitaampi kuin iänmukaisten viitearvojen 2,5. persentiilissä. c. Hoito on lopetettava pysyvästi, jos haittavaikutus uusiutuu. d. Hoito on lopetettava pysyvästi, jos potilas ei siedä kritsotinibia sen jälkeen, kun annosta on pienennetty kahdesti, ellei taulukoissa 5 ja 6 ole muuta ilmoitettu. | |
Maksan vajaatoiminta
Kritsotinibi metaboloituu pääosin maksassa. Kritsotinibihoitoa tulee käyttää varoen, jos potilaalla on maksan vajaatoiminta (ks. taulukot 4 ja 8 ja kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset ja Farmakokinetiikka).
Annosmuutokset aikuispotilaille, joilla on edennyt ALK-positiivinen tai ROS1-positiivinen NSCLC
Kritsotinibin aloitusannoksen muuttamista ei suositella potilaille, joilla on lievä maksan vajaatoiminta (joko ASAT > viitevälin yläraja (ULN) ja bilirubiini ≤ ULN tai mikä tahansa ASAT ja bilirubiini > ULN mutta ≤ 1,5 × ULN) National Cancer Institute (NCI) -luokituksen mukaan. Kritsotinibin suositeltu aloitusannos potilaille, joilla on keskivaikea maksan vajaatoiminta (mikä tahansa ASAT ja bilirubiini > 1,5 × ULN ja ≤ 3 × ULN), on 200 mg kaksi kertaa vuorokaudessa. Kritsotinibin suositeltu aloitusannos potilaille, joilla on vaikea maksan vajaatoiminta (mikä tahansa ASAT ja bilirubiini > 3 × ULN), on 250 mg kerran vuorokaudessa (ks. kohta Farmakokinetiikka). Kritsotinibin annoksen muuttamista ei ole tutkittu potilailla, joiden maksan vajaatoiminnan vaikeusaste on määritelty Child-Pugh-luokituksella.
Annosmuutokset pediatrisille potilaille, joilla on ALK-positiivinen ALCL tai ALK-positiivinen IMT
Annosmuutokset pediatrisille potilaille perustuvat aikuispotilailla tehtyyn kliiniseen tutkimukseen (ks. kohta Farmakokinetiikka). Kritsotinibin aloitusannoksen muuttamista ei suositella potilaille, joilla on lievä maksan vajaatoiminta (joko ASAT > ULN ja bilirubiini ≤ ULN tai mikä tahansa ASAT ja bilirubiini > ULN mutta ≤ 1,5 × ULN). Kritsotinibin suositeltu aloitusannos potilaille, joilla on keskivaikea maksan vajaatoiminta (mikä tahansa ASAT ja bilirubiini > 1,5 × ULN ja ≤ 3 × ULN), on kehon pinta-alaan perustuva ensimmäisen annoslaskun mukainen annos taulukoissa 5 ja 6 esitetyn mukaisesti. Kritsotinibin suositeltu aloitusannos potilaille, joilla on vaikea maksan vajaatoiminta (mikä tahansa ASAT ja bilirubiini > 3 × ULN), on kehon pinta-alaan perustuva toisen annoslaskun mukainen annos taulukoissa 5 ja 6 esitetyn mukaisesti.
Munuaisten vajaatoiminta
Annosmuutokset aikuispotilaille, joilla on edennyt ALK-positiivinen tai ROS1-positiivinen NSCLC
Aloitusannoksen muuttamista ei suositella potilaille, joilla on lievä (eGFR 60 – < 90 ml/min) tai kohtalainen (eGFR 30 – < 60 ml/min) munuaisten vajaatoiminta, koska populaatiofarmakokineettinen analyysi ei viitannut kliinisesti merkittäviin muutoksiin vakaan tilan kritsotinibialtistuksessa näillä potilailla. Kritsotinibin pitoisuus plasmassa voi suurentua vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min) sairastavilla potilailla. Kritsotinibin aloitusannos tulee muuttaa 250 mg:aan kerran vuorokaudessa suun kautta vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla, jotka eivät tarvitse peritoneaalidialyysiä tai hemodialyysiä. Kun hoitoa on jatkettu vähintään 4 viikkoa, annosta voidaan nostaa yksilöllisen turvallisuuden ja siedettävyyden perusteella 200 mg:aan kaksi kertaa vuorokaudessa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Annosmuutokset pediatrisille potilaille, joilla on ALK‑positiivinen ALCL tai ALK‑positiivinen IMT
Annosmuutokset pediatrisille potilaille perustuvat aikuispotilaista saatuihin tietoihin (ks. kohta Farmakokinetiikka). Aloitusannosta ei tarvitse muuttaa potilaille, joilla on Schwartzin kaavan mukaan laskettuna lievä (eGFR 60 – < 90 ml/min) tai kohtalainen (eGFR 30 – < 60 ml/min) munuaisten vajaatoiminta. Kritsotinibin suositeltu aloitusannos potilaille, joilla on vaikea munuaisten vajaatoiminta (eGFR < 30 ml/min) ja jotka eivät tarvitse dialyysia, on kehon pinta-alaan perustuva toisen annoslaskun mukainen annos taulukoissa 5 ja 6 esitetyn mukaisesti. Kun hoitoa on jatkettu vähintään 4 viikkoa, annosta voidaan suurentaa yksilöllisen turvallisuuden ja siedettävyyden perusteella kehon pinta-alaan perustuvaan ensimmäiseen annoslaskun mukaiseen annokseen taulukoissa 5 ja 6 esitetyn mukaisesti.
Iäkkäät
Aloitusannoksen muuttaminen ei ole tarpeen (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Pediatriset potilaat
Kritsotinibin turvallisuutta ja tehoa ALK-positiivista tai ROS1-positiivista ei‑pienisoluista keuhkosyöpää sairastavien pediatristen potilaiden hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Kritsotinibin turvallisuus ja teho on varmistettu 3 – < 18‑vuotiaiden pediatristen potilaiden hoidossa, joilla on uusiutunut tai refraktorinen systeeminen ALK-positiivinen anaplastinen suurisoluinen lymfooma (ALCL), tai 2 – < 18‑vuotiaiden pediatristen potilaiden hoidossa, joilla on leikkaukseen soveltumaton, uusiutunut tai refraktorinen ALK-positiivinen tulehduksellinen myofibroblastituumori (IMT) (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). Kritsotinibihoidon turvallisuutta tai tehoa koskevia tietoja ei ole saatavilla alle 3‑vuotiaista pediatrisista potilaista, joilla on ALK‑positiivinen ALCL, tai alle 2‑vuotiaista pediatrisista potilaista, joilla on ALK-positiivinen IMT.
Antotapa
Suun kautta.
XALKORI-valmiste voidaan ottaa joko ruokailun jälkeen tai paastotilassa. Xalkori-rakeita ei saa sirotella ruokaan. Greippihedelmiä ja greippimehua tulee välttää, koska ne saattavat suurentaa kritsotinibin pitoisuutta plasmassa. Mäkikuismaa tulee välttää, koska se voi pienentää kritsotinibin pitoisuutta plasmassa (ks. kohta Yhteisvaikutukset).
Jos annos jää ottamatta, se tulisi ottaa heti, kun potilas tai hoidosta vastaava henkilö huomaa annoksen unohtuneen. Potilaan ei kuitenkaan pidä ottaa unohtunutta annosta, jos seuraavan hoito-ohjelman mukaisen annoksen ottamisajankohtaan on alle 6 tuntia. Kahta annosta ei pidä ottaa samanaikaisesti yhden unohtuneen annoksen korvaamiseksi.
XALKORI 200 mg ja 250 mg kovat kapselit
XALKORI 200 mg ja 250 mg kovat kapselit pitää niellä kokonaisina mieluiten veden kanssa. Niitä ei saa murskata, liuottaa eikä avata.
XALKORI- rakeet avattavissa kapseleissa
Avattavissa kapseleissa olevia rakeita ei pidä pureskella, murskata eikä sirotella ruokaan. Kapselikuorta ei saa niellä, vaan se on avattava varovasti seuraavalla tavalla:
- Kapselia pidellään siten, että painatus ”Pfizer” on ylhäällä. Kapselia naputellaan, jotta kaikki rakeet ovat varmasti kapselin alaosassa.
- Kapselin alaosaa puristetaan varovasti.
- Kapselin ylä- ja alaosaa kierretään vastakkaisiin suuntiin ja kapseli vedetään auki.
- Kapseli(e)n avaamisen jälkeen rakeet voidaan antaa kahdella vaihtoehtoisella tavalla:
- Sisältö tyhjennetään suoraan potilaan suuhun TAI
- Sisältö tyhjennetään potilaan omaan kuivaan antovälineeseen (esim. lusikkaan, lääkeannostelukuppiin). Sen jälkeen rakeet annetaan antovälineen avulla potilaan suuhun.
- Kummassakin vaihtoehdossa kapselia pitää naputella, jotta varmasti kaikki rakeet annetaan.
Jos koko määrättyä annosta avattavissa kapseleissa olevia rakeita ei voida ottaa kerralla, avattavissa kapseleissa olevat rakeet pitää antaa osissa, kunnes koko määrätty annos on annettu. Välittömästi kunkin osan antamisen jälkeen pitää antaa riittävä määrä vettä, jotta kaikki lääke varmasti niellään. Lääkkeen nielemisen jälkeen voidaan juoda muita nesteitä tai syödä muita ruokia (lukuun ottamatta niitä, jotka mainitaan kohdassa Yhteisvaikutukset, Lääkeaineet, jotka voivat suurentaa kritsotinibin pitoisuutta plasmassa).
Pakkausselosteessa on yksityiskohtaiset piktogrammit siitä, miten avattavissa kapseleissa olevat rakeet annetaan.
Pediatriset potilaat, joilla on ALK‑positiivinen ALCL tai ALK-positiivinen IMT
Pahoinvoinnin ja oksentelun ehkäisemiseksi pahoinvointilääkkeitä suositellaan pediatrisille potilaille, joilla on ALK‑positiivinen ALCL tai ALK positiivinen IMT, ennen kritsotinibihoidon aloittamista ja hoidon aikana. Ruoansulatuskanavaan kohdistuvan toksisuuden hallintaan suositellaan tavanomaisia pahoinvointi- ja ripulilääkkeitä. Tukihoitoa, kuten laskimoon annettavaa tai suun kautta otettavaa nesteytystä sekä elektrolyytti- ja ravintolisiä, suositellaan käyttämään kliinisen tarpeen mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vasta-aiheet
Yliherkkyys kritsotinibille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
ALK- ja ROS1-statuksen määritys
On tärkeää määrittää kasvaimen ALK- tai ROS1-status hyvin validoidulla ja luotettavalla menetelmällä väärien negatiivisten tai väärien positiivisten tulosten välttämiseksi.
Maksatoksisuus
Kliinisissä tutkimuksissa kritsotinibia saaneilla potilailla on raportoitu lääkkeen aiheuttamaa maksatoksisuutta (mukaan lukien aikuispotilaiden kuolemaan johtaneita tapauksia) (ks. kohta Haittavaikutukset). Maksan toimintakokeet, mukaan lukien ALAT, ASAT ja bilirubiini, tulee tehdä kerran viikossa kahden ensimmäisen hoitokuukauden aikana, sen jälkeen kuukausittain ja kliinisen tarpeen mukaan. Määritykset tulee toistaa tiheämmin, jos maksan toimintakokeiden arvot kohoavat vaikeusasteelle 2, 3 tai 4. Potilaan transaminaasien nousun huomioiminen annostelussa, ks. kohta Annostus ja antotapa.
Interstitiaalinen keuhkosairaus / pneumoniitti
Kritsotinibihoitoa saavilla potilailla voi esiintyä vaikea-asteista, hengenvaarallista tai kuolemaan johtavaa interstitiaalista keuhkosairautta (ILD)/pneumoniittia. Potilaita, joilla on ILD:hen/pneumoniittiin viittaavia keuhko-oireita, tulee seurata. Kritsotinibihoito tulee keskeyttää, jos ILD:tä/pneumoniittia epäillään. Erotusdiagnostiikassa on otettava huomioon lääkkeen aiheuttaman ILD:n/pneumoniitin mahdollisuus, jos potilaalla on ILD:n kaltainen tila kuten pneumoniitti, sädepneumoniitti, allerginen alveoliitti, interstitiaalinen pneumoniitti, keuhkofibroosi, aikuisen hengitysvaikeusoireyhtymä (ARDS), alveoliitti, keuhkoinfiltraatio, keuhkokuume, keuhkoödeema, keuhkoahtaumatauti, pleuraeffuusio, aspiraatiokeuhkokuume, bronkiitti, tukkeava bronkioliitti tai keuhkoputkien laajentuma. ILD:n/pneumoniitin muut mahdolliset syyt tulee sulkea pois, ja kritsotinibihoito tulee lopettaa pysyvästi, jos potilaalla todetaan hoitoon liittyvä ILD/pneumoniitti (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
QT-ajan piteneminen
Kliinisissä tutkimuksissa kritsotinibihoitoa saaneilla potilailla on havaittu QTc-ajan pitenemistä (ks. kohdat Haittavaikutukset ja Farmakokinetiikka), mikä voi lisätä kammioperäisten takyarytmioiden (esim. kääntyvien kärkien takykardia, torsade de pointes) tai äkkikuoleman riskiä. Ennen hoidon aloittamista siitä saatava hyöty ja mahdolliset riskit on arvioitava potilailla, joilla on bradykardia tai joilla on aiemmin esiintynyt QTc-ajan pitenemistä tai alttius QTc-ajan pitenemiseen, samoin potilailla, jotka käyttävät samanaikaisesti rytmihäiriölääkkeitä tai tunnetusti QTc-aikaa pidentäviä lääkevalmisteita ja potilailla, joilla on sydänsairaus ja/tai elektrolyyttihäiriöitä. Varovaisuutta tulee noudattaa annettaessa kritsotinibia näille potilaille ja sydänsähkökäyrä (EKG) on rekisteröitävä, elektrolyytit määritettävä ja munuaisten toiminta tutkittava säännöllisin väliajoin. Käytettäessä kritsotinibia EKG tulee rekisteröidä ja elektrolyytit (esim. kalsium, magnesium, kalium) määrittää juuri ennen hoidon aloittamista. Säännöllisin väliajoin toistuvia EKG- ja elektrolyyttitutkimuksia suositellaan erityisesti, jos hoidon alussa esiintyy oksentelua, ripulia, elimistön nestevajausta tai heikentynyttä munuaisten toimintaa. Elektrolyytit tulee korjata tarvittaessa. Jos QTc-aika pitenee ≥ 60 millisekuntia lähtötasosta, mutta on < 500 millisekuntia, kritsotinibihoito tulee keskeyttää ja kardiologia tulee konsultoida. Jos QTc-aika pitenee ≥ 500 millisekuntiin, kardiologia tulee konsultoida välittömästi. Potilaan QTc-ajan piteneminen, ks. kohdat Annostus ja antotapa, Haittavaikutukset ja Farmakokinetiikka.
Bradykardia
Bradykardiaa (syy-yhteydestä riippumatta) raportoitiin kliinisissä tutkimuksissa 13 %:lla kritsotinibilla hoidetuista aikuispotilaista, joilla oli ALK-positiivinen tai ROS1-positiivinen NSCLC, ja 17 %:lla kritsotinibilla hoidetuista pediatrisista potilaista, joilla oli ALK-positiivinen ALCL tai ALK-positiivinen IMT. Kritsotinibia saavilla potilailla voi esiintyä oireista bradykardiaa (esim. pyörtymistä, heitehuimausta ja hypotensiota). Kritsotinibin maksimaalinen vaikutus sykkeen hidastumiseen voi ilmetä vasta useita viikkoja hoidon aloittamisen jälkeen. Kritsotinibin käyttöä yhdessä muiden bradykardiaa aiheuttavien lääkeaineiden (esim. beetasalpaajien, dihydropyridiineihin kuulumattomien kalsiumkanavan salpaajien, kuten verapamiilin ja diltiatseemin, klonidiinin ja digoksiinin) kanssa tulee mahdollisuuksien mukaan välttää, koska samanaikainen käyttö lisää oireisen bradykardian riskiä. Sydämen sykettä ja verenpainetta tulee seurata säännöllisesti. Annostusta ei tarvitse muuttaa, jos bradykardia on oireetonta. Potilaat, joille kehittyy oireinen bradykardia, ks. kohdat ”Annoksen muuttaminen” ja ”Haittavaikutukset” (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Sydämen vajaatoiminta
Kritsotinibin kliinisissä tutkimuksissa ja markkinoille tulon jälkeisessä seurannassa aikuispotilailla on raportoitu haittavaikutuksina vaikeaa, hengenvaarallista tai kuolemaan johtanutta sydämen vajaatoimintaa (ks. kohta Haittavaikutukset).
Kritsotinibia saavia potilaita on seurattava sydämen vajaatoiminnan merkkien ja oireiden varalta (hengenahdistus, turvotus, nesteretentiosta johtuva nopea painonnousu). Seurantaohje koskee sekä potilaita, joilla on aiemmin ollut sydämen toiminnan häiriöitä, että potilaita, joilla sydämen toiminnan häiriöitä ei ole ollut. Annostelun keskeyttämistä, annoksen pienentämistä tai hoidon lopettamista on harkittava tarpeen mukaan, jos edellä mainittuja oireita havaitaan.
Neutropenia ja leukopenia
Kritsotinibin kliinisissä tutkimuksissa ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla aikuispotilailla vaikeusasteen 3 tai 4 neutropeniaa on raportoitu hyvin yleisesti (12 %). Kritsotinibin kliinisissä tutkimuksissa ALK-positiivista ALCL:ää tai ALK-positiivista IMT:tä sairastavilla pediatrisilla potilailla vaikeusasteen 3 tai 4 neutropeniaa on raportoitu hyvin yleisesti (68 %). ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla potilailla vaikeusasteen 3 tai 4 leukopeniaa on raportoitu yleisesti (3 %) ja ALK-positiivista ALCL:ää tai ALK-positiivista IMT:tä sairastavilla pediatrisilla potilailla hyvin yleisesti (24 %) (ks. kohta Haittavaikutukset). Kritsotinibin kliinisissä tutkimuksissa ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla aikuispotilailla alle 0,5 %:lla potilaista esiintyi kuumeista neutropeniaa. ALK-positiivista ALCL:ää tai ALK-positiivista IMT:tä sairastavilla pediatrisilla potilailla kuumeista neutropeniaa on raportoitu yleisesti (yksi potilas, 2,4 %). Täydellistä verenkuvaa, veren valkosolujen erittelylaskenta mukaan lukien, tulee seurata kliinisen tarpeen mukaan. Määrityksiä tulee tehdä tiheämmin, jos havaitaan vaikeusasteen 3 tai 4 poikkeavuuksia tai jos potilaalla esiintyy kuumetta tai infektio (ks. kohta Annostus ja antotapa).
Maha-suolikanavan perforaatio
Kritsotinibin kliinisissä tutkimuksissa raportoitiin maha-suolikanavan perforaatiotapahtumia. Kritsotinibin markkinoille tulon jälkeen on raportoitu kuolemaan johtaneita maha-suolikanavan perforaatiotapahtumia (ks. kohta Haittavaikutukset).
Kritsotinibia tulee käyttää varoen, jos potilaalla on suurentunut riski saada maha-suolikanavan perforaatio (esim. aiempi divertikuliitti, maha-suolikanavan metastaaseja tai jos samanaikaisesti käytetään lääkevalmisteita, joihin tunnetusti liittyy maha-suolikanavan perforaation riski).
Kritsotinibihoito tulee lopettaa, jos potilaalle kehittyy maha-suolikanavan perforaatio. Potilaille tulee kertoa ensimmäisistä maha-suolikanavan perforaatioon viittaavista oireista ja heitä tulee neuvoa hakeutumaan nopeasti lääkäriin, jos näitä oireita esiintyy.
Vaikutukset munuaisten toimintaan
Kritsotinibin kliinisissä tutkimuksissa potilailla havaittiin kohonnutta veren kreatiniinipitoisuutta ja kreatiniinipuhdistuman laskua. Kliinisissä tutkimuksissa ja markkinoille tulon jälkeisessä seurannassa kritsotinibilla hoidetuilla potilailla on raportoitu munuaisten vajaatoimintaa ja akuuttia munuaisten vajaatoimintaa. Myös kuolemaan johtaneita tapauksia, hemodialyysia vaatineita tapauksia sekä vaikeusasteen 4 hyperkalemiaa on todettu aikuispotilailla. Potilaiden munuaisten toiminnan tutkimista ennen kritsotinibihoidon aloittamista ja seurantaa hoidon aikana suositellaan. Erityistä huomiota tulee kiinnittää potilaisiin, joilla on riskitekijöitä tai joilla on aikaisemmin todettu munuaisten toiminnan huononemista (ks. kohta Haittavaikutukset).
Munuaisten vajaatoiminta
Kritsotinibin annostusta tulee muuttaa vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla, jotka eivät tarvitse peritoneaalidialyysiä tai hemodialyysiä (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Vaikutukset näkökykyyn
Kritsotinibin kliinisissä tutkimuksissa ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla aikuispotilailla (n = 1 722) vaikeusasteen 4 näkökenttäpuutosta, johon liittyi näönmenetys, on raportoitu 4 potilaalla (0,2 %). Mahdollisina syinä näönmenetykselle on raportoitu näköhermon surkastumaa ja toimintahäiriötä.
Kritsotinibin kliinisissä tutkimuksissa ALK-positiivista ALCL:ää tai ALK-positiivista IMT:tä sairastavilla pediatrisilla potilailla näköhäiriöitä esiintyi 25 potilaalla 41:stä (61 %) (ks. kohta Haittavaikutukset).
ALCL:ää tai IMT:tä sairastaville pediatrisille potilaille tulee tehdä lähtötilanteen silmätutkimus ennen kritsotinibihoidon aloittamista. Seurantasilmätutkimusta, verkkokalvon tutkimus mukaan lukien, suositellaan 1 kuukauden kuluessa kritsotinibihoidon aloittamisesta ja sen jälkeen 3 kuukauden välein sekä aina uusien näkökykyyn liittyvien oireiden ilmetessä. Terveydenhuollon ammattilaisten tulee kertoa potilaille ja huoltajille silmätoksisuuden oireista ja mahdollisesta näönmenetyksen riskistä. Vaikeusasteen 2 näköhäiriöiden oireita tulee seurata ja niistä pitää ilmoittaa silmälääkärille, ja samalla on harkittava annoksen pienentämistä. Kritsotinibihoito tulee keskeyttää vaikeusasteen 3 tai 4 silmiin liittyvän häiriön arvioinnin ajaksi, ja jos ilmenee vaikeusasteen 3 tai 4 vakava näönmenetys, kritsotinibihoito tulee lopettaa pysyvästi, ellei muuta syytä näönmenetykseen tunnisteta (ks. kohta Annostus ja antotapa, taulukko 8).
Jos potilaalle ilmaantuu uutena oireena vakava näönmenetys (paras korjattu näöntarkkuus alle 6/60 toisessa silmässä tai molemmissa silmissä), kritsotinibihoito tulisi keskeyttää (ks. kohta Annostus ja antotapa). Potilaalle tulisi tehdä silmätutkimuksia, joihin sisältyvät paras korjattu näöntarkkuus, verkkokalvon kuvaus, näkökenttätutkimus, optinen koherenssitomografia (OCT) ja muut uuden näönmenetyksen ja muiden näkökykyyn liittyvien oireiden tutkimukseen tarvittavat arvioinnit kliinisen tarpeen mukaan (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Saatavilla ei ole riittävästi tietoa, jotta kritsotinibihoidon uudelleen aloittamiseen liittyvä riski voitaisiin arvioida potilaalla, jolla ilmenee näkökykyyn liittyviä oireita tai näönmenetys. Päätöksessä kritsotinibihoidon uudelleen aloittamisesta tulisi ottaa huomioon potilaan hoidosta mahdollisesti saama hyöty suhteessa potilaalle aiheutuviin riskeihin.
Silmien tutkimista suositellaan, jos potilaan näköhäiriöt pitkittyvät tai niiden vaikeusaste pahenee (ks. kohta Haittavaikutukset).
Valoherkkyys
Xalkori-hoitoa saaneilla potilailla on raportoitu valoherkkyyttä (ks. kohta Haittavaikutukset). Potilaita tulee neuvoa välttämään pitkäkestoista altistumista auringolle Xalkori-valmisteen käytön aikana ja suojautumaan ulkona auringonvalolta (esim. suojaava vaatetus ja/tai aurinkovoide).
Lääkkeiden yhteisvaikutukset
Kritsotinibin käyttöä samanaikaisesti voimakkaiden CYP3A4:n estäjien tai voimakkaiden ja kohtalaisten CYP3A4:n induktoreiden kanssa on vältettävä (ks. kohta Yhteisvaikutukset).
Kritsotinibin samanakaista käyttöä kapean terapeuttisen indeksin omaavien CYP3A4:n substraattien kanssa on vältettävä (ks. kohta Yhteisvaikutukset). Kritsotinibin käyttöä muiden bradykardiaa aiheuttavien lääkeaineiden, tunnetusti QT-aikaa pidentävien lääkevalmisteiden ja/tai rytmihäiriölääkkeiden kanssa on vältettävä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ”QT‑ajan piteneminen”, ”Bradykardia” ja kohta Yhteisvaikutukset).
Ruoan vaikutus
Greippihedelmiä ja greippimehua tulee välttää kritsotinibihoidon aikana (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Histologia muu kuin adenokarsinooma (NSCLC)
ALK-positiivista ja ROS1-positiivista NSCLC:ää sairastavista potilaista, joiden kasvain on histologialtaan muu kuin adenokarsinooma, mukaan lukien levyepiteelikarsinooma, on vähän tietoja saatavilla (ks. kohta Farmakodynamiikka).
XALKORI 200 mg ja 250 mg kovat kapselit
Natriumin saanti
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 200 mg:n tai 250 mg:n kapseli eli sen voidaan sanoa olevan ”natriumiton”.
XALKORI- rakeet avattavissa kapseleissa
Sakkaroosin saanti
Potilaiden, joilla on harvinainen perinnöllinen fruktoosi-intoleranssi, glukoosi-galaktoosi-imeytymishäiriö tai sakkaroosi-isomaltaasin puutos, ei pidä käyttää tätä lääkettä.
Pediatriset potilaat
Ruoansulatuskanavaan kohdistuva toksisuus
Kritsotinibi voi aiheuttaa vaikeaa ruoansulatuskanavaan kohdistuvaa toksisuutta ALK-positiivista ALCL:ää tai ALK-positiivista IMT:tä sairastaville pediatrisille potilaille. Oksentelua esiintyi 95 %:lla ja ripulia 85 %:lla pediatrisista potilaista, joilla oli ALK-positiivinen ALCL tai ALK-positiivinen IMT.
Pahoinvoinnin ja oksentelun ehkäisemiseksi suositellaan käyttämään pahoinvointilääkkeitä ennen kritsotinibihoidon aloittamista ja sen aikana. Ruoansulatuskanavaan kohdistuvan toksisuuden hallintaan suositellaan tavanomaisia pahoinvointi- ja ripulilääkkeitä. Jos pediatriselle potilaalle kehittyy vaikeusasteen 3 pahoinvointia, joka kestää 3 vuorokautta, tai vaikeusasteen 3 tai 4 ripulia tai oksentelua huolimatta maksimaalisesta lääkehoidosta, on suositeltavaa keskeyttää kritsotinibihoito, kunnes haittavaikutus korjaantuu, ja jatkaa kritsotinibihoitoa sitten yhtä annostasoa pienemmällä annostuksella. Tukihoitoa, kuten nesteytystä sekä elektrolyytti- ja ravintolisiä, suositellaan käyttämään kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa).
Yhteisvaikutukset
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Farmakokineettiset yhteisvaikutukset
Lääkeaineet, jotka voivat suurentaa kritsotinibin pitoisuutta plasmassa
Käytettäessä kritsotinibia samanaikaisesti voimakkaiden CYP3A:n estäjien kanssa on odotettavissa, että kritsotinibin pitoisuus plasmassa suurenee. Kritsotinibin 150 mg:n kerta-annoksen antaminen suun kautta samanaikaisesti voimakkaan CYP3A:n estäjän ketokonatsolin (200 mg kaksi kertaa vuorokaudessa) kanssa suurensi kritsotinibin systeemistä altistusta, jolloin kritsotinibin plasmapitoisuus-aikakäyrän alla oleva pinta-ala nollahetkestä äärettömyyteen (AUCinf) oli noin 3,2-kertainen ja suurin havaittu pitoisuus plasmassa (Cmax) noin 1,4-kertainen verrattuna kritsotinibin antoon yksinään.
Kritsotinibin toistuvien annosten (250 mg kerran vuorokaudessa) samanaikainen anto voimakkaan CYP3A:n estäjän itrakonatsolin toistuvien annosten (200 mg kerran vuorokaudessa) kanssa suurensi kritsotinibin vakaan tilan AUCtau-arvon noin 1,6‑kertaiseksi ja Cmax-arvon noin 1,3‑kertaiseksi verrattuna kritsotinibin antoon yksinään.
Näin ollen voimakkaiden CYP3A:n estäjien (muun muassa atatsanaviirin, ritonaviirin, kobisistaatin, itrakonatsolin, ketokonatsolin, posakonatsolin, vorikonatsolin, klaritromysiinin, telitromysiinin ja erytromysiinin) samanaikaista käyttöä on vältettävä. Niitä voidaan kuitenkin käyttää, jos mahdollinen hyöty potilaalle arvioidaan riskiä suuremmaksi; potilasta on tällöin tarkkailtava tiiviisti kritsotinibin haittavaikutusten varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Fysiologiaan perustuvan farmakokineettisen (PBPK) mallinnuksen avulla on ennustettu, että kritsotinibin vakaan tilan AUC-arvo suurenee 17 % kohtalaisella CYP3A:n estäjällä diltiatseemilla tai verapamiililla tapahtuneen hoidon jälkeen. Siksi on suositeltavaa noudattaa varovaisuutta, kun kritsotinibia annetaan samanaikaisesti kohtalaisten CYP3A:n estäjien kanssa.
Greippihedelmä tai greippimehu voi myös suurentaa kritsotinibin pitoisuutta plasmassa, joten niitä on vältettävä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Lääkeaineet, jotka voivat pienentää kritsotinibin pitoisuutta plasmassa
Kritsotinibin toistuvien annosten (250 mg kaksi kertaa vuorokaudessa) samanaikainen anto voimakkaan CYP3A4:n induktorin rifampisiinin toistuvien annosten (600 mg kerran vuorokaudessa) kanssa pienensi kritsotinibin vakaan tilan AUCtau-arvoa 84 % ja Cmax-pitoisuutta 79 % verrattuna kritsotinibin antoon yksinään. Samanaikaista käyttöä voimakkaiden CYP3A:n induktoreiden, kuten mm. karbamatsepiinin, fenobarbitaalin, fenytoiinin, rifampisiinin ja mäkikuisman kanssa, on vältettävä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kohtalaisten induktorien, kuten mm. efavirentsin tai rifabutiinin, vaikutusta ei ole täysin selvitetty ja siksi niiden yhdistämistä kritsotinibiin on myös vältettävä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Samanaikainen anto mahanesteen pH:ta nostavien lääkevalmisteiden kanssa
Kritsotinibin vesiliukoisuus riippuu pH:sta: matala (hapan) pH parantaa kritsotinibin liukoisuutta.
XALKORI 200 mg ja 250 mg kovat kapselit
Kun kritsotinibia annettiin 250 mg:n kerta-annos kapseleina viiden vuorokauden esomepratsolihoidon (40 mg kerran vuorokaudessa) jälkeen, kritsotinibin kokonaisaltistus (AUCinf) pieneni noin 10 %, mutta huippupitoisuus (Cmax) ei muuttunut. Muutoksen suuruutta kokonaisaltistuksen osalta ei katsottu kliinisesti merkittäväksi.
XALKORI- rakeet avattavissa kapseleissa
Kun kritsotinibia annettiin suun kautta 250 mg:n kerta-annos rakeina avattavissa kapseleissa viiden vuorokauden esomepratsolihoidon (40 mg kerran vuorokaudessa) jälkeen, kritsotinibin kokonaisaltistus (AUCinf) pieneni noin 19 % ja huippupitoisuus (Cmax) pieneni 23 %. Muutoksen suuruutta kokonaisaltistuksen osalta ei katsottu kliinisesti merkittäväksi.
Aloitusannosta ei tarvitse muuttaa, kun kritsotinibia annetaan samanaikaisesti mahanesteen pH:ta nostavien lääkeaineiden (kuten protonipumpun estäjien, H2-salpaajien tai antasidien) kanssa.
Lääkeaineet, joiden pitoisuutta plasmassa kritsotinibi voi muuttaa
Kun kritsotinibia annettiin syöpäpotilaille annostuksella 250 mg kaksi kertaa vuorokaudessa 28 päivän ajan, suun kautta otettavan midatsolaamin AUCinf oli 3,7-kertainen verrattuna midatsolaamin käyttöön yksinään, mikä viittaa siihen, että kritsotinibi on kohtalainen CYP3A:n estäjä. Kritsotinibin käyttöä samanaikaisesti kapean terapeuttisen indeksin omaavien CYP3A:n substraattien (mm. alfentaniili, sisapridi, siklosporiini, torajyväjohdokset, fentanyyli, pimotsidi, kinidiini, sirolimuusi ja takrolimuusi) kanssa on vältettävä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos samanaikainen käyttö on välttämätöntä, potilaita tulee seurata tarkasti.
In vitro -tutkimukset osoittivat, että kritsotinibi on CYP2B6:n estäjä. Kritsotinibi voi siten suurentaa samanaikaisesti käytettyjen CYP2B6:n kautta metaboloituvien lääkevalmisteiden (esim. bupropioni, efavirentsi) pitoisuuksia plasmassa.
In vitro ‑tutkimukset ihmisen hepatosyyteillä viittasivat siihen, että kritsotinibi voi indusoida pregnaani X ‑reseptorin (PXR) ja konstitutiivisen androstaanireseptorin (CAR) säätelemiä entsyymejä (esim. CYP3A4, CYP2B6, CYP2C8, CYP2C9, UGT1A1). Induktiota ei kuitenkaan havaittu in vivo annettaessa kritsotinibia samanaikaisesti CYP3A:n mallisubstraatin midatsolaamin kanssa. Varovaisuutta tulee noudattaa käytettäessä kritsotinibia yhdessä pääasiassa näiden entsyymien välityksellä metaboloituvien lääkevalmisteiden kanssa. On huomioitava, että samanaikaisesti käytettyjen suun kautta otettavien ehkäisyvalmisteiden teho voi heikentyä.
In vitro -tutkimukset osoittivat, että kritsotinibi on heikko uridiinidifosfaattiglukuronosyylitransferaasi (UGT) 1A1:n ja UGT2B7:n estäjä. Kritsotinibi voi siten suurentaa samanaikaisesti käytettyjen pääasiassa UGT1A1:n (esim. raltegraviiri, irinotekaani) tai UGT2B7:n (morfiini, naloksoni) kautta metaboloituvien lääkevalmisteiden pitoisuuksia plasmassa.
In vitro ‑tutkimuksen perusteella kritsotinibin oletetaan estävän suoliston P-glykoproteiinia (P-gp). Kritsotinibin samanaikainen käyttö P-gp:n substraatteina toimivien lääkevalmisteiden (esim. digoksiini, dabigatraani, kolkisiini, pravastatiini) kanssa voi siten voimistaa näiden terapeuttista vaikutusta ja lisätä haittavaikutuksia. Tarkkaa kliinistä seurantaa suositellaan käytettäessä kritsotinibia tällaisten lääkevalmisteiden kanssa.
Kritsotinibi on OCT1:n ja OCT2:n estäjä in vitro. Kritsotinibi saattaa siten suurentaa samanaikaisesti annettujen OCT1:n tai OCT2:n substraatteina toimivien lääkevalmisteiden (esim. metformiini, prokaiiniamidi) pitoisuuksia plasmassa.
Farmakodynaamiset yhteisvaikutukset
Kliinisissä tutkimuksissa havaittiin kritsotinibin käytön yhteydessä QT-ajan pitenemistä. Siksi on harkittava tarkkaan kritsotinibin käyttöä samanaikaisesti QT-aikaa tunnetusti pidentävien lääkevalmisteiden tai kääntyvien kärkien takykardiaa (torsade de pointes) mahdollisesti aiheuttavien lääkevalmisteiden kanssa (esim. luokka IA [kinidiini, disopyramidi] tai luokka III [esim. amiodaroni, sotaloli, dofetilidi, ibutilidi], metadoni, sisapridi, moksifloksasiini, psykoosilääkkeet, jne.). QT-aikaa tulee seurata tällaisten lääkevalmisteiden yhteiskäytössä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kliinisissä tutkimuksissa on raportoitu bradykardiaa. Suurentuneen bradykardiariskin vuoksi kritsotinibia tulee käyttää varoen samanaikaisesti muiden bradykardiaa aiheuttavien lääkeaineiden kanssa (esim. dihydropyridiineihin kuulumattomat kalsiumkanavan salpaajat, kuten verapamiili ja diltiatseemi, beetasalpaajat, klonidiini, guanfasiini, digoksiini, meflokiini, antikoliiniesteraasit, pilokarpiini) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisia, jotka voivat tulla raskaaksi, tulee neuvoa välttämään raskaaksi tuloa XALKORI-hoidon aikana.
Miesten ja naisten ehkäisy
Luotettavaa raskauden ehkäisyä tulee käyttää hoidon aikana ja vähintään 90 päivän ajan hoidon päättymisen jälkeen (ks. kohta Yhteisvaikutukset).
Raskaus
Raskaana olevalle naiselle annettu XALKORI-hoito saattaa vahingoittaa sikiötä. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Ei ole olemassa tietoja kritsotinibin käytöstä raskaana oleville naisille. Tätä lääkevalmistetta ei tule käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa. Jos naispotilas on raskaana tai tulee raskaaksi kritsotinibihoidon aikana tai jos kritsotinibihoitoa saavan miespotilaan kumppani tulee raskaaksi, tulee potilaalle kertoa sikiölle mahdollisesti aiheutuvasta vaarasta.
Imetys
Ei tiedetä, erittyvätkö kritsotinibi ja sen metaboliitit ihmisen rintamaitoon. Äitejä tulee neuvoa välttämään rintaruokintaa XALKORI-hoidon aikana, koska siitä saattaa aiheutua haittaa imeväiselle (ks. kohta Prekliiniset tiedot turvallisuudesta).
Hedelmällisyys
XALKORI-hoito saattaa prekliinisten turvallisuustutkimusten perusteella heikentää miehen ja naisen hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta). Sekä miesten että naisten tulisi saada ennen hoitoa tietoa toimenpiteistä lisääntymiskyvyn säilyttämiseksi.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
XALKORI-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Autoa ajettaessa tai koneita käytettäessä on noudatettava varovaisuutta, koska XALKORI-hoidon aikana potilaalla saattaa esiintyä oireista bradykardiaa (esim. pyörtymistä, heitehuimausta ja hypotensiota), näköhäiriöitä tai väsymystä (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Haittavaikutukset
Yhteenveto edennyttä ALK-positiivista tai ROS1-positiivista ei-pienisoluista keuhkosyöpää (NSCLC) sairastavien aikuispotilaiden turvallisuusprofiilista
Seuraavaksi esitetyt tiedot perustuvat edennyttä ALK-positiivista NSCLC:ää sairastavien potilaiden altistukseen XALKORI-hoidolle (n = 1669) kahdessa satunnaistetussa vaiheen 3 tutkimuksessa (tutkimukset 1007 ja 1014) ja kahdessa yksihaaraisessa tutkimuksessa (tutkimukset 1001 ja 1005) sekä edennyttä ROS1-positiivista NSCLC:ää sairastavien potilaiden altistukseen XALKORI-hoidolle (n = 53) yksihaaraisessa tutkimuksessa 1001. Tiedot ovat siten yhteensä 1722 potilaasta (ks. kohta Farmakodynamiikka). Potilaiden aloitusannos oli 250 mg suun kautta kaksi kertaa vuorokaudessa yhtäjaksoisesti. Tutkimuksessa 1014 kritsotinibihoidon keston mediaani oli 47 viikkoa potilailla, jotka satunnaistettiin kritsotinibihaaraan (n = 171), ja 23 viikkoa potilailla, jotka satunnaistettiin solunsalpaajahaaraan, mutta jotka siirtyivät saamaan kritsotinibia taudin edettyä (cross-over, n = 109). Tutkimuksessa 1007 kritsotinibihoidon keston mediaani oli 48 viikkoa kritsotinibihaaraan satunnaistetuilla potilailla (n = 172). ALK-positiivista NSCLC:ää sairastavilla potilailla tehdyssä tutkimuksessa 1001 (n = 154) kritsotinibihoidon keston mediaani oli 57 viikkoa ja tutkimuksessa 1005 (n = 1063) 45 viikkoa. ROS1-positiivista NSCLC:ää sairastavilla potilailla tehdyssä tutkimuksessa 1001 (n = 53) hoidon keston mediaani oli 101 viikkoa.
Edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla potilailla (n = 1722) vakavimpia haittavaikutuksia olivat maksatoksisuus, interstitiaalinen keuhkosairaus (ILD) / pneumoniitti, neutropenia ja QT-ajan piteneminen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla potilailla yleisimpiä haittavaikutuksia (≥ 25 %) olivat näköhäiriöt, pahoinvointi, ripuli, oksentelu, turvotus, ummetus, transaminaasien nousu, väsymys, heikentynyt ruokahalu, heitehuimaus ja neuropatia.
Yleisimpiä annostelun keskeyttämiseen johtaneita haittavaikutuksia (≥ 3 %, syy-yhteydestä riippumatta) olivat neutropenia (11 %), transaminaasien nousu (7 %), oksentelu (5 %) ja pahoinvointi (4 %). Yleisimpiä annoksen pienentämiseen johtaneita haittavaikutuksia (≥ 3 %, syy-yhteydestä riippumatta) olivat transaminaasien nousu (4 %) ja neutropenia (3 %). Hoidon pysyvään lopettamiseen johtaneita haittatapahtumia (syy-yhteydestä riippumatta) ilmeni 302 potilaalla (18 %). Näistä yleisimpiä (≥ 1 %) olivat ILD (1 %) ja transaminaasien nousu (1 %).
Haittavaikutustaulukko
Taulukossa 9 esitetään kritsotinibia saaneilla edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla potilailla (n = 1722) raportoidut haittavaikutukset kahdesta satunnaistetusta vaiheen 3 tutkimuksesta (1007 ja 1014) ja kahdesta yksihaaraisesta kliinisestä tutkimuksesta (1001 ja 1005) (ks. kohta Farmakodynamiikka).
Taulukossa 9 luetellaan haittavaikutukset elinjärjestelmien ja esiintymistiheyksien mukaan. Esiintymistiheydet on määritelty seuraavan luokituksen mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 9. Kritsotinibin kliinisissä NSCLC-tutkimuksissa (n = 1722) raportoidut haittavaikutukset
| Elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen |
| Veri ja imukudos | Neutropeniaa (22 %) Anemiab (15 %) Leukopeniac (15 %) | ||
| Aineenvaihdunta ja ravitsemus | Heikentynyt ruokahalu (30 %) | Hypofosfatemia (6 %) | |
| Hermosto | Neuropatiad (25 %) Makuaistin häiriöt (21 %) | ||
| Silmät | Näköhäiriöte (63 %) | ||
| Sydän | Heitehuimausf (26 %) Bradykardiag (13 %) | Sydämen vajaatoimintah (1 %) EKG:ssä todettu pidentynyt QT-aika (4 %) Synkopee (3 %) | |
| Hengityselimet, rintakehä ja välikarsina | Interstitiaalinen keuhkosairausi (3 %) | ||
| Ruoansulatuselimistö | Oksentelu (51 %) Ripuli (54 %) Pahoinvointi (57 %) Ummetus (43 %) Vatsakipuj (21 %) | Ruokatorven tulehdusk (2 %) Dyspepsia (8 %) | Maha-suolikanavan perforaatiol (< 1 %) |
| Maksa ja sappi | Transaminaasien nousum (32 %) | Kohonnut veren alkalisen fosfataasin pitoisuus (7 %) | Maksan vajaatoiminta (< 1 %) |
| Iho ja ihonalainen kudos | Ihottuma (13 %) | Valoherkkyys (< 1 %) | |
| Munuaiset ja virtsatiet | Munuaiskystan (3 %) Kohonnut veren kreatiniinipitoisuuso (8 %) | Akuutti munuaisten vajaatoiminta (< 1 %) Munuaisten vajaatoiminta (< 1 %) | |
| Yleisoireet ja antopaikassa todettavat haitat | Turvotusp (47 %) Väsymys (30 %) | ||
| Tutkimukset | Alentunut veren testosteronipitoisuusq (2 %) | Suurentunut veren kreatiinikinaasi-pitoisuus (< 1 %)* |
Samaa lääketieteellistä käsitettä tai sairaustilaa koskevat termit yhdistettiin ryhmiksi ja ne on raportoitu yksittäisenä haittavaikutuksena taulukossa 9. Tiedonkeruun päättymispäivään mennessä tutkimuksessa tosiasiallisesti raportoidut termit, jotka ovat osallisena kyseisessä haittavaikutuksessa, on merkitty alla sulkuihin.
* Kritsotinibin kliinisissä tutkimuksissa kreatiinikinaasi ei ollut tavanomainen laboratoriokoe.
a. Neutropenia (kuumeinen neutropenia, neutropenia, pienentynyt neutrofiilimäärä)
b. Anemia (anemia, hemoglobiinin lasku, hypokrominen anemia)
c. Leukopenia (leukopenia, valkosolumäärän lasku)
d. Neuropatia (poltteleva tunne, tuntohäiriö, formikaatio (tunne muurahaisten kävelemisestä iholla), kävelyhäiriö, lisääntynyt tuntoherkkyys, heikentynyt tunto, hypotonia, motorinen häiriö, lihasatrofia, lihasheikkous, neuralgia, neuriitti, ääreishermojen neuropatia, neurotoksisuus, parestesia, ääreishermojen motorinen neuropatia, ääreishermojen sensomotorinen neuropatia, ääreishermojen sensorinen neuropatia, pohjehermohalvaus, polyneuropatia, tuntohäiriö, poltteleva tunne iholla)
e. Näköhäiriöt (kahtena näkeminen, valorenkaiden näkeminen, valonarkuus, valonvälähdysten näkeminen, näön hämärtyminen, näöntarkkuuden heikkeneminen, näköaistimuksen kirkkaus, näkökyvyn heikkeneminen, jälkikuvat, lasiaiskellujat)
f. Heitehuimaus (tasapainohäiriö, heitehuimaus, asentohuimaus, presynkopee)
g. Bradykardia (bradykardia, hidastunut sydämen syke, sinusbradykardia)
h. Sydämen vajaatoiminta (sydämen vajaatoiminta, kongestiivinen sydämen vajaatoiminta, pienentynyt ejektiofraktio, vasemman kammion vajaatoiminta, keuhkoedeema). Kritsotinibia kliinisissä tutkimuksissa saaneista (n = 1722) 19 potilaalla (1,1 %) todettiin jonkin vaikeusasteen sydämen vajaatoiminta. Sydämen vajaatoiminta oli 8 potilaalla (0,5 %) vaikeusastetta 3 tai 4 ja johti 3 potilaan (0,2 %) kuolemaan.
i. Interstitiaalinen keuhkosairaus (aikuisen hengitysvaikeusoireyhtymä, alveoliitti, interstitiaalinen keuhkosairaus, pneumoniitti)
j. Vatsakipu (epämiellyttävät tuntemukset vatsassa, vatsakipu, alavatsakipu, ylävatsakipu, vatsan arkuus)
k. Ruokatorven tulehdus (ruokatorven tulehdus, ruokatorven haavauma)
l. Maha-suolikanavan perforaatio (maha-suolikanavan perforaatio, suoliston perforaatio, paksusuolen perforaatio)
m. Transaminaasien nousu (kohonnut ALAT-arvo, kohonnut ASAT-arvo, kohonnut glutamyylitransferaasiarvo (GT), maksaentsyymien nousu, maksan toiminnan poikkeavuudet, maksan toimintakokeiden poikkeavuudet, transaminaasien nousu)
n. Munuaiskysta (märkäpesäke munuaisissa, munuaiskysta, hemorraginen munuaiskysta, infektoitunut munuaiskysta)
o. Kohonnut veren kreatiniinipitoisuus (kohonnut veren kreatiniinipitoisuus, munuaisten kreatiniinipuhdistuman pieneneminen)
p. Turvotus (kasvojen turvotus, yleinen turvotus, paikallinen turvotus, paikallisesti rajoittunut turvotus, turvotus, raajojen turvotus, periorbitaalinen turvotus)
q. Alentunut veren testosteronipitoisuus (alentunut veren testosteronipitoisuus, hypogonadismi, sekundaarinen hypogonadismi)
Yhteenveto pediatristen potilaiden turvallisuusprofiilista
Turvallisuusanalyysin potilaspopulaatio koostuu 110 pediatrisesta potilaasta (iältään 1 – < 18 vuotta), joilla oli minkä tahansa tyyppisiä kasvaimia, mukaan lukien 41 potilaasta, joilla oli uusiutunut tai refraktorinen systeeminen ALK-positiivinen anaplastinen suurisoluinen lymfooma (ALCL) tai leikkaukseen soveltumaton, uusiutunut tai refraktorinen ALK-positiivinen tulehduksellinen myofibroblastituumori (IMT). ALCL:ää ja IMT:tä sairastavat potilaat saivat kritsotinibia kahdessa yksihaaraisessa tutkimuksessa, 0912 (n = 36) ja 1013 (n = 5). Tutkimuksessa 0912 potilaat saivat kritsotinibia aloitusannoksella 100 mg/m2, 130 mg/m2, 165 mg/m2, 215 mg/m2, 280 mg/m2 tai 365 mg/m2 kaksi kertaa vuorokaudessa. Tutkimuksessa 1013 kritsotinibia annettiin aloitusannoksella 250 mg kaksi kertaa vuorokaudessa. Kaikkiaan potilaspopulaatioon kuului 25 pediatrista (iältään 3 – < 18 vuotta) potilasta, joilla oli ALK‑positiivinen ALCL, ja 16 pediatrista (iältään 2 – < 18 vuotta) potilasta, joilla oli ALK‑positiivinen IMT. Kokemukset kritsotinibin käytöstä pediatrisille potilaille eri alaryhmissä (ikä, sukupuoli ja etninen tausta) ovat vähäisiä, eikä niiden perusteella voida tehdä lopullisia johtopäätöksiä. Ikään, sukupuoleen ja etniseen taustaan perustuvien eri alaryhmien turvallisuusprofiilit olivat yhdenmukaisia, vaikka kussakin alaryhmässä haittavaikutusten esiintymistiheyksissä oli hieman eroja. Kaikissa alaryhmissä (ikä, sukupuoli ja etninen tausta) yleisimmin raportoituja haittavaikutuksia (≥ 80 %) olivat transaminaasien nousu, oksentelu, neutropenia, pahoinvointi, ripuli ja leukopenia. Yleisin vaikea haittavaikutus (90 %) oli neutropenia.
Kaikilla pediatrisilla potilailla kasvaintyypistä riippumatta hoidon keston mediaani oli 2,8 kuukautta. 11 potilasta (10 %) lopetti hoidon pysyvästi haittavaikutuksen vuoksi. 47 potilaan (43 %) annostelu keskeytettiin ja 15 potilaan (14 %) annosta pienennettiin. Yleisimpiä haittavaikutuksia (> 60 %) olivat transaminaasien nousu, oksentelu, neutropenia, pahoinvointi, ripuli ja leukopenia. Yleisin vaikeusasteen 3 tai 4 haittavaikutus (≥ 40 %) oli neutropenia.
ALK-positiivista ALCL:ää sairastavilla pediatrisilla potilailla hoidon keston mediaani oli 5,1 kuukautta. 1 potilas (4 %) lopetti hoidon pysyvästi haittavaikutuksen vuoksi. 11 potilasta ALK-positiivista ALCL:ää sairastavasta 25 potilaasta (44 %) lopetti kritsotinibihoidon pysyvästi siirtyäkseen hematopoieettisten kantasolujen siirtoon. 17 potilaan (68 %) annostelu keskeytettiin ja 4 potilaan (16 %) annosta pienennettiin. Yleisimpiä haittavaikutuksia (≥ 80 %) olivat ripuli, oksentelu, transaminaasien nousu, neutropenia, leukopenia ja pahoinvointi. Yleisimpiä vaikeusasteen 3 tai 4 haittavaikutuksia (≥ 40 %) olivat neutropenia, leukopenia ja lymfosytopenia.
ALK-positiivista IMT:tä sairastavilla pediatrisilla potilailla hoidon keston mediaani oli 21,8 kuukautta. 4 potilasta (25 %) lopetti hoidon pysyvästi haittavaikutuksen vuoksi. 12 potilaan (75 %) annostelu keskeytettiin ja 4 potilaan (25 %) annosta pienennettiin. Yleisimpiä haittavaikutuksia (≥ 80 %) olivat neutropenia, pahoinvointi ja oksentelu. Yleisin vaikeusasteen 3 tai 4 haittavaikutus (≥ 40 %) oli neutropenia.
Kritsotinibin turvallisuusprofiili ALK-positiivista ALCL:ää tai ALK-positiivista IMT:tä sairastavilla pediatrisilla potilailla oli yleisesti yhdenmukainen edennyttä ALK‑positiivista tai ROS1‑positiivista NSCLC:ää sairastavien aikuispotilaiden turvallisuusprofiilin kanssa, mutta haittavaikutusten esiintymistiheyksissä oli jonkin verran vaihtelua. Vaikeusasteen 3 tai 4 haittavaikutuksista neutropeniaa, leukopeniaa ja ripulia raportoitiin yleisemmin (ero ≥ 10 %) pediatrisilla potilailla, joilla oli ALK‑positiivinen ALCL tai ALK-positiivinen IMT, verrattuna ALK‑positiivista tai ROS1‑positiivista NSCLC:ää sairastaviin aikuispotilaisiin. Nämä kaksi potilaspopulaatiota eroavat toisistaan iän, liitännäissairauksien ja perussairauksien osalta, mikä saattaa selittää erot haittavaikutusten esiintymistiheydessä.
Taulukossa 10 luetellaan pediatristen potilaiden (kaikki kasvaintyypit) haittavaikutukset elinjärjestelmien ja esiintymistiheyksien mukaan. Esiintymistiheydet on määritelty seuraavan luokituksen mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 10. Pediatrisilla potilailla raportoidut haittavaikutukset (n = 110)
Kaikki kasvaintyypit (n = 110) | ||
| Elinjärjestelmä | Hyvin yleinen | Yleinen |
| Veri ja imukudos | Neutropeniaa (71 %) Leukopeniab (63 %) Anemiac (52 %) Trombosytopeniad (21 %) | |
| Aineenvaihdunta ja ravitsemus | Hypofosfatemia (30 %) Heikentynyt ruokahalu (39 %) | |
| Hermosto | Neuropatiae (26 %) Makuaistin häiriöt (10 %) | |
| Silmät | Näköhäiriötf (44 %) | |
| Sydän | Bradykardiag (14 %) Heitehuimaus (16 %) | EKG:ssä todettu pidentynyt QT-aika (4 %) |
| Ruoansulatuselimistö | Oksentelu (77 %) Ripuli (69 %) Pahoinvointi (71 %) Ummetus (31 %) Dyspepsia (10 %) Vatsakipuh (43 %) | Ruokatorven tulehdus (4 %) |
| Maksa ja sappi | Transaminaasien nousui (87 %) Kohonnut veren alkalisen fosfataasin pitoisuus (19 %) | |
| Iho ja ihonalainen kudos | Ihottuma (3 %) | |
| Munuaiset ja virtsatiet | Kohonnut veren kreatiniinipitoisuus (45 %) | |
| Yleisoireet ja antopaikassa todettavat haitat | Turvotusj (20 %) Väsymys (46 %) | |
Tiedonkeruun päättymispäivä: 3. syyskuuta 2019.
Samaa lääketieteellistä käsitettä tai sairaustilaa koskevat termit yhdistettiin ryhmiksi, ja ne on raportoitu yksittäisenä haittavaikutuksena taulukossa 10. Tiedonkeruun päättymispäivään mennessä tutkimuksessa tosiasiallisesti raportoidut termit, jotka ovat osallisena kyseisessä haittavaikutuksessa, on merkitty jäljempänä sulkuihin.
a. Neutropenia (kuumeinen neutropenia, neutropenia, pienentynyt neutrofiilimäärä)
b. Leukopenia (leukopenia, valkosolumäärän lasku)
c. Anemia (anemia, makrosyyttinen anemia, megaloblastianemia, hemoglobiini, hemoglobiinin lasku, hyperkrominen anemia, hypokrominen anemia, hypoplastinen anemia, mikrosyyttinen anemia, normokrominen normosyyttinen anemia)
d. Trombosytopenia (pienentynyt verihiutaleiden määrä, trombosytopenia)
e. Neuropatia (poltteleva tunne, kävelyhäiriö, lihasheikkous, parestesia, ääreishermojen motorinen neuropatia, ääreishermojen sensorinen neuropatia)
f. Näköhäiriöt (valonarkuus, valonvälähdysten näkeminen, näön hämärtyminen, näöntarkkuuden heikkeneminen, näkökyvyn heikkeneminen, lasiaiskellujat)
g. Bradykardia (bradykardia, sinusbradykardia)
h. Vatsakipu (epämiellyttävät tuntemukset vatsassa, vatsakipu, alavatsakipu, ylävatsakipu, vatsan arkuus)
i. Transaminaasien nousu (kohonnut ALAT-arvo, kohonnut ASAT-arvo, kohonnut gammaglutamyylitransferaasiarvo)
j. Turvotus (kasvojen turvotus, paikallinen turvotus, raajojen turvotus, periorbitaalinen turvotus)
Vaikka kaikkia aikuispotilailla tunnistettuja haittavaikutuksia ei ole todettu pediatristen potilaiden kliinisissä tutkimuksissa, aikuispotilailla tunnistetut haittavaikutukset on otettava huomioon myös hoidettaessa pediatrisia potilaita. Aikuispotilaita koskevat varoitukset ja varotoimet on otettava huomioon myös hoidettaessa pediatrisia potilaita.
Valikoitujen haittavaikutusten kuvaus
Maksatoksisuus
Potilaita on seurattava maksatoksisuuden varalta ja hoidettava kohdissa Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet suositellulla tavalla.
Aikuispotilaat, joilla on ei-pienisoluinen keuhkosyöpä (NSCLC)
Kliinisissä tutkimuksissa lääkkeen aiheuttamaa kuolemaan johtanutta maksatoksisuutta on esiintynyt 0,1 %:lla kritsotininbia saaneista aikuispotilaista, joilla on NSCLC (n = 1722). Samanaikaista ALAT- ja/tai ASAT-arvon nousua ≥ 3 kertaa viitevälin ylärajaa suuremmaksi ja bilirubiinin nousua ≥ 2 kertaa viitevälin ylärajaa suuremmaksi ilman merkittävää alkalisen fosfataasin nousua (≤ 2 kertaa viitevälin ylärajaa suuremmaksi) on havaittu alle 1 %:lla kritsotinibia saaneista potilaista.
Vaikeusasteen 3 tai 4 ALAT-arvon nousua havaittiin 187 aikuispotilaalla (11 %) ja vaikeusasteen 3 tai 4 ASAT-arvon nousua 95 aikuispotilaalla (6 %). Transaminaasien nousuun liittyen 17 potilaan (1 %) hoito oli lopetettava pysyvästi, mikä viittaa siihen, että tapahtumat voitiin yleensä hoitaa taulukon 4 mukaisilla annosmuutoksilla (ks. kohta Annostus ja antotapa). Satunnaistetussa vaiheen 3 tutkimuksessa 1014 vaikeusasteen 3 tai 4 ALAT-arvon nousua havaittiin 15 %:lla ja vaikeusasteen 3 tai 4 ASAT-arvon nousua 8 %:lla kritsotinibia saaneista potilaista ja vastaavasti 2 %:lla ja 1 %:lla solunsalpaajahoitoa saaneista potilaista. Satunnaistetussa vaiheen 3 tutkimuksessa 1007 vaikeusasteen 3 tai 4 ALAT-arvon nousua havaittiin 18 %:lla ja vaikeusasteen 3 tai 4 ASAT-arvon nousua 9 %:lla kritsotinibia saaneista potilaista ja vastaavasti 5 %:lla ja < 1 %:lla solunsalpaajahoitoa saaneista potilaista.
Transaminaasien nousu tapahtui yleensä kahden ensimmäisen hoitokuukauden aikana. ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavien aikuispotilaiden kritsotinibitutkimuksissa mediaaniaika vaikeusasteen 1 tai 2 transaminaasien nousun ilmaantumiseen oli 23 vuorokautta. Mediaaniaika vaikeusasteen 3 tai 4 transaminaasien nousun ilmaantumiseen oli 43 vuorokautta.
Vaikeusasteen 3 ja 4 transaminaasien nousu yleensä korjaantui annostelun keskeyttämisen jälkeen. ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavien aikuispotilaiden kritsotinibitutkimuksissa (n = 1722) 76 potilaan (4 %) annosta pienennettiin transaminaasien nousun vuoksi. Hoito oli lopetettava pysyvästi 17 potilaalla (1 %).
Pediatriset potilaat
Kliinisissä tutkimuksissa 110 pediatrisella potilaalla, joilla oli erityyppisiä kasvaimia ja jotka saivat kritsotinibihoitoa, ASAT-arvo nousi 70 %:lla potilaista ja ALAT-arvo nousi 75 %:lla potilaista. Vaikeusasteen 3 tai 4 ASAT-arvon nousua todettiin 7 %:lla potilaista ja vaikeusasteen 3 tai 4 ALAT-arvon nousua 6 %:lla potilaista.
Vaikutukset ruoansulatuselimistöön
Tukihoitoon tulisi sisältyä pahoinvointilääkitys. Katso lisätietoja tukihoidosta pediatrisille potilaille kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Aikuispotilaat, joilla on NSCLC
Pahoinvointi (57 %), ripuli (54 %), oksentelu (51 %) ja ummetus (43 %) olivat yleisimpiä raportoituja ruoansulatuselimistön haittatapahtumia (syy-yhteydestä riippumatta) ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla aikuispotilailla. Useimmat näistä haittatapahtumista olivat vaikeusasteeltaan lieviä tai kohtalaisia. Mediaaniaika pahoinvoinnin ja oksentelun ilmaantumiseen oli 3 vuorokautta, ja niiden esiintyvyys väheni 3 viikon hoidon jälkeen. Mediaaniaika ripulin ilmaantumiseen oli 13 vuorokautta ja ummetuksen ilmaantumiseen 17 vuorokautta. Ripulin oireenmukaiseen hoitoon tulisi sisältyä tavanomainen ripulilääke ja ummetuksen hoitoon tavanomainen ulostuslääke.
Kliinisissä tutkimuksissa NSCLC:ää sairastavilla aikuispotilailla, jotka saivat kritsotinibihoitoa, raportoitiin maha-suolikanavan perforaatiotapahtumia. Kritsotinibin markkinoille tulon jälkeen on raportoitu kuolemaan johtaneita maha-suolikanavan perforaatiotapahtumia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Kliinisissä tutkimuksissa 110 pediatrisella potilaalla, joilla oli erityyppisiä kasvaimia ja jotka saivat kritsotinibihoitoa, oksentelu (77 %), ripuli (69 %), pahoinvointi (71 %), vatsakipu (43 %) ja ummetus (31 %) olivat yleisimmin raportoituja ruoansulatuselimistön haittatapahtumia (syy-yhteydestä riippumatta). Potilaista, joilla on ALK-positiivinen ALCL tai ALK-positiivinen IMT ja jotka saivat kritsotinibihoitoa, oksentelu (95 %), ripuli (85 %), pahoinvointi (83 %), vatsakipu (54 %) ja ummetus (34 %) olivat yleisimmin raportoituja ruoansulatuselimistön haittatapahtumia (syy-yhteydestä riippumatta) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kritsotinibi voi aiheuttaa vaikeaa ruoansulatuskanavaan kohdistuvaa toksisuutta pediatrisille potilaille, joilla on ALCL tai IMT (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
QT-ajan piteneminen
QT‑ajan piteneminen voi johtaa rytmihäiriöihin, ja se on äkkikuoleman riskitekijä. QT‑ajan piteneminen voi kliinisesti ilmetä bradykardiana, heitehuimauksena ja pyörtymisenä. Elektrolyyttihäiriöt, elimistön kuivuminen ja bradykardia voivat edelleen lisätä riskiä QTc‑ajan pitenemiseen, joten säännöllisin väliajoin toistuvia EKG- ja elektrolyyttitutkimuksia suositellaan potilaille, joilla esiintyy ruoansulatuskanavaan kohdistuvaa toksisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Aikuispotilaat, joilla on NSCLC
Edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavien aikuispotilaiden tutkimuksissa QTcF-aika (Fridericia-menetelmällä korjattu QT-aika) oli ≥ 500 millisekuntia 34 potilaalla 1619 potilaasta (2,1 %), joiden EKG rekisteröitiin vähintään kerran lähtötilanteen jälkeen. Suurimmat QTcF-ajan pitenemiset lähtötilanteeseen verrattuna (≥ 60 millisekuntia) todettiin 79 potilaalla 1585 potilaasta (5,0 %), joiden EKG rekisteröitiin lähtötilanteessa ja vähintään kerran sen jälkeen. EKG:ssä todettua vaikeusasteen 3 tai 4 pidentynyttä QT-aikaa (syy-yhteydestä riippumatta) raportoitiin 27 potilaalla 1722 potilaasta (1,6 %) (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakokinetiikka).
Yksihaarainen, aikuispotilaiden EKG-alatutkimus (ks. kohta Farmakokinetiikka) perustui sokkoutettuihin EKG-mittauksiin ja mittaustulosten manuaaliseen arviointiin. Lähtötilanteeseen verrattuna QTcF-aika oli pidentynyt ≥ 30 millisekuntia mutta alle 60 millisekuntia 11 potilaalla (21 %) ja ≥ 60 millisekuntia yhdellä potilaalla (2 %). Yhdelläkään potilaalla QTcF-ajan maksimiarvo ei ollut ≥ 480 millisekuntia. Tilastollisen analyysin (central tendency analysis) perusteella QTcF-ajan suurin keskimääräinen muutos lähtötilanteeseen verrattuna oli 12,3 millisekuntia (95 %:n luottamusväli 5,1−19,5 millisekuntia, varianssianalyysin pienimmän neliösumman keskiarvo), ja se ilmeni 6 tunnin kuluttua lääkeannoksen ottamisesta syklin 2 ensimmäisenä päivänä. Kaikissa syklin 2 ensimmäisen päivän aikapisteissä QTcF-ajan (muutos lähtötilanteesta, pienimmän neliösumman keskiarvo) 90 %:n luottamusvälin ylärajat olivat alle 20 millisekuntia.
Pediatriset potilaat
Kritsotinibin kliinisissä tutkimuksissa 110 pediatrisella potilaalla, joilla oli erityyppisiä kasvaimia, EKG:ssä todettiin pidentynyt QT-aika 4 %:lla potilaista.
Bradykardia
Samanaikaista käyttöä muiden bradykardian kehittymiseen liitettyjen lääkevalmisteiden kanssa on harkittava tarkoin. Potilaat, joille ilmaantuu oireinen bradykardia, ks. suositukset kohdissa ”Annoksen muuttaminen” ja ”Varoitukset ja varotoimet” (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Aikuispotilaat, joilla on NSCLC
Edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavien aikuispotilaiden tutkimuksissa bradykardiaa (syy-yhteydestä riippumatta) esiintyi 219 potilaalla kritsotinibia saaneista 1 722 potilaasta (13 %). Useimmat tapaukset olivat vaikeusasteeltaan lieviä. Yhteensä 259 potilaalla 1 666 potilaasta (16 %), joiden vitaalimerkit arvioitiin vähintään kerran lähtötilanteen jälkeen, sydämen syke oli alle 50 lyöntiä minuutissa.
Pediatriset potilaat
Kritsotinibin kliinisissä tutkimuksissa 110 pediatrisella potilaalla, joilla oli erityyppisiä kasvaimia, bradykardiaa (syy-yhteydestä riippumatta) raportoitiin 14 %:lla potilaista, mukaan lukien vaikeusasteen 3 bradykardiaa 1 %:lla potilaista.
Interstitiaalinen keuhkosairaus / pneumoniitti
Potilaita, joilla on interstitiaaliseen keuhkosairauteen (ILD) / pneumoniittiin viittaavia keuhko-oireita, tulee seurata. ILD:n/pneumoniitin muut mahdolliset syyt pitää sulkea pois (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Aikuispotilaat, joilla on NSCLC
Kritsotinibihoitoa saavilla potilailla voi esiintyä vaikea-asteista, hengenvaarallista tai kuolemaan johtavaa ILD:tä/pneumoniittia. Tutkimuksissa kritsotinibia saaneista edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavista aikuispotilaista (n = 1 722) 50 potilaalla (3 %) todettiin jonkin vaikeusasteen ILD (syy-yhteydestä riippumatta), mukaan lukien 18 potilaan (1 %) vaikeusasteen 3 tai 4 tapaukset ja 8 potilaan (< 1 %) kuolemaan johtaneet tapaukset. Riippumattoman arviointikomitean ALK-positiivista NSCLC:ää sairastavista potilaista (n = 1 669) tekemän arvioinnin mukaan 20 potilaalla (1,2 %) oli ILD/pneumoniitti, mukaan lukien 10 kuolemaan johtanutta tapausta (< 1 %). Nämä tapaukset ilmaantuivat yleensä kolmen ensimmäisen kuukauden aikana hoidon aloittamisesta.
Pediatriset potilaat
Kritsotinibin kliinisissä tutkimuksissa pediatrisilla potilailla, joilla on erityypisiä kasvaimia, ILD:tä/pneumoniittia raportoitiin 1 potilaalla (1 %). Kyseessä oli vaikeusasteen 1 pneumoniitti.
Vaikutukset näkökykyyn
Silmätutkimuksia suositellaan, jos potilaan näköhäiriöt pitkittyvät tai niiden vaikeusaste pahenee. Pediatrisille potilaille tulee tehdä silmätutkimuksia lähtötilanteessa ja seurannan aikana (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Aikuispotilaat, joilla on NSCLC
Kritsotinibin kliinisissä tutkimuksissa edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla aikuispotilailla (n = 1722) vaikeusasteen 4 näkökenttäpuutosta, johon liittyi näönmenetys, on raportoitu 4 potilaalla (0,2 %). Mahdollisina syinä näönmenetykselle on raportoitu näköhermon surkastumaa ja toimintahäiriötä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kaikkien vaikeusasteiden näköhäiriöitä (syy-yhteydestä riippumatta) esiintyi 1084 potilaalla kritsotinibia saaneista 1722 aikuispotilaasta (63 %). Näköhäiriöt ilmenivät useimmiten näkökyvyn heikkenemisenä, valonvälähdysten näkemisenä, näön hämärtymisenä ja lasiaiskellujina. Näköhäiriöitä kokeneista 1084 potilaasta 95 %:lla tapahtumat olivat vaikeusasteeltaan lieviä. Näköhäiriöt liittyivät 7 potilaan (0,4 %) hoidon tilapäiseen keskeyttämiseen ja 2 potilaan (0,1 %) annoksen pienentämiseen. Yhdelläkään kritsotinibia saaneista 1722 potilaasta hoidon pysyvä lopettaminen ei liittynyt näköhäiriöihin.
Näkökykyyn liittyviä oireita kartoitettiin kyselytutkimuksella (Visual Symptom Assessment Questionnaire, VSAQ-ALK). Sen perusteella tutkimuksessa 1007 ja tutkimuksessa 1014 kritsotinibihoitoa saaneilla aikuispotilailla raportoitiin enemmän näköhäiriöitä kuin solunsalpaajahoitoa saaneilla. Näköhäiriöt ilmaantuivat yleensä ensimmäisellä hoitoviikolla. Satunnaistetussa vaiheen 3 tutkimuksessa 1007 ja tutkimuksessa 1014 enemmistö (> 50 %) kritsotinibihaaran potilaista raportoi näköhäiriöitä. VSAQ-ALK-kyselytutkimuksen mukaan niitä esiintyi 4−7 päivänä viikossa, ne kestivät enintään minuutin ajan ja niillä oli lievä vaikutus tai ei lainkaan vaikutusta päivittäisiin toimiin (pisteet 0–3 enimmäispistemäärästä 10).
Oftalmologisessa alatutkimuksessa oli mukana 54 NSCLC:ää sairastavaa aikuispotilasta, jotka saivat kritsotinibia 250 mg kaksi kertaa vuorokaudessa. Tutkimuksessa tehtiin tiettyinä ajankohtina määrättyjä silmätutkimuksia. Hoidon yhteydessä havaittu tai vaikeutunut elinjärjestelmäluokan ”Silmät” haittatapahtuma (syy-yhteydestä riippumatta) ilmeni 38 potilaalla 54 potilaasta (70,4 %), joista 30 potilaalle tehtiin silmätutkimuksia. Näistä 30 potilaasta 14 potilaalla (36,8 %) raportoitiin jokin silmään liittyvä poikkeavuus, kun taas 16 potilaalla (42,1 %) silmälöydöksiä ei tehty. Yleisimmin löydökset liittyivät biomikroskooppi- (21,1 %), silmänpohja- (15,8 %) ja näöntarkkuustutkimuksiin (13,2 %). Usealla potilaalla oli jo ennestään silmään liittyviä poikkeavuuksia ja samanaikaisia sairauksia, jotka saattoivat myötävaikuttaa silmälöydöksiin. Siksi syy-yhteyttä kritsotinibihoitoon ei voitu luotettavasti määrittää. Kammionesteen solujen kokonaismäärään ja silmän etukammion valotiehen liittyviä löydöksiä ei tehty. Kritsotinibiin liittyvät näköhäiriöt eivät näyttäneet olevan yhteydessä parhaan korjatun näöntarkkuuden, lasiaisen, verkkokalvon tai näköhermon muutoksiin.
Jos aikuispotilaalle ilmaantuu uutena oireena vaikeusasteen 4 näönmenetys, kritsotinibihoito tulisi keskeyttää ja tehdä potilaalle silmätutkimuksia.
Pediatriset potilaat
Kritsotinibin kliinisissä tutkimuksissa 110 pediatrisella potilaalla, joilla oli erityyppisiä kasvaimia, näköhäiriöitä raportoitiin 48 potilaalla (44 %). Yleisimpiä näkökykyyn liittyviä oireita olivat näön hämärtyminen (20 %) ja näkökyvyn heikkeneminen (11 %).
Kritsotinibin kliinisissä tutkimuksissa 41 potilaalla, joilla on ALK-positiivinen ALCL tai ALK-positiivinen IMT, näköhäiriöitä raportoitiin 25 potilaalla (61 %). Näköhäiriöitä kokeneista pediatrisista potilaista yhdellä IMT:tä sairastavalla potilaalla ilmeni vaikeusasteen 3 myooppinen näköhermon häiriö, joka oli lähtötilanteessa vaikeusastetta 1. Yleisimpiä näkökykyyn liittyviä oireita olivat näön hämärtyminen (24 %), näkökyvyn heikkeneminen (20 %), valonvälähdysten näkeminen (17 %) ja lasiaiskellujat (15 %). Kaikki olivat joko vaikeusastetta 1 tai 2.
Vaikutukset hermostoon
Aikuispotilaat, joilla on NSCLC
Taulukossa 9 mainittua neuropatiaa (syy-yhteydestä riippumatta) esiintyi 435:lla (25 %:lla) edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavalla potilaalla kritsotinibia saaneista 1 722 aikuispotilaasta. Näissä tutkimuksissa raportoitiin hyvin yleisesti myös makuaistin häiriötä, joka oli pääasiassa vaikeusastetta 1.
Pediatriset potilaat
Kritsotinibin kliinisissä tutkimuksissa 110 pediatrisella potilaalla, joilla oli erityyppisiä kasvaimia, neuropatiaa raportoitiin 26 %:lla potilaista ja makuaistin häiriöitä 9 %:lla potilaista.
Munuaiskysta
Jos potilaalle ilmaantuu munuaiskysta, kuvantamista ja virtsa-analyysiä potilaan säännöllisessä seurannassa tulee harkita.
Aikuispotilaat, joilla on NSCLC
Rakenteeltaan epäsäännöllisiä munuaiskystoja (syy-yhteydestä riippumatta) esiintyi 52:lla (3 %:lla). edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavalla potilaalla kritsotinibia saaneista 1 722 aikuispotilaasta. Joillakin potilailla havaittiin paikallista kystojen leviämistä munuaisen ulkopuolelle.
Pediatriset potilaat
Kritsotinibin kliinisissä tutkimuksissa 110 pediatrisella potilalla, joilla oli erityyppisiä kasvaimia, ei raportoitu munuaiskystoja.
Neutropenia ja leukopenia
Täydellistä verenkuvaa, veren valkosolujen erittelylaskenta mukaan lukien, tulee seurata kliinisen tarpeen mukaan. Määrityksiä tulee tehdä tiheämmin, jos havaitaan vaikeusasteen 3 tai 4 poikkeavuuksia tai jos potilaalla ilmenee kuumetta tai infektio. Jos potilaalle kehittyy hematologisia laboratorioarvojen poikkeavuuksia, ks. kohta Annostus ja antotapa.
Aikuispotilaat, joilla on NSCLC
Tutkimuksissa kritsotinibia saaneista edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavista aikuispotilaista (n = 1 722) vaikeusasteen 3 tai 4 neutropeniaa havaittiin 212 potilaalla (12 %). Mediaaniaika minkä tahansa vaikeusasteen neutropenian ilmaantumiseen oli 89 vuorokautta. Neutropenia liittyi annoksen pienentämiseen 3 %:lla potilaista ja hoidon pysyvään lopettamiseen alle 1 %:lla potilaista. Kritsotinibin kliinisissä tutkimuksissa alle 0,5 %:lla potilaista esiintyi kuumeista neutropeniaa.
Edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavien aikuispotilaiden tutkimuksissa (n = 1 722) vaikeusasteen 3 tai 4 leukopeniaa havaittiin 48:lla (3 %:lla) kritsotinibilla hoidetulla potilaalla. Mediaaniaika minkä tahansa vaikeusasteen leukopenian ilmaantumiseen oli 85 vuorokautta. Leukopenia liittyi annoksen pienentämiseen alle 0,5 %:lla potilaista. Yhdenkään potilaan kritsotinibihoitoa ei lopetettu pysyvästi leukopeniaan liittyen.
Edennyttä ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavien aikuispotilaiden kritsotibitutkimuksissa vaikeusasteen 3 tai 4 leukosyyttien vähenemistä todettiin 4 %:lla potilaista ja vaikeusasteen 3 tai 4 neutrofiilien vähenemistä 13 %:lla potilaista.
Pediatriset potilaat
Kritsotinibin kliinisissä tutkimuksissa 110 pediatrisella potilaalla, joilla oli erityyppisiä kasvaimia, neutropeniaa raportoitiin 71 %:lla potilaista, mukaan lukien 58 potilasta (53 %), joilla todettiin vaikeusasteeen 3 tai 4 neutropenia. Kuumeista neutropeniaa esiintyi 4 potilaalla (3,6 %). Leukopeniaa raportoitiin 63 %:lla potilaista, mukaan lukien 18 potilasta (16 %), joilla todettiin vaikeusasteen 3 tai 4 leukopenia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tämän lääkevalmisteen yliannostuksen hoito käsittää yleiset elintoimintoja tukevat toimenpiteet. XALKORIlle ei ole vasta-ainetta.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Syöpälääkkeet, proteiinikinaasin estäjät. ATC-koodi: L01ED01.
Vaikutusmekanismi
Kritsotinibi on anaplastisen lymfoomakinaasin (ALK) reseptorityrosiinikinaasin ja sen onkogeenisten varianttien (eli ALK-fuusioiden ja tiettyjen ALK-mutaatioiden) selektiivinen, pienimolekyylinen estäjä. Kritsotinibi on myös hepatosyyttikasvutekijän reseptorin (HGFR, c‑Met), ROS1-kinaasin (c‑ros) ja RON (Recepteur d’Origine Nantais) -reseptorin tyrosiinikinaasin estäjä. Kritsotinibin on todettu biokemiallisissa määrityksissä estävän ALK-, ROS1- ja c‑Met-kinaasien aktiivisuutta lääkepitoisuuksista riippuvaisella tavalla ja solupohjaisissa määrityksissä estävän fosforylaatiota ja säätelevän kinaasiriippuvaisia fenotyyppejä. Kritsotinibilla on todettu voimakas ja selektiivinen kasvua estävä aktiivisuus ja sen on todettu indusoivan apoptoosia kasvainsolulinjoissa, jotka ilmentävät ALK-fuusiotapahtumia (echinoderm microtubule‑associated protein‑like 4 [EML4]‑ALK ja nukleofosmiini [NPM]‑ALK mukaan lukien), ROS1-fuusiotapahtumia tai joissa ALK- tai MET-geenilokus on monistunut. Kritsotinibilla todettiin antituumorivaikutus, mukaan lukien huomattava sytoreduktiivinen antituumorivaikutus, hiirillä, joilla oli ALK-fuusioproteiineja ilmentäviä vieraslajikasvainsiirteitä. Kritsotinibin antituumorivaikutus oli annosriippuvainen ja korreloi ALK-fuusioproteiinien (EML4‑ALK ja NPM‑ALK mukaan lukien) fosforylaation farmakodynaamiseen estymiseen kasvaimissa in vivo. Kritsotinibilla todettiin merkittävä antituumorivaikutus myös hiiren vieraslajisiirretutkimuksissa. Kasvaimet tuotettiin NIH‑3T3-solulinjasarjassa, jotka oli suunniteltu ilmentämään ihmisen kasvaimista tunnistettuja keskeisiä ROS1-fuusioita. Kritsotinibin antituumorivaikutus oli annosriippuvainen ja sen osoitettiin korreloivan ROS1:n fosforylaation estymiseen in vivo. In vitro ‑tutkimukset, jotka koskivat kahta anaplastisesta suurisoluisesta lymfoomasta johdettua solulinjaa (SU‑DHL‑1 ja Karpas‑299, joihin molempiin sisältyy NPM‑ALK:ta), osoittivat, että kritsotinibilla oli kyky indusoida apoptoosia, ja Karpas‑299-soluissa kritsotinibi esti solujen proliferaation sekä ALK-välitteisen signaloinnin kliinisesti saavutettavissa olevilla annoksilla. Karpas‑299-mallista in vivo saadut tiedot osoittivat, että annostuksella 100 mg/kg kerran vuorokaudessa saavutettiin kasvaimen täydellinen regressio.
Kliiniset tutkimukset
Aiemmin hoitamaton edennyt ALK-positiivinen ei-pienisoluinen keuhkosyöpä (NSCLC) (ensilinjan hoito) – satunnaistettu vaiheen 3 tutkimus 1014
Kritsotinibin teho ja turvallisuus metastasoituneen ALK-positiivisen NSCLC:n ensilinjan hoidossa osoitettiin monikansallisessa, satunnaistetussa, avoimessa tutkimuksessa 1014. Potilaat eivät olleet saaneet aikaisempaa systeemistä hoitoa edenneeseen tautiin.
Koko analyysipopulaatio käsitti 343 potilasta, joilla oli edennyt ALK-positiivinen NSCLC. ALK-positiivisuus oli todettu FISH (Fluorescence In Situ Hybridization) -menetelmällä ennen satunnaistamista. 172 potilasta satunnaistettiin saamaan kritsotinibia ja 171 potilasta satunnaistettiin saamaan solunsalpaajaa (pemetreksedi ja karboplatiini tai sisplatiini; enintään 6 hoitosykliä). Koko tutkimuspopulaation demografisten ja sairauteen liittyvien tietojen mukaan 62 % potilaista oli naisia, mediaani ikä oli 53 vuotta, lähtötilanteen Eastern Cooperative Oncology Group (ECOG) -suorituskykyluokka oli 95 %:lla potilaista 0 tai 1, 51 % oli valkoihoisia ja 46 % aasialaisia, 4 % tupakoi edelleen, 32 % oli aiemmin tupakoinut ja 64 % ei ollut koskaan tupakoinut. Kasvain oli metastasoitunut 98 %:lla tutkimuspotilaista, 92 % syövistä luokiteltiin histologialtaan adenokarsinoomiksi ja 27 %:lla tutkimuspotilaista oli aivometastaaseja.
Potilaiden saamaa kritsotinibihoitoa voitiin jatkaa vielä kiinteässä kasvaimessa todetun vasteen arviointikriteerien (Response Evaluation Criteria in Solid Tumours, RECIST) mukaisen taudin etenemisen jälkeen, jos tutkija katsoi potilaan saavan hoidosta kliinistä hyötyä. Kritsotinibilla hoidetuista 89 potilaasta 65 potilasta (73 %) ja solunsalpaajahoitoa saaneista 132 potilaasta 11 potilasta (8,3 %) jatkoivat hoitoa vähintään 3 viikkoa objektiivisen taudin etenemisen jälkeen. Solunsalpaajahoitohaaran potilaiden oli mahdollista siirtyä saamaan kritsotinibia (cross-over), kun riippumaton radiologinen arviointi oli vahvistanut RECIST-kriteerien mukaisen taudin etenemisen. Solunsalpaajahaaran potilaista 144 (84 %) siirtyi saamaan kritsotinibihoitoa.
Riippumattoman radiologisen arvioinnin mukaan kritsotinibi pidensi merkitsevästi taudin etenemisestä vapaata elinaikaa (PFS) solunsalpaajahoitoon verrattuna, mikä oli tutkimuksen ensisijainen päätetapahtuma. Kritsotinibilla saavutettu PFS-hyöty oli johdonmukainen kaikissa lähtötilanteen tietoihin perustuvissa potilaiden alaryhmissä, mukaan lukien ikä, sukupuoli, etninen tausta, tupakointitausta, diagnoosista tutkimushoidon aloittamiseen kulunut aika, ECOG-suorituskykyluokka ja aivometastaasien esiintyminen. Kritsotinibilla hoidetuilla potilailla kokonaiselinaika oli numeerisesti parempi, mutta muutos ei ollut tilastollisesti merkitsevä. Satunnaistetun vaiheen 3 tutkimuksen 1014 tehoa koskevien tietojen yhteenveto on esitetty taulukossa 11 ja taudin etenemisestä vapaan elinajan todennäköisyyttä kuvaavat Kaplan-Meierin käyrät kuvassa 1. Kokonaiselinajan todennäköisyyttä kuvaavat Kaplan-Meierin käyrät on esitetty kuvassa 2.
Taulukko 11. Tehoa koskevat tulokset satunnaistetusta vaiheen 3 tutkimuksesta 1014 (koko analyysipopulaatio) edennyttä ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla, jotka eivät olleet saaneet aikaisempaa hoitoa edenneeseen tautiin*
| Vastemuuttuja | Kritsotinibi n = 172 | Solunsalpaaja n = 171 |
| Taudin etenemisestä vapaa elinaika (riippumattoman radiologisen arvioinnin perusteella) | ||
| Tapahtumien lukumäärä, n (%) | 100 (58 %) | 137 (80 %) |
| Taudin etenemisestä vapaan elinajan mediaani, kk (95 %:n luottamusväli) | 10,9 (8,3, 13,9) | 7,0a (6,8, 8,2) |
| HR (95 %:n luottamusväli)b | 0,45 (0,35, 0,60) | |
| p-arvoc | < 0,0001 | |
| Kokonaiselinaikad | ||
| Kuolemien lukumäärä, n (%) | 71 (41 %) | 81 (47 %) |
| Kokonaiselinajan mediaani, kk (95 %:n luottamusväli) | NR (45,8, NR) | 47,5 (32,2, NR) |
| HR (95 %:n luottamusväli)b | 0,76 (0,55, 1,05) | |
| p-arvoc | 0,0489 | |
| 12 kuukauden eloonjäämisen todennäköisyysd, % (95 %:n luottamusväli) | 83,5 (77,0, 88,3) | 78,4 (71,3, 83,9) |
| 18 kuukauden eloonjäämisen todennäköisyysd, % (95 %:n luottamusväli) | 71,5 (64,0, 77,7) | 66,6 (58,8, 73,2) |
| 48 kuukauden eloonjäämisen todennäköisyysd, % (95 %:n luottamusväli) | 56,6 (48,3, 64,1) | 49,1 (40,5, 57,1) |
| Objektiivisen vasteen saaneiden osuus (riippumattoman radiologisen arvioinnin perusteella) | ||
| Objektiivisen vasteen saaneiden osuus, % (95 %:n luottamusväli) | 74 % (67, 81) | 45 %e (37, 53) |
| p-arvof | < 0,0001 | |
| Vasteen kesto | ||
| kkg (95 %:n luottamusväli) | 11,3 (8,1, 13,8) | 5,3 (4,1, 5,8) |
Lyhenteet: HR (Hazard Ratio) = riskitiheyksien suhde; n = potilaiden lukumäärä; NR (not reached) = ei saavutettu
* Taudin etenemisestä vapaa elinaika, objektiivisen vasteen saaneiden osuus ja vasteen kesto perustuvat tiedonkeruun katkaisuajankohtaan 30. marraskuuta 2013; kokonaiselinaika perustuu viimeisen potilaan viimeisen tutkimuskäynnin päivämäärään 30. marraskuuta 2016 ja seuranta-aikaan, jonka mediaani on noin 46 kuukautta.
a. Taudin etenemisestä vapaan elinajan mediaani oli 6,9 kuukautta (95 %:n luottamusväli: 6,6, 8,3) käytettäessä pemetreksedin ja sisplatiinin yhdistelmää (HR = 0,49; p-arvo < 0,0001 kritsotinibille verrattuna pemetreksedin ja sisplatiinin yhdistelmään) ja 7,0 kuukautta (95 %:n luottamusväli: 5,9, 8,3) käytettäessä pemetreksedin ja karboplatiinin yhdistelmää (HR = 0,45; p-arvo < 0,0001 kritsotinibille verrattuna pemetreksedin ja karboplatiinin yhdistelmään).
b. Perustuu Coxin suhteellisten riskitiheyksien mallin ositettuun analyysiin.
c. Perustuu ositettuun log-rank-testiin (yksitahoiseen).
d. Päivitetty kokonaiselinajan lopullisen analyysin perusteella. Kokonaiselinajan analyysiä ei vakioitu cross-overista mahdollisesti aiheutuvien sekoittavien tekijöiden varalta (solunsalpaajahaaran potilaista 144 [84 %] siirtyi saamaan kritsotinibihoitoa).
e. Objektiivisen vasteen saaneiden potilaiden osuus oli 47 % (95 %:n luottamusväli: 37, 58) käytettäessä pemetreksedin ja sisplatiinin yhdistelmää (p-arvo < 0,0001 verrattuna kritsotinibiin) ja 44 % (95 %:n luottamusväli: 32, 55) käytettäessä pemetreksedin ja karboplatiinin yhdistelmää (p-arvo < 0,0001 verrattuna kritsotinibiin).
f. Perustuu ositettuun Cochran-Mantel-Haenszelin testiin (kaksitahoiseen).
g. Arvioitu Kaplan-Meierin menetelmällä.
Kuva 1. Kaplan-Meierin käyrät taudin etenemisestä vapaalle elinajalle (riippumattoman radiologisen arvion mukaan) tutkimushaaroittain satunnaistetussa vaiheen 3 tutkimuksessa 1014 (koko analyysipopulaatio) edennyttä ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla, jotka eivät olleet saaneet aikaisempaa hoitoa edenneeseen tautiin
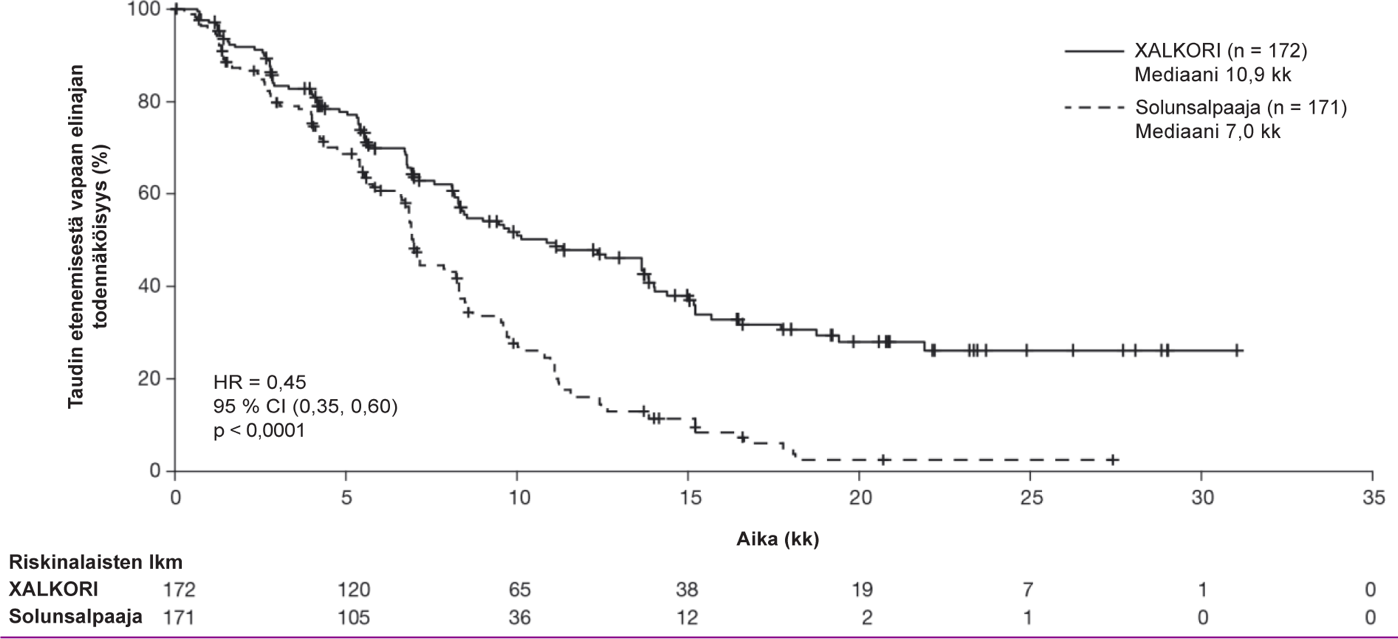
Lyhenteet: HR (Hazard Ratio) = riskitiheyksien suhde; CI (Confidence Interval) = luottamusväli;
p = p-arvo; n = potilaiden lukumäärä
Kuva 2. Kaplan-Meierin käyrät kokonaiselinajalle tutkimushaaroittain satunnaistetussa vaiheen 3 tutkimuksessa 1014 (koko analyysipopulaatio) edennyttä ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla, jotka eivät olleet saaneet aikaisempaa hoitoa edenneeseen tautiin
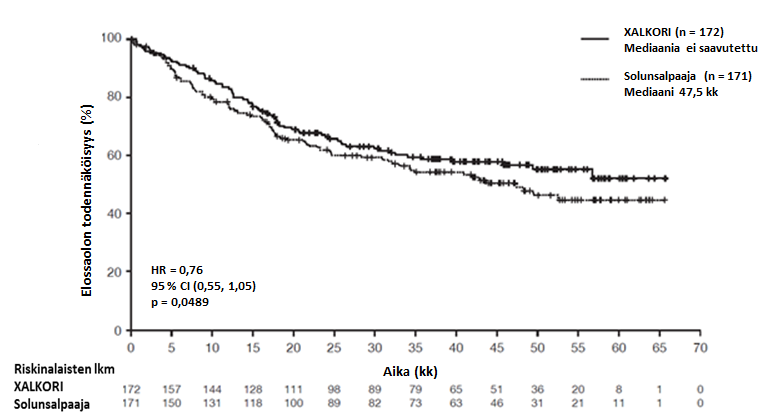
Lyhenteet: HR = riskitiheyksien suhde; CI = luottamusväli; p = p-arvo; n = potilaiden lukumäärä
Potilailla, joilla oli lähtötilanteessa aiemmin hoidettuja aivometastaaseja, mediaaniaika taudin intrakraniaaliseen etenemiseen (IC-TTP) oli 15,7 kuukautta kritsotinibihaarassa (n = 39) ja 12,5 kuukautta solunsalpaajahaarassa (n = 40) (HR = 0,45; 95 %:n luottamusväli: 0,19, 1,07; yksitahoinen p-arvo = 0,0315). Potilailla, joilla ei ollut lähtötilanteessa aivometastaaseja, mediaaniaikaa taudin intrakraniaaliseen etenemiseen ei saavutettu kritsotinibihaarassa (n = 132) eikä solunsalpaajahaarassa (n = 131) (HR = 0,69; 95 %:n luottamusväli: 0,33, 1,45; yksitahoinen p-arvo = 0,1617).
Potilaiden raportoimien oireiden ja yleistä elämänlaatua koskevien tietojen keräämiseen käytettiin EORTC QLQ-C30 -kyselylomaketta ja sen keuhkosyöpää koskevaa osiota EORTC QLQ-LC13. Yhteensä 166 kritsotinibihaaran ja 163 solunsalpaajahaaran potilaista vastasi EORTC QLQ-C30- ja LC13 -kyselylomakkeisiin lähtötilanteessa ja ainakin yhdellä vastaanottokäynnillä tutkimuksen aloituksen jälkeen. Kritsotinibihaarassa yleinen elämänlaatu parani merkitsevästi verrattuna solunsalpaajahaaraan (kokonaisero muutokselle lähtötilanteen pisteistä 13,8; p-arvo < 0,0001).
Aika oireiden pahenemiseen (Time to Deterioration, TTD) oli ennalta määritelty ajankohdaksi, jolloin oireiden muutosta kuvaavat pisteet ensimmäisen kerran nousisivat lähtötilanteesta ≥ 10 pisteellä rintakivun, yskän tai hengenahdistuksen osalta EORTC QLQ-LC13 -kyselylomakkeella arvioituna.
Solunsalpaajahoitoon verrattuna kritsotinibi johti hyötyihin oireiden hallinnassa pidentämällä merkitsevästi aikaa oireiden pahenemiseen (mediaani 2,1 kk vs 0,5 kk; HR = 0,59; 95 %:n luottamusväli: 0,45, 0,77; Hochbergin vakioitu kaksitahoinen log-rank-testi, p-arvo = 0,0005).
Aiemmin hoidettu edennyt ALK-positiivinen ei-pienisoluinen keuhkosyöpä (NSCLC) − satunnaistettu vaiheen 3 tutkimus 1007
Kritsotinibin teho ja turvallisuus metastasoituneen ALK-positiivisen NSCLC:n hoidossa potilailla, jotka olivat saaneet aikaisempaa systeemistä hoitoa taudin edettyä, on osoitettu monikansallisessa, satunnaistetussa ja avoimessa tutkimuksessa 1007.
Koko analyysipopulaatio käsitti 347 potilasta, joilla oli edennyt ALK-positiivinen NSCLC. ALK-positiivisuus oli todettu FISH-menetelmällä ennen satunnaistamista. 173 potilasta satunnaistettiin saamaan kritsotinibia ja 174 potilasta satunnaistettiin saamaan solunsalpaajaa (pemetreksedi tai dosetakseli). Koko tutkimuspopulaation demografisten ja sairauteen liittyvien tietojen mukaan 56 % potilaista oli naisia, mediaani ikä oli 50 vuotta, lähtötilanteen ECOG-suorituskykyluokka oli 39 %:lla potilaista 0 ja 52 %:lla potilaista 1, 52 % oli valkoihoisia ja 45 % aasialaisia, 4 % tupakoi edelleen, 33 % oli aiemmin tupakoinut ja 63 % ei ollut koskaan tupakoinut, kasvain oli metastasoitunut 93 %:lla potilaista ja 93 % syövistä luokiteltiin histologialtaan adenokarsinoomiksi.
Potilaiden saamaa tutkimushoitoa voitiin jatkaa vielä RECIST-kriteerien mukaisen taudin etenemisen jälkeen, jos tutkija katsoi potilaan saavan hoidosta kliinistä hyötyä. Kritsotinibia saaneista 84 potilaasta 58 potilasta (69 %) ja solunsalpaajahoitoa saaneista 119 potilaasta 17 potilasta (14 %) jatkoi hoitoa vähintään kolmen viikon ajan objektiivisen taudin etenemisen jälkeen. Solunsalpaajahoitohaaran potilaiden oli mahdollista siirtyä saamaan kritsotinibia (cross-over), kun riippumaton radiologinen arviointi oli vahvistanut RECIST-kriteerien mukaisen taudin etenemisen.
Riippumattoman radiologisen arvioinnin mukaan kritsotinibi pidensi merkitsevästi taudin etenemisestä vapaata elinaikaa (PFS) solunsalpaajahoitoon verrattuna, mikä oli tutkimuksen ensisijainen päätetapahtuma. Kritsotinibilla saavutettu PFS-hyöty oli johdonmukainen kaikissa lähtötilanteen tietoihin perustuvissa potilaiden alaryhmissä, mukaan lukien ikä, sukupuoli, etninen tausta, tupakointitausta, diagnoosista tutkimushoidon aloittamiseen kulunut aika, ECOG-suorituskykyluokka, aivometastaasien esiintyminen ja aiempi EGFR-tyrosiinikinaasin estäjän käyttö.
Tutkimuksen 1007 tehoa koskevien tietojen yhteenveto on esitetty taulukossa 12 ja taudin etenemisestä vapaan elinajan todennäköisyytta kuvaavat Kaplan-Meierin käyrät kuvassa 3. Kokonaiselinajan todennäköisyytta kuvaavat Kaplan-Meierin käyrät on esitetty kuvassa 4.
Taulukko 12. Tehoa koskevat tulokset satunnaistetusta vaiheen 3 tutkimuksesta 1007 (koko analyysipopulaatio) edennyttä ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla, jotka olivat saaneet aikaisempaa hoitoa edenneeseen tautiin*
| Vastemuuttuja | Kritsotinibi n = 173 | Solunsalpaaja n = 174 |
| Taudin etenemisestä vapaa elinaika (riippumattoman radiologisen arvioinnin perusteella) | ||
| Tapahtumien lukumäärä, n (%) | 100 (58 %) | 127 (73 %) |
| Tapahtuma, n (%) | ||
| Taudin eteneminen | 84 (49 %) | 119 (68 %) |
| Kuolema ilman objektiivista taudin etenemistä | 16 (9 %) | 8 (5 %) |
| Taudin etenemisestä vapaan elinajan mediaani, kk (95 %:n luottamusväli) | 7,7 (6,0, 8,8) | 3,0a (2,6, 4,3) |
| HR (95 %:n luottamusväli)b | 0,49 (0,37, 0,64) | |
| p-arvoc | < 0,0001 | |
| Kokonaiselinaikad | ||
| Kuolemien lukumäärä, n (%) | 116 (67 %) | 126 (72 %) |
| Kokonaiselinajan mediaani, kk (95 %:n luottamusväli) | 21,7 (18,9, 30,5) | 21,9 (16,8, 26,0) |
| HR (95 %:n luottamusväli)b | 0,85 (0,66, 1,10) | |
| p-arvoc | 0,1145 | |
| 6 kuukauden eloonjäämisen todennäköisyyse, % (95 %:n luottamusväli) | 86,6 (80,5, 90,9) | 83,8 (77,4, 88,5) |
| 1 vuoden eloonjäämisen todennäköisyyse, % (95 %:n luottamusväli) | 70,4 (62,9, 76,7) | 66,7 (59,1, 73,2) |
| Objektiivisen vasteen saaneiden osuus (riippumattoman radiologisen arvioinnin perusteella) | ||
| Objektiivisen vasteen saaneiden osuus, % (95 %:n luottamusväli) | 65 % (58, 72) | 20 %f (14, 26) |
| p-arvog | < 0,0001 | |
| Vasteen kesto | ||
| Mediaanie, kk (95 %:n luottamusväli) | 7,4 (6,1, 9,7) | 5,6 (3,4, 8,3) |
Lyhenteet: HR (Hazard Ratio) = riskitiheyksien suhde; n = potilaiden lukumäärä
* Taudin etenemisestä vapaa elinaika, objektiivisen vasteen saaneiden osuus ja vasteen kesto perustuvat tiedonkeruun päättymispäivään 30. maaliskuuta 2012. Kokonaiselinaika perustuu tiedonkeruun päättymispäivään 31. elokuuta 2015.
a. Taudin etenemisestä vapaan elinajan mediaani oli 4,2 kuukautta (95 %:n luottamusväli: 2,8, 5,7) käytettäessä pemetreksediä (HR = 0,59; p-arvo = 0,0004 kritsotinibille verrattuna pemetreksediin) ja 2,6 kuukautta (95 %:n luottamusväli: 1,6, 4,0) käytettäessä dosetakselia (HR = 0,30; p-arvo < 0,0001 kritsotinibille verrattuna dosetakseliin).
b. Perustuu Coxin suhteellisten riskitiheyksien mallin ositettuun analyysiin.
c. Perustuu ositettuun log-rank-testiin (yksitahoiseen).
d. Päivitetty kokonaiselinajan lopullisen analyysin mukaisesti. Kokonaiselinajan lopullista analyysia ei vakioitu
cross-overista mahdollisesti aiheutuvien sekoittavien tekijöiden varalta (154 potilasta (89 %) siirtyi
kritsotinibihoitoon).
e. Arvioitu Kaplan-Meierin menetelmällä.
f. Objektiivisen vasteen saaneiden potilaiden osuus oli 29 % (95 %:n luottamusväli: 21, 39) käytettäessä pemetreksediä (p-arvo < 0,0001 verrattuna kritsotinibiin) ja 7 % (95 %:n luottamusväli: 2, 16) käytettäessä dosetakselia (p-arvo < 0,0001 verrattuna kritsotinibiin).
g. Perustuu ositettuun Cochran-Mantel-Haenszelin testiin (kaksitahoiseen).
Kuva 3. Kaplan-Meierin käyrät taudin etenemisestä vapaalle elinajalle (riippumattoman radiologisen arvion mukaan) tutkimushaaroittain satunnaistetussa vaiheen 3 tutkimuksessa 1007 (koko analyysipopulaatio) edennyttä ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla, jotka olivat saaneet aikaisempaa hoitoa edenneeseen tautiin
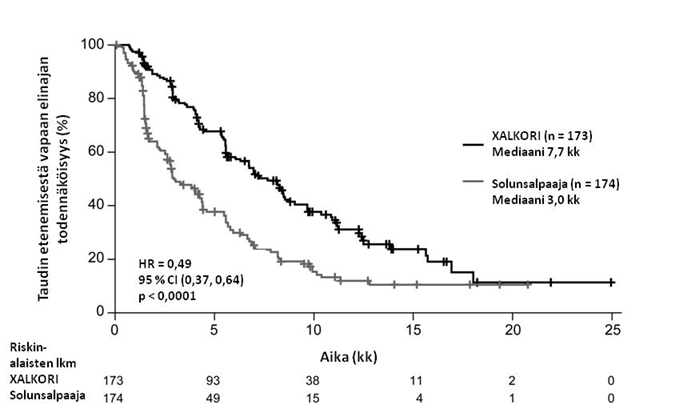
Lyhenteet: HR = riskitiheyksien suhde; CI = luottamusväli; p = p-arvo; n = potilaiden lukumäärä
Kuva 4. Kaplan-Meierin käyrät kokonaiselinajalle tutkimushaaroittain satunnaistetussa vaiheen 3 tutkimuksessa 1007 (koko analyysipopulaatio) edennyttä ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla, jotka olivat saaneet aikaisempaa hoitoa edenneeseen tautiin
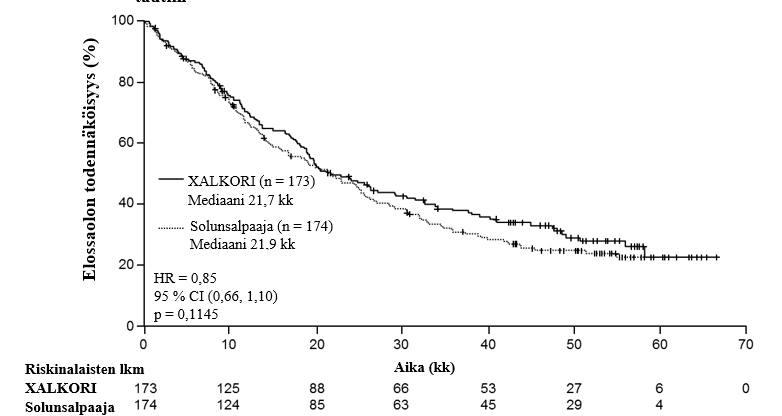
Lyhenteet: n = potilaiden lukumäärä; HR = riskitiheyksien suhde; CI = luottamusväli; p = p-arvo
Satunnaistetussa vaiheen 3 tutkimuksessa 1007 kritsotinibihoitoa sai 52 potilasta ja solunsalpaajahoitoa 57 potilasta, joilla oli aiemmin hoidettuja tai hoitamattomia oireettomia aivometastaaseja. Viikolla 12 tauti oli intrakraniaalisesti hallinnassa (IC-DCR) 65 %:lla kritsotinibia ja 46 %:lla solunsalpaajaa saaneista potilaista.
Potilaiden raportoimien oireiden ja yleistä elämänlaatua koskevien tietojen keräämiseen käytettiin EORTC QLQ-C30 -kyselylomaketta ja sen keuhkosyöpää koskevaa osiota EORTC QLQ-LC13. Tiedot kerättiin lähtötilanteessa (syklin 1 päivänä 1) ja jokaisen seuraavan hoitosyklin päivänä 1. Yhteensä 162 kritsotinibihaaran ja 151 solunsalpaajahaaran potilasta vastasi EORTC QLQ-C30‑ ja LC-13 -kyselylomakkeisiin lähtötilanteessa ja vähintään yhdellä vastaanottokäynnillä tutkimuksen aloituksen jälkeen.
Solunsalpaajahoitoon verrattuna kritsotinibi johti hyötyihin oireiden hallinnassa pidentämällä merkitsevästi aikaa potilaiden raportoimien oireiden (rintakivun, hengenahdistuksen ja yskän) pahenemiseen (mediaani 4,5 kk vs 1,4 kk; HR = 0,50; 95 %:n luottamusväli: 0,37, 0,66; Hochbergin vakioitu kaksitahoinen log-rank-testi, p-arvo < 0,0001).
Kritsotinibia käytettäessä seuraavat lähtötilanteen oireet paranivat merkitsevästi enemmän kuin solunsalpaajaa käytettäessä: hiustenlähtö (syklit 2−15; p-arvo < 0,05), yskä (syklit 2−20; p-arvo < 0,0001), hengenahdistus (syklit 2−20; p-arvo < 0,0001), veriyskä (syklit 2−20; p-arvo < 0,05), yläraajojen tai hartioiden kipu (syklit 2−20; p-arvo < 0,0001), rintakipu (syklit 2−20; p-arvo < 0,0001) ja muiden kehon osien kipu (syklit 2−20; p-arvo < 0,05). Kritsotinibia käytettäessä seuraavat lähtötilanteen oireet pahenivat merkitsevästi vähemmän kuin solunsalpaajahoitoa käytettäessä: raajojen neuropatia (syklit 6−20; p-arvo < 0,05), nielemisvaikeudet (syklit 5−11; p-arvo < 0,05) ja suun arkuus (syklit 2−20; p-arvo < 0,05).
Kaiken kaikkiaan kritsotinibia käytettäessä lähtötilanteen yleinen elämänlaatu parani merkitsevästi enemmän kuin solunsalpaajaa käytettäessä (syklit 2−20; p-arvo < 0,05).
Edenneen ALK-positiivisen ei-pienisoluisen keuhkosyövän (NSCLC) yksihaaraiset tutkimukset
Kritsotinibin käyttöä ainoana lääkkeenä edenneen ALK-positiivisen NSCLC:n hoidossa tutkittiin kahdessa monikansallisessa, yksihaaraisessa tutkimuksessa (tutkimukset 1001 ja 1005). Seuraavassa kuvataan näihin tutkimuksiin mukaan otetuista potilaista ne, jotka olivat aiemmin saaneet systeemistä hoitoa paikallisesti edenneen tai metastasoituneen taudin hoitoon. Kummankin tutkimuksen ensisijainen tehon päätetapahtuma oli RECIST-kriteereihin perustuva objektiivisen vasteen saaneiden potilaiden osuus (ORR).
Tutkimukseen 1001 oli PFS:n ja ORR:n analysointiajankohtana otettu mukaan yhteensä 149 edennyttä ALK-positiivista NSCLC:ää sairastavaa potilasta, joista 125 oli saanut aiempaa hoitoa edenneeseen NSCLC:ään. Demografisten ja sairauteen liittyvien tietojen mukaan 50 % potilaista oli naisia, mediaani ikä oli 51 vuotta, lähtötilanteen ECOG-suorituskykyluokka oli 32 %:lla potilaista 0 ja 55 %:lla potilaista 1, 61 % oli valkoihoisia ja 30 % aasialaisia, alle 1 % tupakoi edelleen, 27 % oli aiemmin tupakoinut ja 72 % ei ollut koskaan tupakoinut, kasvain oli metastasoitunut 94 %:lla potilaista ja 98 % syövistä luokiteltiin histologialtaan adenokarsinoomiksi. Hoidon keston mediaani oli 42 viikkoa.
Tutkimuksen 1005 PFS:n ja ORR:n analysointiajankohtana yhteensä 934 edennyttä ALK-positiivista NSCLC:ää sairastavaa potilasta oli saanut kritsotinibihoitoa. Demografisten ja sairauteen liittyvien tietojen mukaan 57 % potilaista oli naisia, mediaani ikä oli 53 vuotta, lähtötilanteen ECOG-suorituskykyluokka oli 82 %:lla potilaista 0 tai 1 ja 18 %:lla potilaista 2 tai 3, 52 % oli valkoihoisia ja 44 % aasialaisia, 4 % tupakoi edelleen, 30 % oli aiemmin tupakoinut ja 66 % ei ollut koskaan tupakoinut, kasvain oli metastasoitunut 92 %:lla potilaista ja 94 % syövistä luokiteltiin histologialtaan adenokarsinoomiksi. Hoidon keston mediaani oli 23 viikkoa. Potilaiden saamaa tutkimushoitoa voitiin jatkaa vielä RECIST-kriteerien mukaisen taudin etenemisen jälkeen tutkijan harkinnan mukaan. 77 potilasta 106 potilaasta (73 %) jatkoi kritsotinibihoitoa vähintään kolmen viikon ajan objektiivisen taudin etenemisen jälkeen.
Tutkimusten 1001 ja 1005 tehoa koskevat tiedot on esitetty taulukossa 13.
Taulukko 13. Tehoa koskevat tulokset tutkimuksista 1001 ja 1005 edennyttä ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla
| Tehon muuttuja | Tutkimus 1001 | Tutkimus 1005 |
| n = 125a | n = 765a | |
| Objektiivisen vasteen saaneiden osuusb [% (95 %:n luottamusväli)] | 60 (51, 69) | 48 (44, 51) |
| Aika kasvaimessa todettuun vasteeseen [mediaani (vaihteluväli)], vko | 7,9 (2,1, 39,6) | 6,1 (3, 49) |
| Vasteen kestoc [mediaani (95 %:n luottamusväli)], vko | 48,1 (35,7, 64,1) | 47,3 (36, 54) |
| Taudin etenemisestä vapaa elinaikac [mediaani (95 %:n luottamusväli)], kk | 9,2 (7,3, 12,7) | 7,8 (6,9, 9,5)d |
| n = 154e | n = 905e | |
| Kuolemien lukumäärä, n (%) | 83 (54 %) | 504 (56 %) |
| Kokonaiselinaikac [mediaani (95 %:n luottamusväli)], kk | 28,9 (21,1, 40,1) | 21,5 (19,3, 23,6) |
Lyhenne: n = potilaiden lukumäärä
a. Tiedonkeruu katkaistu 1. kesäkuuta 2011 (tutkimus 1001) ja 15. helmikuuta 2012 (tutkimus 1005).
b. 1001-tutkimuksessa 3 potilaan ja 1005-tutkimuksessa 42 potilaan vaste ei ollut arvioitavissa.
c. Arvioitu Kaplan-Meierin menetelmällä.
d. Tutkimuksen 1005 taudin etenemisestä vapaata elinaikaa koskevissa tiedoissa oli mukana 807 turvallisuusanalyysissä mukana ollutta potilasta, joiden kasvaimen ALK-positiivisuus oli todettu FISH-määrityksellä (tiedonkeruu katkaistu 15. helmikuuta 2012).
f. Tiedonkeruu katkaistu 30. marraskuuta 2013.
Edennyt ROS1-positiivinen ei-pienisoluinen keuhkosyöpä (NSCLC)
Kritsotinibin käyttöä ainoana lääkkeenä edenneen ROS1-positiivisen NSCLC:n hoidossa tutkittiin monikansallisessa, yksihaaraisessa monikeskustutkimuksessa 1001. Tutkimukseen oli tulosten analysointiajankohtana otettu mukaan yhteensä 53 edennyttä ROS1-positiivista NSCLC:ää sairastavaa potilasta, joista 46 oli saanut aiempaa hoitoa edenneeseen ROS1-positiiviseen NSCLC:ään. Pieni joukko potilaita (n = 7) ei ollut aiemmin saanut systeemistä hoitoa. Tutkimuksen ensisijainen tehon päätetapahtuma oli RECIST-kriteereihin perustuva objektiivisen vasteen saaneiden potilaiden osuus (ORR). Toissijaisia päätetapahtumia olivat aika kasvaimessa todettuun vasteeseen (TTR), vasteen kesto (DoR), taudin etenemisestä vapaa elinaika (PFS) ja kokonaiselinaika (OS). Potilaat saivat 250 mg kritsotinibia suun kautta kaksi kertaa vuorokaudessa.
Demografisten tietojen mukaan 57 % potilaista oli naisia, mediaani ikä oli 55 vuotta, lähtötilanteen ECOG-suorituskykyluokka oli 98 %:lla potilaista 0 tai 1 ja 2 %:lla potilaista 2, 57 % oli valkoihoisia ja 40 % aasialaisia, 25 % oli aiemmin tupakoinut ja 75 % ei ollut koskaan tupakoinut. Sairauteen liittyvien tietojen mukaan kasvain oli metastasoitunut 94 %:lla potilaista, 96 % syövistä luokiteltiin histologisesti adenokarsinoomiksi ja 13 %:ssa tapauksista metastasoitunutta syöpää ei ollut aiemmin hoidettu systeemisellä lääkehoidolla.
Tutkimuksessa 1001 potilailla edellytettiin olevan edennyt ROS1-positiivinen NSCLC ennen kuin heidät otettiin mukaan kliiniseen tutkimukseen. Useimpien potilaiden ROS1-positiivinen NSCLC todettiin FISH-menetelmällä. Hoidon keston mediaani oli 22,4 kuukautta (95 %:n luottamusväli: 15,0, 35,9). Täydellinen vaste todettiin 6 potilaalla ja osittainen vaste 32 potilaalla, joten objektiivisen vasteen saaneiden potilaiden osuus (ORR) oli 72 % (95 %:n luottamusväli: 58 %, 83 %). Vasteen keston (DoR) mediaani oli 24,7 kuukautta (95 %:n luottamusväli: 15,2, 45,3). Objektiivisista kasvaimissa todetuista vasteista 50 % saavutettiin ensimmäisten 8 hoitoviikon aikana. Taudin etenemisestä vapaan elinajan (PFS) mediaani tulosten analysointiajankohtana oli 19,3 kuukautta (95 %:n luottamusväli: 15,2, 39,1). Kokonaiselinajan mediaani oli tulosten analysointiajankohtana 51,4 kuukautta (95 %:n luottamusväli: 29,3, ei saavutettu).
Tehoa koskevat tulokset tutkimuksesta 1001 edennyttä ROS1-positiivista NSCLC:ää sairastavilla potilailla on esitetty taulukossa 14.
Taulukko 14. Tehoa koskevat tulokset tutkimuksesta 1001 edennyttä ROS1-positiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla
| Tehon muuttuja | Tutkimus 1001 n = 53a |
| Objektiivisen vasteen saaneiden potilaiden osuus (ORR) [% (95 %:n luottamusväli)] | 72 (58, 83) |
| Aika kasvaimessa todettuun vasteeseen (TTR) [mediaani (vaihteluväli)] vko | 8 (4, 104) |
| Vasteen kesto (DR)b [mediaani (95 %:n luottamusväli)] kk | 24,7 (15,2, 45,3) |
| Taudin etenemisestä vapaa elinaika (PFS)b [mediaani (95 %:n luottamusväli)] kk | 19,3 (15,2, 39,1) |
| Kokonaiselinaika (OS)b [mediaani (95 %:n luottamusväli)] kk | 51,4 (29,3, NR) |
Lyhenteet: n = potilaiden lukumäärä; NR (not reached) = ei saavutettu OS perustuu seuranta-aikaan, jonka mediaani oli noin 63 kuukautta. a. Tiedonkeruu katkaistu 30. kesäkuuta 2018. b. Arvioitu Kaplan-Meierin menetelmällä. | |
Histologia muu kuin adenokarsinooma
Satunnaistettuun vaiheen 3 tutkimukseen 1014 otettiin mukaan 21 potilasta, joiden aiemmin hoitamaton edennyt ALK-positiivinen NSCLC oli histologialtaan muu kuin adenokarsinooma. Satunnaistettuun vaiheen 3 tutkimukseen 1007 otettiin mukaan 12 potilasta, joiden aiemmin hoidettu edennyt ALK-positiivinen NSCLC oli histologialtaan muu kuin adenokarsinooma. Näiden tutkimusten alaryhmät olivat liian pieniä luotettavien johtopäätösten tekemiseen. On huomattava, että tutkimuksen 1007 kritsotinibihaaraan ei satunnaistettu potilaita, joiden syöpä oli histolgialtaan levyepiteelikarsinooma, eikä näitä potilaita otettu mukaan myöskään tutkimukseen 1014, koska vertailuna käytettiin pemetreksedipohjaista hoitoa.
Tutkimuksesta 1005 on saatavissa tietoja vain 45 potilaasta, joiden aiemmin hoidettu NSCLC oli histologialtaan muu kuin adenokarsinooma (mukaan lukien 22 potilaista, joiden syöpä oli histologialtaan levyepiteelikarsioooma) ja joiden vastetta voitiin arvioida. Osittainen vaste todettiin 20 potilaalla 45 potilaasta, joiden NSCLC oli histologialtaan muu kuin adenokarsinooma (objektiivisen vasteen saaneiden potilaiden osuus 44 %), ja 9 potilaalla 22 potilaasta, joiden NSCLC oli histologialtaan levyepiteelikarsinooma (objektiivisen vasteen saaneiden potilaiden osuus 41 %). Näissä potilasryhmissä objektiivisen vasteen saaneiden potilaiden osuus oli pienempi kuin tutkimuksen 1005 koko potilasjoukossa raportoitu (54 %).
Uusintahoito kritsotinibilla
Turvallisuuteen ja tehoon liittyviä tietoja ei ole saatavilla kritsotinibin käytöstä uudelleen potilaille, jotka ovat saaneet kritsotinibia edeltävissä hoitolinjoissa.
Iäkkäät
Satunnaistetun vaiheen 3 tutkimuksen 1014 kritsotinibilla hoidetuista ALK-positiivista NSCLC:ää sairastavasta 171 potilaasta 22 potilasta (13 %) oli vähintään 65 vuoden ikäisiä. Solunsalpaajahaarasta kritsotinibihoitoon siirtyneistä (cross-over) 109 ALK-positiivisesta potilaasta 26 potilasta (24 %) oli vähintään 65 vuoden ikäisiä. Satunnaistetun vaiheen 3 tutkimuksen 1007 kritsotinibilla hoidetuista 172 ALK-positiivisesta potilaasta 27 potilasta (16 %) oli vähintään 65 vuoden ikäisiä. Yksihaaraisen 1001-tutkimuksen ALK-positiivista NSCLC:ää sairastavasta 154 potilaasta 22 potilasta (14 %) ja yksihaaraisen 1005-tutkimuksen ALK-positiivista NSCLC:ää sairastavasta 1063 potilaasta 173 potilasta (16 %) oli vähintään 65 vuoden ikäisiä. Haittavaikutusten esiintymistiheydet olivat yleisesti samanlaisia ALK-positiivista NSCLC:ää sairastavilla alle 65-vuotiailla potilailla verrattuna 65-vuotiaisiin ja sitä vanhempiin potilaisiin. Poikkeuksena olivat turvotus ja ummetus, joita raportoitiin tutkimuksessa 1014 yleisemmin (≥ 15 % ero) kritsotinibilla hoidetuilla vähintään 65 vuoden ikäisillä potilailla. Satunnaistettujen vaiheen 3 tutkimusten 1007 ja 1014 sekä yksihaaraisen tutkimuksen 1005 kritsotinibihoitohaaroissa ei ollut mukana yhtään 85-vuotiasta tai sitä vanhempaa potilasta. Yksihaaraisen 1001-tutkimuksen 154 ALK-positiivisesta potilaasta yksi oli yli 85-vuotias (ks. myös kohdat Annostus ja antotapa ja Farmakokinetiikka). Yksihaaraisen 1001-tutkimuksen ROS1-positiivista NSCLC:ää sairastavasta 53 potilaasta 15 potilasta (28 %) oli 65-vuotiaita tai vanhempia. Tutkimuksessa 1001 ei ollut mukana yhtään yli 85-vuotiasta ROS1-positiivista potilasta.
Pediatriset potilaat
Kritsotinibin turvallisuus ja teho on varmistettu 3 – < 18‑vuotiaiden pediatristen potilaiden hoidossa, joilla on uusiutunut tai refraktorinen systeeminen ALK-positiivinen anaplastinen suurisoluinen lymfooma (ALCL), tai 2 – < 18‑vuotiaiden pediatristen potilaiden hoidossa, joilla on leikkaukseen soveltumaton, uusiutunut tai refraktorinen ALK-positiivinen tulehduksellinen myofibroblastituumori (IMT). Kritsotinibihoidon turvallisuutta tai tehoa koskevia tietoja ei ole saatavissa alle 3‑vuotiaista pediatrisista potilaista, joilla on ALK-positiivinen ALCL, tai alle 2‑vuotiaista pediatrista potilaista, joilla on ALK-positiivinen IMT.
Pediatriset potilaat, joilla on ALK-positiivinen ALCL (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka)
Kritsotinibin käyttöä ainoana lääkkeenä hoidettaessa pediatrisia potilaita, joilla on uusiutunut tai refraktorinen systeeminen ALK‑positiivinen ALCL, selvitettiin tutkimuksessa 0912 (n = 22). Kaikki tutkimukseen osallistuneet potilaat olivat saaneet sairauteensa aiempaa systeemistä hoitoa: 14 oli saanut aiemmin 1 hoitolinjan systeemistä hoitoa, 6 oli saanut aiemmin 2 hoitolinjaa systeemistä hoitoa ja 2 oli saanut aiemmin enemmän kuin 2 hoitolinjaa systeemistä hoitoa. Tutkimukseen 0912 osallistuneista 22 potilaasta 2 oli saanut aiemmin luuydinsiirteen. Tällä hetkellä ei ole saatavissa kliinisiä tietoja pediatrisista potilaista, jotka saavat kritsotinibihoidon jälkeen hematopoieettisen kantasolusiirteen. Tutkimukseen ei otettu mukaan potilaita, joilla on primaarisia tai metastasoituneita keskushermoston kasvaimia. Tutkimukseen 0912 osallistuneet 22 potilasta saivat kritsotinibia seuraavilla aloitusannostuksilla: 280 mg/m2 (16 potilasta) tai 165 mg/m2 (6 potilasta) kaksi kertaa vuorokaudessa. Tutkimuksen 0912 tehon päätetapahtumia olivat objektiivisen vasteen saaneiden osuus (ORR), aika kasvaimessa todettuun vasteeseen (TTR) ja vasteen kesto (DoR), ja ne määritettiin riippumattoman arvioinnin perusteella. Seurannan keston mediaani oli 5,5 kuukautta.
Demografisten tietojen mukaan 23 % oli naisia, mediaani-ikä oli 11 vuotta, 50 % oli valkoihoisia ja 9 % aasialaisia. Lähtötilanteessa suorituskykyä mittaavat Lansky Play Score ‑pisteet (≤ 16‑vuotiaille potilaille) tai Karnofsky Performance Score ‑pisteet (> 16‑vuotiaille potilaille) olivat 100 (50 % potilaista) tai 90 (27 % potilaista). Tutkimukseen osallistuneiden potilaiden ikäjakauma oli seuraava: 4 iältään 3 – < 6‑vuotiasta potilasta, 11 iältään 6 – < 12‑vuotiasta potilasta ja 7 iältään 12 – < 18‑vuotiasta potilasta. Tutkimukseen ei otettu mukaan alle 3‑vuotiaita potilaita.
Taulukossa 15 on esitetty riippumattomaan arviointiin perustuvat tehoa koskevat tiedot.
Taulukko 15. Tehoa koskevat tulokset tutkimuksesta 0912 systeemistä ALK‑positiivista
ALCL:ää sairastavilla potilailla
| Tehon muuttujaa | n = 22b |
Objektiivisen vasteen saaneiden osuus [% (95 %:n luottamusväli)]c Täydellinen vaste, n (%) Osittainen vaste, n (%) | 86 (67, 95) 17 (77) 2 (9) |
Aika kasvaimessa todettuun vasteeseend Mediaani (vaihteluväli), kk | 0,9 (0,8, 2,1) |
Vasteen kestod,e Mediaani (vaihteluväli), kk | 3,6 (0,0, 15,0) |
Lyhenne: n = potilaiden lukumäärä a. Riippumattoman arviointikomitean arvio perustuu vastetta kuvaaviin Lugano Classification ‑kriteereihin. b. Tiedonkeruu katkaistu 19. tammikuuta 2018. c. 95 %:n luottamusväli, perustuu Wilsonin pistemenetelmään. d. Arvioitu käyttämällä kuvailevia tilastomenetelmiä. e. 10 potilasta 19:stä (53 %) sai hematopoieettisen kantasolusiirteen objektiivisen vasteen ilmenemisen jälkeen. Siirteen saaneiden potilaiden vasteen kestoa koskevat tiedot jätettiin pois tutkimustuloksista, kun kasvainta arvioitiin viimeisen kerran ennen siirteen saamista. | |
Pediatriset potilaat, joilla on ALK-positiivinen IMT (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka)
Kritsotinibin käyttöä ainoana lääkkeenä hoidettaessa pediatrisia potilaita, joilla on leikkaukseen soveltumaton, uusiutunut tai refraktorinen ALK‑positiivinen IMT, selvitettiin tutkimuksessa 0912 (n = 14). Suurin osa tutkimukseen osallistuneista potilaista (12 potilasta 14:stä) oli saanut sairauteensa leikkaushoitoa (8 potilasta) tai aiempaa systeemistä hoitoa (7 potilasta: 5 oli saanut aiemmin 1 hoitolinjan systeemistä hoitoa, 1 oli saanut aiemmin 2 hoitolinjaa systeemistä hoitoa ja 1 oli saanut aiemmin enemmän kuin 2 hoitolinjaa systeemistä hoitoa). Tutkimukseen ei otettu mukaan potilaita, joilla on primaarisia tai metastasoituneita keskushermoston kasvaimia. Tutkimukseen 0912 osallistuneet 14 potilasta saivat kritsotinibia seuraavilla aloitusannostuksilla: 280 mg/m2 (12 potilasta) tai 165 mg/m2 (1 potilas) tai 100 mg/m2 (1 potilas) kaksi kertaa vuorokaudessa. Tutkimuksen 0912 tehon päätetapahtumia olivat objektiivisen vasteen saaneiden osuus (ORR), aika kasvaimessa todettuun vasteeseen (TTR) ja vasteen kesto (DoR), ja ne määritettiin riippumattoman arvioinnin perusteella. Seurannan keston mediaani oli 17,6 kuukautta.
Demografisten tietojen mukaan 64 % oli naisia, mediaani-ikä oli 6,5 vuotta ja 71 % oli valkoihoisia. Lähtötilanteessa suorituskykyä mittaavat Lansky Play Score ‑pisteet (≤ 16‑vuotiaille potilaille) tai Karnofsky Performance Score ‑pisteet (> 16‑vuotiaille potilaille) olivat 100 (71 % potilaista), 90 (14 % potilaista) tai 80 (14 % potilaista). Tutkimukseen osallistuneiden potilaiden ikäjakauma oli seuraava: 4 iältään 2 – < 6‑vuotiasta potilasta, 8 iältään 6 – < 12‑vuotiasta potilasta ja 2 iältään 12 – < 18‑vuotiasta potilasta. Tutkimukseen ei otettu alle 2‑vuotiaita potilaita.
Taulukossa 16 on esitetty riippumattomaan arviointiin perustuvat tehoa koskevat tiedot.
Taulukko 16. Tehoa koskevat tulokset tutkimuksesta 0912 ALK-positiivista IMT:tä
sairastavilla potilailla
| Tehon muuttujaa | n = 14b |
Objektiivisen vasteen saaneiden osuus [% (95 %:n luottamusväli)]c Täydellinen vaste, n (%) Osittainen vaste, n (%) | 86 (60, 96) 5 (36) 7 (50) |
Aika kasvaimessa todettuun vasteeseend Mediaani (vaihteluväli), kk | 1,0 (0,8, 4,6) |
Vasteen kestod,e Mediaani (vaihteluväli), kk | 14,8 (2,8, 48,9) |
Lyhenne: n = potilaiden lukumäärä a. Riippumattoman arviointikomitean arvio. b. Tiedonkeruu katkaistu 19. tammikuuta 2018. c. 95 %:n luottamusväli, perustuu Wilsonin pistemenetelmään. d. Arvioitu käyttämällä kuvailevia tilastomenetelmiä. e. Yhdelläkään 12 potilaasta, joiden kasvaimessa todettiin objektiivinen vaste, sairaus ei edennyt seurannan aikana, ja heitä koskevat tiedot vasteen kestosta poistettiin, kun kasvainta arvioitiin viimeisen kerran. | |
Pediatriset potilaat, joilla on ALK‑positiivinen tai ROS1‑positiivinen NSCLC
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset XALKORIn käytöstä ei-pienisoluisen keuhkosyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Kritsotinibin farmakokineettisiä ominaisuuksia on selvitetty aikuisilla, ellei pediatrisista potilaista ole erityisesti muuta mainittu.
Imeytyminen
XALKORI 200 mg ja 250 mg kovat kapselit
Paastotilassa suun kautta otetun kritsotinibin kerta-annoksen jälkeen mediaaniaika plasman huippupitoisuuden saavuttamiseen on 4–6 tuntia. Vakaa tila (steady-state) saavutettiin kaksi kertaa vuorokaudessa tapahtuneessa annostelussa 15 vuorokauden kuluessa. Kritsotinibin absoluuttiseksi hyötyosuudeksi määritettiin 43 % suun kautta otetun 250 mg:n kerta-annoksen jälkeen.
Runsasrasvainen ateria vähensi kritsotinibin AUCinf- ja Cmax-arvoja noin 14 %, kun terveille vapaaehtoisille koehenkilöille annettiin 250 mg:n kerta-annos. Kritsotinibi voidaan ottaa aterian yhteydessä tai tyhjään mahaan (ks. kohta Annostus ja antotapa).
XALKORI- rakeet avattavissa kapseleissa
Paastotilassa suun kautta kerta-annoksena otetut kritsotinibirakeet avattavissa kapseleissa ovat bioekvivalentteja kritsotinibikapselien kanssa.
Suun kautta otettavien kritsotinibirakeiden avattavissa kapseleissa antaminen runsasrasvaisen/runsaskalorisen aterian yhteydessä pienensi kritsotinibin kokonaisaltistusta (AUCinf) noin 15 % ja huippupitoisuutta (Cmax) noin 23 % verrattuna saman valmistemuodon ottamiseen paastotilassa. Kritsotinibirakeet avattavissa kapseleissa voidaan ottaa aterian yhteydessä tai ilman ateriaa (ks. kohta Annostus ja antotapa).
Jakautuminen
Laskimoon annetun 50 mg:n annoksen jälkeen kritsotinibin jakautumistilavuuden (VSS) geometrinen keskiarvo oli 1772 litraa, mikä viittaa laajaan jakautumiseen plasmasta kudoksiin.
Kritsotinibi sitoutuu 91-prosenttisesti ihmisen plasman proteiineihin in vitro riippumatta lääkevalmisteen pitoisuudesta. In vitro ‑tutkimukset viittaavat siihen, että kritsotinibi on P-glykoproteiinin (P-gp) substraatti.
Biotransformaatio
In vitro ‑tutkimukset osoittivat, että CYP3A4/5 olivat kritsotinibin metaboliseen puhdistumaan osallistuvat pääasialliset entsyymit. Pääasiallinen metaboliareitti ihmisellä oli piperidiinirenkaan oksidaatio kritsotinibilaktaamiksi ja O-dealkylaatio, jonka jälkeen tapahtuu O-dealkyloituneiden metaboliittien vaiheen 2 konjugaatio.
In vitro ‑tutkimukset ihmisen maksan mikrosomeilla osoittivat, että kritsotinibi on aikariippuvainen CYP2B6:n ja CYP3A:n estäjä (ks. kohta Yhteisvaikutukset). In vitro ‑tutkimusten perusteella on epätodennäköistä, että kritsotinibi aiheuttaisi lääkeainemetabolian estymisen seurauksena kliinisiä yhteisvaikutuksia lääkeaineiden kanssa, jotka ovat CYP1A2:n, CYP2C8:n, CYP2C9:n, CYP2C19:n tai CYP2D6:n substraatteja.
In vitro -tutkimukset osoittivat, että kritsotinibi on heikko UGT1A1:n ja UGT2B7:n estäjä (ks. kohta Yhteisvaikutukset). In vitro ‑tutkimusten perusteella on kuitenkin epätodennäköistä, että kritsotinibi aiheuttaisi lääkeainemetabolian estymisen seurauksena kliinisiä yhteisvaikutuksia lääkevalmisteiden kanssa, jotka ovat UGT1A4:n, UGT1A6:n tai UGT1A9:n substraatteja.
Ihmisen hepatosyyteillä in vitro tehtyjen tutkimusten perusteella on epätodennäköistä, että kritsotinibi aiheuttaisi lääkeainemetabolian induktion seurauksena kliinisiä yhteisvaikutuksia lääkeaineiden kanssa, jotka ovat CYP1A2:n substraatteja.
Eliminaatio
Kritsotinibin näennäinen terminaalinen puoliintumisaika potilaan plasmassa on kritsotinibin kerta-annoksen jälkeen 42 tuntia.
Terveille vapaaehtoisille koehenkilöille annetun radioaktiivisesti merkityn 250 mg:n kritsotinibin kerta-annoksen jälkeen ulosteessa havaittiin 63 % ja virtsassa 22 % annetusta annoksesta. Ulosteessa havaittu muuttumaton kritsotinibi edusti noin 53 % ja virtsassa havaittu muuttumaton kritsotinibi edusti noin 2,3 % annetusta annoksesta.
Samanaikainen käyttö lääkevalmisteiden kanssa, jotka ovat kuljettajaproteiinien substraatteja
Kritsotinibi on P-glykoproteiinin (P-gp) estäjä in vitro. Kritsotinibi saattaa siksi suurentaa samanaikaisesti annettujen lääkevalmisteiden pitoisuutta plasmassa, jos ne ovat P-gp:n substraatteja (ks. kohta Yhteisvaikutukset).
Kritsotinibi on OCT1:n ja OCT2:n estäjä in vitro. Kritsotinibi saattaa siten suurentaa samanaikaisesti annettujen OCT1:n tai OCT2:n substraatteina toimivien lääkevalmisteiden pitoisuuksia plasmassa (ks. kohta Yhteisvaikutukset).
Kritsotinibi ei estänyt kliinisesti merkittävinä pitoisuuksina in vitro ihmisen maksan kuljettajaproteiineja OATP1B1 tai OATP1B3 (orgaanisten anionien kuljettajapolypeptidi, OATP) tai munuaisten kuljettajaproteiineja OAT1 tai OAT3 (orgaanisten anionien kuljettajaproteiini, OAT). On siten epätodennäköistä, että kritsotinibi aiheuttaisi kliinisiä lääkkeiden yhteisvaikutuksia perustuen näiden kuljettajaproteiinien substraatteina olevien lääkkeiden maksa- tai munuaissoluihin oton estymiseen.
Vaikutus muihin kuljettajaproteiineihin
Kritsotinibi ei ole kliinisesti merkittävinä pitoisuuksina in vitro sappihappopumpun (BSEP) estäjä.
Farmakokinetiikka erityispotilasryhmillä
Maksan vajaatoiminta
Kritsotinibi metaboloituu pääosin maksassa. Avoimeen, ei-satunnaistettuun kliiniseen tutkimukseen (tutkimus 1012) otettiin mukaan potilaita, joiden maksan vajaatoiminta oli lievä (joko ASAT > ULN ja bilirubiini ≤ ULN tai mikä tahansa ASAT ja bilirubiini > ULN mutta ≤ 1,5 × ULN), keskivaikea (mikä tahansa ASAT ja bilirubiini > 1,5 × ULN ja ≤ 3 × ULN) tai vaikea (mikä tahansa ASAT ja bilirubiini > 3 × ULN). Lievää ja keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden verrokeiksi otettiin mukaan myös potilaita, joilla oli normaali maksan toiminta (ASAT ja bilirubiini ≤ ULN). Maksan vajaatoiminnan luokittelussa käytettiin NCI-kriteereitä.
Lievää maksan vajaatoimintaa sairastavien potilaiden (n = 10) kritsotinibin systeeminen altistus annostuksella 250 mg kaksi kertaa vuorokaudessa vakaassa tilassa oli samaa luokkaa kuin potilailla, joilla oli normaali maksan toiminta (n = 8); vuorokausi-AUC‑arvon (plasman pitoisuus-aikakuvaajan alla oleva pinta-ala päivittäisessä altistuksessa vakaassa tilassa) geometrisen keskiarvon suhdeluku oli 91,1 % ja Cmax‑arvon geometrisen keskiarvon suhdeluku oli 91,2 %. Aloitusannoksen muuttamista ei suositella potilaille, joilla on lievä maksan vajaatoiminta.
Keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden (n = 8) kritsotinibin systeeminen altistus annostuksella 200 mg kaksi kertaa vuorokaudessa oli suurempi kuin samalla annostuksella todettu altistus potilailla, joilla oli normaali maksan toiminta (n = 9); vuorokausi-AUC‑arvon geometrisen keskiarvon suhdeluku oli 150 % ja Cmax‑arvon geometrisen keskiarvon suhdeluku oli 144 %. Keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden kritsotinibin systeeminen altistus annostuksella 200 mg kaksi kertaa vuorokaudessa oli kuitenkin verrannollinen annostuksella 250 mg kaksi kertaa vuorokaudessa todettuun altistukseen potilailla, joilla oli normaali maksan toiminta; vuorokausi-AUC‑arvon geometrisen keskiarvon suhdeluku oli 114 % ja Cmax‑arvon geometrisen keskiarvon suhdeluku oli 109 %.
Vaikeaa maksan vajaatoimintaa sairastavilla potilailla (n = 6) kritsotinibin systeemisen altistuksen parametrit (vuorokausi-AUC ja Cmax) annostuksella 250 mg kerran vuorokaudessa olivat noin 64,7 % ja 72,6 % vastaavista parametreistä potilailla, joilla oli normaali maksan toiminta ja jotka saivat kritsotinibia annostuksella 250 mg kaksi kertaa vuorokaudessa.
Kritsotinibiannoksen muuttamista suositellaan annettaessa kritsotinibia potilaille, joiden maksan vajaatoiminta on joko keskivaikea tai vaikea (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Yksihaaraisiin tutkimuksiin 1001 ja 1005 otettiin mukaan potilaita, joilla oli lievä (eGFR 60 – < 90 ml/min) tai kohtalainen (eGFR 30 – < 60 ml/min) munuaisten vajaatoiminta. Tutkimuksissa arvioitiin munuaisten toiminnan (lähtötason kreatiniinipuhdistuma) vaikutusta havaittuihin kritsotinibin vakaan tilan jäännöspitoisuuksiin (Ctrough,ss). Tutkimuksessa 1001 plasman Ctrough, ss-arvon korjattu geometrinen keskiarvo oli lievää munuaisten vajaatoimintaa sairastavilla potilailla (n = 35) 5,1 % korkeampi ja kohtalaista vajaatoimintaa sairastavilla potilailla (n = 8) 11 % korkeampi kuin potilailla, joiden munuaisten toiminta oli normaali. Tutkimuksessa 1005 kritsotinibin Ctrough, ss-arvon korjattu geometrinen keskiarvo oli lievää munuaisten vajaatoimintaa sairastavilla potilailla (n = 191) 9,1 % korkeampi ja kohtalaista vajaatoimintaa sairastavilla potilailla (n = 65) 15 % korkeampi kuin potilailla, joiden munuaisten toiminta oli normaali. Lisäksi tutkimusten 1001, 1005 ja 1007 tiedoista tehty populaatiofarmakokineettinen analyysi osoitti, ettei kreatiniinipuhdistumalla ollut kliinisesti merkittävää vaikutusta kritsotinibin farmakokinetiikkaan. Koska kritsotinibialtistus kasvoi vain vähän (5–15 %), aloitusannoksen muuttamista ei suositella potilaille, joilla on lievä tai kohtalainen munuaisten vajaatoiminta.
Annettaessa 250 mg:n kerta-annos vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min) sairastaville koehenkilöille, jotka eivät tarvinneet peritoneaalidialyysiä tai hemodialyysiä, kritsotinibin AUCinf suureni 79 % ja Cmax 34 % verrattuna koehenkilöihin, joiden munuaisten toiminta oli normaali. Kritsotinibin annostuksen muuttamista suositellaan vaikeaa munuaisten vajaatoimintaa sairastaville potilaille, jotka eivät tarvitse peritoneaalidialyysiä tai hemodialyysiä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat, joilla on syöpä
Käytettäessä kritsotinibia annostuksella 280 mg/m2 kaksi kertaa vuorokaudessa (noin kaksinkertainen verrattuna aikuisille suositeltuun annokseen) ennen seuraavan annoksen antamista havaittu kritsotinibin jäännöspitoisuus (Ctrough) vakaassa tilassa oli samankaltainen kehon painon kvartiilista riippumatta. Pediatristen potilaiden keskimääräinen Ctrough-arvo vakaassa tilassa annostuksella 280 mg/m2 kaksi kertaa vuorokaudessa oli 482 ng/ml, kun taas useissa eri kliinisissä tutkimuksissa aikuisten syöpää sairastavien potilaiden keskimääräinen Ctrough-arvo vakaassa tilassa annostuksella 250 mg kaksi kertaa vuorokaudessa vaihteli välillä 263–316 ng/ml.
Pediatrisilla potilailla painolla on merkittävä vaikutus kritsotinibin farmakokinetiikkaan, sillä painavammilla potilailla on havaittu pienempiä kritsotinibialtistuksia.
Ikä
Tutkimusten 1001, 1005 ja 1007 aikuisia koskevista tiedoista tehdyn populaatiofarmakokineettisen analyysin perusteella iällä ei ole vaikutusta kritsotinibin farmakokinetiikkaan (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Paino ja sukupuoli
Tutkimusten 1001, 1005 ja 1007 aikuisia koskevista tiedoista tehdyn populaatiofarmakokineettisen analyysin perusteella painolla tai sukupuolella ei ollut kliinisesti merkittävää vaikutusta kritsotinibin farmakokinetiikkaan.
Etninen tausta
Tutkimusten 1001, 1005 ja 1007 tiedoista tehdyn populaatiofarmakokineettisen analyysin perusteella ennustettu plasmapitoisuus-aikakäyrän alla oleva pinta-ala vakaassa tilassa (AUCss) (95 %:n luottamusväli) oli aasialaisilla potilailla (n = 523) 23−37 % suurempi kuin muun etnisen taustan omaavilla potilailla (n = 691).
Edennyttä ALK-positiivista NSCLC:ää sairastavien potilaiden tutkimuksissa (n = 1669) seuraavien haittavaikutusten raportoinnissa oli ≥ 10 %:n absoluuttinen ero aasialaisten potilaiden (n = 753) ja etniseltä taustaltaan muiden potilaiden (n = 916) välillä: transaminaasien nousu, heikentynyt ruokahalu, neutropenia ja leukopenia. Mitään haittavaikutusta ei raportoitu ≥ 15 % absoluuttisella erolla.
Iäkkäät potilaat
Iäkkäiden potilaiden hoidosta on vähän tietoja saatavilla (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka). Tutkimusten 1001, 1005 ja 1007 tiedoista tehdyn populaatiofarmakokineettisen analyysin perusteella iällä ei ole vaikutusta kritsotinibin farmakokinetiikkaan.
Sydämen sähköfysiologia
Kritsotinibin vaikutusta QT-ajan pitenemiseen tutkittiin ALK-positiivista tai ROS1-positiivista NSCLC:ää sairastavilla potilailla, jotka saivat 250 mg kritsotinibia kaksi kertaa vuorokaudessa. Kolmen EKG-rekisteröinnin sarja kerättiin kerta-annoksen jälkeen ja vakaassa tilassa. QTcF-aika oli ≥ 500 millisekuntia 34 potilaalla 1619 potilaasta (2,1 %), joiden EKG rekisteröitiin vähintään kerran lähtötilanteen jälkeen. Lähtötilanteeseen verrattuna QTcF-aika oli pidentynyt ≥ 60 millisekuntia 79 potilaalla 1585 potilaasta (5,0 %), joiden EKG rekisteröitiin lähtötilanteessa ja vähintään kerran sen jälkeen EKG:n automaattisessa analyysissä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sokkoutetussa EKG-alatutkimuksessa, jossa mittaustulokset arvioitiin manuaalisesti, oli mukana 52 ALK-positiivista NSCLC:ää sairastavaa potilasta. Potilaat saivat kritsotinibia 250 mg kaksi kertaa vuorokaudessa. Lähtötilanteeseen verrattuna QTcF-aika oli pidentynyt ≥ 30 millisekuntia mutta alle 60 millisekuntia 11 potilaalla (21 %) ja ≥ 60 millisekuntia yhdellä potilaalla (2 %). Yhdelläkään potilaalla QTcF-ajan maksimiarvo ei ollut ≥ 480 millisekuntia. Tilastollisen analyysin (central tendency analysis) perusteella kaikissa syklin 2 ensimmäisen päivän aikapisteissä QTcF-ajan (muutos lähtötilanteesta, pienimmän neliösumman keskiarvo) 90 %:n luottamusvälin ylärajat olivat alle 20 millisekuntia. Farmakokineettinen/farmakodynaaminen analyysi viittasi yhteyteen kritsotinibin plasmapitoisuuden ja QTc-ajan välillä. Lisäksi sydämen sykkeen hidastumisen todettiin olevan yhteydessä suurentuneeseen kritsotinibin pitoisuuteen plasmassa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet); suurimmillaan keskimääräinen lasku oli 17,8 lyöntiä minuutissa syklin 2 ensimmäisenä päivänä 8 tunnin kuluttua lääkeannoksen ottamisesta.
Prekliiniset tiedot turvallisuudesta
Rotalla ja koiralla toistuvan altistuksen toksisuutta selvittäneissä enintään 3 kuukautta kestäneissä tutkimuksissa pääasialliset kohde-elinvaikutukset liittyivät ruoansulatuselimistöön (oksentelu, ulosteiden muutokset, verentungos), hematopoieesiin (luuytimen soluniukkuus), sydämeen ja verenkiertoelimistöön (sekamuotoinen ionikanavan salpaus, hidastunut sydämen syketaajuus ja alentunut verenpaine, kohonnut vasemman kammion loppudiastolinen paine [LVEDP], QRS- ja PR-ajan piteneminen ja alentunut sydänlihaksen supistuvuus) tai lisääntymiseen (pakyteenivaiheen spermatosyyttien rappeutuminen kiveksissä, munarakkuloiden solukuolema). Näiden löydösten haitaton vaikutustaso (NOAEL-arvo) oli joko subterapeuttinen tai AUC-arvon perusteella enintään 1,3-kertainen ihmisen kliiniseen altistukseen verrattuna. Muita löydöksiä olivat maksavaikutukset (maksan transaminaasien kohoaminen), vaikutukset verkkokalvon toimintaan sekä mahdollinen fosfolipidoosi useissa elimissä ilman korreloivaa toksisuutta.
Kritsotinibi ei ollut mutageeninen in vitro bakteerien mutaatiomäärityksessä (Ames). Kritsotinibi aiheutti aneuploidiaa kiinanhamsterin munasarjasoluilla tehdyssä mikrotumatestissä in vitro sekä ihmisen lymfosyyttien kromosomipoikkemamäärityksessä in vitro. Ihmisen lymfosyyteissä havaittiin sytotoksisten pitoisuuksien yhteydessä rakenteellisten kromosomipoikkeamien vähäistä lisääntymistä. Aneuploidian suhteen suurin annos, jolla ei saada merkitsevää vastetta (No Observed Effect Level, NOEL), oli AUC-arvon perusteella noin 1,8–2,1-kertainen ihmisen kliiniseen altistukseen verrattuna.
Kritsotinibilla ei ole tehty karsinogeenisuustutkimuksia.
Kritsotinibilla ei ole tehty erityisiä eläinkokeita hedelmällisyyteen kohdistuvien vaikutusten tutkimiseksi. Kritsotinibin katsotaan kuitenkin toistuvan altistuksen toksisuutta rotilla selvittäneiden tutkimusten perusteella mahdollisesti heikentävän ihmisen lisääntymiskykyä ja hedelmällisyyttä. Urosrotan lisääntymiselimissä havaittuja löydöksiä oli mm. pakyteenivaiheen spermatosyyttien rappeutuminen kiveksissä, kun rotille annettiin annoksia ≥ 50 mg/kg/vrk 28 vuorokauden ajan (AUC-arvon perusteella noin 1,1–1,3-kertainen altistus ihmisen kliiniseen altistukseen verrattuna). Naaraiden lisääntymiselimissä havaittuja löydöksiä olivat munarakkuloiden solukuolema, kun rotille annettiin annoksia 500 mg/kg/vrk 3 vuorokauden ajan.
Kritsotinibin ei todettu olevan teratogeeninen tiineille rotille ja kaniineille. Rotalla todettiin haittavaikutuksena implantaation jälkeisten alkiokuolemien lisääntymistä annoksilla ≥ 50 mg/kg/vrk (AUC-arvon perusteella noin 0,4–0,5-kertainen annos ihmisen suositeltuun annokseen verrattuna), ja rotalla ja kaniinilla todettiin sikiön painon alenemista annoksilla 200 mg/kg/vrk (rotta) ja 60 mg/kg/vrk (kaniini) (AUC-arvon perusteella noin 1,2–2,0-kertainen altistus ihmisen kliiniseen altistukseen verrattuna).
Kasvuikäisille rotille kerran päivässä 28 päivän ajan annettujen annosten 150 mg/kg/vrk (AUC-arvon perusteella noin 3,3–3,9-kertainen altistus ihmisen kliiniseen altistukseen verrattuna) yhteydessä havaittiin luunmuodostuksen vähenemistä kasvavissa pitkissä luissa. Muuta pediatristen potilaiden hoidon kannalta huolestuttavaa toksisuutta ei ole tutkittu nuorilla eläimillä.
Fototoksisuustutkimuksesta in vitro saadut tulokset osoittivat, että kritsotinibi saattaa olla fototoksinen.
Farmaseuttiset tiedot
Apuaineet
XALKORI 200 mg ja 250 mg kovat kapselit
Kapselin sisältö
Vedetön kolloidinen piidioksidi
Mikrokiteinen selluloosa
Vedetön kalsiumvetyfosfaatti
Natriumtärkkelysglykolaatti (tyyppi A)
Magnesiumstearaatti
Kapselikuori
Liivate
Titaanidioksidi (E 171)
Punainen rautaoksidi (E 172)
Painomuste
Shellakka (E 904)
Propyleeniglykoli (E 1520)
Kaliumhydroksidi (E 525)
Musta rautaoksidi (E 172)
XALKORI 20 mg, 50 mg ja 150 mg rakeet avattavissa kapseleissa
Rakeiden sisältö
Stearyylialkoholi
Poloksameeri
Sakkaroosi
Talkki (E 553b)
Hypromelloosi (E 464)
Makrogoli (E 1521)
Glyseryylimonostearaatti (E 471)
Keskipitkäketjuiset triglyseridit
Kapselikuori
Liivate
Titaanidioksidi (E 171)
Briljanttisininen (E 133) tai musta rautaoksidi (E 172)
Painomuste
Shellakka (E 904)
Propyleeniglykoli (E 1520)
Kaliumhydroksidi (E 525)
Musta rautaoksidi (E 172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
XALKORI 200 mg ja 250 mg kovat kapselit
4 vuotta
XALKORI 20 mg, 50 mg ja 150 mg rakeet avattavissa kapseleissa
2 vuotta
Säilytys
XALKORI 200 mg ja 250 mg kovat kapselit
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
XALKORI 20 mg, 50 mg ja 150 mg rakeet avattavissa kapseleissa
Säilytä alle 25°C.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
XALKORI kapseli, kova
200 mg (L:ei) 60 fol (4005,44 €)
250 mg (L:ei) 60 fol (4889,99 €)
PF-selosteen tieto
XALKORI 200 mg ja 250 mg kovat kapselit
60 kovaa kapselia sisältävä HDPE-purkki, jossa on polypropeenisuljin.
PVC/folioläpipainoliuskat, joissa on 10 kovaa kapselia.
Yksi kartonkikotelo sisältää 60 kovaa kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
XALKORI 20 mg, 50 mg ja 150 mg rakeet avattavissa kapseleissa
XALKORI-rakeet on pakattu suurtiheyspolyeteenistä (HDPE) valmistettuihin purkkeihin, joissa on polypropeeninen turvasuljin ja alumiinifoliosta/polyeteenistä valmistettu kuumainduktiosinetti. Purkit sisältävät 60 avattavaa kapselia.
Valmisteen kuvaus:
Kapseli, kova
XALKORI 200 mg kovat kapselit
Valkoinen läpinäkymätön ja vaaleanpunainen läpinäkymätön kova kapseli, jonka kansiosaan on painettu ”Pfizer” ja runko-osaan ”CRZ 200”.
XALKORI 250 mg kovat kapselit
Vaaleanpunainen läpinäkymätön kova kapseli, jonka kansiosaan on painettu ”Pfizer” ja runko-osaan ”CRZ 250”.
Rakeet avattavissa kapseleissa
Rakeet ovat valkoisia tai luonnonvalkoisia, ja ne ovat läpinäkymättömän kovan kapselin sisällä.
XALKORI 20 mg rakeet avattavissa kapseleissa
Vaaleansininen kansiosa, johon on painettu mustalla musteella ”Pfizer”, ja valkoinen runko-osa, johon on painettu mustalla musteella ”CRZ 20”
XALKORI 50 mg rakeet avattavissa kapseleissa
Harmaa kansiosa, johon on painettu mustalla musteella ”Pfizer”, ja vaaleanharmaa runko-osa, johon on painettu mustalla musteella ”CRZ 50”.
XALKORI 150 mg rakeet avattavissa kapseleissa
Vaaleansininen kansiosa, johon on painettu mustalla musteella ”Pfizer”, ja vaaleansininen runko-osa, johon on painettu mustalla musteella ”CRZ 150”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte, esim. avattavien kapseleiden rakeita sisältävät kapselikuoret, on hävitettävä paikallisten vaatimusten mukaisesti. Tyhjät XALKORI-rakeita sisältäneet kapselikuoret pitää hävittää talousjätteen mukana.
Korvattavuus
XALKORI kapseli, kova
200 mg 60 fol
250 mg 60 fol
- Ylempi erityiskorvaus (100 %). Kritsotinibi: Edenneen ei-pienisoluisen keuhkosyövän sekä anaplastisen suurisoluisen lymfooman ja tulehduksellisen myofibroblastituumorin hoito erityisin edellytyksin (169).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Kritsotinibi: Edenneen ei-pienisoluisen keuhkosyövän hoito erityisin edellytyksin (363).
ATC-koodi
L01ED01
Valmisteyhteenvedon muuttamispäivämäärä
05.11.2024
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com