
SOMAVERT injektiokuiva-aine ja liuotin, liuosta varten 10 mg, 15 mg, 20 mg, 25 mg, 30 mg
Vaikuttavat aineet ja niiden määrät
Somavert 10 mg injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää 10 mg pegvisomanttia.
Käyttökuntoon saattamisen jälkeen 1 ml liuosta sisältää 10 mg pegvisomanttia.*
Apuaine, jonka vaikutus tunnetaan
Lääkevalmisteen 10 mg:n vahvuus sisältää 0,4 mg natriumia per kuiva-ainepullo.
Somavert 15 mg injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää 15 mg pegvisomanttia.
Käyttökuntoon saattamisen jälkeen 1 ml liuosta sisältää 15 mg pegvisomanttia.*
Apuaine, jonka vaikutus tunnetaan
Lääkevalmisteen 15 mg:n vahvuus sisältää 0,4 mg natriumia per kuiva-ainepullo.
Somavert 20 mg injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää 20 mg pegvisomanttia.
Käyttökuntoon saattamisen jälkeen 1 ml liuosta sisältää 20 mg pegvisomanttia.*
Apuaine, jonka vaikutus tunnetaan
Lääkevalmisteen 20 mg:n vahvuus sisältää 0,4 mg natriumia per kuiva-ainepullo.
Somavert 25 mg injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää 25 mg pegvisomanttia.
Käyttökuntoon saattamisen jälkeen 1 ml liuosta sisältää 25 mg pegvisomanttia.*
Apuaine, jonka vaikutus tunnetaan
Lääkevalmisteen 25 mg:n vahvuus sisältää 0,5 mg natriumia per kuiva-ainepullo.
Somavert 30 mg injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää 30 mg pegvisomanttia.
Käyttökuntoon saattamisen jälkeen 1 ml liuosta sisältää 30 mg pegvisomanttia.*
Apuaine, jonka vaikutus tunnetaan
Lääkevalmisteen 30 mg:n vahvuus sisältää 0,6 mg natriumia per kuiva-ainepullo.
*Valmistettu yhdistelmä-DNA-tekniikalla Escherichia coli -soluissa.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten (injektiokuiva-aine)
Kliiniset tiedot
Käyttöaiheet
Sellaisten aikuisten akromegaliapotilaiden hoitoon, jotka eivät ole vastanneet leikkaukseen ja/tai sädehoitoon riittävällä tavalla ja joilla asianmukainen lääketieteellinen hoito somatostatiinianalogeilla ei normalisoinut IGF-I-pitoisuuksia tai ei ollut siedetty.
Ehto
Hoito on aloitettava sellaisen lääkärin valvonnassa, jolla on kokemusta akromegalian hoidosta.
Annostus ja antotapa
Hoito on aloitettava sellaisen lääkärin valvonnassa, jolla on kokemusta akromegalian hoidosta.
Annostus
Pegvisomantin 80 mg:n induktioannos annetaan ihon alle hoitohenkilökunnan valvonnassa. Tämän jälkeen potilaalle injisoidaan ihon alle SOMAVERT-valmistetta 10 mg 1 ml:ssa liuotinta kerran vuorokaudessa.
Sopiva annos on määritettävä seerumin IGF-I-pitoisuuksien perusteella. Seerumin IGF-I-pitoisuudet on mitattava 4–6 viikon välein ja annosta on sovitettava asianmukaisesti aina kerralla 5 mg/vrk, jotta seerumin IGF-I-pitoisuus saataisiin pysymään ikään suhteutetulla normaalilla vaihteluvälillä optimaalisen hoitovasteen säilyttämiseksi.
Maksaentsyymien lähtöpitoisuuksien arviointi ennen SOMAVERT-hoidon aloittamista
Potilaalta on määritettävä lähtötilanteen maksa-arvot (seerumin alaniiniaminotransferaasi [ALAT], aspartaattiaminotransferaasi [ASAT], seerumin kokonaisbilirubiini ja alkalinen fosfataasi [AFOS]) ennen SOMAVERT-hoidon aloittamista. Kohdassa 4.4 Varoitukset ja varotoimet olevassa taulukossa A on suosituksia SOMAVERT-hoidon aloittamisesta lähtötilanteen maksakokeiden perusteella ja maksa-arvojen seurannasta SOMAVERT-hoidon aikana.
Enimmäisannos ei saa olla yli 30 mg/vrk.
Seuraavat vahvuudet ovat saatavina erilaisia annostusohjelmia varten: Somavert 10 mg, Somavert 15 mg, Somavert 20 mg, SOMAVERT 25 mg ja SOMAVERT 30 mg.
Pediatriset potilaat
SOMAVERT-valmisteen turvallisuutta ja tehoa 0–17 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Iäkkäät
Annosta ei tarvitse sovittaa.
Munuaisten tai maksan vajaatoiminta
SOMAVERT-valmisteen turvallisuutta ja tehoa potilailla, joilla on munuaisten tai maksan vajaatoiminta, ei ole osoitettu.
Antotapa
Pegvisomantti annetaan ihonalaisena injektiona.
Pistoskohtaa on vaihdeltava päivittäin lipohypertrofian estämiseksi.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Kasvuhormonia erittävät kasvaimet
Koska kasvuhormonia erittävät aivolisäkkeen kasvaimet voivat joskus suurentua ja aiheuttaa vakavia komplikaatioita (esim. näkökenttäpuutoksia), tulee kaikkia potilaita ehdottomasti seurata tarkoin. Jos merkkejä kasvaimen suurentumisesta ilmenee, vaihtoehtoiset toimenpiteet voivat olla suositeltavia.
Seerumin IGF-I-pitoisuuksien seuranta
Pegvisomantti on kasvuhormonin vaikutuksen potentti estäjä. Tämän lääkevalmisteen anto voi johtaa kasvuhormonin puutostilaan seerumin koholla olevista kasvuhormonitasoista huolimatta. Seerumin IGF-I-pitoisuuksia on seurattava ja ne on säilytettävä ikään suhteutetulla normaalilla vaihteluvälillä sovittamalla pegvisomanttiannosta.
Kohonneet ALAT- tai ASAT-arvot
Potilaalta on määritettävä lähtötilanteen maksa-arvot (seerumin alaniiniaminotransferaasi [ALAT], aspartaattiaminotransferaasi [ASAT], seerumin kokonaisbilirubiini ja alkalinen fosfataasi [AFOS]) ennen SOMAVERT-hoidon aloittamista.
Ahtauttavan sappitietaudin mahdollisuus on poissuljettava potilailla, joilla on kohonneet ALAT‑ ja ASAT-arvot tai joita on aiemmin hoidettu jollakin somatostatiinianalogilla. Pegvisomantin anto on keskeytettävä, jos merkkejä maksasairaudesta ilmenee.
Katso taulukosta A suosituksia SOMAVERT-hoidon aloittamisesta lähtötilanteen maksakokeiden perusteella ja maksa-arvojen seurannasta SOMAVERT-hoidon aikana.
Taulukko A: Suosituksia SOMAVERT-hoidon aloittamisesta lähtötilanteen maksakokeiden perusteella ja maksa-arvojen säännöllisestä seurannasta SOMAVERT-hoidon aikana
Maksa-arvot lähtötilanteessa | Suositukset |
Normaalit |
|
Kohonneet, mutta enintään 3-kertaiset normaalin vaihteluvälin ylärajaan verrattuna |
|
Yli 3-kertaiset normaalin vaihteluvälin ylärajaan verrattuna |
|
Lyhenteet: ALAT = alaniiniaminotransferaasi; ASAT = aspartaattitransaminaasi.
Jos potilaan maksa-arvot kohoavat tai potilaalle kehittyy muita maksan toimintahäiriön merkkejä tai oireita SOMAVERT-hoidon aikana, suositellaan seuraavan taulukon B mukaisia hoitosuosituksia.
Taulukko B. Kliiniset suositukset, jotka perustuvat SOMAVERT-hoidon aikaisiin epänormaaleihin maksa-arvoihin
Maksa-arvot ja kliiniset merkit/oireet | Suositukset |
Kohonneet, mutta enintään 3-kertaiset normaalin vaihteluvälin ylärajaan verrattuna |
|
Yli 3‑kertaiset mutta alle 5‑kertaiset normaalin vaihteluvälin ylärajaan verrattuna (ei maksatulehduksen tai muun maksavaurion merkkejä/oireita eikä seerumin kokonaisbilirubiinin kohoamista) |
|
Vähintään 5‑kertaiset normaalin vaihteluvälin ylärajaan verrattuna tai transaminaasiarvon kohoaminen vähintään 3-kertaiseksi vaihteluvälin ylärajaan verrattuna ja siihen liittyvä minkä tahansa suuruinen seerumin kokonaisbilirubiinin kohoaminen (johon voi liittyä maksatulehduksen tai muun maksavaurion merkkejä/oireita) |
|
Maksatulehdukseen tai muuhun maksavaurioon viittaavat merkit tai oireet (esim. keltaisuus, bilirubinuria, väsymys, pahoinvointi, oksentelu, kipu vatsan oikealla yläneljänneksellä, askites, selittämätön turvotus, mustelmaherkkyys) |
|
Hypoglykemia
Pegvisomanttitutkimus diabetespotilailla, joita hoidettiin joko insuliinilla tai suun kautta otettavilla diabeteslääkkeillä, paljasti hypoglykemian riskin tässä potilasryhmässä. Siksi diabetes mellitusta sairastavan akromegaliapotilaan insuliini- tai suun kautta otettavan diabeteslääkkeen annosta on ehkä pienennettävä (ks. kohta Yhteisvaikutukset).
Parantunut hedelmällisyys
Terapeuttiset hyödyt IGF-I-pitoisuuden pienenemisestä ja siten potilaan kliinisen tilan paranemisesta saattavat myös parantaa naispotilaiden hedelmällisyyttä (ks. kohta Raskaus ja imetys).
Raskaus
Akromegalia saattaa olla raskauden aikana paremmassa hoitotasapainossa. Pegvisomantin käyttöä raskauden aikana ei suositella (ks. kohta Raskaus ja imetys). Jos pegvisomanttia käytetään raskauden aikana, IGF-I-pitoisuutta pitää seurata tarkoin ja pegvisomanttiannosta saattaa olla tarpeen säätää (ks. kohta Annostus ja antotapa) IGF-I-arvojen perusteella.
Natriumsisältö
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”. Potilaille, joilla on ruokavalion natriumrajoitus, voidaan sanoa, että tämä lääkevalmiste on ”natriumiton”.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. On harkittava, jatketaanko somatostatiinianalogeilla annettavaa hoitoa. Tämän lääkkeen käyttöä yhdessä muiden akromegalian hoitoon käytettävien lääkevalmisteiden kanssa ei ole tutkittu laajalti.
Insuliinin tai suun kautta annettavien diabeteslääkkeiden annosta on ehkä pienennettävä näitä vaikuttavia aineita saavalla potilaalla, koska pegvisomantti vaikuttaa insuliiniherkkyyteen (katso kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pegvisomantti on rakenteeltaan merkitsevästi samankaltainen kuin kasvuhormoni, mikä aiheuttaa ristireaktioita kaupallisissa kasvuhormonitesteissä. Koska tämän lääkkeen terapeuttisesti tehokkaiden annosten pitoisuudet seerumissa ovat yleensä 100–1000-kertaisia verrattuna akromegaliapotilailla mitattuihin varsinaisiin kasvuhormonipitoisuuksiin seerumissa, kaupalliset seerumista tehtävät kasvuhormonipitoisuustestit antavat vääriä tuloksia. Siksi pegvisomanttihoitoa ei tulisi seurata tai muuttaa tällaisista testeistä saatavien seerumin kasvuhormonipitoisuuksien perusteella.
Raskaus ja imetys
Raskaus
On vain vähän tietoja pegvisomantin käytöstä raskaana oleville naisille. Riittäviä eläinkokeita lisääntymistoksisuuden selvittämiseksi ei ole tehty (katso kohta Prekliiniset tiedot turvallisuudesta).
SOMAVERT-valmisteen käyttöä ei suositella raskauden aikana eikä annettavaksi naisille, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Jos pegvisomanttia käytetään raskauden aikana, IGF-I-pitoisuutta pitää seurata tarkoin, etenkin ensimmäisen raskauskolmanneksen aikana. Pegvisomanttiannosta voi olla tarpeen säätää raskauden aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Imetys
Pegvisomantin erittymistä rintamaitoon ei ole tutkittu eläimillä. Kliiniset tiedot ovat liian vähäisiä (yksi raportoitu tapaus), jotta voitaisiin tehdä johtopäätöksiä pegvisomantin erittymisestä ihmisen rintamaitoon. Tämän vuoksi pegvisomanttia ei saa käyttää rintaruokinnan aikana. Rintaruokintaa voidaan kuitenkin jatkaa, jos hoito tällä lääkkeellä keskeytetään. Tätä päätöstä tehtäessä on otettava huomioon pegvisomanttihoidosta koituvat hyödyt äidille ja rintaruokinnasta aiheutuvat hyödyt lapselle.
Hedelmällisyys
Saatavilla ei ole tietoja pegvisomantin vaikutuksesta hedelmällisyyteen.
Terapeuttiset hyödyt IGF-I-pitoisuuden pienenemisestä ja siten potilaan kliinisen tilan paranemisesta saattavat myös parantaa naispotilaiden hedelmällisyyttä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia valmisteen vaikutuksista ajokykyyn tai koneidenkäyttökykyyn ei ole tehty.
Haittavaikutukset
Yhteenveto haittavaikutusprofiilista
Alla on lueteltu SOMAVERT-valmisteen kliinisissä tutkimuksissa ilmenneet haittavaikutukset.
Kliinisissä tutkimuksissa pegvisomantilla hoidetuilla potilailla (n = 550) suurin osa pegvisomantin aiheuttamista haittareaktioista oli vaikeusasteeltaan lieviä tai kohtalaisia, lyhytkestoisia eivätkä edellyttäneet hoidon lopettamista.
Kliinisissä tutkimuksissa yleisimmin raportoituja haittavaikutuksia, joita esiintyi > 10 prosentilla pegvisomantilla hoidetuista akromegaliapotilaista, olivat päänsärky 25 %:lla, artralgia 16 %:lla ja ripuli 13 %:lla.
Taulukko haittavaikutuksista
Alla on lueteltu kliinisissä tutkimuksissa ilmenneet tai spontaanisti raportoidut haittavaikutukset luokiteltuna elinjärjestelmän ja esiintymistiheyden mukaan.
Haittavaikutukset on lueteltu seuraavien luokitusten mukaan:
Hyvin yleinen: > 1/10
Yleinen: > 1/100, < 1/10
Melko harvinainen: > 1/1 000, < 1/100
Tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin)
Elinjärjestelmä- luokka | Hyvin yleinen (> 1/10) | Yleinen (> 1/100, < 1/10) | Melko harvinainen (> 1/1000, < 1/100) | Tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin) |
Veri ja imukudos | trombosytopenia, leukopenia, leukosytoosi, verenvuototaipumus | |||
Immuunijärjestelmä | yliherkkyysreaktiotb | anafylaktinen reaktiob, anafylaktoidi-nen reaktiob | ||
Aineenvaihdunta ja ravitsemus | hyperkolestero-lemia, hyperglykemia, hypoglykemia, painonnousu | hypertriglyseridemia | ||
Psyykkiset häiriöt | poikkeavat unet | paniikkikohtaus, lyhytkestoinen muistinmenetys, apatia, sekavuus, unihäiriö, sukupuolivietin voimistuminen | vihaisuus | |
Hermosto | päänsärky | uneliaisuus, vapina, heitehuimaus, hypoestesia | narkolepsia, migreeni, dysgeusia | |
Silmät | silmäkipu | astenopia | ||
Kuulo ja tasapainoelin | Ménièren tauti | |||
Sydän | perifeerinen turvotus | |||
Verisuonisto | hypertensio | |||
Hengityselimet, rintakehä ja välikarsina | hengenahdistus | kurkunpään kouristusb | ||
Ruoansulatuselimistö | ripuli | oksentelu, ummetus, pahoinvointi, vatsan pingotus, ruoansulatus-häiriöt, ilmavaivat | peräpukamat, syljen liikaeritys, suun kuivuminen, hammashäiriöt | |
Maksa ja sappi | epänormaalit maksan toimintakoearvot (esim. kohonneet transaminaasi-arvot) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | |||
Iho ja ihonalainen kudos | liikahikoilu, ruhjevammat, kutinab, ihottumab | kasvojen turvotus, ihon kuivuminen, mustelmataipumus, yöhikoilu, eryteemab, urtikariab | angioedeemab | |
Luusto, lihakset ja sidekudos | nivelkipu | lihaskipu, artriitti | ||
Munuaiset ja virtsatiet | hematuria | proteinuria, polyuria, heikentynyt munuaistoiminta | ||
Yleisoireet ja antopaikassa todettavat haitat | reaktio pistoskohdassa (mukaan lukien yliherkkyys pistoskohdassa), mustelma tai verenvuoto pistoskohdassa, hypertrofia pistoskohdassa (esim. lipohypertrofia)a, influenssaa muistuttava sairaus, väsymys, voimattomuus, kuume | epänormaali olo, paranemisen heikentyminen, nälkä |
a Katso ”Valittujen haittavaikutusten kuvaus” alla.
b Yliherkkyyteen liittyvät haittavaikutukset.
Valittujen haittavaikutusten kuvaus
Useimpia pistoskohdan reaktioita luonnehdittiin paikalliseksi punoitukseksi ja aristukseksi, ja ne hävisivät itsestään oireenmukaisella paikallishoidolla pegvisomanttihoidon jatkuessa. Pistoskohdassa on havaittu hypertrofiaa, myös lipohypertrofiaa.
Yksittäisten kasvuhormonia estävien vasta-aineiden kehittymistä pieninä määrinä havaittiin 16,9 prosentilla pegvisomantilla hoidetuista potilaista. Näiden vasta-aineiden kliinistä merkitystä ei tunneta.
Systeemisiä yliherkkyysreaktioita mukaan lukien anafylaktisia/anafylaktoidisia reaktioita, kurkunpään kouristusta, angioedeemaa ja laajalle levinneitä ihoreaktioita (ihottumaa, eryteemaa, kutinaa, urtikariaa) on raportoitu valmisteen markkinoille tulon jälkeen. Osa potilaista on tarvinnut sairaalahoitoa. Kaikilla potilailla oireet eivät ole ilmaantuneet uudelleen annettaessa valmistetta toistamiseen.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kokemukset pegvisomantin yliannoksesta ovat vähäisiä. Ainoassa raportoidussa akuutissa yliannostapauksessa potilas sai 80 mg/vrk seitsemän vuorokauden ajan, jolloin hänellä ilmeni väsymyksen lievää lisääntymistä ja suun kuivumista. Hoidon lopettamista seuranneella viikolla todettuja haittavaikutuksia olivat unettomuus, lisääntynyt väsymys, perifeerinen turvotus, vapina ja painonnousu. Kahden viikon kuluttua hoidon lopettamisesta havaittiin leukosytoosia ja kohtalaista verenvuotoa pistoskohdasta ja laskimon punktiokohdista; näitä vaikutuksia pidettiin mahdollisesti pegvisomanttiin liittyvinä.
Yliannostuksen sattuessa tämän lääkkeen anto on lopetettava, eikä sitä saa aloittaa uudestaan, ennen kuin IGF-I-tasot ovat palautuneet normaalille vaihteluvälille tai sen yläpuolelle.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut aivolisäkkeen etulohkon hormonit ja analogit, ATC-koodi: H01AX01
Vaikutusmekanismi
Pegvisomantti on ihmisen kasvuhormonin analogi, joka on geneettisesti muunnettu kasvuhormonireseptorin antagonistiksi. Pegvisomantti sitoutuu solujen pinnalla kasvuhormonireseptoreihin, joissa se estää kasvuhormonin sitoutumisen ja siten häiritsee kasvuhormonin signaalin transduktiota solun sisällä. Pegvisomantti on erittäin selektiivinen kasvuhormonireseptorin suhteen, eikä sillä ole ristireaktioita muiden sytokiinireseptorien kanssa, mukaan lukien prolaktiini.
Farmakodynaamiset vaikutukset
Kasvuhormonin vaikutuksen esto pegvisomantilla pienentää insuliininkaltaisen kasvutekijä I:n (IGF-I) pitoisuuksia seerumissa. Se pienentää myös muiden kasvuhormoniin vastaavien seerumin proteiinien, kuten sitoutumattoman IGF‑I:n, IGF-I:n happolabiilin alayksikön (ALS) ja insuliininkaltaista kasvutekijää sitovan proteiini-3:n (IGFBP-3) pitoisuuksia.
Kliininen teho ja turvallisuus
Eräässä 12-viikkoisessa satunnaistetussa kaksoissokkoutetussa monikeskustutkimuksessa verrattiin lumelääkettä ja pegvisomanttia 112 akromegaliapotilaalla. Pegvisomanttiryhmissä havaittiin kaikilla lähtötilannetta seuranneilla käynneillä, että IGF-I:n (p < 0,0001), sitoutumattoman IGF-I:n (p < 0,05), IGFBP-3:n (p < 0,05) ja ALS:n (p < 0,05) keskimääräiset pitoisuudet pienenivät annoksesta riippuvaisesti ja tilastollisesti merkitsevästi. Seerumin IGF-I normalisoitui tutkimuksen lopussa (viikko 12) 9,7 %:lla lumelääkkeellä hoidetuista potilaista ja 38,5 %:lla 10 mg/vrk pegvisomanttia saaneista, 75 %:lla 15 mg/vrk pegvisomanttia saaneista ja 82 %:lla 20 mg/vrk pegvisomanttia saaneista potilaista.
Lumelääkkeeseen verrattuna kaikissa annosryhmissä havaittiin tilastollisesti merkitseviä eroja (p < 0,05) kaikkien merkkien ja oirepisteiden paranemisessa.
Eräässä pitkäaikaisessa avoimessa annostitraustutkimuksessa, joka kesti vähintään 12 perättäistä kuukautta (keskiarvo = 55 viikkoa), seurattiin 38 akromegaliapotilasta, joille annettiin päivittäin pegvisomanttia. Tässä kohortissa keskimääräinen IGF-I-pitoisuus pieneni pegvisomanttia saaneilla arvosta 917 ng/ml arvoon 299 ng/ml ja 92 %:lla saavutettiin normaali (ikään suhteutettu) IGF-I-pitoisuus.
Eri tutkimuksissa ja myös Acrostudy-tutkimuksessa pegvisomantti normalisoi IGF-I-pitoisuuden suurella prosenttiosuudella potilaista (> 70 %) ja pienensi merkittävästi plasman paastoglukoosipitoisuutta sekä plasman paastoinsuliinipitoisuutta.
Pegvisomantti parantaa myös insuliiniherkkyyttä todennäköisesti salpaamalla kasvuhormonireseptoreita kudoksissa, pääasiassa maksassa ja myös rasvakudoksessa, munuaisissa ja luustolihaksissa, ja poistaa siten kasvuhormonin haitallista vaikutusta insuliinin signalointiin, lipolyysiin ja glukoneogeneesiin. Näiden vaikutusten vaikutusmekanismia ei kuitenkaan tunneta varmasti. Diabetesta sairastavien akromegaliapotilaiden insuliiniannosta tai veren glukoosipitoisuutta alentavan lääkevalmisteen annosta voi olla tarpeen pienentää (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Farmakokinetiikka
Imeytyminen
Ihon alle annetun pegvisomantin imeytyminen on hidasta ja pitkittynyttä. Pegvisomantin huippupitoisuudet seerumissa saavutetaan yleensä vasta 33–77 tunnin kuluttua annosta. Ihonalaisesta annoksesta imeytyy keskimäärin 57 % verrattuna laskimonsisäiseen annokseen.
Jakautuminen
Pegvisomantin näennäinen jakautumistilavuus on suhteellisen pieni (7–12 l).
Biotransformaatio
Pegvisomantin metaboliaa ei ole tutkittu.
Eliminaatio
Pegvisomantin keskimääräiseksi elimistön systeemiseksi kokonaispuhdistumaksi toistuvan annon jälkeen on arvioitu 28 ml/h, kun ihonalainen annos on 10–20 mg/vrk. Pegvisomantin munuaispuhdistuma on olematon; se on alle 1 % elimistön kokonaispuhdistumasta. Pegvisomantti eliminoituu seerumista hitaasti, ja sen arvioitu keskimääräinen puoliintumisaika joko kerta-annoksen tai toistuvan annon jälkeen on yleensä 74–172 tuntia.
Lineaarisuus/ei-lineaarisuus
Pegvisomantin ihonalaisen kerta-annon jälkeen ei havaita lineaarisuutta suurenevilla 10, 15 ja 20 mg:n annoksilla. Farmakokineettisissä väestötutkimuksissa tehtyjen havaintojen mukaan farmakokinetiikka on lähes lineaarinen vakaassa tilassa. Kun kahdessa pitkäaikaistutkimuksessa annettiin 145 potilaalle päivittäin 10, 15 tai 20 mg:n annos, pegvisomantin keskimääräiset pitoisuudet seerumissa (+ SD) olivat näillä annoksilla noin 8 800+6 300, 13 200+8 000 ja 15 600+10 300 ng/ml.
Pegvisomantin farmakokinetiikka on samankaltainen normaaleilla terveillä vapaaehtoisilla koehenkilöillä ja akromegaliapotilailla. Painavammilla yksilöillä pegvisomantin kokonaispuhdistuma elimistöstä tosin on yleensä pienempi kuin kevyemmillä yksilöillä. Tämän vuoksi painavammat yksilöt saattavat tarvita suurempia pegvisomanttiannoksia.
Prekliiniset tiedot turvallisuudesta
Rotilla ja apinoilla tehtyjen toistuvan altistuksen aiheuttamaa toksisuutta koskevien tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Koska farmakologinen vaste oli apinalla kuitenkin huomattava, sellaisia systeemisiä altistuksia, jotka ylittävät potilailla hoitoannoksilla saavutetut, ei ole tutkittu.
Rotilla tehdyssä karsinogeenisuustutkimuksessa havaittiin uroksilla injektiokohdassa pahanlaatuisia fibroottisia histiosytoomia, joihin liittyi fibroosia ja histiosyyttitulehdusta. Havainnot tehtiin kolme kertaa ihmisen altistusta vastaavilla altistusannoksilla perustuen plasmakonsentraatioiden keskiarvoon kahdessa pitkäaikaistutkimuksessa, joissa käytettiin 30 mg päiväannosta. Tämän löydöksen merkitystä ihmisille ei tällä hetkellä tiedetä. Injektiokohdan kasvainten lisääntynyt ilmaantuvuus johtui todennäköisimmin ärsytyksestä ja rottien merkittävästä herkkyydestä ihon alle toistuvasti annetuille injektioille.
Tiineillä kaniineilla tehtiin alkion varhaisvaiheen kehitystä sekä alkion ja sikiön kehitystä koskevia tutkimuksia ihon alle annetuilla pegvisomanttiannoksilla 1, 3 ja 10 mg/kg/vrk. Pegvisomantin antoon organogeneesin aikana ei havaittu liittyneen teratogeenisia vaikutuksia. Kummassakin tutkimuksessa havaittiin annoksella 10 mg/kg/vrk (kehon pinta-alan perusteella 6-kertainen ihmisen suurimpaan hoitoannokseen nähden) alkiokatoa implantaation jälkeen. Hedelmällisyyttä koskevia tutkimuksia ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine:
Glysiini
Mannitoli (E421)
Dinatriumfosfaatti, vedetön
Natriumdivetyfosfaatti, monohydraatti
Liuotin:
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta.
Käyttökuntoon saattamisen jälkeen valmiste on käytettävä heti.
Säilytys
Säilytä kuiva-ainepullot jääkaapissa (2–8 °C). Ei saa jäätyä. Pidä injektiopullot kotelossa. Herkkä valolle.
SOMAVERT kuiva-ainepulloja sisältäviä koteloita voidaan säilyttää huoneenlämmössä (enintään 25 °C:ssa) yhden, enintään 30 päivän pituisen jakson ajan. Viimeinen käyttöajankohta (enintään 30 päivää jääkaapista ottamisen jälkeen) pitää merkitä koteloon. Injektiopullot on suojattava valolta, eikä niitä saa laittaa takaisin jääkaappiin. SOMAVERT kuiva-ainepullot on hävitettävä, jos niitä ei käytetä 30 päivän kuluessa siitä, kun ne on otettu huoneenlämpöön, tai koteloon painettuun viimeiseen käyttöpäivämäärään mennessä, sen mukaan, kumpi päivämääristä on aiemmin.
Säilytä esitäytetyt ruiskut alle 30 °C tai jääkaapissa (2–8 °C). Ei saa jäätyä.
Käyttökuntoon saattamisen jälkeen:
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SOMAVERT injektiokuiva-aine ja liuotin, liuosta varten
10 mg (L:ei) 30 x 1 kpl (turvaneula, liuotin esitäytetyssä ruiskussa) (2167,52 €)
15 mg (L:ei) 30 x 1 kpl (turvaneula, liuotin esitäytetyssä ruiskussa) (3161,16 €)
20 mg (L:ei) 30 x 1 kpl (turvaneula, liuotin esitäytetyssä ruiskussa) (4154,68 €)
25 mg (L:ei) 30 x 1 kpl (turvaneula, liuotin esitäytetyssä ruiskussa) (5073,68 €)
30 mg (L:ei) 30 x 1 kpl (turvaneula, liuotin esitäytetyssä ruiskussa) (6052,29 €)
PF-selosteen tieto
10 mg, 15 mg, 20 mg, 25 mg tai 30 mg pegvisomanttia kuiva-aineena injektiopullossa (tyypin I piilasia), jossa on suljin (klorobutyylikumia). 1 ml liuotinta (injektionesteisiin käytettävä vesi) esitäytetyssä ruiskussa (tyypin I borosilikaattilasia), jossa männän tulppa (bromobutyylikumia) ja ruiskun kärkisuojus (bromobutyylikumia). Muovisen suojakorkin väri riippuu valmisteen vahvuudesta.
SOMAVERT 10 mg ja 15 mg
Pakkauskoko: 30 injektiopulloa, esitäytettyä ruiskua ja turvaneulaa.
SOMAVERT 20 mg, 25 mg ja 30 mg
Pakkauskoot: 1 injektiopullo, esitäytetty ruisku ja turvaneula; 30 injektiopulloa, esitäytettyä ruiskua ja turvaneulaa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kuiva-aine on valkoinen tai lähes valkoinen.
Käyttö- ja käsittelyohjeet
Pistoksen antamiseen käytettävä ruisku ja turvaneula toimitetaan lääkevalmisteen mukana.
Ruiskun korkki on poistettava esitäytetystä ruiskusta ennen kuin pakkauksen mukana toimitettu turvaneula kiinnitetään. Tämä tapahtuu napsauttamalla korkki pois. Ruisku on pidettävä pystyasennossa vuodon estämiseksi. Ruiskun pää ei saa koskea mihinkään.
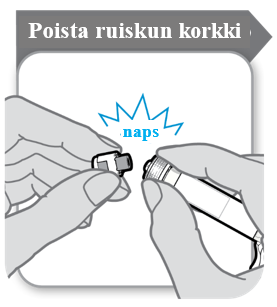
Kuiva-aine on saatettava käyttökuntoon 1 ml:lla liuotinta. Kun liuotin lisätään ruiskusta, injektiopullo ja ruisku on pidettävä tietyssä kulmassa alla olevan kuvan mukaisesti.
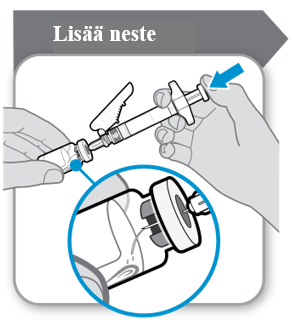
Lisää liuotin kuiva-ainetta sisältävään injektiopulloon. Liuotin on tyhjennettävä injektiopulloon hitaasti, jotta vältetään vaahdon muodostuminen. Vaahto tekee lääkevalmisteen käyttökelvottomaksi. Liuota injektiokuiva-aine varovasti kääntelemällä injektiopulloa hitaasti. Älä ravista voimakkaasti, koska se voi denaturoida vaikuttavan aineen.
Ennen lääkkeen antoa käyttövalmis liuos on tarkastettava silmämääräisesti ylimääräisten/vieraiden hiukkasten ja liuoksen ulkonäön muutosten varalta. Jos tällaista havaitaan, hävitä lääkevalmiste.
Ennen liuotetun SOMAVERT-injektionesteen vetämistä injektiopullosta, käännä injektiopullo ylösalaisin ruiskun ollessa edelleen kiinnitettynä siihen, ja varmista, että sulkimessa oleva aukko on näkyvissä alla olevan kuvan mukaisesti.
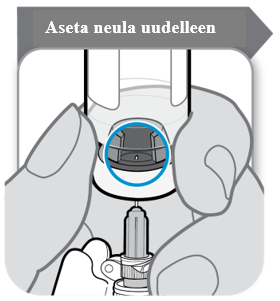
Vedä neulaa alaspäin, niin että neulan kärki on mahdollisimman alhaalla nesteessä. Vedä ruiskun mäntää hitaasti taaksepäin, jotta lääke saadaan injektiopullosta. Jos ruiskussa näkyy ilmaa, naputtele säiliötä kuplien saamiseksi ruiskun yläosaan ja työnnä kuplat sitten varovasti pois ruiskusta injektiopulloon.
Ennen ruiskun ja neulan hävittämistä käännä turvasuojus neulan päälle ja varmista, että se napsahtaa paikoilleen. Ruiskua ja neulaa ei saa koskaan käyttää uudelleen.
Vain kertakäyttöön. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SOMAVERT injektiokuiva-aine ja liuotin, liuosta varten
10 mg 30 x 1 kpl
15 mg 30 x 1 kpl
20 mg 30 x 1 kpl
25 mg 30 x 1 kpl
30 mg 30 x 1 kpl
- Peruskorvaus (40 %).
ATC-koodi
H01AX01
Valmisteyhteenvedon muuttamispäivämäärä
16.06.2022
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com