
SIMPONI injektioneste, liuos, esitäytetty kynä 50 mg/0,5 ml, injektioneste, liuos, esitäytetty ruisku 50 mg/0,5 ml
Vaikuttavat aineet ja niiden määrät
Simponi 50 mg injektioneste, liuos, esitäytetty kynä
Yksi 0,5 ml esitäytetty kynä sisältää 50 mg golimumabia*.
Simponi 50 mg injektioneste, liuos, esitäytetty ruisku
Yksi 0,5 ml esitäytetty ruisku sisältää 50 mg golimumabia*.
* Ihmisen IgG1κ monoklonaalinen vasta-aine, joka on tuotettu rekombinantti DNA-teknologialla hiiren hybridoomasolulinjassa.
Apuaineet, joiden vaikutus tunnetaan
Yksi esitäytetty kynä sisältää 20,5 mg sorbitolia 50 mg:n annosta kohti.
Yksi esitäytetty ruisku sisältää 20,5 mg sorbitolia 50 mg:n annosta kohti.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos, esitäytetty kynä (injektioneste), SmartJect
Injektioneste, liuos, esitäytetty ruisku (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Nivelreuma
Simponi, yhdessä metotreksaatin kanssa, on tarkoitettu:
- kohtalaisen tai vaikean, aktiivisen nivelreuman hoitoon aikuisille, kun tautiprosessia hidastavilla reumalääkkeillä (DMARD-lääkkeillä), mukaan lukien metotreksaatti, on saatu riittämätön vaste.
- vaikean, aktiivisen ja etenevän nivelreuman hoitoon aikuisille, joita ei ole aiemmin hoidettu metotreksaatilla.
Simponin, yhdessä metotreksaatin kanssa, on röntgenkuvien perusteella osoitettu hidastavan nivelvaurioiden etenemisnopeutta sekä parantavan fyysistä toimintakykyä.
Juveniili idiopaattinen artriitti
Idiopaattinen juveniili polyartriitti (pJIA)
Simponi, yhdessä metotreksaatin kanssa, on tarkoitettu idiopaattisen juveniilin polyartriitin hoitoon 2 vuotta täyttäneille lapsille, kun metotreksaatilla saatu vaste on ollut riittämätön.
Nivelpsoriaasi
Simponi, yksin tai yhdessä metotreksaatin kanssa, on tarkoitettu aktiivisen ja etenevän nivelpsoriaasin hoitoon aikuisille, kun DMARD-lääkkeillä on saatu riittämätön vaste. Simponin on röntgenkuvien perusteella osoitettu hidastavan perifeerisen nivelvaurion etenemistä nivelpsoriaasipotilailla, joilla on symmetrinen moniniveltulehdus (ks. kohta Farmakodynamiikka), sekä parantavan fyysistä toimintakykyä.
Aksiaalinen spondylartriitti
Selkärankareuma
Simponi on tarkoitettu vaikean, aktiivisen selkärankareuman hoitoon aikuisille, kun tavanomaisilla hoitomuodoilla saatu vaste on ollut riittämätön.
Röntgennegatiivinen aksiaalinen spondylartriitti
Simponi on tarkoitettu vaikean, aktiivisen röntgennegatiivisen aksiaalisen spondylartriitin hoitoon aikuisille, joilla on selviä tulehduslöydöksiä, jotka näkyvät suurentuneena C-reaktiivisen proteiinin (CRP) pitoisuutena ja/tai magneettitutkimuksessa, silloin kun tulehduskipulääkkeillä (NSAIDit) ei ole saatu riittävää hoitovastetta tai kun potilas ei siedä niitä.
Haavainen koliitti
Simponi on tarkoitettu kohtalaisen tai vaikean aktiivisen haavaisen koliitin (haavaisen paksusuolitulehduksen) hoitoon aikuisille potilaille, kun tavanomaisilla hoitomuodoilla, kuten kortikosteroideilla ja 6-merkaptopuriinilla tai atsatiopriinilla, saatu vaste on ollut riittämätön tai potilas ei siedä näitä hoitoja tai lääketieteellinen vasta-aihe estää niiden käytön.
Ehto
Hoidon saavat aloittaa valmisteyhteenvedossa mainittuun indikaatioon perehtyneet lääkärit.
Annostus ja antotapa
Hoidon aloittavan ja sitä valvovan lääkärin pitää olla perehtynyt nivelreuman, idiopaattisen juveniilin polyartriitin, nivelpsoriaasin, selkärankareuman, röntgennegatiivisen aksiaalisen spondylartriitin tai haavaisen koliitin diagnosointiin ja hoitoon. Simponi-hoitoa saaville potilaille annetaan erityinen potilaskortti.
Annostus
Nivelreuma
50 mg Simponia annetaan kerran kuukaudessa, aina samana päivänä kuukaudesta.
Simponi annetaan yhdessä metotreksaatin kanssa.
Nivelpsoriaasi, selkärankareuma tai röntgennegatiivinen aksiaalinen spondylartriitti
50 mg Simponia annetaan kerran kuukaudessa, aina samana päivänä kuukaudesta.
Kaikissa yllä mainituissa käyttöaiheissa käytettävissä olevien tietojen mukaan kliininen vaste saavutetaan yleensä 12 – 14 viikon kuluessa hoidon aloituksesta (3 – 4 annoksen jälkeen). Hoidon jatkamista pitää harkita uudelleen niille potilaille, joilla ei nähdä merkkejä hoidollisesta hyödystä tänä aikana.
Potilaat, jotka painavat yli 100 kg
Kaikissa yllä mainituissa käyttöaiheissa yli 100 kg painaville potilaille, joilla on nivelreuma, nivelpsoriaasi, selkärankareuma tai röntgennegatiivinen aksiaalinen spondylartriitti ja joilla ei saavuteta riittävää kliinistä vastetta 3 – 4 annoksen jälkeen, golimumabiannoksen nostamista tasolle 100 mg kerran kuukaudessa voidaan harkita, ottaen huomioon 100 mg:n annokseen liittyvien tiettyjen vakavien haittavaikutusten riskin suurenemisen verrattuna 50 mg:n annokseen (ks. kohta Haittavaikutukset). On tarpeen harkita uudelleen hoidon jatkamista niille potilaille, joilla ei 3–4:n 100 mg:n lisäannoksen jälkeen nähdä merkkejä hoidollisesta hyödystä.
Haavainen koliitti
Alle 80 kg painavat potilaat
Simponin aloitusannos on 200 mg, jonka jälkeen annetaan 100 mg viikolla 2. Potilaille, joilla vaste on riittävä, annetaan 50 mg viikolla 6 ja sen jälkeen 4 viikon välein. Potilaat, joilla vaste ei ole riittävä, saattavat hyötyä hoidon jatkamisesta 100 mg:n annoksella viikolla 6 ja sen jälkeen 4 viikon välein (ks. kohta Farmakodynamiikka).
80 kg tai enemmän painavat potilaat
Simponin aloitusannos on 200 mg, jonka jälkeen annetaan 100 mg viikolla 2 ja sen jälkeen 100 mg 4 viikon välein (ks. kohta Farmakodynamiikka).
Ylläpitohoidon aikana kortikosteroidiannosta voidaan pienentää asteittain hoitosuositusten mukaisesti.
Käytettävissä olevien tietojen mukaan kliininen vaste saavutetaan yleensä 12–14 viikon kuluessa hoidon aloituksesta (4 annoksen jälkeen). Hoidon jatkamista pitää harkita uudelleen niille potilaille, joilla ei nähdä merkkejä hoidollisesta hyödystä tänä aikana.
Annoksen unohtaminen
Jos potilas unohtaa pistää Simponi-annoksen suunniteltuna päivänä, unohtunut annos pistetään heti kun potilas sen muistaa. Potilasta pitää neuvoa, että hän ei saa ottaa kaksinkertaista annosta korvatakseen unohtamansa annoksen.
Seuraava annos otetaan alla olevien ohjeiden mukaan:
- jos annos on vähemmän kuin 2 viikkoa myöhässä, potilas pistää unohtuneen annoksen ja jatkaa alkuperäisen aikataulun mukaan.
- jos annos on enemmän kuin 2 viikkoa myöhässä, potilas pistää unohtuneen annoksen ja tästä pistospäivästä alkaen laaditaan uusi aikataulu.
Erityiset potilasryhmät
Iäkkäät potilaat (≥ 65-vuotiaat)
Annoksen muuttaminen iäkkäille potilaille ei ole tarpeen.
Munuaisten ja maksan toiminnan heikkeneminen
Simponilla ei ole tehty tutkimuksia näissä potilasryhmissä. Annossuosituksia ei voida antaa.
Pediatriset potilaat
Simponin turvallisuutta ja tehoa alle 18 vuoden ikäisten potilaiden hoidossa ei ole varmistettu muissa käyttöaiheissa kuin idiopaattisessa juveniilissa polyartriitissa.
Idiopaattinen juveniili polyartriitti
50 mg Simponia annetaan kerran kuukaudessa, aina samana päivänä kuukaudesta, vähintään 40 kg painaville lapsille. Alle 40 kg painaville idiopaattista juveniilia polyartriittia sairastaville lapsille on saatavana 45 mg/0,45 ml esitäytetty kynä.
Käytettävissä olevien tietojen mukaan kliininen vaste saavutetaan yleensä 12‑14 viikon kuluessa hoidon aloituksesta (3‑4 annoksen jälkeen). Hoidon jatkamista pitää harkita uudelleen niille lapsille, joilla ei nähdä merkkejä hoidollisesta hyödystä tänä aikana.
Antotapa
Simponi annetaan ihon alle. Kun potilas on saanut riittävän opetuksen pistotekniikasta, hän voi itse pistää lääkkeen, jos lääkäri pitää sitä tarkoituksenmukaisena ja potilaan tilaa seurataan tarpeen mukaan. Potilasta neuvotaan pistämään koko Simponi-määrä pakkausselosteessa olevien yksityiskohtaisten käyttöohjeiden mukaan. Jos tarvitaan useita pistoksia, ne on pistettävä kehon eri alueille.
Ohjeet lääkkeen antamisesta, ks. kohta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiivinen tuberkuloosi tai jokin muu vaikea infektio kuten sepsis, ja opportunistiset infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kohtalainen tai vaikea sydämen vajaatoiminta (NYHA:n luokat III/IV) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Potilaita on tarkkailtava huolellisesti infektioiden, mukaan lukien tuberkuloosin, varalta ennen golimumabihoitoa, sen aikana ja sen päätyttyä. Koska golimumabin eliminaatio saattaa kestää jopa 5 kuukautta, potilaan seurantaa jatketaan koko tämän ajan. Golimumabihoitoa ei enää jatkossa saa antaa, jos potilaalle kehittyy vakava infektio tai sepsis (ks. kohta Vasta-aiheet).
Golimumabia ei pidä antaa potilaille, joilla on kliinisesti merkittävä, aktiivinen infektio. Varovaisuutta on noudatettava, jos golimumabin käyttöä harkitaan potilaille, joilla on krooninen infektio tai joilla on ollut toistuva infektio. Potilaita pitää neuvoa välttämään altistumista mahdollisille infektion riskitekijöille tilanteen mukaan.
Potilaat, jotka käyttävät TNF-estäjiä, ovat alttiimpia saamaan vakavia infektioita.
Golimumabia saaneilla potilailla on raportoitu bakteeri-infektioita (mukaan lukien sepsis ja keuhkokuume), mykobakteeri-infektioita (mukaan lukien tuberkuloosi), invasiivisia sieni-infektioita sekä opportunistisia infektioita, myös kuolemaan johtaneita. Joitain näistä vakavista infektioista on ilmennyt potilailla, jotka ovat saaneet samanaikaista immunosuppressiivista hoitoa, mikä taustalla olevan sairauden lisäksi saattaa altistaa heidät infektioille. Potilaita, joille kehittyy uusi infektio golimumabihoidon aikana, pitää tarkkailla huolellisesti ja heille tehdään täydellinen diagnostinen arviointi. Golimumabihoito pitää keskeyttää, jos potilaalle kehittyy uusi vakava infektio tai sepsis, ja hänelle aloitetaan asianmukainen antimikrobinen tai antifungaalinen hoito, kunnes infektio on saatu hallintaan.
Potilailla, jotka ovat asuneet tai matkustaneet alueilla, joilla invasiivisia sieni-infektioita kuten histoplasmoosia, kokkidioidomykoosia tai blastomykoosia esiintyy endeemisenä, pitää golimumabihoidon hyötyjä ja riskejä pohtia huolellisesti ennen hoidon aloitusta. Jos golimumabihoitoa saaneille riskiryhmän potilaille kehittyy vakava systeeminen sairaus, on syytä epäillä invasiivista sieni-infektiota. Diagnosointi ja empiirisen antifungaalisen hoidon antaminen näille potilaille pitää tehdä konsultoimalla invasiivisten sieni-infektioiden hoitoon erikoistunutta lääkäriä, mikäli mahdollista.
Tuberkuloosi
Golimumabia saaneilla potilailla on raportoitu tuberkuloosia. Merkille pantavaa on, että suurimmassa osassa näistä raporteista tuberkuloosi oli muualle kuin keuhkoihin paikantuvaa, ilmeten joko paikallisena tai laajalle levinneenä tautina.
Ennen golimumabihoidon aloitusta kaikilta potilailta on arvioitava sekä aktiivinen että inaktiivinen (latentti) tuberkuloosi. Tähän arviointiin pitää kuulua yksityiskohtainen selvitys potilaan aiemmista sairauksista sekä henkilökohtainen tuberkuloositausta tai mahdolliset aiemmat tuberkuloosikontaktit sekä aiempi ja/tai nykyinen immunosuppressiivinen hoito. Kaikille potilaille pitää tehdä (paikallisia suosituksia soveltaen) asianmukaiset seulontakokeet, ts. tuberkuliini-ihokoe tai -verikoe ja keuhkoröntgen. On suositeltavaa, että näiden testien suorittaminen kirjataan potilaalle annettavaan erityiseen potilaskorttiin. Lääkettä määräävän lääkärin on syytä muistaa, että tuberkuliini-ihokokeesta on mahdollista saada vääriä negatiivisia tuloksia, erityisesti vaikeasti sairailla potilailla tai immunosuppressoiduilla potilailla.
Jos potilaalla todetaan aktiivinen tuberkuloosi, golimumabihoitoa ei saa aloittaa (ks. kohta Vasta-aiheet).
Jos epäillään latenttia tuberkuloosia, on syytä konsultoida tuberkuloosin hoitoon perehtynyttä lääkäriä. Kaikissa alla kuvatuissa tilanteissa on golimumabihoidon hyöty/haitta -suhdetta arvioitava erittäin huolellisesti.
Jos todetaan inaktiivinen (latentti) tuberkuloosi, latentin tuberkuloosin hoitoon on aloitettava tuberkuloosilääkitys paikallisten hoitosuositusten mukaisesti ennen golimumabihoidon aloitusta.
Potilaille, joilla on useita tai merkittäviä tuberkuloosin riskitekijöitä ja joilla tulos latentin tuberkuloosin seulontakokeesta on negatiivinen, on syytä harkita tuberkuloosilääkitystä ennen golimumabihoidon aloitusta. Tuberkuloosilääkitystä on syytä harkita myös ennen golimumabihoidon aloitusta potilaille, joilla on aiemmin ollut latentti tai aktiivinen tuberkuloosi, ja joilla asianmukaisen lääkekuurin antoa ei voida varmistaa.
Aktiivista tuberkuloosia on ilmaantunut golimumabihoitoa saaville potilaille hoidettaessa latenttia tuberkuloosia tai sen jälkeen. Golimumabia saavia potilaita on tarkkailtava huolellisesti, jotta havaitaan aktiivisen tuberkuloosin merkit ja oireet. Tämä koskee myös potilaita, joilla latentin tuberkuloosin testi oli negatiivinen, sekä potilaita, joita hoidetaan latentin tuberkuloosin vuoksi ja potilaita, jotka ovat aiemmin saaneet hoitoa tuberkuloosi-infektion vuoksi.
Kaikkia potilaita pitää neuvoa ottamaan yhteyttä terveydenhoitohenkilökuntaan, jos golimumabihoidon aikana tai sen jälkeen ilmenee mahdollisia tuberkuloosin merkkejä ja/tai oireita (esimerkiksi itsepintaista yskää, kuihtumista/painon alenemista, lievää kuumetta).
Hepatiitti B -viruksen reaktivaatio
Hepatiitti B:n reaktivaatiota on ilmennyt potilailla, jotka ovat saaneet TNF-antagonistia mukaan lukien golimumabia ja jotka ovat tämän viruksen kroonisia kantajia (ts. pinta-antigeenipositiivisia). Jotkut tapauksista ovat johtaneet kuolemaan.
Potilailta pitää testata HBV-infektio ennen golimumabihoidon aloitusta. Jos HBV-infektiotestin tulos on positiivinen, on suositeltavaa konsultoida hepatiitti B:n hoitoon erikoistunutta lääkäriä.
HBV:n kantajia, joille golimumabihoito on tarpeen, pitää tarkkailla huolellisesti aktiivisen HBV-infektion merkkien ja oireiden varalta hoidon ajan sekä useita kuukausia hoidon lopettamisen jälkeen. Ei ole olemassa riittäviä tietoja HBV:tä kantavien potilaiden HBV-reaktivaation estosta käytettäessä viruslääkkeitä samanaikaisesti TNF-antagonistihoidon kanssa. Jos potilaalle kehittyy HBV-infektion reaktivaatio, golimumabihoito lopetetaan ja potilaalle aloitetaan tehokas antiviraalinen lääkitys sekä asianmukainen supportiivinen hoito.
Maligniteetit ja lymfoproliferatiiviset sairaudet
TNF-estäjien mahdollista osuutta maligniteettien kehittymisessä ei tiedetä. Nykytiedon perusteella lymfoomien, leukemian tai muiden maligniteettien kehittymisen mahdollisuutta ei voi sulkea pois TNF-antagonistia saaneilla potilailla. Varovaisuutta on syytä noudattaa harkittaessa TNF-estäjähoitoa potilaille, joilla on aiemmin todettu maligniteetti tai kun harkitaan hoidon jatkamista potilaille, joille kehittyy maligniteetti.
Pediatrinen maligniteetti
TNF-estäjiä saaneilla lapsilla, nuorilla ja nuorilla aikuisilla (22-vuotiailla ja sitä nuoremmilla) on markkinoillaolon aikaisessa seurannassa raportoitu maligniteetteja, joista osa on johtanut kuolemaan. Potilaat olivat aloittaneet hoidon viimeistään 18 vuoden iässä. Noin puolet tapauksista oli lymfoomia. Muut olivat erilaisia maligniteetteja ja niihin kuului harvinaisia maligniteetteja, jotka tavallisesti liittyvät immunosuppressioon. Maligniteettien kehittymisen mahdollisuutta ei voi sulkea pois TNF-estäjiä saaneilla lapsilla ja nuorilla.
Lymfooma ja leukemia
TNF-estäjillä, mukaan lukien golimumabi, tehtyjen kliinisten tutkimusten vertailevissa osioissa on TNF-estäjiä saaneilla potilailla todettu enemmän lymfoomia kuin verrokkiryhmän potilailla. Simponilla tehdyissä faasi IIb:n ja faasi III:n kliinisissä nivelreuma-, nivelpsoriaasi- ja selkärankareumatutkimuksissa lymfoomien ilmaantuvuus golimumabia saaneilla potilailla oli suurempi kuin normaaliväestössä on odotettavissa. Golimumabihoitoa saaneilla potilailla on raportoitu leukemiaa. Taustalla oleva lymfooma- ja leukemiariski on suurentunut nivelreumapotilailla, joilla on pitkäaikainen, hyvin aktiivinen ja tulehduksellinen tauti, mikä vaikeuttaa riskin arviointia.
Muita TNF-estäjiä saaneilla potilailla on markkinoille tulon jälkeen raportoitu harvoin hepatospleenistä T-solulymfoomaa (HSTCL) (ks. kohta Haittavaikutukset). Tämän harvinaisen T-solulymfoomatyypin taudinkulku on hyvin aggressiivinen ja johtaa yleensä kuolemaan. Suurin osa tapauksista on esiintynyt teini-ikäisillä ja nuorilla aikuisilla miehillä, joista lähes kaikki saivat samanaikaisesti atsatiopriinia (AZA) tai 6-merkaptopuriinia (6-MP) tulehduksellisen suolistosairauden hoitoon. AZA- tai 6-MP-hoidon ja golimumabin samanaikaisen käytön mahdollisia riskejä on punnittava huolellisesti. Hepatospleenisen T-solulymfooman riskiä ei voida sulkea pois TNF-estäjiä käytettäessä.
Maligniteetit, muut kuin lymfooma
Simponilla tehdyissä faasi IIb:n ja faasi III:n kliinisten tutkimusten vertailevissa osioissa nivelreumassa, nivelpsoriaasissa, selkärankareumassa ja haavaisessa koliitissa muiden maligniteettien kuin lymfooman ilmaantuvuus (pois lukien ei-melanooma ihosyöpä) oli sama golimumabiryhmässä ja verrokeilla.
Paksusuolen dysplasia / syöpä
Ei tiedetä, vaikuttaako golimumabihoito dysplasian tai paksusuolisyövän kehittymisen riskiin. Seulontatutkimuksia dysplasian toteamiseksi on tehtävä säännöllisin välein ennen hoidon aloittamista ja koko taudin keston ajan kaikille haavaista koliittia sairastaville potilaille, joilla on suurentunut dysplasian tai paksusuolisyövän riski (esimerkiksi potilaille, joilla on pitkäaikainen haavainen koliitti tai primaarinen sklerosoiva kolangiitti) tai joilla on aiemmin todettu dysplasia tai paksusuolisyöpä. Näihin seurantatutkimuksiin kuuluvat kolonoskopia ja biopsiat paikallisten suositusten mukaisesti. Jos golimumabia saavalla potilaalla on hiljattain diagnosoitu dysplasia, hoidon riskejä ja hyötyjä on punnittava tarkoin ja harkittava, pitäisikö potilaan hoitoa jatkaa.
Eksploratiivisessa kliinisessä tutkimuksessa, jossa arvioitiin golimumabin käyttöä potilaille, joilla on vaikea itsepintainen astma, raportoitiin enemmän maligniteetteja golimumabia saaneilla potilailla kuin verrokeilla (ks. kohta Haittavaikutukset). Tämän havainnon merkitystä ei tiedetä.
Eksploratiivisessa kliinisessä tutkimuksessa, jossa arvioitiin toisen anti-TNF-aineen infliksimabin käyttöä keskivaikeaa tai vaikeaa kroonista keuhkoahtaumatautia (COPD) sairastavilla potilailla, infliksimabia saaneilla raportoitiin enemmän maligniteetteja, lähinnä keuhkoissa tai pään ja kaulan alueella, kuin verrokkiryhmässä. Kaikki potilaat olivat tupakoineet runsaasti. Varovaisuutta on siksi noudatettava käytettäessä mitä tahansa TNF-antagonistia COPD-potilaille, kuten myös potilaille, joilla maligniteettiriski on kohonnut runsaan tupakoinnin vuoksi.
Ihosyövät
Melanoomaa ja merkelinsolukarsinoomaa on raportoitu TNF-salpaajahoitoa, mukaan lukien golimumabia, saaneilla potilailla (ks. kohta Haittavaikutukset). On suositeltavaa tehdä säännöllinen ihotutkimus, erityisesti potilaille, joilla on ihosyövän riskitekijöitä.
Sydämen vajaatoiminta
TNF-estäjiä, mukaan lukien golimumabia, saaneilla potilailla on raportoitu sydämen vajaatoiminnan pahenemista sekä sydämen vajaatoiminnan uusia ilmaantumisia. Jotkut tapaukset ovat johtaneet kuolemaan. Kliinisessä tutkimuksessa toisella TNF-antagonistilla havaittiin sydämen vajaatoiminnan lisääntymistä sekä sydämen vajaatoiminnasta johtuvan kuolleisuuden lisääntymistä. Golimumabia ei ole tutkittu potilailla, joilla on sydämen vajaatoiminta. Golimumabia on käytettävä varoen potilaille, joilla on lievä sydämen vajaatoiminta (NYHA:n luokat I/II). Potilaiden tilaa pitää seurata tarkoin ja golimumabihoito on keskeytettävä niillä potilailla, joille ilmaantuu uusia tai pahenevia sydämen vajaatoiminnan oireita (ks. kohta Vasta-aiheet).
Neurologiset tapahtumat
TNF-estäjien käyttöön, mukaan lukien golimumabi, on liittynyt keskushermoston demyelinisoivien sairauksien, mukaan lukien multippeli skleroosin ja perifeerisen demyelinisoivan sairauden, ilmaantumisia tai kliinisten oireiden ja/tai röntgenologisten löydösten pahenemisia. Jos potilaalla on aiempi tai äskettäin puhjennut demyelinisoiva sairaus, anti-TNF-hoidon hyödyt ja haitat on arvioitava huolellisesti ennen golimumabihoidon aloitusta. Golimumabihoidon lopettamista pitää harkita, jos näitä sairauksia ilmaantuu (ks. kohta Haittavaikutukset).
Kirurgiset toimenpiteet
Golimumabihoitoa saaneille potilaille tehdyistä kirurgisista toimenpiteistä, mukaan lukien artroplastia, on vain rajoitetusti turvallisuustietoa. Pitkä puoliintumisaika pitää ottaa huomioon, jos suunnitellaan kirurgista toimenpidettä. Kirurgista hoitoa tarvitsevaa potilasta, joka saa golimumabia, on tarkkailtava huolellisesti infektioiden varalta. Tilanteen mukaan on ryhdyttävä asianmukaisiin toimenpiteisiin.
Immunosuppressio
On mahdollista, että TNF-estäjät, mukaan lukien golimumabi, vaikuttavat puolustusmekanismeihin infektioita ja maligniteetteja vastaan, sillä TNF toimii välittäjänä tulehduksessa ja muuntaa soluvälitteisiä immuunivasteita.
Autoimmuuniprosessi
Anti-TNF-hoidon aiheuttama suhteellinen TNFα-vaje saattaa johtaa autoimmuuniprosessin käynnistymiseen. Jos golimumabihoitoa saaneelle potilaalle kehittyy lupuksen kaltaiseen oireyhtymään viittaavia oireita ja jos potilaalle kehittyy vasta-aineita kaksisäikeiselle DNA:lle, golimumabihoito pitää lopettaa (ks. kohta Haittavaikutukset).
Hematologiset reaktiot
TNF-estäjiä, mukaan lukien golimumabi, saaneilla potilailla on raportoitu pansytopeniaa, leukopeniaa, neutropeniaa, agranulosytoosia, aplastista anemiaa ja trombosytopeniaa. Kaikkia potilaita pitää neuvoa hakeutumaan välittömästi lääkärin hoitoon, jos heille kehittyy veren dyskrasiaan viittaavia merkkejä ja oireita (esim. itsepintaista kuumetta, mustelmia, verenvuotoa, kalpeutta). Golimumabihoidon lopettamista pitää harkita potilaille, joilla on varmistettu olevan merkittäviä hematologisia poikkeamia.
TNF-estäjien ja anakinran samanaikainen anto
Kliinisissä tutkimuksissa todettiin vakavia infektioita ja neutropeniaa käytettäessä samanaikaisesti anakinraa ja toista TNF-estäjää, etanerseptiä, eikä yhdistelmähoidolla saavutettu kliinistä lisähyötyä. Tällä yhdistelmähoidolla havaittujen haittavaikutusten luonteen huomioiden samanlaista toksisuutta voi olla seurauksena käytettäessä anakinraa myös yhdessä muiden TNF-estäjien kanssa. Golimumabin antamista samanaikaisesti anakinran kanssa ei suositella.
TNF-estäjien ja abataseptin samanaikainen anto
Kliinisissä tutkimuksissa TNF-estäjien ja abataseptin samanaikaiseen käyttöön on liittynyt infektioiden, mukaan lukien vakavien infektioiden, riskin lisääntyminen verrattuna hoitoon pelkillä TNF-estäjillä, eikä yhdistelmähoidolla saavutettu kliinistä lisähyötyä. Golimumabin antamista samanaikaisesti abataseptin kanssa ei suositella.
Muiden biologisten lääkkeiden samanaikainen anto
Golimumabin käytöstä samanaikaisesti muiden, samaan käyttöaiheeseen kuin golimumabi tarkoitettujen biologisten lääkkeiden kanssa ei ole riittävästi tietoa. Golimumabin samanaikaista käyttöä näiden biologisten lääkkeiden kanssa ei suositella johtuen infektioriskin suurenemisen mahdollisuudesta sekä muista mahdollisista farmakologisista yhteisvaikutuksista.
Vaihtaminen biologisesta DMARD-lääkkeestä toiseen
Varovaisuutta on noudatettava ja potilaiden seurantaa jatkettava, kun vaihdetaan biologisesta lääkevalmisteesta toiseen, sillä biologisten aktiivisuuksien päällekkäisyys saattaa entisestään lisätä haittavaikutusten, muun muassa infektioiden, riskiä.
Rokotukset / hoidolliset tartunnanaiheuttajat
Golimumabihoitoa saaville potilaille voidaan antaa samanaikaisesti rokotuksia eläviä rokotteita lukuun ottamatta (ks. kohdat Yhteisvaikutukset sekä Raskaus ja imetys). Anti-TNF-hoitoa saavista potilaista on vain rajallisesti tietoa vaikutuksista rokotevasteeseen elävillä rokotteilla tai näiden rokotteiden sisältämien mikrobien aiheuttamista sekundääri-infektioista. Elävien rokotteiden käyttö voi johtaa kliinisiin infektioihin, mukaan lukien laajalle levinneisiin infektioihin.
Hoidollisten tartunnanaiheuttajien, kuten elävien heikennettyjen bakteerien, muunlainen käyttö (esim. BCG-rokote virtsarakon instillaatioon syövän hoidossa) voi johtaa kliinisiin infektioihin, mukaan lukien laajalle levinneisiin infektioihin. On suositeltavaa, että hoidollisia tartunnanaiheuttajia ei anneta samanaikaisesti golimumabin kanssa.
Allergiset reaktiot
Markkinoillaolon aikaisessa käytössä on golimumabin antamisen jälkeen raportoitu vakavia systeemisiä yliherkkyysreaktioita (mukaan lukien anafylaktinen reaktio). Jotkut näistä reaktioista ilmenivät ensimmäisen golimumabiannoksen jälkeen. Jos ilmenee anafylaktinen reaktio tai muita vakavia allergisia reaktioita, golimumabin antaminen pitää keskeyttää välittömästi ja aloittaa asianmukainen hoito.
Herkkyys lateksille
Esitäytetyn kynän tai esitäytetyn ruiskun neulan suojus on valmistettu kuivasta luonnonkumista, joka sisältää lateksia. Se saattaa aiheuttaa allergisen reaktion lateksille herkistyneillä henkilöillä.
Erityiset potilasryhmät
Iäkkäät potilaat (≥ 65-vuotiaat)
Nivelreumassa, nivelpsoriaasissa, selkärankareumassa ja haavaisessa koliitissa tehdyissä faasi III:n tutkimuksissa ei havaittu kokonaiseroja haittatapahtumissa, vakavissa haittatapahtumissa tai vakavissa infektioissa 65-vuotiailla ja sitä vanhemmilla potilailla, jotka saivat golimumabia, kun heitä verrattiin nuorempiin potilaisiin. Varovaisuutta on kuitenkin syytä noudattaa hoidettaessa iäkkäitä potilaita ja on kiinnitettävä erityistä huomiota infektioiden ilmaantumiseen. Röntgennegatiivista aksiaalista spondylartriittia koskevaan tutkimukseen osallistui vain alle 45-vuotiaita potilaita.
Munuaisten ja maksan toiminnan heikkeneminen
Golimumabilla ei ole tehty erityisiä tutkimuksia potilailla, joilla munuaisten tai maksan toimintakyky on heikentynyt. Golimumabia on käytettävä varoen potilaille, joilla maksan toiminta on heikentynyt (ks. kohta Annostus ja antotapa).
Pediatriset potilaat
Rokotukset
On suositeltavaa, että lapsipotilaiden rokotukset saatetaan mahdollisuuksien mukaan rokotussuositusten mukaisesti ajan tasalle ennen golimumabihoidon aloittamista (ks. edellä kohta Rokotukset / hoidolliset tartunnanaiheuttajat).
Apuaineet
Simponi sisältää sorbitolia (E420). Samanaikaisesti käytettävien sorbitolia (tai fruktoosia) sisältävien valmisteiden ja ravinnosta saatavan sorbitolin (tai fruktoosin) additiivinen vaikutus on otettava huomioon hoidettaessa potilaita, joilla on harvinainen perinnöllinen fruktoosi‑intoleranssi (ks. kohta Vaikuttavat aineet ja niiden määrät).
Lääkitysvirheen mahdollisuus
Simponilla on myyntilupa kahdelle ihon alle annettavalle vahvuudelle, 50 mg ja 100 mg. On tärkeää, että käytetään oikeaa vahvuutta, jotta saadaan annostusohjeen mukainen oikea annos (ks. kohta Annostus ja antotapa). On noudatettava huolellisuutta, että potilas saa oikean vahvuuden, jotta varmistetaan, ettei hän saa ali- tai yliannosta.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Samanaikainen käyttö muiden biologisten lääkkeiden kanssa
Yhdistelmähoitoa golimumabilla ja muilla golimumabin kanssa samaan käyttöaiheeseen annettavilla biologisilla lääkkeillä, mukaan lukien anakinra ja abatasepti, ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Elävät rokotteet / hoidolliset tartunnanaiheuttajat
Eläviä rokotteita ei pidä antaa samanaikaisesti golimumabihoidon kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet sekä Raskaus ja imetys).
Hoidollisia tartunnanaiheuttajia ei pidä antaa samanaikaisesti golimumabin kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Metotreksaatti
Vaikka metotreksaatin samanaikainen käyttö nostaa golimumabin vakaan tilan pienimpiä (trough) pitoisuuksia nivelreuma-, nivelpsoriaasi- ja selkärankareumapotilaissa, käytettävissä olevien tietojen mukaan ei ole tarvetta golimumabin tai metotreksaatin annosten muuttamiseen (ks. kohta Farmakokinetiikka).
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset
Hedelmällisessä iässä olevien naisten on käytettävä asianmukaista raskauden ehkäisyä ja jatkettava sen käyttöä vähintään 6 kuukauden ajan viimeisen golimumabihoidon jälkeen.
Raskaus
Raskauden aikaisesta altistumisesta golimumabille on olemassa kohtalaisen laajat tiedot. Saatavilla on prospektiivisesti kerätyt tiedot noin 400 raskaudesta, jotka päättyivät elävän lapsen syntymään ja joissa lopputulos on tiedossa. Näistä raskauksista 220:ssä altistuminen oli tapahtunut ensimmäisellä raskauskolmanneksella. Pohjois-Euroopassa tehdyssä väestöpohjaisessa tutkimuksessa, jossa arvioitiin tietoja 131 raskaudesta (ja 134 lapsesta), merkittäviä synnynnäisiä epämuodostumia todettiin kuudella 134:stä Simponi-valmisteelle kohdussa altistuneesta lapsesta (4,5 %:lla). Ei-biologiselle systeemiselle hoidolle kohdussa altistuneilla lapsilla vastaava osuus oli 599 / 10 823 (5,5 %) ja tutkimuksen taustaväestössä 4,6 %. Sekoittavien tekijöiden suhteen korjattu kerroinsuhde oli 0,79 (95 %:n luottamusväli 0,35–1,81) verrattaessa Simponi-valmisteelle altistuneita lapsia ei-biologiselle systeemiselle hoidolle altistuneisiin lapsiin ja 0,95 (95 %:n luottamusväli 0,42–2,16) verrattaessa Simponi-valmisteelle altistuneita lapsia taustaväestöön.
Johtuen TNF-inhibitiosta, raskauden aikana annettu golimumabi saattaa vaikuttaa vastasyntyneen normaaliin immuunivasteeseen. Eläinkokeiden perusteella ei ole saatu tietoa suorista tai epäsuorista haitallisista vaikutuksista raskauteen, alkion/sikiön kehitykseen, synnytykseen tai postnataaliseen kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta). Kliinistä kokemusta on vain vähän. Golimumabia pitäisi käyttää raskauden aikana vain, jos se on selvästi tarpeen.
Golimumabi läpäisee istukan. Kun TNF:n toimintaa estävää monoklonaalista vasta-ainetta on annettu raskauden aikana, vasta-ainetta on havaittu hoitoa saaneille naisille syntyneiden lasten seerumista jopa 6 kuukauden ajan. Näillä lapsilla saattaa infektioriski täten olla suurentunut. Elävien rokotteiden antamista lapsille, jotka ovat altistuneet golimumabille kohdussa, ei suositella 6 kuukauteen äidin viimeisen raskaudenaikaisen golimumabi-injektion jälkeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet sekä Yhteisvaikutukset).
Imetys
Ei tiedetä, erittyykö golimumabi rintamaitoon tai imeytyykö se rintamaitoa nauttineen lapsen elimistöön. Golimumabin osoitettiin kulkeutuvan rintamaitoon apinoilla ja koska humaani-immunoglobuliinit erittyvät rintamaitoon, nainen ei saa imettää golimumabihoidon aikana eikä vähintään 6 kuukauteen golimumabihoidon jälkeen.
Hedelmällisyys
Golimumabilla ei ole tehty hedelmällisyystutkimuksia eläimillä. Hedelmällisyystutkimuksessa, joka tehtiin hiirillä käyttäen analogista vasta-ainetta, joka selektiivisesti estää hiirten TNF-α:n funktionaalista aktiviteettia, ei havaittu merkittävää vaikutusta hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Simponi-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Simponin annon jälkeen saattaa kuitenkin ilmetä heitehuimausta (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Ylähengitystieinfektio oli yleisin raportoitu haittavaikutus nivelreuman, nivelpsoriaasin, selkärankareuman, röntgennegatiivisen aksiaalisen spondylartriitin ja haavaisen koliitin keskeisten tutkimusten vertailevassa vaiheessa. Sitä ilmeni 12,6 %:lla golimumabia saaneista potilaista ja 11,0 %:lla verrokkiryhmän potilaista. Vakavimpia golimumabilla raportoituja haittavaikutuksia ovat vakavat infektiot (mukaan lukien sepsis, keuhkokuume, tuberkuloosi, invasiiviset sieni-infektiot ja opportunistiset infektiot), demyelinisoivat sairaudet, B-hepatiitin reaktivaatio, sydämen vajaatoiminta, autoimmuuniprosessit (lupuksen kaltainen oireyhtymä), hematologiset reaktiot, vakavat systeemiset yliherkkyysreaktiot (mukaan lukien anafylaktinen reaktio), vaskuliitti, lymfooma ja leukemia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Taulukossa 1 on lueteltu haittavaikutukset, joita ilmeni kliinisissä tutkimuksissa ja joita on raportoitu golimumabin markkinoillaolon aikaisessa maailmanlaajuisessa käytössä. Haittavaikutukset on lueteltu elinjärjestelmäluokkien mukaan käyttäen seuraavanlaista esiintymistiheyden luokittelua: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Kaikissa esiintymistiheysryhmissä haittavaikutukset on lueteltu vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1
Taulukoitu luettelo haittavaikutuksista
Infektiot | |
Hyvin yleinen: | Ylähengitystieinfektio (nasofaryngiitti, nielutulehdus, kurkunpäätulehdus ja riniitti) |
Yleinen: | Bakteeri-infektiot (kuten selluliitti), alahengitystieinfektio (kuten keuhkokuume), virusinfektiot (kuten influenssa ja herpes), keuhkoputkitulehdus, sinuiitti, pinnalliset sieni-infektiot, absessi |
Melko harvinainen: | Sepsis mukaan lukien septinen sokki, pyelonefriitti |
Harvinainen: | Tuberkuloosi, opportunistiset infektiot (kuten invasiiviset sieni-infektiot [histoplasmoosi, kokkidioidomykoosi, pneumokystoosi], bakteeri-infektiot, epätyypilliset mykobakteeri-infektiot ja alkueläinperäiset infektiot), B-hepatiitin reaktivaatio, bakteerin aiheuttama niveltulehdus, infektiivinen bursiitti |
Hyvän- ja pahanlaatuiset kasvaimet | |
Melko harvinainen: | Neoplasmat (kuten ihosyöpä, okasolusyöpä ja melanosyyttiluomi) |
Harvinainen: | Lymfooma, leukemia, melanooma, merkelinsolukarsinooma |
Tuntematon: | Hepatospleeninen T-solulymfooma*, Kaposin sarkooma |
Veri ja imukudos | |
Yleinen: | Leukopenia (mukaan lukien neutropenia), anemia |
Melko harvinainen: | Trombosytopenia, pansytopenia |
Harvinainen: | Aplastinen anemia, agranulosytoosi |
Immuunijärjestelmä | |
Yleinen: | Allergiset reaktiot (bronkospasmi, yliherkkyys, urtikaria), positiivinen autovasta-aine |
Harvinainen: | Vakava systeeminen yliherkkyysreaktio (mukaan lukien anafylaktinen reaktio), vaskuliitti (systeeminen), sarkoidoosi |
Umpieritys | |
Melko harvinainen: | Kilpirauhasen häiriö (kuten kilpirauhasen vajaatoiminta, kilpirauhasen liikatoiminta ja struuma) |
Aineenvaihdunta ja ravitsemus | |
Melko harvinainen: | Veren glukoosiarvon kohoaminen, lipidiarvon kohoaminen |
Psyykkiset häiriöt | |
Yleinen: | Masennus, unettomuus |
Hermosto | |
Yleinen: | Heitehuimaus, päänsärky, tuntoharha |
Melko harvinainen: | Tasapainohäiriöt |
Harvinainen: | Demyelinisoiva sairaus (sentraalinen sekä perifeerinen), makuhäiriö |
Silmät | |
Melko harvinainen: | Näköhäiriöt (kuten näön sumentuminen ja heikentynyt näöntarkkuus), sidekalvotulehdus, silmäallergia (kuten kutina ja ärsytys) |
Sydän | |
Melko harvinainen: | Rytmihäiriö, iskeeminen sepelvaltimosairaus |
Harvinainen: | Sydämen vajaatoiminta (uusi tai paheneva) |
Verisuonisto | |
Yleinen: | Kohonnut verenpaine |
Melko harvinainen: | Tromboosi (kuten laskimon tai aortan), punastuminen |
Harvinainen: | Raynaud’n oireyhtymä |
Hengityselimet, rintakehä ja välikarsina | |
Yleinen: | Astma ja vastaavanlaiset oireet (kuten hengityksen vinkuminen ja keuhkoputkien supistusherkkyys) |
Melko harvinainen: | Interstitiaali keuhkosairaus |
Ruoansulatuselimistö | |
Yleinen: | Dyspepsia, maha-suolikanavan sekä vatsan kipu, pahoinvointi, maha-suolikanavan tulehdukselliset sairaudet (kuten mahatulehdus ja koliitti), suutulehdus |
Melko harvinainen: | Ummetus, gastroesofageaalinen refluksitauti |
Maksa ja sappi | |
Yleinen: | Alaniiniaminotransferaasiarvon kohoaminen, aspartaattiaminotransferaasiarvon kohoaminen |
Melko harvinainen: | Sappikivitauti, maksan häiriöt |
Iho ja ihonalainen kudos | |
Yleinen: | Kutina, ihottuma, alopesia, ihotulehdus |
Melko harvinainen: | Rakkulainen ihoreaktio, psoriaasi (uusi tai olemassa olevan psoriaasin paheneminen, kämmenten/jalkapohjien sekä märkärakkulainen), urtikaria |
Harvinainen: | Jäkälää muistuttavat reaktiot, ihon eksfoliaatio, vaskuliitti (kutaaninen) |
| Tuntematon: | Dermatomyosiitin oireiden paheneminen |
Luusto, lihakset ja sidekudos | |
Harvinainen: | Lupuksen kaltainen oireyhtymä |
Munuaiset ja virtsatiet | |
Harvinainen: | Virtsarakon häiriöt, munuaiseen liittyvät häiriöt |
Sukupuolielimet ja rinnat | |
Melko harvinainen: | Rintoihin liittyvät häiriöt, kuukautisiin liittyvät häiriöt |
Yleisoireet ja antopaikassa todettavat haitat | |
Yleinen: | Kuume, voimattomuus, pistokohdan reaktio (kuten pistokohdan punoitus, urtikaria, kovettuma, kipu, mustelmat, kutina, ärsytys ja tuntoharha), epämukava tunne rinnassa |
Harvinainen: | Paranemisen hidastuminen |
Vammat ja myrkytykset | |
Yleinen: | Luunmurtumat |
* Havaittu muilla TNF-estäjillä. | |
Tässä osiossa golimumabin käyttöaika esitetään seuranta-ajan keskilukuna (mediaani, noin 4 vuotta) erittelemättä golimumabin annoksia. Jos golimumabin käyttöä kuvataan annoksittain, seuranta-ajan mediaani vaihtelee (noin 2 vuotta, kun annos on 50 mg, ja noin 3 vuotta, kun annos on 100 mg), sillä potilaat ovat saattaneet siirtyä annoksesta toiseen.
Valikoitujen haittavaikutusten kuvaus
Infektiot
Keskeisten tutkimusten vertailevassa vaiheessa ylähengitystieinfektio oli yleisin haittavaikutus, jota raportoitiin 12,6 %:lla golimumabia saaneista potilaista (ilmaantuvuus 100 potilasvuotta kohti: 60,8; 95 %:n luottamusväli: 55,0; 67,1) ja verrokkiryhmässä 11,0 %:lla (ilmaantuvuus 100 potilasvuotta kohti: 54,5; 95 %:n luottamusväli: 46,1; 64,0). Tutkimusten vertailevissa sekä ei‑vertailevissa osioissa, joissa seuranta‑ajan mediaani oli noin 4 vuotta, ylähengitystieinfektioiden ilmaantuvuus 100 potilasvuotta kohti oli 34,9 tapahtumaa; 95 %:n luottamusväli: 33,8; 36,0 golimumabia saaneilla potilailla.
Keskeisten tutkimusten vertailevassa vaiheessa infektioita havaittiin 23,0 %:lla golimumabia saaneista potilaista (ilmaantuvuus 100 potilasvuotta kohti: 132,0; 95 %:n luottamusväli: 123,3; 141,1) ja verrokkiryhmässä 20,2 %:lla (ilmaantuvuus 100 potilasvuotta kohti: 122,3; 95 %:n luottamusväli: 109,5; 136,2). Tutkimusten vertailevissa sekä ei‑vertailevissa osioissa, joissa seuranta‑ajan mediaani oli noin 4 vuotta, infektioiden ilmaantuvuus 100 potilasvuotta kohti oli 81,1 tapahtumaa; 95 %:n luottamusväli: 79,5; 82,8 golimumabia saaneilla potilailla.
Keskeisten nivelreumaa, nivelpsoriaasia, selkärankareumaa ja röntgennegatiivista aksiaalista spondylartriittia koskevien tutkimusten vertailevassa vaiheessa vakavia infektioita havaittiin 1,2 %:lla golimumabia saaneista potilaista ja 1,2 %:lla verrokkiryhmässä. Vakavien infektioiden ilmaantuvuus seuranta‑ajan 100 potilasvuotta kohti nivelreumaa, nivelpsoriaasia, selkärankareumaa ja röntgennegatiivista aksiaalista spondylartriittia koskevien tutkimusten vertailevassa vaiheessa oli 7,3; 95 %:n luottamusväli: 4,6; 11,1 golimumabia 100 mg saaneiden ryhmässä, 2,9; 95 %:n luottamusväli: 1,2; 6,0 golimumabia 50 mg saaneiden ryhmässä ja 3,6; 95 %:n luottamusväli: 1,5; 7,0 plaseboryhmässä. Haavaisen koliitin golimumabi‑induktiohoidosta tehtyjen tutkimusten vertailevassa vaiheessa vakavia infektioita todettiin 0,8 %:lla golimumabihoitoa saaneista potilaista ja 1,5 %:lla verrokkiryhmässä. Golimumabia saaneilla potilailla todettuja vakavia infektioita olivat tuberkuloosi, bakteeri‑infektiot mukaan lukien sepsis ja keuhkokuume, invasiiviset sieni‑infektiot ja muut opportunistiset infektiot. Jotkut näistä infektioista ovat johtaneet kuolemaan. Keskeisten tutkimusten vertailevissa sekä ei‑vertailevissa osioissa, joissa seuranta‑ajan mediaani oli enintään 3 vuotta, 100 mg golimumabia saaneiden ryhmässä vakavien infektioiden, mukaan lukien opportunistiset infektiot ja tuberkuloosi, ilmaantuvuus oli suurempi kuin 50 mg golimumabia saaneiden ryhmässä. Kaikkien vakavien infektioiden ilmaantuvuus 100 potilasvuotta kohti oli 4,1; 95 %:n luottamusväli: 3,6; 4,5 golimumabia 100 mg saaneilla potilailla ja 2,5; 95 %:n luottamusväli: 2,0; 3,1 golimumabia 50 mg saaneilla potilailla.
Maligniteetit
Lymfooma
Lymfoomien ilmaantuvuus oli keskeisissä tutkimuksissa golimumabia saaneilla potilailla suurempi kuin normaaliväestössä on odotettavissa. Näiden tutkimusten vertailevissa sekä ei-vertailevissa osioissa, joissa seurantavaiheen mediaani oli enintään 3 vuotta, lymfooman ilmaantuvuus oli suurempi 100 mg golimumabia saaneiden potilaiden ryhmässä kuin 50 mg golimumabia saaneiden potilaiden ryhmässä. Lymfooma todettiin 11 potilaalla (yhdellä 50 mg golimumabia saaneiden ryhmässä ja kymmenellä 100 mg golimumabia saaneiden ryhmässä). Ilmaantuvuus (95 %:n luottamusväli) seuranta-ajan 100 potilasvuotta kohti oli 0,03 tapahtumaa (0,00; 0,15) 50 mg golimumabia saaneilla ja 0,13 tapahtumaa (0,06; 0,24) 100 mg golimumabia saaneilla sekä 0,00 tapahtumaa (0,00; 0,57) plasebolla. Pääosa lymfoomista ilmaantui GO-AFTER-tutkimuksessa, jossa tutkimukseen otettavat potilaat olivat aiemmin saaneet TNF-estäjiä ja joilla sairaus oli pitkäkestoisempi ja huonommin hoitoon reagoiva (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maligniteetit, muut kuin lymfooma
Keskeisten tutkimusten vertailevassa vaiheessa sekä noin 4 vuoden pituisena seuranta-aikana muiden maligniteettien kuin lymfoomien (pois lukien ei-melanooma ihosyöpä) ilmaantuvuus oli sama golimumabi- ja verrokkiryhmissä. Noin 4 vuoden seuranta-aikana muiden maligniteettien kuin lymfoomien (pois lukien ei-melanooma ihosyöpä) ilmaantuvuus oli sama kuin väestössä keskimäärin.
Keskeisten tutkimusten vertailevissa ja ei-vertailevissa vaiheissa, joissa seuranta-ajan mediaani oli enintään 3 vuotta, ei-melanooma ihosyöpää todettiin 5 potilaalla plaseboryhmässä, 10 potilaalla 50 mg golimumabia saaneiden ryhmässä ja 31 potilaalla 100 mg golimumabia saaneiden ryhmässä. Ilmaantuvuus (95 %:n luottamusväli) seuranta-ajan 100 potilasvuotta kohti oli 0,36 (0,26; 0,49) yhdistetyssä golimumabia saaneiden ryhmässä ja 0,87 (0,28; 2,04) plasebolla.
Keskeisten tutkimusten vertailevassa ja ei-vertailevassa vaiheessa, jossa seuranta-ajan mediaani oli enintään 3 vuotta, muita maligniteetteja kuin melanoomaa, ei-melanooma ihosyöpää ja lymfoomia, todettiin 5 potilaalla plaseboryhmässä, 21 potilaalla 50 mg golimumabia saaneiden ryhmässä ja 34 potilaalla 100 mg golimumabia saaneiden ryhmässä. Ilmaantuvuus (95 %:n luottamusväli) seuranta-ajan 100 potilasvuotta kohti oli 0,48 (0,36; 0,62) yhdistetyssä golimumabia saaneiden ryhmässä ja 0,87 (0,28; 2,04) plasebolla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kliinisissä astmatutkimuksissa raportoidut tapahtumat
Eksploratiivisessa kliinisessä tutkimuksessa vaikeaa, itsepintaista astmaa sairastavat potilaat saivat latausannoksen golimumabia (150 % sovitusta hoitoannoksesta) ihon alle viikolla 0 ja sen jälkeen 200 mg golimumabia, 100 mg golimumabia tai 50 mg golimumabia joka 4. viikko ihon alle viikolle 52 asti. Yhdistetyissä golimumabiryhmissä (n = 230) raportoitiin 8 maligniteettia ja plaseboa saaneilla ei yhtään (n = 79). Lymfooma raportoitiin 1 potilaalla, ei-melanooma ihosyöpä 2 potilaalla ja muita maligniteetteja 5 potilaalla. Mitään erityistä kasautumista tietyntyyppisiin maligniteetteihin ei todettu.
Tutkimuksen plasebovertaillussa osiossa maligniteettien ilmaantuvuus (95 %:n luottamusväli) 100 seuranta-ajan potilasvuotta kohti oli 3,19 (1,38; 6,28) golimumabia saaneiden ryhmässä. Tässä tutkimuksessa lymfoomien ilmaantuvuus 100 seuranta-ajan potilasvuotta kohti (95 %:n luottamusväli) oli golimumabia saaneiden ryhmässä 0,40 (0,01; 2,20), ei-melanooma ihosyöpien ilmaantuvuus 0,79 (0,10; 2,86) ja muiden maligniteettien 1,99 (0,64; 4,63). Plaseboa saaneilla potilailla näiden maligniteettien ilmaantuvuus (95 %:n luottamusväli) 100 seuranta-ajan potilasvuotta kohti oli 0,00 (0,00; 2,94). Tämän havainnon merkitystä ei tiedetä.
Neurologiset tapahtumat
Keskeisten tutkimusten vertailevissa ja ei-vertailevissa vaiheissa, joissa seuranta-ajan mediaani oli enintään 3 vuotta, demyelinaation ilmaantuvuus oli suurempi 100 mg golimumabia saaneilla potilailla kuin 50 mg golimumabia saaneilla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksaentsyymien kohoaminen
Keskeisten nivelreuma- ja nivelpsoriaasitutkimusten vertailevassa vaiheessa tapahtui lievää ALAT-arvojen kohoamista (> 1 ja < 3 x normaalin ylärajan) samassa laajuudessa golimumabia saaneilla sekä verrokeilla nivelreuma- ja nivelpsoriaasitutkimuksissa (22,1-27,4 %:lla potilaista). Selkärankareumaa ja röntgennegatiivista aksiaalista spondylartriittia koskevassa tutkimuksessa golimumabia saaneilla potilailla esiintyi enemmän lievää ALAT-arvojen kohoamista (26,9 %) kuin verrokeilla (10,6 %). Keskeisten nivelreuma- ja nivelpsoriaasitutkimusten vertailevissa ja ei-vertailevissa vaiheissa, joissa seuranta-ajan mediaani oli noin 5 vuotta, lievän ALAT-arvojen kohoamisen ilmaantuvuus oli yhtä suuri golimumabia saaneilla potilailla kuin verrokeilla nivelreumassa ja nivelpsoriaasissa tehdyissä tutkimuksissa. Haavaisen koliitin keskeisten golimumabi-induktiotutkimusten vertailevassa vaiheessa, lievää ALAT-arvojen kohoamista (> 1 ja < 3 x normaalin ylärajan) esiintyi samassa määrin golimumabia saaneilla (8,0 %) ja vertailuryhmän potilailla (6,9 %). Haavaisen koliitin keskeisten tutkimusten vertailevissa ja ei-vertailevissa vaiheissa, joissa seuranta-ajan mediaani oli noin 2 vuotta, lievää ALAT-arvojen kohoamista esiintyi 24,7 %:lla potilaista, jotka saivat golimumabia haavaisen koliitin tutkimuksen ylläpitovaiheessa.
Keskeisten nivelreuma- ja selkärankareumatutkimusten vertailevassa vaiheessa ≥ 5 x normaalin ylärajan yli kohonneet ALAT-arvot olivat melko harvinaisia ja niitä todettiin useammin golimumabia saaneilla potilailla (0,4–0,9 %) kuin verrokeilla (0,0 %). Tätä suuntausta ei havaittu nivelpsoriaasipotilaiden populaatiossa. Keskeisten nivelreuma-, nivelpsoriaasi- ja selkärankareumatutkimusten vertailevissa ja ei-vertailevissa vaiheissa, joissa seuranta-ajan mediaani oli 5 vuotta, ALAT-arvojen kohoamista ≥ 5 x normaalin ylärajan yli todettiin saman verran golimumabia saaneilla kuin verrokeilla. Yleensä nämä kohoamiset olivat oireettomia ja poikkeamat vähenivät tai hävisivät joko jatkamalla golimumabin antoa tai keskeyttämällä se tai muuttamalla muuta samanaikaista lääkitystä. Röntgennegatiivista aksiaalista spondylartriittia koskevan tutkimuksen vertailevissa ja ei-vertailevissa vaiheissa (enintään 1 vuosi) ei havaittu yhtään tapausta. Haavaisen koliitin keskeisten golimumabi-induktiohoitotutkimusten vertailevissa vaiheissa, ALAT-arvojen kohoamista ≥ 5 x normaalin ylärajan yli esiintyi samassa määrin golimumabia saaneilla (0,3 %) ja plaseboa saaneilla (1,0 %) potilailla. Haavaisen koliitin keskeisten tutkimusten vertailevissa ja ei-vertailevissa vaiheissa, joissa seuranta-ajan mediaani oli noin 2 vuotta, ALAT-arvojen kohoamista ≥ 5 x normaalin ylärajan yli todettiin 0,8 %:lla golimumabia saaneista potilaista haavaisen koliitin tutkimuksen ylläpito-osiossa.
Keskeisissä nivelreumaa, nivelpsoriaasia, selkärankareumaa ja röntgennegatiivista aksiaalista spondylartriittia koskevissa tutkimuksissa yhdelle golimumabia saaneelle nivelreumatutkimukseen osallistuneelle potilaalle, jolla oli aiempia poikkeamia maksassa sekä tilannetta sekoittava lääkitys, kehittyi ei-infektioosi fataali maksatulehdus ja keltatauti. Golimumabin roolia tähän myötävaikuttavana tai tätä pahentavana tekijänä ei voi sulkea pois.
Pistokohdan reaktiot
Keskeisten tutkimusten vertailevissa vaiheissa 5,4 % golimumabia saaneista potilaista ja 2,0 % verrokkiryhmän potilaista sai pistokohdan reaktion. Golimumabin vasta-aineiden esiintyminen saattaa lisätä pistokohdan reaktioiden riskiä. Suurin osa pistokohdan reaktioista oli lieviä tai kohtalaisia, ja yleisin ilmenemismuoto oli pistokohdan punoitus. Pistokohdan reaktiot eivät yleensä johtaneet lääkevalmisteen käytön lopettamiseen.
Vertailevissa faasi IIb:n ja/tai faasi III:n nivelreumaa, nivelpsoriaasia, selkärankareumaa, röntgennegatiivista aksiaalista spondylartriittia sekä vaikeaa itsepintaista astmaa koskevissa tutkimuksissa ja faasi II/III:n haavaista koliittia koskevissa tutkimuksissa yhdellekään golimumabia saaneista potilaista ei ilmaantunut anafylaktisia reaktioita.
Autoimmuunivasta-aineet
Keskeisten tutkimusten vertailevissa ja ei-vertailevissa vaiheissa yhden vuoden pituisena seuranta-aikana 3,5 % golimumabia saaneista potilaista ja 2,3 % verrokkiryhmän potilaista tuli ANA-positiivisiksi (tiitterit 1:160 tai enemmän). Anti-dsDNA-vasta-aineita esiintyi vuoden pituisena seuranta-aikana 1,1 prosentilla potilaista, jotka olivat lähtötilanteessa anti-dsDNA-negatiivisia.
Pediatriset potilaat
Idiopaattinen juveniili polyartriitti
Golimumabin turvallisuutta on tutkittu faasi III:n tutkimuksessa, johon osallistui 173 iältään 2−17-vuotiasta idiopaattista juveliinia polyartriittia sairastavaa potilasta. Keskimääräinen seuranta-aika oli noin kaksi vuotta. Tässä tutkimuksessa raportoitujen haittavaikutusten laatu ja esiintymistiheys olivat yleensä samanlaisia kuin on raportoitu aikuisten nivelreumatutkimuksissa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisessä tutkimuksessa on annettu jopa 10 mg/kg:n suuruisia kerta-annoksia laskimoon ilman annosta rajoittavaa toksisuutta. Yliannostustapauksessa on suositeltavaa, että potilasta tarkkaillaan haittavaikutusten merkkien ja oireiden varalta sekä aloitetaan välittömästi asiaankuuluva oireenmukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, tuumorinekroositekijä alfan (TNF-α) estäjät, ATC-koodi: L04AB06
Vaikutusmekanismi
Golimumabi on ihmisen monoklonaalinen vasta-aine, joka sitoutuu suurella affiniteetilla bioaktiivisiin ihmisen TNF-α:n liukoisiin ja transmembraanisiin muotoihin ja muodostaa pysyviä komplekseja niiden kanssa, mikä estää TNF-α:n sitoutumisen reseptoreihinsa.
Farmakodynaamiset vaikutukset
On osoitettu, että sitoutuessaan ihmisen TNF:ään golimumabi neutraloi TNF-α:n aiheuttamaa adheesiomolekyylien E-selektiinin, VCAM-1:n (vascular cell adhesion molecule-1) ja ICAM-1:n (intercellular adhesion molecule-1) ilmentymistä solujen pinnassa ihmisen endoteelisoluissa. In vitro golimumabi esti myös TNF:n indusoimaa interleukiini-6:n (IL-6:n), IL-8:n ja granulosyytti-makrofagikasvutekijän (GM-CSF) vapautumista ihmisen endoteelisoluissa.
C-reaktiivisen proteiinin (CRP) arvot paranivat plaseboryhmiin verrattuina, ja Simponi-hoito laski merkitsevästi seerumin IL-6:n, ICAM-1:n, matriksimetalloproteinaasi-3:n (MMP-3:n) ja verisuonten endoteelikasvutekijän (VEGF) pitoisuuksia lähtöarvosta vertailuhoitoon verrattuna. Lisäksi TNF-α-pitoisuus pieneni nivelreuma- ja selkärankareumapotilailla ja IL-8:n pitoisuus nivelpsoriaasipotilailla. Muutokset todettiin ensimmäisessä arvioinnissa (viikolla 4) ensimmäisen Simponi-annoksen jälkeen, ja ne säilyivät yleensä viikkoon 24 asti.
Kliininen teho
Nivelreuma
Simponin teho osoitettiin kolmessa plasebokontrolloidussa, satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa. Tutkimuksissa oli mukana yli 1500 potilasta, jotka olivat ≥ 18-vuotiaita ja joilla oli vähintään 3 kuukautta ennen seulontaa diagnosoitu kohtalaisen aktiivinen tai hyvin aktiivinen nivelreuma ACR:n (American College of Rheumatology) kriteerien perusteella. Potilailla oli vähintään 4 turvonnutta ja 4 aristavaa niveltä. Simponi tai plasebo annettiin injektioina ihon alle 4 viikon välein.
GO-FORWARD-tutkimuksessa arvioitiin 444 potilasta, joilla oli aktiivinen nivelreuma vakioannoksina (vähintään 15 mg/viikko) annetusta metotreksaattihoidosta huolimatta ja jotka eivät olleet aikaisemmin saaneet TNF-estäjiä. Potilaat saivat satunnaistetusti plaseboa + metotreksaattia, Simponia 50 mg + metotreksaattia, Simponia 100 mg + metotreksaattia tai Simponia 100 mg + plaseboa. Plaseboa + metotreksaattia saaneille potilaille vaihdettiin viikon 24 jälkeen hoidoksi Simponi 50 mg + metotreksaatti. Viikolla 52 potilaat aloittivat avoimen pitkäaikaisen jatkotutkimuksen.
GO-AFTER-tutkimuksessa arvioitiin 445 potilasta, jotka olivat aikaisemmin saaneet yhtä tai useampaa TNF-estäjää, adalimumabia, etanerseptiä tai infliksimabia. Potilaat saivat satunnaistetusti plaseboa, Simponia 50 mg tai Simponia 100 mg. Potilaat saivat jatkaa tautiprosessia hidastavien reumalääkkeiden (DMARD) metotreksaatin, sulfasalatsiinin ja/tai hydroksiklorokiinin samanaikaista käyttöä tutkimuksen aikana. Aikaisemman TNF-estäjähoidon lopettamisen syiksi ilmoitettiin tehon puuttuminen (58 %), potilas ei sietänyt hoitoa (13 %) ja/tai muut kuin hoidon turvallisuuteen tai tehoon liittyvät syyt (29 %, pääasiassa taloudelliset syyt).
GO-BEFORE-tutkimuksessa arvioitiin 637 potilasta, joilla oli aktiivinen nivelreuma ja jotka eivät olleet saaneet metotreksaattia ja joita ei ollut aiemmin hoidettu TNF-estäjillä. Potilaat satunnaistettiin saamaan plaseboa + metotreksaattia, Simponia 50 mg + metotreksaattia, Simponia 100 mg + metotreksaattia tai Simponia 100 mg + plaseboa. Viikolla 52 potilaat aloittivat avoimen jatkovaiheen, jossa niille potilaille, jotka olivat saaneet plaseboa + metotreksaattia ja joilla oli vähintään yksi arka tai turvonnut nivel, vaihdettiin hoidoksi Simponi 50 mg + metotreksaatti.
GO-FORWARD-tutkimuksessa toinen ensisijainen päätetapahtuma oli ACR20-vasteen saavuttaneiden potilaiden osuus viikolla 14 ja toinen HAQ-toimintakykyindeksin (Health Assessment Questionnaire, HAQ) paraneminen lähtötilanteeseen verrattuna viikolla 24. GO-AFTER-tutkimuksessa ensisijainen päätetapahtuma oli ACR20-vasteen saavuttaneiden potilaiden osuus viikolla 14.
GO-BEFORE-tutkimuksessa ensisijaiset päätetapahtumat olivat ACR50-vasteen saavuttaneiden potilaiden osuus viikolla 24 ja muutos lähtötilanteesta van der Heijde-modifioidulla Sharpin asteikolla (vdH-S) viikolla 52. Ensisijaisten päätetapahtumien lisäksi arvioitiin Simponi-hoidon vaikutusta niveltulehduksen merkkeihin ja oireisiin, röntgenologiseen vasteeseen, fyysiseen toimintakykyyn sekä terveyteen liittyvään elämänlaatuun.
Tehoa kuvaavissa tuloksissa ei havaittu yleisiä kliinisesti merkittäviä eroja 50 mg:n ja 100 mg:n Simponi-annosten välillä, kun niitä oli annettu yhdessä metotreksaatin kanssa 104 viikon ajan GO-FORWARD- ja GO-BEFORE-tutkimuksissa ja 24 viikon ajan GO-AFTER-tutkimuksessa. Jokaisessa nivelreumatutkimuksessa tutkimusasetelman mukaisesti potilaat saivat jatkovaiheessa vaihdellen 50 mg:n tai 100 mg:n Simponi-annoksia tutkijalääkärin harkinnan mukaan.
Merkit ja oireet
Taulukossa 2 ovat tärkeimmät Simponin 50 mg:n annoksella viikoilla 14, 24 ja 52 GO-FORWARD-, GO-AFTER- ja GO-BEFORE-tutkimuksissa saavutetut ACR-tulokset, joita kuvataan seuraavassa. Vasteet todettiin ensimmäisessä arvioinnissa (viikolla 4) ensimmäisen Simponi-annoksen jälkeen.
GO-FORWARD-tutkimuksessa 89 koehenkilöstä, jotka satunnaistettiin saamaan Simponia 50 mg + metotreksaattia, 48 sai tätä hoitoa edelleen viikolla 104. Näistä 40 potilasta sai ACR20-vasteen, 33 ACR50-vasteen ja 24 ACR70-vasteen viikolla 104. Niillä potilailla, jotka pysyivät tutkimuksessa ja saivat Simponia, havaitut ACR20/50/70-vasteet olivat samanlaisia viikoilla 104–256.
GO-AFTER-tutkimuksessa ACR20-vasteen saavuttaneiden potilaiden osuus oli suurempi Simponi-hoitoa kuin plaseboa saaneessa ryhmässä riippumatta siitä, mikä oli ilmoitettu syyksi yhden tai useamman aikaisemman TNF-estäjähoidon lopettamiseen.
Taulukko 2
GO-FORWARD-, GO-AFTER- ja GO-BEFORE-tutkimusten vertailevien osioiden tärkeimmät tehoa kuvaavat tulokset
| GO-FORWARD Aktiivinen nivelreuma meto- treksaattihoidosta (MTX) huolimatta | GO-AFTER Aktiivinen nivelreuma, saaneet aikaisemmin yhtä tai useampaa TNF-estäjää | GO-BEFORE | ||||
| Plasebo + MTX | Simponi 50 mg + MTX | Plasebo | Simponi 50 mg | Plasebo | Simponi 50 mg + MTX | |
| na | 133 | 89 | 150 | 147 | 160 | 159 |
| Hoitovaste, % potilaista | ||||||
| ACR 20 | ||||||
Viikko 14 | 33 % | 55 %* | 18 % | 35 %* | NA | NA |
Viikko 24 | 28 % | 60 %* | 16 % | 31 % p = 0,002 | 49 % | 62 % |
| Viikko 52 | NA | NA | NA | NA | 52 % | 60 % |
| ACR 50 | ||||||
Viikko 14 | 10 % | 35 %* | 7 % | 15 % p = 0,021 | NA | NA |
Viikko 24 | 14 % | 37 %* | 4 % | 16 %* | 29 % | 40 % |
| Viikko 52 | NA | NA | NA | NA | 36 % | 42 % |
| ACR 70 | ||||||
Viikko 14 | 4 % | 14 % p = 0,008 | 2 % | 10 % p = 0,005 | NA | NA |
Viikko 24 | 5 % | 20 %* | 2 % | 9 % p = 0,009 | 16 % | 24 % |
| Viikko 52 | NA | NA | NA | NA | 22 % | 28% |
a n tarkoittaa satunnaistettuja potilaita; kutakin päätetapahtumaa varten arvioitavissa | ||||||
GO-BEFORE-tutkimuksessa ensisijainen analyysi potilailla, joilla oli kohtalainen tai vaikea nivelreuma (yhdistetyt Simponi 50 ja 100 mg + metotreksaatti -ryhmät vs. pelkkä metotreksaatti ACR50-vasteelle) ei ollut tilastollisesti merkitsevä viikolla 24 (p = 0,053). Viikolla 52 kokonaispopulaatiossa ACR-vasteen saaneiden potilaiden osuus Simponia 50 mg + metotreksaattia saaneiden ryhmässä oli yleisesti korkeampi, mutta ei tilastollisesti merkitsevästi erilainen kuin pelkkää metotreksaattia saaneilla (ks. taulukko 2). Lisäanalyysejä tehtiin sellaisten potilaiden alaryhmissä, joilla oli vaikea, aktiivinen ja etenevä nivelreuma. Simponi 50 mg + metotreksaatti -hoidolla oli yleisesti suurempi teho kuin pelkällä metotreksaatilla indikoidussa populaatiossa verrattuna kokonaispopulaatioon.
GO-FORWARD- ja GO-AFTER-tutkimuksissa todettiin kliinisesti merkittävät ja tilastollisesti merkitsevät vasteet taudin aktiivisuutta mittaavalla DAS28-asteikolla (Disease Activity Scale 28) kumpanakin etukäteen määriteltynä arviointiajankohtana, viikolla 14 ja viikolla 24 (p ≤ 0,001). Potilailla, jotka pysyivät siinä Simponi-hoidossa, jota saamaan heidät satunnaistettiin tutkimuksen alussa, DAS28-vasteet säilyivät viikolle 104 asti. Niillä potilailla, jotka pysyivät tutkimuksessa ja saivat Simponia, DAS28-vasteet olivat samanlaisia viikoilla 104–256.
GO-BEFORE-tutkimuksessa mitattiin merkittävä kliininen vaste, joka määritettiin ACR70-vasteen säilymisenä yhtäjaksoisesti 6 kuukauden ajan. Viikolla 52 15 % potilaista Simponia 50 mg + metotreksaattia saaneiden ryhmässä ja vastaavasti 7 % potilaista plaseboa + metotreksaattia saaneiden ryhmässä sai merkittävän kliinisen vasteen (p = 0,018). 159 potilaasta, jotka oli satunnaistettu saamaan Simponia 50 mg + metotreksaattia, 96 sai tätä hoitoa edelleen viikolla 104. Näistä 85 potilasta sai ACR20-vasteen, 66 sai ACR50-vasteen ja 53 sai ACR70-vasteen viikolla 104. Niillä potilailla, jotka pysyivät tutkimuksessa ja saivat Simponia, havaitut ACR20/50/70-vasteet olivat samanlaisia viikoilla 104–256.
Röntgenologinen vaste
GO-BEFORE-tutkimuksessa arvioitiin käsien/ranteiden ja jalkojen rakenteellisia vaurioita vdH-S-pistemäärän muutoksella lähtötilanteesta, jonka pisteytys koostuu röntgenologisista mittauksista eroosioiden määrästä ja koosta ja nivelvälin kaventumisesta. Taulukossa 3 on esitetty Simponin 50 mg:n annoksella viikolla 52 saadut tärkeimmät tulokset.
Niiden potilaiden lukumäärä, joilla ei havaittu uusia eroosioita tai joilla vdH-S-kokonaispistemäärän muutos lähtötilanteesta oli ≤ 0, oli merkittävästi suurempi Simponia saaneiden ryhmässä kuin verrokkiryhmässä (p = 0,003). Viikolla 52 havaitut röntgenologiset vaikutukset säilyivät viikolle 104 asti. Niillä potilailla, jotka pysyivät tutkimuksessa ja saivat Simponia, röntgenologiset vaikutukset olivat samanlaisia viikoilla 104–256.
Taulukko 3
Keskimääräiset röntgenologiset muutokset lähtötilanteesta (keskihajonta, SD) kokonaispistemääränä vdH-S -asteikolla viikolla 52 GO-BEFORE-tutkimuksen kokonaispopulaatiossa
| Plasebo + MTX | Simponi 50 mg + MTX | |
| na | 160 | 159 |
| Kokonaispistemäärä | ||
| Lähtötilanne | 19,7 (35,4) | 18,7 (32,4) |
| Muutos lähtötilanteesta | 1,4 (4,6) | 0,7 (5,2)* |
| Eroosioiden määrä | ||
| Lähtötilanne | 11,3 (18,6) | 10,8 (17,4) |
| Muutos lähtötilanteesta | 0,7 (2,8) | 0,5 (2,1) |
| Nivelvälin kaventuminen | ||
| Lähtötilanne | 8,4 (17,8) | 7,9 (16,1) |
| Muutos lähtötilanteesta | 0,6 (2,3) | 0,2 (2,0)** |
| a n tarkoittaa satunnaistettuja potilaita * p = 0,015 ** p = 0,044 | ||
Fyysinen toimintakyky ja terveyteen liittyvä elämänlaatu
GO-FORWARD- ja GO-AFTER-tutkimuksissa fyysistä toimintakykyä ja toiminnanvajavuutta (disability) arvioitiin erillisenä päätetapahtumana HAQ DI -toimintakykyindeksiä käyttäen. Näissä tutkimuksissa Simponi paransi HAQ DI -indeksiä kliinisesti merkittävästi ja tilastollisesti merkitsevästi lähtöarvosta vertailuryhmään verrattuna viikkoon 24 mennessä. Potilailla, jotka pysyivät siinä Simponi-hoidossa, jota saamaan heidät satunnaistettiin tutkimuksen alussa, HAQ DI -indeksin paraneminen säilyi viikolle 104 asti. Niillä potilailla, jotka pysyivät tutkimuksessa ja saivat Simponia, HAQ-toimintakykyindeksin paraneminen oli samanlaista viikoilla 104–256.
GO-FORWARD-tutkimuksessa todettiin Simponi-hoitoa saaneiden potilaiden terveyteen liittyvän elämänlaadun kliinisesti merkittävä ja tilastollisesti merkitsevä paraneminen SF-36-asteikon fyysistä toimintakykyä mittaavalla osiolla plaseboa saaneisiin potilaisiin verrattuna viikolla 24. Potilailla, jotka pysyivät siinä Simponi-hoidossa, jota saamaan heidät satunnaistettiin tutkimuksen alussa, SF-36-asteikon fyysistä toimintakykyä mittaavalla osiolla havaittu paraneminen säilyi viikolle 104 asti. Niillä potilailla, jotka pysyivät tutkimuksessa ja saivat Simponia, SF–36-asteikon fyysistä toimintakykyä mittaavalla osiolla havaittu paraneminen oli samanlaista viikoilla 104–256.
GO-FORWARD- ja GO-AFTER-tutkimuksissa todettiin väsymyksen tilastollisesti merkitsevä paraneminen väsymystä mittaavalla FACIT-F-asteikolla (Functional Assessment of Chronic Illness Therapy - Fatigue).
Nivelpsoriaasi
Simponin tehoa ja turvallisuutta verrattiin plaseboon satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa (GO-REVEAL) 405 aikuispotilaalla, joilla oli aktiivinen nivelpsoriaasi (≥ 3 turvonnutta ja ≥ 3 aristavaa niveltä) tulehduskipulääkkeiden (NSAID) tai tautiprosessia hidastavien reumalääkkeiden (DMARD) käytöstä huolimatta. Tämän tutkimuksen potilaiden nivelpsoriaasi oli diagnosoitu vähintään 6 kuukautta aikaisemmin, ja heillä oli vähintään lievä psoriaattinen tauti. Mukaan otettiin potilaita kustakin nivelpsoriaasityypistä, joita olivat moniniveltulehdus, johon ei liittynyt reumakyhmyjä (43 %), epäsymmetrinen perifeerinen niveltulehdus (30 %), kärkinivelten (DIP-nivelten) tulehdus (15 %), spondyliitti, johon liittyi perifeerinen artriitti (11 %) ja arthritis mutilans -niveltulehdus (1 %). Aikaisempaa hoitoa TNF-estäjillä ei sallittu. Simponi tai plasebo annettiin injektioina ihon alle 4 viikon välein. Potilaat saivat satunnaistetusti plaseboa, Simponia 50 mg tai Simponia 100 mg. Plaseboa saaneet potilaat siirrettiin viikon 24 jälkeen Simponia 50 mg saavaan ryhmään. Viikolla 52 potilaat siirtyivät avoimeen pitkäaikaiseen jatkotutkimukseen. Noin 48 % potilaista jatkoi metotreksaattihoitoa vakioannostuksella (≤ 25 mg/viikko). Ensisijaiset päätetapahtumat olivat ACR20-vasteen saavuttaneiden potilaiden osuus viikolla 14 ja nivelpsoriaasiin mukautetulla vdH-S-asteikolla saadun kokonaispistemäärän muutos lähtötilanteesta viikolla 24.
Tehoa kuvaavissa tuloksissa ei havaittu yleisiä kliinisesti merkittäviä eroja 50 mg:n ja 100 mg:n Simponi-annosten välillä viikolle 104 asti. Tutkimusasetelman mukaisesti potilaat saivat jatkovaiheessa vaihdellen 50 mg:n tai 100 mg:n Simponi-annoksia tutkijalääkärin harkinnan mukaan.
Merkit ja oireet
Taulukossa 4 ovat tärkeimmät 50 mg:n annoksella saavutetut tulokset viikoilla 14 ja 24, ja niitä kuvataan alla.
Taulukko 4
GO-REVEAL-tutkimuksen tärkeimmät tehoa kuvaavat tulokset
| Plasebo | Simponi 50 mg* | |
| na | 113 | 146 |
| Hoitovaste, % potilaista | ||
| ACR 20 | ||
Viikko 14 | 9 % | 51 % |
Viikko 24 | 12 % | 52 % |
| ACR 50 | ||
| Viikko 14 | 2 % | 30 % |
| Viikko 24 | 4 % | 32 % |
| ACR 70 | ||
| Viikko 14 | 1 % | 12 % |
| Viikko 24 | 1 % | 19 % |
| PASIb 75c | ||
| Viikko 14 | 3 % | 40 % |
| Viikko 24 | 1 % | 56 % |
* p < 0,05 kaikissa vertailuissa | ||
Vaste havaittiin ensimmäisessä arvioinnissa (viikolla 4) ensimmäisen Simponi-annoksen jälkeen. Samanlaiset ACR20-vasteet todettiin viikolla 14 nivelpsoriaasin alatyypeissä moniniveltulehdus, johon ei liittynyt reumakyhmyjä, ja epäsymmetrinen perifeerinen niveltulehdus. Muissa nivelpsoriaasin alatyypeissä potilasmäärät olivat niin pieniä, ettei pätevää arviota voitu tehdä. Simponi-hoitoa saaneissa ryhmissä vasteet olivat samansuuruisia riippumatta siitä, saivatko potilaat samanaikaisesti metotreksaattia vai eivät. Simponia 50 mg:n annoksina saaneeseen ryhmään satunnaistetuista 146 potilaasta 70 jatkoi edelleen tätä hoitoa viikolla 104. Näistä 70 potilaasta ACR20-vaste todettiin 64 potilaalla, ACR50-vaste 46 potilaalla ja ACR70-vaste 31 potilaalla. Niillä potilailla, jotka pysyivät tutkimuksessa ja saivat Simponia, havaitut ACR20/50/70-vasteet olivat samanlaisia viikoilla 104–256.
Myös DAS28-asteikolla todettiin tilastollisesti merkitsevä vaste viikoilla 14 ja 24 (p < 0,05).
Viikolla 24 Simponi-hoitoa saaneilla potilailla havaittiin paranemista nivelpsoriaasille tyypillistä perifeeristä aktiivisuutta mittaavissa parametreissä (esim. turvonneiden nivelten lukumäärä, kipeiden/aristavien nivelten lukumäärä, daktyliitti ja entesiitti). Simponi-hoito paransi merkitsevästi fyysistä toimintakykyä HAQ DI -toimintakykyindeksillä arvioituna. Myös terveyteen liittyvä elämänlaatu parani merkitsevästi SF-36-asteikon fyysisen ja psyykkisen osion yhdistettyjen tulosten perusteella. DAS28- ja HAQ DI -vasteet säilyivät viikolle 104 asti niiden potilaiden ryhmässä, jotka jatkoivat samaa Simponi-hoitoa, johon heidät oli tutkimuksen alussa satunnaistettu. Niillä potilailla, jotka pysyivät tutkimuksessa ja saivat Simponia, DAS28-vasteet ja HAQ-toimintakykyindeksin paraneminen olivat samanlaisia viikoilla 104–256.
Röntgenologinen vaste
Molempien käsien ja jalkojen rakenteellisia vaurioita arvioitiin radiologisesti nivelpsoriaasiin mukautetulla vdH-S-asteikolla, jossa olivat mukana myös sormien kärkinivelet, käyttämällä kriteerinä pistemäärän muutosta lähtötilanteesta.
Simponi-hoito 50 mg:n annoksina hidasti perifeerisen nivelvaurion etenemistä plaseboon verrattuna viikolla 24 tehtyjen mittausten perusteella, kun kriteerinä oli modifioidulla vdH-S-asteikolla saadun kokonaispistemäärän muutos lähtöarvosta (pisteiden keskiarvo ± SD oli plaseboryhmässä 0,27 ± 1,3 ja Simponi-ryhmässä -0,16 ± 1,3; p = 0,011). Niistä 146 potilaasta, jotka oli satunnaistettu 50 mg:n Simponi-annoksia saavaan ryhmään, 52 viikon röntgentutkimusten tulokset olivat käytettävissä 126 potilaasta, joista 77 prosentilla tauti ei ollut edennyt lähtötilanteeseen verrattuna. Viikolla 104 röntgentutkimusten tulokset olivat käytettävissä 114 potilaasta, ja heistä 77 prosentilla tauti ei ollut edennyt lähtötilanteeseen verrattuna. Potilaista, jotka pysyivät tutkimuksessa ja saivat Simponia, niiden osuus, joilla tauti ei ollut edennyt lähtötilanteesta viikoilla 104–256, oli samansuuruinen.
Aksiaalinen spondylartriitti
Selkärankareuma
Simponin tehoa ja turvallisuutta verrattiin plaseboon satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa (GO-RAISE) 356 aikuispotilaalla, joilla oli aktiivinen selkärankareuma (Bath Ankylosing Spondylitis Disease Activity Index [BASDAI] pistearvo ≥ 4 ja kipujanalla (VAS) mitattu kokoselän kipu ≥ 4 asteikolla 0–10 cm). Tutkimukseen otetuilla potilailla oli aktiivinen tauti tämänhetkisestä tai aikaisemmasta tulehduskipulääkityksestä (NSAID) tai tautiprosessia hidastavasta reumalääkityksestä (DMARD) huolimatta, eivätkä he olleet saaneet aikaisemmin TNF-estäjiä. Simponi tai plasebo annettiin injektioina ihon alle 4 viikon välein. Potilaille annettiin satunnaistetusti plaseboa, Simponia 50 mg tai Simponia 100 mg, ja he saivat jatkaa samanaikaista tautiprosessia hidastavaa reumalääkitystä (DMARD) (metotreksaattia, sulfasalatsiinia ja/tai hydroksiklorokiinia). Ensisijainen päätetapahtuma oli ASAS20-vasteen (Ankylosing Spondylitis Assessment Study Group) saavuttaneiden potilaiden osuus viikolla 14. Plasebovertailuun perustuvat tehoa kuvaavat tulokset kerättiin ja analysoitiin 24 viikon ajalta.
Taulukossa 5 ovat tärkeimmät 50 mg:n annoksella saadut tulokset ja niitä kuvataan alla. Tehoa kuvaavissa tuloksissa ei havaittu yleisiä kliinisesti merkittäviä eroja 50 mg:n ja 100 mg:n Simponi-annosten välillä viikolle 24 asti. Tutkimusasetelman mukaisesti potilaat saivat jatkovaiheessa vaihdellen 50 mg:n tai 100 mg:n Simponi-annoksia tutkijalääkärin harkinnan mukaan.
Taulukko 5
GO-RAISE-tutkimuksen tärkeimmät tehoa kuvaavat tulokset
| Plasebo | Simponi 50 mg* | |
| na | 78 | 138 |
| Hoitovaste, % potilaista | ||
| ASAS 20 | ||
| Viikko 14 | 22 % | 59 % |
| Viikko 24 | 23 % | 56 % |
| ASAS 40 | ||
| Viikko 14 | 15 % | 45 % |
| Viikko 24 | 15 % | 44 % |
| ASAS 5/6 | ||
| Viikko 14 | 8 % | 50 % |
| Viikko 24 | 13 % | 49 % |
* p ≤ 0,001 kaikissa vertailuissa | ||
Potilaista, jotka pysyivät tutkimuksessa ja saivat Simponia, ASAS20- ja ASAS40-vasteen saavuttaneiden potilaiden osuus oli samansuuruinen viikoilla 24–256.
Viikoilla 14 ja 24 todettiin myös tilastollisesti merkitsevät vasteet BASDAI-50, -70 ja -90-vasteen (p ≤ 0,017) perusteella. Tärkeimmissä taudin aktiivisuutta mittaavissa tuloksissa havaittiin paranemista ensimmäisessä arvioinnissa (viikolla 4) ensimmäisen Simponi-annoksen jälkeen, ja muutokset säilyivät viikkoon 24 asti. Niillä potilailla, jotka pysyivät tutkimuksessa ja saivat Simponia, havaitut BASDAI-vasteen muutokset lähtötilanteesta olivat samansuuruisia viikoilla 24–256. Viikolla 14 todettuun ASAS20-vasteeseen perustuva teho oli yhdenmukainen riippumatta tautiprosessia hidastavien reumalääkkeiden (DMARD) (metotreksaatin, sulfasalatsiinin ja/tai hydroksiklorokiinin) käytöstä, HLA-B27-antigeenistatuksesta tai lähtötilanteen CRP-arvosta.
Simponi-hoito johti merkittävään fyysisen toimintakyvyn paranemiseen arvioituna BASFI-indeksin muutoksena lähtötilanteeseen verrattuna viikoilla 14 ja 24. Terveyteen liittyvä elämänlaatu oli myös parantunut merkitsevästi SF-36-asteikon fyysisen osion pistearvon perusteella viikoilla 14 ja 24. Niillä potilailla, jotka pysyivät tutkimuksessa ja saivat Simponia, fyysisen toimintakyvyn ja terveyteen liittyvän elämänlaadun paraneminen olivat samansuuruisia viikoilla 24–256.
Röntgennegatiivinen aksiaalinen spondylartriitti
GO-AHEAD
Simponin turvallisuutta ja tehoa verrattiin plaseboon satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa (GO-AHEAD) 197 aikuispotilaalla, joilla oli vaikea aktiivinen röntgennegatiivinen aksiaalinen spondylartriitti (eli potilailla, jotka täyttivät ASAS-luokittelun kriteerit aksiaaliselle spondylartriitille, mutta eivät täyttäneet modifioituja New York -kriteerejä selkärankareumalle). Tähän tutkimukseen otetuilla potilailla oli aktiivinen tauti (BASDAI ≥ 4 ja kipujanalla (VAS) mitattu kokoselän kipu ≥ 4 asteikolla 0–10 cm) senhetkisestä tai aikaisemmasta tulehduskipulääkityksestä huolimatta, eivätkä he olleet saaneet aikaisemmin biologisia lääkkeitä, kuten TNF-estäjiä. Potilaat satunnaistettiin saamaan plaseboa tai Simponia 50 mg ihon alle 4 viikon välein. Viikolla 16 potilaat aloittivat avoimen vaiheen, jossa kaikki potilaat saivat Simponia 50 mg ihon alle 4 viikon välein viikolle 48 asti, ja tehon arviointia jatkettiin viikolle 52 ja turvallisuusseurantaa viikolle 60 asti. Noin 93 % potilaista, jotka saivat Simponia avoimen vaiheen alussa (viikolla 16), jatkoi hoitoa tutkimuksen loppuun (viikolle 52) asti. Analyysit tehtiin sekä kaikkien hoidettujen potilaiden joukolle (N = 197) että niiden potilaiden joukolle, joilla oli objektiivisia tulehduslöydöksiä (N = 158, määriteltiin lähtötilanteessa suurentuneena C-reaktiivisen proteiinin (CRP) pitoisuutena ja/tai magneettitutkimuksessa näkyvänä risti-suoliluunivelen tulehduksena). Plasebovertailuun perustuvat tehoa kuvaavat tulokset kerättiin ja analysoitiin 16 viikon ajalta. Ensisijainen päätetapahtuma oli ASAS 20 -vasteen saavuttaneiden potilaiden osuus viikolla 16. Keskeiset tulokset on esitetty taulukossa 6 ja kuvattu jäljempänä.
Taulukko 6
GO-AHEAD-tutkimuksen tärkeimmät tehoa kuvaavat tulokset viikolla 16
Oireiden ja merkkien paraneneminen | ||||
| Kaikki hoidetut potilaat
| Potilaat, joilla oli objektiivisia tulehduslöydöksiä | ||
Plasebo | Simponi 50 mg | Plasebo | Simponi 50 mg | |
na | 100 | 97 | 80 | 78 |
Hoitovaste, % potilaista | ||||
ASAS 20 | 40 % | 71 %** | 38 % | 77 %** |
ASAS 40 | 23 % | 57 %** | 23 % | 60 %** |
ASAS 5/6 | 23 % | 54 %** | 23 % | 63 %** |
ASAS osittainen remissio | 18 % | 33 %* | 19 % | 35 %* |
ASDAS-C b < 1,3 | 13 % | 33 %* | 16 % | 35 %* |
BASDAI 50 | 30 % | 58 %** | 29 % | 59 %** |
Risti-suoliluunivelen tulehduksen esto magneettitutkimuksella mitattuna | ||||
| Plasebo | Simponi 50 mg | Plasebo | Simponi 50 mg |
n C | 87 | 74 | 69 | 61 |
Risti-suoliluunivelen SPARCCd MRI ‑pistemäärän keskimääräinen muutos | ‑0,9 | ‑5,3** | ‑1,2 | ‑6,4** |
a n kuvaa satunnaistettuja ja hoitoa saaneita potilaita | ||||
Vaikea-asteisen aktiivisen röntgennegatiivisen aksiaalisen spondylartriitin oireissa ja merkeissä osoitettiin viikolla 16 plaseboon verrattuna tilastollisesti merkitseviä parannuksia potilailla, jotka olivat saaneet Simponia 50 mg:n annoksella (taulukko 6). Parannuksia todettiin ensimmäisessä arvioinnissa (viikolla 4) Simponin ensimmäisen annoksen jälkeen. Magneettitutkimuksella mitatussa SPARCC-pistemäärässä todettiin viikolla 16 plaseboon verrattuna tilastollisesti merkitsevä risti-suoliluunivelen tulehduksen väheneminen potilailla, jotka olivat saaneet Simponia 50 mg:n annoksella (taulukko 6). Myös kokoselän kipuna ja yöllisenä selkäkipuna kipujanalla (VAS) arvioidussa kivussa sekä ASDAS-C-pisteinä mitatussa taudin aktiivisuudessa osoitettiin viikolla 16 plaseboon verrattuna tilastollisesti merkitsevä parannus lähtötilanteesta potilailla, jotka olivat saaneet Simponia 50 mg:n annoksella (p < 0,0001).
Selkärangan liikkuvuudessa BASMI-indeksillä (Bath Ankylosing Spondylitis Metrology Index) arvioituna ja fyysisessä toimintakyvyssä BASFI-indeksillä arvioituna osoitettiin tilastollisesti merkitseviä parannuksia Simponia 50 mg:n annoksella saaneilla potilailla verrattuna plaseboa saaneisiin potilaisiin (p < 0,0001). ASQoL- ja EQ-5D -mittarilla sekä SF-36-mittarin fyysisinä ja psyykkisinä komponentteina arvioitu terveyteen liittyvä elämänlaatu parani Simponia saaneilla potilailla huomattavasti enemmän kuin plaseboa saaneilla. WPAI-kyselyllä mitattuna myös tuottavuus parani Simponia saaneilla merkitsevästi verrattuna plaseboa saaneisiin, kun sitä arvioitiin kokonaistyökyvyn ja aktiivisuuden paranemisena.
Kaikkien edellä kuvattujen päätemuuttujien suhteen tilastollisesti merkitsevät tulokset osoitettiin viikolla 16 myös siinä potilasjoukossa, jolla oli objektiivisia tulehduslöydöksiä.
Sekä kaikkien hoidettujen potilaiden potilasjoukossa että niiden joukossa, joilla oli objektiivisia tulehduslöydöksiä, löydösten ja oireiden, selkärangan liikkuvuuden, fyysisen toimintakyvyn, elämänlaadun ja tuottavuuden paraneminen, joka havaittiin Simponia 50 mg saaneessa ryhmässä viikolla 16, jatkui edelleen potilailla, jotka pysyivät tutkimuksessa viikolle 52 asti.
GO-BACK
Golimumabihoidon jatkamisen (tavanomaisella tai harvennetulla antotiheydellä) tehoa ja turvallisuutta verrattuna hoidon lopettamiseen arvioitiin aikuisilla (18–45‑vuotiailla) potilailla, joilla oli aktiivinen röntgennegatiivinen aksiaalinen spondylartriitti ja joiden tauti oli pitkäkestoisessa remissiossa, kun avointa Simponi-hoitoa oli annettu kerran kuukaudessa 10 kuukauden ajan (GO-BACK). Tutkimukseen soveltuneet potilaat (jotka saavuttivat kliinisen vasteen kuukauteen 4 mennessä ja joiden tauti oli inaktiivinen [ASDAS-pisteet < 1,3] sekä kuukauden 7 että kuukauden 10 kohdalla), jotka siirtyivät kaksoissokkoutettuun hoidon lopettamisen vaiheeseen, satunnaistettiin jatkamaan kerran kuukaudessa annettavaa Simponi-hoitoa (täysiannoksinen hoito, N = 63), kahden kuukauden välein annettavaa Simponi-hoitoa (harvennettu hoito, N = 63) tai kerran kuukaudessa annettavaa plasebohoitoa (hoidon lopettaminen, N = 62) enintään noin 12 kuukauteen asti.
Ensisijainen tehoa koskeva päätetapahtuma oli niiden potilaiden osuus, joilla ei ilmennyt taudin aktiivisuuden pahenemisvaihetta. Potilaat, joilla ilmeni pahenemisvaihe eli joilla todettiin kahdessa peräkkäisessä ASDAS-arvioinnissa molemmissa joko absoluuttinen pistemäärä ≥ 2,1 tai hoidon lopettamisen jälkeen ≥ 1,1 pistettä suurempi pistemäärä kuin kuukauden 10 kohdalla (avoimen vaiheen lopussa), aloittivat uudelleen kerran kuukaudessa annettavan Simponi-hoidon avoimessa uusintahoitovaiheessa kliinisen vasteen selvittämiseksi.
Kliininen vaste hoidon kaksoissokkoutetun lopettamisen jälkeen
Niistä 188 potilaasta, joiden tauti oli inaktiivinen ja jotka saivat vähintään yhden annoksen kaksoissokkoutettua hoitoa, merkitsevästi (p < 0,001) suuremmalla osalla potilaista ei ilmennyt taudin pahenemisvaihetta, kun Simponi-hoitoa jatkettiin joko täysiannoksisena hoitona (84,1 %) tai harvennettuna hoitona (68,3 %), verrattuna hoidon lopettaneisiin potilaisiin (33,9 %) (taulukko 7).
Taulukko 7
Analyysi niiden osallistujien osuudesta, joilla ei ilmennyt pahenemisvaihettaa
Koko analyysijoukon populaatio (vaihe 2 – kaksoissokkoutettu)
|
|
| Ero, %, vs. plasebo | |
Hoito | n/N | % | Arvio (95 %:n luottamusväli)b | p‑arvob |
Golimumabi ihon alle kerran kuukaudessa | 53/63 | 84,1 | 50,2 (34,1; 63,6) | < 0,001 |
Golimumabi ihon alle kahden kuukauden välein | 43/63 | 68,3 | 34,4 (17,0; 49,7) | < 0,001 |
Plasebo | 21/62 | 33,9 |
|
|
Koko analyysijoukkoon kuuluvat kaikki satunnaistetut osallistujat, joiden tauti muuttui inaktiiviseksi vaiheessa 1 ja jotka saivat vähintään yhden annoksen sokkoutettua tutkimushoitoa. a Määritelmä: kahdella peräkkäisellä käynnillä tehdyissä ASDAS-arvioinneissa molemmissa joko absoluuttinen pistemäärä ≥ 2,1 tai pistemäärä suurentunut hoidon lopettamisen jälkeen ≥ 1,1 pisteellä verrattuna kuukauden 10 (käynnin 23) tietoihin. | ||||
Hoidon lopettaneen ryhmän ja kahden Simponi-hoitoryhmän väliset erot ajassa ensimmäiseen pahenemisvaiheeseen on esitetty kuvassa 1 (log rank -testin p < 0,0001 molempien vertailujen kohdalla). Plaseboryhmässä pahenemisvaiheet alkoivat noin 2 kuukauden kuluttua Simponi-hoidon lopettamisesta, ja suurin osa pahenemisvaiheista ilmeni 4 kuukauden aikana hoidon lopettamisen jälkeen (kuva 1).
Kuva 1: Kaplan–Meier-analyysi ajasta ensimmäiseen pahenemisvaiheeseen
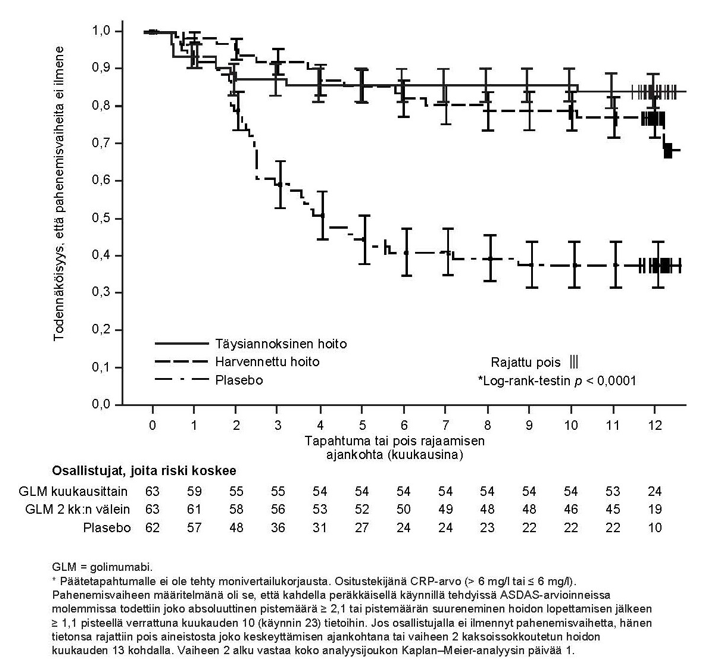
Kliininen vaste taudin pahenemisvaiheen vuoksi annettuun uusintahoitoon
Kliininen vaste määriteltiin BASDAI-pistearvon paranemiseksi ≥ 2 pistettä tai ≥ 50 % verrattuna taudin pahenemisvaiheeseen liittyneiden kahden peräkkäisen BASDAI-pistearvon keskiarvoon. Niistä 53 osallistujasta, jotka saivat harvennettua hoitoa tai joiden hoito lopetettiin ja joilla todettiin vahvistettu taudin pahenemisvaihe, 51 osallistujaa (96,2 %) saavutti kliinisen vasteen Simponi-hoidolle uusintahoidon ensimmäisten 3 kuukauden aikana, joskin vaste säilyi koko 3 kuukauden ajan pienemmällä osalla potilaista (71,7 %).
Haavainen koliitti
Simponin tehoa arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, plasebokontrolloidussa kliinisessä tutkimuksessa aikuisten potilaiden hoidossa.
Induktiotutkimuksessa (PURSUIT-Induction) oli mukana kohtalaista tai vaikeaa aktiivista haavaista koliittia (pisteet Mayo-asteikolla 6–12, pisteet endoskopia-osiossa ≥ 2) sairastavia potilaita, joiden vaste tavanomaiseen hoitoon oli riittämätön tai jotka eivät sietäneet tavanomaisia hoitoja tai olivat riippuvaisia kortikosteroideista. Tutkimuksen annoksenmääritysosassa 761 potilasta jaettiin satunnaistetusti ryhmiin, jotka saivat 400 mg Simponia ihon alle viikolla 0 ja 200 mg viikolla 2 tai 200 mg Simponia ihon alle viikolla 0 ja 100 mg viikolla 2 tai plaseboa ihon alle viikoilla 0 ja 2. Oraalisten aminosalisylaattien, kortikosteroidien ja/tai immunomoduloivien lääkkeiden samanaikainen käyttö oli sallittua vakioannoksina. Tässä tutkimuksessa arvioitiin Simponin tehoa viikolle 6 asti.
Ylläpitohoitotutkimuksen (PURSUIT-Maintenance) tulokset perustuivat niiden 456 potilaan arviointiin, jotka saavuttivat kliinisen hoitovasteen aikaisemmalla Simponi-induktiolla. Potilaat jaettiin satunnaistetusti ryhmiin, jotka saivat 50 mg Simponia, 100 mg Simponia tai plaseboa ihonalaisina injektioina 4 viikon välein. Oraalisten aminosalisylaattien ja/tai immunomoduloivien lääkkeiden samanaikainen käyttö oli sallittua vakioannoksina. Kortikosteroideja oli tarkoitus vähentää asteittain ylläpitohoitotutkimuksen alkaessa. Tässä tutkimuksessa arvioitiin Simponin tehoa viikolle 54 asti. Potilaat, jotka olivat mukana ylläpitohoitotutkimuksen loppuun asti eli viikolle 54, jatkoivat hoitoa tutkimuksen jatkovaiheessa, jossa hoitotehoa arvioitiin viikolle 216 asti. Tehon arviointi perustui tutkimuksen jatkovaiheessa muutoksiin kortikosteroidien käytössä, lääkärin yleisarvioon taudin aktiivisuudesta sekä elämänlaadun paranemiseen IBDQ-kyselyllä (inflammatory bowel disease questionnaire) mitattuna.
Taulukko 8
Tärkeimmät tehoa mittaavat tulokset PURSUIT-Induction- ja PURSUIT-Maintenance-tutkimuksissa
| PURSUIT-Induction | |||
| Plasebo N = 251 | Simponi 200/100 mg N = 253 | ||
| Prosenttia (%) potilaista | |||
| Kliininen hoitovaste viikolla 6a | 30 % | 51 %** | |
| Kliininen remissio viikolla 6b | 6 % | 18 %** | |
| Parantunut limakalvo viikolla 6c | 29 % | 42 %* | |
| PURSUIT-Maintenance | |||
| Plasebod N = 154 | Simponi 50 mg N = 151 | Simponi 100 mg N = 151 | |
| Prosenttia (%) potilaista | |||
| Hoitovasteen säilyminen (kliininen vaste säilyi viikolle 54)e | 31 % | 47 %* | 50 %** |
| Pitkäkestoinen remissio (kliininen remissio sekä viikolla 30 että viikolla 54) f | 16 % | 23 %g | 28 %* |
| N = Potilaiden lukumäärä ** p ≤ 0,001 * p ≤ 0,01 a Mayo-asteikolla saatu pistearvo pienentynyt ≥ 30 % ja ≥ 3 pistettä lähtöarvosta sekä verenvuoto peräsuolesta -osion pistearvo pienentynyt ≥ 1 pistettä tai verenvuoto peräsuolesta -osion pistearvo 0 tai 1. b Pistearvo Mayo-asteikolla ≤ 2 eikä minkään yksittäisen osion pistearvo ole > 1. c Mayo-asteikon endoskopia-osion pistearvo 0 tai 1. d Pelkkä Simponi-induktio. e Haavaisen koliitin aktiivisuus arvioitiin osittaisella Mayo-asteikolla 4 viikon välein (hoitovasteen puuttuminen vahvistettiin endoskopian avulla). Potilaalla, jonka hoitovaste säilyi, oli siten jatkuva kliininen vaste jokaisessa arvioinnissa viikolle 54 asti. f Kestävän remission edellytyksenä oli, että tauti oli remissiossa sekä viikolla 30 että viikolla 54 (vasteen heikkenemistä ei saanut esiintyä minään ajankohtana viikolle 54 asti). g Alle 80 kg painavien potilaiden ryhmässä suuremmalla osalla 50 mg:n ylläpitoannosta saaneista potilaista todettiin pitkäkestoinen kliininen remissio plaseboa saaneisiin potilaisiin verrattuna. | |||
Limakalvon pitkäkestoinen paraneminen (parantunut limakalvo sekä viikolla 30 että viikolla 54) todettiin suuremmalla osalla potilaista Simponia 50 mg:n annoksina saaneessa ryhmässä (42 %, nimellinen p < 0,05) ja 100 mg:n annoksina saaneessa ryhmässä (42 %, p < 0,005) kuin plaseboryhmässä (27 %).
PURSUIT-Maintenance-tutkimuksen alkaessa 54 % potilaista sai samanaikaisesti kortikosteroideja (247/456). Tässä ryhmässä niiden potilaiden osuus, joiden hoitovaste säilyi viikolle 54 asti ja jotka eivät käyttäneet kortikosteroideja viikolla 54, oli suurempi 50 mg:n annoksia saaneessa ryhmässä (38 %, 30/78) ja 100 mg:n annoksia saaneessa ryhmässä (30 %, 25/82) kuin plaseboryhmässä (21 %, 18/87). Niiden potilaiden osuus, jotka lopettivat kortikosteroidihoidon viikkoon 54 mennessä, oli suurempi 50 mg:n annoksia saaneessa ryhmässä (41 %, 32/78) ja 100 mg:n annoksia saaneessa ryhmässä (33 %, 27/82) kuin plaseboryhmässä (22 %, 19/87). Tutkimuksen jatkovaiheeseen siirtyneessä potilasjoukossa niiden potilaiden osuus, jotka eivät tarvinneet kortikosteroideja, pysyi pääosin samana viikolle 216 asti.
Potilaille, joilla ei ollut saavutettu kliinistä vastetta viikolla 6 PURSUIT-Induction-tutkimuksissa, annettiin Simponia 100 mg 4 viikon välein PURSUIT-Maintenance-tutkimuksessa. Viikolla 14 näistä potilaista 28 %:lla oli saavutettu vaste, joka määriteltiin osittaisella Mayo-asteikolla saadun pistearvon perusteella (pienentynyt ≥ 3 pistettä induktion alkuun verrattuna). Viikolla 54 näillä potilailla todetut kliiniset tulokset olivat samanlaisia kuin kliiniset tulokset, jotka ilmoitettiin potilailla, joilla oli saavutettu kliininen vaste viikolla 6.
Viikolla 6 Simponi oli parantanut elämänlaatua merkittävästi, kun kriteerinä oli tautispesifisellä mittarilla, IBDQ-kyselyllä (inflammatory bowel disease questionnaire), saatujen tulosten muutos lähtötasosta. Potilailla, jotka saivat Simponia ylläpitohoitona, IBDQ-kyselyn tuloksiin perustuva elämänlaadun paraneminen säilyi viikolle 54 asti.
Noin 63 % potilaista, jotka saivat Simponia tutkimuksen jatkovaiheen alussa (viikolla 56), jatkoi hoitoa tutkimuksen loppuun asti (viimeinen golimumabiannos viikolla 212).
Immunogeenisuus
Golimumabin vasta-aineita todettiin entsyymi-immunologisella menetelmällä (EIA) kaikissa faasi III:n nivelreuma-, nivelpsoriaasi- ja selkärankareumatutkimuksissa 52 viikon aikana 5 prosentilla (105/2062) golimumabia saaneista potilaista ja testatuista lähes kaikki vasta-aineet olivat neutraloivia in vitro. Vastaavia lukuja on saatu kaikissa reumatologisissa käyttöaiheissa. Golimumabin vasta-aineita todettiin pienemmällä osalla niistä potilaista, jotka saivat samanaikaisesti metotreksaattia (noin 3 % [41/1235]), kuin potilaista, jotka saivat golimumabia ilman metotreksaattia (8 % [64/827]).
Röntgennegatiivisessa aksiaalisessa spondylartriitissa golimumabin vasta-aineita todettiin EIA-menetelmällä 7 %:lla (14/193) golimumabia saaneista potilaista viikkoon 52 mennessä.
Golimumabin vasta-aineita todettiin EIA-menetelmällä haavaisen koliitin faasi II:n ja III:n tutkimuksissa 54 viikon aikana 3 prosentilla (26/946) golimumabia saaneista potilaista. Vasta-aineita saaneista potilaista 68 prosentilla (21/31) vasta-aineet olivat neutraloivia in vitro. Golimumabin vasta-aineita todettiin pienemmällä osalla niistä potilaista, jotka saivat samanaikaisesti immunomoduloivia lääkkeitä (atsatiopriinia, 6-merkaptopuriinia tai metotreksaattia) (1 % [4/308]) kuin potilaista, jotka saivat golimumabia ilman immunomoduloivia lääkkeitä (3 % [22/638]). Tutkimuksen jatkovaiheeseen siirtyneistä potilaista, joilta saatiin arvioitavissa olevat näytteet viikolle 228 asti, golimumabin vasta-aineita todettiin 4 prosentilla (23/604) golimumabia saaneista potilaista. Vasta-aineita saaneista potilaista 82 prosentilla (18/22) vasta-aineet olivat neutraloivia in vitro.
Idiopaattisen juveniilin polyartriitin tutkimuksessa käytettiin lääketoleranttia (drug-tolerant) EIA-menetelmää golimumabin vasta-aineiden havaitsemiseen. Suuremman herkkyyden ja paremman lääketoleranssin vuoksi oli odotettavissa, että golimumabin vasta-aineita löytyisi enemmän lääketolerantilla EIA-menetelmällä kuin EIA-menetelmällä. Faasi III:n idiopaattisen juveniilin polyartriitin tutkimuksessa golimumabin vasta-aineita löytyi viikkoon 48 mennessä lääketolerantilla EIA-menetelmällä 40 %:lla (69/172) golimumabia saaneista lapsista, joista suurimmalla osalla titteri oli alle 1:1000. Vaikutus seerumin golimumabipitoisuuteen näkyi > 1:100 ‑tittereissä, kun taas vaikutus tehoon havaittiin vasta > 1:1000 ‑tittereissä. Niiden lasten määrä, joilla titteri oli > 1:1000, oli alhainen (N = 8). Neutraloivia vasta-aineita oli 39 %:lla (25/65) niistä lapsista, joille kehittyi golimumabin vasta-aineita. Lääketolerantilla EIA-menetelmällä saadulla vasta-aineiden suuremmalla esiintymistiheydellä ei ollut selvästi havaittavaa vaikutusta lääkepitoisuuksiin, tehoon ja turvallisuuteen, koska vasta-ainetitterit olivat enimmäkseen matalia, eikä uusia turvallisuuteen liittyviä signaaleja täten ilmennyt.
Golimumabin vasta-aineiden ilmaantuminen saattaa lisätä pistokohdan reaktioiden riskiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Potilaita, joille kehittyi golimumabin vasta-aineita, oli liian vähän, jotta voitaisiin tehdä varmoja johtopäätöksiä golimumabivasta-aineiden suhteesta kliiniseen tehoon tai turvallisuuteen.
Koska immunogeenisuustutkimukset ovat valmiste- ja analyysispesifisiä, vasta-aineiden esiintymistä ei voida verrata muiden valmisteiden vastaaviin lukuihin.
Pediatriset potilaat
Idiopaattinen juveniili polyartriitti
Simponin tehoa ja turvallisuutta tutkittiin satunnaistetussa, kaksoissokkoutetussa, plasebokontrolloidussa tutkimuksessa (GO-KIDS), johon osallistui 173 lasta (2–17-vuotiaita), joilla oli aktiivinen idiopaattinen juveniili polyartriitti ja vähintään 5 tulehtunutta niveltä eikä metotreksaatilla saatu riittävää vastetta. Tutkimukseen otettiin lapsia, joilla oli idiopaattinen juveniili polyartriitti (reumatekijäpositiivinen tai ‑negatiivinen polyartriitti, moniniveltaudiksi muuttuva oligoartriitti, juveniili nivelpsoriaasi tai systeeminen lastenreuma (JIA), mutta ei sillä hetkellä systeemisiä oireita). Tulehtuneiden nivelten määrän mediaani oli lähtötilanteessa 12 ja CRP-pitoisuuden mediaani 1,7 mg/l (0,17 mg/dl).
Tutkimuksen 1. osa koostui 16 viikon avoimesta vaiheesta, jossa 173 lasta sai Simponia 30 mg/m2 (enintään 50 mg) ihon alle 4 viikon välein sekä metotreksaattia. 154 lasta, joilla saavutettiin ACR Ped 30 ‑vaste viikolla 16, siirtyivät tutkimuksen 2. osan satunnaistettuun vaiheeseen, jossa he saivat Simponia 30 mg/m2 (enintään 50 mg) ja metotreksaattia tai plaseboa ja metotreksaattia 4 viikon välein. Taudin pahentuessa lapset saivat Simponia 30 mg/m2 (enintään 50 mg) ja metotreksaattia. Viikolla 48 aloitettiin avoin jatkotutkimus.
Tutkimukseen osallistuneilla lapsilla saavutettiin ACR Ped 30, 50, 70 ja 90 vasteita viikosta 4 lähtien.
Viikolla 16 vasteet olivat: 87 %:lla lapsista ACR Ped 30, 79 %:lla lapsista ACR Ped 50, 66 %:lla lapsista ACR Ped 70 ja 36 %:lla lapsista ACR Ped 90. Viikolla 16 lapsista 34 %:lla sairaus oli inaktiivinen, joka määriteltiin seuraavasti: ei aktiivista niveltulehdusta; ei kuumetta, ihottumaa, serosiittia, splenomegaliaa, hepatomegaliaa tai lastenreumasta johtuvaa yleistä lymfadenopatiaa; ei aktiivista uveiittia; normaali lasko-arvo (< 20 mm/h) tai CRP-pitoisuus (< 10 mg/l, [< 1,0 mg/dl]); lääkärin kokonaisarvio sairauden aktiivisuudesta (≤ 5 mm VAS-janalla); aamujäykkyys < 15 minuuttia.
Viikolla 16 kaikki ACR Ped ‑osatekijät osoittivat kliinisesti merkittävää paranemista lähtötilanteeseen verrattuna (ks. taulukko 9).
Taulukko 9
ACR Ped ‑osatekijöiden paraneminen lähtötilanteeseen verrattuna viikolla 16a
| Paranemisprosentti, mediaani |
| Simponi 30 mg/m2 nb = 173 |
Lääkärin kokonaisarvio sairaudesta (VASc 0‑10 cm) | 88 % |
Koehenkilön/vanhemman kokonaisarvio yleisestä voinnista (VAS 0‑10 cm) | 67 % |
Tulehtuneet nivelet | 92 % |
Nivelet, joiden liikkuvuus rajoittunut | 80 % |
Fyysinen toimintakyky CHAQd:n perusteella | 50 % |
Lasko (mm/h)e | 33 % |
a lähtötilanne = viikko 0 | |
Ensisijaista vastemuuttujaa eli niiden lasten suhteellista osuutta, jotka olivat saaneet ACR Ped 30 ‑vasteen viikolla 16, ja joille ei tullut sairauden pahenemisvaihetta viikkojen 16 ja 48 välillä, ei saavutettu. Suurimmalle osalle lapsista pahenemisvaihetta ei tullut viikkojen 16 ja 48 välillä (59 % Simponi ja metotreksaatti ‑ryhmässä ja 53 % plasebo ja metotreksaatti ‑ryhmässä, p = 0,41).
Ensisijaisen vastemuuttujan suhteen tehtiin ennaltamäärätty alaryhmäanalyysi lähtötilanteen CRP-pitoisuuden (≥ 10 mg/l vs. < 10 mg/l) mukaan. Tässä analyysissä plasebo + metotreksaatti -hoitoa saaneilla havaittiin useammin taudin paheneminen verrattuna Simponi + metotreksaatti –hoitoa saaneisiin (87 % vs. 40 %, p = 0,0068), kun lähtötilanteessa potilaiden CRP-pitoisuus oli ≥ 10 mg/l.
48. viikolla 53 % Simponi + metotreksaatti ‑ryhmän ja 55 % plasebo + metotreksaatti ‑ryhmän lapsista oli saavuttanut ACR Ped 30 ‑vasteen, ja 40 %:lla Simponi + metotreksaatti ‑ryhmän ja 28 %:lla plasebo + metotreksaatti ‑ryhmän lapsista sairaus oli inaktiivisessa tilassa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Simponin käytöstä haavaisen koliitin hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Terveille vapaaehtoisille tai nivelreumapotilaille annetun ihonalaisen kerta-annoksen jälkeen golimumabin maksimipitoisuus seerumissa (Tmax) saavutettiin 2–6 vuorokauden (mediaani) kuluttua. Terveille vapaaehtoisille ihonalaisena injektiona annetun 50 mg:n golimumabiannoksen jälkeen maksimipitoisuuden (Cmax) keskiarvo ± keskihajonta oli 3,1 ± 1,4 μg/ml.
Golimumabi imeytyi samalla tavoin olkavarren, vatsan ja reiden ihon alle annetun 100 mg:n kerta-annoksen jälkeen, ja absoluuttisen hyötyosuuden keskiarvo oli 51 %. Koska ihon alle annetun golimumabin farmakokinetiikka oli jokseenkin suoraan annoksesta riippuvainen, 50 mg:n tai 200 mg:n golimumabiannoksen absoluuttisen hyötyosuuden oletetaan olevan samanlainen.
Jakautuminen
Laskimoon annetun kerta-annoksen jälkeen jakautumistilavuuden keskiarvo oli 115 ± 19 ml/kg.
Eliminaatio
Golimumabin systeemisen puhdistuman arvioitiin olevan 6,9 ± 2,0 ml/vrk/kg. Terminaalisen puoliintumisajan arvioitiin olevan noin 12 ± 3 vuorokautta terveillä vapaaehtoisilla, ja vastaavia arvoja saatiin nivelreumaa, nivelpsoriaasia, selkärankareumaa tai haavaista koliittia sairastavilla potilailla.
Vakaan tilan pitoisuus seerumissa saavutettiin viikkoon 12 mennessä, kun nivelreumaa, nivelpsoriaasia tai selkärankareumaa sairastaville potilaille annettiin golimumabia 50 mg:n annoksina ihon alle 4 viikon välein. Kun golimumabia annettiin 50 mg:n annoksina ihon alle 4 viikon välein samanaikaisesti metotreksaatin kanssa, pienimpien vakaan tilan aikaisten golimumabipitoisuuksien (trough) keskiarvo (± keskihajonta) seerumissa oli noin 0,6 ± 0,4 μg/ml nivelreumapotilailla, joiden nivelreuma oli aktiivinen metotreksaattihoidosta huolimatta, noin 0,5 ± 0,4 μg/ml aktiivista nivelpsoriaasia sairastavilla potilailla ja noin 0,8 ± 0,4 μg/ml selkärankareumapotilailla. Seerumin keskimääräiset golimumabin jäännöspitoisuudet (trough) vakaassa tilassa olivat röntgennegatiivista aksiaalista spondylartriittia sairastavilla potilailla samansuuruisia kuin selkärankareumapotilailla, kun golimumabia annettiin 50 mg ihon alle 4 viikon välein.
Nivelreuma-, nivelpsoriaasi- tai selkärankareumapotilailla, jotka eivät saaneet samanaikaisesti metotreksaattia, vakaan tilan aikaiset pienimmät golimumabipitoisuudet (trough) olivat noin 30 % pienempiä kuin potilailla, jotka saivat golimumabia samanaikaisesti metotreksaatin kanssa. Suppealla määrällä nivelreumapotilaita, jotka saivat golimumabia ihon alle yli 6 kuukauden ajan, samanaikainen metotreksaattihoito pienensi golimumabin puhdistumaa noin 36 %.
Populaatiofarmakokineettinen analyysi osoitti kuitenkin, että tulehduskipulääkkeiden (NSAID), suun kautta otettavien kortikosteroidien tai sulfasalatsiinin samanaikainen käyttö ei vaikuttanut golimumabin puhdistumaan.
Kun haavaista koliittia sairastaville potilaille annettiin golimumabia induktioannoksina 200 mg viikolla 0 ja 100 mg viikolla 2 ja sen jälkeen ylläpitoannoksina 50 mg tai 100 mg ihon alle 4 viikon välein, vakaan tilan golimumabipitoisuus seerumissa saavutettiin noin 14 viikon kuluttua hoidon alkamisesta. Kun golimumabia annettiin ylläpitovaiheessa 50 mg:n tai 100 mg:n annoksina ihon alle 4 viikon välein, vakaan tilan aikaisten pienimpien golimumabipitoisuuksien (trough) keskiarvo seerumissa oli 50 mg annoksia saaneilla potilailla noin 0,9 ± 0,5 μg/ml ja 100 mg:n annoksia saaneilla noin 1,8 ± 1,1 μg/ml.
Immunomoduloivien lääkkeiden samanaikainen käyttö ei vaikuttanut oleellisesti vakaan tilan aikaisiin pienimpiin golimumabipitoisuuksiin haavaista koliittia sairastavilla potilailla, jotka saivat golimumabia 50 mg tai 100 mg ihon alle 4 viikon välein.
Potilailla, joille kehittyi vasta-aineita golimumabille, vakaan tilan aikaiset pienimmät golimumabipitoisuudet (trough) olivat yleensä pieniä (ks. kohta Farmakodynamiikka).
Lineaarisuus
Kerta-annoksena laskimoon annetun golimumabin farmakokinetiikka oli nivelreumapotilailla jokseenkin suoraan annoksesta riippuvainen annosalueella 0,1-10,0 mg/kg. Myös terveille tutkittaville annetun ihonalaisen kerta-annoksen jälkeen farmakokinetiikka oli jokseenkin suoraan annoksesta riippuvainen annosalueella 50–400 mg.
Painon vaikutus farmakokinetiikkaan
Tutkimuksessa golimumabin puhdistuma näytti korreloivan positiivisesti potilaan painon kanssa (puhdistuma suurin painavimmilla potilailla) (ks. kohta Annostus ja antotapa).
Pediatriset potilaat
Golimumabin farmakokinetiikka määritettiin 173:lla 2−17-vuotiaalla idiopaattista juveniilia polyartriittia sairastavalla lapsella. pJIA-tutkimuksessa niillä lapsilla, jotka saivat golimumabia 30 mg/m2 (enintään 50 mg) ihon alle 4 viikon välein, vakaan tilan keskimääräiset golimumabipitoisuudet olivat samalla tasolla eri ikäryhmissä ja lisäksi samalla tasolla tai hieman korkeampia kuin aikuisilla reumapotilailla, jotka olivat saaneet 50 mg golimumabia 4 viikon välein.
Populaatiofarmakokineettinen/farmakodynaaminen mallinnus ja simulointi pJIA-lapsilla vahvistivat seerumin lääkepitoisuuden perusteella todetun golimumabialtistuksen ja kliinisen tehon välisen yhteyden ja puoltavat sitä, että 50 mg:n golimumabiannos 4 viikon välein vähintään 40 kg painaville pJIA-lapsille saa aikaan samanlaisen lääkealtistuksen, joka on aikuisilla osoitettu tehokkaaksi.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta sekä lisääntymis‑ ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Golimumabilla ei ole tehty mutageenisuustutkimuksia, hedelmällisyystutkimuksia eläimillä eikä pitkäaikaisia karsinogeenisuustutkimuksia.
Hiirien hedelmällisyyttä ja yleisiä lisääntymistoimintoja kartoittavassa tutkimuksessa tiineiden hiirien määrä pieneni, kun käytettiin analogista vasta-ainetta, joka estää selektiivisesti hiiren TNF-α:n toiminnallista aktiivisuutta. Ei tiedetä, johtuiko tämä löydös uros- vai naarashiiriin vai molempiin kohdistuneista vaikutuksista. Yksilönkehitykseen kohdistuvaa toksisuutta selvittävässä tutkimuksessa annettiin hiirille samaa analogista vasta-ainetta ja cynomolgus-apinoille golimumabia. Tutkimuksessa ei havaittu viitteitä emoon tai alkioon kohdistuvista toksisista vaikutuksista eikä teratogeenisuudesta.
Farmaseuttiset tiedot
Apuaineet
Sorbitoli (E 420)
Histidiini
Histidiinihydrokloridimonohydraatti
Polysorbaatti 80
Injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
2 vuotta
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Ei saa jäätyä.
Pidä esitäytetty kynä tai esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Simponi voidaan säilyttää korkeintaan 25 °C:n lämpötilassa yksittäisen korkeintaan 30 vuorokauden pituisen ajanjakson ajan. Säilytysaika ei saa ylittää alkuperäistä koteloon painettua viimeistä käyttöpäivämäärää. Uusi viimeinen käyttöpäivämäärä on merkittävä koteloon (korkeintaan 30 vuorokautta päivästä, jolloin tuote on otettu pois jääkaapista).
Huoneenlämmössä säilytettyä Simponia ei saa palauttaa takaisin jääkaappisäilytykseen. Simponi on hävitettävä, jos sitä ei ole käytetty 30 vuorokauden huoneenlämpösäilytyksen aikana.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SIMPONI injektioneste, liuos, esitäytetty kynä
50 mg/0,5 ml 0,5 ml (SmartJect) (951,13 €)
SIMPONI injektioneste, liuos, esitäytetty ruisku
50 mg/0,5 ml 0,5 ml (951,13 €)
PF-selosteen tieto
Simponi 50 mg injektioneste, liuos, esitäytetty kynä
0,5 ml liuosta esitäytetyssä kynässä olevassa esitäytetyssä ruiskussa (tyyppi I lasia), johon on kiinnitetty neula (ruostumatonta terästä) sekä neulan suojus (lateksia sisältävää kumia). Simponi on saatavana 1 esitäytetyn kynän sisältävänä pakkauksena sekä kerrannaispakkauksena, jossa on 3 esitäytettyä kynää (3 yhden kynän sisältävää pakkausta).
Simponi 50 mg injektioneste, liuos, esitäytetty ruisku
0,5 ml liuosta esitäytetyssä ruiskussa (tyyppi I lasia), johon on kiinnitetty neula (ruostumatonta terästä) sekä neulan suojus (lateksia sisältävää kumia). Simponi on saatavana 1 esitäytetyn ruiskun sisältävänä pakkauksena sekä kerrannaispakkauksena, jossa on 3 esitäytettyä ruiskua (3 yhden ruiskun sisältävää pakkausta).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Liuos on kirkas tai hieman opaalinhohtoinen, väritön tai vaaleankeltainen.
Käyttö- ja käsittelyohjeet
Simponi on saatavana kertakäyttöön tarkoitetussa esitäytetyssä kynässä, jota kutsutaan nimellä SmartJect, tai kertakäyttöön tarkoitetussa esitäytetyssä ruiskussa. Jokaisessa pakkauksessa on käyttöohjeet, joissa kerrotaan yksityiskohtaisesti kynän tai ruiskun käytöstä. Kun esitäytetty kynä tai esitäytetty ruisku on otettu jääkaapista, odotetaan 30 minuuttia, jotta se lämpenee huoneenlämpöiseksi ennen Simponin pistämistä. Kynää tai ruiskua ei saa ravistaa.
Liuos on kirkas tai hieman opaalinhohtoinen, väritön tai vaaleankeltainen ja se saattaa sisältää muutamia pieniä, läpikuultavia tai valkoisia proteiinihiukkasia. Tämä ei ole harvinaista liuoksille, jotka sisältävät proteiinia. Simponia ei pidä käyttää, jos liuos on värjäytynyt, samea tai se sisältää silmin nähtäviä vieraita hiukkasia.
Pakkausselosteessa annetaan yksityiskohtaiset ohjeet Simponin esitäytetyn kynän tai esitäytetyn ruiskun valmistelusta ja pistoksen ottamisesta.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SIMPONI injektioneste, liuos, esitäytetty kynä
50 mg/0,5 ml 0,5 ml
SIMPONI injektioneste, liuos, esitäytetty ruisku
50 mg/0,5 ml 0,5 ml
- Alempi erityiskorvaus (65 %). Abatasepti, adalimumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli ja tosilitsumabi (tulehdukselliset reumasairaudet): Nivelreuman, juveniilin polyartriitin, psoriaasiin liittyvän niveltulehduksen, selkärankareuman tai edellä mainittuja niveltulehduksia läheisesti muistuttavan niveltulehduksen hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman hoito erityisin edellytyksin (281).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli, tosilitsumabi ja ustekinumabi (tulehdukselliset reumasairaudet): Eräiden reumasairauksien hoito erityisin edellytyksin / Adalimumabi: Uveiitin hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman ja jättisoluarteriitin hoito erityisin edellytyksin (313), Adalimumabi, golimumabi, infliksimabi, risankitsumabi ja ustekinumabi (tulehdukselliset suolistosairaudet): Eräiden suolistosairauksien hoito erityisin edellytyksin (326).
ATC-koodi
L04AB06
Valmisteyhteenvedon muuttamispäivämäärä
09.11.2023
Yhteystiedot
 MSD FINLAND OY
MSD FINLAND OY Keilaniementie 1, PL 46
02151 Espoo
09 804 650
www.msd.fi
info@msd.fi