
VOLIBRIS tabletti, kalvopäällysteinen 5 mg, 10 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Tärkeää turvallisuuteen liittyvää tietoa potilaille, jotka käyttävät Volibris-valmistetta (ambrisentaani).
Vaikuttavat aineet ja niiden määrät
Volibris 2,5 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää 2,5 mg ambrisentaania.
Apuaine(et), joiden vaikutus tunnetaan:
Yksi tabletti sisältää noin 92,6 mg laktoosia (monohydraattina) ja noin 0,25 mg lesitiiniä (soija) (E322).
Volibris 5 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää 5 mg ambrisentaania.
Apuaine(et), joiden vaikutus tunnetaan:
Yksi tabletti sisältää noin 90,3 mg laktoosia (monohydraattina), noin 0,25 mg lesitiiniä (soija) (E322) ja noin 0,11 mg allurapunaista AC (E129).
Volibris 10 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää 10 mg ambrisentaania.
Apuaine(et), joiden vaikutus tunnetaan:
Yksi tabletti sisältää noin 85,5 mg laktoosia (monohydraattina), noin 0,25 mg lesitiiniä (soija) (E322) ja noin 0,45 mg allurapunaista AC (E129).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti)
Kliiniset tiedot
Käyttöaiheet
Volibris on tarkoitettu WHO:n toimintakykyluokkiin II ja III kuuluvien aikuispotilaiden pulmonaalihypertension hoitoon, mukaan lukien käyttö yhdistelmähoitona (ks. kohta Farmakodynamiikka). Teho on osoitettu idiopaattisessa ja sidekudossairauteen liittyvässä pulmonaalihypertensiossa.
Volibris on tarkoitettu WHO:n toimintakykyluokkiin II ja III kuuluvien lasten ja nuorten (8 – < 18-vuotiaiden) pulmonaalihypertension hoitoon, mukaan lukien käyttö yhdistelmähoitona. Teho on osoitettu idiopaattisessa, familiaalisessa, korjatussa synnynnäisessä ja sidekudossairauteen liittyvässä pulmonaalihypertensiossa (ks. kohta Farmakodynamiikka).
Ehto
Hoidon saa aloittaa vain pulmonaalihypertension hoitoon perehtynyt lääkäri.
Annostus ja antotapa
Hoidon saa aloittaa vain pulmonaalihypertension hoitoon perehtynyt lääkäri.
Annostus
Aikuiset
Ambrisentaanimonoterapia
Volibris otetaan suun kautta. Aloitusannos on 5 mg kerran vuorokaudessa. Annos voidaan suurentaa 10 mg:aan vuorokaudessa kliinisestä vasteesta ja siedettävyydestä riippuen.
Ambrisentaani yhdessä tadalafiilin kanssa
Kun Volibris-valmistetta käytetään yhdessä tadalafiilin kanssa, Volibris-annos nostetaan 10 mg:aan kerran vuorokaudessa.
AMBITION-tutkimuksessa potilaat saivat 5 mg ambrisentaania vuorokaudessa ensimmäisten 8 hoitoviikon ajan, minkä jälkeen annos suurennettiin 10 mg:aan siedettävyydestä riippuen (ks. kohta Farmakodynamiikka). Kun lääkettä käytettiin yhdessä tadalafiilin kanssa, aloitusannos oli 5 mg ambrisentaania ja 20 mg tadalafiilia. Siedettävyydestä riippuen tadalafiiliannos suurennettiin 40 mg:aan 4 viikon kuluttua ja ambrisentaaniannos suurennettiin 10 mg:aan 8 viikon kuluttua. Tämä toteutui yli 90 %:lla potilaista. Annoksia voitiin myös pienentää siedettävyydestä riippuen.
Käytettävissä oleva tieto viittaa siihen, että ambrisentaani-hoidon äkilliseen keskeyttämiseen ei liity pulmonaalihypertension pahenemista.
Ambrisentaani yhdessä siklosporiini A:n kanssa
Aikuisilla ambrisentaaniannos on rajoitettava 5 mg:aan kerran vuorokaudessa, kun sitä annetaan samanaikaisesti siklosporiini A:n kanssa. Potilaan tilaa on seurattava huolellisesti (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Pediatriset potilaat (8 – < 18-vuotiaat)
Ambrisentaani ainoana lääkkeenä tai yhdessä muiden pulmonaalihypertensiolääkkeiden kanssa
Volibris otetaan suun kautta alla esitetyn annosohjelman mukaisesti:
Paino (kg) | Aloitusannos kerran vuorokaudessa (mg) | Myöhempi annostitraus kerran vuorokaudessa (mg)a |
≥ 50 | 5 | 10 |
≥ 35–< 50 | 5 | 7,5 |
≥ 20–< 35 | 2,5 | 5 |
aRiippuu kliinisestä vasteesta ja siedettävyydestä (ks. kohta Farmakodynamiikka). | ||
Ambrisentaani yhdessä siklosporiini A:n kanssa
Pediatrisilla potilailla ambrisentaaniannos on rajoitettava ≥ 50 kg painavilla potilailla määrään 5 mg kerran vuorokaudessa ja ≥ 20 – < 50 kg painavilla potilailla määrään 2,5 mg kerran vuorokaudessa, kun sitä annetaan samanaikaisesti siklosporiini A:n kanssa. Potilaan tilaa on seurattava huolellisesti (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Erityispotilasryhmät
Iäkkäät potilaat
Yli 65-vuotiaiden potilaiden annostusta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Potilaat, joilla on munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavien potilaiden annostusta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka). Kokemukset ambrisentaanin käytöstä potilaille, joilla on vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma <30 ml/min), ovat vähäiset. Hoito on aloitettava varovasti näille potilaille ja potilaista on huolehdittava erityisesti, jos ambrisentaaniannosta suurennetaan määrään 10 mg.
Potilaat, joilla on maksan vajaatoiminta
Ambrisentaania ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla (joilla on maksakirroosi tai ei ole sitä). Maksan vajaatoiminnan voidaan olettaa suurentavan ambrisentaanialtistusta (Cmax ja AUC) johtuen siitä, että päämetaboliareitit ovat glukuronidaatio ja oksidaatio sekä näitä seuraava eliminaatio sappinesteen mukana. Siksi ambrisentaanihoitoa ei saa aloittaa potilaille, joilla on vaikea maksan vajaatoiminta tai kliinisesti merkittävät, kohonneet maksan aminotransferaasiarvot (yli kolminkertaiset viitearvojen ylärajaan [upper normal limit, ULN] verrattuna, ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Ambrisentaanin tehoa ja turvallisuutta alle 8 vuoden ikäisten lasten hoidossa ei ole varmistettu.
Kliinisiä tietoja ei ole saatavilla (katso kohta Prekliiniset tiedot turvallisuudesta saatavilla olevista tiedoista nuorilla eläimillä).
Antotapa
Volibris otetaan suun kautta. Tabletin nielemistä kokonaisena suositellaan ja se voidaan ottaa joko ruoan kanssa tai ilman ruokaa. On suositeltavaa, että tablettia ei jaeta, murskata eikä pureskella.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle, soijalle tai kohdassa Apuaineet mainituille apuaineille.
Raskaus (ks. kohta Raskaus ja imetys).
Käyttö naisille, jotka voivat tulla raskaaksi ja jotka eivät käytä luotettavaa ehkäisymenetelmää (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys).
Imetys (ks. kohta Raskaus ja imetys).
Vaikea maksan vajaatoiminta (maksakirroosiin liittyen tai ilman sitä) (ks. kohta Annostus ja antotapa).
Maksan aminotransferaasien lähtöarvot (aspartaattiaminotransferaasi ASAT ja/tai alaniininaminotransferaasi ALAT) > 3 x ULN (viitearvojen ylärajan) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Idiopaattinen keuhkofibroosi (IPF), johon voi liittyä sekundaarinen pulmonaalihypertensio (ks. kohta Farmakodynamiikka).
Varoitukset ja käyttöön liittyvät varotoimet
Ambrisentaania ei ole tutkittu riittävästi hyöty/haitta-suhteen arvioimiseksi WHO:n toimintakykyluokkaan I kuuluvien pulmonaalihypertensiota sairastavien potilaiden osalta.
Ambrisentaanin tehoa ainoana lääkkeenä ei ole varmistettu WHO:n toimintakykyluokkaan IV kuuluvien pulmonaalihypertensiota sairastavien potilaitten osalta. Hoitoa, jota suositellaan taudin vaikeassa vaiheessa (esim. epoprostenolia), on harkittava, jos potilaan kliininen tila huononee.
Maksan toiminta
Pulmonaalihypertensiopotilailla on esiintynyt maksan toiminnan poikkeavuuksia. Autoimmuunihepatiittiin liittyviä tapauksia, kuten mahdollista olemassa olevan autoimmuunihepatiitin pahenemista, maksavaurioita ja mahdollisesti hoitoon liittyvää maksaentsyymiarvojen nousua, on havaittu ambrisentaania käytettäessä (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). Siksi maksan aminotransferaasipitoisuudet (ALAT ja ASAT) on mitattava ennen ambrisentaanihoidon aloittamista ja hoitoa ei saa aloittaa potilaille, joilla ALAT- ja/tai ASAT-lähtöarvot ovat > 3 x ULN (viitearvojen ylärajan; ks. kohta Vasta-aiheet).
Maksavaurion merkkejä on seurattava potilailta ja ALAT- ja ASAT-arvojen kuukausittaista seurantaa suositellaan. Jos potilaalla ilmenee pysyvä selittämätön kliinisesti merkitsevä ALAT- ja/tai ASAT-arvojen nousu, tai jos ALAT- ja/tai ASAT-arvojen nousuun liittyy maksavaurion merkkejä tai oireita (esim. keltaisuus), ambrisentaanihoito on keskeytettävä.
Potilailla, joilla ei ilmene maksavaurion kliinisiä oireita tai keltaisuutta, ambrisentaanin aloittamista uudelleen voidaan harkita maksaentsyymiarvojen palauduttua normaalille tasolle. On suositeltavaa kääntyä maksasairauksien hoitoon perehtyneen erikoislääkärin puoleen.
Hemoglobiinipitoisuus
Endoteliinireseptoriantagonistihoitoon (ERA), myös ambrisentaanin käyttöön, on liittynyt hemoglobiinipitoisuuden ja hematokriitin pienemistä. Suurin osa pienentymisistä havaittiin ensimmäisten neljän hoitoviikon aikana. Tämän jälkeen hemoglobiinipitoisuus yleensä vakiintui. Hemoglobiinipitoisuuden keskiarvoinen lasku lähtötasoon verrattuna (vaihteluväli 9–12 g/l [0,9–1,2 g/dl]) kesti pisimmillään neljänteen vuoteen asti avoimessa pitkäaikaisessa faasin III kliinisessä tutkimuksessa. Kliinisen käytön aikana on raportoitu verisolujen siirtoa vaatineita anemiatapauksia (ks. kohta Haittavaikutukset).
Ambrisentaanihoidon aloittamista ei suositella potilaille, joilla on kliinisesti merkitsevä anemia. Hemoglobiini- ja/tai hematokriittiarvojen mittaamista suositellaan ambrisentaanihoidon aikana esimerkiksi kuukauden kuluttua hoidon aloittamisesta, kolmen kuukauden kuluttua ja sen jälkeen määräajoin vallitsevan kliinisen käytännön mukaan. Jos hemoglobiini- tai hematokriittiarvoissa havaitaan kliinisesti merkitsevää pienenemistä, ja muut syyt poissuljetaan, annoksen pienentämistä tai hoidon keskeyttämistä tulee harkita. Anemian ilmaantuvuus lisääntyi, kun ambrisentaania annettiin yhdessä tadalafiilin kanssa (haittatapahtumia esiintyi 15 %:lla). Ambrisentaanimonoterapian yhteydessä anemian ilmaantuvuus oli 7 % ja tadalafiilimonoterapian yhteydessä 11 %.
Nesteen kerääntyminen elimistöön
Endoteliinireseptoriantagonistien kuten ambrisentaanin on havaittu aiheuttaneen perifeeristä turvotusta. Kliinisissä tutkimuksissa ambrisentaanilla havaittu perifeerinen turvotus oli useimmissa tapauksissa luonteeltaan lievää tai kohtalaista. Sitä saattaa esiintyä enemmän ja vakavampana 65-vuotiailla ja sitä vanhemmilla potilailla. Lyhytkestoisissa kliinisissä tutkimuksissa perifeeristä turvotusta raportoitiin useammin 10 mg:n ambrisentaaniannokseen liittyen (ks. kohta Haittavaikutukset).
Markkinoille tulon jälkeen on saatu raportteja nesteen kerääntymisestä tai sydämen vajaatoiminnan dekompensaatiosta viikkojen kuluttua ambrisentaanihoidon aloittamisesta. Joissakin tapauksissa on tarvittu hoitoa diureetilla tai nesteytyksen hoitoa sairaalassa. Jos potilaalla on aikaisempi nesteylimäärä, se on hoidettava vakiintuneen hoitokäytännön mukaisesti ennen ambrisentaanihoidon aloittamista.
Jos nestettä kertyy kliinisesti merkitsevästi ambrisentaanihoidon aikana riippumatta siitä, liittyykö siihen painonnousua vai ei, lisäarviointi syyn (kuten ambrisentaani tai taustalla oleva sydämen vajaatoiminta) selvittämiseksi on aloitettava. Mahdollinen erityishoidon tarve tai ambrisentaanihoidon keskeyttämisen tarve tulee arvioida. Perifeerisen turvotuksen ilmaantuvuus lisääntyi, kun ambrisentaania annettiin yhdessä tadalafiilin kanssa (haittatapahtumia esiintyi 45 %:lla). Ambrisentaanimonoterapian yhteydessä perifeerisen turvotuksen ilmaantuvuus oli 38 % ja tadalafiilimonoterapian yhteydessä 28 %. Perifeeristä turvotusta esiintyi tavallisimmin ensimmäisen hoitokuukauden aikana.
Naiset, jotka voivat tulla raskaaksi
Volibris-hoitoa ei saa aloittaa naisille, jotka voivat tulla raskaaksi ellei hoitoa edeltäneen raskaustestin tulos ole negatiivinen ja käytössä ole luotettavaa ehkäisyä. On harkittava kääntymistä naistentautien hoitoon perehtyneen erikoislääkärin puoleen, jos on epäilystä siitä, millaista ehkäisyneuvontaa yksittäiselle potilaalle tulisi antaa. Ambrisentaanihoidon aikana suositellaan raskaustestiä kuukausittain (ks. kohdat Vasta-aiheet ja Raskaus ja imetys).
Keuhkolaskimoahtauma
Keuhkoedeemaa on raportoitu keuhkolaskimoahtaumaa sairastavilla potilailla vasodilatoivien lääkevalmisteiden esim. endoteelireseptorin antagonistien käytön yhteydessä. Siksi mahdollista keuhkolaskimoahtaumaa on epäiltävä, jos potilaille kehittyy akuutti keuhkoedeema ambrisentaanihoidon aikana.
Samanaikainen käyttö muiden lääkkeiden kanssa
Ambrisentaanihoitoa saavia potilaita on seurattava huolellisesti, kun rifampisiinihoito aloitetaan (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Apuaineet
Volibris 2,5 mg, 5 mg ja 10 mg kalvopäällysteiset tabletit
Laktoosi
Tämä lääkevalmiste sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi‑imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Lesitiini (soija)
Tämä lääkevalmiste sisältää soijasta eristettyä lesitiiniä. Potilaan, joka on allerginen soijalle, ei tule käyttää ambrisentaania (ks. kohta Vasta-aiheet).
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Volibris 5 mg ja 10 mg kalvopäällysteiset tabletit
Allurapunainen AC
Volibris 5 mg ja 10 mg ‑tabletit sisältävät atsoväriainetta allurapunainen AC (E129), joka saattaa aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Ambrisentaani ei inhiboi eikä indusoi I tai II vaiheen lääkeaineita metaboloivia entsyymejä kliinisesti merkityksellisillä pitoisuuksilla prekliinisissä in vitro ja in vivo-tutkimuksissa. Siksi ambrisentaanin uskotaan muuttavan näiden reittien kautta metaboloituvien lääkevalmisteiden kinetiikkaa vain vähän.
Ambrisentaanin mahdollista kykyä indusoida CYP3A4:n aktiivisuutta tutkittiin terveillä vapaaehtoisilla. Tulokset viittasivat siihen, ettei ambrisentaani indusoi CYP3A4 isoentsyymiä.
Siklosporiini A
Kun ambrisentaanin annossa terveille vapaaehtoisille saavutettiin vakaa tila ja sitä annettiin samanaikaisesti siklosporiini A:n kanssa, tämä johti kaksinkertaiseen altistumiseen ambrisentaanille. Tämä voi johtua siitä, että siklosporiini A estää ambrisentaanin farmakokinetiikkaan osallistuvia transporttereita ja metaboloivia entsyymejä. Siksi ambrisentaaniannos on rajoitettava aikuispotilailla tai ≥ 50 kg painavilla pediatrisilla potilailla 5 mg:aan kerran vuorokaudessa ja ≥ 20–< 50 kg painavilla pediatrisilla potilailla 2,5 mg:aan kerran vuorokaudessa, kun ambrisentaania annetaan samanaikaisesti siklosporiini A:n kanssa (ks. kohta Annostus ja antotapa). Toistuvilla ambrisentaaniannoksilla ei ollut vaikutusta altistumiseen siklosporiini A:lle ja siklosporiini A:n annosta ei ole aiheellista muuttaa.
Rifampisiini
Rifampisiinin (orgaanisen anionikuljettajaproteiinin [OATP:n] estäjä, voimakas CYP3A- ja 2C19-entsyymien indusoija, Pgp:n ja uridiinidifosfoglukuronosyylitransferaasien [UGT:en] indusoija) samanaikaiseen antoon liittyi ohimenevä (noin kaksinkertainen) ambrisentaanin altistuksen kasvu, kun terveille vapaaehtoisille annettiin aloitusannokset. Kahdeksanteen päivään mennessä rifampisiinin annolla vakaassa tilassa ei kuitenkaan ollut kliinisesti merkitsevää vaikutusta ambrisentaanin altistukseen. Ambrisentaanihoitoa saavia potilaita on seurattava huolellisesti, kun rifampisiinihoito aloitetaan (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Fosfodiesteraasiestäjät
Ambrisentaanin samanaikainen käyttö fosfodiesteraasiestäjän joko sildenafiilin tai tadalafiilin kanssa (molemmat ovat CYP3A4:n substraatteja) terveillä vapaaehtoisilla ei vaikuttanut fosfodiesteraasinestäjän tai ambrisentaanin farmakokinetiikkaan merkitsevästi (ks. kohta Farmakokinetiikka).
Muut kohdennetut pulmonaalihypertensiohoidot
Yhdessä muiden pulmonaalihypertension hoitoon käytettävien lääkkeiden (esim. prostanoidien ja liukoisen guanylaattisyklaasin stimulaattorien) kanssa, ambrisentaanin tehoa ja turvallisuutta ei ole erikseen tutkittu kontrolloiduissa kliinisissä tutkimuksissa pulmonaalihypertensiopotilailla (ks. kohta Farmakodynamiikka). Tiedossa olevien biotransformaatiotietojen perusteella ambrisentaanin ja liukoisen guanylaattisyklaasin stimulaattorien tai prostanoidien välillä ei ole odotettavissa erityisiä yhteisvaikutuksia (ks. kohta Farmakokinetiikka). Näillä lääkevalmisteilla ei kuitenkaan ole tehty erityisiä yhteisvaikutustutkimuksia. Samanaikaisessa käytössä suositellaan varovaisuutta.
Oraaliset ehkäisyvalmisteet
Kun ambrisentaania annettiin 10 mg kerran vuorokaudessa kliinisessä tutkimuksessa terveille vapaaehtoisille, joilla vakaa tila oli saavutettu, sillä ei ollut kliinisesti merkityksellistä vaikutusta kerta-annoksena annetun oraalisen ehkäisyvalmisteen (yhdistelmävalmisteena etinyyliestradiolia ja noretindronia) farmakokinetiikkaan (ks. kohta Farmakokinetiikka). Tämän farmakokineettisen tutkimuksen perusteella ambrisentaanin ei odoteta merkitsevästi vaikuttavan altistumiseen estrogeenia tai progestogeenia sisältäville ehkäisyvalmisteille.
Varfariini
Ambrisentaani ei vaikuttanut varfariinin vakaan tilan farmakokinetiikkaan tai varfariinin antikoagulaatioaktiivisuuteen terveillä vapaaehtoisilla tehdyssä tutkimuksessa (ks. kohta Farmakokinetiikka). Myöskään varfariinillä ei ollut kliinisesti merkitsevää vaikutusta ambrisentaanin farmakokinetiikkaan. Lisäksi ambrisentaanilla ei ollut kokonaisvaikutusta varfariinityyppisen antikoagulantin viikkoannokseen, protrombiiniaikaan (PT) ja INR-arvoon (international normalized ratio) potilailla.
Ketokonatsoli
Kun ketokonatsolia (voimakas CYP3A4 inhibiittori) annettiin ja vakaa tila oli saavutettu, se ei johtanut kliinisesti merkitsevään altistumisen lisääntymiseen ambrisentaanille (ks. kohta Farmakokinetiikka).
Ambrisentaanin vaikutus ksenobioottien transporttereihin
Kliinisesti merkityksellisillä pitoisuuksilla annettuna ambrisentaani ei vaikuta inhiboivasti ihmisen transporttereihin kuten P-glykoproteiiniin (Pgp), rintasyöpäresistenssiproteiiniin (BCRP), monilääkeresistenssiproteiinin isoentsyymi-2:een (MRP2), sappisuolojen kuljettajaan (BSEP), orgaanisten anionien kuljettaproteiinehin (OATP1B1 ja OATP1B3) tai natrium riippuvaiseen taurokolaatti ko-transportteriin (NTCP ) in vitro.
Ambrisentaani on Pgp-välitteisen effluksin substraatti.
In vitro-tutkimukset rotan maksasoluilla osoittivat myös, että ambrisentaani ei indusoinut Pgp-, BSEP- tai MRP2-proteiinien ekspressiota.
Kun ambrisentaania annettiin terveille vapaaehtoisille vakaan tilan tutkimuksessa, sillä ei ollut kliinisesti merkityksellisiä vaikutuksia kerta-annoksena annetun digoksiinin (Pgp:n substraatti) farmakokinetiikkaan (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Ambrisentaanihoitoa ei saa aloittaa naisille, jotka voivat tulla raskaaksi ellei raskaustestin tulos ole negatiivinen ja käytössä ole luotettavaa ehkäisyä. Ambrisentaanihoidon aikana suositellaan raskaustestiä kuukausittain.
Raskaus
Ambrisentaani on vasta-aiheista raskauden aikana (ks. kohta Vasta-aiheet). Eläinkokeet ovat osoittaneet ambrisentaanin olevan teratogeenista. Kokemusta ihmisisten hoidosta ei ole.
Ambrisentaania käyttäville naisille on kerrottava sikiövaurion riskistä. Vaihtoehtoinen hoito on aloitettava, jos raskaus todetaan (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Prekliiniset tiedot turvallisuudesta).
Imetys
Ei tiedetä, erittyykö ambrisentaani ihmisen rintamaitoon. Ambrisentaanin erittymistä rintamaitoon ei ole tutkittu eläimillä. Siksi imetys on vasta-aiheista ambrisentaania saaville naisille (ks. kohta Vasta-aiheet).
Miesten hedelmällisyys
Pitkäkestoiseen endoteliinireseptoriantagonistihoitoon, myös ambrisentaaniin, on yhdistetty tubulaarisen atrofian kehittyminen kiveksiin eläimille (ks. kohta Prekliiniset tiedot turvallisuudesta). Vaikka ARIES-E-tutkimuksessa ei saatu selvää näyttöä ambrisentaanin haitallisesta vaikutuksesta siemennesteen määrään pitkäkestoisessa altistuksessa, jatkuvaan ambrisentaanin antoon liittyi muutoksia spermatogeneesin merkkiaineissa. Plasman inhibiini-B-pitoisuuden pienenemistä ja plasman FSH-pitoisuuden suurenemista havaittiin. Vaikutusta miehen hedelmällisyyteen ei tunneta, mutta spermatogeneesin heikkenemistä ei voida poissulkea. Kliinisissä tutkimuksissa pitkäkestoiseen ambrisentaanin antoon ei liittynyt muutosta plasman testosteronipitoisuudessa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ambrisentaanilla on vähäinen tai kohtalainen vaikutus ajokykyyn tai koneiden käyttökykyyn. Potilaan kliininen tila ja ambrisentaanin haittavaikutukset (kuten verenpaineen lasku, huimaus, heikotus, uupumus) on pidettävä mielessä, kun arvioidaan potilaan suoritumista tehtävissä, jotka vaativat arvostelukykyä, motorisia tai kognitiivisia taitoja (ks. kohta Haittavaikutukset). Potilaiden on tiedostettava kuinka ambrisentaani vaikuttaa heihin ennen autolla ajamista tai koneiden käyttöä.
Haittavaikutukset
Yhteenveto turvallisuudesta
Perifeerinen turvotus (37 %) ja päänsärky (28 %) olivat yleisimmät ambrisentaanin käytön aikana havaituista haittavaikutuksista. Suuremmalla 10 mg:n annoksella näitä haittoja ilmeni useammin ja perifeerinen turvotus oli lyhytkestoisissa kliinisissä tutkimuksissa vaikeampaa 65-vuotiailla ja sitä vanhemmilla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ambrisentaanin käyttöön liittyviä vakavia haittavaikutuksia ovat mm. anemia (pienentynyt hemoglobiiniarvo, pienentynyt hematokriittiarvo) ja maksatoksisuus.
ERA‑hoitoon, myös ambrisentaanin käyttöön, on liittynyt hemoglobiinipitoisuuden ja hematokriitin pienenemistä (10 %). Suurin osa pienentymisistä havaittiin ensimmäisten neljän hoitoviikon aikana. Tämän jälkeen hemoglobiinipitoisuus yleensä vakiintui (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksaentsyymiarvojen suurenemista (2 %), maksavaurioita ja autoimmuunihepatiittia (myös perussairauden pahenemista) on havaittu ambrisentaanin käytön yhteydessä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Taulukointi haittavaikutuksista
Ilmaantuvuus on määritelty: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Annosriippuvaisissa haittavaikutuksissa ilmaantuvuusluokka on suuremman ambrisentaaniannoksen mukainen. Kussakin ilmaantuvuusryhmässä haittavaikutukset on esitetty alenevassa järjestyksessä vakavuuden perusteella.
Elinjärjestelmä | Yleisyys | Haittavaikutukset |
Veri ja imukudos | Hyvin yleinen | Anemia (pienentynyt hemoglobiiniarvo, pienentynyt hematokriittiarvo)1 |
Immuunijärjestelmä | Yleinen | Yliherkkyysreaktiot (esim. angioedeema, ihottuma, kutina) |
Hermosto | Hyvin yleinen | Päänsärky (ml. sinuspäänsärky, migreeni)2, huimaus |
Silmät | Yleinen | Näön hämärtyminen, näön heikkeneminen |
Kuulo ja tasapainoelin | Yleinen | Tinnitus3 |
Melko harvinainen | Äkillinen kuulon menetys3 | |
Sydän | Hyvin yleinen | Sydämentykytys |
Yleinen | Sydämen vajaatoiminta4 | |
Verisuonisto | Hyvin yleinen | Punastuminen5 |
Yleinen | Hypotensio, synkopee | |
Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Hengenahdistus6, ylähengitysteiden (esim. nenä, sinus) tukkoisuus7, nenänielun tulehdus7 |
Yleinen | Nenäverenvuoto, nuha7, sinuiitti7 | |
Ruoansulatuselimistö | Hyvin yleinen | Pahoinvointi, ripuli, oksentelu5 |
Yleinen | Vatsakipu, ummetus | |
Maksa ja sappi | Yleinen | Kohonneet maksan transaminaasiarvot |
Melko harvinainen | Maksavaurio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), autoimmuunihepatiitti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | |
Iho ja ihonalainen kudos | Yleinen | Ihottuma8 |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Perifeerinen turvotus, nesteen kertyminen elimistöön, rintakipu / epämiellyttävä tuntemus rinnassa5, uupumus |
Yleinen | Voimattomuus |
1 Katso kohta ”Kuvaus valituista haittavaikutuksista”.
2 Päänsärkyä esiintyi useammin, kun ambrisentaania annettiin 10 mg.
3 Tapauksia havaittiin vain ambrisentaani- ja tadalafiiliyhdistelmähoitoa tarkastelleessa lumekontrolloidussa kliinisessä tutkimuksessa.
4 Suurimpaan osaan raportoiduista sydämen vajaatoiminta tapauksista liittyi nesteretentiota.
5 Yleisyys todettiin ambrisentaani- ja tadalafiiliyhdistelmähoitoa tarkastelleessa lumekontrolloidussa kliinisessä tutkimuksessa. Ambrisentaanimonoterapian yhteydessä ilmaantuvuus oli pienempi.
6 Tapauksia hengenahdistuksen pahenemisesta, joiden etiologia on epäselvä, on raportoitu pian ambrisentaanihoidon aloittamisen jälkeen.
7 Nenän tukkoisuuden ilmaantuvuus riippui ambrisentaaniannoksesta hoidon aikana.
8 Ihottuma-termi kattaa punoittavan ihottuman, yleistyneen ihottuman, papulaarisen ihottuman ja kutisevan ihottuman.
Kuvaus valituista haittavaikutuksista
Pienentynyt hemoglobiiniarvo
Kliinisen käytön aikana on raportoitu verisolujen siirtoa vaatineita anemiatapauksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Hemoglobiiniarvon pienenemistä (anemiaa) ilmeni useammin 10 mg:n ambrisentaaniannoksella. 12 viikkoa kestäneiden III-vaiheen kliinisten tutkimusten aikana hemoglobiinin keskiarvot pienenivät ambrisentaaniryhmien potilailla. Tämä havaittiin jo neljännellä viikolla -8,3 g/l (0,83 g/dl). Keskimääräiset muutokset lähtötasolta näyttivät tasoittuvan seuraavien kahdeksan viikon aikana. Kokonaisuudessaan 17 potilaalla (6,5 %) ambrisentaanihoitoryhmässä hemoglobiiniarvo laski ≥ 15 % lähtötasolta ja alle normaalin arvon.
Pediatriset potilaat
Ambrisentaanin turvallisuutta pulmonaalihypertensiota sairastavilla 8 – < 18-vuotiailla pediatrisilla potilailla arvioitiin vaiheen 2b avoimessa tutkimuksessa 41 potilaalla, jotka saivat 2,5 mg tai 5 mg ambrisentaania kerran vuorokaudessa (pienen annoksen ryhmä) tai 2,5 mg tai 5 mg ambrisentaania kerran vuorokaudessa, mistä annos titrattiin 5 mg:aan, 7,5 mg:aan tai 10 mg:aan painon perusteella (suuren annoksen ryhmä), ainoana lääkkeenä tai yhdessä muiden pulmonaalihypertensiolääkkeiden kanssa 24 viikon ajan. Turvallisuutta on arvioitu lisäksi pitkäaikaisessa jatkotutkimuksessa 38:lla näistä 41 tutkittavasta. Haittavaikutukset, joiden arvioitiin liittyneen ambrisentaaniin, olivat yhdenmukaisia aikuispotilailla kontrolloiduissa tutkimuksissa havaittujen haittavaikutusten kanssa. Yleisimmät haittavaikutukset olivat päänsärky (15 %, 6 tutkittavalla 41:stä 24-viikkoisessa vaiheen 2b avoimessa tutkimuksessa, ja 8 %, 3 tutkittavalla 38:sta pitkäaikaisessa jatkotutkimuksessa) ja nenän tukkoisuus (7 %, 3 tutkittavalla 41:stä 24-viikkoisessa vaiheen 2b avoimessa tutkimuksessa).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteenhyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Terveillä vapaaehtoisilla 50 mg:n tai 100 mg:n kerta-annokseen (5–10-kertainen suositusten mukaiseen annokseen verrattuna) liittyi päänsärkyä, punastumista, huimausta, pahoinvointia ja nenän tukkoisuutta.
Vaikutusmekanismista johtuen ambrisentaanin yliannostus voisi mahdollisesti aiheuttaa hypotensiota (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollinen voimakas hypotensio voi vaatia aktiivista kardiovaskulaarista tukihoitoa. Spesifistä antidoottia ei ole saatavilla.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: verenpainelääkkeet, muut verenpainelääkkeet, ATC-koodi: C02KX02
Vaikutusmekanismi
Ambrisentaani on propionihappojohdannainen, joka suun kautta otettuna on aktiivinen ETA-selektiivinen endoteliinireseptoriantagonisti. Endoteliinillä on merkittävä osa pulmonaalihypertension patofysiologiassa.
Ambrisentaani on ETA-reseptorin antagonisti (selektiivisyys ETA-reseptoriin noin 4000-kertainen verrattuna ETB-reseptoriin). Ambrisentaani salpaa ETA-reseptorialatyyppiä. Näitä reseptoreita on pääasiassa verisuonten sileissä lihassoluissa ja sydänlihassoluissa. Salpaus estää endoteliinin välittämän toisiolähettijärjestelmän aktivoitumisen. Aktivoituminen johtaa vasokonstriktioon ja sileiden lihassolujen lisääntymiseen. Ambrisentaanin selektiivisyys ETA-reseptoreihin on suurempi kuin ETB-reseptoreihin. Siksi ETB-reseptoreiden välittämän vasodilataattoreiden (typpioksidin ja prostasykliinin) tuotannon odotetaan säilyvän.
Kliininen teho ja turvallisuus
Teho osoitettiin kahdessa keskeisessä satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa, III:nnen faasin monikeskustutkimuksessa (ARIES-1 ja ‑2). ARIES 1-tutkimuksessa, johon osallistui 201 potilasta, verrattiin ambrisentaanin 5 mg:n ja 10 mg:n annosta lumelääkkeeseen. ARIES‑2:ssa, jossa oli mukana 192 potilasta, verrattiin ambrisentaania annoksilla 2,5 mg ja 5 mg lumelääkkeeseen. Kummassakin tutkimuksessa ambrisentaani lisättiin potilaiden tuki-/peruslääkkeisiin, joihin saattoi kuulua digoksiini, antikoagulantteja, diureetteja, happea ja vasodilaattoreita (kalsiumkanavan salpaajia, ACE:n estäjiä). Tutkimukseen osallistuneilla potilailla oli idiopaattinen tai sidekudossairauteen liittyvä pulmonaalihypertensio (PAH-CTD). Suurimmalla osalla potilaista oli WHO:n toimintakykyluokan II (38,4 %) tai luokan III (55,0 %) oireita. Potilaita, joilla oli maksasairaus (maksakirroosi tai kliinisesti merkittävästi nousseet aminotransferaasiarvot) tai muu hoito pulmonaalihypertensioon (esim. prostanoidihoito), ei otettu tutkimukseen. Näissä tutkimuksissa ei arvioitu hemodynaamisia parametreja.
Faasin III tutkimuksissa ensisijaiseksi päätetapahtumaksi määriteltiin kuormituksen siedon parantuminen lähtötilanteesta viikkoon 12. Se arvioitiin kuuden minuutin kävelymatkan pituudessa (6MWD) tapahtuneena muutoksena. Molemmissa tutkimuksissa ambrisentaanihoito paransi merkitsevästi kuuden minuutin kävelytestin tuloksia kaikilla ambrisentaaniannoksilla.
Verrattuna lähtötasoon viikolla 12, lumekorjattu paraneminen kuuden minuutin kävelymatkan keskiarvoissa oli 30,6 m (95 %:n luottamusväli 2,9–58,3, p = 0,008) ARIES‑1-tutkimuksessa ja 59,4 m (95 %:n luottamusväli 29,6–89,3 p < 0,001) 5 mg:aa saaneilla potilailla ARIES‑2-tutkimuksessa. ARIES‑1-tutkimuksessa 10 mg:n annosta saaneilla potilailla viikolla 12 kuuden minuutin kävelymatkan pituudessa tapahtunut lumekorjattu paraneminen oli keskimäärin 51,4 m (95 %:n luottamusväli 26,6–76,2, p < 0,001).
Faasin III tutkimuksista tehtiin ennalta määritetty yhdistetty analyysi (ARIES-C). Kävelymatkan pituudessa tapahtunut lumekorjattu piteneminen oli 44,6 m (95 %:n luottamusväli 24,3–64,9 m, p < 0,001) 5 mg:n annoksella ja 52,5 m (95 %:n luottamusväli 28,8–76,2 m, p < 0,001) 10 mg:n annoksella.
ARIES-2:ssa (yhdistetyt annosryhmät) ambrisentaanihoito viivästytti merkittävästi kliinisen tilan huononemista pulmonaalihypertensiota sairastavilla potilailla lumelääkkeeseen verrattuna (p < 0,001). Riskisuhteessa nähtiin 80 %:n lasku (95 %:n luottamusväli 47 %:sta 92 %:iin). Kliinisen tilan huononemiseen otettiin mukaan mm. kuolema, keuhkonsiirto, sairaalaan joutuminen pulmonaalihypertension vuoksi, atriaalinen septostomia, muiden pulmonaalihypertension hoitoon tarkoitettujen lääkkeiden lisääminen ja ennenaikainen hoidon keskeyttämistarve. Fyysisen suorituskyvyn SF-36 Health Survey ‑asteikon mukaan yhdistetyissä annosryhmissä havaittiin tilastollisesti merkitsevä kasvu (3,41 ± 6,96) verrattuna lumelääkkeeseen (-0,20 ± 8,14, p = 0,005). Ambrisentaanihoito johti tilastollisesti merkitsevään Borg Dyspnea Indeksin (BDI) paranemiseen viikolla 12 (lumekorjattu BDI-arvo oli -1,1 [95 %:n luottamusväli -1,8–0,4, p = 0,019, yhdistetyt annosryhmät]).
Pitkäaikaistulokset
ARIES-1- ja ‑2-tutkimuksiin osallistuneet potilaat saivat halutessaan osallistua avoimeen pitkäaikaiseen ARIES-E-tutkimukseen (n = 383). Yhdistetty keskimääräinen altistus kesti noin 145 ± 80 viikkoa. Pitkäaikaisin altistus kesti noin 295 viikkoa. Tämän tutkimuksen ensisijaiset päätetapahtumat olivat ambrisentaanin pitkäaikaiseen altistukseen liittyvien haittavaikutusten ilmeneminen ja vakavuus, mukaan lukien maksan toimintakokeiden arvot seerumissa. Tässä pitkäaikaisessa ambrisentaanialtistustutkimuksessa havaitut turvallisuuslöydökset olivat yhdenmukaisia 12 viikkoa kestäneiden lumekontrolloitujen tutkimusten havaintojen kanssa.
Havaittu eloonjäämisen todennäköisyys ambrisentaania saaneilla tutkittavilla (yhdistetyt ambrisentaaniannosryhmät) oli ensimmäisenä vuotena 93 %, toisena vuotena 85 % ja kolmentena vuotena 79 %.
Avoimessa tutkimuksessa (AMB222) arvioitiin ambrisentaanin vaikutusta seerumin aminotransferaasipitoisuuksien nousuun 36 potilaalla, jotka olivat aiemmin lopettaneet hoidon toisella endoteliinireseptoriantagonistilla aminotransferaasiarvojen poikkeavuuksien vuoksi. Keskimäärin 53 ambrisentaanihoitoviikon aikana kenellekään osallistuneista potilaista ei vahvistettu seerumin ALAT-arvon olevan yli kolme kertaa viitearvojen ylärajan, joka olisi vaatinut pysyvää hoidon lopettamista. Hoidon aikana ambrisentaanin annosta lisättiin 5 mg:sta 10 mg:aan 50 %:lla potilaista.
Kaikissa faasin II ja III tutkimuksissa (ja niihin liittyvissä avoimissa jatkotutkimuksissa) seerumin aminotransferaasien poikkeavuuksia yli kolme kertaa viitearvojen ylärajan esiintyi 17:lla 483 tutkimuspotilaasta, kun keskimääräinen altistusaika oli 79,5 viikkoa. Tämä vastaa 2,3 tapausta 100 ambrisentaanipotilasvuotta kohti. Avoimessa pitkäaikaisessa ARIES-E-tutkimuksessa kahden vuoden aikana seerumin aminotransferaasiarvojen nousun kehittymisriski yli kolme kertaa viitearvojen ylärajan (> 3 x ULN) oli 3,9 % ambrisentaanilla hoidetuilla potilailla.
Muu kliininen tieto
Faasin II tutkimuksessa (AMB220) havaittiin hemodynaamisten parametrien paranemista 29:llä pulmonaalihypertensiota sairastavalla potilaalla 12 viikon jälkeen. Keskimääräinen sydänindeksi nousi ja keuhkovaltimon keskipaine ja keskimääräinen keuhkovaltimoresistenssi pienenivät ambrisentaanihoidolla.
Systolisen ja diastollisen verenpaineen laskua on raportoitu ambrisentaanihoidossa. Lumekontrolloiduissa 12 viikkoa kestäneissä kliinisissä tutkimuksissa systolinen verenpaine laski lähtötasolta hoidon päättymiseen 3 mmHg ja diastolinen verenpaine 4,2 mmHg. Ambrisentaanilla hoidettaessa systolisen ja diastolisen verenpaineen keskimääräinen lasku säilyi pisimmillään neljä vuotta avoimessa pitkäaikaisessa ARIES-E-tutkimuksessa.
Terveillä vapaaehtoisilla tehdyssä interaktiotutkimuksessa ei ambrisentaanin tai sildenafiilin todettu vaikuttavan kliinisesti merkitsevästi toistensa farmakokinetiikkaan. Lääkeyhdistelmä oli hyvin siedetty. Potilaita, jotka saivat samanaikaisesti ambrisentaania ja sildenafiilia, oli 22 (5,7 %) ARIES-E-tutkimuksessa ja 17 (47 %) AMB222-tutkimuksessa. Muitakaan turvallisuusongelmia ei tunnistettu näiltä potilailta.
Kliininen teho yhdessä tadalafiilin kanssa
Kaksoissokkoutetussa, aktiivikontrolloidussa, tapahtumalähtöisessä faasin 3 monikeskustutkimuksessa (AMB112565/AMBITION) verrattiin ensilinjan hoitona käytetyn ambrisentaani- ja tadalafiiliyhdistelmähoidon tehoa ambrisentaanimonoterapian ja tadalafiilimonoterapian tehoon. Tutkimuksen 500 aiemmin hoitamatonta pulmonaalihypertensiopotilasta satunnaistettiin näihin hoitoryhmiin suhteessa 2:1:1. Kukaan ei saanut pelkkää lumelääkettä. Ensisijaisessa analyysissä yhdistelmähoitoryhmää verrattiin yhdistettyihin monoterapiaryhmiin. Lisäksi tehtiin tutkimusta tukevia vertailuja, joissa yhdistelmähoitoryhmää verrattiin erikseen kumpaankin monoterapiaryhmään. Potilaat, joilla oli merkittävä anemia, nesteretentiota tai harvinaisia verkkokalvosairauksia, suljettiin pois tutkijoiden arvioinnin mukaisesti. Potilaat, joiden ALAT- ja ASAT-arvot olivat lähtötilanteessa yli 2 x ULN (viitearvojen ylärajan), suljettiin myös pois.
Lähtötilanteessa 96 % potilaista ei ollut saanut aiemmin mitään kohdennettua pulmonaalihypertension hoitoa. Mediaaniaika diagnoosista tutkimukseen ottoon oli 22 vuorokautta. Potilaat saivat aluksi 5 mg ambrisentaania ja 20 mg tadalafiilia. Jos siedettävyysongelmia ei ollut, tadalafiiliannos titrattiin 40 mg:aan viikolla 4 ja ambrisentaaniannos 10 mg:aan viikolla 8. Kaksoissokkoutetun yhdistelmähoidon mediaanikesto oli yli 1,5 vuotta.
Ensisijainen päätetapahtuma oli aika ensimmäiseen kliiniseen hoidon epäonnistumiseen, joka määriteltiin seuraavasti:
- kuolema tai
- sairaalahoito pulmonaalihypertension pahenemisen vuoksi,
- taudin eteneminen,
- riittämätön pitkän aikavälin kliininen hoitovaste.
Potilaiden keski-ikä oli 54 vuotta (keskihajonta 15; vaihteluväli 18–75 vuotta). Potilaiden WHO:n toimintakykyluokka oli lähtötilanteessa II (31 %) tai III (69 %). Tutkimusaineistossa pulmonaalihypertension yleisin syy oli idiopaattinen tai perinnöllinen pulmonaalihypertensio (56 %). Seuraavaksi yleisimmät syyt olivat sidekudossairauteen liittyvä pulmonaalihypertensio (37 %), lääkkeisiin, päihteisiin tai toksiineihin liittyvä pulmonaalihypertensio (3 %), korjattuun yksinkertaiseen synnynnäiseen sydänvikaan liittyvä pulmonaalihypertensio (2 %) ja HIV (2 %). Potilailla, joiden WHO:n toimintakykyluokka oli II tai III, 6 minuutin kävelymatkan keskiarvo oli lähtötilanteessa 353 m.
Hoidon päätetapahtumat
Yhdistelmähoito pienensi riskiä hoidon epäonnistumiseen viimeiseen arviointikäyntiin mennessä 50 % (riskisuhde [HR] 0,502; 95 %:n luottamusväli 0,348–0,724; p = 0,0002) verrattuna yhdistettyihin monoterapiaryhmiin [kuva 1 ja taulukko 1]. Hoidon vaikutus näkyi etenkin sairaalahoitojen vähenemisenä 63 %:lla yhdistelmähoitoryhmässä. Vaikutus tuli esiin varhain ja oli pitkäkestoinen. Yhdistelmähoidon teho ensisijaiseen päätetapahtumaan oli johdonmukainen verrattuna yksittäisiin monoterapiaryhmiin sekä iän, etnisen taustan, maantieteellisen alueen ja etiologian (idiopaattinen/perinnöllinen pulmonaalihypertensio ja sidekudossairauteen liittyvä pulmonaalihypertensio) mukaan määritellyissä alaryhmissä. Vaikutus oli merkitsevä sekä toimintakykyluokassa II että luokassa III.
Kuva 1: Aika kliiniseen hoidon epäonnistumiseen
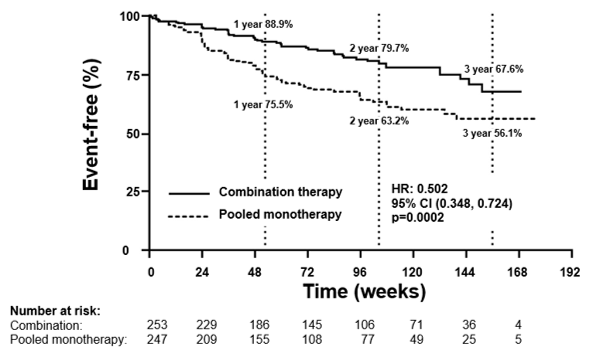
Taulukko 1
Ambrisentaani + tadalafiili (N = 253) | Yhdistetyt monoterapiaryhmät (N = 247) | Ambrisentaanimonoterapia (N = 126) | Tadalafiilimonoterapia (N = 121) | |
| Aika ensimmäiseen kliiniseen hoidon epäonnistumistapahtumaan (vahvistetut tapahtumat) | ||||
| Kliininen hoidon epäonnistuminen, määrä (%) | 46 (18) | 77 (31) | 43 (34) | 34 (28) |
| Riskisuhde (95 %:n luottamusväli) | 0,502 (0,348–0,724) | 0,477 (0,314–0,723) | 0,528 (0,338–0,827) | |
| p-arvo, log-rank-testi | 0,0002 | 0,0004 | 0,0045 | |
| Ensimmäinen kliininen hoidon epäonnistumistapahtuma tapahtumatyypeittäin (vahvistetut tapahtumat) | ||||
| Kuolema (kaikki syyt) | 9 (4 %) | 8 (3 %) | 2 (2 %) | 6 (5 %) |
| Sairaalahoito pulmonaalihypertension pahenemisen vuoksi | 10 (4 %) | 30 (12 %) | 18 (14 %) | 12 (10 %) |
| Taudin eteneminen | 10 (4 %) | 16 (6 %) | 12 (10 %) | 4 (3 %) |
| Riittämätön pitkän aikavälin kliininen hoitovaste | 17 (7 %) | 23 (9 %) | 11 (9 %) | 12 (10 %) |
| Aika ensimmäiseen sairaalahoitoon pulmonaalihypertension pahenemisen vuoksi (vahvistetut tapahtumat) | ||||
| Ensimmäinen sairaalahoito, määrä (%) | 19 (8 %) | 44 (18 %) | 27 (21 %) | 17 (14 %) |
| Riskisuhde (95 %:n luottamusväli) | 0,372 | 0,323 | 0,442 | |
| p-arvo, log-rank-testi | 0,0002 | < 0,0001 | 0,0124 | |
Toissijaiset päätetapahtumat
Toissijaiset päätetapahtumat testattiin:
Taulukko 2
| Toissijaiset päätetapahtumat (muutos lähtötilanteesta viikolle 24) | Ambrisentaani + tadalafiili | Yhdistetyt monoterapiaryhmät | Ero ja luottamusväli | p-arvo |
| NT-proBNP (arvon alenema, %) | -67,2 | -50,4 | ero, % -33,8; 95 %:n luottamusväli: -44,8–(-20,7) | p < 0,0001 |
| Tyydyttävä kliininen vaste viikolla 24, % potilaista | 39 | 29 | Ristitulosuhde (OR) 1,56: 95 %:n luottamusväli: 1,05–2,32 | p = 0,026 |
| 6 minuutin kävelymatka (m, mediaanimuutos) | 49,0 | 23,8 | 22,75 m; 95 %:n luottamusväli: 12,00–33,50 | p < 0,0001 |
Idiopaattinen kehkofibroosi
On tehty tutkimus 492:lla idiopaattista keuhkofibroosia (IPF) sairastavalla potilaalla (ambrisentaani N = 329, lume N = 163), joista 11 %:lla oli sekundaarinen pulmonaalihypertensio (WHO:n ryhmä 3). Tutkimus lopettiin alkuvaiheessa, kun huomattiin, että primaarista päätetapahtumaa ei voitaisi saavuttaa (ARTEMIS-IPF-tutkimus). Ambrisentaaniryhmässä havaittiin 90 (27 %) IPF:n pahenemista (mukaan lukien sairaalahoitoon ottamiset hengitysvaikeuksien takia) tai kuolemaa verrattuna 28 tapahtumaan (17 %) lumeryhmässä. Siksi ambrisentaanin käyttö on vasta-aiheista IPF:a sairastaville potilaille, joilla sairauteen voi liittyä sekundaarinen pulmonaalihypertensio (ks. kohta Vasta-aiheet).
Pediatriset potilaat
AMB112529-tutkimus
Kerran vuorokaudessa otettavan ambrisentaanin turvallisuutta ja siedettävyyttä arvioitiin 24 viikon ajan avoimessa kontrolloimattomassa tutkimuksessa 41:llä pulmonaalihypertensiota sairastavalla 8 – < 18‑vuotiaalla (mediaani 13 vuotta) pediatrisella potilaalla. Pulmonaalihypertension etiologia oli idiopaattinen (n = 26; 63 %), synnynnäinen korjausleikkauksesta huolimatta persistoiva (n = 11; 27 %), sidekudossairauteen liittyvä (n = 1; 2 %) tai familiaalinen (n = 3; 7,3 %). Niistä 11:stä tutkittavasta, joilla oli synnynnäinen sydänsairaus, 9:llä oli kammioväliseinän aukko, 2:lla eteisväliseinän aukko ja 1:llä persistoiva avoin tiehyt. Potilaat kuuluivat tutkimushoidon alkaessa WHO:n toimintakykyluokkaan II (n = 32; 78 %) tai III (n = 9; 22 %). Tutkimukseenottohetkellä potilailla oli käytössä pulmonaalihypertensiolääkkeitä (useimmilla PDE5:n estäjä monoterapiana [n = 18; 44 %], PDE5:n estäjä ja prostanoidi yhdistelmähoitona [n = 8; 20 %] tai prostanoidi monoterapiana [n = 1; 2 %]), ja tätä pulmonaalihypertensiohoitoa jatkettiin tutkimuksen ajan. Potilaat jaettiin kahteen annosryhmään: 2,5 mg tai 5 mg ambrisentaania kerran vuorokaudessa (pienen annoksen ryhmä, n = 21) tai 2,5 mg tai 5 mg ambrisentaania kerran vuorokaudessa, mistä annos titrattiin 5 mg:aan, 7,5 mg:aan tai 10 mg:aan painon perusteella (suuren annoksen ryhmä, n = 20). Yhteensä 20 potilaan annos molemmista annosryhmistä titrattiin 2 viikon kohdalla kliinisen vasteen ja siedettävyyden perusteella; 37 potilasta oli tutkimuksessa mukana loppuun asti; 4 potilasta jättäytyi pois tutkimuksesta.
Ambrisentaanin vaikutuksessa tärkeimpään tehon tulosmuuttujaan eli kuormituksen sietoon (6 minuutin kävelymatkan pituus, 6MWD) ei havaittu annokseen liittyvää trendiä. Keskimääräinen 6MWD:n muutos lähtötilanteesta viikon 24 kohdalla (mittaus lähtötilanteessa ja viikolla 24) oli +55,14 m (95 %:n luottamusväli 4,32–105,95) 18 potilaalla pienen annoksen ryhmässä ja +26,25 m (95 %:n luottamusväli -4,59–57,09) 18 potilaalla suuren annoksen ryhmässä. Yhteensä 36 potilaan (molempien annosryhmien yhdistetty) keskimääräinen 6MWD:n muutos lähtötilanteesta viikon 24 kohdalla oli +40,69 m (95 %:n luottamusväli 12,08–69,31). Tulokset olivat yhdenmukaisia aikuisilla havaittujen tulosten kanssa. Viikon 24 kohdalla 95 %:lla pienen annoksen ryhmän potilaista ja 100 %:lla suuren annoksen ryhmän potilaista tila oli pysynyt stabiilina (toimintakykyluokka oli pysynyt samana tai parantunut). Kaplan–Meier‑estimaatti elossaololle ilman pulmonaalihypertension pahenemistapahtumia (kuolema [kaikki syyt], keuhkonsiirto tai pulmonaalihypertension pahenemisesta tai pulmonaalihypertensioon liittyvästä tilan heikkenemisestä johtuva sairaalahoito) viikon 24 kohdalla oli pienen annoksen ryhmässä 86 % ja suuren annoksen ryhmässä 85 %.
Hemodynamiikkaa mitattiin 5 potilaalla (pienen annoksen ryhmässä). Sydänindeksi suureni lähtötilanteesta keskimäärin 0,94 l/min/m2, keuhkovaltimon keskipaine pieneni keskimäärin 2,2 mmHg ja keuhkovaltimoresistenssi pieneni keskimäärin 277 dyn s/cm5 (3,46 mmHg/l/min).
Pulmonaalihypertensiota sairastavilla, ambrisentaania 24 viikon ajan saaneilla pediatrisilla potilailla NT-proBNP-pitoisuuksien geometrisen keskiarvon pienenemä lähtötasosta oli pienen annoksen ryhmässä (2,5 mg ja 5 mg) 31 % ja suuren annoksen ryhmässä (5 mg, 7,5 mg ja 10 mg) 28 %.
AMB114588-tutkimus
Pitkäaikaistuloksia saatiin 38:lta ambrisentaania 24-viikkoisessa satunnaistetussa tutkimuksessa saaneesta 41 pulmonaalihypertensiota sairastaneesta vähintään 8-vuotiaasta ja alle 18-vuotiaasta pediatrisesta potilaasta. Useimmilla näistä pitkäkestoiseen vaiheeseen siirtyneistä tutkittavista oli idiopaattinen tai perinnöllinen pulmonaalihypertensio (68 %) AMB112529-tutkimuksen lähtötilanteen mukaisesti. Ambrisentaanihoidolle altistumisen keskimääräinen kesto (± keskihajonta) oli 4,0 ± 2,5 vuotta (vaihteluväli 3 kk –10,0 vuotta). Avoimessa jatkotutkimuksessa potilaiden oli mahdollista saada pulmonaalihypertensioon lisähoitoa tarpeen mukaan, ja ambrisentaaniannosta voitiin muuttaa 2,5 mg:n annosvälein. Kaiken kaikkiaan 66 %:lla jatkotutkimuksessa jatkaneista ambrisentaaniannos pysyi samana kuin AMB112529-tutkimuksessa.
Kliininen paheneminen oli määritelty seuraavasti: kuolema (kaikki syyt), jonoon asettaminen keuhkonsiirtoa tai atriaalista septostomiaa varten, sellainen pulmonaalihypertension paheneminen, joka johtaa sairaalahoitoon, ambrisentaaniannoksen muutokseen, käytössä olevan kohdennetun pulmonaalihypertensiolääkkeen annoksen muutokseen tai tällaisen lääkkeen käytön aloittamiseen tai WHO:n toimintakykyluokan suurenemiseen, 6MWD‑tuloksen 20 % pieneneminen tai sydämen oikean puolen vajaatoiminnan oireet/löydökset. Tarkasteluajankohtina kaikkiaan 71 %:lla potilaista ei todettu pulmonaalihypertension pahenemista, ja kaikista neljästä annosryhmästä yhteensä 11:llä (29 %) potilaalla todettiin pulmonaalihypertension paheneminen vähintään yhden kriteerin perusteella; 5 potilaalla 11:stä (45 %) täyttyi useampi kuin yksi kriteeri. Elossaolon Kaplan–Meier‑estimaatti oli 94,74 % 3 vuoden ja 92,11 % 4 vuoden kohdalla hoidon aloituksesta.
Muutoksena AMB112529-tutkimuksen lähtötilanteesta jatkotutkimuksen loppuun todettiin 6MWD-tuloksen keskimääräinen suureneminen 58,4 ± 88 metriä (17 % paraneminen lähtötilanteeseen verrattuna) kaikissa annosryhmissä.
AMB114588-tutkimuksen tutkimukseenottovaiheessa kaikki neljä WHO:n toimintakykyluokkaa (I, II, III ja IV) olivat edustettuina, ja yli puolella osallistujista toimintakykyluokka oli II (n = 22; 58 %) ja lopuilla toimintakykyluokka oli I (n = 9; 24 %), III (n = 6; 16 %) tai IV (n = 1; 3 %). Muutoksena AMB112529-tutkimuksen lähtötilanteesta jatkotutkimuksen loppuun (N = 29) todettiin WHO:n toimintakykyluokan paraneminen (45 %) tai ei muutosta (55 %) eikä huononemista, sekä 6MWD-tuloksen keskimäärin 17,0 %:n suureneminen.
Farmakokinetiikka
Imeytyminen
Ambrisentaani imeytyy ihmisellä nopeasti. Suun kautta otettuna ambrisentaanin huippupitoisuus plasmassa (Cmax) saavutetaan tyypillisesti noin 1,5 tunnissa sekä paastotilassa että ruokailun jälkeen. Cmax- ja pitoisuuden plasmassa aikakäyrän alle jäävä pinta-ala (AUC) kasvavat annosriippuvaisesti yli terapeuttisen alueen annoksen kasvaessa. Vakaa tila saavutetaan yleensä neljässä vuorokaudessa toistuvassa annostelussa.
Ruoan vaikutusta tutkittiin terveillä vapaaehtoisilla siten että ambrisentaania annettiin sekä paastotilassa että runsaasti rasvaa sisältävän aterian yhteydessä. Cmax-arvo pieneni 12 % kun taas AUC-arvo pysyi muuttumattomana. Tämä huippupitoisuuden pieneneminen ei ole kliinisesti merkitsevää, joten ambrisentaanin voi ottaa ruoan kanssa tai ilman.
Jakautuminen
Ambrisentaani sitoutuu voimakkaasti plasman proteiineihin. In vitro keskimäärin 98,8 % ambrisentaanista sitoutui plasman proteiineihin pitoisuudesta riippumatta pitoisuuden ollessa 0,2–20 mikrog/ml välillä. Ambrisentaani sitoutuu pääasiassa albumiiniin (96,5 %) ja vähemmässä määrin happamaan alfa1-glykoproteiiniin.
Punaisissa verisoluissa ambrisentaania on vähän; keskimääräinen veri-plasma-suhde on miehillä 0,57 ja naisilla 0,61.
Biotransformaatio
Ambrisentaani on endoteliinireseptoriantagonisti (propionihappojohdos), joka ei kuulu sulfonamideihin.
Ambrisentaani glukuronisoituu useiden UGT-isoentsyymien (UGT1A9S, UGT2B7S ja UGT1A3S) vaikutuksesta muodostaen ambrisentaaniglukuronidia (13 %). Ambrisentaani metaboloituu myös oksidatiivisesti CYP3A4:n ja vähemässä määrin CYP3A5- ja CYP2C19- entsyymien vaikutuksesta muodostaen 4-hydroksimetyyliambrisentaania (21 %), joka glukuronisoituu edelleen 4-hydroksimetyyliambrisentaaniglukuronidiksi (5 %). 4-hydroksimetyyliambrisentaanin affiniteetti ihmisen endoteliinireseptoreihin on 65 kertaa pienempi kuin ambrisentaanilla. Siksi plasmassa havaituilla 4-hydroksimetyyliambrisentaanin pitoisuuksilla (noin 4 % verrattuna kanta-aineeseen) ei odoteta olevan osuutta ambrisentaanin farmakologiseen vaikutukseen.
In vitro-tutkimustiedot osoittavat, että 300 mikromoolin pitoisuus ambrisentaania aiheutti enintään 50%:sen inhibiition UGT1A1-, UGT1A6-, UGT1A9-, UGT2B7- (enintään 30 %) entsyymien tai sytokromi P450 entsyymien 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ja 3A4 (enintään 25 %) aktiivisuuteen. Ambrisentaani ei vaikuta kliinisesti merkityksellisillä pitoisuuksilla inhiboivasti humaanitransporttereihin kuten Pgp, BCRP, MRP2, BSEP, OATP1B1, OATP1B3 tai NTCP in vitro. Ambrisentaani ei myöskään indusoinut MRP, Pgp tai BSEP proteiinien ekspressiota rotan maksasoluissa. Kaiken kaikkiaan in vitro ‑tutkimustiedot viittaavat siihen, että kliinisesti merkityksellisillä pitoisuuksilla (plasman Cmax-pitoisuus enintään 3,2 mikromooolia) ambrisentaanilla ei olisi vaikutusta UGT1A1-, UGT1A6-, UGT1A9-, UGT2B7-entsyymeihin tai sytokromi P450 entsyymeihin 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4 tai BSEP, BCRP, Pgp, MRP2, OATP1B1/3, NTCP-transporttereihin.
Ambrisentaanin (10 mg kerran vuorokaudessa) vaikutusta vakaassa tilassa varfariinin farmakokinetiikkaan ja farmakodynamiikkaan tutkittiin 20 terveessä vapaaehtoisessa. Tutkimuksessa annettiin kerta-annos (25 mg) varfariinia ja mitattiin protrombiiniaika (PT) ja INR. Ambrisentaanilla ei ollut kliinisesti merkitsevää vaikutusta varfariinin farmakokinetiikkaan tai ‑dynamiikkaan. Varfariinin samanaikainen anto ei vaikuttanut puolestaan ambrisentaanin farmakokinetiikkaan (ks. kohta Yhteisvaikutukset).
Sildenafiilin (20 mg kolme kertaa vuorokaudessa 7 vuorokauden ajan) vaikutusta ambrisentaanin (kerta-annoksena) farmakokinetiikkaan ja ambrisentaanin (10 mg kerran vuorokaudessa 7 vuorokauden ajan) vaikutusta sildenafiilin (kerta-annoksena) farmakokinetiikkaan tutkittiin 19 terveessä vapaaehtoisessa. Sildenafiilin Cmax-arvo nousi 13 % kun sitä annettiin samanaikaisesti ambrisentaanin kanssa. Sildenafiilin, N-desmetyylisildenafiilin ja ambrisentaanin farmakokineettisissä parametreissä ei havaittu muita muutoksia. Cmax-arvon lievän nousun ei katsota katsota olevan kliinisesti merkityksellistä (ks. kohta Yhteisvaikutukset).
Ambrisentaanin (10 mg kerran vuorokaudessa) vaikutusta vakaassa tilassa kerta-annoksena annetun tadalafiilin farmakokinetiikkaan ja tadalafiilin (40 mg kerran vuorokaudessa) vaikutusta vakaassa tilassa kerta-annoksena annetun ambrisentaanin farmakokinetiikkaan tutkittiin 23 terveessä vapaaehtoisessa. Ambrisentaanilla ei ollut kliinisesti merkitsevää vaikutusta tadalafiilin farmakokinetiikkaan. Tadalafiilin samanaikainen anto ei vaikuttanut puolestaan ambrisentaanin farmakokinetiikkaan (ks. kohta Yhteisvaikutukset).
Ketokonatsolin (400 mg kerran vuorokaudessa) toistuvan annostuksen vaikutusta ambrisentaanin (10 mg kerta-annoksena) farmakokinetiikkaan tutkittiin 16 terveessä vapaaehtoisessa. Altistuminen ambrisentaanille mitattiin AUC(0-inf)- ja Cmax -arvoina, jotka kasvoivat 35 % ja 20 %. On epätodennäköistä, että altistuksen muutoksella olisi kliinistä merkitystä. Siksi ambrisentaania voi käyttää ketokonatsolin kanssa samanaikaisesti.
Siklosporiini A:n toistuvan annostuksen (100–150 mg kaksi kertaa vuorokaudessa) vaikutusta ambrisentaanin (5 mg kerran vuorokaudessa) farmakokinetiikkaan, kun vakaa tila oli saavutettu sekä ambrisentaanin toistuvan annostuksen (5 mg kerran vuorokaudessa) vaikutusta siklosporiini A:n (100–150 mg kaksi kertaa vuorokaudessa) vakaan tilan farmakokinetiikkaan tutkittiin terveillä vapaaehtoisilla. Ambrisentaanin Cmax-arvo suureni 48 % ja AUC(0-t) -arvo suureni 121 % toistuvien siklosporiiniA ‑annosten annon yhteydessä. Näiden muutosten perusteella ambrisentaaniannos on rajoitettava 5 mg:aan kerran vuorokaudessa aikuisilla ja ≥ 50 kg painavilla pediatrisilla potilailla ja 2,5 mg:aan kerran vuorokaudessa ≥ 20–< 50 kg painavilla pediatrisilla potilailla, kun ambrisentaania annetaan yhdessä siklosporiini A:n kanssa (ks. kohta Annostus ja antotapa). Toistuvilla ambrisentaanin annoksilla ei ollut kliinisesti merkityksellistä vaikutusta altistumiseen siklosporiini A:lle ja siklosporiini A:n annosta ei ole tarpeen muuttaa.
Rifampisiinin (600 mg kerran vuorokaudessa) akuutin ja toistuvan annostuksen vaikutusta ambrisentaanin (10 mg kerran vuorokaudessa) vakaan tilan farmakokinetiikkaan tutkittiin terveissä vapaaehtoisissa. Rifampisiinin aloitusannosten jälkeen havaittiin ohimenevä ambrisentaanin AUC(0-τ)-arvon nousu (rifampisiinin ensimmäisten annosten jälkeen 121 % ja 116 % toisten annosten jälkeen), mikä luultavasti johtuu rifampisiinin aiheuttamasta OATP:n estymisestä. Tällä ei kuitenkaan ollut kliinisesti merkitsevää vaikutusta ambrisentaanin altistukseen kahdeksanteen päivään mennessä, kun rifampisiinia annettiin toistuvasti. Ambrisentaanihoitoa saavia potilaita on seurattava huolellisesti, kun rifampisiinihoito aloitetaan (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Ambrisentaanin (10 mg) toistuvan annostuksen vaikutusta digoksiinin (kerta-annoksen) farmakokinetiikkaan tutkittiin 15 terveessä vapaaehtoisessa. Toistuvat ambrisentaaniannokset johtivat lievään digoksiinin AUC0-last-arvojen ja minimipitoisuuksien nousuun sekä digoksiinin Cmax-arvon kasvuun 29 %:lla. Digoksiinialtistuksen lisääntymisellä toistuvien ambrisentaanin annosten annon yhteydessä ei katsota olevan kliinistä merkitystä ja digoksiinin annosta ei ole aiheellista muuttaa (ks. kohta Yhteisvaikutukset).
Ambrisentaanin (10 mg kerran vuorokaudessa 12 vuorokauden ajan) vaikutusta kerta-annoksena annetun etinyyliestradiolia (35 mikrog) ja noretindronia (1 mg) sisältävän oraalisen ehkäisyvalmisteen farmakokinetiikkaan tutkittiin terveillä vapaaehtoisilla naisilla. Etinyyliestradiolin Cmax- ja AUC(0-∞)-arvot pienenivät hiukan (8 % ja 4 %). Noretindronin vastaavat arvot puolestaan suurenivat 13 % ja14 %. Altistumisen muutokset etinyyliestradiolille tai noretindronille olivat pieniä. On epätodennäköistä, että muutoksilla olisi kliinistä merkitystä (ks. kohta Yhteisvaikutukset).
Eliminaatio
Ambrisentaani ja sen metaboliitit eliminoituvat erittymällä pääasiassa sappeen metaboloiduttuaan ensin maksassa ja/tai ekstrahepaattisesti. Noin 22 % suun kautta annetusta annoksesta on todettavissa virtsasta. 3,3 % annoksesta on muuttumatonta ambrisentaania. Puoliintumisaika plasmassa vaihtelee 13,6 tunnista 16,5 tuntiin.
Erityispotilasryhmät
Aikuiset (sukupuoli, ikä)
Terveillä vapaaehtoisilla ja pulmonaalihypertensiota sairastavilla potilailla tehdyn populaatiofarmakokineettisen analyysin mukaan sukupuoli tai ikä ei merkittävästi vaikuttanut ambrisentaanin farmakokinetiikkaan (ks. kohta Annostus ja antotapa).
Pediatriset potilaat
Pediatrisista potilaista on saatavilla vain vähän farmakokineettisiä tietoja. Farmakokinetiikkaa tutkittiin 8 – < 18-vuotiailla pediatrisilla tutkittavilla yhdessä kliinisessä tutkimuksessa (AMB112529).
Suun kautta annetun ambrisentaanin farmakokinetiikka 8 – < 18-vuotiailla pulmonaalihypertensiota sairastavilla tutkittavilla oli yleisesti ottaen yhdenmukainen aikuisten farmakokinetiikan kanssa painon huomioimisen jälkeen. Mallista johdetut pediatristen tutkittavien vakaan tilan altistukset (AUCss) pienillä ja suurilla annoksilla kaikissa painoryhmissä asettuivat historiallisen aikuisten altistuksen 5. persentiiliin (pieni annos, 5 mg) ja 95. persentiiliin (suuri annos, 10 mg).
Heikentynyt munuaisten toiminta
Ambrisentaani ei metaboloidu merkitsevästi munuaisissa tai erity munuaisten kautta. Populaatiofarmakokineettisessä analyysissä kreatiniinipuhdistuman todettiin olevan tilastollisesti merkitsevä kovariantti, joka vaikuttaa ambrisentaanin oraaliseen puhdistumaan. Oraalisen puhdistuman pieneneminen on kuitenkin vaatimatonta (20–40 %) potilailla, joilla on kohtalainen munuaisten vajaatoiminta. Siksi sillä ei ole todennäköisesti kliinistä merkitystä. Varovaisuutta on kuitenkin noudatettava potilailla, joilla on vaikea munuaisten vajaatoiminta (ks. kohta Annostus ja antotapa).
Heikentynyt maksan toiminta
Ambrisentaanin päämetaboliareitit ovat glukuronisaatio ja oksidaatio sekä näitä seuraava eliminaatio sappeen. Siksi maksan vajaatoiminta voi lisätä altistumista (Cmax ja AUC) ambrisentaanille. Populaatiofarmakokineettisessä analyysissä oraalisen puhdistuman osoitettiin vähenevän bilirubiinipitoisuuden kohotessa. Bilirubiinipitoisuuden vaikutuksen suuruus on kuitenkin kohtalainen (verrattuna tyypilliseen potilaaseen, jolla bilirubiini on 10,3 mikromol/l [0,6 mg/dl], ja potilaalla, jolla bilirubiini on noussut arvoon 77,0 mikromol/l [4,5 mg/dl], olisi noin 30 % pienempi ambrisentaanin oraalinen puhdistuma). Ambrisentaanin farmakokinetiikkaa potilaissa, joilla on maksan vajaatoiminta (maksakirroosiin liittyen tai ilman sitä), ei ole tutkittu. Siksi ambrisentaanihoitoa ei tule aloittaa potilaille, joilla on vaikea maksan toiminnanvajaus tai kliinisesti merkitsevä maksan aminotransferaasiarvojen nousu (> 3 x ULN, viitearvojen ylärajan, ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Prekliiniset tiedot turvallisuudesta
Ensisijaisesta farmakologisesta luokkavaikutuksesta johtuen suuri kerta-annos ambrisentaania (esim. yliannostus) saattaa laskea valtimopainetta ja mahdollisesti aiheuttaa hypotensiota ja oireita, jotka liittyvät vasodilataatioon.
Ambrisentaani ei inhiboi sappihappojen kuljetusta eikä ole ilmeisen maksatoksista.
Inflammaatioita ja muutoksia nenäontelon epiteelissä havaittiin annettaessa ambrisentaania pitkäaikaisesti jyrsijöille altistuksella, joka oli ihmisen terapeuttista altistusta pienempi. Lievää inflammatorista vastetta havaittiin koirissa annettaessa ambrisentaania pitkäaikaisesti 20 kertaa suuremmalla altistuksella kuin potilailla havaitut altistukset.
Ambrisentaanilla hoidetuilla rotilla havaittiin seulalokeroston luun hyperplasiaa altistustasoilla, jotka vastasivat kolmekertaista altistusta kliiniseen AUC:hen verrattuna. Ambrisentaania saaneilla hiirillä tai koirilla ei havaittu nenän luiden hyperplasiaa. Muista yhdisteistä saadun kokemuksen perusteella seulaluiden hyperplasia on rotalla tunnettu vaste nenätulehdukselle.
In vitro ‑testeissä nisäkkään soluilla ambrisentaani oli klastogeenista suurilla pitoisuuksilla. Ambrisentaanin mutageenisuudesta tai genotoksisista vaikutuksista ei saatu viitteitä bakteereilla tehdyssä testeissä tai kahdessa jyrsijöille tehdyssä in vivo ‑tutkimuksessa.
Karsinogeenisesta potentiaalista ei saatu viitteitä rotilla ja hiirillä kaksi vuotta kestäneissä oraalisissa tutkimuksissa. Rintarauhasen hyvänlaatuiset fibroadenoomat lisääntyivät hieman vain suurimmilla annoksilla urosrotilla. Urosrotilla systeeminen altistuminen ambrisentaanille tällä annoksella (perustuu vakaan tilan AUC-arvoon) oli kuusinkertainen siihen nähden, mikä saavutetiin kliinisessä käytössä ihmisille antamalla 10 mg vuorokaudessa.
Oraalisissa toistuvien annosten toksisuus- ja hedelmällisyystutkimuksissa koirasrotilla ja -hiirillä havaittiin tubulaarista atrofiaa kiveksissä, joskus aspermiaan liittyen ilman turvallisuusmariginaalia. Muutokset kiveksissä eivät parantuneet täysin arvioitujen lääkkeettömien ajanjaksojen aikana. Koirilla tehdyissä 39 viikkoa kestäneissä tutkimuksissa ei kuitenkaan havaittu muutoksia kiveksissä, joissa altistus vastasi 35-kertaista altistumista ihmisen AUC-arvoon nähden. Rottauroksilla ambrisentaanin ei havaittu vaikuttavan siittiöiden liikkuvuuteen kaikilla tutkituilla annoksilla (ad 300 mg/kg/vuorokausi). Morfologisesti normaalien siittiöiden määrässä havaittiin pientä (< 10 %) laskua annoksella 300 mg/kg/vuorokausi, mutta ei annoksella 100 mg/kg/vuorokausi (yli yhdeksänkertainen altistus verrattuna normaalialtistukseen annoksella 10 mg/vuorokausi). Ambrisentaanin vaikutusta miehen hedelmällisyyteen ei tunneta.
Ambrisentaanin on osoitettu olevan teratogeenista rotille ja kaneille. Alaleukaluun, kielen ja/tai suulaen epämuodostumia havaittiin kaikilla tutkituilla annoksilla. Rotilla tehdyssä tutkimuksessa todettiin suurempi ilmaantuvuus koskien kammioväliseinäaukkoja, vartalon verisuonien kehityshäiriöitä, kilpirauhasen ja kateenkorvan poikkeavuuksia, kitaluun luutumista sekä napavaltimon sijaintia virtsarakon vasemmalla puolella oikean puolen sijasta. Teratogeenisuuden epäillään olevan endoteliinireseptoriantagonistien luokkavaikutus.
Ambrisentaanin anto naarasrotille tiineyden loppuvaiheesta imetyksen loppuun asti aiheutti haittavaikutuksia emon käyttäytymisessä ja jälkeläisten heikentynyttä lisääntymiskykyä (pienet kivekset ruumiinavauksessa) sekä lisäsi poikaskuolleisuutta. Annettu annos vastasi kolmekertaista altistusta AUC-arvoon nähden korkeimmalla ihmiselle suositellulla annoksella.
Nuorilla rotilla abrisentaania annosteltiin suun kautta kerran päivässä syntymänjälkeisinä päivinä 7–26, –36 tai –62 (vastaten ihmisen kehittymistä vastasyntyneestä myöhäiseen nuoruuteen). Aivojen painon pienenemistä (-3 % – -8 %), johon ei liittynyt morfologisia tai neurobehavioraalisia muutoksia, esiintyi hengitysäänien, apnean tai hypoksian havaitsemisen jälkeen. Nämä vaikutukset esiintyivät AUC-arvoilla, jotka olivat noin 1,8–7-kertaisia ihmislasten 10 mg:n altistukseen verrattuna. Toisessa tutkimuksessa, jossa hoidettiin 5 viikon ikäisiä rottia (joka vastaa noin 8 vuoden ikää ihmisillä), aivojen painon laskua havaittiin vain hyvin suurella annoksella, ainoastaan uroksilla. Saatavilla oleva ei-kliininen tieto ei anna täyttä ymmärrystä tämän löydöksen kliinisestä merkityksestä alle 8-vuotiailla lapsilla.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Laktoosimonohydraatti
Mikrokiteinen selluloosa
Kroskarmelloosinatrium
Magnesiumstearaatti
Kalvopäällyste
Volibris 2,5 mg kalvopäällysteiset tabletit
Polyvinyylialkoholi
Talkki
Titaanidioksidi (E171)
Makrogoli
Lesitiini (soija) (E322)
Volibris 5 mg ja 10 mg kalvopäällysteiset tabletit
Polyvinyylialkoholi
Talkki
Titaanidioksidi (E171)
Makrogoli Lesitiini (soija) (E322)
Allurapunainen AC (E129)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Volibris 2,5 mg kalvopäällysteiset tabletit
2 vuotta
Volibris 5 mg ja 10 mg kalvopäällysteiset tabletit
5 vuotta.
Säilytys
Tämä lääke ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VOLIBRIS tabletti, kalvopäällysteinen
5 mg (L:kyllä) 30 fol (936,79 €)
10 mg (L:kyllä) 30 fol (857,61 €)
PF-selosteen tieto
Volibris 2,5 mg kalvopäällysteiset tabletit
Läpinäkymätön, valkoinen HDPE‑purkki, jossa on polypropeenista valmistettu turvasuljin ja polyeteenipinnoitettu, induktiokuumasaumattu tiiviste.
Purkissa on 30 kalvopäällysteistä tablettia.
Volibris 5 mg ja 10 mg kalvopäällysteiset tabletit
PVC/PVDC/alumiini-läpipainopakkaus.
Kerta-annosläpipainopakkaus 10 × 1 tai 30 × 1 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Volibris 2,5 mg kalvopäällysteiset tabletit
Valkoinen, 7 mm:n pyöreä, kupera, kalvopäällysteinen tabletti, jossa toisella puolella on merkintä ˮGSˮ ja toisella ˮK11ˮ.
Volibris 5 mg kalvopäällysteiset tabletit
Hennon vaaleanpunainen, 6,6 mm:n neliönmuotoinen, kupera, kalvopäällysteinen tabletti, jossa toisella puolella on merkintä ˮGSˮ ja toisella ˮK2Cˮ.
Volibris 10 mg kalvopäällysteiset tabletit
Voimakkaan vaaleanpunainen, 9,8 mm × 4,9 mm:n ovaalinmuotoinen, kupera, kalvopäällysteinen tabletti, jossa toisella puolella on merkintä ˮGSˮ ja toisella ˮKE3ˮ.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VOLIBRIS tabletti, kalvopäällysteinen
5 mg 30 fol
10 mg 30 fol
- Alempi erityiskorvaus (65 %). Krooninen verenpainetauti (205).
- Peruskorvaus (40 %).
ATC-koodi
C02KX02
Valmisteyhteenvedon muuttamispäivämäärä
16.04.2026
Yhteystiedot
 GLAXOSMITHKLINE OY
GLAXOSMITHKLINE OY Porkkalankatu 20 A
00180 Helsinki
010 303 030
www.glaxosmithkline.fi