
NEORECORMON injektioneste, liuos, esitäytetty ruisku 500 IU, 6000 IU, 10000 IU
Vaikuttavat aineet ja niiden määrät
NeoRecormon 500 KY injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty 0,3 ml:n ruisku injektionestettä sisältää 500 kansainvälistä yksikköä (KY) vastaten 4,15 mikrogrammaa epoetiini beetaa* (rekombinantti ihmisen erytropoietiini).
Yksi ml injektionestettä sisältää 1 667 KY epoetiini beetaa.
NeoRecormon 2 000 KY injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty 0,3 ml:n ruisku injektionestettä sisältää 2 000 kansainvälistä yksikköä (KY) vastaten 16,6 mikrogrammaa epoetiini beetaa* (rekombinantti ihmisen erytropoietiini).
Yksi ml injektionestettä sisältää 6 667 KY epoetiini beetaa.
NeoRecormon 3 000 KY injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty 0,3 ml:n ruisku injektionestettä sisältää 3 000 kansainvälistä yksikköä (KY) vastaten 24,9 mikrogrammaa epoetiini beetaa* (rekombinantti ihmisen erytropoietiini).
Yksi ml injektionestettä sisältää 10 000 KY epoetiini beetaa.
NeoRecormon 4 000 KY injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty 0,3 ml:n ruisku injektionestettä sisältää 4 000 kansainvälistä yksikköä (KY) vastaten 33,2 mikrogrammaa epoetiini beetaa* (rekombinantti ihmisen erytropoietiini).
Yksi ml injektionestettä sisältää 13 333 KY epoetiini beetaa.
NeoRecormon 5 000 KY injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty 0,3 ml:n ruisku injektionestettä sisältää 5 000 kansainvälistä yksikköä (KY) vastaten 41,5 mikrogrammaa epoetiini beetaa* (rekombinantti ihmisen erytropoietiini).
Yksi ml injektionestettä sisältää 16 667 KY epoetiini beetaa.
NeoRecormon 6 000 KY injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty 0,3 ml:n ruisku injektionestettä sisältää 6 000 kansainvälistä yksikköä (KY) vastaten 49,8 mikrogrammaa epoetiini beetaa* (rekombinantti ihmisen erytropoietiini).
Yksi ml injektionestettä sisältää 20 000 KY epoetiini beetaa.
NeoRecormon 10 000 KY injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty 0,6 ml:n ruisku injektionestettä sisältää 10 000 kansainvälistä yksikköä (KY) vastaten 83 mikrogrammaa epoetiini beetaa* (rekombinantti ihmisen erytropoietiini).
Yksi ml injektionestettä sisältää 16 667 KY epoetiini beetaa.
NeoRecormon 20 000 KY injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty 0,6 ml:n ruisku injektionestettä sisältää 20 000 kansainvälistä yksikköä (KY) vastaten 166 mikrogrammaa epoetiini beetaa* (rekombinantti ihmisen erytropoietiini).
Yksi ml injektionestettä sisältää 33 333 KY epoetiini beetaa.
NeoRecormon 30 000 KY injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty 0,6 ml:n ruisku injektionestettä sisältää 30 000 kansainvälistä yksikköä (KY) vastaten 250 mikrogrammaa epoetiini beetaa* (rekombinantti ihmisen erytropoietiini).
Yksi ml injektionestettä sisältää 50 000 KY epoetiini beetaa.
* tuotetaan Kiinan hamsterin munasarjan soluissa (CHO-solulinjassa) rekombinantti-DNA-tekniikalla.
Apuaineet , joiden vaikutus tunnetaan:
Fenyylialaniini (enintään 0,3 mg/ruisku)
Natrium (alle 1 mmol/ruisku)
Polysorbaatti 20 (0,034 mg/ruiskun 0,3 ml:n nimellistilavuus ja 0,063 mg/ruiskun 0,6 ml:n nimellistilavuus)
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
NeoRecormon on tarkoitettu:
Oireisen anemian hoito kroonista munuaisten vajaatoimintaa sairastavilla aikuis- ja lapsipotilailla.
- Anemian ennaltaehkäisy syntymäpainoltaan 750–1 500 g:n keskosilla, jotka ovat syntyneet ennen 34. raskausviikkoa.
- Oireisen anemian hoito solunsalpaajia saavilla aikuispotilailla, joilla on ei-myeloidinen syöpä..
-
Autologisesti luovutettavan veren saannon lisääminen ennen leikkausta.
NeoRecormon-valmisteen käyttöä tässä indikaatiossa on punnittava suurentunutta tromboembolisten komplikaatioiden riskiä vastaan. Hoitoa tulisi antaa vain lievästi aneemisille potilaille (Hb 100–130 g/l, [6,21–8,07 mmol/l], ei raudanpuutetta), jos verta säästäviä menetelmiä ei ole käytettävissä tai ne eivät ole riittäviä silloin, kun elektiivisessä leikkauksessa tarvittava verimäärä on suuri (naisilla vähintään 4 ja miehillä vähintään 5 yksikköä). Ks. kohta Farmakodynamiikka
Ehto
Hoidon saavat aloittaa lääkärit, joilla on kokemusta valmisteyhteenvedossa mainituista indikaatioista. Koska yksittäistapauksissa on todettu anafylaktisia reaktioita, ensimmäinen annos tulisi antaa lääkärin valvonnassa.
Annostus ja antotapa
NeoRecormon-valmisteella hoidon saavat aloittaa lääkärit, joilla on kokemusta edellä mainituista indikaatioista. Koska yksittäistapauksissa on todettu anafylaktoidisia reaktioita, suositellaan ensimmäisen annoksen antamista lääkärin valvonnassa.
Annostus
Oireisen anemian hoito kroonista munuaisten vajaatoimintaa sairastavilla aikuis- ja lapsipotilailla
Anemian oireet ja seuraukset voivat vaihdella iän, sukupuolen ja yleisen tautitaakan mukaan: Lääkärin arvio yksittäisen potilaan taudin kulusta ja kliinisestä tilasta on välttämätön. NeoRecormonia annetaan joko ihon alle tai laskimoon hemoglobiiniarvon nostamiseksi korkeintaan 120 g:aan/l (7,45 mmol/l). Potilaille, jotka eivät saa hemodialyysihoitoa, suositellaan ihonalaista injektiota perifeeristen laskimopistosten välttämiseksi. Laskimoon valmiste annetaan noin kaksi minuuttia kestävänä injektiona, hemodialyysipotilailla esimerkiksi AV-suntin kautta dialyysin lopussa.
Potilaskohtaisen vaihtelevuuden vuoksi yksittäisen henkilön havaittu hemoglobiiniarvo voi satunnaisesti olla yli tai alle hemoglobiiniarvon tavoitetason. Hemoglobiiniarvon vaihtelevuuteen on puututtava annosta muuttamalla huomioiden hemoglobiiniarvon tavoitetaso 100–120 g/l (6,21 mmol/l – 7,45 mmol/l). Jatkuvaa yli 120 g/l (7,45 mmol/l) hemoglobiinitasoa tulee välttää. Alla esitetään ohjeistus sopivan annoksen säätämiseksi, kun hemoglobiiniarvo ylittää 120 g/l (7,45 mmol/l).
Hemoglobiiniarvon nousua yli 20 g/l (1,25 mmol/l) neljän viikon jakson aikana on vältettävä. Sellaisessa tapauksessa annosta on muutettava ohjeiden mukaisesti. Jos hemoglobiinipitoisuus suurenee enemmän kuin 20 g/l (1,25 mmol/l) kuukauden aikana tai jos hemoglobiini nousee lähelle arvoa 120 g/l (7,45 mmol/l), annosta on pienennettävä noin 25 %. Jos hemoglobiiniarvo nousee edelleen, hoito on keskeytettävä, kunnes hemoglobiini alkaa laskea, jolloin hoito aloitetaan uudelleen annoksella, joka on noin 25 % pienempi kuin aikaisemmin annettu annos.
Potilaita on seurattava huolella, jotta voidaan varmistaa pienimmän mahdollisen tehokkaan NeoRecormon-annoksen käyttö anemiaoireiden hoidossa siten, että hemoglobiinipitoisuus pysyy samalla enintään arvossa 12 g/dl (120 g/l) (7,45 mmol/l).
NeoRecormon-annoksen suurentamisessa pitää olla varovainen, jos potilaalla on krooninen munuaisten vajaatoiminta. Jos potilaan hemoglobiinipitoisuuden vaste NeoRecormon-hoitoon on huono, muut mahdolliset syyt huonoon vasteeseen on otettava huomioon (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Hemoglobiinin viikoittainen nousu ja tavoitearvo tulee määrittää yksilöllisesti kliinisen tilan perusteella potilailla, joilla on kohonnut verenpaine tai sydänsairaus, aivo- tai perifeerinen verisuonisairaus.
Hoito NeoRecormon-valmisteella on kaksivaiheinen.
1. Korjausvaihe
-
Ihonalainen anto:
Aloitusannos on 3 x 20 KY painokiloa kohti viikossa. Annosta voidaan suurentaa neljän viikon välein 3 x 20 KY:llä/kg viikossa, jos hemoglobiini nousee riittämättömästi (< 2,5 g:aa/l viikossa).
Viikkoannos voidaan myös jakaa päivittäisiksi annoksiksi. -
Laskimonsisäinen anto:
Aloitusannos on 3 x 40 KY/kg viikossa. Neljän viikon kuluttua annos voidaan nostaa 80 KY:hyn/kg kolmesti viikossa. Tarvittaessa sitä voidaan edelleen nostaa annoksella 20 KY/kg kolmesti viikossa kuukauden välein.
Maksimiannos ei kummassakaan antotavassa saa ylittää 720 KY:tä/kg viikossa.
2. Ylläpitovaihe
Hemoglobiiniarvon pitämiseksi tasolla 100–120 g/l annosta lasketaan aluksi puoleen aiemmasta. Sen jälkeen annos sovitetaan yksilöllisesti yhden tai kahden viikon välein (ylläpitoannos).
Annettaessa NeoRecormon ihonalaisesti, viikkoannos voidaan pistää kerran viikossa tai jaettuna kolmeen tai seitsemään annokseen viikossa. Potilaat, jotka ovat hyvässä hoitotasapainossa kerran viikossa annostellulla lääkkeellä, voidaan vaihtaa annosteluun kerran kahdessa viikossa. Tässä tapauksessa annoksen lisääminen saattaa olla välttämätöntä.
Lapsilla tehdyt kliiniset tutkimukset ovat osoittaneet, että tarvittava NeoRecormon-annos on yleensä sitä suurempi, mitä nuoremmasta potilaasta on kyse. Suositeltua annostusta on kuitenkin noudatettava, koska yksilöllistä vastetta ei voida ennustaa.
Hoito NeoRecormon-valmisteella on yleensä pitkäaikaista. Hoito voidaan kuitenkin tarvittaessa keskeyttää milloin tahansa. Tiedot kerran viikossa annostelusta perustuvat 24 viikkoa kestäneisiin kliinisiin hoitotutkimuksiin.
Keskosten anemian ehkäisy
Injektionesteen annostus on 3 x 250 KY painokiloa kohti viikossa ihon alle. Keskoset, jotka jo ovat saaneet verensiirron ennen NeoRecormon-hoidon aloittamista, eivät todennäköisesti hyödy hoidosta yhtä paljon kuin ne, jotka eivät ole saaneet verensiirtoa. Hoidon kestoksi suositellaan 6 viikkoa.
Syöpäpotilaiden kemoterapian aiheuttaman oireisen anemian hoito
NeoRecormonia annetaan anemiapotilaiden (esim. hemoglobiinipitoisuus ≤ 100 g/l (6,21 mmol/l) ihon alle. Anemian oireet ja seuraukset voivat vaihdella iän, sukupuolen ja yleisen tautitaakan mukaan: Lääkärin arvio yksittäisen potilaan taudin kulusta ja kliinisestä tilasta on välttämätön.
Viikkoannos voidaan pistää kerran viikossa tai jaettuna 3–7 annokseen viikossa.
Suositeltu aloitusannos on 30 000 KY:tä viikossa (vastaten noin 450 KY/kg/viikko keskipainoisella potilaalla).
Potilaskohtaisen vaihtelevuuden vuoksi yksittäisen henkilön havaittu hemoglobiiniarvo voi satunnaisesti olla yli tai alle hemoglobiiniarvon tavoitetason. Hemoglobiiniarvon vaihtelevuuteen on puututtava annosta muuttamalla huomioiden hemoglobiiniarvon tavoitetaso 100–120 g/l (6,21 mmol/l – 7,45 mmol/l). Jatkuvaa yli 120 g/l (7,45 mmol/l) hemoglobiinitasoa tulee välttää. Alla esitetään ohjeistus sopivan annoksen säätämiseksi, kun hemoglobiiniarvo ylittää 120 g/l (7,45 mmol/l).
Jos hemoglobiiniarvo neljän viikon hoidon jälkeen on noussut vähintään 10 g:lla/l (0,62 mmol/l), jatketaan hoitoa samalla annoksella. Jos hemoglobiiniarvo ei ole noussut vähintään 10 g:lla/l (0,62 mmol/l), voidaan harkita annoksen kaksinkertaistamista. Jos hemoglobiiniarvo ei vielä kahdeksankaan viikon jälkeen ole noussut vähintään 10 g:lla/l (0,62 mmol/l), hoitovaste on epätodennäköinen ja hoito tulee lopettaa.
Hoitoa tulisi jatkaa kemoterapian jälkeen vielä 4 viikkoa.
Maksimiannos ei saa ylittää 60 000 KY:tä viikossa.
Kun potilaan terapeuttinen tavoite on saavutettu, annosta vähennetään 25 %:sta 50 %:lla, jotta hemoglobiini saadaan ylläpidettyä tällä tasolla. Sopivan annoksen titrausta on harkittava.
Jos hemoglobiini nousee yli 120 g:aan/l (7,45 mmol/l), annosta on pienennettävä noin 25 % – 50 %. NeoRecormon-hoito on tilapäisesti keskeytettävä, jos hemoglobiinitaso ylittää 130 g:aa/l (8,1 mmol/l). Kun hemoglobiini on laskenut 120 g:aan/l (7,45 mmol/l) tai alle, hoito on aloitettava uudelleen noin 25 % aikaisempaa annosta pienemmällä annoksella.
Jos hemoglobiini nousee enemmän kuin 20 g/l (1,3 mmol/l) 4 viikossa, pitää annosta pienentää 25 % – 50 %.
Potilaita on seurattava huolella, jotta voidaan varmistaa pienimmän mahdollisen NeoRecormon-annoksen käyttö anemiaoireiden hoidossa.
Autologisesti luovutetun veren saannon lisääminen
Injektioneste annetaan noin kaksi minuuttia kestävänä injektiona laskimoon tai ihon alle.
NeoRecormon-valmistetta annetaan kahdesti viikossa neljän viikon ajan. Silloin kun potilaan hematokriittitaso mahdollistaa veren talteenoton eli on vähintään 33 %, NeoRecormon-valmistetta annetaan verenluovutuksen päätteeksi.
Hematokriittiarvoa 48 % ei pidä ylittää hoidon aikana.
Annoksen määrää leikkausryhmä kullekin potilaalle yksilöllisesti ottaen huomioon ennen leikkausta luovutettavan veren tarpeen ja potilaan oman punasoluvarannon:
1. Ennen leikkausta talteen otettavan veren määrä riippuu arvioidusta verenhukasta leikkauksen aikana, mahdollisesta verta säästävien menetelmien käytöstä sekä potilaan kunnosta.
Verta otetaan talteen määrä, jonka arvioidaan olevan riittävä homologisen verensiirron välttämiseksi.
Talteen otettava verimäärä ilmaistaan yksikköinä siten, että yksi yksikkö nomogrammissa vastaa 180 ml:aa punasoluja.
2. Mahdollisuus veren luovutukseen riippuu ensisijassa potilaan veritilavuudesta ja lähtötilanteen hematokriittiarvosta. Nämä muuttujat määrittelevät punasoluvarannon, joka voidaan laskea seuraavan kaavan mukaan:
potilaan punasoluvaranto = veritilavuus [ml] x (HKR - 33): 100
naiset: veritilavuus [ml] = 41 [ml/kg] x kehon paino [kg] + 1200 [ml]
miehet: veritilavuus [ml] = 44 [ml/kg] x kehon paino [kg] + 1600 [ml]
(kehon paino ≥ 45 kg)
NeoRecormon-hoidon aiheellisuus ja kerta-annos arvioidaan talteen otettavan verimäärän ja potilaan oman punasoluvarannon perusteella seuraavien kuvaajien mukaisesti.
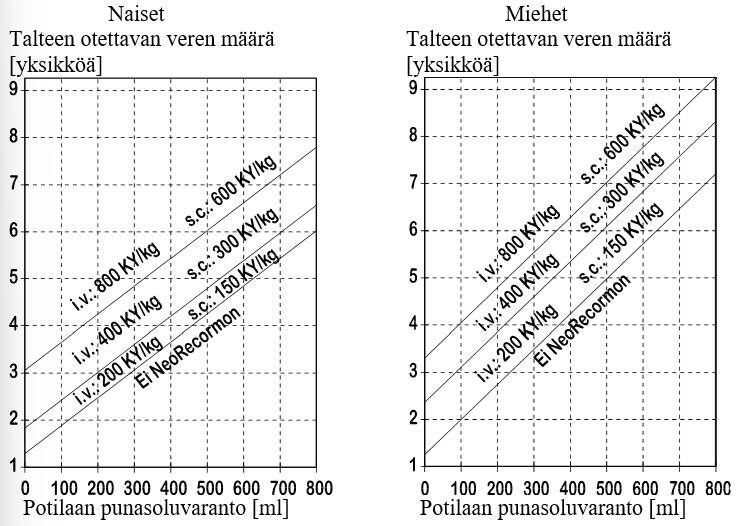
Näin määritetty kerta-annos annetaan kahdesti viikossa neljän viikon ajan. Maksimiannos ei saa ylittää 1 600 KY:tä painokiloa kohti viikossa laskimoon tai 1 200 KY:tä/kg viikossa ihon alle annettuna.
Antotapa
NeoRecormon on käyttövalmis. Vain kirkasta tai hieman opalisoivaa väritöntä ja käytännöllisesti katsoen hiukkasetonta liuosta saa injisoida.
NeoRecormon esitäytetyssä ruiskussa on steriili, mutta säilytysaineeton. Samasta ruiskusta ei saa koskaan antaa enempää kuin yhden annoksen. Lääkevalmiste on kertakäyttöinen.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Huonossa hoitotasapainossa oleva verenpainetauti.
Indikaatiossa "autologisesti luovutetun veren saannon lisääminen": sydäninfarkti tai aivohalvaus kuukautta ennen hoitoa, epästabiili angina pectoris, lisääntynyt riski saada syvä laskimoveritulppa (esim. potilaat, joilla on aikaisemmin ollut laskimopuolen tromboembolisia sairauksia).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Potilaalle annetun valmisteen kauppanimi ja eränumero on tallennettava selkeästi potilastietoihin biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi.
NeoRecormon-valmisteen käytössä on noudatettava varovaisuutta, jos potilaalla on refraktäärinen anemia, johon liittyy liiallinen blastitransformaatio, epilepsia, trombosytoosi tai krooninen maksan vajaatoiminta. Foolihapon ja B12-vitamiinin puutostilat on suljettava pois, koska ne heikentävät NeoRecormon-valmisteen tehoa.
NeoRecormon-annoksen suurentamisessa pitää olla varovainen, jos potilaalla on krooninen munuaisten vajaatoiminta, koska suuriin kumulatiivisiin epoetiiniannoksiin saattaa liittyä suurempi kuolleisuuden, vakavien sydän- ja verisuonitapahtumien sekä aivoverisuonitapahtumien riski. Jos potilaan hemoglobiinipitoisuuden vaste epoetiinihoitoon on huono, muut mahdolliset syyt huonoon vasteeseen on otettava huomioon (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Tehokkaan erytropoieesin varmistamiseksi kaikkien potilaiden rautastatus on tarkistettava ennen hoitoa ja hoidon aikana sekä tarvittaessa lisärautahoito terapeuttisten ohjeistojen mukaan voi olla tarpeen.
Munuaisten vajaatoiminnan hoidosta johtuva vaikea alumiinikuormitus voi heikentää NeoRecormon-valmisteen tehoa.
NeoRecormon-hoidon tarve tulee määrittää yksilöllisesti nefroskleroottisilla potilailla, jotka eivät vielä ole dialyysihoidossa, koska munuaisten vajaatoiminnan etenemisen mahdollista nopeutumista ei voida varmuudella sulkea pois.
Puhdas punasoluaplasia (PRCA)
Rekombinanttien erytropoieettisten proteiinien tavoin myös NeoRecormonin käyttöön on liittynyt neutraloivien erytropoietiinin vasta-aineiden aiheuttamaa puhdasta punasoluaplasiaa. Näiden vasta-aineiden on todettu reagoivan ristiin kaikkien erytropoieettisten proteiinien kanssa. Siksi NeoRecormon-hoitoon ei pidä siirtyä, jos potilaalla epäillään olevan tai on todettu neutraloivia erytropoietiinin vasta-aineita (ks. kohta Haittavaikutukset).
PRCA C-hepatiittia sairastavilla potilailla
Jos hemoglobiiniarvo laskee odottamattomasti ja potilaille kehittyy vaikea anemia, johon liittyy retikulosyyttien vähyyttä, epoetiinihoito on keskeytettävä heti ja tehtävä erytropoietiinin vasta-ainetestaus. Tapauksia on raportoitu interferonilla ja ribaviriinilla hoidetuilla C-hepatiittia sairastavilla potilailla, joille on samanaikaisesti annettu epoetiineja. Epoetiineja ei ole hyväksytty C-hepatiittiin liittyvän anemian hoitoon.
Verenpaineen seuranta
Verenpaineen nousu tai jo olemassa olevan verenpainetaudin paheneminen varsinkin silloin, kun hematokriittiarvo kohoaa nopeasti. Verenpainetta voidaan tällöin alentaa lääkkeillä. Jos verenpaineen nousua ei saada hallituksi lääkehoidolla, suositellaan NeoRecormon-hoidon tilapäistä keskeyttämistä. Varsinkin hoidon alussa verenpainetta tulisi seurata säännöllisesti myös dialyysien välillä. Hypertensiivinen kriisi, johon liittyy enkefalopatiaa muistuttavia oireita, on mahdollinen ja vaatii välitöntä lääkärin huomiota ja tehohoitoa. Huomiota on kiinnitettävä erityisesti äkilliseen, viiltävään, migreenin kaltaiseen päänsärkyyn, joka voi olla varoitusoire.
Epoetiinihoidon yhteydessä on ilmoitettu vakavia ihoon kohdistuvia haittavaikutuksia, mukaan lukien Stevens-Johnsonin oireyhtymää ja toksista epidermaalista nekrolyysiä, jotka voivat olla hengenvaarallisia tai kuolemaan johtavia. Vakavampia tapauksia on havaittu pitkävaikutteisten epoetiinien yhteydessä. Lääkettä määrättäessä potilaille on kerrottava oireista ja ihoreaktioita on seurattava tarkasti. Jos näihin reaktioihin viittaavia oireita ilmenee, NeoRecormon-valmisteen käyttö on lopetettava heti ja vaihtoehtoista hoitoa on harkittava. Jos potilaalla on vakava ihoon kohdistuva reaktio, kuten NeoRecormon-valmisteen käytöstä johtuva Stevens-Johnsonin oireyhtymä tai toksinen epidermaalinen nekrolyysi, hoitoa erytropoieesia stimuloivilla aineilla ei saa aloittaa kyseisellä potilaalla uudelleen missään vaiheessa.
Krooninen munuaisten vajaatoiminta
NeoRecormon-hoidon aikana kroonista munuaisten vajaatoimintaa sairastavien potilaiden veren trombosyyttimäärä voi annosriippuvaisesti jonkin verran lisääntyä viitealueen puitteissa varsinkin laskimoon annon jälkeen. Tämä korjautuu hoidon jatkuessa. Trombosyyttitasoja suositellaan seurattavaksi säännöllisesti hoidon ensimmäisten kahdeksan viikon aikana.
Hemoglobiinipitoisuus
Kroonista munuaisten vajaatoimintaa sairastavilla potilailla hemoglobiinipitoisuus ei saa ylläpitohoidon aikana ylittää kohdassa Annostus ja antotapa. suositellun tavoitearvon ylärajaa. Kliinisissä tutkimuksissa havaittiin suurentunut kuoleman ja vakavien sydän- tai aivoverisuonitapahtumien riski, mukaan lukien aivohalvaus, annosteltaessa erytropoieesia stimuloivia aineita yli hemoglobiinin tavoitetason 120 g/l (7,45 mmol/l).
Kontrolloiduista kliinisistä tutkimuksista ei ole osoitettu saatavan merkittävää lisähyötyä, kun hemoglobiiniarvo nostetaan epoetiinilla yli tason, jolla anemian oireet pysyvät hallinnassa ja verensiirrot vältetään.
Keskosten trombosyyttiarvoja on seurattava säännöllisesti, koska ne voivat nousta jonkin verran varsinkin ensimmäisten 12–14 elinpäivän aikana.
Vaikutus kasvaimen kasvuun
Epoetiinit ovat kasvutekijöitä, jotka stimuloivat ensisijaisesti punasolujen tuotantoa. Erytropoietiinireseptoreita voi ilmetä erityyppisten kasvainsolujen pinnalla. Huolena on, että erytropoietiinin, kuten kaikkien kasvutekijöiden, oletetaan voivan stimuloida kasvaimen kasvua. Useissa kontrolloiduissa kliinisissä tutkimuksissa epoetiinien ei ole osoitettu pidentävän aneemisten syöpäpotilaiden kokonaiselinaikaa eikä pienentävän syövän etenemisen riskiä.
Kontrolloiduissa kliinisissä tutkimuksissa NeoRecormonin ja muiden erytropoieesia stimuloivien lääkeaineiden käytön on osoitettu:
- lyhentävän kasvaimen progressioaikaa sädehoitoa saavilla potilailla, joilla on edennyt pään tai niskan alueen syöpä, kun hemoglobiinin tavoitetaso oli yli 140 g/l (8,69 mmol/l).
- lyhentävän kokonaiselinaikaa ja lisäävän neljän kuukauden kohdalla sairauden etenemisestä johtuvia kuolemia solunsalpaajahoitoa saavilla metastasoitunutta rintasyöpää sairastavilla potilailla, kun hemoglobiinin tavoitetaso oli 120–140 g/l (7,45–8,69 mmol/l),
- lisäävän kuoleman riskiä syöpää sairastavilla potilailla, jotka eivät saaneet solunsalpaajia eivätkä sädehoitoa, kun hemoglobiinin tavoitetaso oli 120 g/l (7,45 mmol/l). Erytropoieesia stimuloivia lääkeaineita ei ole tarkoitettu tälle potilasryhmälle.
Edellä mainituiden syiden vuoksi verensiirtoa pidetään joissakin kliinisissä tilanteissa parempana syöpäpotilaiden anemian hoitomenetelmänä. Päätöksen rekombinanttierytropoietiinin antamisesta on perustuttava yksittäisen potilaan riski-hyötysuhteen arvioimiseen, mikä tulee huomioida kussakin kliinisessä tapauksessa. Arvioinnissa huomioon otettavat tekijät liittyvät kasvaimeen ja sen levinneisyysasteeseen, anemia-asteeseen, odotettavissa olevaan elinikään, potilaan hoitoympäristöön sekä potilaan valintaan (ks. kohta Farmakodynamiikka).
Verenpaineen nousua, jota voidaan hoitaa lääkkeillä, voi esiintyä. Siksi verenpaineen seurantaa suositellaan syöpäpotilailla varsinkin hoidon alkuvaiheessa.
Syöpäpotilaiden trombosyytti- ja hemoglobiiniarvot tulee tarkastaa säännöllisin väliajoin.
Potilailla, jotka ovat autologisessa verensiirto-ohjelmassa, trombosyyttiarvo voi nousta, useimmiten kuitenkin viitealueen rajoissa. Siksi trombosyytit tulisi näillä potilailla määrittää vähintään kerran viikossa. Jos trombosyyttiarvo nousee enemmän kuin 150 x 109/l tai ylittää viitealueen ylärajan, NeoRecormon-hoito pitäisi lopettaa.
Keskosilla ei voida sulkea pois erytropoietiinista aiheutuvan retinopatian mahdollista riskiä, joten hoidossa on oltava varovainen. Päätettäessä hoidon antamisesta keskosille hoidon mahdolliset hyödyt ja riskit pitää arvioida ja huomioida muut käytettävissä olevat hoitovaihtoehdot.
Hematokriitin nousun seurauksena hepariiniannosta on hemodialyysin aikana usein suurennettava kroonista munuaisten vajaatoimintaa sairastavilla potilailla, jotka saavat NeoRecormon-hoitoa. Dialyysijärjestelmä voi tukkeutua, ellei heparinisaatio ole optimaalinen.
Kroonista munuaisten vajaatoimintaa sairastavilla potilailla, joilla on riski sunttitromboosien esiintymiseen, suositellaan fistelin varhaista korjaamista ja tromboosiprofylaksiaa esimerkiksi asetyylisalisyylihapolla.
Seerumin kalium- ja fosfaattitasot on määritettävä säännöllisesti NeoRecormon-hoidon aikana. Muutamilla NeoRecormon-hoitoa saaneilla ureemisilla potilailla on todettu suurentuneita kaliumpitoisuuksia, joskaan syy-yhteyttä hoitoon ei ole vahvistettu. Jos kaliumpitoisuuden todetaan olevan suurentunut tai suurenevan, on harkittava NeoRecormon-hoidon keskeyttämistä, kunnes kaliumpitoisuus on korjautunut.
Käytettäessä NeoRecormon-valmistetta autologisen veren talteenoton yhteydessä on noudatettava virallisia, verenluovutusta koskevia ohjeita ja otettava huomioon erityisesti seuraavat seikat:
- ainoastaan potilaat, joiden hematokriitti on ≥ 33 % (hemoglobiini ≥ 110 g/l [6,83 mmol/l]), voivat luovuttaa verta,
- alle 50 kg:n painoisten potilaiden kohdalla on noudatettava erityistä varovaisuutta,
- kerralla talteen otettu verimäärä saa olla korkeintaan noin 12 % potilaan arvioidusta veritilavuudesta.
Hoitoa tulisi antaa vain sellaisille potilaille, joilla homologisen verensiirron välttämistä pidetään erityisen tärkeänä ottaen huomioon homologisten verensiirtojen riski/hyötyarvio.
Väärinkäyttö
Valmisteen väärinkäyttö voi terveillä ihmisillä johtaa hematokriitin liialliseen nousuun. Tähän voi liittyä hengenvaarallisia kardiovaskulaarisia komplikaatioita.
Apuaineet
Yksi NeoRecormon esitäytetty ruisku sisältää enintään 0,3 mg fenyylialaniinia apuaineena. Tämä on huomioitava potilailla, joilla on vaikea fenyyliketonuria.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per ruisku eli sen voidaan sanoa olevan ”natriumiton”.
Tämä lääkevalmiste sisältää polysorbaattia 20 (0,034 mg/ruiskun 0,3 ml:n nimellistilavuus ja 0,063 mg/ruiskun 0,6 ml:n nimellistilavuus). Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Kliinisestä käytöstä tähän mennessä saadut tulokset eivät ole osoittaneet NeoRecormon-valmisteella olevan yhteisvaikutuksia muiden lääkevalmisteiden kanssa.
Eläinkokeet ovat osoittaneet, että epoetiini beeta ei lisää sytostaattien, kuten etoposidin, sisplatiinin, syklofosfamidin tai fluorourasiilin myelotoksisuutta.
Raskaus ja imetys
Raskaus
Epoetiini beetalle altistuneista raskauksista ei ole kliinisiä tietoja saatavana.
Varovaisuutta on noudatettava määrättäessä valmistetta raskaana oleville naisille.
Imetys
Ei tiedetä, erittyykö epoetiini beeta ihmisen rintamaitoon. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko epoetiini beeta ‑hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja epoetiini beeta ‑hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Eläinkokeissa ei ole havaittu suoria tai epäsuoria haitallisia vaikutuksia raskauteen, alkion/sikiön kehitykseen, synnytykseen tai postnataaliseen kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
NeoRecormonilla ei ole haitallista vaikutusta ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Haittavaikutusprofiilin yhteenveto
Kliinisten tutkimusten (n = 1725) mukaan NeoRecormon-hoidossa olevista potilaista 8 %:n voidaan olettaa kokevan haittavaikutuksia.
Anemiapotilaat, joilla on krooninen munuaisten vajaatoiminta
Yleisin NeoRecormon-hoidon aikana esiintyvä haittavaikutus on verenpaineen nousu tai jo koholla olevan verenpainetaudin paheneminen varsinkin silloin, kun hematokriittiarvo kohoaa nopeasti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Hypertensiivinen kriisi, johon liittyy enkefalopatiaa muistuttavia oireita (esim. päänsärky ja sekavuus, sensomotoriset häiriöt, kuten puhehäiriöt ja kävelyvaikeus, sekä toonis-klooniset kohtaukset), on mahdollinen ja sitä voi esiintyä myös yksittäisillä potilailla, joilla muutoin on normaali tai alhainen verenpaine (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sunttitrombooseja voi esiintyä erityisesti sellaisilla potilailla, joilla on taipumusta hypotensioon tai joiden AV-fistelissä ilmenee komplikaatioita (esim. stenoosit, aneurysmat), ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Useimmilla potilailla seerumin ferritiinipitoisuus laskee samalla kun hematokriittiarvo nousee (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Yksittäistapauksissa on lisäksi todettu seerumin kalium- ja fosfaattipitoisuuksien tilapäistä nousua (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
NeoRecormon-hoidon yhteydessä on yksittäisissä tapauksissa esiintynyt neutraloivien erytropoietiinin vasta-aineiden aiheuttamaa punasoluaplasiaa (PRCA). Jos punasoluaplasia todetaan, NeoRecormon-hoito on lopetettava eikä hoitoa saa vaihtaa toiseen rekombinanttiin erytropoieettiseen proteiiniin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutukset luetellaan jäljempänä taulukossa 1.
Syöpäpotilaat
Epoetiini beeta -hoitoon liittyvää lääkkeillä hoidettavaa päänsärkyä ja verenpaineen nousua esiintyy yleisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Joillakin potilailla seerumin rautaparametrit laskevat (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kliinisissä tutkimuksissa tromboembolisten tapahtumien ilmaantuvuuden on havaittu lisääntyneen NeoRecormonilla hoidetuilla syöpäpotilailla verrattuna hoitamattomiin verrokkipotilaisiin tai plaseboa saaneisiin potilaisiin. NeoRecormonilla hoidetuilla potilailla tapahtumien ilmaantuvuus oli 7 % ja kontrolliryhmässä 4 %; tähän ei liittynyt tromboembolista kuolleisuuden kasvua.
Haittavaikutukset luetellaan jäljempänä taulukossa 2.
Autologisessa verensiirto-ohjelmassa olevat potilaat
Potilailla, joilta on otettu talteen autologista verta, on raportoitu tromboembolisten tapahtumien lievää lisääntymistä. Kuitenkaan syy-seuraussuhdetta NeoRecormon-hoitoon ei ole voitu varmistaa.
Plasebokontrolloiduissa tutkimuksissa esiintyi ohimenevää raudan puutosta enemmän NeoRecormon-potilailla kuin verrokeilla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutukset luetellaan jäljempänä taulukossa 3.
Epoetiinihoidon yhteydessä on ilmoitettu vakavia ihoon kohdistuvia haittavaikutuksia, mukaan lukien Stevens-Johnsonin oireyhtymää ja toksista epidermaalista nekrolyysiä, jotka voivat olla hengenvaarallisia tai kuolemaan johtavia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Haittavaikutukset on lueteltu MedDRA-elinjärjestelmä- ja ‑yleisyysluokituksen mukaisesti.
Yleisyysluokat on määritelty seuraavan esitystavan mukaisesti:
hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
Taulukko 1. Kroonista munuaissairautta sairastavilla potilailla tehdyissä kontrolloiduissa kliinisissä tutkimuksissa NeoRecormon-hoitoon liittyneet haittavaikutukset
| Elinjärjestelmä | Haittavaikutus | Esiintyvyys |
| Verisuonisto | Hypertensio | Yleinen |
| Hypertensiivinen kriisi | Melko harvinainen | |
| Hermosto | Päänsärky | Yleinen |
| Veri ja imukudos | Sunttitromboosi Trombosytoosi | Harvinainen Hyvin harvinainen |
Taulukko 2. Syöpäpotilailla tehdyissä kontrolloiduissa kliinisissä tutkimuksissa NeoRecormon-hoitoon liittyneet haittavaikutukset
| Elinjärjestelmä | Haittavaikutus | Esiintyvyys |
| Verisuonisto | Hypertensio | Yleinen |
| Veri ja imukudos | Tromboembolinen tapahtuma | Yleinen |
| Hermosto | Päänsärky | Yleinen |
Taulukko 3. Autologisessa verensiirto-ohjelmassa mukana olleilla potilailla tehdyissä kontrolloiduissa kliinisissä tutkimuksissa NeoRecormon-hoitoon liittyneet haittavaikutukset
| Elinjärjestelmä | Haittavaikutus | Esiintyvyys |
| Hermosto | Päänsärky | Yleinen |
Keskoset
Seerumin ferritiinipitoisuuden lasku on hyvin yleistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Valikoitujen haittavaikutusten kuvaus
Harvinaisina epoetiinihoitoon liittyvinä ihoreaktioina on raportoitu ihottumaa, kutinaa, urtikariaa tai reaktioita injektiokohdassa. Epoetiinihoitoon liittyviä anafylaktoidisia reaktioita on todettu hyvin harvoin. Kuitenkaan kontrolloiduissa kliinisissä tutkimuksissa yliherkkyysreaktioiden ei ole todettu lisääntyneen.
Epoetiinihoitoon liittyviä flunssan tapaisia oireita, kuten kuumetta, vilunväristyksiä, päänsärkyä, kipua jäsenissä, huonovointisuutta ja/tai luukipua on raportoitu hyvin harvoin ja etenkin hoidon alussa. Nämä reaktiot ovat olleet lieviä tai keskivaikeita ja rauhoittuneet parissa tunnissa, viimeistään parissa päivässä
Epoetiini alfa ja darbepoetiini alfa -valmisteita koskevassa kontrolloidussa kliinisessä tutkimuksessa raportoitiin aivohalvaus esiintyvyydeltään yleiseksi haittavaikutukseksi.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
NeoRecormon-valmisteen terapeuttinen alue on hyvin leveä. Myrkytysoireita ei ole todettu erittäin suurillakaan seerumin erytropoietiinitasoilla.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: anemialääkkeet, ATC-koodi: BO3XA01
Vaikutusmekanismi
Erytropoietiini on glykoproteiini, joka stimuloi punasolun muodostusta sen esiasteista. Se toimii mitoosia ja solujen erilaistumista stimuloivana hormonina.
NeoRecormonin vaikuttavan aineen, epoetiini beetan, aminohappo- ja hiilihydraattikoostumus on identtinen aneemisten potilaiden virtsasta eristetyn erytropoietiinin kanssa.
Epoetiini beetan biologinen teho laskimonsisäisen ja ihonalaisen annon jälkeen on osoitettu useissa eläinmalleissa in vivo (normaaleilla ja ureemisilla rotilla, polysyteemisillä hiirillä, koirilla). Epoetiini beetan antamisen jälkeen punasolujen määrä, hemoglobiiniarvo ja retikulosyyttiarvo suurenevat, samoin 59Fe-inkorporaatio.
Epoetiini beetan inkubaation jälkeen on todettu 3H-tymidiinin lisääntynyttä inkorporaatiota erytroidisiin tumallisiin pernasoluihin in vitro (hiiren pernasoluviljelmä).
Ihmisen luuydinsoluviljelmillä tehdyt tutkimukset osoittivat, että epoetiini beeta stimuloi spesifisesti erytropoieesia, mutta ei vaikuta leukopoieesiin. Epoetiini beetalla ei todettu sytotoksisia vaikutuksia luuytimeen eikä ihmisen ihosoluihin.
Kerta-annos epoetiini beetaa ei vaikuttanut hiirien käyttäytymiseen ja liikkeisiin eikä koirien verenkierto- ja hengityselimistön toimintaan.
Kliininen teho ja turvallisuus
Satunnaistettuun, kaksoissokkoutettuun, plasebokontrolloituun tutkimukseen osallistui 4038 kroonista munuaisten vajaatoimintaa sairastavaa diabetespotilasta, jotka eivät olleet dialyysihoidossa. Potilaiden hemoglobiinitaso oli ≤ 110 g/l ja heitä hoidettiin joko darbepoetiini alfalla (hemoglobiinin tavoitetaso 130 g/l) tai plasebolla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Tutkimuksella ei saavutettu ensisijaista tavoitetta, joka oli kokonaiskuolleisuuden, kardiovaskulaarisen sairastuvuuden tai loppuvaiheen munuaissairauden (ESRD) riskin pieneneminen. Yhdistetyn lopputapahtuman yksittäisten osatekijöiden analysointi osoitti seuraavat riskisuhteet (95 %:n luottamusväli): kuolema 1,05 (0,92 ; 1,21), aivohalvaus 1,92 (1,38 ; 2,68), sydämen vajaatoiminta 0,89 (0,74 ; 1,08), sydäninfarkti 0,96 (0,75 ; 1,23), sydänlihasiskemiasta johtuva sairaalahoito 0,84 (0,55 ; 1,27), ESRD 1,02 (0,87 ; 1,18).
NeoRecormon-annoksen suurentamisessa pitää olla varovainen, jos potilaalla on krooninen munuaisten vajaatoiminta, koska suuriin kumulatiivisiin epoetiiniannoksiin saattaa liittyä suurempi kuolleisuuden, vakavien sydän- ja verisuonitapahtumien sekä aivoverisuonitapahtumien riski. Jos potilaan hemoglobiinipitoisuuden vaste epoetiinihoitoon on huono, muut mahdolliset syyt huonoon vasteeseen on otettava huomioon (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Erytropoietiini on kasvutekijä, joka stimuloi ensisijaisesti punasolujen tuotantoa. Erytropoietiinireseptorit voivat ilmetä erityyppisten kasvainsolujen pinnalla.
Elinaikaa ja syövän etenemistä on tutkittu viidessä laajassa kontrolloidussa tutkimuksessa, joihin osallistui kaikkiaan 2833 potilasta. Näistä tutkimuksista neljä oli kaksoissokkoutettuja, plasebokontrolloituja tutkimuksia ja yksi avoin tutkimus. Kahteen tutkimukseen rekrytoitiin kemoterapialla hoidettavia potilaita ja näiden tutkimusten hemoglobiinipitoisuuden tavoitearvo oli >130 g/l. Lopuilla kolmella tutkimuksella hemoglobiinipitoisuuden tavoitearvo oli 120–140 g/l. Avoimessa tutkimuksessa havaittiin, ettei kokonaiselinajassa ollut eroja rekombinanttihumaanierytropoietiinilla hoidettujen potilaiden ryhmän ja vertailuryhmän välillä. Neljässä plasebokontrolloidussa tutkimuksessa kokonaiselinajan riskisuhteet sijoittuivat välille 1,25−2,47 vertailuryhmän potilaiden eduksi. Nämä tutkimukset osoittavat tilastollisesti merkitsevästi selittämättömän kuolleisuuden yhdenmukaisen kasvun rekombinanttihumaanierytropoietiinia saavilla eri syöpiä sairastavilla anemiapotilailla vertailuryhmän potilaisiin verrattuna. Tutkimusten kokonaiselinaikatuloksia ei voida tyydyttävästi selittää rekombinanttihumaanierytropoietiinin ja vertailuryhmän eroilla tromboosin esiintyvyydessä eikä niihin liittyvissä komplikaatioissa.
Yksittäisten potilaiden tietoihin perustuva meta-analyysi kaikkien 12 kontrolloidun kliinisen tutkimuksen tuloksista NeoRecormonilla hoidetuilla aneemisilla syöpäpotilailla (n = 2 301) osoitti kokonaiselinajan riski-hyötysuhteen pistearvioksi 1,13 kontrolliryhmän eduksi (95 %:n luottamusväli: 0,87–1,46). Potilailla (n = 899), joiden lähtötason hemoglobiini oli ≤ 100 g/l, kokonaiselinajan riski-hyötysuhteen pistearvio oli 0,98 (95 %:n luottamusväli: 0,68–1,40). Koko tutkimusjoukossa havaittiin lisääntynyt suhteellisten tromboembolisten tapahtumien riski (suhteellinen riski (RR) 1,62: 95:n luottamusväli: 1,13–2,31).
Lisäksi on tehty potilastason analyysi 53 kontrolloituun kliiniseen tutkimukseen osallistuneista yli 13 900 syöpäpotilaasta. Tutkimuksissa käytettiin useampia epoetiineja ja potilaita hoidettiin joko kemoterapialla, sädehoidolla tai molemmilla tai heille ei annettu hoitoa. Kokonaiselinaika-aineiston meta-analyysi tuotti riskisuhteen piste-arvioksi 1,06 vertailuryhmän eduksi (95 %:n luottamusväli: 1,00–1,12; 53 tutkimusta ja 13 933 potilasta). Kemoterapiaa saaneilla syöpäpotilailla kokonaiselinajan riskisuhteen pistearvio oli 1,04 (95 %:n luottamusväli: 0,97–1,11; 38 tutkimusta ja 10 441 potilasta). Meta-analyysit osoittavat myös johdonmukaisesti, että tromboembolisten tapahtumien suhteellinen riski lisääntyy huomattavasti rekombinanttia humaanierytropoietiinia saavilla syöpäpotilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Erittäin harvoissa tapauksissa neutraloivia antierytropoietiinivasta-aineita, joko varhaisen punasoluniukkuuden (PRCA) kanssa tai ilman, on esiintynyt rHuEPO-terapian yhteydessä.
Farmakokinetiikka
Terveillä vapaaehtoisilla koehenkilöillä ja ureemisilla potilailla tehdyt farmakokineettiset tutkimukset osoittavat, että laskimoon annetun epoetiini beetan puoliintumisaika on 4–12 tuntia ja että sen jakautumistilavuus on 1–2 kertaa plasman tilavuus. Vastaavia tuloksia on saatu ureemisilla ja normaaleilla rotilla tehdyissä kokeissa.
Annettaessa epoetiini beetaa ihonalaisesti ureemisille potilaille hidastunut imeytyminen aiheuttaa tasannevaiheen seerumipitoisuuden nousussa, ja huippupitoisuus saavutetaan keskimäärin 12−28 tunnin kuluttua. Terminaalinen puoliintumisaika on pitempi kuin laskimoon annettaessa, keskimäärin 13–28 tuntia.
Ihonalaisesti annetun epoetiini beetan biologinen hyötyosuus on 23–42 % laskimonsisäiseen annokseen suhteutettuna.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, geenitoksisuutta, karsinogeenisuutta sekä lisääntymistoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Hiirillä tehdyssä karsinogeenisuustutkimuksessa ei ole todettu viitteitä siitä, että homologinen erytropoietiini lisäisi solunjakautumista tai aiheuttaisi kasvaimia.
Farmaseuttiset tiedot
Apuaineet
Urea
Natriumkloridi
Polysorbaatti 20
Natriumdivetyfosfaatti
Dinatriumfosfaattidodekahydraatti
Kalsiumklorididihydraatti
Glysiini
L-leusiini
L-isoleusiini
L-treoniini
L-glutamiinihappo
L-fenyylialaniini
Injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
2 vuotta.
Säilytys
Säilytä jääkaapissa (2°C – 8°C).
Pidä esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Avohoitokäytössä potilas voi säilyttää valmistetta jääkaapin ulkopuolella huoneenlämmössä (enintään +25 °C) ainoastaan kerran korkeintaan kolmen vuorokauden pituisen jakson ajan.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
NEORECORMON injektioneste, liuos, esitäytetty ruisku
500 IU (L:ei) 6 x 0,3 ml (32,82 €)
6000 IU (L:ei) 6 x 0,3 ml (314,54 €)
10000 IU (L:ei) 6 x 0,6 ml (464,14 €)
PF-selosteen tieto
Esitäytetty ruisku (tyypin I lasia), jossa on kärjen suojus ja männän tulppa (teflonoitua kumia).
NeoRecormon 500 KY, 2 000 KY, 3 000 KY, 4 000 KY, 5 000 KY ja 6 000 KY: yksi esitäytetty ruisku sisältää 0,3 ml liuosta.
NeoRecormon 10 000 KY, 20 000 KY ja 30 000 KY: yksi esitäytetty ruisku sisältää 0,6 ml liuosta.
NeoRecormon-injektionestettä, liuosta, on saatavana seuraavina pakkauskokoina:
NeoRecormon 500 KY
1 esitäytetty ruisku ja yksi neula (30 G 1/2) tai
6 esitäytettyä ruiskua ja 6 neulaa (30 G 1/2).
NeoRecormon 2 000 KY, 3 000 KY, 4 000 KY, 5 000 KY, 6 000 KY, 10 000 KY ja 20 000 KY
1 esitäytetty ruisku ja yksi neula (27 G 1/2) tai
6 esitäytettyä ruiskua ja 6 neulaa (27 G 1/2).
NeoRecormon 30 000 KY
1 esitäytetty ruisku ja yksi neula (27 G 1/2) tai
4 esitäytettyä ruiskua ja 4 neulaa (27 G 1/2).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Väritön, kirkas tai hieman opalisoiva liuos.
Käyttö- ja käsittelyohjeet
Pese ensin kädet!
- Ota ruisku pakkauksestaan ja tarkista, että liuos on kirkas, väritön ja käytännöllisesti katsoen hiukkaseton. Poista ruiskun kärjen suojus.
- Ota neula samasta pakkauksen osasta, kiinnitä se ruiskuun ja poista neulan suojus.
- Poista ilma ruiskusta ja neulasta pitämällä ruiskua pystyssä ja työntämällä varovasti mäntää ylöspäin. Työnnä mäntää, kunnes NeoRecormon-valmisteen määrä ruiskussa on lääkemääräyksen mukainen.
- Puhdista injektiokohdan iho alkoholipyyhkeellä. Purista kevyesti ihopoimu peukalon ja etusormen väliin. Tartu ruiskun säiliöön neulan läheltä ja työnnä neula ihopoimuun nopealla, napakalla liikkeellä. Ruiskuta NeoRecormon-valmiste. Vedä neula ulos nopeasti ja paina pistokohtaa kuivalla, steriilillä tupolla.
Lääkevalmiste on kertakäyttöinen. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
NEORECORMON injektioneste, liuos, esitäytetty ruisku
6000 IU 6 x 0,3 ml
- Ylempi erityiskorvaus (100 %). Munuaisten vajaatoimintaan liittyvä vaikea anemia (138).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Epoetiini, darbepoetiini alfa ja metoksipolyetyleeniglykoliepoetiini beeta: Krooniseen munuaissairauteen liittyvän anemian hoito erityisin edellytyksin / Epoetiini ja darbepoetiini alfa: Syöpäsairauteen tai solunsalpaajahoitoihin liittyvän anemian hoito sekä valmistauduttaessa autologiseen verensiirtoon (306).
NEORECORMON injektioneste, liuos, esitäytetty ruisku
10000 IU 6 x 0,6 ml
- Ylempi erityiskorvaus (100 %). Rintasyöpä (115), Eturauhassyöpä (116), Leukemiat, muut pahanlaatuiset veri- ja luuydintaudit sekä pahanlaatuiset imukudostaudit (117), Gynekologiset syövät (128), Pahanlaatuiset kasvaimet, joita ei ole edellä erikseen mainittu (130), Munuaisten vajaatoimintaan liittyvä vaikea anemia (138).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Epoetiini, darbepoetiini alfa ja metoksipolyetyleeniglykoliepoetiini beeta: Krooniseen munuaissairauteen liittyvän anemian hoito erityisin edellytyksin / Epoetiini ja darbepoetiini alfa: Syöpäsairauteen tai solunsalpaajahoitoihin liittyvän anemian hoito sekä valmistauduttaessa autologiseen verensiirtoon (306).
NEORECORMON injektioneste, liuos, esitäytetty ruisku
500 IU 6 x 0,3 ml
- Ei korvausta.
ATC-koodi
B03XA01
Valmisteyhteenvedon muuttamispäivämäärä
01.05.2025
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com