
RELVAR ELLIPTA inhalaatiojauhe, annosteltu 184/22 mikrog/annos
Vaikuttavat aineet ja niiden määrät
Yksi inhaloitava annos (suukappaleesta vapautuva annos) sisältää 184 mikrogrammaa flutikasonifuroaattia ja 22 mikrogrammaa vilanterolia (trifenataattina). Vastaava esipakattu annos on 200 mikrogrammaa flutikasonifuroaattia ja 25 mikrogrammaa vilanterolia (trifenataattina).
Apuaine(et), joiden vaikutus tunnetaan
Yksi inhaloitu annos sisältää noin 25 mg laktoosimonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Inhalaatiojauhe, annosteltu
Kliiniset tiedot
Käyttöaiheet
Astma
Relvar Ellipta on tarkoitettu astman säännölliseen hoitoon aikuisille ja yli 12-vuotiaille nuorille silloin, kun lääkitys yhdistelmälääkkeellä (pitkävaikutteinen beeta2-agonisti ja inhaloitava kortikosteroidi) on tarkoituksenmukaista:
- potilaat, joiden oireita ei ole saatu riittävästi hallintaan inhaloitavilla kortikosteroideilla ja ”tarvittaessa” inhaloitavilla lyhytvaikutteisilla beeta2-agonisteilla.
- potilaat, joiden oireet ovat jo riittävästi hallinnassa käytettäessä sekä inhaloitavaa kortikosteroidia että pitkävaikutteista beeta2-agonistia.
Annostus ja antotapa
Annostus
Astma
Astmapotilaille tulee antaa Relvar Elliptaa, jonka flutikasonifuroaattivahvuus (FF) vastaa heidän sairautensa vaikeusastetta. Lääkäreiden on hyvä tietää, että astmapotilaiden hoidossa 100 mikrogrammaa flutikasonifuroaattia (FF) kerran vuorokaudessa vastaa noin 250 mikrogrammaa flutikasonipropionaattia (FP) kahdesti vuorokaudessa, ja 200 mikrogrammaa flutikasonifuroaattia (FF) kerran vuorokaudessa vastaa noin 500 mikrogrammaa flutikasonipropionaattia (FP) kahdesti vuorokaudessa.
Aikuiset ja yli 12-vuotiaat nuoret
Relvar Elliptan aloitusannosta yksi inhalaatio 92/22 mikrogrammaa kerran vuorokaudessa suositellaan aikuisille ja yli 12-vuotiaille nuorille, jotka tarvitsevat pientä tai keskisuurta inhaloitavaa kortikosteroidiannosta yhdistettynä pitkävaikutteiseen beeta2-agonistiin. Ellei astman oireita saada riittävän hyvin hallintaan Relvar Ellipta ‑annoksella 92/22 mikrogrammaa, annoksen nostaminen tasolle 184/22 mikrogrammaa saattaa parantaa oireiden hallintaa.
Potilaiden tulee käydä säännöllisesti lääkärin arvioinnissa, jotta heidän flutikasonifuroaatti- ja vilanteroliannoksensa vahvuus pysyy optimaalisena eikä annosta muuteta neuvottelematta lääkärin kanssa. Potilaalle tulee antaa pienin annos, joka tarvitaan tehokkaaseen oireiden hallintaan.
Relvar Elliptan annosta 184/22 mikrogrammaa suositellaan aikuisille ja yli 12-vuotiaille nuorille, jotka tarvitsevat suurempaa inhaloitavaa kortikosteroidiannosta yhdistettynä pitkävaikutteiseen beeta2-agonistiin.
Potilaat huomaavat keuhkojen toiminnan paranemista yleensä jo 15 minuutin kuluessa Relvar Ellipta ‑inhalaatiosta. Potilaalle on kuitenkin kerrottava, että Relvar Elliptan säännöllinen päivittäinen käyttö on tarpeellista, jotta astman oireet pysyvät hallinnassa, ja käyttöä on jatkettava silloinkin, kun potilas tuntee itsensä oireettomaksi.
Nopeaan oireiden lievitykseen on käytettävä inhaloitavaa lyhytvaikutteista beeta2-agonistia, jos oireita ilmaantuu annosten välillä.
Relvar Elliptan suositeltu enimmäisannos on 184 mikrog/22 mikrogrammaa kerran vuorokaudessa.
Alle 12-vuotiaat lapset
Relvar Elliptan turvallisuutta ja tehoa alle 12-vuotiaiden lasten astman hoidossa ei ole varmistettu.
Relvar Ellipta ‑valmistetta ei tule käyttää alle 12-vuotiaille lapsille. Saatavissa olevat tiedot on kuvattu kohdissa Farmakodynamiikka ja Farmakokinetiikka.
Erityisryhmät
Iäkkäät
Annostusta ei tarvitse muuttaa ≥ 65-vuotiaille potilaille (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annostusta ei tarvitse muuttaa näille potilaille (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Lievää, kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavien potilaiden tutkimuksissa havaittiin systeemisen flutikasonifuroaattialtistuksen (sekä Cmax- että AUC-arvon) suurenemista (ks. kohta Farmakokinetiikka).
Varovaisuutta on noudatettava annettaessa Relvar Elliptaa potilaille, joilla on maksan vajaatoiminta, sillä he saattavat olla alttiimpia kortikosteroidien systeemisille haittavaikutuksille.
Kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavien potilaiden enimmäisannos on 92/22 mikrogrammaa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Antotapa
Relvar Ellipta on tarkoitettu vain inhalaatioon suun kautta.
Relvar Ellipta otetaan samaan aikaan päivästä joka päivä.
Lääkäri päättää siitä, otetaanko annos illalla vai aamulla.
Potilaiden on huuhdeltava suu vedellä inhalaation jälkeen. Vettä ei saa niellä.
Jos annos unohtuu, seuraava annos otetaan tavanomaiseen aikaan seuraavana päivänä.
Jos inhalaattoria säilytetään jääkaapissa, sen on annettava lämmetä huoneenlämmössä vähintään tunnin ajan ennen käyttöä.
Laitteen toimintaa ei tarvitse tarkistaa eikä sitä tarvitse saattaa käyttökuntoon millään erityisellä tavalla ennen ensimmäistä käyttökertaa. Vaiheittaisia käyttöohjeita on noudatettava.
Ellipta-laite on pakkauksessa, jonka sisällä on kuivatusainetta sisältävä pussi. Kuivatusainepussi on hävitettävä. Sitä ei saa avata, niellä, eikä sen sisältöä saa joutua hengitysteihin.
Potilasta tulee neuvoa, että pakkausta ei saa avata, ennen kuin potilas on valmis inhaloimaan annoksen.
Laite on suljettuna, kun se otetaan pois pakkauksesta. ”Käytettävä ennen -päiväys” tulisi kirjoittaa sille varattuun tilaan laitteen etiketissä. ”Käytettävä ennen -päiväys” on kuusi viikkoa pakkauksen avaamisesta.
Tämän päivämäärän jälkeen laitetta ei tulisi enää käyttää. Pakkaus voidaan hävittää avaamisen jälkeen.
Alla olevat 30 annoksen Ellipta-laitteen (30 päivän tarvetta vastaava määrä) vaiheittaiset käyttöohjeet koskevat myös 14 annoksen Ellipta-laitetta (14 päivän tarvetta vastaava määrä).
Käyttöohjeet
1. Lue tämä ennen kuin aloitat
Jos laitteen kansi avataan ja suljetaan lääkevalmistetta ottamatta, annos menetetään. Menetetty annos jää laitteen sisälle, mutta sitä ei voida enää käyttää.
Yhdellä inhalaatiokerralla ei voi ottaa vahingossa liikaa lääkevalmistetta eikä kaksinkertaista annosta.
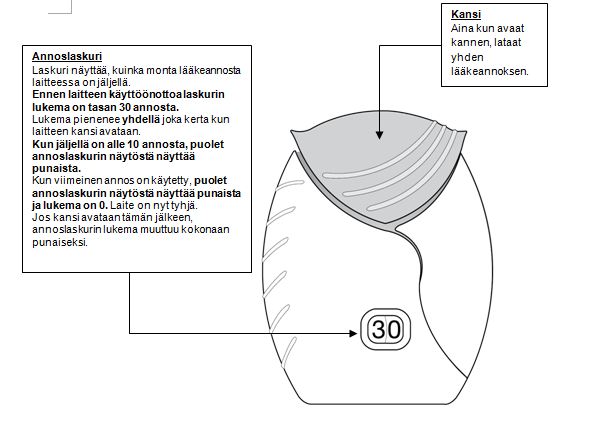
2. Valmistele annos
Avaa laitteen kansi vasta, kun olet valmis inhaloimaan annoksen. Laitetta ei saa ravistaa.
Liu’uta kansi laitteen sivulle, kunnes kuuluu naksahdus (’klik’). Lääkevalmiste on nyt valmis otettavaksi.
Laitteen laskurin lukema pienenee yhdellä. Ellei laskurin lukema pienene naksahduksen jälkeen, laitteesta ei vapaudu annosta. Laite on tällöin palautettava apteekkiin, mistä saa lisäohjeita.
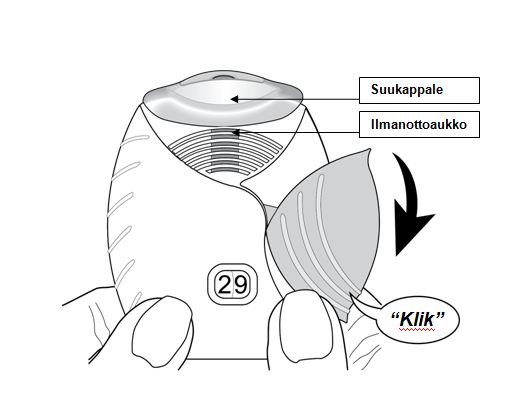
3. Lääkevalmisteen ottaminen
Laite pidetään poissa suun edestä, ja ulos hengitetään niin pitkään kuin se vaivatta onnistuu. Laitteeseen ei saa hengittää.
Suukappale viedään huulien väliin ja huulet puristetaan tiukasti sen ympärille. Ilmanottoaukkoja ei saa tukkia käytön aikana sormilla.
- Hengitä sisään tasaisesti ja syvään. Hengitystä on pidätettävä niin pitkään kuin mahdollista (vähintään 3–4 sekuntia).
- Ota suukappale pois suusta.
- Hengitä ulos hitaasti ja rauhallisesti.
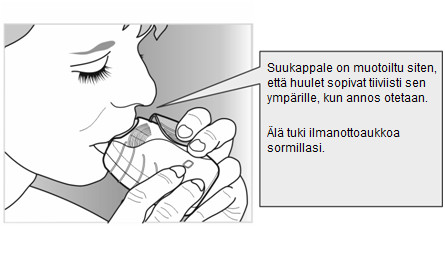
Lääkevalmiste ei välttämättä tunnu tai maistu miltään, vaikka laitetta käytetään oikein.
Suukappale voidaan puhdistaa kuivalla paperipyyhkeellä ennen kannen sulkemista.
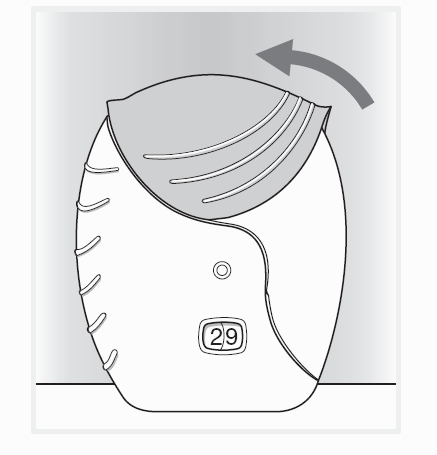
4. Sulje laitteen kansi ja huuhtele suu
Liu’uta kantta ylöspäin niin pitkälle kuin se menee, kunnes se peittää suukappaleen.
Huuhtele suu vedellä lääkkeenoton jälkeen, älä niele.
Tämä vähentää mahdollista lääkkeen aiheuttamaa suun tai kurkun ärsytystä.
Vasta-aiheet
Yliherkkyys vaikuttaville aineille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Taudin vaikeutuminen
Flutikasonifuroaatti-vilanterolivalmistetta ei ole tarkoitettu akuuttien astmaoireiden pahenemisvaiheiden hoitoon. Akuuttien astmaoireiden ja keuhkoahtaumataudin pahenemisvaiheiden laukaisemiseen tarvitaan lyhytvaikutteista bronkodilataattoria. Oireiden hoitoon tarvittavien lyhytvaikutteisten bronkodilataattoreiden käytön lisääntyminen on merkki taudin vaikeutumisesta ja sen hallinnan huonontumisesta, jolloin potilaan tila vaatii lääkärin arviointia.
Astmaa sairastavien potilaiden ei pidä lopettaa flutikasonifuroaatti-vilanterolihoitoa ilman lääkärin valvontaa, sillä oireet saattavat uusiutua hoidon lopettamisen jälkeen.
Astmaan liittyviä haittatapahtumia ja pahenemisvaiheita saattaa esiintyä flutikasonifuroaatti-vilanterolihoidon aikana. Potilaita on kehotettava jatkamaan hoitoa, mutta ottamaan yhteyttä lääkäriin, jos astmaoireet eivät lievity tai jos ne pahenevat Relvar Ellipta ‑hoidon aloittamisen jälkeen.
Paradoksaalinen bronkospasmi
Relvar Elliptaa käytettäessä voi ilmetä paradoksaalinen bronkospasmi, jossa hengityksen vinkuna lisääntyy heti lääkkeen annon jälkeen. Tämä on hoidettava heti lyhytvaikutteisella inhaloitavalla bronkodilataattorilla. Relvar Elliptan käyttö on lopetettava heti, potilas arvioitava ja jokin muu vaihtoehtoinen hoito on aloitettava tarpeen mukaan.
Kardiovaskulaariset vaikutukset
Muiden sympatomimeettien tavoin Relvar Ellipta voi aiheuttaa kardiovaskulaarisia vaikutuksia, kuten sydämen rytmihäiriöitä, esim. supraventrikulaarista takykardiaa ja sydämen lisälyöntisyyttä. Lumekontrolloidussa tutkimuksessa, joka tehtiin keskivaikeaa keuhkoahtaumatautia sairastavilla potilailla, joilla oli sydän- ja verisuonitautihistoria tai suurentunut riski sairastua sydän- ja verisuonitauteihin, ei havaittu suurentunutta riskiä sydän- ja verisuonitapahtumiin flutikasonifuroaatti-vilanterolivalmistetta saavilla potilailla verrattuna lumevalmisteeseen. Flutikasonifuroaatti-vilanterolivalmisteen käytössä on kuitenkin noudatettava varovaisuutta hoidettaessa potilaita, joilla on jokin vaikea sydän- tai verisuonisairaus tai sydämen rytmihäiriöitä, tyreotoksikoosi, korjaamaton hypokalemia tai potilaita, joilla on taipumusta alhaisiin seerumin kaliumpitoisuuksiin.
Maksan vajaatoiminta
Kohtalaista tai vaikeaa maksan vajaatoimintaa sairastaville potilaille on annettava 92/22 mikrogramman annoksia, ja heidän tilaansa on seurattava kortikosteroidien systeemisten haittavaikutusten varalta (ks. kohta Farmakokinetiikka).
Systeemiset kortikosteroidivaikutukset
Inhaloitavat kortikosteroidit voivat aiheuttaa systeemivaikutuksia, varsinkin jos niitä käytetään suurina annoksina pitkiä aikoja. Systeemivaikutusten esiintyminen on kuitenkin vähemmän todennäköistä kuin käytettäessä nieltäviä kortikosteroideja. Mahdollisia kortikosteroidien systeemivaikutuksia ovat Cushingin oireyhtymä, Cushingin oireyhtymään liittyvät oireet, lisämunuaiskuoren vajaatoiminta, luiden mineraalitiheyden väheneminen, harmaakaihi ja glaukooma, sekä harvemmin psykologiset ja käyttäytymiseen liittyvät vaikutukset kuten psykomotorinen hyperaktiivisuus, unihäiriöt, ahdistuneisuus, masentuneisuus tai aggressiivinen käyttäytyminen (erityisesti lapsilla).
Erityinen varovaisuus on tarpeen annettaessa flutikasonifuroaatti-vilanterolihoitoa potilaille, joilla on keuhkotuberkuloosi tai kroonisia tai hoitamattomia infektioita.
Näköhäiriö
Systeemisten ja paikallisten kortikosteroidien käytön yhteydessä voi esiintyä näköhäiriöitä. Jos potilaalla ilmenee näön hämärtymistä tai muita näköhäiriöitä, tulisi harkita potilaan lähettämistä silmälääkärin arvioitavaksi syiden selvittämiseksi. Mahdollisia syitä voivat olla kaihi, glaukooma tai harvinaiset sairaudet, kuten sentraalinen seroosi korioretinopatia (CSCR), joita on raportoitu systeemisten ja paikallisten kortikosteroidien käytön yhteydessä.
Hyperglykemia
Veren glukoosipitoisuuden nousua on raportoitu. Tämä on syytä ottaa huomioon määrättäessä valmistetta potilaille, joilla on diabetes mellitus.
Keuhkokuume keuhkoahtaumatautipotilailla
Inhaloitavia kortikosteroideja saavilla keuhkoahtaumatautipotilailla on havaittu keuhkokuumeen ilmaantuvuuden lisääntymistä, myös sairaalahoitoa vaativaa keuhkokuumetta. Keuhkokuumeriskin kasvusta steroidiannoksen kasvun myötä on jonkin verran näyttöä, mutta tätä ei ole voitu varmasti osoittaa kaikissa tutkimuksissa.
Luotettavaa näyttöä keuhkokuumeriskin suuruuden luokansisäisistä eroista eri inhaloitavien kortikosteroidivalmisteiden välillä ei ole.
Lääkärien on seurattava keuhkoahtaumatautipotilaiden tilaa valppaasti keuhkokuumeen mahdollisen kehittymisen varalta, sillä näiden infektioiden kliiniset piirteet ovat samankaltaisia keuhkoahtaumataudin pahenemisvaiheen oireiden kanssa.
Keuhkoahtaumatautipotilailla keuhkokuumeen riskitekijöitä ovat mm. tupakoinnin jatkuminen, korkea ikä, alhainen painoindeksi ja vaikea keuhkoahtaumatauti.
Keuhkokuume astmaa sairastavilla potilailla
Astmapotilailla keuhkokuumetta esiintyi yleisesti suuremmalla annoksella. Keuhkokuumetta tavattiin numeerisesti enemmän astmapotilailla, jotka käyttivät flutikasonifuroaatti-vilanterolia 184/22 mikrogramman annoksina, kuin potilailla, jotka saivat 92/22 mikrogramman annoksia tai lumevalmistetta (ks. kohta Haittavaikutukset). Mitään riskitekijöitä ei todettu.
Apuaineet
Tämä lääkevalmiste sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Yhteisvaikutukset
Flutikasonifuroaatti-vilanteroli ei todennäköisesti aiheuta kliinisesti merkittäviä yhteisvaikutuksia muiden lääkeaineiden kanssa kliinisinä annoksina käytettynä, koska plasman lääkeainepitoisuudet ovat pieniä inhaloidun annoksen jälkeen.
Yhteisvaikutukset beetasalpaajien kanssa
Beeta2-salpaajat voivat heikentää tai estää beeta2-agonistien vaikutusta. Sekä epäselektiivisten että selektiivisten beeta2-salpaajien samanaikaista käyttöä on vältettävä, ellei siihen ole pakottavia syitä.
Yhteisvaikutukset CYP3A4-estäjien kanssa
Flutikasonifuroaatti ja vilanteroli poistuvat nopeasti elimistöstä voimakkaan ensikierron metabolian aikana maksan CYP3A4-entsyymin välityksellä.
Varovaisuutta on noudatettava voimakkaiden CYP3A:n estäjien (esim. ketokonatsolin, ritonaviirin, kobisistaattia sisältävien valmisteiden) samanaikaisessa käytössä, koska se saattaa suurentaa systeemistä flutikasonifuroaatti- ja vilanterolialtistusta. Yhteiskäyttöä on vältettävä, ellei hoidosta saatava hyöty ole suurempi kuin systeemiseen kortikosteroidialtistukseen liittyvien haittavaikutusten suurentunut riski, jolloin potilaan tilaa on seurattava kortikosteroidien systeemisten haittavaikutusten varalta. CYP3A4-yhteisvaikutustutkimus tehtiin terveille tutkittaville flutikasonifuroaatti-vilanteroliyhdistelmän (184/22 mikrogrammaa) ja voimakkaan CYP3A4-estäjän (ketokonatsoli 400 mg) toistuvilla annoksilla. Yhteiskäyttö suurensi flutikasonifuroaatin AUC(0‑24)-arvojen keskiarvoa 36 % ja Cmax-arvojen keskiarvoa 33 %. Flutikasonifuroaattialtistuksen suureneminen laski seerumin kortisolin 0–24 tunnin painotettua keskiarvoa 27 %. Yhteiskäyttö suurensi vilanterolin AUC(0-t)-arvojen keskiarvoa 65 % ja Cmax-arvojen keskiarvoa 22 %. Vilanterolialtistuksen suureneminen ei lisännyt beeta2-agonistien systeemisiä vaikutuksia sydämen sykkeeseen, veren kaliumpitoisuuteen eikä QTcF-väliin.
Yhteisvaikutukset P-glykoproteiinin estäjien kanssa
Flutikasonifuroaatti ja vilanteroli ovat molemmat P-glykoproteiinin (P-gp:n) substraatteja. Kliinis-farmakologisessa tutkimuksessa, jossa terveille tutkittaville annettiin samanaikaisesti vilanterolia sekä voimakasta P-gp:n ja kohtalaisen voimakasta CYP3A4:n estäjää, verapamiilia, ei havaittu merkittäviä vilanterolin farmakokinetiikkaan kohdistuvia vaikutuksia. Flutikasonifuroaatilla ja spesifisellä P-gp:n estäjällä ei ole tehty kliinis-farmakologisia tutkimuksia.
Sympatomimeetit
Muiden sympatomimeettien samanaikainen käyttö (joko yksinään tai yhdistelmävalmisteissa) saattaa voimistaa flutikasonifuroaatti-vilanterolivalmisteen haittavaikutuksia. Relvar Elliptaa ei tule käyttää yhdessä muiden pitkävaikutteisten beeta2-agonistien eikä pitkävaikutteisia beeta2-agonisteja sisältävien lääkevalmisteiden kanssa.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Eläinkokeissa on havaittu lisääntymistoksisuutta altistustasoilla, jotka eivät ole kliinisesti merkityksellisiä (ks. kohta Prekliiniset tiedot turvallisuudesta). Ei ole olemassa tietoja tai on vain vähän tietoja flutikasonifuroaatin ja vilanterolitrifenataatin käytöstä raskaana oleville naisille.
Flutikasonifuroaatti-vilanterolivalmisteen käyttöä tulisi harkita raskauden aikana vain, jos hoidon odotettu hyöty äidille on suurempi kuin mahdollinen sikiölle aiheutuva vaara.
Imetys
Ei ole riittävästi tietoa flutikasonifuroaatin tai vilanterolitrifenataatin ja/tai metaboliittien erittymisestä ihmisen rintamaitoon. Muiden kortikosteroidien ja beeta2-agonistien on kuitenkin havaittu erittyvän ihmisen rintamaitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Äidinmaitoa saavaan vastasyntyneeseen/imeväiseen kohdistuvaa riskiä ei voida poissulkea.
On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko flutikasonifuroaatti-vilanterolihoito, ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Ihmisiä koskevia hedelmällisyystietoja ei ole. Eläinkokeissa flutikasonifuroaatti-vilanterolitrifenataatti ei vaikuttanut hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Flutikasonifuroaatilla tai vilanterolilla ei ole haitallista vaikutusta ajokykyyn eikä koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuustiedoista
Flutikasonifuroaatti-vilanterolivalmisteen haittavaikutusten yleisyyden arvioinnissa käytettiin laajojen kliinisten astma- ja keuhkoahtaumatautitutkimusten tietoja. Astmaa koskevan kliinisen kehitysohjelman yhdistetyssä haittavaikutusarvioinnissa oli mukana yhteensä 7034 potilasta. Keuhkoahtaumatautia koskevan kliinisen kehitysohjelman yhdistetyssä haittavaikutusarvioinnissa oli mukana yhteensä 6237 potilasta.
Flutikasonifuroaatin ja vilanterolin yleisimmin raportoidut haittavaikutukset olivat päänsärky ja nenänielun tulehdus. Keuhkokuumetta ja luunmurtumia lukuun ottamatta turvallisuusprofiili oli samanlainen astma- ja keuhkoahtaumatautipotilailla. Kliinisissä tutkimuksissa havaittiin useammin keuhkokuumetta ja luunmurtumia keuhkoahtaumatautia sairastavilla potilailla.
Haittavaikutustaulukko
Haittavaikutukset on lueteltu elinjärjestelmän ja yleisyyden mukaan. Yleisyysluokituksessa on noudatettu seuraavaa käytäntöä: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1000, < 1/100), harvinainen (≥ 1/10 000, < 1/1000), hyvin harvinainen (< 1/10 000).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Elinjärjestelmä | Haittavaikutus (-vaikutukset) | Yleisyys |
Infektiot | Keuhkokuume* Ylähengitystieinfektio Bronkiitti Influenssa Suun ja nielun hiivasieni-infektio | Yleinen |
Immuunijärjestelmä | Yliherkkyysreaktiot, mukaan lukien anafylaksia, angioedeema, ihottuma ja urtikaria | Harvinainen |
Aineenvaihdunta ja ravitsemus | Hyperglykemia | Melko harvinainen |
Psyykkiset häiriöt | Levottomuus | Harvinainen |
Hermosto | Päänsärky Vapina | Hyvin yleinen Harvinainen |
Silmät | Näön hämärtyminen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Melko harvinainen |
Sydän | Lisälyönnit Sydämentykytys Takykardia | Melko harvinainen Harvinainen Harvinainen |
Hengityselimet, rintakehä ja välikarsina | Nenänielun tulehdus | Hyvin yleinen |
Suunielun kipu Sivuontelotulehdus Nielutulehdus Nuha Yskä Dysfonia | Yleinen | |
| Paradoksaalinen bronkospasmi | Harvinainen | |
Ruoansulatuselimistö | Vatsakipu | Yleinen |
Luusto, lihakset ja sidekudos | Nivelkipu Selkäkipu Luunmurtumat** Lihasspasmit | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | Kuume | Yleinen |
*, ** Katso alta valikoitujen haittavaikutusten kuvaus
Valikoitujen haittavaikutusten kuvaus
*Keuhkokuume (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.)
Kahden samanlaisen, vuoden kestäneen keskivaikean tai vaikean keuhkoahtaumataudin tutkimuksen yhdistetyssä analyysissä (bronkodilataation jälkeisen FEV1-arvon keskiarvo seulontakäynnillä 45 % viitearvosta, keskihajonta [SD] 13 %), jossa potilailla oli pahenemisvaihe tutkimusta edeltävän vuoden aikana (n= 3255), keuhkokuumetapausten määrät olivat 97,9/1000 potilasvuotta 184/22 mikrog flutikasonifuroaatti-vilanteroliryhmässä, 85,7/1000 potilasvuotta 92/22 mikrog flutikasonifuroaatti-vilanteroliryhmässä ja 42,3/1000 potilasvuotta 22 mikrog vilanteroliryhmässä. Vaikeiden keuhkokuumetapausten määrät 1000 potilasvuotta kohden olivat vastaavasti 33,6; 35,5 ja 7,6, ja edelleen vakavien keuhkokuumetapausten vastaavat määrät olivat 35,1/1000 potilasvuotta 184/22 mikrog flutikasonifuroaatti-vilanteroliryhmässä, 42,9/1000 potilasvuotta 92/22 mikrog flutikasonifuroaatti-vilanteroliryhmässä ja 12,1/1000 potilasvuotta 22 mikrog vilanterolia saaneessa ryhmässä. Kuolemaan johtaneiden keuhkokuumetapausten altistukseen suhteutettu määrä oli 8,8 184/22 mikrog flutikasonifuroaatti-vilanteroliryhmässä, 1,5 92/22 mikrog flutikasonifuroaatti-vilanteroliryhmässä ja 0 22 mikrog vilanteroliryhmässä.
Lumekontrolloidussa tutkimuksessa (SUMMIT), jossa tutkittiin keskivaikeaa keuhkoahtaumatautia sairastavia potilaita (bronkodilataation jälkeisen prosentuaalisen FEV1-arvon keskiarvo seulontakäynnillä 60 %, SD 6 %), joilla oli sydän- ja verisuonitautihistoria tai suurentunut riski sairastua sydän- ja verisuonitauteihin, keuhkokuumetapausten ilmaantuvuus oli flutikasonifuroaatti-vilanterolilla, flutikasonifuroaatilla , vilanterolilla ja lumevalmisteella seuraava: haittatapahtumat (6 %, 5 %, 4 %, 5 %), vakavat haittatapahtumat (3 %, 4 %, 3 %, 3 %), vahvistetut hoidon aikaiset keuhkokuumeesta johtuneet
kuolemantapaukset (0,3 %, 0,2 %, 0,1 %, 0,2 %). Vastaavasti altistukseen suhteutetut ilmaantuvuudet (1 000:ta potilasvuotta kohden) olivat: haittatapahtumat (39,5; 42,4; 27,7; 38,4), vakavat haittatapahtumat (22,4; 25,1; 16,4; 22,2), vahvistetut hoidon aikaiset keuhkokuumeesta johtuneet kuolemantapaukset (1,8; 1,5; 0,9; 1,4).
Yhdentoista astmatutkimuksen yhdistetyssä analyysissä (7034 potilasta) keuhkokuumetapausten ilmaantuvuus 1000 potilasvuotta kohden oli 18,4 184/22 mikrog flutikasonifuroaatti-vilanteroliryhmässä, 9,6 92/22 mikrog flutikasonifuroaatti-vilanteroliryhmässä ja lumeryhmässä 8,0.
**Luunmurtumat
Kahdessa 12 kuukauden tutkimuksessa, joissa oli mukana yhteensä 3255 keuhkoahtaumatautia sairastavaa potilasta, luunmurtumien kokonaisilmaantuvuus oli kaikissa hoitoryhmissä pieni, mutta se oli kaikissa Relvar Ellipta ‑ryhmissä suurempi (2 %) kuin pelkkää vilanterolia (22 mikrogrammaa) saaneessa ryhmässä (< 1 %). Vaikka luunmurtumia esiintyi enemmän Relvar Ellipta ‑ryhmissä kuin vilanterolia (22 mikrog) saaneessa ryhmässä, tyypillisiä kortikosteroidihoitoon liittyviä murtumia (esim. selkärangan kompressiomurtumia / rinta- ja lannenikamien murtumia, lonkka- ja lonkkamaljamurtumia) esiintyi < 1 %:lla potilaista sekä Relvar Ellipta- että vilanterolihoitoryhmissä.
SUMMIT-tutkimuksessa kaikkien murtumatapahtumien ilmaantuvuus oli flutikasonifuroaatti-vilanterolilla, flutikasonifuroaatilla, vilanterolilla ja lumevalmisteella 2 % kussakin hoitoryhmässä. Inhaloitavaan kortikosteroidihoitoon (ICS) yleisesti liittyviä murtumia esiintyi vähemmän kuin 1 % kussakin hoitoryhmässä. Vastaavasti altistukseen suhteutetut ilmaantuvuudet (1 000:ta potilasvuotta kohden) kaikille murtumatapahtumille olivat 13,6; 12,8; 13,2; 11,5. Inhaloitavaan kortikosteroidihoitoon yleisesti liittyvien murtumien esiintyvyys oli vastaavasti 3,4; 3,9; 2,4; 2,1.
Yhdentoista astmatutkimuksen yhdistetyssä analyysissä (7034 potilasta) murtumien ilmaantuvuus oli < 1 %, ja ne olivat yleensä traumaperäisiä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet ja löydökset
Flutikasonifuroaatti-vilanterolivalmisteen yliannostus voi aiheuttaa kummastakin yksittäisestä lääkeaineesta johtuvia oireita ja löydöksiä, jotka voivat olla samankaltaisia kuin muiden beeta2-agonistien yliannostuksen aiheuttamat oireet ja inhaloitavien kortikosteroidien tunnetut vaikutukset (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hoito
Flutikasonifuroaatti-vilanterolivalmisteen yliannostukseen ei ole spesifistä hoitoa. Yliannostustapauksissa on annettava asianmukaista tukihoitoa ja potilaan tilaa on seurattava tarpeen mukaan.
Sydänselektiivisten beetasalpaajien käyttöä on harkittava vain, jos vilanterolin huomattavan yliannostuksen vaikutukset ovat kliinisesti huolestuttavia eikä vastetta saada tukihoitotoimenpiteisiin. Sydänselektiivisten beetasalpaajien käytössä on noudatettava varovaisuutta hoidettaessa potilaita, joilla on ollut bronkospasmeja.
Muuta hoitoa on annettava kliinisen tarpeen mukaan tai noudatettava Myrkytystietokeskuksen ohjeita, mikäli mahdollista.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Adrenergiset lääkeaineet yhd.valmisteina kortikosteroidien tai muiden lääkeaineiden kanssa, ATC-koodi: R03AK10
Vaikutusmekanismi
Flutikasonifuroaatti ja vilanteroli kuuluvat eri lääkeaineryhmiin (synteettinen kortikosteroidi ja selektiivinen pitkävaikutteinen beeta2-reseptoriagonisti).
Farmakodynaamiset vaikutukset
Flutikasonifuroaatti
Flutikasonifuroaatti on synteettinen trifluorinoitu kortikosteroidi, jolla on voimakas anti-inflammatorinen vaikutus. Tarkkaa mekanismia, jonka kautta flutikasonifuroaatti vaikuttaa astman ja keuhkoahtaumataudin oireisiin, ei tunneta. Kortikosteroideilla on osoitettu olevan hyvin monenlaisia vaikutuksia useisiin solutyyppeihin (esim. eosinofiileihin, makrofageihin, lymfosyytteihin) ja välittäjäaineisiin (esim. tulehdusreaktioon osallistuviin sytokiineihin ja kemokiineihin).
Vilanterolitrifenataatti
Vilanterolitrifenataatti on selektiivinen pitkävaikutteinen beeta2-agonisti (LABA).
Beeta2-agonistien vaikuttavien aineiden, myös vilanterolitrifenataatin, farmakologiset vaikutukset johtuvat ainakin osittain solunsisäisen adenylaattisyklaasin stimuloitumisesta. Adenylaattisyklaasi on entsyymi, joka katalysoi adenosiinitrifosfaatin (ATP) muuttumista sykliseksi 3’,5’-adenosiinimonofosfaatiksi (sykliseksi AMP:ksi). Syklisen AMP:n lisääntyminen johtaa keuhkoputkien sileälihaksen relaksaatioon ja hillitsee välittömän yliherkkyysreaktion välittäjäaineiden vapautumista soluista, erityisesti syöttösoluista.
Kortikosteroideilla ja pitkävaikutteisilla beeta2-agonisteilla on molekulaarisia yhteisvaikutuksia. Steroidit aktivoivat beeta2-reseptorigeenin, mikä lisää reseptoreiden määrää ja herkkyyttä, ja pitkävaikutteiset beeta2-agonistit herkistävät glukokortikoidireseptorin steroidista riippuvalle aktivoitumiselle ja tehostavat reseptorin siirtymistä solun tumaan. Nämä synergistiset interaktiot johtavat anti-inflammatorisen vaikutuksen tehostumiseen. Tämä on osoitettu useissa astman ja keuhkoahtaumataudin patofysiologian kannalta merkittävissä tulehdussoluissa in vitro ja in vivo. Keuhkoahtaumatautipotilaiden perifeerisen veren mononukleaarisoluissa havaittiin suurempi anti-inflammatorinen vaikutus flutikasonifuroaatti-vilanterolin yhdistelmällä verrattuna pelkkään flutikasonifuroaattiin pitoisuuksilla, jotka saavutetaan kliinisillä annoksilla. LABA-komponentin aiheuttama lisääntynyt anti-inflammatorinen vaikutus vastasi muilla ICS-LABA yhdistelmillä saatavaa vaikutusta.
Kliininen teho ja turvallisuus
Astma
Kolmessa eripituisessa vaiheen III satunnaistetussa kaksoissokkotutkimuksessa (HZA106827, HZA106829 ja HZA106837) arvioitiin flutikasonifuroaatti-vilanterolivalmisteen turvallisuutta ja tehoa aikuisten ja nuorten potilaiden jatkuvaoireisen astman hoidossa. Kaikki potilaat käyttivät inhaloitavaa kortikosteroidia (ICS) joko yhdessä pitkävaikutteisen beeta2-agonistin kanssa tai ilman sitä, vähintään 12 viikon ajan ennen ensimmäistä tutkimuskäyntiä. HZA106837-tutkimuksessa kaikilla potilailla oli vähintään yksi oraalista kortikosteroidihoitoa vaativa pahenemisvaihe edeltävän vuoden aikana ennen ensimmäistä tutkimuskäyntiä. HZA106827-tutkimus kesti 12 viikkoa, ja siinä verrattiin flutikasonifuroaatin-vilanterolin (92/22 mikrog) [n = 201] ja flutikasonifuroaatin (FF) (92 mikrog) [n = 205] tehoa lumevalmisteeseen [n = 203]. Kaikkia valmisteita annettiin kerran vuorokaudessa. HZA106829-tutkimus kesti 24 viikkoa, ja siinä verrattiin kerran vuorokaudessa annettujen flutikasonifuroaatin-vilanterolin (184/22 mikrog) [n = 197] ja flutikasonifuroaatin (184 mikrog) [n = 194] tehoa flutikasonipropionaattiin (FP), jota annettiin 500 mikrog kahdesti vuorokaudessa [n = 195].
HZA106827/HZA106829-tutkimuksissa ensisijaiset tehoa mittaavat päätetapahtumat olivat kaikkien potilaiden FEV1-jäännösarvon (trough) muutos lähtötasosta (ennen bronkodilataatiokoetta ja ennen lääkeannosta) hoitojakson lopussa, sekä tutkittavien alaryhmästä 0–24 tuntia annoksen jälkeen tehtyjen FEV1:n sarjamittausten painotettu keskiarvo hoitojakson päättyessä. Tutkimuksen voimaan vaikuttava toissijainen päätetapahtuma oli hoidon aikana 24 tunnin kohtauslääkkeettömien jaksojen prosentuaalisen osuuden muutos lähtötasoon verrattuna. Näiden tutkimusten ensisijaisten ja tärkeimpien toissijaisten päätetapahtumien tuloksia kuvataan taulukossa 1.
Taulukko 1 Tutkimusten HZA106827 ja HZA106829 ensisijaisten ja tärkeimpien toissijaisten päätetapahtumien tulokset
| Tutkimuksen n:o | HZA106829 | HZA106827 | ||
FF/VI*-hoitoannos (mikrogrammaa) | FF/VI 184/22 1 x/vrk tai FF 184 1 x/vrk | FF/VI 184/22 1 x/vrk tai FP 500 mg 2 x/vrk | FF/VI 92/22 1 x/vrk tai FF 92 1 x/vrk | FF/VI 92/22 1 x/vrk tai lumevalmiste 1 x/vrk |
| FEV1-jäännösarvon (trough) muutos lähtötasosta - viimeisen havaintoarvon siirto eteenpäin (LOCF) | ||||
Hoitojen ero p-arvo (95 % CI) | 193 ml p < 0,001 (108–277) | 210 ml p < 0,001 (127–294) | 36 ml p = 0,405 (−48–120) | 172 ml p < 0,001 (87–258) |
| 0–24 tuntia annoksesta tehtyjen FEV1-sarjamittausten painotettu keskiarvo | ||||
Hoitojen ero p-arvo (95 % CI) | 136 ml p = 0,048 (1–270) | 206 ml p = 0,003 (73–339) | 116 ml p = 0,06 (−5–236) | 302 ml p < 0,001 (178–426) |
| Kohtauslääkkeettömien 24 tunnin jaksojen prosentuaalisen osuuden muutos lähtötasosta | ||||
Hoitojen ero p-arvo (95 % CI) | 11,7 % p < 0,001 (4,9–18,4) | 6,3 % p = 0,067 (−0,4–13,1) | 10,6 % p < 0,001 (4,3–16,8) | 19,3 % p < 0,001 (13,0–25,6) |
| Oireettomien 24 tunnin jaksojen prosentuaalisen osuuden muutos lähtötasosta | ||||
Hoitojen ero p-arvo (95 % CI) | 8,4 % p = 0,010 (2,0–14,8) | 4,9 % p = 0,137 (−1,6–11,3) | 12,1 % p < 0,001 (6,2–18,1) | 18,0 % p < 0,001 (12,0–23,9) |
| Aamupäivän PEF-arvon (uloshengityksen huippuvirtausarvon) muutos lähtötasosta | ||||
Hoitojen ero p-arvo (95 % CI) | 33,5 l/min p < 0,001 (22,3–41,7) | 32,9 l/min p < 0,001 (24,8–41,1) | 14,6 l/min p < 0,001 (7,9–21,3) | 33,3 l/min p < 0,001 (26,5–40,0) |
| Iltapäivän PEF-arvon muutos lähtötasosta | ||||
Hoitojen ero p-arvo (95 % CI) | 30,7 l/min p < 0,001 (22,5–38,9) | 26,2 l/min p < 0,001 (18,0–34,3) | 12,3 l/min p < 0,001 (5,8–18,8) | 28,2 l/min p < 0,001 (21,7–34,8) |
*FF/VI = flutikasonifuroaatti/vilanteroli
HZA106837-tutkimuksessa hoidon kesto vaihteli (24 viikosta 76 viikkoon, suurin osa potilaista sai hoitoa vähintään 52 viikon ajan). HZA106837-tutkimuksessa potilaat saivat satunnaistetusti joko flutikasonifuroaatti-vilanterolia 92/22 mikrog [n = 1009] tai flutikasonifuroaattia 92 mikrog [n = 1010] kerran vuorokaudessa. HZA106837-tutkimuksen ensisijainen päätetapahtuma oli aika ensimmäiseen vaikeaan astman pahenemisvaiheeseen. Vaikean astman pahenemisvaiheen kriteerinä oli astman vaikeutuminen, joka vaatii vähintään 3 vuorokautta systeemistä kortikosteroidihoitoa, tai sairaalahoitoa tai käyntiä ensiapupoliklinikalla systeemistä kortikosteroidihoitoa vaatineen astman vuoksi. Toissijaisena päätetapahtumana arvioitiin myös FEV1-jäännösarvon (trough) muutoksen (lähtötilanteeseen verrattuna) korjattu keskiarvo.
HZA106837-tutkimuksessa astman vaikean pahenemisvaiheen riski oli flutikasonifuroaatti-vilanterolia 92/22 mikrog saaneilla potilailla 20 % pienempi kuin potilailla, jotka saivat pelkkää flutikasonifuroaattia 92 mikrog (vaarasuhde 0,795, p = 0,036; 95 %:n CI (0,642–0,985)). Astman vaikeiden pahenemisvaiheiden määrä potilasta kohti vuodessa oli FF 92 mikrog ‑ryhmässä 0,19 (noin 1 pahenemisvaihe 5 vuoden aikana) ja flutikasonifuroaatti-vilanteroli 92/22 mikrog ‑ryhmässä 0,14 (noin 1 pahenemisvaihe 7 vuoden aikana). Pahenemisvaiheiden lukumäärien suhde flutikasonifuroaatti-vilanteroli 92/22 mikrog ‑ryhmän ja FF 92 mikrog ‑ryhmän välillä oli 0,755 (95 %:n CI: 0,603–0,945). Tämä vastaa 25 %:n vähenemistä astman vaikeiden pahenemisvaiheiden määrässä flutikasonifuroaatti-vilanteroli 92/22 mikrog ‑ryhmässä FF 92 mikrog ‑ryhmään verrattuna (p = 0,014). Flutikasonifuroaatti-vilanterolin 24 tunnin bronkodilataatiovaikutus säilyi vuoden kestäneen hoitojakson ajan, eikä viitteitä tehon heikkenemisestä havaittu (ei takyfylaksiaa). Flutikasonifuroaatti-vilanteroli 92/22 mikrog paransi johdonmukaisesti FEV1-jäännösarvoa 83–95 ml viikoilla 12, 36 ja 52 ja tutkimuksen päättyessä verrattuna FF 92 mikrog ‑ryhmään (p < 0,001; 95 %:n CI: 52–126 ml tutkimuksen päättyessä). Flutikasonifuroaatti-vilanteroli 92/22 mikrog ‑ryhmän potilaista 44 prosentilla hoitotasapaino oli hyvä (ACQ7 ≤ 0,75) hoidon päättyessä verrattuna 36 prosentin flutikasonifuroaatti 92 mikrog ‑ryhmän potilaihin (p < 0,001; 95 %:n CI: 1,23–1,82).
Vertailututkimukset salmeteroli-flutikasonipropionaattivalmisteiden kanssa
Jatkuvaoireista, huonossa hoitotasapainossa olevaa astmaa sairastavilla aikuisilla ja nuorilla tehdyssä 24 viikon tutkimuksessa (HZA113091) annettiin flutikasonifuroaatti-vilanterolia 92/22 mikrog kerran vuorokaudessa iltaisin ja salmeteroli-flutikasonipropionaattia 50/250 mikrog kaksi kertaa vuorokaudessa. Molemmat hoidot paransivat keuhkojen toimintaa lähtötilanteeseen verrattuna. 0–24 tunnin FEV1-mittausten painotettujen keskiarvojen hoitokohtaisten paranemien (lähtötasoon verrattuna) korjatut keskiarvot, 341 ml (flutikasonifuroaatti-vilanteroli) ja 377 ml (salmeteroli-flutikasonipropionaatti), osoittivat, että keuhkojen toiminta parani yleisesti 24 tunnin ajaksi molemmissa hoitoryhmissä. Korjattujen keskiarvojen ero hoitoryhmien välillä ei ollut tilastollisesti merkitsevä (-37 ml, p = 0,162). FEV1-jäännösarvon osalta flutikasonifuroaatti-vilanteroliryhmässä pienimmän neliösumman (LS) muutos lähtötasoon verrattuna oli 281 ml ja salmeteroli-flutikasonipropionaattiryhmässä 300 ml (korjatun keskiarvon ero, -19 ml (95 %:n CI: −0,073–0,034) ei ollut tilastollisesti merkitsevä (p = 0,485)).
Flutikasonifuroaatti-vilanterolin ja salmeteroli-flutikasonipropionaatin vertailukelpoisuutta (non-inferiority, käyttäen marginaalina -100 ml FEV1-jäännösarvossa) tutkittiin 24 viikon satunnaistetussa, kaksoissokkoutetussa rinnakkaisryhmätutkimuksessa (201378). Tutkimus tehtiin aikuisilla ja nuorilla, joilla astma oli hyvässä hoitotasapainossa 4 viikon avoimen salmeteroli-flutikasonipropionaattihoidon 50/250 mikrog kahdesti vuorokaudessa jälkeen (N = 1504). Potilaat satunnaistettiin saamaan flutikasonifuroaatti-vilanterolia 92/22 mikrog kerran vuorokaudessa tai salmeteroli-flutikasonipropionaattia 50/250 mikrog kahdesti vuorokaudessa. Verrattuna potilaisiin, jotka saivat salmeteroli-flutikasonipropionaattia kahdesti vuorokaudessa, keuhkojen toiminta säilyi yhdenvertaisena flutikasonifuroaatti-vilanterolia kerran vuorokaudessa saaneilla potilailla (ero FEV1-jäännösarvossa +19 ml (95 %:n CI: -11–49)).
Sellaisia vertailevia tutkimuksia salmeteroli-flutikasonipropionaattiin tai muihin ICS/LABA-yhdistelmiin ei ole tehty, joissa olisi asianmukaisesti vertailtu vaikutuksia astman pahenemisvaiheisiin.
Flutikasonifuroaatti ainoana lääkkeenä
Flutikasonifuroaatin (FF) ja flutikasonipropionaatin (FP) tehoa ja turvallisuutta verrattiin 24 viikon satunnaistetussa kaksoissokkoutetussa lumevertailututkimuksessa (FFA112059) jatkuvaoireista astmaa sairastavien aikuisten ja nuorten potilaiden hoidossa. Potilaat saivat flutikasonifuroaattia 92 mikrog kerran vuorokaudessa [n = 114] tai flutikasonipropionaattia 250 mikrog kahdesti vuorokaudessa [n = 114] tai lumevalmistetta [n = 115]. Kaikkien potilaiden oli pitänyt käyttää inhaloitavia kortikosteroideja muuttumattomalla annostuksella vähintään 4 viikon ajan ennen ensimmäistä tutkimuskäyntiä (seulontakäynti), ja pitkävaikutteisten beeta2-agonistien käyttö oli kiellettyä ensimmäistä käyntiä edeltävien 4 viikon aikana. Ensisijainen tehoa mittaava päätetapahtuma oli tutkimuskäynnillä (ennen bronkodilataatiokoetta ja ennen lääkeannosta) mitatun FEV1-jäännösarvon (trough) muutos lähtötasoon verrattuna hoitojakson päättyessä. Tutkimuksen voimaan vaikuttava toissijainen päätetapahtuma oli 24 tunnin kohtauslääkkeettömien jaksojen prosentuaalisen osuuden muutos lähtötasoon verrattuna 24 viikon hoitojakson aikana. 24 viikon kuluttua FEV1-jäännösarvo oli suurentunut FF ‑ryhmässä 146 ml (95 %:n CI: 36–257 ml, p = 0,009) ja FP-ryhmässä 145 ml (95 %:n CI: 33–257 ml, p = 0,011) lumeryhmään verrattuna. FF suurensi 24 tunnin kohtauslääkkeettömien jaksojen osuutta 14,8 % (95 %:n CI: 6,9–22,7, p < 0,001) ja FP 17,9 % (95 %:n CI: 10,0–25,7, p < 0,001).
Allergeenialtistustutkimus
Flutikasonifuroaatti-vilanterolivalmisteen (92/22 mikrog) keuhkoputkia suojaavaa tehoa inhaloidun allergeenin aiheuttamaan varhaiseen ja viivästyneeseen astmareaktioon arvioitiin toistuvilla annoksilla tehdyssä nelisuuntaisessa vaihtovuoroisessa lumevertailututkimuksessa (HZA113126), jossa oli mukana lievää astmaa sairastavia potilaita. Potilaat jaettiin satunnaistetusti ryhmiin, jotka saivat flutikasonifuroaatti-vilanterolivalmistetta 92/22 mikrog, flutikasonifuroaattia 92 mikrog, vilanterolia 22 mikrog tai lumevalmistetta kerran vuorokaudessa 21 vuorokauden ajan, minkä jälkeen tehtiin allergeenialtistus 1 tunnin kuluttua viimeisestä annoksesta. Allergeenina oli pölypunkki, kissan hilse tai koivun siitepöly. Valikoima perustui yksilöllisiin seulontakokeisiin. FEV1-sarjamittausten tuloksia verrattiin arvoihin, jotka mitattiin ennen allergeenialtistusta keittosuolaliuosinhalaation jälkeen (lähtötaso). Suurin vaikutus varhaiseen astmareaktioon oli flutikasonifuroaatti-vilanterolivalmisteella (92/22 mikrog) pelkkään flutikasonifuroaattiin (92 mikrog) tai pelkkään vilanteroliin (22 mikrog) verrattuna. Sekä flutikasonifuroaatti-vilanteroli 92/22 mikrog että flutikasonifuroaatti 92 mikrog estivät viivästyneen astmareaktion lähes täysin pelkkään vilanteroliin verrattuna. Flutikasonifuroaatti-vilanteroli 92/22 mikrog antoi merkitsevästi paremman suojan allergeenin aiheuttamaa keuhkoputkien hyperreaktiivisuutta vastaan pelkkään flutikasonifuroaattiin tai vilanteroliin verrattuna 22. päivänä tehdyn metakoliinialtistuskokeen perusteella.
Tutkimus, jossa selvitettiin keuhkoputkia suojaavia vaikutuksia ja vaikutuksia hypotalamus-aivolisäke-lisämunuaisakselin (HPA-akseli) toimintaan
Flutikasonifuroaatin keuhkoputkia suojaavia vaikutuksia ja vaikutuksia HPA-akselin toimintaan flutikasonipropionaattiin tai budesonidiin verrattuna arvioitiin nousevaa annosta käyttävässä, lumekontrolloidussa vaihtovuoroisessa tutkimuksessa (203162). Tutkimukseen osallistui 54 aikuista astmapotilasta, joilla oli todettu hengitysteiden hyperreaktiivisuus ja FEV1 ≥ 65 % viitearvosta. Potilaat satunnaistettiin yhteen tai kahteen hoitojaksoon, joissa annosta nostettiin viisi kertaa 7 vuorokauden ajaksi flutikasonifuroaatilla (25, 100, 200, 400, 800 mikrogrammaa/vuorokaudessa), flutikasonipropionaatilla (50, 200, 500, 1 000, 2 000 mikrogrammaa/vuorokaudessa), budesonidilla (100, 400, 800, 1 600, 3 200 mikrogrammaa/vuorokaudessa) tai lumelääkkeellä. Jokaisen hoitojakson jälkeen arvioitiin keuhkoputkia suojaavaa vaikutusta mittaamalla hengitysteiden hyperreaktiivisuutta adenosiini-5'-monofosfaatti (AMP) -altistuksessa (provosoiva konsentraatio, joka aiheuttaa FEV1:ssä ≥ 20 % vähenemän [AMP PC20]) sekä 24 tunnin painotetun keskimääräisen plasman kortisolin keskiarvo.
Astman hoitoon hyväksytyillä terapeuttisilla annosalueilla AMP PC20 (mg/ml) ja kortisolin supressio (%) olivat 81–116 mg/ml ja 7–14 % flutikasonifuroaatilla (100–200 mikrogrammaa/vuorokaudessa), 20–76 mg/ml ja 7–50 % flutikasonipropionaatilla (200–2 000 mikrogrammaa/vuorokaudessa) ja 24–54 mg/ml ja 13–44 % budesonidilla (400–1 600 mikrogrammaa/vuorokaudessa).
Pediatriset potilaat
Astma
Kerran vuorokaudessa annetun flutikasonifuroaatti/vilanterolin (FF/VI) tehoa ja turvallisuutta verrattiin kerran vuorokaudessa annettuun flutikasonifuroaattiin astman hoidossa 5–11-vuotiailla pediatrisilla potilailla satunnaistetussa, kaksoissokkoutetussa kliinisessä monikeskustutkimuksessa, jonka kesto oli 24 viikkoa ja jota seurasi viikon kestänyt seurantavaihe (HZA107116). Tutkimukseen osallistui 673 potilasta, joiden astma ei ollut hoitotasapainossa ja jotka käyttivät inhaloitavia kortikosteroideja.
Kaikki tutkittavat olivat käyttäneet vakaa-annoksista astmalääkitystä (inhaloitava lyhytvaikutteinen beeta-agonisti tai lyhytvaikutteinen muskariiniantagonisti ja lisänä inhaloitava kortikosteroidi) vähintään 4 viikon ajan ennen käyntiä 1. Potilailla oli oireita aiemmasta astmahoidosta huolimatta (eli astma ei ollut hoitotasapainossa).
Tutkittavat saivat flutikasonifuroaatti/vilanteroli 46/22 mikrog ‑hoitoa (337 potilasta) tai flutikasonifuroaatti 46 mikrog ‑hoitoa (336 potilasta). Kahta potilasta (yksi per hoitoryhmä) ei voitu arvioida tehon osalta.
Ensisijainen päätetapahtuma oli ennen lääkkeenottoa mitatun aamupäivän PEF-arvon (uloshengityksen huippuvirtausarvon; jäännösarvo) muutos lähtötasosta laskettuna hoitoviikkojen 1–12 keskiarvona. PEF-arvot kirjattiin päivittäin sähköiseen potilaspäiväkirjaan (FF/VI vs. FF). Kohtauslääkkeettömien 24 tunnin jaksojen prosenttiosuuden muutos lähtötasosta hoitoviikkojen 1–12 aikana oli 5–11-vuotiaiden populaatiossa tutkimuksen voimaan vaikuttava toissijainen päätetapahtuma. FF/VI 46/22 mikrog ‑hoidon ja FF 46 mikrog ‑hoidon tehoissa ei ollut eroja (taulukko 2). Tutkimuksen aikana ei havaittu uusia turvallisuushuolia.
Tutkimuksen HZA107116 päätyttyä kahdessa tutkimuspaikassa havaittiin tutkimuksen suorittamiseen liittyviä huolenaiheita koskien yhteensä neljää satunnaistettua potilasta (FF/VI 46/22 mikrogrammaa n = 1, FF 46 mikrogrammaa n = 3). Tutkimuksen jälkeen tehtiin lisäanalyysi, josta suljettiin pois nämä neljä potilasta. Tämän analyysin tulokset (taulukko 2) ovat yhdenmukaisia ennalta määritellyn analyysin tulosten kanssa.
Taulukko 2 Ensisijaisten päätetapahtumien ja toissijaisten päätetapahtumien, joilla riittävä tilastollinen voima, tulokset (tutkimuksen jälkeen tehty lisäanalyysi)
| Viikot 1–12 | Flutikasonifuroaatti/vilanteroli* n = 335 | Flutikasonifuroaatti* n = 332 |
| Ensisijainen päätetapahtuma | ||
| Aamupäivän PEF-arvon muutos lähtötasosta (l/min) | ||
| Keskimuutos, pienin neliösumma (keskivirhe) | 12,1 (1,86) | 8,6 (1,87) |
Hoitojen ero (FF/VI vs. FF) (95 % lv), p-arvo | 3,5 (−1,7, 8,7), p = 0,188 | |
| Toissijainen päätetapahtuma, jolla riittävä tilastollinen voima | ||
| Kohtauslääkkeettömien 24 tunnin jaksojen prosentuaalisen osuuden muutos lähtötasosta | ||
| Keskimuutos, pienin neliösumma (keskivirhe) | 27,1 (1,75) | 26,0 (1,76) |
Hoitojen ero (FF/VI vs. FF) (95 % lv), p-arvo | 1,1 (−3,8, 6,0), p = 0,659 | |
*Potilaat saivat kerran vuorokaudessa joko FF/VI 46/22 mikrog ‑hoitoa tai FF 46 mikrog ‑hoitoa
lv = luottamusväli, n = analyysiin osallistuneiden määrä (ITT-populaatio: FF/VI-ryhmässä 337 ja FF-ryhmässä 336)
Farmakokinetiikka
Imeytyminen
Flutikasonifuroaatti ja vilanteroli-inhalaationa annetun flutikasonifuroaatin absoluuttinen hyötyosuus oli keskimäärin 15,2 % ja vilanterolin 27,3 %. Sekä flutikasonifuroaatin että vilanterolin oraalinen hyötyosuus oli pieni, flutikasonifuroaatin keskimäärin 1,26 % ja vilanterolin < 2 %. Ottaen huomioon pienen oraalisen hyötyosuuden, systeeminen flutikasonifuroaatti- ja vilanterolialtistus inhaloidun annoksen jälkeen johtuu pääasiassa inhaloidusta annoksesta keuhkoihin päätyvän osan imeytymisestä.
Jakautuminen
Flutikasonifuroaatti ja vilanteroli jakautuvat elimistössä voimakkaasti laskimoon annetun annoksen jälkeen. Flutikasonifuroaatin keskimääräinen jakautumistilavuus vakaan tilan aikana on 661 litraa ja vilanterolin 165 litraa.
Sekä flutikasonifuroaatin että vilanterolin sitoutuminen punasoluihin on vähäistä. In vitro flutikasonifuroaatti ja vilanteroli sitoutuivat voimakkaasti ihmisen plasman proteiineihin, flutikasonifuroaatin sitoutumisaste oli > 99,6 % ja vilanterolin 93,9 %. Munuaisten tai maksan vajaatoiminta ei vähentänyt lääkeaineiden sitoutumista plasman proteiineihin in vitro.
Flutikasonifuroaatti ja vilanteroli ovat P-glykoproteiinin (P-gp:n) substraatteja. Flutikasonifuroaatti-vilanterolin ja P-gp:n estäjien samanaikainen käyttö ei todennäköisesti kuitenkaan vaikuta systeemiseen flutikasonifuroaatti- tai vilanterolialtistukseen, sillä molemmat ovat hyvin imeytyviä molekyylejä.
Biotransformaatio
In vitro saatujen tietojen perusteella sekä flutikasonifuroaatin että vilanterolin metaboloituminen ihmiselimistössä tapahtuu pääasiassa CYP3A4-entsyymin välityksellä.
Flutikasonifuroaatin metaboloituminen perustuu pääasiassa S-fluorometyylikarbotioaattiryhmän hydrolysoitumiseen metaboliiteiksi, joilla on huomattavasti heikentynyt kortikosteroidivaikutus. Vilanteroli metaboloituu pääasiassa O-dealkylaation kautta useiksi metaboliiteiksi, joilla on huomattavasti heikentynyt β1- ja β2-agonistivaikutus.
Eliminaatio
Oraalisesti annettu flutikasonifuroaatti poistui ihmiselimistöstä pääasiassa metaboloitumalla, ja metaboliitit erittyivät lähes yksinomaan ulosteeseen. Alle 1 % radioaktiivisesti merkitystä annoksesta erittyi virtsaan.
Vilanteroli poistui ihmiselimistöstä oraalisesti annetun radioaktiivisesti merkityn annoksen jälkeen pääasiassa metaboloitumalla, ja noin 70 % radioaktiivisesta annoksesta erittyi metaboliitteina virtsaan ja 30 % ulosteeseen. Vilanterolin näennäinen eliminoitumisen puoliintumisaika plasmassa oli flutikasonifuroaatti-vilanterolivalmisteen inhaloidun kerta-annoksen jälkeen keskimäärin 2,5 tuntia. Vilanterolin kumuloitumisen efektiivinen puoliintumisaika, määritettynä inhalaationa annetuista toistuvista 25 mikrogramman vilanteroliannoksista, on astmapotilailla 16,0 tuntia ja keuhkoahtaumatautipotilailla 21,3 tuntia.
Pediatriset potilaat
Annostusta ei tarvitse muuttaa nuoria (vähintään 12-vuotiaita) potilaita hoidettaessa.
Flutikasonifuroaatti-vilanterolivalmisteen farmakokinetiikkaa, turvallisuutta ja tehoa on tutkittu 5–11-vuotiailla lapsilla, mutta suosituksia annostuksesta ei voida antaa (ks. kohta Annostus ja antotapa). Flutikasonifuroaatti-vilanterolin farmakokinetiikkaa, turvallisuutta ja tehoa alle 5-vuotiaiden lasten hoidossa ei ole varmistettu.
Erityisryhmät
Iäkkäät
Iän vaikutus flutikasonifuroaatin ja vilanterolin farmakokinetiikkaan määritettiin vaiheen III tutkimuksissa keuhkoahtaumataudin ja astman hoidossa. Astmapotilailla ei havaittu viitteitä iän (12–84 vuotta) vaikutuksista flutikasonifuroaatin ja vilanterolin farmakokinetiikkaan.
Iäkkäiden astmapotilaiden ja iäkkäiden keuhkoahtaumatautipotilaiden annostusta ei tarvitse muuttaa.
Munuaisten vajaatoiminta
Flutikasonifuroaatti-vilanterolivalmisteen kliinis-farmakologinen tutkimus osoitti, ettei vaikeaan munuaisten vajaatoimintaan (kreatiniinipuhdistuma < 30 ml/min) liittynyt merkittävää flutikasonifuroaatti- tai vilanterolialtistuksen suurenemista, eikä systeemisten kortikosteroidi- tai beeta2-agonistivaikutusten voimistumista terveisiin tutkittaviin verrattuna.
Annosta ei tarvitse sovittaa munuaisten vajaatoimintaa sairastavia potilaita hoidettaessa.
Hemodialyysin vaikutuksia ei ole tutkittu.
Maksan vajaatoiminta
Maksan vajaatoimintaa (Child-Pugh A, B tai C) sairastavien potilaiden systeeminen flutikasonifuroaattialtistus suureni (enintään kominkertaiseksi AUC(0–24)-arvon perusteella) terveisiin tutkittaviin verrattuna, kun flutikasonifuroaatti-vilanterolivalmistetta annettiin toistuvina annoksina 7 vuorokauden ajan. Kohtalaista maksan vajaatoimintaa sairastavien potilaiden systeemisen flutikasonifuroaattialtistuksen suurenemiseen (Child-Pugh B; flutikasonifuroaatti-vilanteroli 184/22 mikrogrammaa) liittyi keskimäärin 34 %:n seerumin kortisolipitoisuuden lasku terveisiin tutkittaviin verrattuna. Annoksen suhteen normalisoitu systeeminen flutikasonifuroaattialtistus oli samanlainen kohtalaista ja vaikeaa maksan vajaatoimintaa sairastavilla potilailla (Child-Pugh B tai C).
Lievää, kohtalaista tai vaikeaa maksan vajaatoimintaa (Child-Pugh A, B tai C) sairastavien potilaiden systeeminen vilanterolialtistus (Cmax ja AUC) ei suurentunut merkittävästi, kun flutikasonifuroaatti-vilanterolivalmistetta annettiin toistuvina annoksina 7 vuorokauden ajan.
Flutikasonifuroaatti-vilanterolivalmisteella ei ollut kliinisesti merkittäviä systeemisiä (sydämen sykkeeseen tai seerumin kaliumpitoisuuteen kohdistuvia) beeta-adrenergisia vaikutuksia lievää tai kohtalaista maksan vajaatoimintaa (vilanteroli, 22 mikrogrammaa) tai vaikeaa maksan vajaatoimintaa (vilanteroli, 12,5 mikrogrammaa) sairastavia potilaita hoidettaessa verrattuna terveisiin tutkittaviin.
Muut erityisryhmät
Flutikasonifuroaatin AUC(0-24)-arvon estimaatit olivat itäaasialaisilla, japanilaisilla ja kaakkoisaasialaisilla astmapotilailla (12–13 % potilaista) 33–53 % suurempia kuin muissa etnisissä ryhmissä. Ei kuitenkaan havaittu viitteitä siitä, että suuremmalla systeemisellä altistuksella olisi ollut voimakkaampi vaikutus virtsasta määritettyyn 24 tunnin kortisolieritykseen tässä potilasjoukossa. Vilanterolin Cmax-arvon ennustetaan olevan keskimäärin 220–287 % suurempi ja AUC(0-24)-arvon samansuuruinen aasialaista syntyperää olevilla potilailla kuin muissa etnisissä ryhmissä. Vilanterolin suurempaan Cmax-arvoon ei ole kuitenkaan havaittu liittyvän kliinisesti merkittäviä sydämen sykkeeseen kohdistuvia vaikutuksia.
Sukupuoli, paino ja painoindeksi
Sukupuolen, painon tai painoindeksin (BMI) ei havaittu vaikuttavan flutikasonifuroaatin farmakokinetiikkaan vaiheen III tutkimustuloksista tehdyn populaatiofarmakokineettisen analyysin perusteella. Analyysissä oli mukana 1213 astmapotilasta (naisia 712).
Sukupuolen, painon tai painoindeksin (BMI) ei havaittu vaikuttavan vilanterolin farmakokinetiikkaan 856 astmapotilaasta (naisia 500) tehdyn populaatiofarmakokineettisen analyysin perusteella.
Annostuksen muuttaminen ei ole tarpeen sukupuolen, painon eikä painoindeksin perusteella.
Prekliiniset tiedot turvallisuudesta
Prekliinisissä tutkimuksissa havaitut flutikasonifuroaatin tai vilanterolin farmakologiset ja toksikologiset vaikutukset olivat joko glukokortikoideille tai beeta2-agonisteille tyypillisiä vaikutuksia. Flutikasonifuroaatin antaminen yhdessä vilanterolin kanssa ei aiheuttanut merkittäviä uusia toksisia vaikutuksia.
Genotoksisuus ja karsinogeenisuus
Flutikasonifuroaatti
Flutikasonifuroaatti ei ollut genotoksinen standarditesteissä, eikä karsinogeeninen rottien ja hiirten koko eliniän kestäneissä inhalaatiotutkimuksissa altistustasoilla, jotka vastasivat ihmisille suositellun enimmäisannoksen aikaansaamaa altistusta AUC-arvon perusteella.
Vilanterolitrifenataatti
Vilanteroli (alfa-fenyylisinnamaattina) ja trifenyylietikkahappo eivät olleet genotoksisia geneettisissä toksisuustutkimuksissa, mikä osoittaa, ettei vilanteroli (trifenataattina) aiheuta genotoksista vaaraa ihmisille.
Vilanterolitrifenataatti aiheutti proliferatiivisia vaikutuksia naarasrottien ja ‑hiirten lisääntymiselimissä ja aivolisäkkeessä elinikäisissä inhalaatiotutkimuksissa, mikä vastasi muista beeta2-agonisteista tehtyjä havaintoja. Kasvainten ilmaantuvuus ei lisääntynyt rotilla eikä hiirillä, kun altistus oli hiirillä 1,2-kertainen ja rotilla 30-kertainen verrattuna ihmisille suositellun enimmäisannoksen aikaansaamaan altistukseen AUC-arvon perusteella.
Lisääntymis- ja kehitystoksisuus
Flutikasonifuroaatti
Rotille inhalaationa yhdessä vilanterolin kanssa annetun flutikasonifuroaatin vaikutukset olivat samanlaisia kuin pelkän flutikasonifuroaatin vaikutukset.
Flutikasonifuroaatti ei aiheuttanut epämuodostumia rotille eikä kaniineille, mutta se viivästytti rottien kehitystä ja aiheutti kaniineille keskenmenoja toksisia annoksia käytettäessä. Rottien kehitykseen kohdistuvia vaikutuksia ei havaittu, kun altistus oli noin kolme kertaa suurempi kuin ihmisille suositellun enimmäisannoksen aikaansaama altistus AUC-arvon perusteella.
Vilanterolitrifenataatti
Vilanterolitrifenataatti ei aiheuttanut epämuodostumia rotille. Kaniineille tehdyissä inhalaatiotutkimuksissa vilanterolitrifenataatti aiheutti samanlaisia vaikutuksia kuin muidenkin beeta2-agonistien on havaittu aiheuttavan (suulakihalkio, avoimet silmäluomet, rintalastan segmenttien yhteenkasvaminen ja raajojen taipuminen/kiertyminen virheasentoon). Ihonalaisilla annoksilla ei ollut vaikutuksia altistuksen ollessa 84 kertaa suurempi kuin ihmisille suositellun enimmäisannoksen aikaansaama altistus AUC-arvon perusteella.
Flutikasonifuroaatilla eikä vilanterolitrifenataatilla ollut haitallisia vaikutuksia rottien hedelmällisyyteen, eikä pre- ja postnataaliseen kehitykseen.
Farmaseuttiset tiedot
Apuaineet
Laktoosimonohydraatti
Magnesiumstearaatti
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
2 vuotta
Käytönaikainen kestoaika pakkauksen avaamisen jälkeen: 6 viikkoa.
Säilytys
Säilytä alle 25 °C. Jos inhalaattoria säilytetään jääkaapissa, sen on annettava lämmetä huoneenlämmössä vähintään tunnin ajan ennen käyttöä.
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Kirjoita laitteen etiketissä varattuun tilaan päivämäärä, johon mennessä lääke on käytettävä. Päivämäärä tulisi lisätä heti, kun laite on poistettu pakkauksesta.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
RELVAR ELLIPTA inhalaatiojauhe, annosteltu
184/22 mikrog/annos (L:kyllä) 30 annosta (49,68 €), 3 x 30 annosta (139,41 €)
PF-selosteen tieto
Ellipta-inhalaattorissa on vaaleanharmaa runko-osa, keltainen suukappaleen suojus ja annoslaskuri. Laite on pakattu foliolaminaattilaatikkoon, jossa on mukana silikageeli-kuivatusainepussi. Foliolaminaattilaatikko on suljettu repäistävällä foliokannella.
Inhalaattori on moniosainen laite, jonka valmistusaineita ovat polypropyleeni, polyetyleeni (HDPE), polyoksimetyleeni, polybutyleenitereftalaatti, akryylinitriilibutadieenistyreeni, polykarbonaatti ja ruostumaton teräs.
Laitteessa on kaksi alumiinifoliolaminaatista valmistettua läpipainoliuskaa, joista saa kaikkiaan 14 tai 30 annosta (14 tai 30 päivän tarvetta vastaava määrä).
Pakkauskoot: 14 tai 30 annosta. Monipakkaus: 3 x 30 annosta.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen jauhe vaaleanharmaassa inhalaattorissa (Ellipta), jossa on keltainen suukappaleen suojus ja annoslaskuri.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
RELVAR ELLIPTA inhalaatiojauhe, annosteltu
184/22 mikrog/annos 30 annosta, 3 x 30 annosta
- Alempi erityiskorvaus (65 %). Krooninen keuhkoastma ja sitä läheisesti muistuttavat krooniset obstruktiiviset keuhkosairaudet (203).
- Peruskorvaus (40 %).
ATC-koodi
R03AK10
Valmisteyhteenvedon muuttamispäivämäärä
12.03.2025
Yhteystiedot
 GLAXOSMITHKLINE OY
GLAXOSMITHKLINE OY Porkkalankatu 20 A
00180 Helsinki
010 303 030
www.glaxosmithkline.fi