
IMNOVID kapseli, kova 1 mg, 2 mg, 3 mg, 4 mg
Vaikuttavat aineet ja niiden määrät
Yksi kova kapseli sisältää 1 mg, 2 mg, 3 mg tai 4 mg pomalidomidia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, kova.
Kliiniset tiedot
Käyttöaiheet
Imnovid bortetsomibiin ja deksametasoniin yhdistettynä on tarkoitettu sellaisten multippelia myeloomaa sairastavien aikuispotilaiden hoitoon, jotka ovat saaneet vähintään yhtä aiempaa hoitoa, mukaan lukien lenalidomidihoito.
Imnovid deksametasoniin yhdistettynä on tarkoitettu sellaisten relapsoitunutta ja refraktorista multippelia myeloomaa sairastavien aikuispotilaiden hoitoon, jotka ovat saaneet vähintään kahta aiempaa hoitoa, mukaan lukien sekä lenalidomidi- että bortetsomibihoito, ja joiden sairaus eteni viimeisimmän hoidon aikana.
Ehto
Hoito tulee aloittaa ja sitä tulee seurata multippelin myelooman (MM) hoitoon perehtyneiden lääkäreiden valvonnassa. Lääkkeen määrääminen ja toimittaminen naisille, jotka voivat tulla raskaaksi, on rajoitettu 30 päivän lääkitykseen yhdellä reseptillä, ja hoidon jatkaminen vaatii uuden lääkemääräyksen. Naisille, jotka voivat tulla raskaaksi, kirjoitettu lääkemääräys on voimassa seitsemän (7) päivää lääkemääräyksen kirjoittamisesta.
Annostus ja antotapa
Hoito tulee aloittaa ja sitä tulee seurata multippelin myelooman hoitoon perehtyneiden lääkäreiden valvonnassa.
Antoa jatketaan tai muutetaan kliinisten ja laboratoriolöydösten mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Pomalidomidi bortetsomibiin ja deksametasoniin yhdistettynä
Pomalidomidin suositeltu aloitusannos on 4 mg, joka otetaan suun kautta kerran vuorokaudessa 21 vuorokauden pituisten toistuvien hoitosyklien vuorokausina 1–14.
Pomalidomidia annetaan bortetsomibin ja deksametasonin kanssa taulukon 1 mukaisesti.
Bortetsomibin suositeltu aloitusannos on 1,3 mg/m2 laskimoon tai ihon alle kerran vuorokaudessa taulukon 1 mukaisina päivinä. Deksametasonin suositeltu annos on 20 mg, joka otetaan suun kautta kerran vuorokaudessa taulukon 1 mukaisina päivinä.
Hoitoa pomalidomidilla yhdessä bortetsomibin ja deksametasonin kanssa tulee jatkaa taudin etenemiseen tai liiallisen toksisuuden ilmenemiseen saakka.
Taulukko 1. Pomalidomidin suositeltu annostusohjelma bortetsomibiin ja deksametasoniin yhdistettynä
Hoitosyklit 1–8 | Vuorokausi (21 vrk:n hoitosyklissä) | |||||||||||||||||||||||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | |||||||||||||||||||
Pomalidomidi (4 mg) | • | • | • | • | • | • | • | • | • | • | • | • | • | • |
|
|
|
|
|
|
| |||||||||||||||||||
Bortetsomibi (1,3 mg/m2) | • |
|
| • |
|
|
| • |
|
| • |
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||
Deksametasoni (20 mg) * | • | • |
| • | • |
|
| • | • |
| • | • |
|
|
|
|
|
|
|
|
| |||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||
Hoitosykli 9 ja sen jälkeiset hoitosyklit | Vuorokausi (21 vrk:n hoitosyklissä) | |||||||||||||||||||||||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | |||||||||||||||||||
Pomalidomidi (4 mg) | • | • | • | • | • | • | • | • | • | • | • | • | • | • |
|
|
|
|
|
|
| |||||||||||||||||||
Bortetsomibi (1,3 mg/m2) | • |
|
|
|
|
|
| • |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||
Deksametasoni (20 mg) * | • | • |
|
|
|
|
| • | • |
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||
* > 75-vuotiaat potilaat, ks. Erityiset potilasryhmät.
Pomalidomidiannoksen muuttaminen tai hoidon keskeyttäminen
Jotta uusi pomalidomidisykli voidaan aloittaa, neutrofiilimäärän on oltava ≥ 1 x 109/l ja trombosyyttimäärän ≥ 50 x 109/l.
Ohjeet hoidon keskeyttämiseen tai annoksen pienentämiseen pomalidomidiin liittyvien haittavaikutusten yhteydessä on esitetty alla olevassa taulukossa 2; annostasot on määritetty taulukossa 3.
Taulukko 2. Ohjeet pomalidomidiannoksen muuttamiseen∞
Toksisuus | Annosmuutos |
| Neutropenia* | |
ANC** < 0,5 x 109/l tai kuumeinen neutropenia (kuumetta ≥ 38,5 °C ja ANC < 1 x 109/l) | Keskeytä pomalidomidihoito hoitosyklin jäljellä olevaksi ajaksi. Seuraa TVK:ta*** viikoittain. |
ANC palaa arvoon ≥ 1 x 109/l | Jatka pomalidomidihoitoa annoksella, joka on yhtä annostasoa pienempi kuin edellinen annos. |
Tämän jälkeen aina, kun arvo pienenee < 0,5 x 109/l | Keskeytä pomalidomidihoito. |
ANC palaa arvoon ≥ 1 x 109/l | Jatka pomalidomidihoitoa annoksella, joka on yhtä annostasoa pienempi kuin edellinen annos. |
| Trombosytopenia | |
Trombosyyttimäärä < 25 x 109/l | Keskeytä pomalidomidihoito hoitosyklin jäljellä olevaksi ajaksi. Seuraa TVK:ta*** viikoittain. |
Trombosyyttimäärä palaa arvoon ≥ 50 x 109/l | Jatka pomalidomidihoitoa annoksella, joka on yhtä annostasoa pienempi kuin edellinen annos |
Tämän jälkeen aina, kun arvo pienenee < 25 x 109/l | Keskeytä pomalidomidihoito. |
Trombosyyttimäärä palaa arvoon ≥ 50 x 109/l | Jatka pomalidomidihoitoa annoksella, joka on yhtä annostasoa pienempi kuin edellinen annos. |
| Ihottuma | |
Ihottuma = 2. tai 3. asteen | Harkitse pomalidomidihoidon keskeyttämistä tai lopettamista. |
Ihottuma = 4. asteen tai rakkulainen (mukaan lukien angioedeema, anafylaktinen reaktio, hilseilevä tai rakkulainen ihottuma tai epäilty Stevens-Johnsonin oireyhtymä (SJS), toksinen epidermaalinen nekrolyysi (TEN) tai yleisoireinen eosinofiilinen oireyhtymä (DRESS)). | Lopeta hoito pysyvästi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). |
| Muut | |
Muut ≥ 3. asteen pomalidomidiin liittyvät haittavaikutukset | Keskeytä pomalidomidihoito hoitosyklin jäljellä olevaksi ajaksi. Jatka hoitoa seuraavassa hoitosyklissä annoksella, joka on yhtä annostasoa pienempi kuin edellinen annos (haittavaikutuksen tulee olla hävinnyt tai lieventynyt ≤ 2. asteeseen, ennen kuin hoitoa jatketaan). |
∞Tämän taulukon ohjeet annoksen muuttamiseen koskevat pomalidomidia bortetsomibiin ja deksametasoniin yhdistettynä sekä pomalidomidia deksametasoniin yhdistettynä.
* Neutropenian ilmaantuessa lääkärin on harkittava kasvutekijöiden käyttöä.
** ANC – absoluuttinen neutrofiilimäärä
*** TVK – täydellinen verenkuva
Taulukko 3. Pomalidomidiannoksen pienentäminen∞
Annostaso | Suun kautta annettava pomalidomidiannos |
Aloitusannos | 4 mg |
Annostaso -1 | 3 mg |
Annostaso -2 | 2 mg |
Annostaso -3 | 1 mg |
∞ Tämän taulukon ohjeet annoksen pienentämiseen koskevat pomalidomidia bortetsomibiin ja deksametasoniin yhdistettynä sekä pomalidomidia deksametasoniin yhdistettynä.
Jos haittavaikutuksia esiintyy sen jälkeen, kun annos on pienennetty 1 mg:aan asti, hoito on lopetettava.
Voimakkaat CYP1A2:n estäjät
Jos voimakkaita CYP1A2:n estäjiä (kuten siprofloksasiinia, enoksasiinia ja fluvoksamiinia) annetaan samanaikaisesti pomalidomidin kanssa, pomalidomidiannosta pitää pienentää 50 % (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Bortetsomibiannoksen muuttaminen tai hoidon keskeyttäminen
Ks. ohjeet hoidon keskeyttämiseen tai annoksen pienentämiseen bortetsomibiin liittyvien haittavaikutusten yhteydessä bortetsomibin valmisteyhteenvedosta.
Deksametasoniannoksen muuttaminen tai hoidon keskeyttäminen
Ohjeet hoidon keskeyttämiseen ja annoksen pienentämiseen pieniannoksiseen deksametasonihoitoon liittyvien haittavaikutusten yhteydessä on esitetty alla olevissa taulukoissa 4 ja 5. Hoidon keskeyttämisen ja jatkamisen tulee kuitenkin perustua lääkärin harkintaan ja voimassa olevaan valmisteyhteenvetoon.
Taulukko 4. Deksametasoniannoksen muuttaminen
Toksisuus | Annosmuutos |
| Dyspepsia = 1. tai 2. asteen | Älä muuta annosta, ja hoida histamiinireseptorin (H2) salpaajalla tai vastaavalla. Jos oireet jatkuvat, pienennä annosta yhdellä annostasolla. |
Dyspepsia ≥ 3. asteen | Keskeytä hoito, kunnes oireet ovat hallinnassa. Lisää H2-salpaaja tai vastaava, ja jatka annoksella, joka on yhtä annostasoa pienempi kuin edellinen annos. |
Turvotus ≥ 3. asteen | Pienennä annosta yhdellä annostasolla, ja käytä tarpeen mukaan diureetteja. |
Sekavuus tai mielialan vaihtelut ≥ 2. asteen | Keskeytä hoito, kunnes oireet häviävät. Jatka annoksella, joka on yhtä annostasoa pienempi kuin edellinen annos. |
Lihasheikkous ≥ 2. asteen | Keskeytä hoito, kunnes lihasheikkous ≤ 1. asteen. Jatka annoksella, joka on yhtä annostasoa pienempi kuin edellinen annos. |
Hyperglykemia ≥ 3. asteen | Pienennä annosta yhdellä annostasolla. Hoida tarvittaessa insuliinilla tai suun kautta annettavilla verensokeria alentavilla lääkkeillä. |
Akuutti haimatulehdus | Keskeytä deksametasonihoito. |
Muut ≥ 3. asteen deksametasoniin liittyvät haittavaikutukset | Keskeytä deksametasonihoito, kunnes haittavaikutus lievenee ≤ 2. asteeseen. Jatka annoksella, joka on yhtä annostasoa pienempi kuin edellinen annos. |
Jos toksisuuden häviäminen kestää yli 14 vuorokautta, deksametasonihoitoa tulee jatkaa annoksella, joka on yhtä annostasoa pienempi kuin edellinen annos.
Taulukko 5. Deksametasoniannoksen pienentäminen
Annostaso | ≤ 75-vuotiaat | > 75-vuotiaat |
Aloitusannos | 20 mg | 10 mg |
Annostaso -1 | 12 mg | 6 mg |
Annostaso -2 | 8 mg | 4 mg |
Deksametasonihoito tulee ≤ 75-vuotiailla potilailla lopettaa, jos potilas ei siedä 8 mg:n annosta, ja > 75‑vuotiailla potilailla, jos potilas ei siedä 4 mg:n annosta.
Jos hoidon minkä tahansa komponentin antaminen lopetetaan, hoidon jatkaminen jäljellä olevilla lääkevalmisteilla on lääkärin harkinnan varassa.
Pomalidomidi deksametasoniin yhdistettynä
Pomalidomidin suositeltu aloitusannos on 4 mg pomalidomidia, joka otetaan suun kautta kerran vuorokaudessa kunkin 28 vuorokauden pituisen syklin vuorokausina 1–21.
Deksametasonin suositeltu annos on 40 mg, joka otetaan suun kautta kerran vuorokaudessa kunkin 28 vuorokauden syklin vuorokausina 1, 8, 15 ja 22.
Hoitoa pomalidomidilla yhdessä deksametasonin kanssa tulee jatkaa taudin etenemiseen tai liiallisen toksisuuden ilmenemiseen saakka.
Pomalidomidiannoksen muuttaminen tai hoidon keskeyttäminen
Ohjeet pomalidomidihoidon keskeyttämiseen ja pomalidomidiannoksen pienentämiseen haittavaikutusten yhteydessä on esitetty taulukoissa 2 ja 3.
Deksametasoniannoksen muuttaminen tai hoidon keskeyttäminen
Ohjeet annoksen muuttamiseen deksametasoniin liittyvien haittavaikutusten yhteydessä on esitetty taulukossa 4. Ohjeet annoksen pienentämiseen deksametasoniin liittyvien haittavaikutusten yhteydessä on esitetty alla olevassa taulukossa 6. Hoidon keskeyttämisen ja jatkamisen tulee kuitenkin perustua lääkärin harkintaan ja voimassa olevaan valmisteyhteenvetoon.
Taulukko 6. Deksametasoniannoksen pienentäminen
Annostaso | ≤ 75-vuotiaat | > 75-vuotiaat |
Aloitusannos | 40 mg | 20 mg |
Annostaso -1 | 20 mg | 12 mg |
Annostaso -2 | 10 mg | 8 mg |
Deksametasonihoito tulee ≤ 75-vuotiailla potilailla lopettaa, jos potilas ei siedä 10 mg:n annosta, ja > 75‑vuotiailla potilailla, jos potilas ei siedä 8 mg:n annosta.
Erityiset potilasryhmät
Iäkkäät potilaat
Pomalidomidiannoksen säätäminen ei ole tarpeen.
Pomalidomidi bortetsomibiin ja deksametasoniin yhdistettynä
Deksametasonin aloitusannos > 75-vuotiaille potilaille on:
- Hoitosyklit 1–8: 10 mg kerran vuorokaudessa kunkin 21 vuorokauden pituisen hoitosyklin vuorokausina 1, 2, 4, 5, 8, 9, 11 ja 12
- Hoitosykli 9 ja sen jälkeiset hoitosyklit: 10 mg kerran vuorokaudessa kunkin 21 vuorokauden pituisen hoitosyklin vuorokausina 1, 2, 8 ja 9.
Pomalidomidi deksametasoniin yhdistettynä
Deksametasonin aloitusannos > 75-vuotiaille potilaille on
- 20 mg kerran vuorokaudessa kunkin 28 vuorokauden pituisen syklin vuorokausina 1, 8, 15 ja 22.
Maksan vajaatoiminta
Kliinisiin tutkimuksiin ei otettu mukaan potilaita, joiden kokonaisbilirubiinipitoisuus seerumissa oli > 1,5 x viitealueen yläraja. Maksan vajaatoiminnalla on vähäinen vaikutus pomalidomidin farmakokinetiikkaan (ks. kohta Farmakokinetiikka). Pomalidomidin aloitusannoksen säätäminen ei ole tarpeen, jos potilaalla on Child-Pugh-kriteerien mukainen maksan vajaatoiminta. Maksan vajaatoimintaa sairastavia potilaita on kuitenkin seurattava huolellisesti haittavaikutusten varalta, ja annosta tulee pienentää tai pomalidomidihoito keskeyttää tarvittaessa.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavien potilaiden pomalidomidiannoksen säätäminen ei ole tarpeen. Hemodialyysipäivinä pomalidomidiannos tulee ottaa vasta hoidon jälkeen.
Pediatriset potilaat
Ei ole asianmukaista käyttää pomalidomidia 0–17 vuoden ikäisten lasten multippelin myelooman hoitoon.
Pomalidomidia on tutkittu hyväksyttyjen käyttöaiheidensa ulkopuolella 4–18-vuotiailla lapsilla, joilla on uusiutuneita tai eteneviä aivokasvaimia. Tutkimustulosten perusteella ei kuitenkaan ollut mahdollista päätellä, että tällaisen käytön hyödyt ylittäisivät riskit. Tällä hetkellä saatavana olevat tiedot on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka.
Antotapa
Suun kautta.
Imnovid kovat kapselit otetaan suun kautta joka päivä samaan aikaan päivästä. Kapseleita ei saa avata, rikkoa eikä pureskella (ks. kohta Käyttö- ja käsittelyohjeet). Kapselit on nieltävä kokonaisena, mieluiten veden kanssa, joko ruokailun yhteydessä tai tyhjään mahaan. Jos potilas jonakin päivänä unohtaa ottaa pomalidomidiannoksen, hänen tulee ottaa seuraavana päivänä normaali määrätty annos aikataulun mukaisesti. Potilaat eivät saa säätää annosta edellisinä päivinä unohdettujen annosten korvaamiseksi.
On suositeltavaa, että kapselia läpipainopakkauksesta poistettaessa painetaan vain kapselin toisesta päästä sen deformoitumisen tai rikkoutumisen riskin vähentämiseksi.
Vasta-aiheet
- Raskaus
- Naiset, jotka voivat tulla raskaaksi, elleivät kaikki raskaudenehkäisyohjelman ehdot täyty (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys)
- Miespotilaat, jotka eivät kykene noudattamaan edellytettyjä raskaudenehkäisytoimenpiteitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Teratogeenisuus
Pomalidomidia ei saa ottaa raskauden aikana, koska sillä oletetaan olevan teratogeeninen vaikutus. Pomalidomidi muistuttaa rakenteeltaan talidomidia. Talidomidin tiedetään olevan ihmiselle teratogeeninen aine, joka aiheuttaa vaikea-asteisia hengenvaarallisia syntymävaurioita. Pomalidomidin todettiin olevan teratogeeninen sekä rotille että kaniineille, kun sitä annettiin tärkeimmän organogeneesivaiheen aikana (ks. kohta Prekliiniset tiedot turvallisuudesta).
Kaikkien potilaiden on täytettävä raskaudenehkäisyohjelmassa mainitut ehdot, ellei ole luotettavaa näyttöä siitä, että potilas ei voi tulla raskaaksi.
Kriteerit naisille, jotka eivät voi tulla raskaaksi
Naispotilaan tai miespotilaan naispuolisen kumppanin ei katsota olevan hedelmällinen, jos hän täyttää vähintään yhden seuraavista kriteereistä:
- ikä ≥ 50 vuotta ja luonnollisesti amenorreeinen ≥ 1 vuoden (amenorrea syöpähoidon jälkeen tai imetyksen aikana ei sulje pois raskauden mahdollisuutta)
- ennenaikainen munasarjojen vajaatoiminta, jonka gynekologian erikoislääkäri on varmistanut
- aiemmin suoritettu salpingo-ooforektomia tai hysterektomia
- XY-genotyyppi, Turnerin oireyhtymä, kohdun puuttuminen.
Neuvonta
Pomalidomidi on vasta-aiheinen naisilla, jotka voivat tulla raskaaksi, elleivät kaikki seuraavat kriteerit täyty:
- hän ymmärtää sikiölle odotettavasti aiheutuvan teratogeenisen riskin
- hän ymmärtää keskeytyksettä vähintään 4 viikkoa ennen hoidon aloittamista, koko hoidon ajan ja vähintään 4 viikkoa hoidon päätyttyä käytettävän luotettavan ehkäisyn välttämättömyyden
- vaikka naisella, joka voi tulla raskaaksi, olisi amenorrea, hänen on noudatettava kaikkia luotettavasta ehkäisystä annettuja ohjeita
- hänen on kyettävä käyttämään luotettavia ehkäisymenetelmiä
- hän on tietoinen ja ymmärtää raskauden mahdolliset seuraukset ja tarpeen nopeaan konsultointiin raskauden mahdollisuuden yhteydessä
- hän ymmärtää hoidon aloittamisen tarpeen heti pomalidomidin määräämisen jälkeen, raskaustestin ollessa negatiivinen
- hän ymmärtää ja hyväksyy vähintään 4 viikon välein tehtävän raskaustestin välttämättömyyden lukuun ottamatta tapauksia, joissa munanjohdinsterilisaatio on varmistettu
- hän myöntää ymmärtävänsä pomalidomidin käyttöön liittyvät vaarat ja välttämättömät varotoimet.
Lääkkeen määräävän lääkärin on varmistettava niiden naisten osalta, jotka voivat tulla raskaaksi, että:
- potilas täyttää raskaudenehkäisyohjelman asettamat vaatimukset ja potilas ymmärtää asian riittävästi
- potilas on hyväksynyt edellä mainitut hoidon ehdot.
Pomalidomidia käyttävien miespotilaiden osalta farmakokineettiset tiedot ovat osoittaneet, että pomalidomidia esiintyy ihmisen siemennesteessä hoidon aikana. Varotoimena, ja ottaen huomioon erityiset väestöryhmät, joilla on mahdollisesti pidentynyt eliminaatioaika, esim. maksan vajaatoimintaa sairastavat, kaikkien pomalidomidia käyttävien miespotilaiden on täytettävä seuraavat ehdot:
- hänen on ymmärrettävä odotettava teratogeeninen vaara, mikäli hän on sukupuolisessa kanssakäymisessä raskaana olevan naisen tai sellaisen naisen kanssa, joka voi tulla raskaaksi
- hänen on ymmärrettävä kondomin käytön välttämättömyys, mikäli hän on sukupuolisessa kanssakäymisessä raskaana olevan naisen tai sellaisen naisen kanssa, joka voi tulla raskaaksi, ja joka ei käytä tehokasta raskaudenehkäisyä, koko hoidon ja hoitotaukojen aikana sekä 7 päivän ajan hoitotauon alkamisesta ja/tai hoidon loppumisesta. Tämä koskee myös miehiä, joille on tehty vasektomia, ja heidän on käytettävä kondomia ollessaan yhdynnässä raskaana olevan naisen kanssa tai sellaisen naisen kanssa, joka voi tulla raskaaksi, sillä siemenneste voi siittiöiden puuttumisesta huolimatta sisältää pomalidomidia.
- hänen on ymmärrettävä, että jos hänen naispuolinen kumppaninsa tulee raskaaksi pomalidomidihoidon aikana tai 7 päivän kuluessa pomalidomidihoidon lopettamisen jälkeen, hänen on ilmoitettava siitä välittömästi hoitavalle lääkärille, ja että tällöin on suositeltavaa, että naispuolinen kumppani saa arviointia ja ohjeita varten lähetteen teratologiaan erikoistuneelle tai siitä kokemusta saaneelle lääkärille.
Ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä vähintään yhtä luotettavaa ehkäisymenetelmää vähintään 4 viikkoa ennen hoitoa, hoidon aikana ja vähintään 4 viikkoa pomalidomidihoidon jälkeen ja jopa mahdollisen hoitotauon aikana, ellei potilas sitoudu täydelliseen ja jatkuvaan pidättymiseen sukupuolisesta kanssakäymisestä, mikä varmistetaan kuukausittain. Mikäli potilas ei aiemmin ole käyttänyt luotettavaa ehkäisyä, hänet on ohjattava asianomaiselle lääkärille ehkäisyneuvontaa varten, jotta potilas voi aloittaa ehkäisyn.
Seuraavassa on esimerkkejä sopivista ehkäisymenetelmistä:
- implantti
- levonorgestreeliä vapauttava kohdunsisäinen ehkäisin
- medroksiprogesteroniasetaattia sisältävä depotvalmiste
- munanjohdinsterilisaatio
- sukupuoliyhdyntä ainoastaan vasektomialla steriloitujen miespuolisten kumppaneiden kanssa; vasektomia on vahvistettava kahdella negatiivisella siemennestetutkimuksella
- ovulaation estävät pelkkää progestiinia sisältävät ehkäisytabletit (eli desogestreeli).
Pomalidomidia ja deksametasonia käyttävien multippelia myeloomaa sairastavien potilaiden suurentuneen laskimotromboemboliariskin vuoksi suun kautta otettavia yhdistelmäehkäisytabletteja ei suositella (ks. myös kohta Yhteisvaikutukset). Jos potilas käyttää suun kautta otettavaa yhdistelmäehkäisyvalmistetta, potilaan tulee siirtyä käyttämään jotakin edellä mainituista luotettavista menetelmistä. Laskimotromboembolia on mahdollinen 4–6 viikkoa suun kautta otettavan yhdistelmäehkäisyvalmisteen käytön lopettamisen jälkeen. Ehkäisyyn käytettävien steroidien teho saattaa heikentyä, jos niitä käytetään samanaikaisesti deksametasonin kanssa (ks. kohta Yhteisvaikutukset).
Implantteihin ja levonorgestreeliä vapauttaviin kohdunsisäisiin ehkäisimiin liittyy lisääntynyt infektiovaara paikalleen asetuksen yhteydessä sekä epäsäännöllinen emätinverenvuoto. Antibioottiprofylaksiaa tulee harkita erityisesti neutropeniaa sairastavilla potilailla.
Kuparia vapauttavien kohdunsisäisten ehkäisimien asettamista ei yleisesti suositella paikoilleen asettamisen yhteydessä esiintyvän mahdollisen infektiovaaran ja kuukautisverenvuodon vuoksi, mikä saattaa aiheuttaa haittaa vaikea-asteista neutropeniaa tai vaikea-asteista trombosytopeniaa sairastaville potilaille.
Raskaustesti
Paikallisen käytännön mukaisesti naisille, jotka voivat tulla raskaaksi, on tehtävä lääketieteellisesti valvottuja raskaustestejä 25 mIU/ml:n vähimmäisherkkyydellä seuraavien ohjeiden mukaan. Tämä vaatimus koskee myös naisia, jotka voivat tulla raskaaksi ja jotka ovat sitoutuneet täydelliseen ja jatkuvaan pidättymiseen sukupuolisesta kanssakäymisestä. Raskaustestin, lääkkeen määräämisen ja lääkkeen annon tulisi tapahtua mieluiten samana päivänä. Pomalidomidi tulisi antaa naisille, jotka voivat tulla raskaaksi, 7 vuorokauden kuluessa lääkkeen määräämisestä.
Ennen hoidon aloittamista
Lääketieteellisesti valvottu raskaustesti on tehtävä neuvonnan aikana pomalidomidin määräämisen yhteydessä tai lääkettä määräävän lääkärin vastaanotolla käyntiä edeltävän kolmen vuorokauden aikana, kun potilas on käyttänyt luotettavaa raskaudenehkäisyä vähintään 4 viikkoa. Testin on vahvistettava, että potilas ei ole raskaana aloittaessaan pomalidomidihoidon.
Seuranta ja hoidon päättäminen
Lääketieteellisesti valvottu raskaustesti on toistettava vähintään 4 viikon välein, mukaan lukien vähintään 4 viikkoa hoidon päättymisen jälkeen lukuun ottamatta tapauksia, joissa munanjohdinsterilisaatio on varmistettu. Nämä raskaustestit on tehtävä sinä päivänä, jolloin potilas käy lääkettä määräävän lääkärin vastaanotolla tai tätä edeltävän 3 vuorokauden aikana.
Lisävarotoimet
Potilaita on neuvottava, ettei tätä lääkevalmistetta saa koskaan antaa toiselle henkilölle ja että käyttämättömät kapselit on palautettava apteekkiin hoidon päätyttyä.
Potilaat eivät saa luovuttaa verta, siittiöitä tai spermaa hoidon aikana (hoitotauot mukaan lukien) eikä vähintään 7 päivään pomalidomidihoidon päättymisen jälkeen.
Terveydenhuollon ammattilaisten ja huoltajien on läpipainolevyjä tai kapseleita käsitellessään käytettävä kertakäyttöisiä käsineitä. Naisten, jotka ovat raskaana tai epäilevät olevansa raskaana, ei pidä käsitellä läpipainolevyjä tai kapseleita (ks. kohta Käyttö- ja käsittelyohjeet).
Koulutusmateriaali, lääkkeen määräämiseen ja antoon liittyvät rajoitukset
Myyntiluvan haltija toimittaa terveydenhuollon ammattilaisille koulutusmateriaalia, joka sisältää varoituksia pomalidomidin odotettavasta teratogeenisuudesta, neuvoja raskaudenehkäisystä ennen hoidon alkamista ja tietoa raskaustestien tekemisen välttämättömyydestä, auttaakseen potilaita välttämään sikiön altistumista pomalidomidille. Lääkkeen määräävän lääkärin on kerrottava potilaalle odotettavissa olevasta teratogeenisesta riskistä ja raskaudenehkäisyyn liittyvistä tarkoin noudatettavista toimenpiteistä, jotka on määritetty raskaudenehkäisyohjelmassa, sekä annettava potilaalle asianmukainen koulutusesite, potilaskortti ja/tai vastaava kansallisen toimivaltaisen viranomaisen kanssa sovitun mukaisesti. Kunkin maan kansallisen toimivaltaisen viranomaisen kanssa sovitusti on otettu käyttöön kontrolloitu käyttöönoton ohjelma, joka käsittää potilaskortin ja/tai vastaavan menetelmän käytön lääkkeen määräämisen ja/tai toimittamisen kontrollia varten sekä käyttöaihetta koskevien tietojen keräämisen kyseisellä kansallisella alueella tapahtuvan käyttöaiheesta poikkeavan käytön seuraamista varten. Raskaustestin, lääkkeen määräämisen ja lääkkeen toimittamisen tulisi mieluiten tapahtua samana päivänä. Naisille, jotka voivat tulla raskaaksi, pomalidomidi tulee toimittaa 7 vuorokauden kuluessa lääkkeen määräämisestä ja lääkärin valvoman raskaustestin negatiivisen tuloksen jälkeen. Lääkettä saa määrätä naisille, jotka voivat tulla raskaaksi, enintään 4 viikon hoitojaksoa varten hyväksyttyjen käyttöaiheiden annostusohjelmien mukaisesti (ks. kohta Annostus ja antotapa). Muille potilaille lääkettä saa määrätä enintään 12 viikon hoitojaksoa varten.
Hematologiset tapahtumat
Neutropenia oli yleisimmin raportoitu 3. tai 4. asteen hematologinen haittavaikutus potilailla, joilla oli relapsoitunut/refraktorinen multippeli myelooma. Seuraavaksi yleisimmin raportoituja olivat anemia ja trombosytopenia. Potilaita tulee seurata hematologisten haittavaikutusten, etenkin neutropenian, havaitsemiseksi. Potilaita on kehotettava raportoimaan kuumejaksoista välittömästi. Lääkärin on tarkkailtava potilasta verenvuodon, mukaan lukien nenäverenvuodon, havaitsemiseksi, etenkin jos muiden samanaikaisesti annettavien lääkevalmisteiden tiedetään lisäävän verenvuodon riskiä (ks. kohta Haittavaikutukset). Täydellistä verenkuvaa on seurattava lähtötilanteessa, viikoittain ensimmäisten 8 viikon ajan sekä kuukausittain sen jälkeen. Annoksen säätäminen saattaa olla tarpeen (ks. kohta Annostus ja antotapa). Potilaat saattavat tarvita verivalmisteita ja/tai kasvutekijöitä.
Tromboemboliset tapahtumat
Pomalidomidia joko yhdessä bortetsomibin ja deksametasonin tai yhdessä deksametasonin kanssa saaville potilaille on kehittynyt laskimotromboembolisia tapahtumia (pääasiassa syviä laskimotrombooseja ja keuhkoemboliaa) ja valtimotromboositapahtumia (sydäninfarkteja ja aivoverisuonitapahtumia) (ks. kohta Haittavaikutukset). Jos potilaalla tiedetään olevan tromboembolian riskitekijöitä, mukaan lukien aiempi tromboosi, potilasta on seurattava huolellisesti. Muutettavissa olevien riskitekijöiden, kuten tupakoinnin, hypertension ja hyperlipidemian, minimoimiseksi on ryhdyttävä toimenpiteisiin. Potilaan ja lääkärin on tarkkailtava tromboembolian oireita ja löydöksiä. Potilasta on neuvottava hakeutumaan hoitoon, jos hänelle kehittyy oireita, kuten hengenahdistusta, rintakipua tai raajojen turvotusta. Antikoagulanttihoitoa (ellei vasta-aiheinen) suositellaan (esim. asetyylisalisyylihappoa, varfariinia, hepariinia tai klopidrogreeliä), varsinkin jos potilailla on lisäksi muita tromboottisia riskitekijöitä. Päätös profylaktisista antitromboottisista toimenpiteistä tulee tehdä yksittäisen potilaan taustalla olevien riskitekijöiden huolellisen arvioinnin jälkeen. Kliinisissä tutkimuksissa potilaat saivat profylaktisesti asetyylisalisyylihappoa tai vaihtoehtoista antitromboottista hoitoa. Erytropoieettisten lääkeaineiden käyttö lisää tromboottisten tapahtumien, kuten tromboembolian, riskiä. Erytropoieettisia lääkeaineita tai muita tromboembolisten tapahtumien riskiä lisääviä lääkeaineita on siksi käytettävä varoen.
Kilpirauhassairaudet
Kilpirauhasen vajaatoimintatapauksia on raportoitu. Kilpirauhasen toimintaan vaikuttavien samanaikaisten sairauksien saamista optimaaliseen hoitotasapainoon suositellaan ennen hoidon aloittamista. Kilpirauhasen toiminnan seurantaa suositellaan hoitoa aloitettaessa ja hoidon aikana.
Perifeerinen neuropatia
Potilaita, joilla oli parhaillaan ≥ 2. asteen perifeerinen neuropatia, ei otettu mukaan pomalidomidilla tehtyihin kliinisiin tutkimuksiin. Kun pomalidomidihoitoa harkitaan tälle potilasryhmälle, on noudatettava asianmukaista varovaisuutta.
Merkityksellinen sydämen toimintahäiriö
Potilaita, joilla oli merkityksellinen sydämen toimintahäiriö (kongestiivinen sydämen vajaatoiminta [New York Heart Association ‑luokka III tai IV], sydäninfarkti 12 kuukauden sisällä tutkimuksen alkamisesta tai epästabiili tai huonossa hoitotasapainossa oleva angina pectoris), ei otettu mukaan pomalidomidilla tehtyihin kliinisiin tutkimuksiin. Sydäntapahtumia, mukaan lukien kongestiivista sydämen vajaatoimintaa, keuhkoedeemaa ja eteisvärinää (ks. kohta Haittavaikutukset), on raportoitu, pääasiassa potilailla, joilla oli jo ennestään sydänsairaus tai sydänsairauden riskitekijöitä. Kun pomalidomidihoitoa harkitaan tälle potilasryhmälle, on noudatettava asianmukaista varovaisuutta, muun muassa seuraamalla potilaan tilaa säännöllisesti sydäntapahtumien löydösten tai oireiden havaitsemiseksi.
Tuumorilyysioireyhtymä
Tuumorilyysioireyhtymälle altteimpia ovat potilaat, joilla on suuri kasvaintaakka ennen hoitoa. Näitä potilaita on seurattava huolellisesti, ja asianmukaisiin varotoimenpiteisiin on ryhdyttävä.
Muut primaarisyövät
Pomalidomidia saavilla potilailla on raportoitu muita primaarisyöpiä, kuten ei-melanoottisiaihosyöpiä (ks. kohta Haittavaikutukset). Lääkärin on sekä ennen hoitoa että hoidon aikana tutkittava potilas huolellisesti muiden primaarisyöpien havaitsemiseksi tavanomaisen syöpäseulonnan avulla ja aloitettava tarvittaessa asianmukainen hoito.
Allergiset reaktiot ja vaikeat ihoreaktiot
Angioedeemaa, anafylaktisia reaktioita ja vaikea-asteisia ihoreaktioita, mukaan lukien Stevens-Johnsonin oireyhtymää (SJS), toksista epidermaalista nekrolyysiä (TEN) ja yleisoireista eosinofiilista oireyhtymää (DRESS), on raportoitu esiintyneen pomalidomidin käytön yhteydessä (ks. kohta Haittavaikutukset). Lääkkeen määrääjien on kerrottava potilaille näiden reaktioiden merkeistä ja oireista, ja potilaita on neuvottava hakeutumaan lääkäriin heti, jos heille ilmaantuu tällaisia oireita. Pomalidomidihoito on lopetettava, jos potilaalle ilmaantuu hilseilevää tai rakkulaista ihottumaa tai jos epäillään Stevens-Johnsonin oireyhtymää, toksista epidermaalista nekrolyysiä tai yleisoireista eosinofiilista oireyhtymää. Hoitoa ei saa aloittaa uudelleen näiden reaktioiden häviämisen jälkeen. Talidomidi- tai lenalidomidihoidon yhteydessä aiemmin vakavia allergisia reaktioita saaneita potilaita ei otettu mukaan kliinisiin tutkimuksiin. Tällaiset potilaat voivat olla alttiimpia yliherkkyysreaktioille, minkä vuoksi heille ei saa antaa pomalidomidia. Jos potilaalle ilmaantuu asteen 2–3 ihottumaa, pomalidomidihoidon keskeyttämistä tai lopettamista pitää harkita. Jos potilaalle ilmaantuu angioedeema tai anafylaktinen reaktio, pomalidomidihoito on lopetettava pysyvästi.
Huimaus ja sekavuus
Pomalidomidin yhteydessä on raportoitu huimausta ja sekavuutta. Potilaiden on vältettävä tilanteita, joissa huimauksesta tai sekavuudesta voi aiheutua vaaraa, eivätkä he saa käyttää muita lääkevalmisteita, joista saattaa aiheutua huimausta tai sekavuutta, elleivät he ole ensin kysyneet lääkäriltä neuvoa.
Interstitiaalinen keuhkosairaus
Pomalidomidihoidon yhteydessä on todettu interstitiaalista keuhkosairautta ja siihen liittyviä tapahtumia, mukaan lukien keuhkotulehdustapauksia. Jos potilaalla on akuutisti alkaneita tai selittämättömästi pahentuneita keuhko-oireita, hänet on tutkittava huolellisesti, jotta interstitiaalinen keuhkosairaus voidaan sulkea pois. Pomalidomidihoito on keskeytettävä oireiden tutkimisen ajaksi, ja jos interstitiaalinen keuhkosairaus varmistuu, sen asianmukainen hoito on aloitettava. Pomalidomidihoidon saa aloittaa uudelleen vasta hyötyjen ja riskien huolellisen arvioinnin jälkeen.
Maksan toiminnan häiriöt
Pomalidomidihoitoa saaneilla potilailla on todettu alaniiniaminotransferaasi- ja bilirubiiniarvojen huomattavaa kohoamista (ks. kohta Haittavaikutukset). Myös pomalidomidihoidon lopettamiseen johtaneita maksatulehdustapauksia on esiintynyt. Maksan toiminnan säännöllinen tarkkailu on suositeltavaa ensimmäisten 6 hoitokuukauden aikana sekä tämän jälkeen kliinisen tarpeen mukaan.
Infektiot
Pomalidomidia yhdistelmänä deksametasonin kanssa saaneilla potilailla, joilla on aiemmin ollut hepatiitti B -virus (HBV) -infektio, on raportoitu harvinaisina tapauksina B-hepatiitin aktivoitumista uudelleen. Osa näistä tapauksista on edennyt akuutiksi maksan vajaatoiminnaksi ja johtanut pomalidomidihoidon lopettamiseen. Potilaan HBV-status pitää selvittää ennen pomalidomidihoidon aloittamista. Jos potilaan HBV-testitulos on positiivinen, on suositeltavaa konsultoida B-hepatiitin hoitoon perehtynyttä lääkäriä. Kun pomalidomidia annetaan yhdistelmänä deksametasonin kanssa potilaille, joilla on aiemmin ollut HBV-infektio, mukaan lukien potilaat, jotka ovat anti-HBc-positiivisia, mutta HBsAg-negatiivisia, potilaan hoidossa on oltava varovainen. Näitä potilaita pitää tarkkailla huolellisesti koko hoidon ajan aktiivisen HBV-infektion oireiden ja löydösten havaitsemiseksi.
Progressiivinen multifokaalinen leukoenkefalopatia (PML)
Pomalidomidin käytön yhteydessä on ilmoitettu progressiivisista multifokaalisista leukoenkefalopatiatapauksista (PML), mukaan lukien kuolemantapaukset. Progressiivisesta multifokaalisesta leukoenkefalopatiasta on raportoitu useita kuukausia tai vuosia pomalidomidihoidon aloittamisen jälkeen. Tapauksia on yleensä raportoitu potilailla, jotka ovat käyttäneet samanaikaisesti deksametasonia tai, jotka ovat saaneet aiemmin jotakin muuta immunosuppressiivista solunsalpaajahoitoa. Lääkäreiden on seurattava potilaita säännöllisesti ja otettava PML huomioon tehdessään erotusdiagnoosia potilaille, joilla on uusia tai pahenevia neurologisia oireita tai kognitiivisia tai käyttäytymiseen liittyviä merkkejä tai oireita. Lisäksi potilaita on neuvottava kertomaan hoidostaan kumppanilleen tai hoitajilleen, sillä he saattavat huomata oireita, joista potilas ei itse ole tietoinen.
Progressiivisen multifokaalisen leukoenkefalopatian määrittämisen tulee perustua neurologiseen tutkimukseen, aivojen magneettikuvaukseen sekä JC-viruksen DNA:n analyysiin aivo-selkäydinnesteestä polymeraasiketjureaktiomenetelmällä (PCR:llä) tai aivobiopsiaan JCV:n testaamiseksi. Negatiivinen tulos PCR-tutkimuksesta JC-viruksen osalta ei sulje PML:ää pois. Lisäseuranta ja -arviointi voivat olla tarpeen, jos vaihtoehtoista diagnoosia ei voida vahvistaa.
Jos progressiivista multifokaalista leukoenkefalopatiaa epäillään, hoito on keskeytettävä siihen saakka, kunnes PML on suljettu pois. Jos progressiivinen multifokaalinen leukoenkefalopatia diagnosoidaan, pomalidomidihoito on lopetettava pysyvästi.
Natriumsisältö
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per kapseli eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Pomalidomidin vaikutus muihin lääkevalmisteisiin
Pomalidomidi ei oletettavasti aiheuta kliinisesti merkittäviä farmakokineettisiä yhteisvaikutuksia P450-isoentsyymin eston tai induktion tai kuljettajien eston seurauksena, jos sitä annetaan yhdessä näiden entsyymien tai kuljettajien substraattien kanssa. Tällaisten yhteisvaikutusten mahdollisuutta, mukaan lukien pomalidomidin mahdollista vaikutusta yhdistelmäehkäisytablettien farmakokinetiikkaan, ei ole arvioitu kliinisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet; Teratogeenisuus).
Muiden lääkevalmisteiden vaikutus pomalidomidiin
Pomalidomidi metaboloituu osittain CYP1A2:n ja CYP3A4/5:n välityksellä. Se on myös P-glykoproteiinin substraatti. Pomalidomidin anto yhdessä voimakkaan CYP3A4/5:n ja P-gp:n estäjän ketokonatsolin tai voimakkaan CYP3A4/5:n indusoijan karbamatsepiinin kanssa ei vaikuttanut kliinisesti merkityksellisesti pomalidomidialtistukseen. Pomalidomidin anto yhdessä voimakkaan CYP1A2:n estäjän fluvoksamiinin kanssa ketokonatsolin käytön aikana suurensi keskimääräistä altistusta pomalidomidille 107 % (90 %:n luottamusväli [91–124 %]) verrattuna pelkkään pomalidomidin ja ketokonatsolin yhdistelmäkäyttöön. Toisessa tutkimuksessa, jossa arvioitiin pelkän CYP1A2:n estäjän osuutta metabolian muutoksiin, pelkän fluvoksamiinin anto yhdessä pomalidomidin kanssa lisäsi keskimääräistä pomalidomidialtistusta 125 % (90 %:n luottamusväli [98–157 %]) verrattuna pelkkään pomalidomidiin. Jos voimakkaita CYP1A2:n estäjiä (kuten siprofloksasiinia, enoksasiinia ja fluvoksamiinia) annetaan samanaikaisesti pomalidomidin kanssa, pomalidomidiannosta pitää pienentää 50 %.
Deksametasoni
Useiden pomalidomidiannosten antaminen multippelia myeloomaa sairastaville potilaille 4 mg:n annoksiin saakka yhdessä 20–40 mg:n deksametasoniannoksen (heikko tai kohtalaisen voimakas usean CYP-entsyymin mukaan lukien CYP3A:n indusoija) kanssa ei vaikuttanut pomalidomidin farmakokinetiikkaan verrattuna pomalidomidin antamiseen yksinään.
Deksametasonin vaikutusta varfariiniin ei tunneta. Varfariinipitoisuutta suositellaan seuraamaan huolellisesti hoidon aikana.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset / ehkäisy miehille ja naisille
Hedelmällisessä iässä olevien naisten on käytettävä tehokasta ehkäisymenetelmää. Jos pomalidomidihoitoa saava nainen tulee raskaaksi, hoito on lopetettava ja potilas lähetettävä teratologiaan erikoistuneen tai siitä kokemusta saaneen lääkärin vastaanotolle tutkimuksia ja neuvontaa varten. Jos pomalidomidia saavan miespotilaan naispuolinen kumppani tulee raskaaksi, on suositeltavaa lähettää tämä teratologiaan erikoistuneen tai siitä kokemusta saaneen lääkärin vastaanotolle tutkimuksia ja neuvontaa varten. Pomalidomidia on hoidon aikana ihmisen siemennesteessä. Kaikkien pomalidomidia käyttävien miespotilaiden on varotoimena käytettävä kondomia koko hoidon ajan mukaan lukien hoitotaukojen aikana sekä 7 vuorokauden ajan hoidon päättymisen jälkeen, mikäli heidän kumppaninsa on raskaana tai voi tulla raskaaksi eikä käytä ehkäisyä (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Pomalidomidilla on oletettavasti ihmiselle teratogeeninen vaikutus. Pomalidomidi on vasta-aiheista raskauden aikana sekä naisille, jotka voivat tulla raskaaksi, paitsi jos kaikki raskaudenehkäisyä koskevat ehdot täyttyvät (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Imetys
Ei tiedetä, erittyykö pomalidomidi ihmisen rintamaitoon. Pomalidomidia havaittiin imettävien rottien maidossa, kun valmistetta oli annettu emolle. Pomalidomidin rintaruokittaviin lapsiin kohdistuvien haittavaikutusten mahdollisuuden vuoksi on päätettävä lopetetaanko rintaruokinta vai lopetetaanko lääkehoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Pomalidomidin havaittiin vaikuttavan haitallisesti hedelmällisyyteen ja olevan eläimillä teratogeeninen. Pomalidomidi läpäisi istukan ja sitä havaittiin sikiön veressä, kun valmistetta annettiin tiineille kaniineille (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Pomalidomidilla on vähäinen tai kohtalainen vaikutus ajokykyyn ja koneiden käyttökykyyn. Pomalidomidin käytön yhteydessä on raportoitu väsymystä, tajunnan tason alenemista, sekavuutta ja heitehuimausta. Jos tällaisia vaikutuksia ilmenee pomalidomidihoidon aikana, potilaita on kehotettava olemaan ajamatta autoa, käyttämättä koneita tai suorittamatta vaaraa aiheuttavia tehtäviä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Pomalidomidi bortetsomibiin ja deksametasoniin yhdistettynä
Yleisimmin raportoidut veri- ja imukudos -elinjärjestelmäluokkaan kuuluvat haittavaikutukset olivat neutropenia (54,0 %), trombosytopenia (39,9 %) ja anemia (32,0 %). Muita yleisimmin raportoituja haittavaikutuksia olivat muun muassa perifeerinen sensorinen neuropatia (48,2 %), väsymys (38,8 %), ripuli (38,1 %), ummetus (38,1 %) ja raajojen turvotus (36,3 %). Yleisimmin raportoidut 3. tai 4. asteen haittavaikutukset kuuluivat veri- ja imukudos -elinjärjestelmäluokkaan ja ne olivat neutropenia (47,1 %), trombosytopenia (28,1 %) ja anemia (15,1 %). Yleisimmin raportoitu vakava haittavaikutus oli keuhkokuume (12,2 %). Muita raportoituja vakavia haittavaikutuksia olivat kuume (4,3 %), alahengitystieinfektio (3,6 %), influenssa (3,6 %), keuhkoembolia (3,2 %), eteisvärinä (3,2 %) ja akuutti munuaisvaurio (2,9 %).
Pomalidomidi deksametasoniin yhdistettynä
Kliinisissä tutkimuksissa yleisimmin raportoidut haittavaikutukset ovat kuuluneet elinjärjestelmäluokkiin veri- ja imukudos: anemia (45,7 %), neutropenia (45,3 %) ja trombosytopenia (27 %); yleisoireet ja antopaikassa todettavat haitat: väsymys (28,3 %), kuume (21 %) ja raajojen turvotus (13 %); ja infektiot: keuhkokuume (10,7 %). Perifeeristä neuropatiaa raportoitiin 12,3 %:lla potilaista ja laskimoiden tromboembolisia tapahtumia (VTE) 3,3 %:lla potilaista. Yleisimmin raportoidut 3. tai 4. asteen haittavaikutukset ovat kuuluneet elinjärjestelmäluokkiin veri ja imukudos: neutropenia (41,7 %), anemia (27 %) ja trombosytopenia (20,7 %): infektiot: keuhkokuume (9 %): ja yleisoireet ja antopaikassa todettavat haitat: väsymys (4,7 %), kuume (3 %) ja raajojen turvotus (1,3 %). Yleisimmin raportoitu vakava haittavaikutus oli keuhkokuume (9,3 %). Muita raportoituja vakavia haittavaikutuksia olivat kuumeinen neutropenia (4,0 %), neutropenia (2,0 %), trombosytopenia (1,7 %) ja laskimoiden tromboemboliset tapahtumat (1,7 %).
Haittavaikutuksia esiintyi tyypillisesti useammin kahden ensimmäisen pomalidomidihoitosyklin aikana verrattuna myöhempiin sykleihin.
Haittavaikutusten yhteenvetotaulukko
Kaikki haittavaikutukset ja 3. tai 4. asteen haittavaikutukset, joita on todettu pomalidomidin, bortetsomibin ja deksametasonin yhdistelmällä ja pomalidomidin ja deksametasonin yhdistelmällä hoidetuilla potilailla sekä markkinoille tulon jälkeisessä valvonnassa, on lueteltu jäljempänä taulukossa 7 elinjärjestelmän ja yleisyyden mukaan.
Yleisyydet on määritetty voimassa olevien ohjeistojen mukaan seuraavasti: hyvin yleinen (≥1/10), yleinen (≥1/100, <1/10), melko harvinainen (≥1/1 000, <1/100) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 7. Kliinisissä tutkimuksissa ja markkinoille tulon jälkeen raportoidut haittavaikutukset
Yhdistelmähoito | Pomalidomidi/ bortetsomibi/deksametasoni | Pomalidomidi/ deksametasoni | ||
Elinjärjestelmä /Suositeltava MedDRA-termi
| Kaikki haitta-vaikutukset
| 3.−4. asteen haitta-vaikutukset | Kaikki haitta-vaikutukset | 3.−4. asteen haitta-vaikutukset |
Infektiot | ||||
Keuhkokuume | Hyvin yleinen | Hyvin yleinen | - | - |
Keuhkokuume (bakteeri-, virus- ja sieni-infektiot, mukaan lukien opportunistiset infektiot) | - | - | Hyvin yleinen | Yleinen |
Keuhkoputkitulehdus | Hyvin yleinen | Yleinen | Yleinen | Melko harvinainen |
Ylähengitystieinfektio | Hyvin yleinen | Yleinen | Yleinen | Yleinen |
Viruksen aiheuttama ylähengitystieinfektio | Hyvin yleinen | - | - | - |
Sepsis | Yleinen | Yleinen | - | - |
Septinen sokki | Yleinen | Yleinen | - | - |
Neutropeeninen sepsis | - | - | Yleinen | Yleinen |
Clostridium difficile ‑koliitti | Yleinen | Yleinen | - | - |
Pesäkekeuhkokuume | - | - | Yleinen | Yleinen |
Hengitystieinfektio | Yleinen | Yleinen | Yleinen | Yleinen |
Alahengitystieinfektio | Yleinen | Yleinen | - | - |
Keuhkoinfektio | Yleinen | Melko harvinainen | - | - |
Influenssa | Hyvin yleinen | Yleinen | - | - |
Bronkioliitti | Yleinen | Yleinen | - | - |
Virtsatieinfektio | Hyvin yleinen | Yleinen | - | - |
Nenänielun tulehdus | - | - | Yleinen | - |
Vyöruusu | - | - | Yleinen | Melko harvinainen |
B-hepatiitin aktivoituminen uudelleen | - | - | Tuntematon* | Tuntematon* |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | ||||
Tyvisolusyöpä | Yleinen | Melko harvinainen | - | - |
Ihon tyvisolusyöpä | - | - | Melko harvinainen | Melko harvinainen |
Ihon okasolusyöpä | - | - | Melko harvinainen | Melko harvinainen |
Veri ja imukudos | ||||
Neutropenia | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
Trombosytopenia | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
Leukopenia | Hyvin yleinen | Yleinen | Hyvin yleinen | Yleinen |
Anemia | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
Kuumeinen neutropenia | Yleinen | Yleinen | Yleinen | Yleinen |
Lymfopenia | Yleinen | Yleinen | - | - |
Pansytopenia | - | - | Yleinen* | Yleinen* |
Immuunijärjestelmä | ||||
Angioedeema | - | - | Yleinen* | Melko harvinainen* |
Urtikaria | - | - | Yleinen* | Melko harvinainen* |
Anafylaktinen reaktio | Tuntematon* | Tuntematon* | - | - |
Kiinteän elinsiirteen hyljintä | Tuntematon* | - | - | - |
Umpieritys | ||||
Hypotyreoosi | Melko harvinainen* | - | - | -- |
Aineenvaihdunta ja ravitsemus | ||||
Hypokalemia | Hyvin yleinen | Yleinen | - | - |
Hyperglykemia | Hyvin yleinen | Yleinen | - | - |
Hypomagnesemia | Yleinen | Yleinen | - | - |
Hypokalsemia | Yleinen | Yleinen | - | - |
Hypofosfatemia | Yleinen | Yleinen | - | - |
Hyperkalemia | Yleinen | Yleinen | Yleinen | Yleinen |
Hyperkalsemia | Yleinen | Yleinen | - | - |
Hyponatremia | - | - | Yleinen | Yleinen |
Vähentynyt ruokahalu | - | - | Hyvin yleinen | Melko harvinainen |
Hyperurikemia | - | - | Yleinen* | Yleinen* |
Tuumorilyysioireyhtymä | - | - | Melko harvinainen* | Melko harvinainen* |
Psyykkiset häiriöt | ||||
Unettomuus | Hyvin yleinen | Yleinen | - | - |
Masennus | Yleinen | Yleinen | - | - |
Sekavuustila | - | - | Yleinen | Yleinen |
Hermosto | ||||
Perifeerinen sensorinen neuropatia | Hyvin yleinen | Yleinen | Yleinen | Melko harvinainen |
Heitehuimaus | Hyvin yleinen | Melko harvinainen | Yleinen | Melko harvinainen |
Vapina | Hyvin yleinen | Melko harvinainen | Yleinen | Melko harvinainen |
Pyörtyminen | Yleinen | Yleinen | - | - |
Perifeerinen sensomotorinen neuropatia | Yleinen | Yleinen | - | - |
Parestesia | Yleinen | - | - | - |
Makuhäiriö | Yleinen | - | - | - |
Alentunut tajunnan taso | - | - | Yleinen | Yleinen |
Kallonsisäinen verenvuoto | - | - | Yleinen* | Melko harvinainen* |
Aivoverisuonitapahtuma | - | - | Melko harvinainen* | Melko harvinainen* |
Silmät | ||||
Kaihi | Yleinen | Yleinen | - | - |
Kuulo ja tasapainoelin | ||||
Kiertohuimaus | - | - | Yleinen | Yleinen |
Sydän | ||||
Eteisvärinä | Hyvin yleinen | Yleinen | Yleinen* | Yleinen* |
Sydämen vajaatoiminta | - | - | Yleinen* | Yleinen* |
Sydäninfarkti | - | - | Yleinen* | Melko harvinainen* |
Verisuonisto | ||||
Syvä laskimotromboosi | Yleinen | Melko harvinainen | Yleinen | Melko harvinainen |
Hypotensio | Yleinen | Yleinen | - | - |
Hypertensio | Yleinen | Yleinen | - | - |
Hengityselimet, rintakehä ja välikarsina | ||||
Hengenahdistus | Hyvin yleinen | Yleinen | Hyvin yleinen | Yleinen |
Yskä | Hyvin yleinen | - | Hyvin yleinen | Melko harvinainen |
Keuhkoembolia | Yleinen | Yleinen | Yleinen | Melko harvinainen |
Nenäverenvuoto | - | - | Yleinen* | Melko harvinainen* |
Interstitiaalinen keuhkosairaus | - | - | Yleinen* | Melko harvinainen* |
Ruoansulatuselimistö | ||||
Ripuli | Hyvin yleinen | Yleinen | Hyvin yleinen | Yleinen |
Oksentelu | Hyvin yleinen | Yleinen | Yleinen | Yleinen |
Pahoinvointi | Hyvin yleinen | Melko harvinainen | Hyvin yleinen | Melko harvinainen |
Ummetus | Hyvin yleinen | Yleinen | Hyvin yleinen | Yleinen |
Vatsakipu | Hyvin yleinen | Yleinen | - | - |
Ylävatsakipu | Yleinen | Melko harvinainen | - | - |
Suutulehdus | Yleinen | Melko harvinainen | - | - |
Suun kuivuminen | Yleinen | - | - | - |
Vatsan turvotus | Yleinen | Melko harvinainen | - | - |
Maha-suolikanavan verenvuoto | - | - | Yleinen | Melko harvinainen |
Maksa ja sappi | ||||
Hyperbilirubinemia | - | - | Melko harvinainen | Melko harvinainen |
Maksatulehdus | - | - | Melko harvinainen* | - |
Iho ja ihonalainen kudos | ||||
Ihottuma | Hyvin yleinen | Yleinen | Yleinen | Yleinen |
Kutina | - | - | Yleinen | - |
Yleisoireinen eosinofiilinen oireyhtymä | - | - | Tuntematon* | Tuntematon* |
Toksinen epidermaalinen nekrolyysi | - | - | Tuntematon* | Tuntematon* |
Stevens–Johnsonin oireyhtymä | - | - | Tuntematon* | Tuntematon* |
Luusto, lihakset ja sidekudos | ||||
Lihasheikkous | Hyvin yleinen | Yleinen | - | - |
Selkäkipu | Hyvin yleinen | Yleinen | - | - |
Luukipu | Yleinen | Melko harvinainen | Hyvin yleinen | Yleinen |
Lihaskrampit | Hyvin yleinen | - | Hyvin yleinen | Melko harvinainen |
Munuaiset ja virtsatiet | ||||
Akuutti munuaisvaurio | Yleinen | Yleinen | - | - |
Krooninen munuaisvaurio | Yleinen | Yleinen | - | - |
Virtsaumpi | Yleinen | Yleinen | Yleinen | Melko harvinainen |
Munuaisten vajaatoiminta | - | - | Yleinen | Yleinen |
Sukupuolielimet ja rinnat | ||||
Lantiokipu |
|
| Yleinen | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | ||||
Väsymys | Hyvin yleinen | Yleinen | Hyvin yleinen | Yleinen |
Kuume | Hyvin yleinen | Yleinen | Hyvin yleinen | Yleinen |
Raajojen turvotus | Hyvin yleinen | Yleinen | Hyvin yleinen | Yleinen |
Sydämeen liittymätön rintakipu | Yleinen | Yleinen | - | - |
Turvotus | Yleinen | Yleinen | - | - |
Tutkimukset | ||||
Suurentunut alaniiniaminotransferaasi-pitoisuus | Yleinen | Yleinen | Yleinen | Yleinen |
Painon lasku | Yleinen | Yleinen | - | - |
Vähentynyt neutrofiilimäärä | - | - | Yleinen | Yleinen |
Vähentynyt veren valkosolumäärä | - | - | Yleinen | Yleinen |
Vähentynyt trombosyyttimäärä | - | - | Yleinen | Yleinen |
Suurentunut veren virtsahappopitoisuus | - | - | Yleinen* | Melko harvinainen* |
Vammat, myrkytykset ja hoitokomplikaatiot | ||||
Kaatuminen | Yleinen | Yleinen | - | - |
* Raportoitu markkinoille tulon jälkeisessä käytössä.
Valittujen haittavaikutusten kuvaus
Tässä kohdassa kuvatut esiintyvyydet on saatu kliinisistä tutkimuksista potilailla, jotka saivat pomalidomidia yhdistelmähoitona bortetsomibin ja deksametasonin kanssa (Pom + Bor + Deks) tai pomalidomidia yhdistelmähoitona deksametasonin kanssa (Pom + Deks).
Teratogeenisuus
Pomalidomidi muistuttaa rakenteeltaan talidomidia. Talidomidi on ihmiselle tunnetusti teratogeeninen vaikuttava aine, joka aiheuttaa vaikea-asteisia hengenvaarallisia syntymävaurioita. Pomalidomidin todettiin olevan teratogeeninen sekä rotille että kaniineille, kun sitä annettiin tärkeimmän organogeneesivaiheen aikana (ks. kohdat Raskaus ja imetys ja Prekliiniset tiedot turvallisuudesta). Jos pomalidomidia käytetään raskauden aikana, pomalidomidilla oletettavasti on teratogeeninen vaikutus ihmiseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Neutropenia ja trombosytopenia
Neutropeniaa esiintyi enintään 54,0 %:lla potilaista (Pom + Bor + Deks), ja 3. tai 4. asteen neutropeniaa esiintyi 47,1 %:lla potilaista (Pom + Bor + Deks). Neutropenia oli harvoin vakavaa, ja se johti 0,7 %:lla potilaista hoidon lopettamiseen.
Kuumeista neutropeniaa raportoitiin 3,2 %:lla potilaista (Pom + Bor + Deks) ja 6,7 %:lla potilaista (Pom + Deks), ja se oli vakavaa 1,8 %:lla potilaista (Pom + Bor + Deks) ja 4,0 %:lla potilaista (Pom + Deks) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Trombosytopeniaa esiintyi 39,9 %:lla potilaista (Pom + Bor + Deks) ja 27,0 %:lla potilaista (Pom + Deks). Trombosytopenia raportoitiin 3. tai 4. asteen haittavaikutuksena 28,1 %:lla potilaista (Pom + Bor + Deks) ja 20,7 %:lla potilaista (Pom + Deks), se johti pomalidomidihoidon lopettamiseen 0,7 %:lla potilaista (Pom + Bor + Deks) ja 0,7 %:lla potilaista (Pom + Deks), ja se oli vakavaa 0,7 %:lla potilaista (Pom + Bor + Deks) ja 1,7 %:lla potilaista (Pom + Deks) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Neutropeniaa ja trombosytopeniaa esiintyi tyypillisesti useammin kahden ensimmäisen pomalidomidihoitosyklin aikana verrattuna myöhempiin sykleihin käytettäessä pomalidomidia yhdistelmähoitona bortetsomibin ja deksametasonin tai pelkän deksametasonin kanssa.
Infektiot
Infektio oli yleisin ei-hematologinen toksisuus.
Infektioita esiintyi 83,1 %:lla potilaista (Pom + Bor + Deks) ja 55,0%:lla potilaista (Pom + Deks) (näistä 3. tai 4. asteen haittavaikutuksina raportoitiin 34,9 % Pom + Bor + Deks ‑ryhmässä ja 24,0 % Pom + Deks ‑ryhmässä). Keuhkokuume ja ylähengitystieinfektiot olivat yleisimmin ilmenneet infektiot. Kuolemaan johtaneita infektioita (5. aste) ilmeni 4,0 %:lla potilaista (Pom + Bor +Deks) ja 2,7 %:lla potilaista (Pom + Deks). Infektiot johtivat pomalidomidihoidon lopettamiseen 3,6 %:lla potilaista Pom + Bor + Deks ‑ryhmässä ja 2,0 %:lla potilaista Pom + Deks ‑ryhmässä.
Tromboemboliset tapahtumat
Estohoito asetyylisalisyylihapolla (ja suuren riskin potilailla muilla antikoagulanteilla) oli kliinisissä tutkimuksissa pakollista kaikille potilaille. Antikoagulanttihoitoa (ellei se ole vasta-aiheista) suositellaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Laskimoiden tromboembolisia tapahtumia (VTE) esiintyi 12,2 %:lla potilaista (Pom + Bor + Deks) ja 3,3 %:lla potilaista (Pom + Deks) (3. tai 4. asteen tapahtumia esiintyi 5,8 %:lla Pom + Bor + Deks ‑hoitoa saaneista potilaista ja 1,3 %:lla Pom + Dex -hoitoa saaneista potilaista). Vakava laskimon tromboembolinen tapahtuma raportoitiin 4,7 %:lla potilaista (Pom + Bor + Deks) ja 1,7 %:lla potilaista (Pom + Deks). Kuolemaan johtaneita tapahtumia ei raportoitu, ja laskimon tromboemboliset tapahtumat johtivat pomalidomidihoidon lopettamiseen enintään 2,2 %:lla potilaista (Pom + Bor + Deks).
Perifeerinen neuropatia – pomalidomidi bortetsomibiin ja deksametasoniin yhdistettynä
Potilaita, joilla oli jatkuvaa ≥ 2. asteen perifeeristä neuropatiaa, johon liittyi kipua, satunnaistamista edeltävien 14 vuorokauden aikana, ei otettu mukaan kliinisiin tutkimuksiin. Perifeeristä neuropatiaa esiintyi 55,4 %:lla potilaista (10,8 %:lla 3. asteen ja 0,7 %:lla 4. asteen). Altistuksen mukaan korjatut esiintyvyydet olivat verrannollisia hoitoryhmien kesken. Noin 30 %:lla potilaista, joilla esiintyi perifeeristä neuropatiaa, oli lähtötilanteen tietojen mukaan aiemmin esiintynyt neuropatiaa. Perifeerinen neuropatia johti bortetsomibihoidon lopettamiseen noin 14,4 %:lla potilaista, pomalidomidihoidon lopettamiseen 1,8 %:lla potilaista ja deksametasonihoidon lopettamiseen 1,8 %:lla Pom + Bor + Deks ‑ryhmän potilaista ja 8,9 %:lla Bor + Deks ‑ryhmän potilaista potilaista.
Perifeerinen neuropatia – pomalidomidi deksametasoniin yhdistettynä
Potilaita, joilla oli jatkuvaa ≥ 2. asteen perifeeristä neuropatiaa, ei otettu mukaan kliinisiin tutkimuksiin. Perifeeristä neuropatiaa esiintyi 12,3 %:lla potilaista (1,0 %:lla 3. tai 4. asteen). Vakavia perifeerisen neuropatian tapauksia ei raportoitu, ja perifeerinen neuropatia johti hoidon lopettamiseen 0,3 %:lla potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Verenvuoto
Pomalidomidin käytön yhteydessä on raportoitu verenvuotohäiriöitä, etenkin potilailla, joilla on riskitekijöitä, kuten samanaikainen verenvuotoriskiä lisäävien lääkevalmisteiden käyttö. Verenvuototapahtumia ovat olleet mm. nenäverenvuoto, kallonsisäinen verenvuoto ja maha-suolikanavan verenvuoto.
Allergiset reaktiot ja vaikeat ihoreaktiot
Angioedeemaa, anafylaktisia reaktioita ja vaikeita ihoreaktioita, mukaan lukien Stevens-Johnsonin oireyhtymää, toksista epidermaalista nekrolyysiä ja yleisoireista eosinofiilista oireyhtymää, on raportoitu esiintyneen pomalidomidin käytön yhteydessä. Potilaille, joilla on aikaisemmin esiintynyt vaikeaa ihottumaa lenalidomidi- tai talidomidihoidon yhteydessä, ei pidä antaa pomalidomidia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Ilmoitetut haittavaikutukset pediatrisilla potilailla (4–18-vuotiaat), joilla oli uusiutuneita tai eteneviä aivokasvaimia, vastasivat pomalidomidin tunnettua turvallisuusprofiilia aikuispotilailla (ks. kohta Farmakodynamiikka).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Terveillä vapaaehtoisilla koehenkilöillä on tutkittu enimmillään 50 mg:n pomalidomidin kerta-annoksia eikä yliannokseen liittyviä vakavia haittavaikutuksia ole raportoitu. Multippelia myeloomaa sairastavilla potilailla on tutkittu toistettuja enimmillään 10 mg:n vuorokausiannoksia kerran päivässä eikä yliannokseen liittyviä vakavia haittavaikutuksia ole raportoitu. Annoskokoa rajoittava toksisuus oli myelosuppressio. Tutkimuksissa pomalidomidin havaittiin poistuvan hemodialyysillä.
Yliannostustapauksessa suositellaan elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, muut immunosuppressantit, ATC-koodi: L04AX06
Vaikutusmekanismi
Pomalidomidilla on suora myeloomakasvaimia tappava vaikutus sekä immunomodulatorista aktiivisuutta, ja se estää multippelin myelooman kasvainsolujen kasvua tukevia stroomasoluja. Pomalidomidi estää erityisesti hematopoieettisten kasvainsolujen proliferaatiota ja indusoi niiden apoptoosia. Pomalidomidi lisäksi estää lenalidomidille resistenttien multippelin myelooman solulinjojen proliferaatiota ja indusoi kasvainsolujen apoptoosia synergistisesti deksametasonin kanssa sekä lenalidomidille herkissä että lenalidomidille resistenteissä solulinjoissa. Pomalidomidi tehostaa T-solun ja luonnollisen tappajasolun (NK-solu) soluvälitteistä immuniteettia ja estää monosyyttejä tuottamasta tulehdusta edistäviä sytokiineja (esim. TNF-α ja IL-6). Pomalidomidi estää myös angiogeneesin salpaamalla endoteelisolujen migraation ja adheesion.
Pomalidomidi sitoutuu suoraan Cereblon-proteiiniin (CRBN), joka on osa deoksiribonukleiinihappoa (DNA), DNA-vaurioita sitovaa proteiinia 1 (DDB1), Cullin 4 -proteiinia (CUL4) ja Cullin 1 ‑proteiinin säätelijää (Roc1) sisältävää E3-ligaasikompleksia, ja se kykenee estämään CRBN:n itseubikitinaatiota tässä kompleksissa. E3-ubikitiiniligaasit vastaavat useiden eri substraattiproteiinien polyubikitinaatiosta, ja ne saattavat osittain selittää pomalidomidihoidon yhteydessä havaitut pleiotrooppiset soluvaikutukset.
Pomalidomidin läsnä ollessa in vitro substraattiproteiinit Aiolos ja Ikaros ubikitinoituvat ja myöhemmin hajoavat, mikä johtaa suoriin sytotoksisiin ja immunomodulatorisiin vaikutuksiin. Pomalidomidihoito in vivo pienensi Ikaros-proteiinin pitoisuutta relapsoitunutta ja lenalidomidille refraktorista multippelia myeloomaa sairastavilla potilailla.
Kliininen teho ja turvallisuus
Pomalidomidi bortetsomibiin ja deksametasoniin yhdistettynä
Pomalidomidin tehoa ja turvallisuutta yhdistelmänä bortetsomibin ja pieniannoksisen deksametasonin kanssa (Pom + Bor + PA deks) verrattiin bortetsomibiin ja pieniannoksiseen deksametasoniin (Bor + PA deks) vaiheen III satunnaistetussa, avoimessa monikeskustutkimuksessa (CC-4047-MM-007) multippelia myeloomaa sairastavilla aikuispotilailla, jotka olivat saaneet vähintään yhtä aiempaa hoitoa ja joiden sairaus oli edennyt viimeisimmän hoidon aikana tai sen jälkeen. Tutkimukseen otettiin mukaan yhteensä 559 potilasta, joista 281 satunnaistettiin Pom + Bor + PA deks -ryhmään ja 278 Bor + PA deks -ryhmään. Potilaista 54 % oli miehiä, ja kaikkien osallistujien iän mediaani oli 68 vuotta (min. 27 vuotta, maks. 89 vuotta). Noin 70 % potilaista oli refraktorisia lenalidomidille (71,2 % Pom + Bor + PA deks ‑ryhmässä, 68,7 % Bor + PA deks -ryhmässä). Noin 40 %:lla potilaista tauti oli uusiutunut ensimmäistä kertaa, ja noin 73 % oli saanut bortetsomibia aiempana hoitona.
Pom + Bor + PA deks ‑ryhmän potilaille annettiin 4 mg pomalidomidia suun kautta kunkin 21 vuorokauden pituisen syklin vuorokausina 1–14. Bortetsomibia (1,3 mg/m2/annos) annettiin kummankin hoitoryhmän potilaille kunkin 21 vuorokauden pituisen syklin vuorokausina 1, 4, 8 ja 11 hoitosykleinä 1–8 sekä kunkin 21 vuorokauden pituisen syklin vuorokausina 1 ja 8 hoitosyklinä 9 ja sen jälkeisinä hoitosykleinä. Pieniannoksista deksametasonia (20 mg/vrk [≤ 75-vuotiaille] tai 10 mg/vrk [> 75-vuotiaille]) annettiin kummankin hoitoryhmän potilaille kunkin 21 vuorokauden pituisen syklin vuorokausina 1, 2, 4, 5, 8, 9, 11 ja 12 hoitosykleinä 1–8 sekä kunkin 21 vuorokauden pituisen syklin vuorokausina 1, 2, 8 ja 9 hoitosyklinä 9 ja sen jälkeisinä hoitosykleinä. Annoksia pienennettiin ja hoito keskeytettiin väliaikaisesti tai lopetettiin tarpeen mukaan toksisuuden hallitsemiseksi (ks. kohta Annostus ja antotapa).
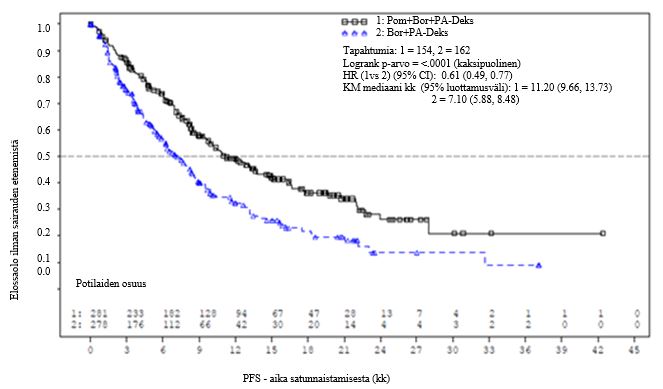
Tehon ensisijainen päätetapahtuma oli riippumattoman tarkkailukomitean (Independent Review Adjudication Committee, IRAC) arvioima, International Myeloma Working Groupin (IMWG) kriteerien mukainen elossaolo ilman taudin etenemistä (Progression Free Survival, PFS) hoitoaikeen mukaisessa (ITT-) potilasjoukossa. Mediaanikestoltaan 15,9 kuukauden pituisen seurannan jälkeen PFS-ajan mediaani oli Pom + Bor + PA deks ‑ryhmässä 11,20 kuukautta (95 %:n luottamusväli: 9,66, 13,73) ja Bor + PA deks ‑ryhmässä 7,1 kuukautta (95 %:n luottamusväli: 5,88, 8,48).
Yhteenveto tehoa koskevista kokonaistiedoista on esitetty taulukossa 8 (tietojen katkaisupiste: 26. lokakuuta 2017). ITT-potilasjoukon PFS-ajan Kaplan-Meier-käyrä on esitetty kuvassa 1.
Taulukko 8. Yhteenveto tehoa koskevista kokonaistiedoista
| Pom + Bor + PA deks | Bor + PA deks |
Elossaolo ilman taudin etenemistä (Progression Free Survival, PFS) (kuukautta) |
| |
Ajan mediaania (95 %:n luottamusväli)b | 11,20 (9,66, 13,73) | 7,10 (5,88, 8,48) |
Riskitiheyksien suhde (Hazard ratio, HR)c (95 %:n luottamusväli), p‑arvod | 0,61 (0,49, 0,77), < 0,0001 | |
Kokonaisvasteosuus, n (%) | 82,2 % | 50,0 % |
Täydellinen vaste lisäehdoin (Stringent complete response, sCR) | 9 (3,2) | 2 (0,7) |
Täydellinen vaste (Complete response, CR) | 35 (12,5) | 9 (3,2) |
Erittäin hyvä osittainen vaste (Very good partial response, VGPR) | 104 (37,0) | 40 (14,4) |
Osittainen vaste (Partial response, PR) | 83 (29,5) | 88 (31,7) |
Kerroinsuhde (Odds ratio, OR)e (95 %:n luottamusväli), p-arvof | 5,02 (3,35, 7,52), < 0,001 | |
Vasteen kesto (Duration of response, DoR) (kuukautta) |
| |
Ajan mediaania (95 %:n luottamusväli)b | 13,7 (10,94, 18,10) | 10,94 (8,11, 14,78) |
Riskitiheyksien suhde (Hazard ratio, HR)c (95 %:n luottamusväli) | 0,76 (0,56, 1,02) | |
Bor = bortetsomibi; PA deks = pieniannoksinen deksametasoni; Pom = pomalidomidi.
a Mediaani perustuu Kaplan-Meier-estimaattiin.
b 95 %:n luottamusväli mediaanille.
c Perustuu Coxin suhteellisten riskitiheyksien malliin.
d p‑arvo perustuu ositettuun log-rank-testiin.
e Kerroinsuhde Pom + Bor + PA deks : Bor + PA deks.
f p-arvo perustuu iän mukaan ositettuun (<= 75 vs. > 75) CMH-testiin, aiempien myeloomahoitojen määrään (1 vs. > 1) ja beeta-2-mikroglobuliinipitoisuuteen seulonnassa (< 3,5 mg/l vs. ≥ 3,5 mg/l, ≤ 5,5 mg/l vs. > 5,5 mg/l).
Hoidon keston mediaani oli 8,8 kuukautta (12 hoitosykliä) Pom + Bor + PA deks -ryhmässä ja 4,9 kuukautta (7 hoitosykliä) Bor + PA deks -ryhmässä.
PFS-hyöty oli suurempi potilailla, jotka olivat saaneet vain yhtä aiempaa hoitoa. PFS-ajan mediaani potilailla, jotka olivat saaneet yhtä aiempaa myeloomahoitoa, oli Pom + Bor + PA deks -ryhmässä 20,73 kuukautta (95 %:n luottamusväli: 15,11, 27,99) ja Bor + PA deks -ryhmässä 11,63 kuukautta (95 %:n luottamusväli: 7,52, 15,74). Pom + Bor + PA deks -hoidossa todettiin 46 %:n riskin vähenemä (riskitiheyksien suhde = 0,54, 95 %:n luottamusväli: 0,36, 0,82).
Kuva 1. IRAC-toimikunnan IMWG:n kriteerien perusteella arvioima elossaoloaika ilman sairauden etenemistä (ositettu log-rank-testi) (ITT-potilasjoukko)
Tietojen katkaisupiste: 26. lokakuuta 2017
Kokonaiselossaoloa (OS) koskevassa lopullisessa analyysissä, jonka tietojen katkaisupiste oli 13. toukokuuta 2022 (seuranta-ajan mediaani 64,5 kuukautta), Kaplan-Meierin estimaatteihin perustuva kokonaiselossaolon mediaani oli 35,6 kuukautta Pom + Bor + PA Deks -ryhmässä ja 31,6 kuukautta Bor + PA deks -ryhmässä (riskitiheyksien suhde = 0,94, 95 %:n luottamusväli: 0,77, 1,15, kokonaistapahtumaosuus 70,0 %). Kokonaiselossaoloa koskevassa analyysissa ei huomioitu tutkimuslääkkeen jälkeistä lääkehoitoa.
Pomalidomidi deksametasoniin yhdistettynä
Pomalidomidin tehoa ja turvallisuutta yhdistelmänä deksametasonin kanssa arvioitiin vaiheen III satunnaistetussa, avoimessa monikeskustutkimuksessa (CC-4047-MM-003), jossa pomalidomidia yhdistelmänä pieniannoksisen deksametasonihoidon (Pom + PA deks) kanssa verrattiin pelkkään suuriannoksiseen deksametasonihoitoon (SA deks) aiemmin hoitoa saaneilla relapsoitunutta ja refraktorista multippelia myeloomaa sairastavilla aikuisilla potilailla, jotka olivat saaneet vähintään kahta aiempaa hoitoa, mukaan lukien sekä lenalidomidia että bortetsomibia, ja joiden sairaus oli edennyt viimeisimmän hoidon aikana. Tutkimukseen otettiin mukaan yhteensä 455 potilasta: 302 Pom + PA deks -ryhmään ja 153 SA deks -ryhmään. Suurin osa potilaista oli miehiä (59 %) ja valkoihoisia (79 %). Kaikkien osallistujien iän mediaani oli 64 vuotta (min. 35 vuotta, maks. 87 vuotta).
Pom + PA deks -ryhmän potilaille annettiin 4 mg pomalidomidia suun kautta kunkin 28 vuorokauden pituisen syklin vuorokausina 1–21. PA deks (40 mg) annettiin kerran vuorokaudessa 28 vuorokauden pituisen syklin vuorokausina 1, 8, 15, ja 22. SA deks -ryhmän potilaille annettiin deksametasonia (40 mg) kerran vuorokaudessa 28 vuorokauden pituisen syklin vuorokausina 1–4, 9–12 ja 17–20. Yli 75-vuotiaat potilaat aloittivat hoidon 20 mg:lla deksametasonia. Hoito jatkui, kunnes potilaiden sairaus eteni.
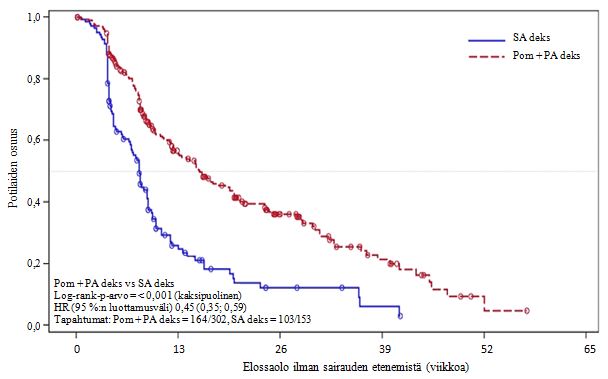
Tehon ensisijainen päätetapahtuma oli International Myeloma Working Groupin (IMWG:n) kriteerien mukainen elossaolo ilman taudin etenemistä (PFS, Progression Free Survival). Tutkimuksen riippumattoman tarkkailukomitean (Independent Review Adjudication Committee, IRAC) IMWG:n kriteerien perusteella tekemän arvion mukaan hoitoaikeen mukaisen (ITT) potilasjoukon PFS-ajan mediaani oli Pom + PA deks ‑ryhmässä 15,7 viikkoa (95 %:n luottamusväli: 13,0; 20,1). Arvioitu 26 viikon tapahtumaton elossaololuku oli 35,99 % (± 3,46). SA deks ‑ryhmässä PFS-ajan mediaani oli 8,0 viikkoa (95 %:n luottamusväli: 7,0; 9,0); arvioitu 26 viikon tapahtumaton elossaololuku oli 12,15 % (± 3,63 %).
PFS-aikaa arvioitiin useassa oleellisessa alaryhmässä: ikä, rotu, ECOG-suorituskyky, ositustekijät (ikä, sairastava potilasjoukko, aiemmat myeloomahoidot [2, > 2], valikoitujen parametrien merkityksellisyys ennusteen kannalta (lähtötilanteen beeta-2-mikroglobuliinipitoisuus, lähtötilanteen albumiinipitoisuudet, munuaisten vajaatoiminta lähtötilanteessa ja sytogeneettinen riski) sekä altistus ja huono reagoivuus aiempiin myeloomahoitoihin. PFS-aika oli arvioidusta alaryhmästä riippumatta useimmiten yhdenmukainen ITT-potilasjoukon PFS-ajan kanssa kummassakin hoitoryhmässä.
PFS-aika ITT-potilasjoukossa on esitetty yhteenvetona taulukossa 9. ITT-potilasjoukon PFS-ajan Kaplan-Meier-käyrä on esitetty kuvassa 2.
Taulukko 9: IRAC-toimikunnan IMWG:n kriteerien perusteella arvioima elossaoloaika ilman sairauden etenemistä (ositettu log-rank-testi) (ITT-potilasjoukko)
| Pom + PA deks (N = 302) | SA deks (N = 153) | |
| Elossaolo ilman sairauden etenemistä (PFS, Progression Free Survival), N | 302 (100,0) | 153 (100,0) |
| Sensuroitu, n (%) | 138 ( 45,7) | 50 ( 32,7) |
| Eteneminen/kuolema, n (%) | 164 ( 54,3) | 103 ( 67,3) |
| Elossaoloaika ilman sairauden etenemistä (viikkoa) | ||
| Mediaania | 15,7 | 8,0 |
| Kaksipuolinen 95 %:n luottamusvälib | [13,0; 20,1] | [7,0; 9,0] |
| Riskitiheyksien suhde (Hazard ratio, HR) (Pom + PA deks: SA deks), kaksipuolinen 95 %:n luottamusvälic | 0,45 [0,35; 0,59] | |
| Log-rank-testin kaksipuolinen p-arvod | < 0,001 | |
IRAC = Independent Review Adjudication Committee; EA = Ei arvioitavissa
a Mediaani perustuu Kaplan-Meier-estimaattiin.
b 95 %:n luottamusväli elossaoloajan ilman sairauden etenemistä mediaanille.
c Perustuu Coxin suhteellisten riskitiheyksien malliin, jossa verrataan hoitoryhmiin liittyviä riskikertoimia ositettuna iän (≤ 75 vs > 75), sairauden tyypin (refraktorinen sekä lenalidomidille että bortetsomibille vs ei refraktorinen kummallekaan lääkkeelle) ja aiempien myeloomahoitojen (= 2 vs. > 2) mukaan.
d p-arvo perustuu ositettuun log-rank-testiin, jonka ositustekijät ovat samat kuin edellä mainitussa Cox-mallissa.
Tietojen katkaisupiste: 7. syyskuuta 2012.
Kuva 2: IRAC-toimikunnan IMWG:n kriteerien perusteella arvioima elossaoloaika ilman sairauden etenemistä (ositettu log-rank-testi) (ITT-potilasjoukko)
Tietojen katkaisupiste: 7. syyskuuta 2012.
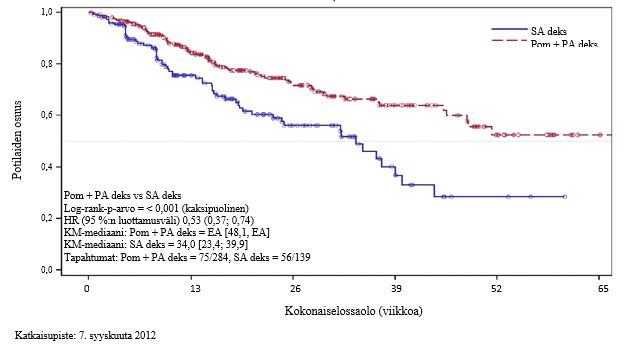
Kokonaiselossaolo oli tärkein toissijainen päätetapahtuma. Pom + PA deks -ryhmän potilaista yhteensä 226 (74,8 %) ja SA deks -ryhmän potilaista 95 (62,1 %) oli elossa tietojen katkaisupäivämääränä (7. syyskuuta 2012). Kaplan-Maier-estimaattien mukaista kokonaiselossaolon mediaania ei ole saavutettu Pom + PA deks -ryhmässä, mutta sen oletetaan olevan vähintään 48 viikkoa (95 %:n luottamusvälin alaraja). SA deks -ryhmän kokonaiselossaolon mediaani oli 34 viikkoa (95 %:n luottamusväli: 23,4; 39,9). Yhden vuoden tapahtumattoman elossaolon esiintyvyys oli Pom + PA deks -ryhmässä 52,6 % (± 5,72 %) ja SA deks -ryhmässä 28,4 % (± 7,51 %). Ero elossaolossa kahden hoitoryhmän välillä oli tilastollisesti merkitsevä (p < 0,001).
ITT-potilasjoukon kokonaiselossaolo on esitetty yhteenvetona taulukossa 10. ITT-potilasjoukon kokonaiselossaolon Kaplan-Meier-käyrä on esitetty kuvassa 3.
Tutkimusta varten perustettu valvontatoimikunta suositteli sekä elossaoloa ilman taudin etenemistä että kokonaiselossaoloa koskevien päätetapahtumien tulosten perusteella, että tutkimus viedään loppuun ja että SA deks -ryhmän potilaat siirretään Pom + PA deks -ryhmään.
Taulukko 10: Kokonaiselossaolo: ITT-potilasjoukko
| Muuttuja | Pom + PA deks (N = 302) | SA deks (N = 153) | ||
| N | 302 (100,0) | 153 (100,0) | ||
| Sensuroitu | n (%) | 226 ( 74,8) | 95 ( 62,1) | |
| Kuollut | n (%) | 76 ( 25,2) | 58 ( 37,9) | |
| Elossaoloaika (viikkoa) | Mediaania | EA | 34,0 | |
| Kaksipuolinen 95 %:n luottamusvälib | [48,1, EA] | [23,4; 39,9] | ||
| Riskitiheyksien suhde (Hazard Ratio, HR) (Pom + PA deks: SA deks) [Kaksipuolinen 95 %:n luottamusvälic] | 0,53 [0,37; 0,74] | |||
| Log-rank-testin kaksipuolinen p-arvod | < 0,001 | |||
EA = Ei arvioitavissa
a Mediaani perustuu Kaplan-Meier-estimaattiin.
b 95 %:n luottamusväli kokonaiselossaoloajan mediaanille.
c Perustuu Coxin suhteellisten riskien malliin, jossa verrataan hoitoryhmiin liittyviä riskikertoimia.
d p-arvo perustuu osittamattomaan log-rank-testiin.
Tietojen katkaisupiste: 7. syyskuuta 2012.
Kuva 3: Kokonaiselossaolon Kaplan-Meier-käyrä (ITT-potilasjoukko)
Pediatriset potilaat
Faasin 1 yksihaaraisessa, avoimessa annosmäärittelytutkimuksessa pomalidomidin suurimmaksi siedetyksi annokseksi (MTD, maximum tolerated dose) ja/tai suositelluksi faasin 2 annokseksi (RP2D) pediatrisille potilaille määriteltiin 2,6 mg/m2/vrk annettuna toistuvan 28 vrk:n syklin päivinä 1-21.
Tehoa ei osoitettu faasin 2 avoimessa rinnakkaisryhmiä käyttäneessä monikeskustutkimuksessa, johon osallistui 52 pomalidomidihoitoa saanutta pediatrista potilasta, jotka olivat iältään 4–18 vuotta ja joilla oli uusiutunut tai etenevä korkean graduksen gliooma, medulloblastooma, ependymooma tai aivorungon diffuusi gliooma (DIPG), joka sijaitsee pääasiassa keskushermostossa.
Faasin 2 tutkimuksessa kaksi korkean graduksen gliooman ryhmän (N = 19) potilasta saavutti tutkimussuunnitelman määrittelemän vasteen. Toinen näistä potilaista saavutti osittaisen vasteen ja toinen saavutti pitkällä aikavälillä vakaan sairauden vasteen (SD, stable disease), minkä tuloksena objektiivisen vasteen (OR) ja pitkällä aikavälillä vakaan sairauden vasteen osuus oli 10,5 % (95 %:n lv: 1,3; 33,1). Yksi ependymoomaryhmän potilaista (N = 9) saavutti pitkällä aikavälillä vakaan sairauden vasteen, minkä tuloksena objektiivisen vasteen ja pitkällä aikavälillä vakaan sairauden vasteen osuus oli 11,1 % (95 %:n lv: 0,3; 48,2). Aivorungon diffuusia gliomaa sairastavien ryhmässä (N = 9) ja medulloblastoomaa sairastavien ryhmässä (N = 9) ei havaittu vahvistettua objektiivista vastetta tai pitkällä aikavälillä vakaan sairauden vastetta yhdelläkään arviointiin kelvanneella potilaalla. Yksikään neljästä tässä faasin 2 tutkimuksessa arvioidusta rinnakkaisryhmästä ei täyttänyt ensisijaiselle päätetapahtumalle määriteltyjä objektiivisen vasteen tai pitkällä aikavälillä vakaan sairauden vasteen osuuksia.
Pediatrisilla potilailla pomalidomidin yleinen turvallisuusprofiili vastasi tunnettua aikuispotilaiden turvallisuusprofiilia. Farmakokineettisiä parametreja arvioitiin integroidussa farmakokineettisessä analyysissä faasin 1 ja 2 tutkimuksissa, eikä niissä todettu merkitseviä eroja verrattuna aikuispotilailla havaittuihin vastaaviin parametreihin (ks. kohta Farmakokinetiikka).
Farmakokinetiikka
Imeytyminen
Pomalidomidi imeytyy siten, että huippupitoisuus (Cmax) plasmassa saavutetaan 2–3 tunnissa. Suun kautta annetusta kerta-annoksesta imeytyy vähintään 73 %. Systeeminen altistus (AUC) pomalidomidille suurenee suunnilleen lineaarisesti ja suhteessa annokseen. Toistettujen annosten jälkeen pomalidomidin elimistöön kertymisen suhde on AUC-arvon perusteella 27–31 %.
Pomalidomidin antaminen yhdessä runsasrasvaisen ja -kalorisen aterian kanssa hidastaa imeytymisnopeutta ja pienentää plasman keskimääräistä Cmax-arvoa noin 27 %, mutta sillä on hyvin vähäinen vaikutus imeytymiseen yleensä (keskimääräinen systeeminen altistus pienenee 8 %). Pomalidomidi voidaan siis antaa ruokailuista riippumatta.
Jakautuminen
Pomalidomidin keskimääräinen näennäinen jakautumistilavuus (Vd/F) vakaassa tilassa on 62–138 l. Pomalidomidia jakautuu terveiden koehenkilöiden siemennesteeseen siten, että pitoisuus on 4 tuntia annon jälkeen (noin Tmax) noin 67 % pitoisuudesta plasmassa, kun pomalidomidia on annettu 2 mg kerran vuorokaudessa 4 vuorokautena. Pomalidomidin enantiomeerien sitoutuminen ihmisen plasman proteiineihin in vitro on 12–44 %. Sitoutuminen ei ole pitoisuudesta riippuvaista.
Biotransformaatio
Pomalidomidi oli pääasiallinen verenkierrossa todettu komponentti (noin 70 % plasman radioaktiivisuudesta) in vivo terveillä koehenkilöillä, jotka saivat kerta-annoksen [14C]-pomalidomidia (2 mg) suun kautta. Minkään metaboliitin osuuden ei havaittu olevan > 10 % verrattuna kanta-aineen radioaktiivisuuteen tai kokonaisradioaktiivisuuteen plasmassa.
Radioaktiivisuuden erittymisen pääasialliset metaboliareitit ovat hydroksylaatio ja sitä seuraava glukuronidaatio tai hydrolyysi. CYP1A2 ja CYP3A4 tunnistettiin pomalidomidin CYP-välitteiseen hydroksylaatioon in vitro osallistuviksi ensisijaisiksi entsyymeiksi. Myös CYP2C19- ja CYP2D6-entsyymien havaittiin osallistuvan vähäisessä määrin. Pomalidomidi on myös P-glykoproteiinin substraatti in vitro. Pomalidomidin samanaikaisella annolla voimakkaan CYP3A4/5:n ja P-glykoproteiinin estäjän ketokonatsolin tai voimakkaan CYP3A4/5:n indusoijan karbamatsepiinin kanssa ei ollut kliinisesti oleellista vaikutusta pomalidomidialtistukseen. Pomalidomidin anto yhdessä voimakkaan CYP1A2:n estäjän fluvoksamiinin kanssa ketokonatsolin käytön aikana suurensi keskimääräistä altistusta pomalidomidille 107 % (90 %:n luottamusväli [91–124 %]) verrattuna pelkkään pomalidomidin ja ketokonatsolin yhdistelmäkäyttöön. Toisessa tutkimuksessa, jossa arvioitiin pelkän CYP1A2:n estäjän osuutta metabolian muutoksiin, pelkän fluvoksamiinin anto yhdessä pomalidomidin kanssa lisäsi keskimääräistä pomalidomidialtistusta 125 % (90 %:n luottamusväli [98–157 %]) verrattuna pelkkään pomalidomidiin. Jos voimakkaita CYP1A2:n estäjiä (kuten siprofloksasiinia, enoksasiinia ja fluvoksamiinia) annetaan samanaikaisesti pomalidomidin kanssa, pomalidomidiannosta pitää pienentää 50 %. Tupakoijille annettuna pomalidomidilla ei ollut kliinisesti merkittävää vaikutusta pomalidomidialtistukseen verrattuna tupakoimattomilla havaittuun pomalidomidialtistukseen, vaikka tupakoinnin tiedetään indusoivan CYP1A2-isoformia.
Pomalidomidi ei ole in vitro -tietojen perusteella sytokromi P-450 -isoentsyymien estäjä eikä indusoija. Se ei myöskään ole minkään tutkitun lääkkeiden kuljettajaproteiinin estäjä. Kliinisesti oleellisia yhteisvaikutuksia ei ennakoida esiintyvän, kun pomalidomidia annetaan samanaikaisesti näiden reittien substraattien kanssa.
Eliminaatio
Pomalidomidi eliminoituu plasmasta siten, että puoliintumisajan mediaani on terveillä koehenkilöillä noin 9,5 tuntia ja multippelia myeloomaa sairastavilla potilailla noin 7,5 tuntia. Pomalidomidin keskimääräinen kokonaispuhdistuma (CL/F) elimistöstä on noin 7–10 l/h.
Terveille koehenkilöille suun kautta annetun [14C]-pomalidomidikerta-annoksen (2 mg) jälkeen noin 73 % radioaktiivisesta annoksesta eliminoitui virtsaan ja noin 15 % ulosteeseen. Annetusta radiohiilestä noin 2 % eliminoitui pomalidomidina virtsaan ja noin 8 % ulosteeseen.
Suuri osa pomalidomidista metaboloituu ennen erittymistä, ja syntyvät metaboliitit eliminoituvat pääasiassa virtsaan. Kolme tärkeintä virtsassa esiintyvää metaboliittia (jotka muodostuvat hydrolyysin tai hydroksylaation ja sen jälkeisen glukuronidaation kautta) käsittävät vastaavasti noin 23 %, 17 %, ja 12 % annoksesta virtsassa.
CYP-riippuvaiset metaboliitit käsittävät noin 43 % erittyneestä kokonaisradioaktiivisuudesta, kun taas CYP-riippumattomat hydrolyyttiset metaboliitit käsittävät siitä 25 % ja erittyminen muuttumattomana pomalidomidina 10 % (2 % virtsaan ja 8 % ulosteeseen).
Populaatiofarmakokinetiikka
Kaksitilamalliin perustuvan populaatiofarmakokineettisen analyysin perusteella näennäinen puhdistuma (CL/F) ja näennäinen sentraalinen jakautumistilavuus (V2/F) olivat verrannollisia terveillä koehenkilöillä ja multippelia myeloomaa sairastavilla potilailla. Perifeerisissä kudoksissa pomalidomidi absorboitui ensisijaisesti kasvaimiin; lääkeaineen näennäinen perifeerinen jakautumispuhdistuma (Q/F) oli potilailla 3,7-kertainen ja näennäinen perifeerinen jakautumistilavuus (V3/F) 8-kertainen terveisiin koehenkilöihin verrattuna.
Pediatriset potilaat
Kun pomalidomidia annettiin yksi annos suun kautta lapsille ja nuorille aikuisille, joilla oli uusiutunut tai etenevä primaarinen aivokasvain, mediaani Tmax oli 2–4 tuntia annoksen jälkeen ja vastasi Cmax ‑arvon (CV-%) arvoa 74,8 ng/ml (59,4 %) annostasolla 1,9 mg/m2, 79,2 ng/ml (51,7 %) annostasolla 2,6 mg/m2 ja 104 ng/ml (18,3 %) annostasolla 3,4 mg/m2. AUC0-24 ja AUC0-inf noudattivat samankaltaisia säännönmukaisuuksia: kahdella pienimmällä annoksella kokonaisaltistus oli noin
700–800 h·ng/ml ja suurella annoksella noin 1 200 h·ng/ml. Arvioidut puoliintumisajat olivat noin
5–7 tuntia.
Suurimmassa siedetyssä annoksessa ei havaittu selkeitä säännönmukaisuuksia iän ja steroidin käytön mukaan ositettuna.
Kaikkiaan aineisto viittaa siihen, että AUC nousi lähes verrannollisesti suhteessa pomalidomidiannoksen suurenemiseen, kun taas Cmax-arvo nousi yleisesti ottaen vähemmän kuin verrannollisesti suhteessa annokseen.
Integroidussa faasin 1 ja 2 tutkimusten analyysissä pomalidomidin farmakokinetiikka määritettiin 70:lle uusiutunutta tai etenevää pediatrista aivokasvainta sairastaville, iältään 4–20-vuotiaalle potilaalle sen jälkeen, kun heille oli annettu suun kautta annoksia, jotka vaihtelivat 1,9 mg/m2/vrk – 3,4 mg/m2/vrk. Pomalidomidin pitoisuus–aika-profiilit kuvailtiin adekvaatisti yhden tilamallin farmakokineettisellä mallilla, jossa imeytyminen ja eliminaatio noudattivat ensimmäisen asteen kinetiikkaa. Pomalidomidin farmakokinetiikka oli lineaarista ja ajasta riippumatonta ja sen vaihtelevuus oli kohtalaista. Pomalidomidin tyypillinen CL/F-arvo oli 3,94 l/h, Vc/F-arvo 43,0, Ka-arvo 1,45 h-1 ja viivearvo 0,454 h. Pomalidomidin lopullisen eliminaation puoliintumisaika oli 7,33 tuntia. Kehon pinta-alaa (BSA) lukuun ottamatta yhdelläkään testatulla kovariaatilla, mukaan lukien ikä ja sukupuoli, ei ollut vaikutusta pomalidomidin farmakokinetiikkaan. Vaikka BSA:n todettiin olevan tilastollisesti merkitsevä pomalidomidin CL/F:n ja Vc/F:n kovariaatti, BSA:n vaikutusta altistuksen parametreihin ei katsottu kliinisesti olennaiseksi.
Yleisesti ottaen pomalidomidin farmakokinetiikassa ei ole merkitsevää eroa lapsi- ja aikuispotilaiden välillä.
Iäkkäät potilaat
Terveillä koehenkilöillä ja multippelia myeloomaa sairastavilla potilailla tehtyjen populaatiofarmakokineettisten analyysien perusteella ikä (19–83 vuotta) ei vaikuttanut merkittävästi suun kautta annetun pomalidomidin puhdistumaan. Kliinisissä tutkimuksissa pomalidomidille altistettujen iäkkäiden (> 65-vuotiaiden) potilaiden annosta ei ollut tarpeen säätää (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Populaatiofarmakokineettisten analyysien mukaan pomalidomidin farmakokineettiset parametrit eivät muuttuneet merkittävästi munuaisten vajaatoimintaa sairastavilla potilailla (määritelty kreatiinipuhdistuman tai arvioidun munuaiskerästen suodattumisnopeuden [eGFR:n] mukaan) verrattuna potilaisiin, joiden munuaiset toimivat normaalisti (CrCl ≥ 60 ml/minuutti). Keskimääräinen normalisoitu käyrän alla oleva altistus pomalidomidille oli 98,2 % (90 %:n luottamusväli [77,4–120,6 %]) keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (eGFR välillä ≥30 ja ≤45 ml/minuutti/1,73 m2) verrattuna potilaisiin, joiden munuaiset toimivat normaalisti. Keskimääräinen normalisoitu käyrän alla oleva altistus pomalidomidille oli 100,2 % (90 %:n luottamusväli [79,7–127,0 %]) potilailla, joilla oli vaikea munuaisten vajaatoiminta, joka ei vaatinut dialyysihoitoa (CrCl < 30 tai eGFR <30 ml/minuutti/1,73 m2) verrattuna potilaisiin, joiden munuaiset toimivat normaalisti. Keskimääräinen normalisoitu käyrän alla oleva altistus pomalidomidille kasvoi 35,8 % (90 %:n luottamusväli [7,5–70,0 %]) dialyysia tarvitsevilla vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (CrCl <30 ml/minuutti, vaatii dialyysia) verrattuna potilaisiin, joiden munuaiset toimivat normaalisti. Pomalidomidialtistuksen keskimääräinen lisääntyminen näissä munuaisten vajaatoimintaryhmissä ei ole niin merkittävä, että se edellyttäisi annoksen säätämistä.
Maksan vajaatoiminta
Maksan vajaatoimintaa (Child-Pugh-kriteerien mukaista) sairastavien potilaiden farmakokineettiset parametrit muuttuivat hieman verrattuna terveisiin koehenkilöihin. Lievää maksan vajaatoimintaa sairastavien potilaiden keskimääräinen pomalidomidialtistus lisääntyi 51 % (90 %:n luottamusväli [9–110 %]) verrattuna terveisiin koehenkilöihin. Keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden keskimääräinen pomalidomidialtistus lisääntyi 58 % (90 %:n luottamusväli [13–119 %]) verrattuna terveisiin koehenkilöihin. Vaikeaa maksan vajaatoimintaa sairastavien potilaiden keskimääräinen pomalidomidialtistus lisääntyi 72 % (90 %:n luottamusväli [24–138 %]) verrattuna terveisiin koehenkilöihin. Pomalidomidialtistuksen keskimääräinen lisääntyminen näissä vajaatoimintaryhmissä ei ole niin merkittävää, että se edellyttäisi annoksen tai antoaikataulun säätämistä (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen toksikologiaa koskevat tutkimukset
Rotat sietivät hyvin kroonisen altistuksen pomalidomidille annoksilla 50, 250 ja 1000 mg/kg/vrk 6 kuukauden ajan. Haittavaikutuksia ei havaittu edes vuorokausiannoksilla 1000 mg/kg (175-kertainen altistus suhteessa 4 mg:n kliiniseen annokseen).
Pomalidomidia tutkittiin apinoilla enimmillään 9 kuukautta kestäneissä toistuvaa altistusta koskeneissa tutkimuksissa. Apinat osoittautuivat näissä tutkimuksissa rottia herkemmiksi pomalidomidille. Apinoilla havaittu ensisijainen toksisuus liittyi hematopoieettiseen/lymforetikulaariseen järjestelmään. Apinoilla tehdyssä 9 kuukauden pituisessa tutkimuksessa, jossa käytetyt annokset olivat 0,05, 0,1 ja 1 mg/kg/vrk, kuudelle annoksen 1 mg/kg/vrk saaneelle eläimelle tehtiin varhaisvaiheessa eutanasia sairastavuuden vuoksi, minkä syynä pidettiin suuren pomalidomidialtistuksen (15-kertainen altistus 4 mg:n kliiniseen annokseen verrattuna) immunosuppressiivisia vaikutuksia (stafylokokki-infektio, ääreisveren lymfosyyttimäärän lasku, krooninen paksusuolitulehdus, histologinen imukudosniukkuus ja luuytimen soluvaje). Nämä immunosuppressiiviset vaikutukset johtivat neljän apinan eutanasiaan varhaisessa vaiheessa heikon terveydentilan vuoksi (vetinen uloste, ruokahaluttomuus, vähentynyt ravinnonotto ja painon lasku); näiden eläinten histopatologisessa tutkimuksessa todettiin paksusuolen krooninen tulehdus ja ohutsuolen suolinukan atrofia. Stafylokokki-infektio todettiin 4 apinalla, joista 3 sai vasteen antibioottihoitoon ja 1 kuoli ilman hoitoa. Lisäksi akuuttiin myelooiseen leukemiaan sopivat löydökset johtivat yhden apinan eutanasiaan; kliiniset havainnot ja kliininen patologia ja/tai kyseisellä eläimellä havaitut luuytimen muutokset olivat yhdenmukaisia immuunisuppression kanssa. Hyvin vähäistä tai lievää sapenjohtimen proliferaatiota ja siihen liittyvää AFOS- ja GGT-arvojen nousua havaittiin myös annoksella 1 mg/kg/vrk. Toipuneiden eläinten tutkiminen osoitti, että kaikki hoitoon liittyvät löydökset olivat korjaantuneet 8 viikon kuluttua hoidon loppumisesta, lukuun ottamatta yhdellä annoksia 1 mg/kg/vrk saaneen ryhmän eläimistä havaittua maksansisäisten sappitiehyiden proliferaatiota. Suurin haitaton annos (NOAEL-arvo) oli 0,1 mg/kg/vrk (0,5-kertainen altistus suhteessa 4 mg:n kliiniseen annokseen).
Geenitoksisuus/karsinogeenisuus
Pomalidomidi ei ollut mutageeninen bakteerien ja nisäkässolujen mutaatiomäärityksissä. Se ei myöskään indusoinut kromosomipoikkeavuuksia ihmisen ääreisveren lymfosyyteissä tai mikrotumien muodostusta rottien luuytimen polykromaattisissa erytrosyyteissä, kun rotille annetut vuorokausiannokset olivat enimmillään 2000 mg/kg. Karsinogeenisuustutkimuksia ei ole tehty.
Hedelmällisyys ja varhainen alkion kehitys
Pomalidomidia annettiin rotilla tehdyssä hedelmällisyyttä ja varhaista alkion kehitystä koskevassa tutkimuksessa uroksille ja naaraille annoksina 25, 250 ja 1000 mg/kg/vrk. Kohdun tutkimuksessa 13. tiineyspäivänä havaittiin elinkykyisten alkioiden keskimääräistä vähenemistä sekä implantaation jälkeisten alkionmenetysten lisääntymistä kaikilla annostasoilla. Siksi NOAEL-arvo näiden havaittujen vaikutusten osalta oli < 25 mg/kg/vrk (AUC24h-arvo tällä pienimmällä tutkitulla annoksella oli 39 960 ng•h/ml (nanogramma•tunti/millilitra) ja altistus 99-kertainen suhteessa 4 mg:n kliiniseen annokseen). Kun tässä tutkimuksessa hoitoa saaneita uroksia pariutettiin hoitoa saamattomien naaraiden kanssa, kaikki kohdun parametrit olivat verrannollisia verrokkiryhmän arvojen kanssa. Havaittujen vaikutusten todettiin näiden tulosten perusteella johtuneen naaraiden saamasta hoidosta.
Alkion ja sikiön kehitys
Pomalidomidin todettiin olevan teratogeeninen sekä rotilla että kaniineilla, kun sitä annettiin tärkeimmän organogeneesivaiheen aikana. Rottien alkion ja sikiön kehitystoksisuutta koskevassa tutkimuksessa havaittiin kaikilla annostasoilla (25, 250 ja 1000 mg/kg/vrk) seuraavia epämuodostumia: virtsarakon puuttuminen, kilpirauhasen puuttuminen sekä lanne- ja rintarangan elementtien (keski- ja/tai nikamankaaren) fuusioituminen ja poikkeava sijainti.
Tässä tutkimuksessa ei havaittu emoon kohdistuvaa toksisuutta. Siksi emon suurin haitaton annos (NOAEL) oli 1000 mg/kg/vrk. NOAEL kehitystoksisuuden osalta oli < 25 mg/kg/vrk (AUC24h-arvo 17. tiineyspäivänä tällä pienimmällä tutkitulla annoksella oli 34340 ng•h/ml ja altistustaso 85-kertainen suhteessa 4 mg:n kliiniseen annokseen). Annokset 10–250 mg/kg aiheuttivat kaniineille alkion ja sikiön kehitykseen liittyviä epämuodostumia. Sydämen poikkeavuuksien havaittiin lisääntyvän kaikilla annoksilla, ja ne lisääntyivät merkittävästi annoksella 250 mg/kg/vrk. Annoksilla 100 ja 250 mg/kg/vrk todettiin implantaation jälkeisten alkionmenetysten vähäistä lisääntymistä ja sikiöiden painon vähäistä laskua. Annoksella 250 mg/kg/vrk havaittiin seuraavia sikiön epämuodostumia: raajojen poikkeavuudet (koukistuneet ja/tai kiertyneet etu- ja/tai takaraajat, irrallinen tai puuttuva varvas) ja niihin liittyvät luuston epämuodostumat (luutumaton kämmenluu, varvasluun ja kämmenluun poikkeava suunta, puuttuva varvas, luutumaton varvasluu ja lyhyt, luutumaton tai taipunut sääriluu); kohtalainen aivojen sivukammion laajentuma, oikean solisvaltimon poikkeava sijainti, keuhkojen keskilohkon puuttuminen, normaalia alempi munuaisten sijainti, poikkeava maksan rakenne, epätäydellinen tai luutumaton lantio, ylimääräisten torakaalisten kylkiluiden keskimääräinen lisääntyminen ja luutuneiden nilkkaluiden keskimääräinen väheneminen. Annoksilla 100 ja 250 mg/kg/vrk havaittiin lievää emon painon nousun vähentymistä, merkityksellistä triglyseridiarvojen pienentymistä ja merkityksellistä pernan absoluuttisen ja suhteellisen painon laskua. Emon NOAEL oli 10 mg/kg/vrk ja alkion/sikiön kehityksen NOAEL < 10 mg/kg/vrk (AUC24h-arvo 19. tiineyspäivänä tällä pienimmällä tutkitulla annoksella oli 418 ng•h/ml, mikä vastasi suurin piirtein 4 mg:n kliinisellä annoksella saatua arvoa).
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö:
Mannitoli (E 421)
Tärkkelys, esigelatinoitu
Natriumstearyylifumaraatti
Kapselin kuori:
Imnovid 1 mg kapselit, kovat
Liivate
Titaanidioksidi (E 171)
Indigotiini (E 132)
Keltainen rautaoksidi (E 172)
Valkoinen ja musta muste
Imnovid 2 mg kapselit, kovat
Liivate
Titaanidioksidi (E 171)
Indigotiini (E 132)
Keltainen rautaoksidi (E 172)
Erytrosiini (E 127)
Valkoinen muste
Imnovid 3 mg kapselit, kovat
Liivate
Titaanidioksidi (E 171)
Indigotiini (E 132)
Keltainen rautaoksidi (E 172)Valkoinen muste
Imnovid 4 mg kapselit, kovat
Liivate
Titaanidioksidi (E 171)
Indigotiini (E 132)
Briljanttisininen FCF (E 133)Valkoinen muste
Painomuste:
Valkoinen muste (Imnovid kovat kapselit, kaikki vahvuudet)
Shellakka
Titaanidioksidi (E 171)
Simetikoni
Propyleeniglykoli (E 1520)
Ammoniumhydroksidi (E 527)
Musta muste (Imnovid 1 mg kapselit, kovat)
Shellakka
Musta rautaoksidi (E 172)
Propyleeniglykoli (E 1520)
Ammoniumhydroksidi (E 527)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
IMNOVID kapseli, kova
1 mg (L:ei) 14 fol (4221,78 €)
2 mg (L:ei) 14 fol (4241,74 €)
3 mg (L:ei) 14 fol (4254,32 €)
4 mg (L:ei) 14 fol (4274,29 €)
PF-selosteen tieto
Kapselit on pakattu polyvinyylikloridista (PVC) / polyklooritrifluorieteenistä (PCTFE) valmistettuihin läpipainopakkauksiin, joissa on alumiininen läpipainokalvo.
Pakkauskoko on 14 tai 21 kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Imnovid 1 mg kapselit, kovat
Tummansininen, läpinäkymätön kansiosa ja keltainen, läpinäkymätön runko-osa, painatus ”POML” valkoisella musteella ja ”1 mg” mustalla musteella, koko 3, kova liivatekapseli.
Imnovid 2 mg kapselit, kovat
Tummansininen, läpinäkymätön kansiosa ja oranssi, läpinäkymätön runko-osa, painatus ”POML 2 mg” valkoisella musteella, koko 1, kova liivatekapseli.
Imnovid 3 mg kapselit, kovat
Tummansininen, läpinäkymätön kansiosa ja vihreä, läpinäkymätön runko-osa, painatus ”POML 3 mg” valkoisella musteella, koko 1, kova liivatekapseli.
Imnovid 4 mg kapselit, kovat
Tummansininen, läpinäkymätön kansiosa ja sininen, läpinäkymätön runko-osa, painatus ”POML 4 mg” valkoisella musteella, koko 1, kova liivatekapseli.
Käyttö- ja käsittelyohjeet
Kapseleita ei saa avata eikä murskata. Jos pomalidomidijauhetta joutuu iholle, iho on pestävä välittömästi ja huolellisesti saippualla ja vedellä. Jos pomalidomidia joutuu limakalvoille, altistunut alue on huuhdeltava huolellisesti vedellä.
Terveydenhuollon ammattilaisten ja huoltajien on läpipainolevyjä tai kapseleita käsitellessään käytettävä kertakäyttöisiä käsineitä. Käsineet on tämän jälkeen riisuttava varovasti ihon altistumisen välttämiseksi ja laitettava suljettavaan polyeteenimuovipussiin, joka hävitetään paikallisten vaatimusten mukaisesti. Kädet on tämän jälkeen pestävä huolellisesti saippualla ja vedellä. Naisten, jotka ovat raskaana tai epäilevät olevansa raskaana, ei pidä käsitellä läpipainolevyjä tai kapseleita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti. Hoidon päättyessä käyttämättä jäävä lääkevalmiste on palautettava apteekkiin.
Korvattavuus
IMNOVID kapseli, kova
1 mg 14 fol
2 mg 14 fol
3 mg 14 fol
4 mg 14 fol
- Ei korvausta.
ATC-koodi
L04AX06
Valmisteyhteenvedon muuttamispäivämäärä
21.08.2025
Yhteystiedot
09 2512 1244
www.bms.com/fi
medinfo.finland@bms.com