
ICLUSIG tabletti, kalvopäällysteinen 15 mg, 30 mg, 45 mg
Vaikuttavat aineet ja niiden määrät
Iclusig 15 mg tabletit, kalvopäällysteiset
Yksi kalvopäällysteinen tabletti sisältää 15 mg ponatinibia (hydrokloridina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 40 mg laktoosimonohydraattia.
Iclusig 30 mg tabletit, kalvopäällysteiset
Yksi kalvopäällysteinen tabletti sisältää 30 mg ponatinibia (hydrokloridina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 80 mg laktoosimonohydraattia.
Iclusig 45 mg tabletit, kalvopäällysteiset
Yksi kalvopäällysteinen tabletti sisältää 45 mg ponatinibia (hydrokloridina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 120 mg laktoosimonohydraattia
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Iclusig on tarkoitettu aikuispotilaille, joilla on
- kroonisen vaiheen, akseleraatiovaiheen tai blastivaiheen krooninen myelooinen leukemia (KML) ja jotka ovat resistenttejä dasatinibille tai nilotinibille; tai jotka eivät siedä dasatinibia tai nilotinibia eikä tämän jälkeinen imatinibihoito ole kliinisesti asianmukaista; tai joilla on T315I‑mutaatio
- Philadelphia‑kromosomipositiivinen akuutti lymfoblastinen leukemia (Ph+ ALL) ja jotka ovat resistenttejä dasatinibille; tai jotka eivät siedä dasatinibia eikä tämän jälkeinen imatinibihoito ole kliinisesti asianmukaista; tai joilla on T315I‑mutaatio.
Iclusig on tarkoitettu yhdessä kevennetyn kemoterapian kanssa aikuispotilaille, joilla on vastadiagnosoitu Ph+ ALL (ks. kohta Farmakodynamiikka).
Kardiovaskulaarisen statuksen arviointi ennen hoidon aloitusta, ks. kohta Annostus ja antotapa; tilanteet, joissa voidaan harkita muuta hoitoa, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Ehto
Hoidon saa aloittaa leukemian diagnosointiin ja hoitoon perehtynyt lääkäri.
Annostus ja antotapa
Hoidon aloittaa leukemiapotilaiden diagnosointiin ja hoitoon perehtynyt lääkäri. Hematologista tukea, kuten verihiutaleiden siirtoa ja hematopoieettisia kasvutekijöitä, voidaan käyttää kliinisesti aiheellisissa tapauksissa hoidon aikana.
Ennen ponatinibihoidon aloitusta on arvioitava potilaan kardiovaskulaarinen status, otettava myös anamneesi ja tehtävä kliininen tutkimus, ja kardiovaskulaariset riskitekijät on hoidettava aktiivisesti. Kardiovaskulaarista statusta on seurattava ja kardiovaskulaariseen riskiin vaikuttavien sairauksien lääketieteellinen ja tukihoito on optimoitava ponatinibihoidon aikana.
Annostus
KML-ja Philadelphia-kromosomipositiivinen akuutti lymfoblastinen leukemia (Ph+ ALL) -potilaat, joita on aiemmin hoidettu muilla tyrosiinikinaasin estäjillä (TKI:t), tai joilla on T315I-mutaatio:
Suositeltava aloitusannos on 45 mg ponatinibia kerran vuorokaudessa. Tavanomaisia annoksia (45 mg kerran vuorokaudessa) varten on saatavilla 45 mg:n kalvopäällysteisiä tabletteja. Hoitoa on jatkettava niin kauan kuin potilaalla ei näy merkkejä taudin etenemisestä eikä sietämätöntä toksisuutta.
Hoitovastetta on seurattava normaalien kliinisten ohjeiden mukaan.
Ponatinibin käytön keskeyttämistä on harkittava, ellei täydellistä hematologista vastetta ole saavutettu 3 kuukauden (90 vuorokauden) kuluessa.
Valtimotukostapahtumien riski on todennäköisesti annoksen suuruudesta riippuvainen. Jos potilaalla on kroonisen vaiheen (CP) KML ja saavutetaan molekulaarinen vaste (MR2 on ≤ 1 % BCR-ABL1IS), tulee harkita Iclusig-annoksen pienentämistä 15 mg:aan. Yksilöllisessä arvioinnissa on otettava huomioon seuraavat seikat: kardiovaskulaarinen riski, ponatinibihoidon haittavaikutukset, aika, jossa vaste saavutettiin, ja BCR‑ABL‑transkriptien määrä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka). Jos annosta pienennetään, vastetta on syytä seurata tarkasti. Jos vaste menetetään, Iclusig-annos voidaan suurentaa uudestaan aiemmin siedettyyn annostukseen (30 mg tai 45 mg suun kautta kerran vuorokaudessa). Iclusig-hoitoa on jatkettava, kunnes uudestaan suurennetulla annoksella saavutettu vaste menetetään tai kunnes ilmaantuu sietämätöntä toksisuutta.
Potilaat, joilla on vastadiagnosoitu Ph+ ALL, yhdessä kemoterapian kanssa:
Suositeltu aloitusannos on 30 mg ponatinibia kerran vuorokaudessa yhdessä kemoterapian kanssa. Annos pienennetään 15 mg:aan kerran vuorokaudessa, jos MRD-negatiivinen täydellinen vaste (≤ 0,01 % BCR-ABL1) saavutetaan induktion päättymiseen mennessä.
Potilailla, joiden MRD-negatiivisuus menetetään, ponatinibiannos voidaan suurentaa uudelleen aiemmin siedetylle annokselle, enintään 30 mg kerran vuorokaudessa. Kun ponatinibin käyttö yhdessä kemoterapian kanssa päättyy, hoitoa jatketaan ponatinibimonoterapiana uudelleen suurennetulla annoksella niin kauan kuin vaste säilyy tai ei ilmene sietämätöntä toksisuutta (ks. kohta Farmakodynamiikka Farmakodynamiikka).
CNS-profylaksi tai hoito, steroidien aloitus, anti-CD20-hoito CD20+-potilailla tai kemoterapia on toteutettava tarpeen mukaan kyseisen valmisteen valmisteyhteenvedon ja normaalien kliinisten ohjeiden mukaisesti.
Ponatinibin lopettamista on harkittava, ellei induktiovaiheen jälkeen saavuteta täydellistä molekulaarista vastetta.
Toksisten vaikutusten hallinta
Iclusig-annoksen muuttamista tai hoidon keskeyttämistä voidaan harkita hematologisen ja ei‑hematologisen toksisuuden hallitsemiseksi. Jos vaikeita haittavaikutuksia ilmenee, hoito on keskeytettävä. Kun Iclusigia annetaan yhdessä kemoterapian kanssa, on noudatettava kemoterapialääkkeiden tavanomaisia annoksen pienentämistä koskevia ohjeita; ks. valmisteiden valmisteyhteenvedot ja normaalit kliiniset ohjeet.
Jos haittavaikutus korjautuu tai lievittyy, Iclusig‑hoito voidaan aloittaa uudelleen ja vuorokausiannoksen suurentamista takaisin ennen haittavaikutusta käytetylle tasolle voidaan harkita, jos se on kliinisesti asianmukaista.
Saatavilla on 15 mg:n ja 30 mg:n kalvopäällysteisiä tabletteja 30 mg tai 15 mg kerran vuorokaudessa ‑annosta varten.
Myelosuppressio
Annoksen muutokset leukemiaan liittymättömän neutropenian (ANC* < 1,0 x 109/l) ja trombosytopenian (verihiutaleet < 50 x 109/l) yhteydessä esitetään yhteenvetona taulukossa 1.
Taulukko 1 Annoksen muutokset myelosuppression takia
ANC* < 1,0 x 109/l tai verihiutaleet < 50 x 109/l | Ensimmäinen kerta:
|
Uusiutuminen 45 mg:n annoksella:
| |
Uusiutuminen 30 mg:n annoksella:
| |
| *ANC = absoluuttinen neutrofiilimäärä | |
Valtimotukos ja laskimotromboembolia
Jos potilaalla epäillään valtimotukostapahtumaa tai laskimotromboemboliaa, Iclusig‑hoito on heti keskeytettävä. Iclusig‑hoidon aloittamisesta uudelleen tapahtuman korjauduttua on päätettävä hyödyn ja riskien arvioinnin perusteella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Korkea verenpaine saattaa lisätä valtimotukostapahtumien riskiä. Iclusig‑hoito on keskeytettävä tilapäisesti, jos hypertension hoitotasapaino on lääkehoidosta huolimatta huono.
Haimatulehdus
Haimaan liittyvien haittavaikutusten yhteydessä suositeltavat muutokset esitetään yhteenvetona taulukossa 2.
Taulukko 2 Annoksen muutokset haimatulehduksen ja lipaasiarvojen kohoamisen yhteydessä
| Asteen 2 haimatulehdus ja/tai asteen 2 lipaasiarvojen nousu (> 1,5–2,0 x IULN tai > 2,0–5,0 x IULN ja oireeton) | Iclusig‑hoitoa jatketaan samalla annoksella |
| Asteen 3 oireeton lipaasiarvojen kohoaminen (> 5,0 x IULN*) | Ilmeneminen 45 mg:n annoksella:
Ilmeneminen 30 mg:n annoksella:
Ilmeneminen 15 mg:n annoksella:
|
| Asteen 3 haimatulehdus tai asteen 3 oireinen lipaasiarvojen nousu (> 2,0–5,0 x IULN) | Ilmeneminen 45 mg:n annoksella:
Ilmeneminen 30 mg:n annoksella:
Ilmeneminen 15 mg:n annoksella:
|
| Asteen 4 haimatulehdus tai asteen 4 lipaasiarvojen nousu (> 5,0 x IULN ja oireinen) | Iclusig‑hoito lopetetaan |
| *IULN = laitoksen viitearvon yläraja | |
Maksatoksisuus
Hoidon keskeyttäminen tai lopettaminen taulukossa 3 kuvatulla tavalla saattaa olla tarpeen.
Taulukko 3 Suositellut annosmuutokset maksatoksisuuden yhteydessä
Maksan transaminaasiarvojen kohoaminen > 3 × ULN* Aste 2, pitkittynyt (yli 7 vrk) Aste 3 tai suurempi | Ilmeneminen 45 mg:n annoksella:
Ilmeneminen 30 mg:n annoksella:
Ilmeneminen 15 mg:n annoksella:
|
| ASAT- tai ALAT-arvon kohoaminen ≥ 3 × ULN ja samanaikainen bilirubiiniarvon kohoaminen > 2 × ULN ja AFOS-arvon kohoaminen < 2 × ULN | Iclusig-hoito lopetetaan |
| *ULN = laboratorioarvon viitealueen yläraja | |
Iäkkäät potilaat
Iclusigin kliinisissä PACE- ja OPTIC-tutkimuksissa, joihon otti osaa 732 potilasta, 191 (26 %) oli ≥ 65 vuotiaita. Alle 65 vuotiaisiin potilaisiin verrattuna vanhemmilla potilailla on todennäköisemmin haittavaikutuksia.
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastaville potilaille voidaan antaa suositeltu aloitusannos. Varovaisuutta suositellaan, jos Iclusigia annetaan potilaille, joilla on maksan vajaatoiminta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Erittyminen munuaisten kautta ei ole ponatinibin tärkeä eliminoitumisreitti. Iclusigia ei ole tutkittu potilailla, joilla on munuaisten vajaatoimintaa. Potilaat, joiden arvioitu kreatiniinin puhdistuma on ≥ 50 ml/min, pystyvät todennäköisesti käyttämään Iclusigia turvallisesti ilman annoksen säätämistä. Varovaisuutta suositellaan annettaessa Iclusigia potilaille, joilla arvioitu kreatiniinin puhdistuma on < 50 ml/min tai joilla on loppuvaiheen munuaistauti.
Pediatriset potilaat
Iclusig-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Iclusig on tarkoitettu otettavaksi suun kautta. Tabletit on nieltävä kokonaisina. Potilaat eivät saa murskata tai liuottaa tabletteja. Iclusig voidaan ottaa ruoan kanssa tai erikseen.
Potilaita on varoitettava nielemästä pullossa olevaa kuivausainepurkkia.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Tärkeät haittavaikutukset
Myelosuppressio
Iclusigiin liittyy vaikeaa (Yhdysvaltain National Cancer Institute ‑syöpäinstituutin Common Terminology Criteria for Adverse Events ‑kriteerien aste 3 tai 4) trombosytopeniaa, neutropeniaa ja anemiaa. Asteen 3 tai 4 trombosyyttiarvon laskun, anemian tai neutropenian kehittyminen tapahtui useimmiten ensimmäisten 3 hoitokuukauden aikana. Nämä tapahtumat ovat yleisempiä potilailla, joilla on akseleraatiovaiheen KML (AP‑KML), blastivaiheen KML (BP‑KML) tai Ph+ ALL, kuin potilailla, joilla on kroonisen vaiheen KML (CP‑KML). Täydellinen verenkuva on määritettävä 2 viikon välein ensimmäisen 3 kuukauden ajan ja sitten kuukausittain tai kliinisen tarpeen mukaan. Myelosuppressio oli yleensä korjautuvaa ja se voitiin yleensä hoitaa keskeyttämällä Iclusig‑hoito tilapäisesti tai alentamalla annosta (ks. kohta Annostus ja antotapa).
Valtimotukos
Iclusig‑hoitoa saaneilla potilailla on todettu valtimotukoksia, kuten kuolemaan johtanut sydäninfarkti, aivohalvaus, verkkokalvon valtimotukoksia, joihin liittyy joissain tapauksissa pysyvä näön heikkeneminen tai menetys, suurten aivovaltimoiden ahtauma, vaikea ääreisverisuonisairaus, munuaisvaltimostenoosi (johon liittyy paheneva, labiili tai hoitoresistentti hypertensio) ja kiireellisen revaskularisaation tarve. Näitä tapahtumia on ilmennyt sekä potilailla, joilla oli kardiovaskulaarisia riskitekijöitä, että potilailla, joilla ei ollut tällaisia riskitekijöitä. Tapahtumia esiintyi myös 50‑vuotiailla ja sitä nuoremmilla. Valtimotukoshaittatapahtumat olivat yleisempiä vanhemmilla potilailla ja niillä, joilla oli anamneesissa iskemia, korkea verenpaine, diabetes tai hyperlipidemia.
Valtimotukostapahtumien riski riippuu todennäköisesti annoksen suuruudesta. (Ks. kohdat Haittavaikutukset ja Farmakodynamiikka).
Kliinisen kehityksen aikana ilmeni haittavaikutuksena (myös vakavana haittavaikutuksena) valtimotukoksia (ks. kohta Haittavaikutukset). Joillakin potilailla oli useampia eri tyyppisiä tapahtumia.
Iclusig‑valmistetta ei pidä käyttää, jos potilaalla on ollut sydäninfarkti, aiempi revaskularisaatio tai aivohalvaus, ellei hoidosta mahdollisesti saatava hyöty ylitä siihen liittyvää riskiä (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Näillä potilailla on ennen ponatinibihoidon aloittamista harkittava myös muita hoitovaihtoehtoja.
Ennen ponatinibihoidon aloitusta on arvioitava potilaan kardiovaskulaarinen status, otettava myös anamneesi ja tehtävä kliininen tutkimus, ja kardiovaskulaariset riskitekijät on hoidettava aktiivisesti. Kardiovaskulaarista statusta on seurattava ja kardiovaskulaariseen riskiin vaikuttavien sairauksien lääketieteellinen ja tukihoito on optimoitava ponatinibihoidon aikana. Ponatinibihoidon turvallisuutta ei ole tutkittu eteisvärinäpotilalla.
Potilasta on seurattava valtimotukoksen merkkien varalta, ja jos potilaalla esiintyy näön heikkenemistä tai hämärtymistä, on tehtävä silmätutkimus (mukaan lukien silmänpohjatutkimus). Valtimotukoksen sattuessa Iclusig‑hoito on heti keskeytettävä. Iclusig‑hoidon aloittamisesta uudelleen on päätettävä hyödyn ja riskien arvioinnin perusteella (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Laskimotromboembolia
Kliinisen kehityksen aikana ilmeni haittavaikutuksena (myös vakavana haittavaikutuksena) laskimotromboemboliaa (ks. kohta Haittavaikutukset).
Potilaita on seurattava tromboembolian merkkien varalta. Jos tromboembolia kehittyy, Iclusig‑hoito on heti keskeytettävä. Iclusig‑hoidon aloittamisesta uudelleen on päätettävä hyöty-riskiarvioinnin perusteella (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Iclusig-hoitoa saaneilla potilailla on esiintynyt verkkokalvon laskimotukoksia, joihin on joissakin tapauksissa liittynyt pysyvä näön heikkeneminen tai näön menetys. Jos näön heikkenemistä tai sumenemista esiintyy, on tehtävä silmätutkimus (mukaan lukien silmänpohjan tutkimus).
Hypertensio
Korkea verenpaine saattaa lisätä valtimotromboositapahtumien riskiä (mukaan lukien munuaisvaltimostenoosi). Verenpainetta on seurattava ja hoidettava jokaisella klinikkakäynnillä Iclusig‑hoidon aikana, ja verenpaine on hoidettava normaaliksi. Iclusig‑hoito on keskeytettävä tilapäisesti, jos hypertension hoitotasapaino on lääkehoidosta huolimatta huono (ks. kohta Annostus ja antotapa).
Jos potilaalla ilmenee merkittävää pahenevaa, labiilia tai hoitoresistenttiä hypertensiota, hoito keskeytetään ja harkitaan potilaan arviointia munuaisvaltimostenoosin varalta.
Iclusig‑hoitoa saaneilla potilailla ilmeni hoidon aikana korkeaa verenpainetta (mukaan lukien hypertensiivistä kriisiä). Nopeat kliiniset toimenpiteet saattavat olla tarpeen, jos korkeaan verenpaineeseen liittyy sekavuutta, päänsärkyä, rintakipua tai hengenahdistusta.
Aneurysmat ja valtimon dissekaatiot
VEGF-reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava tarkoin ennen Iclusig-hoidon aloittamista potilaille, joilla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Kongestiivinen sydämen vajaatoiminta
Iclusig‑hoitoa saavilla potilailla on ilmennyt kuolemaan johtavaa ja vakavaa sydämen vajaatoimintaa ja vasemman kammion toimintahäiriötä sekä aiempiin verisuonitukostapahtumiin liittyviä tapahtumia. Potilaita on seurattava sydämen vajaatoiminnan oireiden ja löydösten varalta ja hoidettava kliinisen tarpeen mukaan tarvittaessa myös keskeyttämällä Iclusig‑hoito. Ponatinibin käytön keskeyttämistä on harkittava, jos potilaalle kehittyy vakava sydämen vajaatoiminta (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Haimatulehdus ja seerumin lipaasi
Iclusigiin liittyy haimatulehdusta. Haimatulehdus on yleisempää ensimmäisten kahden käyttökuukauden aikana. Tarkista seerumin lipaasi 2 viikon välein ensimmäisen 2 kuukauden ajan ja sitten määräajoin. Annostelun keskeyttäminen tai annoksen pienentäminen voi olla tarpeen. Jos kohonneisiin lipaasiarvoihin liittyy vatsavaivoja, Iclusig‑hoito tauotetaan ja potilas arvioidaan haimatulehduksen merkkien varalta (ks. kohta Annostus ja antotapa). Valmistetta on käytettävä varovasti potilailla, joilla on ollut haimatulehdusta tai alkoholin väärinkäyttöä. Jos potilaalla on vaikea tai hyvin vaikea hypertriglyseridemia, asianmukainen hoito on tarpeen haimatulehdusriskin minimoimiseksi.
Maksatoksisuus
Iclusigin käyttäminen voi johtaa kohonneisiin ALAT‑, ASAT‑, bilirubiini‑ ja AFOS‑arvoihin. Suurimmalla osalla potilaista, joille kehittyi maksatoksisuustapahtuma, ensimmäinen tapahtuma todettiin yhden vuoden sisällä hoidon aloittamisesta. Maksan vajaatoimintaa (myös kuolemaan johtaneita tapauksia) on havaittu. Maksan toimintakokeet on suoritettava ennen hoidon aloitusta ja määräajoin kliinisen tarpeen mukaisesti. Maksan toimintaa on tarkkailtava huolellisesti, kun ponatinibia käytetään yhdessä sellaisten kemoterapia-aineiden kanssa, joiden tiedetään myös liittyvän maksan toimintahäiriöön (ks. kohta Haittavaikutukset).
Verenvuodot
Iclusig‑hoitoa saaneilla potilailla on ilmennyt vaikeita verenvuotoja, joista osa on johtanut kuolemaan. Vaikeita verenvuototapahtumia oli enemmän AP‑KML‑, BP‑KML‑ ja Ph+ ALL ‑potilailla. Yleisimmin raportoidut asteen 3/4 verenvuototapahtumat olivat maha‑suolikanavan verenvuoto ja kovakalvonalainen verenpurkauma. Useimmat verenvuototapahtumat, joskaan eivät kaikki, ilmenivät potilailla, joilla oli asteen 3/4 trombosytopenia. Iclusig‑hoito on keskeytettävä ja potilas arvioitava vakavan tai vaikean verenvuodon varalta.
Hepatiitti B:n uudelleen aktivoituminen
Hepatiitti B:n uudelleen aktivoitumista on tapahtunut kyseisen viruksen pysyvillä kantajilla sen jälkeen, kun potilas on saanut BCR-ABL-tyrosiinikinaasin estäjiä. Tämä aiheutti joissakin tapauksissa akuuttia maksan vajaatoimintaa tai fulminanttia hepatiittia, joka johti maksansiirtoon tai kuolemaan.
Potilaat on testattava hepatiitti B -viruksen varalta ennen Iclusig-hoidon aloittamista. Maksasairauksien ja hepatiitti B:n hoitoon perehtyneitä asiantuntijoita on kuultava ennen hoidon aloittamista, jos potilaan hepatiitti B -serologia on positiivinen (mukaan lukien potilaat, joilla sairaus on aktiivinen) ja jos potilas saa positiivisen hepatiitti B -testituloksen hoidon aikana. Hepatiitti B -viruksen kantajia, jotka tarvitsevat Iclusig-hoitoa, on seurattava tarkasti aktiivisen hepatiitti B -infektion oireiden varalta koko hoidon ajan ja useita kuukausia hoidon jälkeen (ks. kohta Haittavaikutukset).
Posteriorinen reversiibeli enkefalopatiaoireyhtymä
Iclusig-hoitoa saavilla potilailla on ilmoitettu markkinoilletulon jälkeen posteriorista reversiibeliä enkefalopatiaoireyhtymää (PRES).
PRES on neurologinen häiriö, jonka oireita ja merkkejä voivat olla esim. kouristuskohtaukset, päänsärky, tarkkaavaisuuden heikentyminen, psyykkisten toimintojen muutokset, näön menetys ja muut näköhäiriöt ja neurologiset häiriöt.
Jos PRES todetaan, Iclusig-hoito on keskeytettävä. Hoito voidaan aloittaa uudelleen vain, kun tapahtuma on korjautunut ja jos hoidon jatkamisesta koituva hyöty ylittää PRES-riskin.
Yhteisvaikutukset muiden lääkevalmisteiden kanssa
On oltava varovainen, jos Iclusigia ja kohtalaisen voimakkaita tai voimakkaita CYP3A:n estäjiä tai kohtalaisen voimakkaita tai voimakkaita CYP3A:n indusoijia käytetään samanaikaisesti (ks. kohta Yhteisvaikutukset)
Ponatinibin käytössä samanaikaisesti veren hyytymistä estävien lääkkeiden kanssa on noudatettava varovaisuutta, jos potilaalla saattaa olla verenvuototapahtumien riski (ks. ”Myelosuppressio” ja ”Verenvuodot”). Muodollisia tutkimuksia ponatinibin käytöstä hyytymistä estävien lääkkeiden kanssa ei ole tehty.
Haittavaikutusten, kuten maksatoksisuuden, luuydinlaman tai muiden, esiintyvyys voi suurentua (ks. kohta Haittavaikutukset) Ph+ ALL -potilailla, kun ponatinibia annetaan yhdessä kemoterapian kanssa (ks. kohta Farmakodynamiikka). Ponatinibin käyttö yhdessä kemoterapian kanssa edellyttää erityisiä varotoimia.
QT‑ajan pidentyminen
Iclusigin mahdollista QT‑aikaa pidentävää vaikutusta tutkittiin 39 leukemiapotilaassa. Mitään kliinisesti merkittävää QT‑ajan pitenemistä ei havaittu (ks. kohta Farmakodynamiikka). Perusteellista QT‑tutkimusta ei kuitenkaan ole tehty. Näin ollen kliinisesti merkittävää vaikutusta QT‑aikaan ei voida sulkea pois.
Erityisryhmät
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastaville potilaille voidaan antaa suositeltu aloitusannos. Varovaisuutta suositellaan, jos Iclusigia annetaan potilaille, joilla on maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Varovaisuutta suositellaan annettaessa Iclusigia potilaille, joilla arvioitu kreatiniinin puhdistuma on < 50 ml/min tai joilla on loppuvaiheen munuaistauti (ks. kohta Annostus ja antotapa).
Laktoosi
Tämä lääkevalmiste sisältää laktoosimonohydraattia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi‑intoleranssi, saamelaisilla esiintyvä laktaasinpuutos tai glukoosi‑galaktoosi‑imeytymishäiriö, ei tule käyttää tätä lääkettä.
Yhteisvaikutukset
Aineet, jotka voivat suurentaa ponatinibin pitoisuutta seerumissa
CYP3A:n estäjät
Ponatinibi metaboloituu CYP3A4:n vaikutuksesta.
Iclusigin suun kautta annettavan 15 mg kerta‑annoksen antaminen yhdessä voimakkaan CYP3A:n estäjän ketokonatsolin kanssa (400 mg vuorokaudessa) sai aikaan systeemisen ponatinibialtistuksen pienehkön lisääntymisen. Ponatinibin AUC0‑∞‑ ja Cmax‑arvot olivat 78 % ja vastaavasti 47 % korkeampia kuin annettaessa ponatinibia yksinään.
On oltava varovainen ja Iclusigin aloitusannoksen pienentämistä 30 mg:aan on harkittava, jos samanaikaisesti käytetään voimakkaita CYP3A:n estäjiä, kuten klaritromysiiniä, indinaviiria, itrakonatsolia, ketokonatsolia, nefatsodonia, nelfinaviiria, ritonaviiria, sakinaviiria, telitromysiiniä, troleandomysiiniä, vorikonatsolia ja greippimehua.
Aineet, jotka voivat pienentää ponatinibin pitoisuutta seerumissa
CYP3A:ta indusoivat aineet
Kun voimakasta CYP3A:n indusoijaa rifampisiinia (600 mg vuorokaudessa) saaville terveille vapaaehtoisille tutkittaville (19 henkilöä) annettiin samanaikaisesti yksi 45 mg:n Iclusig‑annos, ponatinibin AUC0‑∞‑arvot laskivat 62 % ja Cmax‑arvot 42 % verrattuna ponatinibin antoon yksinään.
Voimakkaiden CYP3A4:n indusoijien, kuten karbamatsepiinin, fenobarbitaalin, fenytoiinin, rifabutiinin, rifampisiinin ja mäkikuisman, samanaikaista antoa on vältettävä. CYP3A4:n indusoijille on etsittävä vaihtoehtoja, paitsi jos niiden tuoma hyöty on merkittävämpi kuin liian vähäisen ponatinibialtistuksen mahdollinen riski.
Aineet, joiden pitoisuus seerumissa voi muuttua ponatinibin vaikutuksesta
Kuljettajaproteiinien substraatit
In vitro ponatinibi on P‑gp:n ja BCRP:n estäjä. Näin ollen ponatinibi voi mahdollisesti kohottaa samaan aikaan annettujen P‑gp:n substraattien (esim. digoksiini, dabigatraani, kolkisiini, pravastatiini) tai BCRP:n substraattien (esim. metotreksaatti, rosuvastatiini, sulfasalatsiini) plasmapitoisuuksia ja korostaa niiden terapeuttista vaikutusta ja haittavaikutuksia. Huolellista kliinistä seurantaa suositellaan, kun ponatinibia annetaan näiden lääkevalmisteiden kanssa.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehille ja naisille
Iclusig‑hoitoa saavia naisia, jotka voivat ikänsä puolesta tulla raskaaksi, on kehotettava välttämään raskautta. Iclusig‑hoitoa saavia miehiä on kehotettava välttämään lapsen siittämistä hoidon aikana. Hoidon aikana on käytettävä tehokasta ehkäisymenetelmää. Ei ole tiedossa, heikentääkö ponatinibi systeemisten hormonaalisten ehkäisyvalmisteiden tehokkuutta. Valmistetta saavien potilaiden on käytettävä lisäehkäisyä tai vaihtoehtoista ehkäisymenetelmää.
Raskaus
Käytöstä raskaana oleville naisille on vain vähän tietoja (alle 50 tiedossa olevaa raskautta). Niiden perusteella ponatinibille ensimmäisen raskauskolmanneksen aikana altistuneille naisille syntyneillä lapsilla on ilmoitettu synnynnäistä megakoolonia (Hirschsprungin tautia). Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Iclusigia ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tila edellytä hoitoa ponatinibilla. Jos sitä käytetään raskauden aikana, potilaalle on kerrottava sikiöön kohdistuvasta mahdollisesta riskistä.
Imetys
Ei tiedetä, erittyykö Iclusig ihmisen rintamaitoon. Saatavilla olevat farmakodynaamiset ja toksikologiset tiedot eivät sulje pois mahdollista erittymistä ihmisen rintamaitoon. Imetys tulee keskeyttää Iclusig‑hoidon ajaksi.
Hedelmällisyys
Ponatinibin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Rotalla ponatinibihoito vaikutti naaraiden hedelmällisyyteen mutta ei urosten hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta). Löydösten kliinistä merkitystä ihmisen hedelmällisyydelle ei tunneta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Iclusig-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Iclusig-valmisteeseen liittyy haittavaikutuksia, kuten letargia, huimaus ja näön sumeneminen. Näin ollen suositellaan varovaisuutta ajettaessa tai käytettäessä koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Aiemmin hoidetut KML- tai Ph+ALL -potilaat tai potilaat, joilla T315I-mutaatio (PACE-tutkimus)
Faasin 2 PACE-tutkimuksessa (ks. kohta Farmakodynamiikka) yleisimmät vakavat haittavaikutukset, joiden ilmaantuvuus oli > 2 % (hoidon aikana ilmenneet tapaukset), olivat keuhkokuume (7,3 %), haimatulehdus (5,8 %), vatsakipu (4,7 %), eteisvärinä (4,5 %), kuume (4,5 %), sydäninfarkti (4,0 %), ahtauttava ääreisvaltimotauti (3,8 %), anemia (3,8 %), angina pectoris (3,3 %), verihiutalemäärän pieneneminen (3,1 %), kuumeinen neutropenia (2,9 %), hypertensio (2,9 %), sepelvaltimotauti (2,7 %), kongestiivinen sydämen vajaatoiminta (2,4 %), aivoverenkiertohäiriö (2,4 %), sepsis (2,4 %), selluliitti (2,2 %), akuutti munuaisvaurio (2,0 %), virtsatieinfektio (2,0 %) ja lipaasipitoisuuden suureneminen (2,0 %).
Haittavaikutuksina esiintyi vakavia kardiovaskulaarisia valtimotukoksia 10 prosentilla, aivovaltimoiden tukoksia 7 prosentilla ja ääreisvaltimoiden tukoksia 9 prosentilla Iclusig‑hoitoa saaneista potilaista (hoidon aikana ilmenneet tapaukset). Vakavia laskimotukoksia (hoidon aikana ilmenneet tapaukset) oli 5 prosentilla potilaista.
Haittavaikutuksina esiintyi kardiovaskulaarisia valtimotukoksia 13 prosentilla, aivovaltimoiden tukoksia 9 prosentilla ja ääreisvaltimoiden tukoksia 11 prosentilla Iclusig‑hoitoa saaneista potilaista (hoidon aikana ilmenneet tapaukset). Kaikkiaan valtimotukoksia ilmeni haittavaikutuksina 25 prosentilla Iclusig‑hoitoa saaneista potilaista faasin 2 PACE-tutkimuksessa, jossa seurannan vähimmäiskesto oli 64 kk, ja vakavia haittavaikutuksia oli 20 prosentilla potilaista. Joillakin potilailla oli enemmän kuin yhdenlaisia tapahtumia.
Laskimotromboemboliaa (hoidon aikana ilmenneet tapaukset) esiintyi 6 %:lla potilaista. Tromboembolisten tapahtumien ilmaantuvuus oli suurempi Ph+ ALL- tai BP‑KML-potilailla kuin AP‑KML- tai CP‑KML-potilailla. Mikään laskimotukostapahtuma ei johtanut kuolemaan.
Vähintään 64 kuukauden seurannan jälkeen hoidon keskeyttämiseen johtaneiden haittatapahtumien yleisyydet olivat 20 % CP‑KML:ssä, 11 % AP‑KML:ssä, 15 % BP‑KML:ssä ja 9 % Ph+ ALL:ssa.
Aiemmin hoidetut CP-KML-potilaat (OPTIC-tutkimus)
Faasin 2 OPTIC-tutkimuksessa (ks. kohta Farmakodynamiikka), kun seurannan mediaanikesto oli 77,93 kk, valtimotukoksia ilmeni haittavaikutuksina kaikkiaan 13,8 %:lla Iclusig-hoitoa saaneista potilaista (45 mg:n kohortti) ja 2 näistä johti kuolemaan, ja vakavia haittavaikutuksia ilmeni 8,5 %:lla potilaista (45 mg:n kohortti). Haittavaikutuksina esiintyi kardiovaskulaarisia valtimotukoksia 5,3 %:lla, aivovaltimoiden tukoksia 4,3 %:lla ja ääreisvaltimoiden tukoksia 4,3 %:lla Iclusig-hoitoa saaneista potilaista (45 mg:n kohortti, hoidon aikana ilmenneet tapaukset). 45 mg:n kohortin 94 potilaasta yhdelle ilmaantui laskimotromboembolia (asteen 1 verkkokalvon keskuslaskimotukos).
Potilaat, joilla on vastadiagnosoitu Ph+ ALL (PhALLCON-tutkimus)
Turvallisuusprofiili oli tapahtumien tyypin suhteen yhdenmukainen pelkän ponatinibin turvallisuusprofiilin kanssa kaikilla Ph+ ALL -potilailla, jotka saivat ponatinibia yhdessä kevennetyn kemoterapian kanssa. PhALLCON-tutkimuksessa raportoitiin luuydinlamatapahtumia 83 %:lla ponatinibia saaneista potilaista. Yleisimmin ilmoitetut haittavaikutukset olivat trombosytopenia (47 %), neutropenia (44 %) ja anemia (44 %). Maksatoksisuustapahtumia esiintyi 64 %:lla potilaista. Pelkän ponatinibin käyttöön verrattuna esiintyi kokonaisuudessaan enemmän kemoterapiaan liittyvää luuydinlamaa (kuumeinen neutropenia, pyreksia, pneumonia ja sepsis) sekä perifeeristä sensorista neuropeniaa ja suutulehdusta.
Haittavaikutukset taulukkomuodossa
Haittavaikutusten esiintymistiheydet Iclusig-monoterapiassa perustuvat tietoihin 449:ltä KML- ja Ph+ ALL -potilaalta, jotka saivat ponatinibia faasin 2 PACE-tutkimuksessa, ja 94:ltä KML-potilaalta, jotka saivat ponatinibia (45 mg:n aloitusannos) faasin 2 OPTIC-tutkimuksessa. Ks. kohdasta Farmakodynamiikka tiedot tutkimusten tutkittavien keskeisistä ominaisuuksista. Kaikilla KML‑ ja Ph+ ALL ‑potilailla raportoidut haittavaikutukset on lueteltu taulukossa 4 elinjärjestelmäluokan ja esiintymistiheyden mukaan.
Haittavaikutusten esiintyvyydet Iclusig-hoidossa yhdessä kemoterapian kanssa perustuvat 163:een vastadiagnosoituun Ph+ ALL -potilaaseen, jotka saivat faasin 3 PhALLCON-tutkimuksessa ponatinibia yhdessä kevennetyn kemoterapian kanssa ja sen jälkeen jatkohoitona Iclusig-monoterapiaa. Tutkimukseen osallistuneiden keskeiset ominaisuudet on esitetty kohdassa Farmakodynamiikka. Kaikilla vastadiagnosoiduilla Ph+ ALL -potilailla raportoidut haittavaikutukset on esitetty elinjärjestelmittäin taulukossa 5.
Yleisyyskategoriat ovat: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000) tai hyvin harvinainen (< 1/10 000), sekä tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 4 Aiemmin hoidetuilla KML‑ tai-, Ph+ ALL ‑potilailla ja potilailla, joilla on T315I-mutaatio, havaitut haittavaikutukset – esiintymistiheys perustuu hoidon aikana ilmenneiden tapahtumien ilmaantuvuuteen
| Elinjärjestelmäluokka | Esiintymistiheys | Haittavaikutukset |
| Infektiot | Hyvin yleinen | Ylähengitysteiden infektio |
| Yleinen | Pneumonia, sepsis, follikuliitti, selluliitti, herpes zoster | |
| Veri ja imukudos | Hyvin yleinen | Anemia, verihiutalemäärän pieneneminen, neutrofiilimäärän pieneneminen |
| Yleinen | Pansytopenia, kuumeinen neutropenia, valkosolumäärän pieneneminen, lymfosyyttimäärän pieneneminen, luuydinlama | |
| Umpieritys | Yleinen | Hypotyreoosia |
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Ruokahalun aleneminen, hypertriglyseridemia, hyperkolesterolemia |
| Yleinen | Nestehukka, nesteen kertyminen, hypo-kalsemia, hyperglykemia, hyperurikemia, hypo¬fosfatemia, hypokalemia, painon lasku, hyponatremia, dyslipidemia, heikentynyt glukoosinsieto, suurentunut LDL-kolesteroliarvo, painonnousu, tuumorilyysioireyhtymä | |
| Psyykkiset häiriöt | Hyvin yleinen | Unettomuus |
| Yleinen | Ahdistuneisuus | |
| Hermosto | Hyvin yleinen | Päänsärky, huimaus |
| Yleinen | Aivoverenkiertohäiriö, aivoinfarkti, perifeerinen neuropatia, letargia, migreeni, hyperestesia, hypestesia, parestesia, ohimenevä aivoverenkiertohäiriö, kasvohermon häiriö, kaulavaltimon ahtauma | |
| Melko harvinainen | Aivovaltimon stenoosi, aivoverenvuoto, kallonsisäinen verenvuoto, posteriorinen reversiibeli enkefalopatiaoireyhtymä* | |
| Silmät | Yleinen | Näön sumeneminen, silmän kuivuminen, turvotus silmäkuopan ympärillä, silmäluomien turvotus, sidekalvotulehdus, näön huononeminen, silmäkipu, verkkokalvon laskimotukos |
| Melko harvinainen | Verkkokalvon laskimotromboosi, verkkokalvon valtimotukos | |
| Sydän | Yleinen | Sydämen vajaatoiminta, sydäninfarkti, kongestiivinen sydämen vajaatoiminta, sepelvaltimotauti, angina pectoris, perikardiaalinen effuusio, eteisvärinä, ejektiofraktion pieneneminen, sepelvaltimotautikohtaus, eteislepatus, vasemman kammion toimintahäiriö, vasemman kammion hypertrofia, sinusbradykardia, takykardia, suurentunut B-tyypin natriureettinen N-terminaalinen propeptidiarvo, epävakaa sepelvaltimotauti, sydänlihasiskemia, supraventrikulaariset lisälyönnit, kammiolisälyönnit, pidentynyt sydänsähkökäyrän QT-aika, krooninen sydämen vajaatoiminta, suurentunut B-tyypin natriureettisen peptidin arvo |
| Melko harvinainen | Sydänvaivat, iskeeminen kardiomyopatia, sepelvaltimospasmi | |
| Verisuonisto | Hyvin yleinen | Hypertensio |
| Yleinen | Ahtauttava ääreisvaltimotauti, perifeerinen iskemia, ääreisvaltimoiden ahtauma, katkokävely, syvä laskimotromboosi, kuuma aalto, punastuminen, hypertensiivinen kriisi | |
| Melko harvinainen | Huono ääreisverenkierto, pernainfarkti, laskimoembolia, laskimotromboosi, munuaisvaltimostenoosi | |
| Tuntematon | Aneurysmat ja valtimon dissekaatiot | |
| Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Hengenahdistus, yskä |
| Yleinen | Keuhkoembolia, pleuraeffuusio, nenäverenvuoto, dysfonia, keuhkoverenpainetauti, suunielun kipu, produktiivinen yskä | |
| Ruoansulatuselimistö | Hyvin yleinen | Vatsakipu, ripuli, oksentelu, ummetus, pahoinvointi, lipaasipitoisuuden suureneminen |
| Yleinen | Haimatulehdus, veren amylaasipitoisuuden suureneminen, ruokatorven refluksitauti, suutulehdus, dyspepsia, vatsan turpoaminen, epämukava tunne vatsassa, suun kuivuminen, mahan verenvuoto, mahatulehdus, mahahaava, ikenen verenvuoto | |
| Maksa ja sappi | Hyvin yleinen | Suurentunut alaniiniaminotransferaasipitoisuus, suurentunut aspartaattiaminotransferaasipitoisuus |
| Yleinen | Suurentunut veren bilirubiinipitoisuus, suurentunut alkalisen fosfataasin pitoisuus veressä, suurentunut gammaglutamyylitransferaasipitoisuus, suurentuneet transaminaasiarvot, maksatoksisuus | |
| Melko harvinainen | Maksan vajaatoiminta, ikterus | |
| Iho ja ihonalainen kudos | Hyvin yleinen | Ihottuma, kuiva iho, kutina |
| Yleinen | Kutiseva ihottuma, hilseilevä ihottuma, punoitus, hiustenlähtö, ihon hilseily, yöhikoilut, liikahikoilu, petekiat, mustelma, ihon kipu, eksfoliatiivinen dermatiitti, hyperkeratoosi, ihon hyperpigmentaatio, pannikuliitti (kyhmyruusu mukaan luettuna), dermatiitti, makulopapulaarinen ihottuma, aknea muistuttava dermatiitti, erytematoottinen ihottuma, ekseema, makulaarinen ihottuma, papulaarinen ihottuma, monimuotoinen punavihottuma, allerginen dermatiitti, ihopapillooma, psoriaasinkaltainen dermatiitti | |
| Luusto, lihakset ja sidekudos | Hyvin yleinen | Luukipu, nivelkipu, lihaskipu, raajakipu, selkäkipu, lihasspasmit |
| Yleinen | Lihaksiin ja luustoon liittyvä kipu, niskakipu, lihaksiin ja luustoon liittyvä rintakipu, lihasheikkous, tuki- ja liikuntaelinten jäykkyys, selkärangan kipu, jännetulehdus | |
| Sukupuolielimet ja rinnat | Yleinen | Erektiohäiriö |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Uupumus, astenia, perifeerinen turvotus, kuume, kipu |
| Yleinen | Vilunväristykset, influenssan kaltainen sairaus, sydämeen liittymätön rintakipu, kyhmy antopaikassa, kasvojen turvotus, suurentunut C-reaktiivisen proteiinin arvo, rintakipu |
* Markkinoilletulon jälkeiset spontaanit raportit
a Kilpirauhasen vajaatoiminta sisältää kilpirauhasen vajaatoiminnan ja primaarisen kilpirauhasen vajaatoiminnan
Taulukko 5 Vastadiagnosoiduilla Ph+ ALL -potilailla PhALLCON-tutkimuksessa todetut haittavaikutukset – esiintymistiheys ilmoitettu hoitoon liittyvien tapahtumien esiintyvyyden perusteella
Elinjärjestelmä | Esiintymistiheys | Ponatinibi yhdessä kevennetyn kemoterapian kanssa Haittavaikutukset |
|---|---|---|
Infektiot | Yleinen | Pneumonia, sidekalvotulehdus, sepsis, septinen sokki, neutropeeninen infektio |
Veri ja imukudos | Hyvin yleinen | Trombosytopenia, anemia, neutropenia, kuumeinen neutropenia, leukopenia, leukosytoosi |
Yleinen | Luuydinlama, lymfopenia, sytopenia, agranulosytoosi | |
Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Hypokalemia, hyperglykemia, hypokalsemia, hypofosfatemia, hyperurikemia |
Yleinen | Ruokahalun aleneminen, hypertriglyseridemia, hyponatremia, hypoalbuminemia, hyperkolesterolemia, dyslipidemia, nesteen kertyminen | |
Psyykkiset häiriöt | Hyvin yleinen | Unettomuus |
Hermosto | Hyvin yleinen | Päänsärky, perifeerinen neuropatia, parestesia, perifeerinen sensorinen neuropatia, huimaus |
Yleinen | Hypoestesia | |
Silmät | Yleinen | Sidekalvon verenvuoto |
Melko harvinainen | Verkkokalvon laskimotukos | |
Sydän | Yleinen | Takykardia, sydämentykytykset, sydänpussieffuusio, eteisvärinä, sinusbradykardia, angina pectoris |
Melko harvinainen | Sydämen vajaatoiminta, akuutti sydäninfarkti, kongestiivinen sydämen vajaatoiminta | |
Verisuonisto | Hyvin yleinen | Hypertensio |
Yleinen | Syvä laskimotromboosi, pinnallisen laskimon tromboosi, embolia | |
Melko harvinainen | Ahtauttava ääreisvaltimotauti, ääreisosien kylmyys, tromboosi | |
Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Yskä |
Yleinen | Hengenahdistus, suunielun kipu, pleuraeffuusio, dysfonia, keuhkoembolia | |
Ruoansulatuselimistö | Hyvin yleinen | Ummetus, pahoinvointi, oksentelu, suutulehdus, ripuli, vatsakipu, ylävatsakipu |
Yleinen | Dyspepsia, vatsan turpoaminen, vatsavaivat, haimatulehdus, mahatulehdus, akuutti haimatulehdus | |
Melko harvinainen | Suun verenvuoto | |
Maksa ja sappi | Yleinen | Maksatoksisuus, hyperbilirubinemia, hypertransaminasemia, toksinen maksatulehdus |
Melko harvinainen | Lääkkeen aiheuttama maksavaurio, maksan ja sapen sairaus, maksavaurio | |
Iho ja ihonalainen kudos | Hyvin yleinen | Ihottuma, kuiva iho |
Yleinen | Kutina, hiustenlähtö, makulopapulaarinen ihottuma | |
Luusto, lihakset ja sidekudos | Hyvin yleinen | Selkäkipu, raajakipu, nivelkipu, lihaskipu |
Yleinen | Luukipu, niskakipu, lihasspasmit | |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Pyreksia, uupumus, heikkous, perifeerinen turvotus |
Yleinen | Rintakipu, kipu | |
Tutkimukset | Hyvin yleinen | Suurentunut alaniiniaminotransferaasipitoisuus, lipaasipitoisuuden suureneminen, suurentunut aspartaattiaminotransferaasipitoisuus, suurentunut gammaglutamyylitransferaasipitoisuus, suurentunut veren laktaattidehydrogenaasipitoisuus, amylaasipitoisuuden suureneminen |
Yleinen | Suurentunut alkalisen fosfataasin pitoisuus veressä, suurentunut veren kreatiniinipitoisuus, pienentynyt veren fibrinogeenipitoisuus, suurentunut C-reaktiivisen proteiinin arvo, suurentunut neutrofiilimäärä, pienentynyt kokonaisproteiiniarvo, suurentunut verihiutalemäärä, suurentunut B-tyypin natriureettisen peptidin arvo, suurentunut troponiini 1 -arvo | |
Melko harvinainen | Ejektiofraktion pieneneminen | |
Vammat, myrkytykset ja hoitokomplikaatiot | Melko harvinainen | Subduraalihematooma |
Valikoitujen haittavaikutusten kuvaus
Verisuonitukos (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Iclusig‑hoitoa saaneilla potilailla on todettu vakavia verisuonitukoksia, kuten kardiovaskulaarisia ja aivo‑ sekä ääreisverisuonten tapahtumia ja laskimotromboositapahtumia. Näitä tapahtumia on ilmennyt sekä potilailla, joilla oli kardiovaskulaarisia riskitekijöitä, että potilailla, joilla ei ollut tällaisia riskitekijöitä. Tapahtumia esiintyi myös 50‑vuotiailla ja sitä nuoremmilla. Haittatapahtumat, joihin liittyi valtimotukos, olivat yleisempiä vanhemmilla potilailla ja niillä, joilla oli anamneesissa iskemia, korkea verenpaine, diabetes tai hyperlipidemia.
Faasin 2 PACE-tutkimuksessa (ks. kohta Farmakodynamiikka), kun seurannan vähimmäiskesto oli 64 kk, haittavaikutuksina esiintyi kardiovaskulaarisia valtimotukoksia 13 %:lla, aivovaltimoiden tukoksia 9 %:lla ja ääreisvaltimoiden tukoksia 11 %:lla Iclusig-hoitoa saaneista potilaista (hoidon aikana ilmenneet tapaukset). Valtimotukoksia ilmeni haittavaikutuksina kaikkiaan 25 %:lla faasin 2 PACE-tutkimuksessa Iclusig-hoitoa saaneista potilaista, ja vakavia haittavaikutuksia ilmeni 20 %:lla potilaista. Joillakin potilailla ilmeni useampia eri tyyppisiä tapahtumia. PACE-tutkimuksessa mediaaniaika ensimmäiseen kardiovaskulaariseen valtimotukostapahtumaan oli 351 vrk, ensimmäiseen aivovaltimoiden tukostapahtumaan 611 vrk ja ensimmäiseen ääreisvaltimoiden tukostapahtumaan 605 vrk. Laskimotromboembolisia haittavaikutuksia ilmeni 6 %:lla potilaista (hoidon aikana ilmenneet tapaukset).
Faasin 2 OPTIC-tutkimuksessa (ks. kohta Farmakodynamiikka), kun seurannan mediaanikesto oli 77,9 kk, haittavaikutuksina esiintyi kardiovaskulaarisia valtimotukoksia 5,3 %:lla, aivovaltimoiden tukoksia 4,3 %:lla ja ääreisvaltimoiden tukoksia 4,3 %:lla Iclusig-hoitoa saaneista potilaista (45 mg:n kohortti, hoidon aikana ilmenneet tapaukset). Valtimotukoksia ilmeni haittavaikutuksina kaikkiaan 13,8 %:lla Iclusig-hoitoa saaneista potilaista (45 mg:n kohortti), ja vakavia haittavaikutuksia ilmeni 8,5 %:lla potilaista (45 mg:n kohortti). OPTIC-tutkimuksessa mediaaniaika ensimmäiseen kardiovaskulaariseen valtimotukostapahtumaan oli 473 vrk, ensimmäiseen aivovaltimoiden tukostapahtumaan 356 vrk ja ensimmäiseen ääreisvaltimoiden tukostapahtumaan 108 vrk. OPTIC-tutkimuksen 94 potilaasta (45 mg:n kohortti) yhdelle ilmaantui laskimotromboembolia.
Faasin 3 PhALLCON-tutkimuksessa (ks. kohta Farmakodynamiikka), jossa seuranta-ajan mediaani oli 20,43 kuukautta, kardiovaskulaaristen valtimoiden tukoksia esiintyi haittavaikutuksena 1,2 %:lla, aivoverisuonten tukoksia 0,6 %:lla ja ääreisverenkierron valtimoiden tukoksia 0,6 %:lla potilaista, joita hoidettiin ponatinibilla yhdessä kemoterapian kanssa. Laskimotromboemboliatapahtumia esiintyi 12 %:lla potilaista, jotka saivat PhALLCON-tutkimuksessa ponatinibia yhdessä kemoterapian kanssa.
Luuydinlama
PACE-tutkimuksessa luuydinlamaa raportoitiin yleisesti kaikissa potilasryhmissä. Asteen 3 tai 4 trombosytopenian, neutropenian ja anemian yleisyys oli suurempi potilailla, joilla oli AP‑KML ja BP‑KML/Ph+ ALL, kuin potilailla, joilla oli CP‑KML (ks. taulukko 6). Luuydinlamaa raportoitiin sekä potilailla, joilla oli normaalit lähtötilanteen laboratorioarvot, että potilailla, joiden laboratorioarvot olivat ennestään poikkeavat.
Keskeyttäminen luuydinlaman johdosta oli suhteellisen harvinaista (trombosytopenia 4 %, neutropenia ja anemia < 1 % kumpikin).
Luuydinlamatapahtumia raportoitiin 83 %:lla ponatinibia saaneista potilaista PhALLCON-tutkimuksessa, 63 %:lla ponatinibia saaneista potilaista OPTIC-tutkimuksessa (45 mg:n kohortti) ja 60 %:lla ponatinibia saaneista potilaista PACE-tutkimuksessa.
Maksatoksisuus
Maksatoksisuustapahtumia esiintyi 64 %:lla ponatinibia yhdessä kemoterapian kanssa saaneista potilaista PhALLCON-tutkimuksessa, 28 %:lla ponatinibia saaneista potilaista OPTIC-tutkimuksessa (45 mg:n kohortti) ja 30 %:lla ponatinibia saaneista potilaista PACE-tutkimuksessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hepatiitti B:n uudelleen aktivoituminen
Hepatiitti B:n uudelleen aktivoitumista on ilmoitettu BCR-ABL-tyrosiinikinaasin estäjien käytön yhteydessä. Tämä aiheutti joissakin tapauksissa maksan vajaatoimintaa tai fulminanttia hepatiittia, joka johti maksansiirtoon tai kuolemaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vaikeat ihohaittavaikutukset
Joidenkin BCR-ABL-tyrosiinikinaasin estäjien käytön yhteydessä on ilmoitettu vaikeita ihoreaktioita (kuten Stevens–Johnsonin oireyhtymää). Potilaita on kehotettava ilmoittamaan välittömästi epäillyistä ihoreaktioista etenkin, jos niihin liittyy rakkulamuodostusta, kesimistä, limakalvoaffisiota tai systeemisiä oireita.
Taulukko 6 Kliinisesti relevantit, asteen 3/4* laboratorioarvojen poikkeavuudet, joita esiintyi ≥ 2 %:lla potilaista missä tahansa tautiryhmässä faasin 2 PACE-tutkimuksessa (N = 449): seurannan kesto vähintään 64 kk kaikilla edelleen hoitoa saaneilla potilailla
| Laboratoriokoe | Kaikki potilaat (N=449) (%) | CP‑KML (N=270) (%) | AP‑KML (N=85) (%) | BP‑KML/Ph+ ALL (N=94) (%) |
| Hematologia | ||||
| Trombosytopenia (alentunut verihiutalearvo) | 40 | 35 | 49 | 46 |
| Neutropenia (ANC alentunut) | 34 | 23 | 52 | 52 |
| Leukopenia (valkosolumäärä alentunut) | 25 | 12 | 37 | 53 |
| Anemia (hemoglobiini alentunut) | 20 | 8 | 31 | 46 |
| Lymfopenia | 17 | 10 | 25 | 28 |
| Biokemialliset arvot | ||||
| Lipaasi koholla | 14 | 14 | 13 | 14 |
| Fosfori alentunut | 10 | 10 | 13 | 9 |
| Glukoosi koholla | 7 | 8 | 13 | 1 |
| ALAT koholla | 6 | 4 | 8 | 7 |
| Natrium alentunut | 5 | 6 | 6 | 2 |
| ASAT koholla | 4 | 3 | 5 | 3 |
| Amylaasi koholla | 4 | 4 | 4 | 3 |
| Kalium alentunut | 2 | < 1 | 6 | 2 |
| Kalium koholla | 2 | 2 | 1 | 3 |
| Alkalinen fosfataasi koholla | 2 | 2 | 4 | 2 |
| Bilirubiini | 1 | < 1 | 2 | 1 |
| Kalsium alentunut | 1 | < 1 | 2 | 1 |
ALAT = alaniiniaminotransferaasi, ANC = absoluuttinen neutrofiilimäärä, ASAT = aspartaattiaminotransferaasi. *Raportoitu käyttämällä National Cancer Institute Common Terminology Criteria for Adverse Events kriteerien versiota 4.0. | ||||
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa raportoitiin yksittäisiä, tahattomia Iclusig‑yliannostuksia. 165 mg:n ja arviolta 540 mg:n kerta‑annokset kahdessa potilaassa eivät aiheuttaneet mitään kliinisesti merkittäviä haittavaikutuksia. 90 mg:n vuorokausiannosten toistuva käyttö 12 vuorokauden ajan sai aikaan pneumonian, systeemisen tulehdusvasteen, eteisvärinän ja oireettoman, kohtalaisen perikardiumeffuusion. Hoito keskeytettiin, tapahtumat korjautuivat ja Iclusig aloitettiin uudelleen 45 mg kerran vuorokaudessa ‑annoksella. Iclusig‑yliannoksen yhteydessä potilasta tulee tarkkailla ja antaa asianmukaista tukihoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: solunsalpaajat, proteiinikinaasin estäjät, ATC‑koodi: L01EA05
Ponatinibi on voimakas kaikkien BCR‑ABL‑kinaasien estäjä, jonka rakenteelliset elementit, mm. hiili‑hiili‑kolmoissidos, mahdollistavat natiiviin BCR‑ABL:ään ja ABL‑kinaasin mutanttimuotoihin sitoutumisen korkealla affiniteetilla. Ponatinibi estää ABL:n ja T315I‑mutantti‑ABL:n tyrosiinikinaasiaktiivisuutta IC50‑arvoilla 0,4 nM ja vastaavasti 2,0 nM. Solukokeissa ponatinibi kykeni voittamaan BCR‑ABL‑kinaasidomeenin mutaatioiden välittämän resistenssin imatinibille, dasatinibille ja nilotinibille. Prekliinisissä mutageneesitutkimuksissa todettiin, että 40 nM:n ponatinibipitoisuus riitti estämään > 50 %:lla kaikkia testattuja BCR‑ABL‑mutantteja (T315I mukaan lukien) ilmentävien solujen elinkelpoisuutta ja estämään mutanttikloonien ilmaantumista. Solupohjaisessa nopeutetun mutageneesin kokeessa ei havaittu mitään BCR‑ABL‑mutaatiota, joka voisi antaa resistenssin 40 nM:n ponatinibipitoisuuksille. Ponatinibi sai aikaan kasvaimen kutistumista ja pidensi elinaikaa hiirillä, joiden kasvaimet ilmensivät natiivia tai T315I‑mutantti‑BCR‑ABL:ää.
30 mg:n tai tätä suuremmilla annoksilla ponatinibin vakaan tilan pienimmät pitoisuudet plasmassa ylittävät tyypillisesti arvon 21 ng/ml (40 nM). 15 mg:n tai tätä suuremmilla annoksilla 32 potilaalla 34:stä (94 %:lla) todettiin, että CRKL (CRK-like) fosforylaatio (BCR‑ABL:n estymisen biomarkkeri) väheni perifeerisen veren mononukleaarisoluissa ≥ 50 %. Ponatinibi estää muiden kliinisesti relevanttien kinaasien aktiivisuuden (IC50‑arvo alle 20 nM), ja sillä on osoitettu solutason aktiivisuutta RET:iä, FLT3:a ja KIT:iä vastaan, sekä FGFR‑, PDGFR‑ ja VEGFR‑kinaasiperheiden kinaaseja vastaan.
Kliininen teho ja turvallisuus
Potilaat, joilla on KML ja, Ph+ ALL ja joita on aiemmin hoidettu tyrosiinikinaasin estäjillä (TKI:t) tai joilla on T315I-mutaatio.
PACE-tutkimus
Iclusigin turvallisuus ja teho KML‑ ja Ph+ ALL ‑potilailla, jotka olivat resistenttejä tai intolerantteja aikaisemmalle tyrosiinikinaasin estäjähoidolle (TKI–hoidolle), arvioitiin yksiryhmäisessä, avoimessa, kansainvälisessä monikeskustutkimuksessa. Kaikille potilaille annettiin 45 mg Iclusigia kerran vuorokaudessa, ja annosta voitiin pienentää ja annostelu keskeyttää, jota seurasi annostelun aloittaminen uudelleen ja annoksen koon kasvattaminen uudelleen. Potilaat määrättiin yhteen kuudesta kohortista, perustuen taudin vaiheeseen (CP‑KML; AP‑KML; tai BP‑KML/Ph+ ALL), resistenssiin tai intoleranssiin dasatinibille tai nilotinibille (R/I) ja T315I‑mutaation läsnäoloon.
Resistenssi CP‑KML:ssä määriteltiin kyvyttömyydeksi saavuttaa joko täydellinen hematologinen vaste (3 kk:n kuluessa), vähäinen sytogeneettinen vaste (6 kk:n kuluessa) tai huomattava sytogeneettinen vaste (12 kk:n kuluessa) dasatinibi‑ tai nilotinibihoidon aikana. CP‑KML‑potilaita, joilla hoitovaste katosi tai joille kehittyi kinaasidomeenimutaatio täydellisen sytogeneettisen vasteen puuttuessa tai tauti eteni AP‑KML:ksi tai BP‑KML:ksi missä tahansa dasatinibi‑ tai nilotinibihoidon vaiheessa, pidettiin myös resistentteinä. Resistenssi AP‑KML:ssä ja BP‑KML/Ph+ ALL:ssä määriteltiin joko kyvyttömyydeksi saavuttaa huomattava hematologinen vaste (AP‑KML 3 kk:n kuluessa, BP‑KML/Ph+ ALL 1 kk:n kuluessa), huomattavan hematologisen vasteen katoamiseksi (milloin tahansa) tai kinaasidomeenimutaation kehittymiseksi huomattavan hematologisen vasteen puuttuessa dasatinibi‑ tai nilotinibihoidon aikana.
Intoleranssi määriteltiin dasatinibi‑ tai nilotinibihoidon keskeytymiseksi toksisuuden vuoksi optimaalisesta hoidosta huolimatta täydellisen sytogeneettisen vasteen puuttuessa CP‑KML‑potilailla tai huomattavan hematologisen vasteen puuttuessa AP‑KML‑, BP‑KML‑ tai Ph+ ALL ‑potilailla.
Ensisijainen tehon päätetapahtuma CP‑KML:ssä oli huomattava sytogeneettinen vaste (MCyR), joka sisälsi täydelliset ja osittaiset sytogeneettiset vasteet (CCyR ja PCyR), 12 kk:een mennessä. Toissijaiset tehon päätetapahtumat CP‑KML:ssä olivat täydellinen hematologinen vaste (CHR) ja huomattava molekulaarinen vaste (MMR).
Ensisijainen tehon päätetapahtuma AP‑KML:ssä ja BP‑KML/Ph+ ALL:ssa oli huomattava hematologinen vaste (MaHR), joka määriteltiin joko täydelliseksi hematologiseksi vasteeksi (CHR) tai leukemian merkkien puuttumiseksi (NEL). Toissijaiset tehon päätetapahtumat AP‑KML:ssä ja BP‑KML/Ph+ ALL:ssa olivat MCyR ja MMR.
Kaikilla potilailla toissijaisia tehon lisäpäätetapahtumia olivat vahvistettu MCyR, aika vasteeseen, vasteen kestoaika, elinaika ilman taudin etenemistä ja kokonaiselinaika. Lisäksi tehtiin jälkianalyysejä, joissa arvioitiin lyhyemmän aikavälin sytogeneettisen vasteen (MCyR) ja molekulaarisen vasteen (MMR) yhteyttä pidemmän aikavälin tuloksiin eli etenemättömyysaikaan (PFS) ja kokonaiselossaoloaikaan (OS), vasteen (MCyR ja MMR) säilymistä annoksen pienentämisen jälkeen sekä etenemättömyysaikaa ja kokonaiselossaoloaikaa suhteessa valtimotukostapahtumastatukseen.
Tutkimukseen otettiin 449 potilasta, joista 444 kelpasi analyysiin: 267 CP‑KML‑potilasta (R/I‑kohortti: n=203, T315I‑kohortti: n=64), 83 AP‑KML‑potilasta (R/I‑kohortti: n=65, T315I‑kohortti: n=18), 62 BP‑KML‑potilasta (R/I‑kohortti: n=38, T315I‑kohortti: n=24) ja 32 Ph+ ALL ‑potilasta (R/I‑kohortti: n=10, T315I‑kohortti: n=22). Vain 26 % CP‑KML‑potilaista oli saavuttanut aiemmin MCyR‑tasoisen tai paremman vasteen (MCyR, MMR tai CMR) dasatinibille tai nilotinibille. MaHR‑tasoisen tai paremman vasteen (MaHR, MCyR, MMR tai CMR) oli saavuttanut aiemmin vain 21 % AP‑KML‑potilaista ja 24 % BP‑KML/Ph+ALL‑potilaista. Lähtötilanteen demografiset tiedot esitetään taulukossa 7 alla.
Taulukko 7 Demografiset tiedot ja taudin ominaisuudet PACE-tutkimuksessa
| Potilaiden ominaisuudet tutkimukseen otettaessa | Kokonaisturvallisuuspopulaatio N=449 |
| Ikä | |
| Mediaani, vuotta (vaihteluväli) | 59 (18–94) |
| Sukupuoli, n (%) | |
| Mies | 238 (53 %) |
| Rotu, n (%) | |
| Aasialainen | 59 (13 %) |
| Musta/afroamerikkalainen | 25 (6 %) |
| Valkoinen | 352 (78 %) |
| Muu | 13 (3 %) |
| ECOG‑suorituskykystatus, n (%) | |
| ECOG=0 tai 1 | 414 (92 %) |
| Tautihistoria | |
| Mediaaniaika diagnoosista ensimmäiseen annokseen, vuotta (vaihteluväli) | 6,09 (0,33–28,47) |
| Resistentti aikaisemmalle TKI‑hoidollea*, n (%) | 374 (88 %) |
| Aikaisempi TKI‑hoito – hoito‑ohjelmien lukumäärä, n (%) | |
| 1 | 32 (7 %) |
| 2 | 155 (35 %) |
| ≥ 3 | 262 (58 %) |
| BCR‑ABL‑mutaatioita havaittu tutkimukseen otettaessa, n (%)b | |
| Ei mitään | 198 (44 %) |
| 1 | 192 (43 %) |
| ≥ 2 | 54 (12 %) |
| Samanaikaiset sairaudet | |
| Hypertensio | 159 (35 %) |
| Diabetes | 57 (13 %) |
| Hyperkolesterolemia | 100 (22 %) |
| Anamneesissa iskeeminen sydänsairaus | 67 (15 %) |
a* 427 potilaasta, jotka raportoivat saaneensa aikaisempaa TKI‑hoitoa dasatinibilla tai nilotinibilla b Potilailla, joilla havaittiin yksi tai useampi BCR‑ABL‑kinaasidomeenimutaatio tutkimukseen otettaessa, havaittiin 37 ainutlaatuista mutaatiota. | |
Yhteensä 55 %:lla potilaista oli yksi tai useampi BCR‑ABL‑kinaasidomeenimutaatio tutkimukseen otettaessa, ja yleisimmät mutaatiot olivat T315I (29 %), F317L (8 %), E255K (4 %) ja F359V (4 %). 67 %:lla CP‑KML‑potilaiden R/I‑kohortista ei havaittu mitään mutaatioita tutkimukseen otettaessa.
Tehokkuustulokset esitetään yhteenvetona taulukoissa 8, 9 ja 10.
Taulukko 8 Iclusigin teho resistenteissä tai intoleranteissa kroonisen vaiheen KML‑potilaissa
Kokonaismäärä (N=267) | Resistentti tai intolerantti | ||
R/I‑ kohortti (N=203) | T315I‑ kohortti (N=64) | ||
| Sytogeneettinen vaste | |||
Huomattava (MCyR) a % (95 % lv) | 55 % (49–62) | 51 % (44–58) | 70 % (58–81) |
Täydellinen (CCyR) % (95 % lv) | 46 % (40–52) | 40 % (33–47) | 66 % (53–77) |
Huomattava molekulaarinen vaste b % (95 % lv) | 40 % (35–47) | 35 % (28–42) | 58 % (45–70) |
a Ensisijainen päätetapahtuma CP‑KML‑kohorteissa oli MCyR, joka yhdistää sekä täydelliset (ei havaittavissa olevia Ph+‑soluja) että osittaiset (1 % – 35 % Ph+‑soluja) sytogeneettiset vasteet. b Mitattu perifeerisestä verestä. Määritelmä: BCR‑ABL‑transkriptien ja ABL‑transkriptien suhde ≤ 0,1 % International Scale (IS) ‑asteikolla (ts. ≤ 0,1 % BCR‑ABLIS; potilailla oli oltava b2a2/b3a2 (p210) ‑transkripti) perifeerisessä veressä, mittaustapa qRT‑PCR (kvantitatiivinen käänteistranskriptaasientsyymiä hyödyntävä polymeraasiketjureaktio). Tiedonkeruu tietokannasta päättyi 6.2.2017. | |||
CP‑KML‑potilaat, jotka olivat saaneet vähemmän aikaisempia TKI‑valmisteita, saivat parempia sytogeneettisiä, hematologisia ja molekulaarisia vasteita. CP‑KML‑potilaista, joita oli aikaisemmin hoidettu yhdellä, kahdella, kolmella tai neljällä TKI‑valmisteella, vastaavasti 75 % (12/16), 68 % (66/97), 44 % (63/142) ja 58 % (7/12) sai MCyR:n saadessaan Iclusig‑hoitoa. Annosintensiteetin mediaani oli 28 mg/vrk eli 63 % odotetusta 45 mg:n annoksesta.
Niistä CP‑KML‑potilaista, joilla ei havaittu mutaatiota tutkimukseen otettaessa, 49 % (66/136) saavutti MCyR:n.
Kaikissa BCR‑ABL‑mutaatioissa, joita havaittiin useammassa kuin yhdessä CP‑KML‑potilaassa tutkimukseen otettaessa, saavutettiin MCyR‑vaste Iclusig‑hoidon jälkeen.
CP‑KML‑potilailla, jotka saavuttivat MCyR:n, mediaaniaika MCyR:iin oli 2,8 kk (vaihteluväli: 1,6–11,3 kk); potilailla, jotka saavuttivat MMR:n, mediaaniaika MMR:iin oli 5,5 kk (vaihteluväli: 1,8–55,5 kk). Raporttipäivityksen aikana, jolloin kaikkien edelleen mukana olleiden potilaiden seuranta‑aika oli vähintään 64 kuukautta, MCyR:n ja MMR:n mediaanikestoaikoja ei ollut vielä saavutettu. Kaplan–Meier‑estimaattien perusteella ennustettiin, että 82 % (95 % lv: [74 % – 88 %]) CP‑KML‑potilaista (hoidon mediaanikesto 32,2 kk), jotka saavuttivat MCyR:n, säilyttää tämän vasteen 48 kuukauden kohdalla ja 61 % (95 % lv: [51 % – 70 %]) CP‑KML‑potilaista, jotka saavuttivat MMR:n, säilyttää tämän vasteen 36 kuukauden kohdalla. MCyR- ja MMR-vasteiden säilymistodennäköisyys kaikkien CP-KML-potilaiden kohdalla ei enää muuttunut, kun analyysi pidennettiin 5 vuoden ajalle.
Kun seurannan kesto oli vähintään 64 kk, tauti oli edennyt AP‑KML- tai BP‑KML-vaiheeseen 3,4 %:lla (9/267) CP-KML-potilaista.
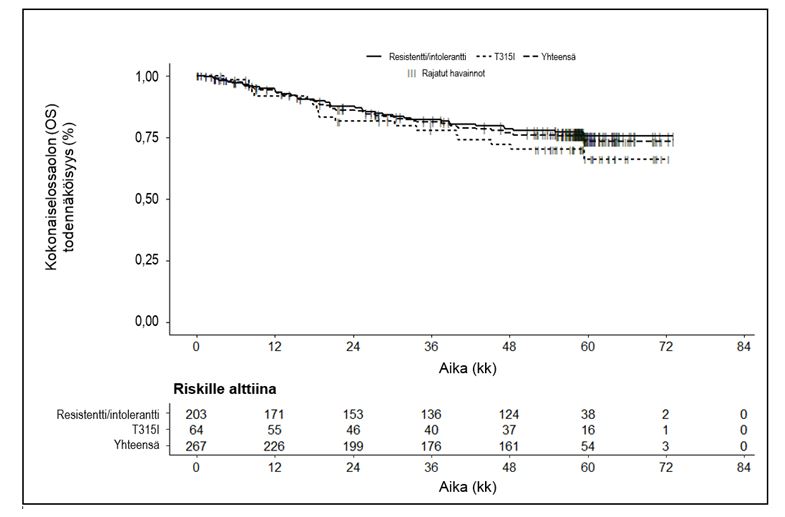
Kokonaiselossaoloajan mediaania ei ole vielä saavutettu kaikkien CP-KML-potilaiden ryhmässä (N = 267), CP-KML-potilaiden R/I-kohortin A potilailla (N = 203) eikä T315I-kohortin B potilailla (N = 64). Koko CP-KML-ryhmässä elossaolon arvioitu todennäköisyys on 2 vuoden kohdalla 86,0 %, 3 vuoden kohdalla 81,2 %, 4 vuoden kohdalla 76,9 % ja 5 vuoden kohdalla 73,3 %, kuten kuvassa 1 esitetään.
Kuva 1 – Kokonaiselossaolon (OS) Kaplan–Meier-estimaatit CP-KML-potilailla (hoidettu populaatio)
CP-KML-potilailla, jotka saavuttivat ensimmäisen hoitovuoden aikana MCyR- tai MMR-vasteen, saavutettiin tilastollisesti merkitsevästi parempi etenemättömyysaika (PFS) ja kokonaiselossaoloaika (OS) kuin potilailla, joilla näitä hoitotavoitteita ei saavutettu. MCyR-vaste 3 kk kohdalla korreloi vahvasti ja tilastollisesti merkitsevästi etenemättömyysaikaan (p < 0,0001) ja kokonaiselossaoloaikaan (p = 0,0006). Etenemättömyysaika ja kokonaiselossaoloaika korreloivat tilastollisesti merkitsevästi MCyR-vasteeseen 12 kk kohdalla (p = < 0,0001 etenemättömyysajan suhteen ja p = 0,0012 kokonaiselossaoloajan suhteen).
Taulukko 9 Iclusigin teho resistenteissä tai intoleranteissa KML‑potilaissa, joilla oli pitkälle edennyt tauti
| Akseleraatiovaiheen KML | Blastivaiheen KML | |||||
Yht. (N=83) | Resistentti tai intolerantti | Yht. (N=62) | Resistentti tai intolerantti | |||
R/I‑kohortti (N=65) | T315I‑kohortti (N=18) | R/I‑ kohortti (N=38) | T315I‑ kohortti (N=24) | |||
| Hematologinen vasteprosentti | ||||||
Huomattavaa (MaHR) % (95 % lv) | 57 % (45–68) | 57 % (44–69) | 56 % (31–79) | 31 % (20–44) | 32 % (18–49) | 29 % (13–51) |
Täydellinenb (CHR) % (95 % lv) | 51 % (39–62) | 49 % (37–62) | 56 % (31–79) | 21 % (12–33) | 24 % (11–40) | 17 % (5–37) |
Huomattava sytogeneettinen vastec % (95 % lv) | 39 % (28–50) | 34 % (23–47) | 56 % (31–79) | 23 % (13–35) | 18 % (8–34) | 29 % (13–51) |
a Ensisijainen päätetapahtuma AP‑KML‑ ja BP‑KML‑/Ph+ ALL ‑kohorteissa oli MaHR, joka yhdistää täydelliset hematologiset vasteet ja leukemian merkkien puuttumisen. b CHR: Valkosolumäärä ≤ hoitolaitoksen ULN, ANC ≥ 1 000/mm3, verihiutaleet ≥ 100 000/mm3, ei blasteja tai promyelosyyttejä perifeerisessä veressä, luuydinblastit ≤ 5 %, myelosyyttejä ja metamyelosyyttejä yhteensä < 5 % perifeerisessä veressä, basofiileja < 5 % perifeerisessä veressä, ei ekstramedullaarista affisiota (mm. ei hepatomegaliaa tai splenomegaliaa). c MCyR yhdistää sekä täydelliset (ei havaittavia Ph+‑soluja) että osittaiset (1 % – 35 % Ph+‑soluja) sytogeneettiset vasteet. Tiedonkeruu tietokannasta päättyi 6.2.2017. | ||||||
AP-KML-potilailla annosintensiteetin mediaani oli 32 mg/vrk.
Taulukko 10 Iclusigin teho resistenteissä tai intoleranteissa Ph+ ALL ‑potilaissa
Yht. (N=32) | Resistentti tai intolerantti | ||
R/I‑ kohortti (N=10) | T315I‑ kohortti (N=22) | ||
| Hematologinen vasteprosentti | |||
Huomattavaa (MaHR) % (95 % lv) | 41 % (24–59) | 50 % (19–81) | 36 % (17–59) |
Täydellinenb (CHR) % (95 % lv) | 34 % (19–53) | 40 % (12–74) | 32 % (14–55) |
Huomattava sytogeneettinen vastec % (95 % lv) | 47 % (29–65) | 60 % (26–88) | 41 % (21–64) |
a Ensisijainen päätetapahtuma AP‑KML‑ ja BP‑KML‑/Ph+ ALL ‑kohorteissa oli MaHR, joka yhdistää täydelliset hematologiset vasteet ja leukemian merkkien puuttumisen. b CHR: Valkosolumäärä ≤ hoitolaitoksen ULN, ANC ≥ 1 000/mm3, verihiutaleet ≥ 100 000/mm3, ei blasteja eikä promyelosyyttejä perifeerisessä veressä, luuydinblastit ≤ 5 %, myelosyyttejä ja metamyelosyyttejä yhteensä < 5 % perifeerisessä veressä, basofiileja < 5 % perifeerisessä veressä, ei ekstramedullaarista affisiota (mm. ei hepatomegaliaa tai splenomegaliaa). c MCyR yhdistää sekä täydelliset (ei havaittavia Ph+‑soluja) että osittaiset (1 % – 35 % Ph+‑soluja) sytogeneettiset vasteet. Tiedonkeruu tietokannasta päättyi 6.2.2017. | |||
BP-KML / Ph+ ALL -potilailla annosintensiteetin mediaani oli 44 mg/vrk.
AP‑KML‑, BP‑KML- ja Ph+ ALL ‑potilailla, jotka saavuttivat MaHR:n, mediaaniaika MaHR:iin oli AP‑KML‑potilailla 0,7 kk (vaihteluväli 0,4–5,8 kk), BP‑KML‑potilailla 1,0 kk (vaihteluväli 0,4–3,7 kk) ja Ph+ ALL ‑potilailla 0,7 kk (vaihteluväli 0,4–5,5 kk). Raporttipäivityksen aikana, jolloin kaikkien edelleen mukana olleiden potilaiden seuranta‑aika oli vähintään 64 kuukautta, MaHR:n mediaanikestoaika AP‑KML‑potilailla (hoidon mediaanikesto 19,4 kk), BP‑KML‑potilailla (hoidon mediaanikesto 2,9 kk) ja Ph+ ALL -potilailla (hoidon mediaanikesto 2,7 kk) oli arviolta 12,9 kk (vaihteluväli: 1,2–68,4 kk), 6,0 kk (vaihteluväli: 1,8–59,6 kk) ja 3,2 kk (vaihteluväli: 1,8‑12,8 kk).
Kun tutkitaan annosintensiteetin ja turvallisuuden suhdetta kaikilla faasin 2 PACE-tutkimuksessa mukana olleilla potilailla, vähintään 3. asteen haittatapahtumat (sydämen vajaatoiminta, valtimotromboosi, korkea verenpaine, trombosytopenia, haimatulehdus, neutropenia, ihottuma, ALAT‑arvon nousu, ASAT‑arvon nousu, lipaasiarvon nousu, myelosuppressio, nivelkipu) lisääntyivät merkitsevästi annosalueella 15−45 mg kerran päivässä.
Analysoitaessa annosintensiteetin ja turvallisuuden suhdetta faasin 2 PACE-tutkimuksessa tultiin siihen tulokseen, että kun kovariaattien vaikutus otettiin huomioon, suurella annosintensiteetillä kaikkiaan on merkitsevä yhteys suurentuneeseen valtimotukoksen riskiin; kerroinsuhde on noin 1,6 kutakin 15 mg:n lisäystä kohti. Lisäksi faasin 1 tutkimuksissa olleiden potilaiden tiedoista tehtyjen logististen regressioanalyysien mukaan valtimotromboositapahtumat näyttävät olevan suhteessa systeemiseen altistukseen (AUC). Annoksen vähentämisen odotetaan siksi vähentävän verisuonitukostapahtumien riskiä, mutta analyysin perusteella suurilla annoksilla saattaa olla viivästevaikutus (carry over effect), jonka vuoksi voi kestää useita kuukausia ennen kuin annoksen vähentäminen vähentää riskiä. Muut kovariaatit, joilla oli tilastollisesti merkitsevä yhteys verisuonitukostapahtumiin tässä analyysissä, olivat lääketieteellinen iskemia‑anamneesi ja ikä.
Annoksen pienentäminen CP‑KML‑potilailla
Faasin 2 PACE-tutkimuksessa annoksen pienentämistä suositeltiin, jos ilmeni haittatapahtumia. Lisäksi tutkimuksessa annettiin verisuonitukostapahtumien riskin vähentämiseksi suositus pienentää prospektiivisesti kaikkien CP‑KML‑potilaiden annoksia, vaikkei haittatapahtumia olisikaan.
Kun seurannan kesto oli vähintään 48 kk ja prospektiivisesta annoksen pienentämissuosituksesta oli kulunut noin 2 vuotta, 110 CP‑KML-potilasta sai edelleen hoitoa. Ilmoitusten mukaan valtaosa näistä edelleen hoitoa saavista potilaista (82/110; 75 % potilaista) sai viimeisenä annoksenaan 15 mg:n annosta; 24/110 (22 % potilaista) sai 30 mg:n annoksia ja 4/110 (4 %) 45 mg:n annoksia. Tutkimuksen päättyessä (seuranta-aika vähintään 64 kk, yli 3 vuoden kuluttua prospektiivisesta annoksen pienentämissuosituksesta), 99 CP-KML-potilasta oli edelleen mukana ja heistä 77 (78 %) sai tutkimuksessa viimeisenä annoksenaan 15 mg:n annosta.
Turvallisuus
Faasin 2 PACE-tutkimuksessa 86 CP‑KML‑potilaalla saavutettiin MCyR annoksen ollessa 45 mg, ja 45 CP‑KML‑potilaalla MCyR saavutettiin, kun annosta oli vähennetty 30 mg:aan, yleensä haittatapahtumien vuoksi.
Verisuonitukostapahtumia oli 44:llä näistä 131 potilaasta. Useimmat tapahtumista ilmenivät käytettäessä sitä annosta, jolla potilas saavutti MCyR:n, harvemmat annoksen vähentämisen jälkeen.
Taulukko 11 Ensimmäiset verisuonitukoshaittatapahtumat CP‑KML‑potilailla, joilla MCyR saavutettiin annoksen ollessa 45 mg tai 30 mg (tiedonhakupäivä 7. huhtikuuta 2014)
| Viimeisin annos ensimmäisen verisuonitukostapahtuman alkaessa | |||
| 45 mg | 30 mg | 15 mg | |
MCyR saavutettu annoksen ollessa 45 mg (N = 86) | 19 | 6 | 0 |
MCyR saavutettu annoksen ollessa 30 mg (N = 45) | 1 | 13 | 5 |
Mediaaniaika ensimmäisen kardiovaskulaarisen valtimotukostapahtuman alkamiseen oli 351 vrk, ensimmäisen aivovaltimoiden tukostapahtuman alkamiseen 611 vrk ja ensimmäisen ääreisvaltimoiden tukostapahtuman alkamiseen 605 vrk. Kun tiedot korjattiin altistuksen suhteen, ensimmäisten valtimotukostapahtumien ilmaantuvuus oli suurin ensimmäisten kahden seurantavuoden aikana ja pieneni vuorokauden annosintensiteetin pienentyessä (prospektiivisen annoksen pienentämissuosituksen jälkeen). Myös muut tekijät kuin annos voivat vaikuttaa osaltaan valtimotukosriskiin.
Teho
Faasin 2 PACE-tutkimuksesta on saatu tiedot hoitovasteen (MCyR ja MMR) säilymisestä kaikilta CP‑KML‑potilailta, joiden annosta vähennettiin, olipa syy vähentämiseen mikä tahansa. Taulukossa 12 on nämä tiedot niistä potilaista, jotka saavuttivat MCyR:n ja MMR:n annoksen ollessa 45 mg. Vastaavat tiedot on saatavana myös potilaista, jotka saavuttivat MCyR:n ja MMR:n annoksen ollessa 30 mg.
Hoitovaste (MCyR ja MMR) on säilynyt tähän mennessä saatavilla olevien seurantatietojen perusteella suurimmalla osalla niistä potilaista, joiden annosta vähennettiin. Osalla potilaista annosta ei yksilöllisen hyöty‑ ja riskiarvion perusteella vähennetty lainkaan.
Taulukko 12 Hoitovasteen säilyminen CP‑KML‑potilailla, jotka saavuttivat MCyR:n tai MMR:n annoksen ollessa 45 mg (tiedonhakupäivä 6.2.2017)
| MCyR saavutettu annoksen ollessa 45 mg (N = 86) | MMR saavutettu annoksen ollessa 45 mg (N = 63) | |||
| Potilaiden määrä | MCyR säilynyt | Potilaiden määrä | MMR säilynyt | |
| Ei annoksen vähennystä | 19 | 13 (68 %) | 18 | 11 (61 %) |
| Annos vähennetty vain 30 mg:aan | 15 | 13 (87 %) | 5 | 3 (60 %) |
| ≥ 3 kk vähennys 30 mg:aan | 12 | 10 (83 %) | 3 | 2 (67 %) |
| ≥ 6 kk vähennys 30 mg:aan | 11 | 9 (82 %) | 3 | 2 (67 %) |
| ≥ 12 kk vähennys 30 mg:aan | 8 | 7 (88 %) | 3 | 2 (67 %) |
| ≥ 18 kk vähennys 30 mg:aan | 7 | 6 (86 %) | 2 | 2 (100 %) |
| ≥ 24 kk vähennys 30 mg:aan | 6 | 6 (100 %) | 2 | 2 (100 %) |
| ≥ 36 kk vähennys 30 mg:aan | 1 | 1 (100 %) | -- | -- |
| Mikä tahansa annoksen vähennys 15 mg:aan | 52 | 51 (98 %) | 40 | 36 (90 %) |
| ≥ 3 kk vähennys 15 mg:aan | 49 | 49 (100 %) | 39 | 36 (92 %) |
| ≥ 6 kk vähennys 15 mg:aan | 47 | 47 (100 %) | 37 | 35 (95 %) |
| ≥ 12 kk vähennys 15 mg:aan | 44 | 44 (100 %) | 34 | 33 (97 %) |
| ≥ 18 kk vähennys 15 mg:aan | 38 | 38 (100 %) | 29 | 29 (100 %) |
| ≥ 24 kk vähennys 15 mg:aan | 32 | 32 (100 %) | 23 | 23 (100 %) |
| ≥ 36 kk vähennys 15 mg:aan | 8 | 8 (100 %) | 4 | 4 (100 %) |
Iclusigin antileukeeminen vaikutus arvioitiin myös faasin 1 annossuurentamistutkimuksessa, joka sisälsi 65 KML‑ ja Ph+ ALL ‑potilasta; tutkimus on päättynyt. 31 CP‑KML‑potilasta 43:sta saavutti MCyR:n seurannan mediaanikestoajan ollessa 55,5 kuukautta (vaihteluväli: 1,7–91,4 kuukautta). Raportointiaikana 25:llä CP‑KML‑potilaalla oli MCyR (MCyR:n mediaanikestoaikaa ei ollut vielä saavutettu).
Faasin 2 satunnaistettu avoin OPTIC-tutkimus
Iclusigin turvallisuutta ja tehoa arvioitiin faasin 2 OPTIC-annosoptimointitutkimuksessa. Tutkimukseen soveltuvilla potilailla oli kroonisen vaiheen KML (CP-KML), ja heidän tautinsa katsottiin olevan resistentti vähintään kahdelle aiemmalle kinaasin estäjälle tai heillä oli T315I-mutaatio. CP-KML:n resistenssiksi määriteltiin aiemman kinaasin estäjähoidon aikana havaittu täydellisen hematologisen vasteen (3 kk:een mennessä), vähäisen sytogeneettisen vasteen (6 kk:een mennessä) tai huomattavan sytogeneettisen vasteen (12 kk:een mennessä) saavuttamattomuus tai uuden BCR-ABL1-kinaasidomeenin mutaation tai uuden klonaalisen evoluution kehittyminen. Potilailla oli oltava tutkimukseenottohetkellä > 1 %:n BCR-ABL1IS-arvo (reaaliaikaisella polymeraasiketjureaktiolla määritettynä). Potilaat saivat yhtä kolmesta aloitusannostuksesta: 45 mg suun kautta kerran vuorokaudessa, 30 mg suun kautta kerran vuorokaudessa tai 15 mg suun kautta kerran vuorokaudessa. Kaikilla potilailla, joiden aloitusannos oli 45 mg tai 30 mg, annosta pienennettiin 15 mg:aan kerran vuorokaudessa, kun he olivat saavuttaneet BCR-ABL1IS-arvon ≤ 1 %. Ensisijainen tehon päätetapahtuma oli molekulaarinen vaste, joka perustui ≤ 1 %:n BCR-ABL1IS -arvon saavuttamiseen 12 kk:n kohdalla. Kaikki potilaat saavuttivat 12 kk:n ajankohdan (ensisijainen päätetapahtuma) ensisijaisen analyysin tiedonkeruun katkaisuun mennessä. 45 mg:n kohortin (N = 94) seurannan mediaanikesto oli 77,9 kk (95 % lv: 72,4–84,0). Alla on kuvattu vain suositellun aloitusannoksen (45 mg) tehotulokset. Iclusigia sai yhteensä 282 potilasta: 94 potilasta sai aloitusannosta 45 mg, 94 potilasta aloitusannosta 30 mg ja 94 potilasta aloitusannosta 15 mg. Aloitusannosta 45 mg saaneiden potilaiden lähtötilanteen demografiset ominaisuudet on kuvattu taulukossa 13.
Taulukko 13 Demografiset tiedot ja taudin ominaisuudet OPTIC-tutkimuksessa
| Potilaiden ominaisuudet tutkimukseen otettaessa | Iclusig 45 mg → 15 mg (N = 94) |
|---|---|
| Ikä | |
| Mediaani, vuotta (vaihteluväli) | 46 (19–81) |
| Sukupuoli, n (%) | |
| Mies | 50 (53 %) |
| Etninen tausta, n (%) | |
| Valkoihoinen | 73 (78 %) |
| Aasialainen | 16 (17 %) |
| Muu / Ei tiedossa | 4 (4 %) |
| Musta/afroamerikkalainen | 1 (1 %) |
| ECOG‑toimintakykyluokka, n (%) | |
| ECOG-luokka 0 tai 1 | 93 (99 %) |
| Tautihistoria | |
| Mediaaniaika diagnoosista ensimmäiseen annokseen, vuotta (vaihteluväli) | 5,5 (1–21) |
| Resistentti aiemmalle kinaasin estäjälle, n (%) | 92 (98 %) |
| Vähintään yksi BCR‑ABL-kinaasidomeenin mutaatio, n (%) | 41 (44 %) |
| Aiempien kinaasin estäjien määrä, n (%) | |
| 1 | 1 (1 %) |
| 2 | 43 (46 %) |
| ≥ 3 | 50 (53 %) |
| T315I-mutaatio lähtötilanteessa | 25 (27 %) |
| Samanaikaiset sairaudet | |
| Hypertensio | 29 (31 %) |
| Diabetes | 5 (5 %) |
| Hyperkolesterolemia | 3 (3 %) |
| Anamneesissa iskeeminen sydänsairaus | 3 (3 %) |
Tehotulosten yhteenveto on esitetty taulukossa 14.
Ensisijainen päätetapahtuma saavutettiin potilailla, jotka saivat aloitusannosta 45 mg.
Kaikkiaan 44 %:lla potilaista oli tutkimukseenottohetkellä vähintään yksi BCR-ABL-kinaasidomeenin mutaatio, joista yleisin oli T315I (27 %). Lähtötilanteen T315I-mutaatiostatukseen perustuvassa alaryhmäanalyysissa 2 kk:n kohdalla niiden potilaiden osuus, joiden BCR-ABL1IS-arvo oli ≤ 1 %, oli T315I-mutaatiopositiivisilla ja T315I-mutaationegatiivisilla potilailla samaa luokkaa (ks. taulukko 14 jäljempänä). Tutkimukseenottohetkellä 54 %:lla 45 mg:n aloitusannosta saaneista potilaista ei todettu mutaatioita.
CP-KML-potilaista, joiden seuranta-ajan mediaani oli 6,5 vuotta, 11,7 %:lla tauti eteni AP-KML-vaiheeseen ja 3,2 %:lla BP-KML-vaiheeseen.
Taulukko 14 Faasin 2 OPTIC-tutkimuksessa Iclusigia 45 mg:n aloitusannoksella saaneiden CP‑KML-potilaiden tehotulokset
| Iclusig 45 mg → 15 mg (N = 93)(a) | |
|---|---|
| Molekulaarinen vaste 12 kk:n kohdalla(b) | |
| Niiden potilaiden kokonaisosuus, joiden BCR‑ABL1IS-arvo oli ≤ 1 % % (n/N) (98,3 % lv)(c) | 44 % (41/93) (32–57 %) |
| T315I-mutaatiopositiiviset potilaat % (n/N) (95 % lv) | 44 % (11/25) (24–65 %) |
| T315I-mutaationegatiiviset potilaat % (n/N) (95 % lv) | 44 % (29/66)(d) (32–57 %) |
| Sytogeneettinen vaste 12 kk:n kohdalla | |
| Huomattava (MCyR)(e) % (n/N) (95 % lv) | 48 % (44/91)(f) (38–59 %) |
| T315I-mutaatiopositiiviset potilaat % (n/N) (95 % lv) | 52 % (13/25) (31–72 %) |
| T315I-mutaationegatiiviset potilaat % (n/N) (95 % lv) | 46 % (30/65)(g) (34–59 %) |
(a) ITT-populaatioksi (N = 93) määriteltiin potilaat, joilla oli BCR-ABL1-transkripti b2a2/b3a2.
(b) Ensisijainen päätetapahtuma oli niiden potilaiden osuus, joiden BCR-ABL1IS-arvo oli 12 kk:n kohdalla ≤ 1 %. Määritelmä: BCR-ABL-transkriptien ja ABL-transkriptien suhde ≤ 1 % International Scale (IS) -asteikolla (ts. ≤ 1 %:n BCR-ABLIS; potilailla oli oltava b2a2/b3a2 (p210) -transkripti) perifeerisessä veressä, mittaustapa qRT-PCR (kvantitatiivinen käänteistranskriptaasientsyymiä hyödyntävä polymeraasiketjureaktio).
(c) 98,3 % lv laskettiin binomiaalisen eksaktin menetelmällä (Clopper–Pearson).
(d) Kahdelle 93 potilaasta ei tehty lähtötilanteessa mutaatioarviointia, ja heidät suljettiin pois mutaatiostatukseen perustuvasta vasteanalyysistä.
(e) Toissijainen päätetapahtuma oli MCyR, joka yhdistää täydelliset (ei havaittavissa olevia Ph+-soluja) ja osittaiset (1–35 % Ph+-soluja vähintään 20 metafaasissa) sytogeneettiset vasteet, 12 kk:n kohdalla.
(f) Analyysi pohjautuu sytogeneettiseen ITT-populaatioon (N = 91). Siihen määriteltiin kuuluviksi potilaat, joille tehtiin lähtötilanteessa sytogeneettinen arviointi, jossa arvioitiin vähintään 20 metafaasia. Analyysista suljettiin pois yksi potilas, jolla oli täydellinen sytogeneettinen vaste lähtötilanteessa.
(g) Yhdelle 91 potilaasta ei tehty lähtötilanteessa mutaatioarviointia, ja hänet suljettiin pois mutaatiostatukseen perustuvasta vasteanalyysistä.
Toissijaisia tehon päätetapahtumia olivat täydellinen sytogeneettinen vaste (CCyR) 12 kk:n kohdalla, huomattava molekulaarinen vaste (MMR) 12 kk:n ja 24 kk:n kohdalla, täydellinen hematologinen vaste 3 kk:n kohdalla, aika vasteeseen, vasteen kesto, vasteen säilyminen, etenemättömyysaika (PFS) ja kokonaiselossaoloaika (OS). Lisäarviointina määritettiin myös ≤ 1 %:n BCR-ABL1IS-arvon saavuttamiseen perustuvat molekulaarisen vasteen osuudet jokaisella potilaskäynnillä 3 kk:n välein 36 kk:n ajan.
- 12 kk:n kohdalla 34 % (31 potilasta 91:stä) saavutti CCyR:n ja 17 % (16 potilasta 93:sta) MMR:n. 24 kk:n kohdalla 34 % (32 potilasta 93:sta) saavutti MMR:n. MMR:n mediaanikestoa ei ollut vielä saavutettu.
- Ponatinibihoidon mediaanikesto oli 31 kk.
- 25:llä (55,6 %) 45 potilaasta, joiden annosta pienennettiin 45 mg:sta 15 mg:aan ≤ 1 %:n BCR-ABL1IS-arvon saavuttamisen jälkeen, vaste säilyi pienennetyllä annoksella vähintään yhden vuoden ajan. Näistä 25 potilaasta 16:lla (64 %) vaste säilyi 15 mg:n annoksella yli 60 kuukauden ajan. Vasteen mediaanikestoa (MR2) ei saavutettu. MR2:n säilymisen todennäköisyys oli 60 kk:n kohdalla 68,8 % (95 % lv: 53,9–79,8).
- Molekulaarisen vasteen osuudet (≤ 1 %:n BCR-ABLIS) olivat 60 kuukauden kohdalla 64,0 % (95 % lv: 42,5–82,0) potilailla, joilla oli T315I-mutaatio, ja 59,1 % (95 % lv: 46,3–71,0) potilailla, joilla ei ollut T315I-mutaatiota.
- Molekulaarisen vasteen osuudet (≤ 1 %:n BCR-ABL1IS) olivat 12 kk:n kohdalla pienemmät enintään kahta aiempaa TKI-hoitoa saaneilla potilailla (40 %) kuin vähintään kolmea aiempaa TKI-hoitoa saaneilla potilailla (48 %).
Potilaat, joilla on vastadiagnosoitu Ph+ ALL
PhALLCON-tutkimus
Iclusigin tehoa yhdessä kevennetyn kemoterapian kanssa ja sitä seuraavana Inclusig-monoterapiahoitona arvioitiin satunnaistetussa, aktiivilääkekontrolloidussa avoimenssa monikeskustutkimuksessa.
Tutkimukseen otetuilla potilailla oli vastadiagnosoitu Ph+ALL. Satunnaistaistamisessa käytettiin ositusperusteena ikää induktiohoitovaiheessa (18 – < 45 vuotta; ≥ 45 – < 60 vuotta ja ≥ 60 vuotta). Potilaat satunnaistettiin (2:1) saamaan Iclusigia 30 mg suun kautta kerran vuorokaudessa tai imatinibia 600 mg suun kautta kerran vuorokaudessa, yhdistettynä 20 sykliin kemoterapiaa. Tämän jälkeen seurasi Iclusig- tai imatinibimonoterapia. Iclusig-annos pienennettiin 15 mg:aan kerran vuorokaudessa induktiovaiheen jälkeen, kun MRD-negatiivinen CR oli saavutettu. Jos potilas menetti MRD-negatiivisuuden milloin tahansa sen jälkeen kun annos oli vasteen perusteella pienennetty 15 mg:aan, annos oli mahdollista suurentaa uudelleen 30 mg:aan kerran vuorokaudessa. Tutkimushoitoa saivat tutkijan harkinnan mukaan jatkaa vain potilaat, joilla saavutettiin CR tai täydellinen vaste ilman täydellistä palautumista (CRi), ja jotka olivat induktiovaiheen päättyessä MRD-negatiivisia.
Tutkimuksen vaiheet ja hoidot
-
Induktiovaihe: Tutkittavat saivat kolme 28 päivän sykliä Iclusig-valmisteen aloitusannosta (30 mg suun kautta kerran vuorokaudessa) tai imatinibin aloitusannosta (600 mg suun kautta kerran vuorokaudessa) annettuna päivästä 1 päivään 28 hoidon sykleinä 1–3 yhdessä seuraavien kanssa:
- Vinkristiini: 1,4 mg/m2, laskimoon annettuna (i.v.), päivät 1 ja 14, enintään 2 mg, ja
- Deksametasoni: tutkittavat, joiden ikä oli < 60 vuotta, saivat 40 mg suun kautta päivinä 1–4 ja päivinä 11–14. Tutkittavat, joiden ikä oli ≥ 60 vuotta: 20 mg suun kautta päivinä 1–4 ja päivinä 11–14.
-
Konsolidatiovaihe (metotreksaatin ja sytarabiinin vuorottelu): Tutkittavat saivat kuusi 28 päivän sykliä Iclusig-valmistetta induktiovaiheen viimeisellä annoksella, mukautettu annos perustui MRD-negatiivisiin CR-tuloksiin, tai imatinibi aloitettiin induktiovaiheen viimeisellä annoksella, annettuna päivästä 1 päivään 28 hoidon sykleinä 4–9 seuraavien kanssa:
- Metotreksaatti: Tutkittavat, joiden ikä oli < 60 vuotta, saivat 1 000 mg/m2 päivänä 1, 24 tunnin laskimoon annettuna (i.v.) infuusiona. Tutkittavat, joiden ikä oli ≥ 60 vuotta, saivat 250 mg/m2, IV, päivänä 1, 24 tunnin infuusiona. Vara (rescue): foliinihappo. Tutkimuksen syklit 4, 6 ja 8.
- Sytarabiini: Tutkittavat, joiden ikä oli < 60 vuotta, saivat 1 000 mg/m2, 2 tunnin laskimoon annettuna (i.v.) infuusiona 12 tunnin välein päivinä 1, 3 ja 5. Tutkittavat, joiden ikä oli ≥ 60 vuotta, saivat 250 mg/m2, IV, 2 tunnin infuusiona 12 tunnin välein päivinä 1, 3 ja 5. Tutkimuksen syklit 5, 7 ja 9.
-
Ylläpitovaihe: Tutkittavat saivat yksitoista 28 päivän sykliä Iclusig-valmistetta konsolidaatiovaiheen viimeisellä annoksella; mukautettu annos perustui MRD-negatiivisiin CR-tuloksiin, tai imatinibi aloitettiina konsolidatiovaiheen viimeisellä annoksella, annettuna päivästä 1 päivään 28 hoidon sykleinä 10–20 yhdessä seuraavien kanssa:
- Vinkristiini: 1,4 mg/m2, annettuna laskimoon (i.v.), injektoituna 1 minuutin kuluessa ylläpitovaiheen kunkin syklin päivänä 1, 1 injektio/kuukausi; enintään 2 mg, ja
- Prednisoni: Tutkittavat, joiden ikä oli < 60 vuotta: 200 mg/vrk, suun kautta päivinä 1–5. Tutkittavat, joiden ikä oli ≥ 60 – 69 vuotta: 100 mg/vrk, suun kautta päivinä 1–5. Tutkittavat, joiden ikä oli ≥ 70 vuotta: 50 mg/vrk, suun kautta päivinä 1–5.
Sen jälkeen kun tutkittavat olivat saaneet 20 sykliä Iclusigia tai imatinibia yhdessä kemoterapian kanssa, he saivat edelleen Iclusigia (21 %) tai imatinibia (9 %) monoterapiana, kunnes tauti uusiutui täydellisen remission (CR) jälkeen, sairaus eteni (PD), tutkittava eteni kantasolusiirtoon (HSCT), tutkittava siirtyi vaihtoehtoiseen hoitoon tai ilmaantui sietämätöntä toksisuutta. Lähtötilanteen demografiset ominaisuudet satunnaistetussa populaatiossa esitetään taulukossa 15.
Taulukko 15 Demografiset tiedot ja sairauden ominaisuudet PhALLCON-tutkimuksessa
| Potilaan ominaisuudet tutkimukseenottohetkellä | Iclusig (N = 164) | Imatinibi (N = 81) |
|---|---|---|
| Ikä (vuotta) | ||
| Mediaani, vuotta (vaihteluväli) | 54 (19–82) | 52 (19–75) |
| Ikäryhmä(a), n (%) | ||
| 18 – < 45 vuotta | 58 (35 %) | 29 (36 %) |
| 45 – < 60 vuotta | 45 (27 %) | 22 (27 %) |
| ≥ 60 vuotta | 61 (37 %) | 30 (37 %) |
| Sukupuoli, n (%) | ||
| Nainen | 90 (55 %) | 43 (53 %) |
| Rotu, n (%) | ||
| Valkoinen | 104 (63 %) | 62 (77 %) |
| Ei ilmoitettu | 28 (17 %) | 2 (3 %) |
| Aasialainen | 20 (12 %) | 11 (14 %) |
| Musta/afroamerikkalainen | 9 (5 %) | 4 (5 %) |
| ECOG-suorituskykystatus, n (%) | ||
| 0 | 72 (44 %) | 33 (41 %) |
| 1 | 85 (52 %) | 43 (53 %) |
| 2 | 7 (4 %) | 5 (6 %) |
| Tautihistoria | ||
| BCR-ABL1:n dominantti variantti p190:n tai p210:n esiintyminen, n (%) | 154 (94 %) | 78 (96 %) |
| Ei ekstramedullaarista tautia, n (%) | 154 (94 %) | 78 (96 %) |
| Mediaani, valkosolumäärä(b) (vaihteluväli) | 4,37 (0,4–197) | 3,21 (0,2–81) |
| Mediaani, leukemiasoluja luuytimessä (%) | 80 % | 75 % |
| Komorbiditeetit, n (%) | ||
| Hypertensio | 58 (35 %) | 30 (37 %) |
| Diabetes | 39 (24 %) | 24 (30 %) |
| Dyslipidemia | 29 (18 %) | 23 (28 %) |
| (a)Satunnaistaistamisessa käytettiin ositusperusteena ikää (18 – < 45 vuotta; ≥ 45 – < 60 vuotta ja ≥ 60 vuotta). (b)Valkosolujen määrä 10^9/l | ||
Tärkein tehon päätetapahtuma oli MRD-negatiivinen CR induktiovaiheen päättyessä. MRD-negatiivisuuden määritelmä oli ≤ 0,01 % BCR-ABL1 keskuslaboratoriossa määritettynä. CR-tilan määritelmä oli < 5 % blasteja luuytimessä ja ei ekstramedullaarista sairautta sekä hematologinen toipuminen vähintään 4 viikon ajan tutkijan arvioimana.
MRD-negatiivisen CR:n ja molekulaarisen vasteen analyysissa tutkittavien populaatioon kuului 232 satunnaistettua tutkittavaa, joilla oli lähtötilanteessa BCR-ABL1:n dominantti variantti p190 tai p210 keskuslaboratoriossa määritettynä (154 tutkittavaa Iclusig-haarassa ja 78 imatinibiryhmässä).
Keskeisen toissijaisen tehon päätetapahtuman, tautitapahtumattoman elossaoloajan (EFS), määritelmänä käytettiin aikaa satunnaistamisesta minkä tahansa seuraavista tapahtumista ilmaantumiseen: CR:n jääminen saavuttamatta induktiovaiheen päättymiseen mennessä, relapsi CR:stä tai mistä tahansa syystä johtuva kuolema. EFS-tutkittavien populaatio perustui ITT-populaation 245:een satunnaistettuun tutkittavaan, joista 164 satunnaistettua tutkittavaa kuului Iclusig-ryhmään (mukaan lukien 1 potilas, joka kuoli COVID-19:ään ennen ensimmäisen annoksen saamista) ja 81 satunnaistettua tutkittavaa kuului imatinibiryhmään, ellei muuta mainita.
Kantasaolusiirtoon (HSCT) etenevien potilaiden kokonaisosuus oli 34 % (56/164) Iclusig-ryhmässä vs. 48 % (39/81) imatinibiryhmässä.
Kokonaiselossaolon seurannan mediaanikesto oli 20,43 kuukautta (95 % lv: [18,39, 23,93] Iclusig-ryhmässä ja 18,14 kuukautta (95 % lv: [13,86, 24,25) imatinibiryhmässä.
Tutkimus osoitti, että Iclusig-ryhmän satunnaistettujen tutkittavien MRD-negatiivinen CR-osuus oli induktiovaiheen lopussa tilastollisesti merkitsevästi suurempi kuin imatinibiryhmässä.
Tiedonkeruun katkaisuun mennessä keskeisen toissijaisen tehon päätetapahtuman EFS:n tulokset eivät olleet kypsiä; lopulliseen analyysiin tarvittavista tapahtumista oli käytettävissä 33,5 % (34/164 tapahtumaa Iclusig-ryhmässä ja 24/81 tapahtumaa imatinibiryhmässä).
Tehoa koskevat tulokset on esitetty taulukossa 16.
Taulukko 16 PhALLCON-tutkimuksen tehotulokset Ph+ ALL potilailla(a)
| Iclusig 30 mg → 15 mg kemoterapian kanssa (N = 154) | Imatinibi 600 mg kemoterapian kanssa (N = 78) | |
| MRD-negatiivinen CR(b) induktion päättyessä | ||
| Saavutettu induktion päättyessä, % (n/N) | 34,4% (53/154) | 16,7% (13/78) |
| Riskiero (95 % lv)(c) | 0,18 (0,06; 0,29) | |
| p-arvo(d) | 0,0021 | |
| Suhteellinen riski (95 % lv)(e) | 2,06 (1,19; 3,56) | |
MRD: minimaalinen jäännöstauti; CR: täydellinen vaste; MR: molekulaarinen vaste; BCR-ABL1: murtumakohta-alueen (BCR) geenin ja Abelson 1-geenin fuusio (breakpoint cluster region-Abelson). (a) Perustuu 232:een satunnaistettuun tutkittavaan, joilla oli lähtötilanteessa BCR-ABL1:n dominantti variantti p190 tai p210 keskuslaboratoriossa määritettynä. | ||
Sydämen elektrofysiologia
Iclusigin mahdollista QT‑aikaa pidentävää vaikutusta tutkittiin 39 leukemiapotilaassa, jotka saivat 30 mg, 45 mg tai 60 mg Iclusigia kerran vuorokaudessa. Kolmen EKG‑tutkimuksen sarjoja otettiin lähtötilanteessa ja vakaassa tilassa, jotta voitiin arvioida ponatinibin vaikutusta QT‑aikoihin. Tutkimuksessa ei havaittu mitään kliinisesti merkittäviä (ts. > 20 ms) muutoksia QTc‑ajan keskiarvossa lähtötilanteeseen verrattuna. Lisäksi farmakokineettis‑farmakodynaamiset mallit eivät osoita altistus‑vaikutussuhdetta, arvioidun keskimääräisen QTcF‑muutoksen ollessa ‑6,4 ms (luottamusvälin yläraja ‑0,9 ms) Cmax‑pitoisuuksien yhteydessä 60 mg:n ryhmässä.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Iclusig-valmisteen käytöstä KML:n ja Ph+ ALL:n hoidossa lapsilla (syntymästä 1 vuoden ikäisiin). Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Iclusig-valmisteen käytöstä KML:n ja Ph+ ALL:n hoidossa pediatrisilla potilailla (1 vuoden ikäisistä alle 18 vuoden ikäisiin) (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Ponatinibin huippupitoisuudet saavutetaan noin 4 tuntia suun kautta antamisen jälkeen. Potilailla arvioidulla, kliinisesti relevantilla annosten vaihteluvälillä (15 mg – 60 mg) ponatinibin Cmax‑ ja AUC‑arvot suurenivat annosriippuvaisesti. Ponatinibin 45 mg:n vuorokausiannoksilla vakaassa tilassa havaitut Cmax‑arvojen ja AUC(0‑τ)‑altistusten geometriset keskiarvot (CV%) olivat 77 ng/ml (50 %) ja 1296 ng•h/ml (48 %). Joko runsasrasvaisen tai vähärasvaisen aterian jälkeen plasman ponatinibialtistukset (Cmax ja AUC) eivät olleet erilaisia kuin paastotilassa. Iclusig voidaan antaa ruoan kanssa tai erikseen. Kun Iclusigia annettiin samanaikaisesti mahahapon erittymistä voimakkaasti estävän valmisteen kanssa, ponatinibin Cmax‑arvo laski hiukan mutta AUC0‑∞ ei laskenut lainkaan.
Jakautuminen
Ponatinibi sitoutuu voimakkaasti (> 99 %) plasmaproteiineihin in vitro. Ponatinibin veri/plasma‑suhde on 0,96. Ibuprofeenin, nifedipiinin, propranololin, salisyylihapon tai varfariinin samanaikainen anto ei syrjäytä ponatinibia. 45 mg:n vuorokausiannoksella näennäisen vakaan tilan jakautumistilavuuden geometrinen keskiarvo (CV%) on 1101 litraa (94 %), mikä viittaa siihen, että ponatinibi jakautuu laajalti ekstravaskulaariseen tilaan. In vitro ‑tutkimukset viittasivat siihen, että ponatinibi joko ei ole P‑gp:n eikä rintasyöpäresistenssiproteiini BCRP:n substraatti tai on niiden heikko substraatti. Ponatinibi ei ole substraatti ihmisen orgaanisia anioneja kuljettaville OATP1B1‑ eikä OATP1B3‑polypeptideille eikä orgaanisia kationeja kuljettavalle OCT‑1:lle.
Biotransformaatio
Ponatinibi metaboloituu inaktiiviseksi karboksyylihapoksi esteraasien ja/tai amidaasien vaikutuksesta. Se metaboloituu CYP3A4:n vaikutuksesta N‑desmetyylimetaboliitiksi, joka on 4 kertaa vähemmän aktiivinen kuin ponatinibi. Karboksyylihappo ja N‑desmetyylimetaboliitti muodostavat 58 % ja 2 % ponatinibin verenkierrossa olevasta määrästä.
Kun seerumin ponatinibipitoisuudet olivat hoitoalueella, ponatinibi ei estänyt OATP1B1‑, OATP1B3‑, OCT1‑ eikä OCT2‑toimintaa, orgaanisten anionien kuljettajia OAT1 eikä OAT3 eikä sappisuolojen poistopumppua (BSEP) in vitro. Näiden kuljettajien substraattien ponatinibivälitteinen esto ei siis todennäköisesti aiheuta kliinisiä lääkeaineyhteisvaikutuksia. In vitro ‑tutkimukset viittaavat siihen, että CYP1A2‑, CYP2B6‑, CYP2C8‑, CYP2C9‑, CYP2C19‑, CYP3A‑ tai CYP2D6‑entsyymien substraattien metabolian ponatinibivälitteinen esto ei todennäköisesti aiheuta kliinisiä lääkeaineyhteisvaikutuksia.
Ihmisen maksasoluissa tehdyn in vitro ‑kokeen tulokset viittaavat siihen, että ponatinibivälitteinen CYP1A2‑, CYP2B6‑ tai CYP3A‑substraattien metabolian induktio ei todennäköisesti aiheuta kliinisiä lääkeaineyhteisvaikutuksia.
Eliminaatio
Iclusigin 45 mg:n kerta‑annosten ja toistuvien 45 mg:n annosten jälkeen ponatinibin terminaalinen eliminaation puoliintumisaika oli 22 tuntia ja vakaa tila saavutetaan tyypillisesti 1 viikon sisällä jatkuvasti annosteltaessa. Kerran vuorokaudessa ‑annostuksella ponatinibin plasma‑altistukset kasvavat noin 1,5‑kertaisiksi ensimmäisen annoksen ja vakaan tilan välillä. Vaikka plasman ponatinibialtistus suurenee jatkuvaa annostusta käytettäessä vakaan tilan tasolle, populaatiofarmakokineettisen analyysin mukaan näennäinen oraalinen puhdistuma suurenee vain vähän ensimmäisten kahden viikon aikana jatkuvaa annostusta käytettäessä, eikä tätä pidetä kliinisesti relevanttina. Ponatinibi eliminoituu enimmäkseen ulosteiden kautta. Suun kautta annetun [14C]‑leimatun ponatinibikerta‑annoksen jälkeen noin 87 % radioaktiivisuudesta erittyy ulosteeseen ja noin 5 % virtsaan. Annetusta annoksesta 24 % erittyi muuttumattomassa muodossa ulosteeseen ja < 1 % virtsaan. Loput annoksesta erittyi metaboliitteina.
Munuaisten vajaatoiminta
Iclusigia ei ole tutkittu munuaisten vajaatoimintapotilailla. Munuaisteitse tapahtuva erittyminen ei kuulu ponatinibin keskeisiin eliminaatioreitteihin, mutta keskivaikean tai vaikean munuaisten vajaatoiminnan mahdollista vaikutusta maksan kautta tapahtuvaan eliminaatioon ei ole määritetty (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Yksi 30 mg:n ponatinibiannos annettiin potilaille, jolla oli lievä, keskivaikea tai vaikea maksan vajaatoiminta, sekä terveille vapaaehtoisille, joilla oli normaali maksan toiminta. Ponatinibin Cmax‑arvot olivat vertailukelpoiset lievää maksan vajaatoimintaa sairastavilla potilailla ja terveillä vapaaehtoisilla, joilla oli normaali maksan toiminta. Keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastavien potilaiden ponatinibin Cmax‑ ja AUC0‑∞‑arvot olivat pienemmät. Ponatinibin plasmaeliminaation puoliintumisaika oli pidempi lievää, keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastavilla potilailla, mutta ei eronnut kliinisesti merkittävällä tavalla terveistä potilaista, joilla oli normaali maksan toiminta.
In vitro ‑tuloksissa ei todettu eroa plasman proteiineihin sitoutumisessa terveiden henkilöiden ja maksan vajaatoimintapotilaiden (lievä, keskivaikea ja vaikea) plasmanäytteiden välillä. Ponatinibin farmakokinetiikassa ei todettu merkittävää eroa terveiden vapaaehtoisten, joilla on normaali maksan toiminta, ja eriasteista maksan vajaatoimintaa sairastavien potilaiden välillä. Iclusigin aloitusannosta ei tarvitse pienentää potilailla, joilla on maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Varovaisuutta suositellaan, jos Iclusigia annetaan maksan vajaatoimintapotilaille (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Yli 30 mg Iclusig-annoksia ei ole tutkittu potilailla, joilla on maksan vajaatoiminta (Child–Pugh-luokat A, B ja C).
Ponatinibin farmakokinetiikkaan vaikuttavat potilaskohtaiset tekijät
Sukupuolen, iän, rodun ja painon vaikutusta ponatinibin farmakokinetiikkaan ei ole arvioitu spesifisissä tutkimuksissa. Sukupuoli, rotu ja paino eivät ennustaneet ponatinibin farmakokinetiikan potilaskohtaista vaihtelua.
Prekliiniset tiedot turvallisuudesta
Iclusig on arvioitu farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, lisääntymistoksisuutta, fototoksisuutta ja karsinogeenisuutta koskevissa tutkimuksissa.
Ponatinibilla ei ilmennyt genotoksisuutta tavanomaisissa in vitro‑ ja in vivo ‑järjestelmissä.
Jäljempänä kuvattavia haittavaikutuksia ei ole todettu kliinisissä tutkimuksissa, mutta niitä on todettu koe‑eläimillä, jotka ovat saaneet hoitoannoksia vastaavia määriä lääkeainetta. Siksi haitoilla voi olla kliinistä merkitystä.
Lymfaattisten elinten depleetiota havaittiin toistuvan altistuksen aiheuttamaa toksisuutta koskevissa tutkimuksissa rotilla ja jaavanmakakeilla. Vaikutusten osoitettiin korjautuvan hoidon lopettamisen jälkeen.
Kasvulevyn kondrosyyttien hyper‑/hypoplastisia muutoksia havaittiin toistuvan altistuksen aiheuttamaa toksisuutta koskevissa tutkimuksissa rotilla.
Pitkäaikaisen annostelun jälkeen rotilla havaittiin esinahka‑ ja klitoraalirauhasissa tulehduksellisia muutoksia, joihin liittyi neutrofiili‑, monosyytti‑, eosinofiili‑ ja fibrinogeeniarvojen nousua.
Ihomuutoksia (rupia, hyperkeratoosia tai punoitusta) havaittiin jaavanmakakeilla tehdyissä toksisuustutkimuksissa. Kuivaa, hilseilevää ihoa havaittiin rotilla tehdyissä toksisuustutkimuksissa.
Rotilla, joita oli hoidettu 5 ja 10 mg/kg ponatinibilla, havaittiin eräässä tutkimuksessa diffuusia sarveiskalvon turvotusta, johon liittyi neutrofiilisolujen infiltraatiota, ja hyperplastisia muutoksia mykiön epiteelissä, mikä viittasi lievään fototoksiseen reaktioon.
Jaavanmakakeilla huomattiin systolisia sydämen sivuääniä, joilla ei ollut makroskooppisia tai mikroskooppisia korrelaatteja, yksittäisillä eläimillä, joita hoidettiin annoksella 5 ja 45 mg/kg kerta‑annostoksisuutta koskevassa tutkimuksessa, sekä annoksella 1, 2,5 ja 5 mg/kg 4 viikon mittaisessa, toistuvan altistuksen aiheuttamaa toksisuutta koskevassa tutkimuksessa. Tämän löydöksen kliinistä merkitystä ei tunneta.
Neljän viikon mittaisessa, toistuvan altistuksen aiheuttamaa toksisuutta koskevassa tutkimuksessa jaavanmakakeilla havaittiin kilpirauhasen follikulaarista atrofiaa, johon useimmiten liittyi T3‑arvojen aleneminen ja taipumus TSH‑arvojen kohoamiseen.
Toistuvan altistuksen aiheuttamaa toksisuutta koskevassa tutkimuksessa jaavanmakakeilla havaittiin ponatinibiin liittyviä mikroskooppisia munasarjamuutoksia (lisääntynyt follikkelien atresia) ja kivesmuutoksia (minimaalista itusolujen degeneroitumista) eläimillä, joita hoidettiin 5 mg/kg ponatinibilla.
Ponatinibi annoksilla 3, 10 ja 30 mg/kg sai aikaan virtsan erityksen ja elektrolyyttien erittymisen lisääntymistä ja mahan tyhjenemisen vähenemistä farmakologista turvallisuutta koskevissa tutkimuksissa rotilla.
Rotilla havaittiin emolle toksisilla annoksilla embryofetaalista toksisuutta (implantaation jälkeisiä alkiokuolemia, sikiöiden painon pienenemistä ja erilaisia pehmytkudoksen ja luuston muutoksia). Erilaisia pehmytkudoksen ja luuston muutoksia havaittiin myös emolle ei‑toksisilla annoksilla.
Uros- ja naarasrotilla tehdyssä hedelmällisyystutkimuksessa naarasrottien hedelmällisyysparametrit huononivat ihmisen kliinistä altistusta vastaavilla annoksilla. Naarasrotilla todettiin näyttöä alkiokuolemista sekä ennen implantaatiota että implantaation jälkeen. Ponatinibi saattaa siis heikentää naaraiden hedelmällisyyttä. Urosrottien hedelmällisyysparametrit eivät muuttuneet. Löydösten kliinistä merkitystä ihmisen hedelmällisyydelle ei tunneta.
Nuorilla rotilla todettiin tulehduksellisiin vaikutuksiin liittyvää kuolevuutta hoitoannoksen ollessa 3 mg/kg/vrk ja painonnousun vähenemistä ennen vieroitusta ja pian vieroituksen jälkeen hoitoannoksen ollessa 0,75, 1,5 tai 3 mg/kg/vrk. Kun toksisuutta tutkittiin nuorilla eläimillä, ponatinibi ei vaikuttanut haitallisesti tärkeisiin kehitysparametreihin.
Kaksivuotisessa karsinogeenisuustutkimuksessa uros- ja naarasrotilla ponatinibin peroraalinen anto (annokset uroksilla 0,05, 0,1 ja 0,2 mg/kg/vrk ja naarailla 0,2 ja 0,4 mg/kg/vrk) ei aiheuttanut tuumorigeenisuutta. Naarailla annos 0,8 mg/kg/vrk tuotti plasman altistuksen, joka oli yleisesti pienempi tai samankaltainen kuin ihmisen altistus annosalueella 15–45 mg/vrk. Kyseisellä annoksella todettiin häpykielirauhasen levyepiteelikarsinooman ilmaantuvuuden suurentuneen tilastollisesti merkitsevästi. Löydöksen kliinistä merkitystä ihmiselle ei tunneta.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Laktoosimonohydraatti, mikrokiteinen selluloosa, natriumtärkkelysglykolaatti, kolloidinen vedetön piidioksidi, magnesiumstearaatti,
Tabletin päällyste
Talkki, makrogoli 4000, poly(vinyylialkoholi), titaanidioksidi (E171)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pullon sisällä on suljettu purkki, joka sisältää molekyyliseulaa kuivausaineena. Säilytä purkki pullossa.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ICLUSIG tabletti, kalvopäällysteinen
15 mg (L:kyllä) 30 kpl (3030,88 €)
30 mg (L:kyllä) 30 kpl (4811,37 €)
45 mg (L:kyllä) 30 kpl (4811,37 €)
PF-selosteen tieto
Iclusig 15 mg tabletit, kalvopäällysteiset
HDPE‑pullot, joissa kierresuljin ja joko 30, 60 tai 180 kalvopäällysteistä tablettia sekä muovipurkki, joka sisältää molekyyliseulaa kuivausaineena.
Iclusig 30 mg tabletit, kalvopäällysteiset
HDPE‑pullot, joissa kierresuljin ja 30 kalvopäällysteistä tablettia sekä muovipurkki, joka sisältää molekyyliseulaa kuivausaineena.
Iclusig 45 mg tabletit, kalvopäällysteiset
HDPE‑pullot, joissa kierresuljin ja joko 30 tai 90 kalvopäällysteistä tablettia sekä muovipurkki, joka sisältää molekyyliseulaa kuivausaineena.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
15 mg: Valkoinen, kaksoiskupera, pyöreä kalvopäällysteinen tabletti, jonka halkaisija on noin 6 mm ja jonka yhdelle puolelle on kaiverrettu ”A5”.
30 mg: Valkoinen, kaksoiskupera, pyöreä kalvopäällysteinen tabletti, jonka halkaisija on noin 8 mm ja jonka yhdelle puolelle on kaiverrettu ”C7”.
45 mg: Valkoinen, kaksoiskupera, pyöreä kalvopäällysteinen tabletti, jonka halkaisija on noin 9 mm ja jonka yhdelle puolelle on kaiverrettu ”AP4”.
Käyttö- ja käsittelyohjeet
Hävittäminen
Ei erityisvaatimuksia hävittämisen suhteen.
Korvattavuus
ICLUSIG tabletti, kalvopäällysteinen
15 mg 30 kpl
30 mg 30 kpl
45 mg 30 kpl
- Ylempi erityiskorvaus (100 %). Ponatinibi: Kroonisen myelooisen ja akuutin lymfoblastisen leukemian hoito erityisin edellytyksin (1529).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Ponatinibi: Kroonisen vaiheen, akseleraatiovaiheen, blastivaiheen kroonisen myelooisen tai Philadelphia-kromosomipositiivisen akuutin lymfoblastisen leukemian hoito aikuispotilailla erityisin edellytyksin (373).
ATC-koodi
L01EA05
Valmisteyhteenvedon muuttamispäivämäärä
13.05.2026
Yhteystiedot
Paasheuvelweg 25
1105 BP Amsterdam
Netherlands
09 7479 0132
eumedinfo@incyte.com