
GIOTRIF tabletti, kalvopäällysteinen 20 mg, 30 mg, 40 mg, 50 mg
Vaikuttavat aineet ja niiden määrät
GIOTRIF 20 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 20 mg afatinibia (dimaleaattina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 118 mg laktoosia (monohydraattina).
GIOTRIF 30 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 30 mg afatinibia (dimaleaattina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 176 mg laktoosia (monohydraattina).
GIOTRIF 40 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 40 mg afatinibia (dimaleaattina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 235 mg laktoosia (monohydraattina).
GIOTRIF 50 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 50 mg afatinibia (dimaleaattina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 294 mg laktoosia (monohydraattina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti).
Kliiniset tiedot
Käyttöaiheet
GIOTRIF‑monoterapia on tarkoitettu
- paikallisesti edenneen tai etäpesäkkeisen ei-pienisoluisen keuhkosyövän hoitoon aikuispotilaille, kun kasvaimessa on vähintään yksi aktivoiva epidermaalisen kasvutekijän reseptorin (EGFR:n) mutaatio ja jos hoidossa ei ole aiemmin käytetty EGFR:n tyrosiinikinaasin estäjiä.
- paikallisesti edenneen tai etäpesäkkeisen, histologialtaan levyepiteeliperäisen ei-pienisoluisen keuhkosyövän hoitoon aikuispotilaille, kun sairaus on edennyt platinapohjaisen solunsalpaajahoidon aikana tai sen jälkeen (ks. kohta Farmakodynamiikka).
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
GIOTRIF‑hoidon aloittaa ja sitä valvoo lääkäri, jolla on kokemusta syöpähoitojen toteuttamisesta.
EGFR‑mutaatiostatus on selvitettävä ennen GIOTRIF‑hoidon aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Suositeltava annos on 40 mg kerran vuorokaudessa.
Tämä lääkevalmiste otetaan ilman ruokaa. Potilaan on oltava syömättä vähintään 3 tuntia ennen lääkevalmisteen ottamista ja vähintään 1 tunti sen ottamisen jälkeen (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
GIOTRIF‑hoitoa jatketaan, kunnes tauti alkaa taas edetä tai kunnes potilas ei enää siedä hoitoa (ks. taulukko 1 alla).
Annoksen suurentaminen
Annosta voidaan suurentaa enintään tasolle 50 mg/vrk, jos potilas sietää aloitusannoksen 40 mg/vrk (ts. ei ripulia, ihottumaa, stomatiittia eikä muita CTCAE‑luokituksen asteen > 1 haittavaikutuksia) ensimmäisen hoitosyklin ajan (21 päivää EGFR-mutaatiopositiivisessa ei-pienisoluisessa keuhkosyövässä ja 28 päivää levyepiteeliperäisessä ei-pienisoluisessa keuhkosyövässä). Annosta ei pidä koskaan suurentaa, jos potilaan annosta on aiemmin pienennetty. Suurin sallittu annos on 50 mg/vrk.
Annoksen muuttaminen haittavaikutusten vuoksi
Oireisia haittavaikutuksia (esim. vaikea/pitkittynyt ripuli tai ihohaitat) voidaan hoitaa tauottamalla lääkkeen käyttö ja pienentämällä annosta tai lopettamalla GIOTRIF‑hoito taulukon 1 mukaisella tavalla (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Taulukko 1: Ohjeet annoksen muuttamisesta haittavaikutusten vuoksi
Haittavaikutukset CTCAE‑luokituksena mukaan | Suositeltava annostelu | |
Aste 1 tai aste 2 | Ei tauotetab | Ei annosmuutosta |
Aste 2 (pitkittynytc tai sietämätön) tai aste ≥ 3 | Tauotetaan, kunnes aste 0/1b | Aloitetaan uudelleen 10 mg pienemmällä annoksellad |
a Yhdysvaltain National Cancer Institute ‑instituutin (NCI) Common Terminology Criteria for Adverse Events ‑haittatapahtumaluokitus
b Jos potilaalla on ripulia, ripulilääkitys (esim. loperamidi) aloitetaan heti. Jos ripuli pitkittyy, ripulilääkkeen käyttöä jatketaan, kunnes ulosteet eivät enää ole löysiä.
c > 48 tuntia kestävä ripuli ja/tai > 7 päivää kestävä ihottuma
d Jos potilas ei siedä annosta 20 mg/vrk, on harkittava GIOTRIF‑hoidon lopettamista pysyvästi
Interstitiaalisen keuhkosairauden mahdollisuutta on harkittava, jos potilaalle kehittyy akuutteja hengitystieoireita tai hengitystieoireet pahenevat. Tässä tapauksessa hoito on tauotettava arvioinnin ajaksi. Jos potilaalla todetaan interstitiaalinen keuhkosairaus, GIOTRIF‑hoito lopetetaan ja aloitetaan asianmukainen hoito tarpeen mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Väliin jäänyt annos
Jos annos jää väliin, se on otettava samana päivänä heti, kun potilas muistaa asian. Jos seuraavaan aikataulun mukaiseen annokseen on kuitenkin alle 8 tuntia, väliin jäänyt annos jätetään ottamatta.
P‑glykoproteiinin (P‑gp) estäjien käyttö
Jos potilaan on käytettävä P‑gp:n estäjiä, ne on otettava käyttäen porrastettua annostelua, eli P‑gp:n estäjän ja GIOTRIF‑annoksen ottamisen välillä on oltava ajallisesti mahdollisimman pitkä väli. Tämä tarkoittaa mieluiten 6 tunnin (jos P‑gp:n estäjä otetaan kahdesti päivässä) tai 12 tunnin (jos P‑gp:n estäjä otetaan kerran päivässä) eroa GIOTRIF‑valmisteen ottoon (ks. kohta Yhteisvaikutukset).
Erityisryhmät
Munuaisten vajaatoimintaa sairastavat potilaat
Afatinibialtistuksen havaittiin nousevan potilailla, jotka sairastivat keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa (ks. kohta Farmakokinetiikka). Aloitusannosta ei ole tarpeen muuttaa potilailla, jotka sairastavat lievää (eGFR 60‒89 ml/min/1,73 m2), keskivaikeaa (eGFR 30‒59 ml/min/1,73 m2) tai vaikeaa (eGFR 15‒29 ml/min/1,73 m2) munuaisten vajaatoimintaa. Tarkkaile vaikeaa munuaisten vajaatoimintaa sairastavia potilaita (eGFR 15‒29 ml/min/1,73 m2) ja muuta GIOTRIF-annosta, jos potilas ei sitä siedä.
GIOTRIF-hoitoa ei suositella potilaille, joilla eGFR on < 15 ml/min/1,73 m2 tai jotka saavat dialyysihoitoa.
Maksan vajaatoimintaa sairastavat potilaat
Afatinibialtistus ei muutu merkitsevästi, jos potilaalla on lievä (Child–Pugh‑luokka A) tai keskivaikea (Child–Pugh‑luokka B) maksan vajaatoiminta (ks. kohta Farmakokinetiikka). Aloitusannosta ei ole tarpeen muuttaa potilaille, joilla on lievä tai keskivaikea maksan vajaatoiminta. Tätä lääkevalmistetta ei ole arvioitu potilailla, joilla on vaikea (Child–Pugh‑luokka C) maksan vajaatoiminta. Hoitoa ei suositella näille potilaille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Ei ole asianmukaista käyttää GIOTRIF‑valmistetta pediatrisille potilaille ei‑pienisoluisen keuhkosyövän hoitoon.
Kliininen tutkimus pediatrisilla potilailla, joilla oli muita sairauksia, ei tukenut lasten tai nuorten GIOTRIF-hoitoa (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka). Turvallisuutta ja tehoa ei ole varmistettu.
Tämän lääkevalmisteen käyttöä lasten ja nuorten hoidossa ei siis suositella.
Antotapa
Tämä lääkevalmiste otetaan suun kautta. Tabletit nielaistaan kokonaisina veden kera. Jos kokonaisia tabletteja ei voida niellä, tabletit voidaan liuottaa noin 100 millilitraan hiilihapotonta juomavettä. Muita nesteitä ei saa käyttää. Tabletti pudotetaan veteen murskaamattomana ja nestettä sekoitetaan silloin tällöin jopa 15 minuutin ajan, kunnes tabletti on hajonnut pienenpieniin hiukkasiin. Dispersio otetaan välittömästi. Lasi huuhdellaan noin 100 millilitralla vettä, joka juodaan. Dispersio voidaan antaa myös mahaletkun kautta.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
EGFR‑mutaatiostatuksen arviointi
On tärkeää käyttää potilaan EGFR‑mutaatiostatuksen arviointiin hyvin validoitua, luotettavaa menetelmää, jotta vältytään vääriltä negatiivisilta ja vääriltä positiivisilta tuloksilta.
Ripuli
GIOTRIF‑hoidon aikana on ilmoitettu ripulia, myös vaikeaa ripulia (ks. kohta Haittavaikutukset). Ripuli voi johtaa nestehukkaan, johon joko liittyy tai ei liity munuaisten vajaatoiminta. Tämä on johtanut harvinaisissa tapauksissa kuolemaan. Ripuli kehittyi yleensä ensimmäisten 2 hoitoviikon aikana. Asteen 3 ripulia esiintyi yleisimmin ensimmäisten 6 hoitoviikon aikana.
Etenkin ensimmäisten 6 hoitoviikon aikana on tärkeää, että heti ripulin ensimerkkien ilmetessä aloitetaan aktiivinen hoito riittävällä nesteytyksellä ja ripulilääkkeillä. Hoidossa käytettävien ripulilääkkeiden (esim. loperamidi) annosta suurennetaan tarvittaessa suurimpaan hyväksyttyyn suositusannokseen asti. Ripulilääkkeitä on oltava välittömästi potilaan saatavilla, jotta hoito voidaan aloittaa heti ripulin ensimerkkien ilmetessä ja sitä voidaan jatkaa, kunnes löysiä ulosteita ei ole esiintynyt 12 tuntiin. Jos potilaalla on vaikea ripuli, GIOTRIF-hoidon tauottaminen ja annoksen pienentäminen tai hoidon lopettaminen voi olla tarpeen (ks. kohta Annostus ja antotapa). Jos potilaalle kehittyy nestehukka, on ehkä tarpeen antaa elektrolyyttejä ja nesteitä laskimoon.
Ihohaitat
Tätä lääkevalmistetta saaneilla potilailla on ilmoitettu ihottumaa/aknea (ks. kohta Haittavaikutukset). Ihottuma ilmenee yleensä lievänä tai keskivaikeana punoittavana ja aknemaisena ihottumana, joka voi kehittyä auringonvalolle altistuneille ihoalueille tai pahentua näillä ihoalueilla. Jos potilas altistuu auringolle, hänen on aiheellista käyttää suojaavaa vaatetusta ja aurinkovoidetta. Ihoreaktioiden varhainen hoito (esim. kosteusvoiteet, antibiootit) voi mahdollistaa GIOTRIF‑hoidon jatkamisen ilman taukoja. Vaikean ihoreaktion yhteydessä voi myös olla tarpeen tauottaa hoito, pienentää annosta (ks. kohta Annostus ja antotapa), ryhtyä muihin hoitotoimiin ja ohjata potilas erikoislääkärille, jolla on kokemusta tällaisten ihoreaktioiden hoidosta.
Potilailla on ilmoitettu vesikelloisia, rakkuloivia ja hilseileviä ihoreaktioita, jotka ovat harvinaisissa tapauksissa voineet viitata Stevens–Johnsonin oireyhtymään ja toksiseen epidermaaliseen nekrolyysiin. Tämän lääkevalmisteen käyttö on tauotettava tai lopetettava, jos potilaalle kehittyy vaikea vesikelloinen, rakkuloiva tai hilseilevä ihoreaktio (ks. kohta Haittavaikutukset).
Naissukupuoli, pienipainoisuus ja munuaisten vajaatoiminta
Afatinibialtistuksen on havaittu suurentuneen naisilla, pienipainoisilla potilailla ja potilailla, joilla on entuudestaan munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka). Tämä voi suurentaa haittavaikutusten, etenkin ripulin, ihottuman/aknen ja stomatiitin riskiä. Jos potilaalla on näitä riskitekijöitä, tiiviimpi seuranta on tarpeen.
Interstitiaalinen keuhkosairaus
Potilailla, jotka ovat saaneet GIOTRIF‑hoitoa ei‑pienisoluisen keuhkosyövän hoitoon, on ilmoitettu interstitiaalista keuhkosairautta tai sen kaltaisia haittavaikutuksia (esim. keuhkoinfiltraatti, pneumoniitti, akuutti hengitysvajaus, allerginen alveoliitti), myös kuolemaan johtaneita tapauksia. Kaikissa kliinisissä tutkimuksissa interstitiaalisen keuhkosairauden kaltaisia haittavaikutuksia ilmoitettiin 0,7 %:lla GIOTRIF-valmistetta saaneista potilaista (mukaan lukien 0,5 %:lla potilaista, joilla oli CTCAE‑luokituksen astetta ≥ 3 edustavia interstitiaalisen keuhkosairauden kaltaisia haittavaikutuksia). Potilaita, joilla on anamneesissa interstitiaalinen keuhkosairaus, ei ole arvioitu.
Jos potilaalle kehittyy akuutisti alkavia keuhko‑oireita (hengenahdistus, yskä, kuume) ja/tai keuhko‑oireet pahenevat selittämättömästi, huolellinen arviointi on aina tarpeen interstitiaalisen keuhkosairauden poissulkemiseksi. Tämän lääkevalmisteen käyttö on tauotettava näiden oireiden arvioinnin ajaksi. Jos potilaalla todetaan interstitiaalinen keuhkosairaus, GIOTRIF‑hoito lopetetaan pysyvästi ja asianmukainen hoito aloitetaan tarpeen mukaan (ks. kohta Annostus ja antotapa).
Vaikea maksan vajaatoiminta
Maksan vajaatoimintaa, myös kuolemantapauksia, on ilmoitettu alle 1 %:lla potilaista lääkehoidon aikana. Näillä potilailla esiintyi sekoittavina tekijöinä aiempaa maksasairautta ja/tai liitännäissairauksia, joilla oli yhteys potilaan syöpätaudin etenemiseen. Säännöllisiä maksan toimintakokeita suositellaan, jos potilaalla on entuudestaan maksasairaus. Keskeisissä tutkimuksissa asteen 3 alaniiniaminotransferaasiarvojen (ALAT) ja aspartaattiaminotransferaasiarvojen (ASAT) suurenemista havaittiin 2,4 %:lla (LUX‑Lung 3) ja 1,6 %:lla (LUX‑Lung 8) potilaista, jotka käyttivät annosta 40 mg/vrk ja joiden maksa‑arvot olivat lähtötilanteessa normaalit. Potilailla, joiden maksa‑arvoissa oli lähtötilanteessa poikkeavuuksia, asteen 3 kohoamat ALAT/ASAT‑arvoissa olivat LUX‑Lung 3 -tutkimuksessa noin 3,5‑kertaisia. LUX‑Lung 8 ‑tutkimuksessa ei havaittu asteen 3 kohoamia ALAT/ASAT-arvoissa potilailla, joiden maksa-arvoissa oli lähtötilanteessa poikkeavuuksia, (ks. kohta Haittavaikutukset). Hoidon tauottaminen voi olla tarpeen, jos potilaan maksatoiminta heikkenee (ks. kohta Annostus ja antotapa). Jos potilaalle kehittyy GIOTRIF‑hoidon aikana vaikea maksan vajaatoiminta, hoito on lopetettava.
Maha-suolikanavan perforaatiot
Maha-suolikanavan perforaatiota, myös kuolemantapauksia, on ilmoitettu GIOTRIF-hoidon aikana 0,2 %:lla potilaista kaikissa satunnaistetuissa, kontrolloiduissa kliinisissä tutkimuksissa. Useimmissa tapauksissa maha-suolikanavan perforaatioon liittyi muita tiedossa olevia riskitekijöitä, kuten muu samanaikainen lääkitys (esim. kortikosteroidit, tulehduskipulääkkeet tai angiogeneesiä estävät lääkkeet), aiempi maha-suolikanavan haavauma, piilevä umpipussitauti, ikä tai suolistometastaasit perforaatiokohdissa. Jos potilaalla ilmenee maha-suolikanavan perforaatio GIOTRIF-hoidon aikana, hoito on lopetettava pysyvästi.
Keratiitti
Jos potilaalla on esim. akuutti tai paheneva silmätulehdus, kyynelvuotoa, valoherkkyyttä, näön sumenemista, silmäkipua ja/tai silmän punoitusta, hänet on ohjattava nopeasti silmätautien erikoislääkärin hoitoon. Jos potilaalla todetaan vahvistetusti haavainen keratiitti, hoito on tauotettava tai lopetettava. Jos potilaalla todetaan keratiitti, hoidon jatkamisen hyötyjä ja riskejä on punnittava tarkoin. Tätä lääkevalmistetta on käytettävä varoen, jos potilaalla on anamneesissa keratiitti, haavainen keratiitti tai vaikeaa silmän kuivuutta. Myös piilolinssien käyttö on keratiitin ja haavaumien riskitekijä (ks. kohta Haittavaikutukset).
Vasemman kammion toiminta
HER2:n estoon on liittynyt vasemman kammion toimintahäiriöitä. Saatavilla olevat kliiniset tutkimustiedot eivät viittaa siihen, että tämä lääkevalmiste vaikuttaisi haitallisesti sydämen supistuvuuteen. Tätä lääkevalmistetta ei kuitenkaan ole tutkittu potilailla, joilla on poikkeava vasemman kammion ejektiofraktio (LVEF) tai muuta merkittävää sydänanamneesissa. Jos potilaalla on sydämeen liittyviä riskitekijöitä tai mahdollisesti LVEF-arvoon vaikuttava tila, on harkittava sydämen toiminnan seurantaa, johon kuuluu LVEF-arviointi lähtötilanteessa ja hoidon aikana. Myös potilaille, joille kehittyy hoidon aikana relevantteja sydänoireita/‑löydöksiä, on harkittava sydäntoiminnan seurantaa, johon kuuluu LVEF-arviointi.
Jos potilaan ejektiofraktio alittaa viitearvojen alarajan, on harkittava sydänkonsultaatiota ja hoidon tauottamista tai lopettamista.
P‑glykoproteiiniin (P‑gp) liittyvät yhteisvaikutukset
Samanaikainen hoito voimakkailla P‑gp:n indusoreilla voi pienentää afatinibialtistusta (ks. kohta Yhteisvaikutukset).
Laktoosi
Tämä lääkevalmiste sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi‑intoleranssi, täydellinen laktaasinpuutos tai glukoosi‑galaktoosi‑imeytymishäiriö, ei pidä käyttää tätä lääkevalmistetta.
Yhteisvaikutukset
Yhteisvaikutukset lääkeaineiden kuljetusjärjestelmien kanssa
P‑glykoproteiinin (P‑gp) ja rintasyöpäresistenssiproteiinin (BCRP) estäjien vaikutukset afatinibiin
In vitro ‑tutkimukset ovat osoittaneet, että afatinibi on P‑gp:n ja BCRP:n substraatti. Kun ritonaviiria, joka on voimakas P‑gp:n ja BCRP:n estäjä, annettiin (200 mg kahdesti vuorokaudessa 3 päivän ajan) 1 tunti ennen 20 mg:n GIOTRIF-kerta‑annosta, afatinibialtistus suureni 48 % (pitoisuus–aika-käyrän alapuolinen pinta-ala [AUC0–∞]) ja 39 % (lääkkeen huippupitoisuus plasmassa [Cmax]). Kun ritonaviiri puolestaan annettiin yhtä aikaa 40 mg:n GIOTRIF-annoksen kanssa tai 6 tuntia sen jälkeen, afatinibin suhteellinen hyötyosuus oli 119 % (AUC0–∞) ja 104 % (Cmax) samanaikaisesti annettaessa ja 111 % (AUC0–∞) ja 105 % (Cmax), kun ritonaviiri annettiin 6 tuntia GIOTRIF-annoksen jälkeen. Sen vuoksi on suositeltavaa antaa voimakkaat P‑gp:n estäjät (mm. ritonaviiri, siklosporiini A, ketokonatsoli, itrakonatsoli, erytromysiini, verapamiili, kinidiini, takrolimuusi, nelfinaviiri, sakinaviiri ja amiodaroni) porrastetusti, mieluiten niin, että aikaa P‑gp:n estäjän annon ja GIOTRIF-valmisteen annon välille jää 6 tai 12 tuntia (ks. kohta Annostus ja antotapa).
P‑gp:n indusorien vaikutukset afatinibiin
Aikaisempi hoito voimakkaasti P‑gp‑toimintaa indusoivalla rifampisiinilla (600 mg kerran vuorokaudessa 7 päivän ajan) pienensi plasman afatinibialtistusta 34 % (AUC0-∞) ja 22 % (Cmax) 40 mg:n GIOTRIF‑kerta‑annoksen jälkeen. Voimakkaat P‑gp:n indusorit (mm. rifampisiini, karbamatsepiini, fenytoiini, fenobarbitaali tai mäkikuisma [Hypericum perforatum]) voivat pienentää afatinibialtistusta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Afatinibin vaikutukset P‑gp:n substraatteihin
In vitro ‑tietojen perusteella afatinibi on kohtalainen P‑gp:n estäjä. Kliinisten tietojen perusteella vaikuttaa kuitenkin epätodennäköiseltä, että GIOTRIF‑hoito muuttaisi muiden P‑gp:n substraattien pitoisuuksia plasmassa.
Yhteisvaikutukset BCRP:n kanssa
In vitro ‑tutkimusten tulokset viittaavat siihen, että afatinibi on BCRP‑kuljettajaproteiinin substraatti ja estäjä. Afatinibi saattaa suurentaa suun kautta annettavien BCRP:n substraattien (mm. rosuvastatiinin ja sulfasalatsiinin) hyötyosuutta.
Ruoan vaikutus afatinibiin
GIOTRIF‑valmisteen otto runsasrasvaisen aterian kanssa pienensi afatinibialtistusta merkitsevästi. Cmax pieneni noin 50 % ja AUC0-∞ noin 39 %. Tämä lääkevalmiste on otettava ilman ruokaa (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisia, jotka voivat tulla raskaaksi, on varmuuden vuoksi kehotettava välttämään raskaaksi tulemista GIOTRIF‑hoidon aikana. Asianmukaista ehkäisyä on käytettävä hoidon aikana ja vähintään yhden kuukauden ajan viimeisen annoksen jälkeen.
Raskaus
Mekanismin perusteella kaikki EGFR‑toimintaan vaikuttavat täsmälääkkeet voivat aiheuttaa haittaa sikiölle.
Afatinibilla tehdyissä eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Eläinkokeissa ei ole havaittu näyttöä teratogeenisuudesta, kun annokset olivat jopa emon kuolemaan johtavia. Haitallisia muutoksia esiintyi vain toksisia annoksia käytettäessä. Eläinten systeeminen altistus oli kuitenkin joko samaa luokkaa kuin potilailla tai tätä pienempi (ks. kohta Prekliiniset tiedot turvallisuudesta).
Ei ole olemassa tietoja tai on vain vähän tietoja tämän lääkevalmisteen käytöstä raskaana oleville naisille. Ihmisille koituvaa riskiä ei siis tunneta. Jos valmistetta käytetään raskauden aikana tai raskaus alkaa GIOTRIF‑hoidon aikana tai sen jälkeen, naiselle on kerrottava sikiöön mahdollisesti kohdistuvista riskeistä.
Imetys
Eläinkokeista saadut farmakokineettiset tiedot ovat osoittaneet, että afatinibi erittyy maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). On siis todennäköistä, että afatinibi erittyy ihmisen rintamaitoon. Imeväiseen kohdistuvia riskejä ei voida poissulkea. Äitejä on kehotettava olemaan imettämättä tämän lääkevalmisteen käytön aikana.
Hedelmällisyys
Afatinibin vaikutusta hedelmällisyyteen ei ole tutkittu ihmisellä. Saatavilla olevat ei‑kliiniset toksikologiset tiedot ovat osoittaneet, että suuremmat annokset vaikuttavat sukuelimiin. Mahdollisuutta, että tämän lääkevalmisteen käyttö vaikuttaa haitallisesti ihmisen hedelmällisyyteen, ei siis voida sulkea pois.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
GIOTRIF‑valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Hoidon aikana joillakin potilailla on ilmoitettu silmähaittoja (konjunktiviitti, kuivasilmäisyys, keratiitti) (ks. kohta Haittavaikutukset), jotka voivat vaikuttaa ajokykyyn tai koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Haittavaikutustyypit liittyivät yleensä afatinibin vaikutustapaan eli EGFR‑toiminnan estoon. Yhteenveto kaikista haittavaikutuksista on esitetty taulukossa 2. Yleisimpiä haittavaikutuksia olivat ripuli ja ihohaitat (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) sekä stomatiitti ja kynsivallintulehdus (ks. myös taulukot 3, 4 ja 5). Yleisesti ottaen annoksen pienentäminen (ks. kohta Annostus ja antotapa) johti yleisten haittavaikutusten esiintymistiheyden pienenemiseen.
Potilailla, jotka olivat saaneet GIOTRIF‑valmistetta 40 mg kerran vuorokaudessa, annosta pienennettiin haittavaikutusten vuoksi 57 %:lla potilaista LUX‑Lung 3 -tutkimuksessa ja 25 %:lla potilaista LUX‑Lung 8 -tutkimuksessa. LUX‑Lung 3 -tutkimuksessa 1,3 % potilaista lopetti hoidon ripulin ja 0 % ihottuman/aknen vuoksi. LUX‑Lung 8 -tutkimuksessa vastaavat luvut olivat 3,8 % ja 2,0 %.
Interstitiaalisen keuhkosairauden kaltaisia haittavaikutuksia ilmoitettiin 0,7 %:lla afatinibihoitoa saaneista potilaista. Potilailla ilmoitettiin vesikelloisia, rakkuloivia ja hilseileviä ihoreaktioita, jotka ovat harvinaisissa tapauksissa voineet viitata Stevens–Johnsonin oireyhtymään ja toksiseen epidermaaliseen nekrolyysiin. Näissä tapauksissa ihoreaktioille oli kuitenkin myös muita vaihtoehtoisia etiologisia syitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Taulukossa 2 esitetään yhteenveto haittavaikutusten esiintymistiheyksistä. Tiedot ovat kaikista sellaisista ei‑pienisoluisen keuhkosyövän tutkimuksista ja markkinoille tulon jälkeen kerätyistä kokemuksista, joissa GIOTRIF‑vuorokausiannos oli 40 mg tai 50 mg ja valmistetta käytettiin monoterapiana. Haittavaikutusten esiintymistiheydet on luokiteltu seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2: Haittavaikutusten yhteenveto yleisyysluokittain
| Elinjärjestelmä | Hyvin yleiset | Yleiset | Melko harvinaiset | Harvinaiset |
| Infektiot | Kynsivallintulehdus1 | Virtsarakkotulehdus | ||
| Aineenvaihdunta ja ravitsemus | Ruokahalun heikkeneminen | Nestehukka Hypokalemia | ||
| Hermosto | Makuaistin muutokset | |||
| Silmät | Konjunktiviitti Kuivasilmäisyys | Keratiitti Poikkeava silmäripsien kasvu | ||
| Hengityselimet, rintakehä ja välikarsina | Nenäverenvuoto | Voimakas nuha | Interstitiaalinen keuhkosairaus | |
| Ruoansulatuselimistö | Ripuli Stomatiitti2 Pahoinvointi Oksentelu | Dyspepsia Huulitulehdus | Pankreatiitti Maha-suolikanavan perforaatio | |
| Maksa ja sappi | ALAT‑arvon suureneminen ASAT‑arvon suureneminen | |||
| Iho ja ihonalainen kudos | Ihottuma3 Aknemainen dermatiitti4 Kutina5 Ihon kuivuus6 | Käsi-jalkaoireyhtymä Kynsisairaudet8 | Stevens-Johnsonin oireyhtymä7 Toksinen epidermaalinen nekrolyysi7 | |
| Luusto, lihakset ja sidekudos | Lihasspasmit | |||
| Munuaiset ja virtsatiet | Munuaistoiminnan heikkeneminen / munuaisten vajaatoiminta | |||
| Yleisoireet ja antopaikassa todettavat haitat | Kuume | |||
| Tutkimukset | Painon lasku |
1 Mukana kynsivallintulehdus, kynsi‑infektio, kynsimarron infektio
2 Mukana stomatiitti, aftainen stomatiitti, limakalvotulehdus, suun haavaumat, suun limakalvon eroosiot, limakalvon eroosiot, limakalvon haavaumat
3 Mukana useita ihottumatyyppejä
4 Mukana akne, pustulaarinen akne, aknemainen dermatiitti
5 Mukana kutina, yleistynyt kutina
6 Mukana ihon kuivuus, ihon rohtuminen
7 Perustuu markkinoille tulon jälkeen kerättyihin kokemuksiin
8 Mukana kynsisairaus, kynnen irtoaminen, kynsitoksisuus, kynnen murtuminen, sisäänkasvanut kynsi, kuoppaiset kynnet, kynsituho, kynsien värjäytyminen, kynsien surkastuminen, harjanteiden muodostuminen kynsiin ja kotkankyntisyys
Tiettyjen haittavaikutusten kuvaus
Taulukoissa 3 ja 4 esitetään yhteenveto haittavaikutuksista, joita esiintyi LUX‑Lung 3- ja LUX-Lung 7 ‑tutkimusten GIOTRIF‑ryhmässä hyvin yleisesti eli vähintään 10 %:lla potilaista. Haittavaikutukset on esitetty Yhdysvaltain NCI:n (National Cancer Institute) Common Toxicity Criteria ‑luokituksen (NCI‑CTC) asteiden mukaisesti.
Taulukko 3: LUX‑Lung 3 ‑tutkimuksessa esiintyneet hyvin yleiset haittavaikutukset
GIOTRIF (40 mg/vrk) N = 229 | Pemetreksedi/ sisplatiini N = 111 | |||||
| NCI‑CTC‑luokituksen mukainen aste | Kaikki asteet | 3 | 4 | Kaikki asteet | 3 | 4 |
| MedDRA‑termi | % | % | % | % | % | % |
| Infektiot | ||||||
| Kynsivallintulehdus1 | 57,6 | 11,4 | 0 | 0 | 0 | 0 |
| Aineenvaihdunta ja ravitsemus | ||||||
| Ruokahalun heikkeneminen | 20,5 | 3,1 | 0 | 53,2 | 2,7 | 0 |
| Hengityselimet, rintakehä ja välikarsina | ||||||
| Nenäverenvuoto | 13,1 | 0 | 0 | 0,9 | 0,9 | 0 |
| Ruoansulatuselimistö | ||||||
| Ripuli | 95,2 | 14,4 | 0 | 15,3 | 0 | 0 |
Stomatiitti2 Huulitulehdus | 69,9 12,2 | 8,3 0 | 0,4 0 | 13,5 0,9 | 0,9 0 | 0 0 |
| Iho ja ihonalainen kudos | ||||||
| Ihottuma3 | 70,3 | 14 | 0 | 6,3 | 0 | 0 |
| Aknemainen dermatiitti4 | 34,9 | 2,6 | 0 | 0 | 0 | 0 |
| Ihon kuivuus5 | 29,7 | 0,4 | 0 | 1,8 | 0 | 0 |
| Kutina6 | 19,2 | 0,4 | 0 | 0,9 | 0 | 0 |
| Tutkimukset | ||||||
| Painon lasku | 10,5 | 0 | 0 | 9,0 | 0 | 0 |
1 Mukana kynsivallintulehdus, kynsi‑infektio, kynsimarron infektio
2 Mukana stomatiitti, aftainen stomatiitti, limakalvotulehdus, suun haavaumat, suun limakalvon eroosiot, limakalvon eroosiot, limakalvon haavaumat
3 Mukana useita ihottumatyyppejä
4 Mukana akne, pustulaarinen akne, aknemainen dermatiitti
5 Mukana ihon kuivuus, ihon rohtuminen
6 Mukana kutina, yleistynyt kutina
Taulukko 4: Hyvin yleiset haittavaikutukset LUX-Lung 7 -tutkimuksessa
GIOTRIF (40 mg/vrk) N=160 | Gefitinibi
N=159 | |||||
| NCI-CTC-luokituksen mukainen aste | Kaikki asteet | 3 | 4 | Kaikki asteet | 3 | 4 |
| MedDRA-termi | % | % | % | % | % | % |
| Infektiot | ||||||
| Kynsivallintulehdus1 | 57,5 | 1,9 | 0 | 17,0 | 0,6 | 0 |
| Virtsarakkotulehdus2 | 11,3 | 1,3 | 0 | 7,5 | 1,3 | 0,6 |
| Aineenvaihdunta ja ravitsemus | ||||||
| Ruokahalun heikkeneminen | 27,5 | 1,3 | 0 | 24,5 | 1,9 | 0 |
| Hypokalemia3 | 10,6 | 2,5 | 1,3 | 5,7 | 1,3 | 0 |
| Hengityselimet, rintakehä ja välikarsina | ||||||
| Voimakas nuha4 | 19,4 | 0 | 0 | 7,5 | 0 | 0 |
| Nenäverenvuoto | 18,1 | 0 | 0 | 8,8 | 0 | 0 |
| Ruoansulatuselimistö | ||||||
| Ripuli | 90,6 | 13,8 | 0,6 | 64,2 | 3,1 | 0 |
| Stomatiitti5 | 64,4 | 4,4 | 0 | 27,0 | 0 | 0 |
| Pahoinvointi | 25,6 | 1,3 | 0 | 27,7 | 1,3 | 0 |
| Oksentelu | 19,4 | 0,6 | 0 | 13,8 | 2,5 | 0 |
| Dyspepsia | 10,0 | 0 | 0 | 8,2 | 0 | 0 |
| Maksa ja sappi | ||||||
| ALAT-arvon suureneminen | 11,3 | 0 | 0 | 27,7 | 8,8 | 0,6 |
| Iho ja ihonalainen kudos | ||||||
| Ihottuma6 | 80,0 | 7,5 | 0 | 67,9 | 3,1 | 0 |
| Ihon kuivuus | 32,5 | 0 | 0 | 39,6 | 0 | 0 |
| Kutina7 | 25,6 | 0 | 0 | 25,2 | 0 | 0 |
| Aknemainen dermatiitti8 | 23,8 | 1,9 | 0 | 32,1 | 0,6 | 0 |
| Yleisoireet ja antopaikassa todettavat haitat | ||||||
| Kuume | 13,8 | 0 | 0 | 6,3 | 0 | 0 |
| Tutkimukset | ||||||
| Painon lasku | 10,0 | 0,6 | 0 | 5,7 | 0,6 | 0 |
1 Mukana kynsivallintulehdus, kynsi‑infektio, kynsimarron infektio
2 Mukana virtsarakkotulehdus, virtsatietulehdus
3 Mukana hypokalemia, veren kaliumpitoisuuden lasku
4 Mukana voimakas nuha, nenän tulehdus
5 Mukana stomatiitti, aftainen stomatiitti, limakalvotulehdus, suun haavaumat, limakalvon eroosiot
6 Mukana useita ihottumatyyppejä
7 Mukana kutina, yleistynyt kutina
8 Mukana aknemainen dermatiitti, akne
Maksan toimintakokeiden poikkeavuudet
40 mg:n GIOTRIF-annoksia käyttäneillä potilailla havaittiin maksan toimintakokeissa poikkeavuuksia, mm. ALAT- ja ASAT-arvojen suurenemista. Arvojen suureneminen oli useimmiten ohimenevää eikä johtanut hoidon lopettamiseen. Asteen 2 (> 2,5–5,0 × viitevälin yläraja [ULN]) ALAT-arvojen suurenemista esiintyi < 8 %:lla tätä lääkevalmistetta käyttäneistä potilaista. Asteen 3 (> 5,0–20,0 × ULN) arvojen suurenemista esiintyi < 4 %:lla GIOTRIF-valmistetta saaneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tiettyjen haittavaikutusten kuvaus
Taulukossa 5 esitetään yhteenveto haittavaikutuksista, joita esiintyi LUX‑Lung 8 ‑tutkimuksen GIOTRIF‑ryhmässä hyvin yleisesti eli vähintään 10 %:lla potilaista. Haittavaikutukset on esitetty Yhdysvaltain NCI:n (National Cancer Institute) Common Toxicity Criteria ‑luokituksen (NCI‑CTC) asteiden mukaisesti.
Taulukko 5: LUX‑Lung 8 ‑tutkimuksessa esiintyneet hyvin yleiset haittavaikutukset*
GIOTRIF (40 mg/vrk) N=392 | Erlotinibi N=395 | |||||
| NCI-CTC -luokituksen mukainen aste | Kaikki asteet | 3 | 4 | Kaikki asteet | 3 | 4 |
| MedDRA-termi | % | % | % | % | % | % |
| Infektiot | ||||||
| Kynsivallintulehdus1 | 11,0 | 0,5 | 0 | 5,1 | 0,3 | 0 |
| Aineenvaihdunta ja ravitsemus | ||||||
| Ruokahalun heikkeneminen | 24,7 | 3,1 | 0 | 26,1 | 2,0 | 0 |
| Ruoansulatuselimistö | ||||||
| Ripuli | 74,7 | 9,9 | 0,8 | 41,3 | 3,0 | 0,3 |
| Stomatiitti2 | 30,1 | 4,1 | 0 | 10,6 | 0,5 | 0 |
| Pahoinvointi | 20,7 | 1,5 | 0 | 16,2 | 1,0 | 0,3 |
| Iho ja ihonalainen kudos | ||||||
| Ihottuma3 | 60,7 | 5,4 | 0 | 56,7 | 8,1 | 0 |
| Aknemainen dermatiitti4 | 14,0 | 1,3 | 0 | 18,0 | 2,5 | 0 |
* Potilailla kaikista syistä ilmenneiden haittavaikutusten yleisyys
1 Mukana kynsivallintulehdus, kynsi‑infektio, kynsimarron infektio
2 Mukana stomatiitti, aftainen stomatiitti, limakalvotulehdus, suun haavaumat, suun limakalvon eroosiot, limakalvon eroosiot, limakalvon haavaumat
3 Mukana useita ihottumatyyppejä
4 Mukana akne, pustulaarinen akne, aknemainen dermatiitti
Maksan toimintakokeiden poikkeavuudet
40 mg:n GIOTRIF‑annoksia käyttäneillä potilailla havaittiin maksan toimintakokeissa poikkeavuuksia, mm. ALAT- ja ASAT‑arvojen suurenemista. Arvojen suureneminen oli useimmiten ohimenevää eikä johtanut hoidon lopettamiseen. Asteen 2 ALAT‑arvojen suurenemista esiintyi 1 %:lla ja asteen 3 arvojen suurenemista 0,8 %:lla GIOTRIF‑valmistetta saaneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet
Suurin afatinibiannos, jota arvioitiin rajallisessa potilasjoukossa vaiheen I kliinisissä tutkimuksissa, oli 160 mg kerran vuorokaudessa 3 päivän ajan ja 100 mg kerran vuorokaudessa 2 viikon ajan. Näillä annoksilla havaitut haittavaikutukset olivat lähinnä ihoreaktioita (ihottuma/akne) ja ruoansulatuskanavan oireita (etenkin ripulia). Kaksi tervettä nuorta sai yliannostuksen, jossa kumpikin otti suun kautta 360 mg afatinibia (ja lisäksi muita lääkkeitä). Tähän liittyviä haittatapahtumia olivat pahoinvointi, oksentelu, voimattomuus, huimaus, päänsärky, vatsakipu ja amylaasipitoisuuden suureneminen (< 1,5 x ULN). Molemmat toipuivat haittatapahtumistaan.
Hoito
Tämän lääkevalmisteen yliannostukselle ei ole spesifistä vastalääkettä. Epäillyissä yliannostustapauksissa GIOTRIF tauotetaan ja aloitetaan elintoimintoja tukeva hoito.
Imeytymätön afatinibi voidaan tarvittaessa eliminoida oksennuttamalla tai mahahuuhtelulla.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: syöpälääkkeet, proteiinikinaasin estäjät, ATC‑koodi: L01EB03.
Vaikutusmekanismi
Afatinibi on voimakas ja selektiivinen irreversiibeli ErbB‑perheen salpaaja. Afatinibi sitoutuu kovalenttisesti kaikkiin ErbB‑perheen molekyylien EGFR (ErbB1), HER2 (ErbB2), ErbB3 ja ErbB4 muodostamiin homo- ja heterodimeereihin ja estää irreversiibelisti niiden signaloinnin.
Farmakodynaamiset vaikutukset
Reseptorimutaatioiden ja/tai -amplifikaation ja/tai reseptorin ligandin yli‑ilmentymisen aiheuttama poikkeava ErbB‑signalointi vaikuttaa syöpätaudin fenotyyppiin. EGFR‑mutaatio tuottaa keuhkosyövän tietyn molekylaarisen alatyypin.
Ei‑kliinisissä tautimalleissa, joissa ErbB‑reitin säätely on häiriintynyt, afatinibin käyttö monoterapiana salpaa tehokkaasti ErbB‑reseptorin signalointia, mikä estää kasvaimen kasvua tai johtaa kasvaimen regressioon. Erityisen herkkiä afatinibihoidolle sekä kliinisesti että ei‑kliinisesti ovat ei‑pienisoluisen keuhkosyövän kasvaimet, joissa on yleisiä aktivoivia EGFR‑mutaatioita (Del 19, L858R) ja useita harvinaisempia EGFR‑mutaatioita eksonissa 18 (G719X) ja eksonissa 21 (L861Q). Ei‑pienisoluisissa keuhkosyöpäkasvaimissa, joissa on eksonin 20 insertiomutaatioita, on todettu rajallista ei‑kliinistä ja/tai kliinistä tehoa.
Sekundaarisen T790M-mutaation synty on merkittävä hankinnaisen afatinibiresistenssin mekanismi, ja T790M:n sisältävän alleelin geenimäärä korreloi resistenssin asteen kanssa in vitro. T790M-mutaatio löytyy noin 50 %:sta potilaiden kasvaimia, kun sairaus etenee afatinibihoidossa, jolloin seuraavan linjan hoitovaihtoehdoksi voidaan harkita T790M-kohdennettua EGFR:n tyrosiinikinaasin estäjää. Prekliinisissä tutkimuksissa on havaittu muita mahdollisia afatinibiresistenssin mekanismeja ja sen lisäksi kliinisesti on havaittu MET-geenin amplifikaatiota.
Kliininen teho ja turvallisuus
GIOTRIF-hoito EGFR-mutaatiopositiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla
LUX‑Lung 3
GIOTRIF-valmisteen tehoa ja turvallisuutta EGFR-mutaatiopositiivisen paikallisesti edenneen tai etäpesäkkeisen ei‑pienisoluisen keuhkosyövän (aste IIIB tai IV) ensilinjan hoitona arvioitiin satunnaistetussa, avoimessa maailmanlaajuisessa monikeskustutkimuksessa. Potilaat seulottiin 29:n eri EGFR-mutaation suhteen PCR-pohjaisella menetelmällä (TheraScreen®: EGFR29 Mutation Kit, Qiagen Manchester Ltd). Potilaat satunnaistettiin (suhteessa 2:1) saamaan 40 mg:n GIOTRIF-annoksia kerran vuorokaudessa tai enintään 6 hoitojaksoa pemetreksedi-/sisplatiinihoitoa. Satunnaistetuista potilaista 65 % oli naisia, mediaani-ikä oli 61 vuotta ja lähtötilanteen ECOG-suorituskykyluokka oli 0 (39 %) tai 1 (61 %). Potilaista 26 % oli valkoihoisia ja 72 % aasialaisia. Potilaista 89 %:lla esiintyi yleisiä EGFR-mutaatioita (Del 19 tai L858R).
Ensisijainen päätetapahtuma oli riippumattomaan arvioon perustuva etenemisvapaa elinaika (PFS); toissijaisia päätetapahtumia olivat kokonaiselinaika ja objektiivinen vaste. Analyysihetkellä 14.11.2013 esiintyi 176 potilaalla (76,5 %) afatinibiryhmässä ja 70 potilaalla (60,9 %) solunsalpaajaryhmässä jokin sellainen tapahtuma, joka vaikutti PFS-analyysiin, eli keskitetyn riippumattoman arvion mukainen sairauden eteneminen tai kuolema. Tehoa kuvaavat tulokset on esitetty kuvassa 1 sekä taulukoissa 6 ja 7.
LUX-Lung 6
GIOTRIF-valmisteen tehoa ja turvallisuutta EGFR-mutaatiopositiivista paikallisesti edennyttä tai etäpesäkkeistä keuhkojen adenokarsinoomaa (aste IIIB/IV) sairastavilla aasialaisilla potilailla arvioitiin satunnaistetussa, avoimessa monikeskustutkimuksessa. Kuten LUX‑Lung 3 -tutkimuksessa, ei-pienisoluista keuhkosyöpää sairastavien potilaiden, jotka eivät olleet saaneet aiempaa hoitoa, EGFR-mutaatiot seulottiin TheraScreen®-menetelmällä (EGFR29 Mutation Kit, Qiagen Manchester Ltd). Satunnaistetuista potilaista 65 % oli naisia, mediaani-ikä oli 58 vuotta ja kaikki potilaat olivat etniseltä alkuperältään aasialaisia. Tutkimuspopulaation potilaista 89 %:lla esiintyi yleisiä EGFR-mutaatioita.
Ensisijainen päätetapahtuma oli keskitettyyn riippumattomaan arvioon perustuva etenemisvapaa elinaika; toissijaisia päätetapahtumia olivat kokonaiselinaika ja objektiivinen vaste.
Etenemisvapaa elinaika parani EGFR-mutaatiopositiivisilla GIOTRIF-hoitoa saaneilla potilailla solunsalpaajahoitoa saaneisiin potilaisiin verrattuna merkitsevästi kummassakin tutkimuksessa. Tehoa kuvaavat tulokset on koottu yhteen kuvassa 1 (LUX‑Lung 3) ja taulukoissa 6 ja 7 (LUX‑Lung 3 ja 6). Taulukossa 7 on esitetty tulokset alaryhmittäin potilailla, joilla oli kaksi yleistä EGFR-mutaatiota, Del 19 ja L858R.
Kuva 1: Riippumattomaan arvioon perustuvat etenemisvapaan elinajan (PFS) Kaplan–Meier‑käyrät hoitoryhmittäin LUX‑Lung 3 ‑tutkimuksessa (koko populaatio)
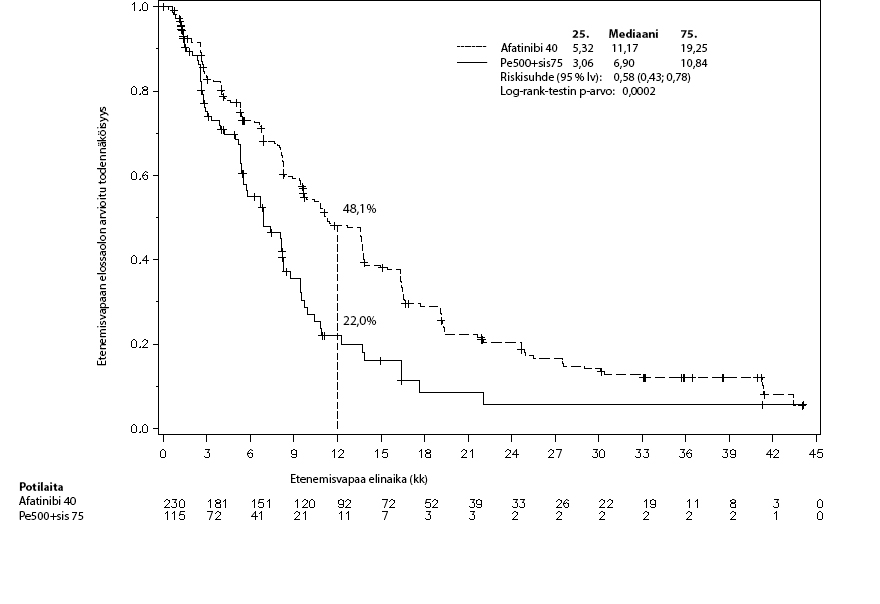
Taulukko 6: Tehotulokset GIOTRIF- ja pemetreksedi/sisplatiiniryhmissä (LUX‑Lung 3) sekä GIOTRIF- ja gemsitabiini/sisplatiiniryhmissä (LUX‑Lung 6) (riippumaton arvio)
LUX-Lung 3 | LUX-Lung 6 | |||
GIOTRIF (N = 230) | Pemetreksedi/ sisplatiini (N = 115) | GIOTRIF (N = 242) | Gemsitabiini/ sisplatiini (N = 122) | |
Etenemisvapaa elinaika kk (mediaani) | 11,2 | 6,9 | 11,0 | 5,6 |
Riskisuhde (HR) (95 %:n lv) | 0,58 (0,43–0,78) | 0,28 (0,20–0,39) | ||
p-arvo1 | 0,0002 | < 0,0001 | ||
1 vuoden PFS-osuus | 48,1 % | 22,0 % | 46,7 % | 2,1 % |
Objektiivinen vaste (CR + PR)2 | 56,5 % | 22,6 % | 67,8 % | 23,0 % |
Ristitulosuhde (OR) (95 %:n lv) | 4,80 (2,89–8,08) | 7,57 (4,52–12,68) | ||
p-arvo1 | < 0,0001 | < 0,0001 | ||
Kokonaiselinaika (OS) kk (mediaani) | 28,2 | 28,2 | 23,1 | 23,5 |
Riskisuhde (HR) (95 %:n lv) | 0,88 (0,66–1,17) | 0,93 (0,72–1,22) | ||
p-arvo1 | 0,3850 | 0,6137 | ||
1 Etenemisvapaata elinaikaa / kokonaiselinaikaa koskeva p-arvo perustuu ositettuun log rank -testiin; objektiivista vastetta koskeva p-arvo perustuu logistiseen regressioon
2 CR = täydellinen vaste, PR = osittainen vaste
Taulukko 7: Etenemisvapaan elinajan ja kokonaiselinajan tehotulokset GIOTRIF- ja pemetreksedi/sisplatiiniryhmissä (LUX‑Lung 3) sekä GIOTRIF- ja gemsitabiini/sisplatiiniryhmissä (LUX‑Lung 6) ennalta määritellyissä EGFR-mutaatioalaryhmissä Del 19 ja L858R (riippumaton arvio)
LUX-Lung 3 | LUX-Lung 6 | |||
Del 19 | GIOTRIF (N = 112) | Pemetreksedi/ sisplatiini (N = 57) | GIOTRIF (N = 124) | Gemsitabiini/ sisplatiini (N = 62) |
Etenemisvapaa elinaika kk (mediaani) | 13,8 | 5,6 | 13,1 | 5,6 |
Riskisuhde (HR) (95 %:n lv) | 0,26 (0,17–0,42) | 0,20 (0,13–0,33) | ||
p-arvo1 | < 0,0001 | < 0,0001 | ||
Kokonaiselinaika (OS) kk (mediaani) | 33,3 | 21,1 | 31,4 | 18,4 |
Riskisuhde (HR) (95 %:n lv) | 0,54 (0,36–0,79) | 0,64 (0,44–0,94) | ||
p-arvo1 | 0,0015 | 0,0229 | ||
L858R | GIOTRIF (N = 91) | Pemetreksedi/ sisplatiini (N = 47) | GIOTRIF (N = 92) | Gemsitabiini/ sisplatiini (N = 46) |
Etenemisvapaa elinaika kk (mediaani) | 10,8 | 8,1 | 9,6 | 5,6 |
Riskisuhde (HR) (95 %:n lv) | 0,75 (0,48–1,19) | 0,31 (0,19–0,52) | ||
p-arvo1 | 0,2191 | < 0,0001 | ||
Kokonaiselinaika (OS) kk (mediaani) | 27,6 | 40,3 | 19,6 | 24,3 |
Riskisuhde (HR) (95 %:n lv) | 1,30 (0,80–2,11) | 1,22 (0,81–1,83) | ||
p-arvo1 | 0,2919 | 0,3432 | ||
1 Etenemisvapaata elinaikaa / kokonaiselinaikaa koskeva p-arvo perustuu ositettuun log rank -testiin.
Ennalta määritellyssä alaryhmässä, jonka potilailla oli yleisiä mutaatioita (yhdistetty Del 19 ja L858R), etenemisvapaan elinajan mediaani oli LUX‑Lung 3 -tutkimuksessa GIOTRIF-hoidolla 13,6 kk ja solunsalpaajahoidolla 6,9 kk (HR 0,48; 95 %:n lv 0,35–0,66; p < 0,0001; N = 307) ja LUX‑Lung 6 -tutkimuksessa GIOTRIF-hoidolla 11,0 kk ja solunsalpaajahoidolla 5,6 kk (HR 0,24; 95 %:n lv 0,17–0,35; p < 0,0001; N = 324).
Etenemisvapaan elinajan paranemisen lisäksi tautiin liittyvät oireet lievittyivät ja taudin pahenemiseen kulunut aika piteni (ks. taulukko 8). GIOTRIF-ryhmä saavutti merkitsevästi paremmat keskipisteet suhteessa aikaan kokonaiselämänlaadun, yleisen terveydentilan, fyysisen toimintakyvyn, roolitoiminnan sekä kognitiivisten, sosiaalisten ja tunneperäisten toimintojen suhteen.
Taulukko 8: GIOTRIF- ja solunsalpaajahoitojen vaikutukset oireisiin LUX‑Lung 3 ja LUX‑Lung 6 ‑tutkimuksissa (EORTC QLQ‑C30 ja EORTC QLQ‑LC13)
LUX-Lung 3 | |||
Yskä | Hengenahdistus | Kipu | |
Oire lievittyi, %-osuus potilaistaa | 67 % ja 60 %; p = 0,2133 | 65 % ja 50 %; p = 0,0078 | 60 % ja 48 %; p = 0,0427 |
Viive mediaaniajassa oireiden pahenemiseen (kk) a,b | 27,0 ja 8,0 HR 0,60; p = 0,0062 | 10,4 ja 2,9 HR 0,68; p = 0,0129 | 4,2 ja 3,1 HR 0,83; p = 0,1882 |
LUX-Lung 6 | |||
Yskä | Hengenahdistus | Kipu | |
Oire lievittyi, %-osuus potilaistaa | 76 % ja 55 %; p = 0,0003 | 71 % ja 48 %; p < 0,0001 | 65 % ja 47 %; p = 0,0017 |
Viive mediaaniajassa oireiden pahenemiseen (kk) a,b | 31,1 ja 10,3 HR 0,46; p = 0,0001 | 7,7 ja 1,7 HR 0,53; p < 0,0001 | 6,9 ja 3,4 HR 0,70; p = 0,0220 |
a Esitetyt arvot koskevat GIOTRIF- ja solunsalpaajahoitoa (tässä järjestyksessä), p-arvo perustuu logistiseen regressioon.
b p-arvo koskee oireiden pahenemiseen kulunutta aikaa ja perustuu ositettuun log rank –testiin
LUX‑Lung 2
LUX‑Lung 2 oli yksiryhmäinen vaiheen II tutkimus, johon osallistuneet 129 potilasta eivät olleet saaneet aiemmin EGFR:n tyrosiinikinaasin estäjähoitoa ja joilla oli asteen IIIB tai IV keuhkojen adenokarsinooma, jossa oli EGFR‑mutaatio. Kyseessä oli joko ensilinjan hoito (N = 61) tai toisen linjan hoito (ts. yksi aiempi solunsalpaajahoito oli epäonnistunut; N = 68). 61 potilasta sai valmistetta ensilinjan hoitona. Riippumattoman arvion mukaan vahvistettu objektiivinen vaste oli tässä ryhmässä 65,6 % ja taudin hallinta 86,9 %. Etenemisvapaan elinajan mediaani oli riippumattoman arvion mukaan 12,0 kk. Teho oli hyvä myös aiempaa solunsalpaajahoitoa saaneessa potilasryhmässä (N = 68, objektiivinen vaste 57,4 %, etenemisvapaan elinajan mediaani riippumattoman arvion mukaan 8 kk). Päivitettyjen tietojen mukaan kokonaiselinajan mediaani oli ensilinjan hoitoryhmässä 31,7 kk ja toisen linjan hoitoryhmässä 23,6 kk.
LUX-Lung 7
LUX‑Lung 7 on satunnaistettu, maailmanlaajuinen, avoin vaiheen IIb tutkimus, joka tutkii GIOTRIF-valmisteen tehoa ja turvallisuutta ensilinjan hoitona paikallisesti edennyttä tai etäpesäkkeistä keuhkojen adenokarsinoomaa (aste IIIB tai IV) sairastavilla potilailla, joilla on EGFR-mutaatioita. Potilaat seulottiin aktivoivien EGFR-mutaatioiden suhteen (Del 19 ja/tai L858R) TheraScreen® EGFR RGQ PCR Kit ‑menetelmällä (Qiagen Manchester Ltd.). Potilaat (N = 319) satunnaistettiin (1:1) saamaan GIOTRIF-valmistetta 40 mg suun kautta kerran vuorokaudessa (N = 160) tai gefitinibiä 250 mg suun kautta kerran vuorokaudessa (N = 159). Satunnaistaminen ositettiin EGFR‑mutaatiostatuksen (Del 19, L858R) ja aivometastaasitilanteen mukaisesti (kyllä, ei).
Satunnaistetuista potilaista 62 % oli naisia, mediaani-ikä oli 63 vuotta, 16 %:lla potilaista oli aivometastaaseja, lähtötilanteen ECOG-suorituskykyluokka oli 0 (31 %) tai 1 (69 %), 57 % oli aasialaisia ja 43 % muita kuin aasialaisia. Potilailta otettu tuumorinäyte luokiteltiin EGFR-mutaation perusteella joko eksonin 19 deleetion tapauksessa Del 19-ryhmään (58 %) tai eksonin 21 substituution tapauksessa L858R-ryhmään (42 %).
Ensisijaisia päätetapahtumia olivat riippumattomalla tavalla arvioitu etenemisvapaa elinaika (PFS) ja kokonaiselinaika (OS). Toissijaisia päätetapahtumia olivat objektiivinen vaste (ORR) ja taudinhallinta (DCR). GIOTRIF paransi merkitsevästi PFS- ja ORR-arvoja EGFR-mutaatiopositiivisilla potilailla gefitinibiin verrattuna. Tehoa kuvaavat tulokset on koottu taulukkoon 9.
Taulukko 9: GIOTRIF-valmistetta ja gefitinibiä koskevat tehoa kuvaavat tulokset (LUX-Lung 7) elokuussa 2015 tehdyn ensimmäisen analyysin perusteella.
| GIOTRIF (N = 160) | Gefitinibi (N = 159) | Riskisuhde (HR)/ Ristitulosuhde (OR) (95 %:n lv) p-arvo2 |
PFS-arvon mediaani (kk), Koko tutkimuspopulaatio
18 kk:n PFS-arvo 24 kk:n PFS-arvo | 11,0
27 % 18 % | 10,9
15 % 8 % | HR 0,73 (0,57;0,95) 0,0165
|
OS-arvon mediaani (kk)1, Koko tutkimuspopulaatio
Elossa 18 kk:n kohdalla Elossa 24 kk:n kohdalla | 27,9
71 % 61 % | 24,5
67 % 51 % | HR 0,86 (0,66–1,12) 0,2580
|
Objektiivinen vaste
| 70 %
| 56 %
| OR 1,87 (1,12–2,99) 0,0083 |
1Kokonaiselinaikaa koskevat tulokset perustuvat ensimmäiseen OS-analyysiin, joka tehtiin huhtikuussa 2016, kun tapahtumia oli 109 (68,1 %) GIOTRIF-ryhmässä ja 117 (73,6 %) gefitinibiryhmässä.
2 Etenemisvapaata elinaikaa / kokonaiselinaikaa koskeva p‑arvo perustuu ositettuun log rank ‑testiin, objektiivista vastetta koskeva p‑arvo perustuu ositettuun logistiseen regressioon.
3CR=täydellinen vaste; PR=osittainen vaste
Etenemisvapaan elinajan riskisuhde niillä potilailla, joilla oli Del 19-mutaatioita ja L858-mutaatioita, oli afatinibilla 0,76 (95 %:n lv [0,55; 1,06], p=0,1071) ja gefitinibillä 0,71 (95 %:n lv [0,47; 1,06], p=0,0856).
Analyysi GIOTRIF-valmisteen tehosta potilailla, jotka eivät olleet saaneet aiemmin EGFR:n tyrosiinikinaasin estäjähoitoa ja joiden kasvaimissa oli harvinaisia EGFR-mutaatioita (LUX‑Lung 2, LUX‑Lung 3 ja LUX‑Lung 6)
Kolmessa GIOTRIF-valmisteella tehdyssä kliinisessä tutkimuksessa, joissa käytettiin prospektiivista kasvaimen genotyypitystä (vaiheen 3 tutkimukset LUX‑Lung 3 ja LUX‑Lung 6 sekä yksiryhmäinen vaiheen 2 tutkimus LUX‑Lung 2), tehtiin analyysi yhteensä 75 sellaisen potilaan tiedoista, jotka eivät olleet saaneet aiemmin tyrosiinikinaasin estäjähoitoa ja joiden pitkälle levinneeseen (asteen IIIb–IV) keuhkojen adenokarsinoomaan liittyi harvinaisia EGFR-mutaatioita. Tällaisiksi mutaatioiksi katsottiin kaikki muut paitsi Del 19- ja L858R‑mutaatiot. Potilaille annettiin GIOTRIF-hoitoa 40 mg:n annoksella (kaikissa kolmessa tutkimuksessa) tai 50 mg:n annoksella (LUX‑Lung 2 -tutkimuksessa) suun kautta kerran vuorokaudessa.
Potilailla, joiden kasvaimissa oli G719X-substituutiomutaatio (N = 18), vahvistettu objektiivinen vaste oli 72,2 % ja vasteen keston mediaani 13,2 kuukautta. Potilailla, joilla oli L861Q-substituutiomutaatio (N = 16), vahvistettu objektiivinen vaste oli 56,3 % ja vasteen keston mediaani 12,9 kuukautta. Potilailla, joilla oli S768I-substituutiomutaatio (N = 8), vahvistettu objektiivinen vaste oli 75,0 % ja vasteen keston mediaani 26,3 kuukautta.
Potilailla, joiden kasvaimissa oli eksonin 20 insertioita (N = 23), vahvistettu objektiivinen vaste oli 8,7 % ja vasteen keston mediaani 7,1 kuukautta. Potilailla, joiden kasvaimissa oli de novo T790M ‑mutaatioita (N = 14), vahvistettu objektiivinen vaste oli 14,3 % ja vasteen keston mediaani 8,3 kuukautta.
GIOTRIF-hoito histologialtaan levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastavilla potilailla
GIOTRIF-valmisteen tehoa ja turvallisuutta toisen linjan hoitona tutkittiin edennyttä, histologialtaan levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastavilla potilailla satunnaistetussa, avoimessa, maailmanlaajuisessa vaiheen 3 LUX‑Lung 8 ‑tutkimuksessa. Potilaat, jotka saivat ensilinjan hoitona vähintään neljä hoitosykliä platinapohjaista hoitoa, satunnaistettiin 1:1 saamaan päivittäin joko 40 mg GIOTRIF-valmistetta tai 150 mg erlotinibia, kunnes sairaus alkoi taas edetä. Satunnaistaminen ositettiin rodun mukaan (itäaasialaiset vs. ei‑itäaasialaiset). Ensisijainen päätetapahtuma oli etenemisvapaa elinaika, tärkein toissijainen päätetapahtuma oli kokonaiselinaika. Muita toissijaisia päätetapahtumia olivat objektiivinen vaste, taudinhallinta, muutos kasvaimen koossa sekä terveyteen liittyvä elämänlaatu.
Satunnaistetuista 795 potilaasta suurin osa oli miehiä (84 %), valkoisia (73 %) ja edelleen tupakoivia tai aiemmin tupakoineita (95 %), ja joiden lähtötilanteen ECOG-suorituskykyluokka oli joko 1 (67 %) tai 0 (33 %).
Toisen linjan GIOTRIF-hoito paransi merkitsevästi etenemisvapaata elinaikaa ja kokonaiselinaikaa histologialtaan levyepiteeliperäistä ei-pienisoluista keuhkosyöpää sairastavilla potilailla verrattuna erlotinibiin. Tehoa kuvaavat tulokset kokonaiselinajan ensisijaisesta analyysistä kaikilla satunnaistetuilla potilailla on esitetty kuvassa 2 ja taulukossa 10.
Taulukko 10: GIOTRIF-valmisteen tehoa kuvaavat tulokset erlotinibiin verrattuna LUX‑Lung 8 -tutkimuksessa kokonaiselinajan ensisijaiseen analyysiin perustuen kaikilla satunnaistetuilla potilailla
GIOTRIF (N=398) | Erlotinibi (n=397) | Riskisuhde (HR) / Ristitulosuhde (OR) (95 %:n lv) | p‑arvo2 | |
Etenemisvapaa elinaika kk (mediaani)
| 2,63 | 1,94 |
HR 0,81 (0,69–0,96) | 0,0103 |
Kokonaiselinaika kk (mediaani)
Elossa 12 kk:n kohdalla Elossa 18 kk:n kohdalla |
7,92
36,4 % 22,0 % |
6,77
28,2 % 14,4 % |
HR 0,81 (0,69–0,95)
| 0,0077
|
Objektiivinen vaste (CR+PR)1 | 5,5 %
| 2,8 %
| OR 2,06 (0,98–4,32) | 0,0551
|
Vasteen kesto kk (mediaani) | 7,29
| 3,71
|
1CR = täydellinen vaste, PR = osittainen vaste
2 Etenemisvapaata elinaikaa / kokonaiselinaikaa koskeva p‑arvo perustuu ositettuun log rank ‑testiin; objektiivista vastetta koskeva p‑arvo perustuu logistiseen regressioon.
Kokonaiselinajan riskisuhde oli alle 65-vuotiailla potilailla 0,68 (95 %:n lv 0,55–0,85) ja 65‑vuotiailla ja tätä vanhemmilla potilailla 0,95 (95 %:n lv 0,76–1,19).
Kuva 2: Kokonaiselinajan Kaplan-Meier -käyrä hoitoryhmittäin LUX‑Lung 8 -tutkimuksessa

Etenemisvapaan elinajan paranemisen lisäksi tautiin liittyvät oireet lievittyivät ja sairauden pahenemiseen kulunut aika piteni (ks. taulukko 11).
Taulukko 11: GIOTRIF-valmisteen ja erlotibinin vaikutukset oireisiin LUX‑Lung 8 -tutkimuksessa (EORTC QLQ‑C30 ja EORTC QLQ‑LC13)
Yskä | Dyspnea | Kipu | |
Oire lievittyi, %-osuus potilaistaa,c | 43 % ja 35 % p=0,0294 | 51 % ja 44 % p=0,0605 | 40 % ja 39 %; p=0,7752 |
Viive ajassa oireiden pahenemiseen (kk)b,c | 4,5 ja 3,7 HR 0,89; p=0,2562 | 2,6 ja 1,9 HR 0,79; p=0,0078 | 2,5 ja 2,4 HR 0,99; p=0,8690 |
a Esitettävät arvot koskevat GIOTRIF-valmistetta ja erlotinibia (tässä järjestyksessä), p‑arvo perustuu logistiseen regressioon.
b p-arvo koskee oireiden pahenemiseen kulunutta aikaa ja perustuu ositettuun log rank -testiin.
c p-arvoja ei sovitettu kertoimien mukaan.
Tehoa ei ole osoitettu EGFR-negatiivisissa kasvaimissa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset tämän lääkevalmisteen käytöstä ei‑pienisoluisen keuhkosyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa). Pediatrista kehitystyötä on kuitenkin tehty pediatrisilla potilailla, joilla oli muita sairauksia.
Vaiheen I/II avoimessa monikeskustutkimuksessa, jossa annosta suurennettiin, arvioitiin GIOTRIF-valmisteen turvallisuutta ja tehoa 2 – < 18-vuotiailla pediatrisilla potilailla. Potilailla oli uusiutuneita tai hoitoon huonosti vastaavia neuroektodermaalisia kasvaimia, rabdomyosarkooma ja/tai muita kiinteitä kasvaimia, joihin tiedetään liittyvän ErbB-reitin säätelyhäiriö kasvaimen histologiasta riippumatta. Annoksen määritysvaiheessa hoitoa annettiin yhteensä 17 potilaalle. Tutkimuksen laajennusvaiheessa, jossa selvitettiin suurinta siedettyä annosta (MTD), GIOTRIF-hoitoa annettiin annoksena 18 mg/m²/vrk 39 potilaalle, jotka valittiin ErbB-reitin säätelyhäiriön biomarkkerien perusteella. Tässä laajennusvaiheessa 38 potilaalla ei todettu mitään objektiivista vastetta. Näistä 6 potilaalla oli hoitoon huonosti vastaava korkea-asteinen gliooma (HGG), 4 potilaalla oli diffuusi aivorungon sisäinen gliooma (DIPG), 8 potilaalla oli ependymooma ja 20 potilaalla oli histologialtaan muun tyyppisiä kasvaimia. Yhdellä potilaalla, jolla oli neuraalis-gliaalinen aivokasvain ja CLIP2-EGFR-geenin fuusio, vahvistettiin osittainen vaste (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa). GIOTRIF-valmisteen haittavaikutusprofiili pediatrisilla potilailla oli samankaltainen kuin aikuisilla todettu haittavaikutusprofiili.
Farmakokinetiikka
Imeytyminen
Afatinibin Cmax saavutettiin noin 2–5 tunnin kuluttua GIOTRIF-annoksen ottamisesta suun kautta. Käytettäessä 20–50 mg:n GIOTRIF‑annoksia Cmax- ja AUC0–∞-arvot suurenivat hiukan enemmän kuin suhteessa annokseen. Systeeminen afatinibialtistus pienenee 50 % (Cmax) ja 39 % (AUC0–∞), jos lääke otetaan runsasrasvaisen aterian kanssa eikä tyhjään mahaan. Eri kasvaintyyppejä koskeneiden kliinisten tutkimusten populaatiofarmakokineettisten tietojen perusteella AUCτ,ss pieneni keskimäärin 26 %, jos potilas söi ruokaa GIOTRIF-valmisteen ottamista edeltävien 3 tunnin kuluessa tai 1 tunnin kuluessa annoksen ottamisen jälkeen. Potilaan on siis oltava syömättä vähintään 3 tuntia ennen GIOTRIF-annosta ja vähintään 1 tunti GIOTRIF-annoksen jälkeen (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Jakautuminen
In vitro afatinibi sitoutuu noin 95‑prosenttisesti ihmisplasman proteiineihin. Afatinibi sitoutuu proteiineihin sekä ei‑kovalenttisesti (tavanomainen proteiineihin sitoutuminen) että kovalenttisesti.
Biotransformaatio
Entsyymien katalysoimilla metaboliareaktioilla on hyvin vähäinen merkitys afatinibin kohdalla in vivo. Kovalentit proteiiniadduktit olivat afatinibin tärkeimpiä verenkierrossa esiintyviä metaboliitteja.
Eliminaatio
Ihmisillä afatinibi erittyy lähinnä ulosteeseen. Kun 15 mg afatinibia annettiin oraaliliuoksena, 85,4 % annoksesta erittyi ulosteeseen ja 4,3 % virtsaan. Erittyneestä annoksesta 88 % oli kanta aineen eli afatinibin muodossa. Afatinibin eliminaation todellinen puoliintumisaika on noin 37 tuntia. Siten vakaan tilan afatinibipitoisuudet plasmassa saavutettiin 8 päivässä, kun afatinibia otettiin toistuvina annoksina. Kumuloituminen oli 2,77‑kertaista (AUC0-∞) ja 2,11‑kertaista (Cmax). Afatinibihoitoa pidempään kuin kuusi kuukautta saaneilla potilailla terminaalisen puoliintumisajan arvioitiin olevan 344 tuntia.
Erityisryhmät
Munuaisten vajaatoiminta
Alle 5 % afatinibin kerta-annoksesta erittyy munuaisten kautta. Afatinibialtistusta verrattiin 40 mg:n GIOTRIF-kerta-annoksen jälkeen munuaisten vajaatoimintaa sairastavien henkilöiden ja terveiden vapaaehtoisten välillä. Keskivaikeaa munuaisten vajaatoimintaa sairastavilla henkilöillä (n = 8; Modification of Diet in Renal Disease [MDRD] kaavan mukaisesti laskettu eGFR 30‑59 ml/min/1,73 m2) altistus oli 101 % (Cmax) ja 122 % (AUC0–tz) verrattuna terveisiin verrokkeihin. Vaikeaa munuaisten vajaatoimintaa sairastavilla tutkittavilla (n = 8; ‑MDRD-kaavan mukaisesti laskettu eGFR 15‒29 ml/min/1,73 m2) altistus oli 122 % (Cmax) ja 150 % (AUC0–tz) verrattuna terveisiin verrokkeihin. Tämän tutkimuksen ja erilaisia kasvaintyyppejä tarkastelleiden kliinisten tutkimusten tietojen pohjalta tehdyn populaatiofarmakokineettisen analyysin perusteella todetaan, että aloitusannosta ei ole tarpeen muuttaa lievää (eGFR 60‒89 ml/min/1,73 m²), keskivaikeaa (eGFR 30‒59 ml/min/1,73 m²) tai vaikeaa (eGFR 15‒29 ml/min/1,73 m²) munuaisten vajaatoimintaa sairastavilla potilailla, mutta vaikeaa munuaisten vajaatoimintaa sairastavia potilaita on tarkkailtava (ks. ”Populaatiofarmakokinetiikan analyysi erityisryhmissä” jäljempänä ja kohta Annostus ja antotapa). GIOTRIF-valmistetta ei ole tutkittu potilailla, joilla eGFR on < 15 ml/min/1,73 m2 tai jotka saavat dialyysihoitoa.
Maksan vajaatoiminta
Afatinibi eliminoituu lähinnä erittymällä sappeen/ulosteeseen. Altistus 50 mg:n GIOTRIF‑kerta‑annoksen jälkeen oli samaa luokkaa sekä terveillä vapaaehtoisilla että potilailla, joilla oli lievä (Child–Pugh‑luokka A) tai keskivaikea (Child–Pugh‑luokka B) maksan vajaatoiminta. Tämä vastaa eri kasvaintyyppejä koskeneiden kliinisten tutkimusten populaatiofarmakokineettisiä tietoja (ks. ”Populaatiofarmakokinetiikan analyysi erityisryhmissä” jäljempänä). Aloitusannosta ei nähtävästi tarvitse muuttaa potilailla, joilla on lievä tai keskivaikea maksan vajaatoiminta (ks. kohta Annostus ja antotapa). Afatinibin farmakokinetiikkaa ei ole arvioitu vaikeaa (Child–Pugh‑luokka C) maksan vajaatoimintaa sairastavilla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Populaatiofarmakokinetiikan analyysi erityisryhmissä
Populaatiofarmakokinetiikan analyysiin otettiin 927 syöpäpotilasta, jotka saivat GIOTRIF‑monoterapiaa. Heistä 764 sairasti ei‑pienisoluista keuhkosyöpää. Aloitusannoksen muuttamista ei pidetty tarpeellisena seuraavien tutkittujen kovariaattien kohdalla.
Ikä
Iän (vaihteluväli 28–87 vuotta) ei havaittu vaikuttavan merkitsevästi afatinibin farmakokinetiikkaan.
Paino
Plasman altistus (AUCτ,ss) oli 42 kg painavilla potilailla (2,5. persentiili) 26 % suurempi ja 95 kg painavilla (97,5. persentiili) 22 % pienempi kuin 62 kg painavilla (koko potilaspopulaation mediaanipaino).
Sukupuoli
Plasman altistus (AUCτ,ss, korjattu painon suhteen) oli naispotilailla 15 % suurempi kuin miehillä.
Rotu
Populaatiofarmakokineettisen analyysin mukaan rotu ei vaikuttanut afatinibin farmakokinetiikkaan. Analyysissä oli mukana aasialaisia, valkoihoisia ja mustaihoisia potilaita. Mustaihoisista potilaista oli rajallisesti tietoa.
Munuaisten vajaatoiminta
Afatinibialtistus suureni kohtalaisesti (Cockcroft–Gault‑kaavalla lasketun) kreatiniinipuhdistuman pienentyessä. Kreatiniinipuhdistuman ollessa 60 ml/min afatinibialtistus (AUCτ,ss) oli 13 % suurempi ja kreatiniinipuhdistuman ollessa 30 ml/min taas 42 % suurempi kuin potilailla, joiden kreatiniinipuhdistuma oli 79 ml/min (koko analyysipopulaation potilaiden kreatiniinipuhdistuman mediaani). Kun kreatiniinipuhdistuma oli 90 ml/min, altistus pieneni 6 %, ja kun se oli 120 ml/min, altistus pieneni 20 %.
Maksan vajaatoiminta
Maksan toimintakokeiden poikkeavien testitulosten perusteella todettuun lievään tai keskivaikeaan maksan vajaatoimintaan ei liittynyt merkitseviä muutoksia afatinibialtistuksessa. Keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastavista potilaista oli rajallisesti tietoa.
Potilaiden muut piirteet/tekijät
Muita potilaiden piirteitä/tekijöitä, joilla oli merkitsevä vaikutus afatinibialtistukseen, olivat ECOG‑suorituskykypisteet, laktaattidehydrogenaasipitoisuus, alkalisen fosfataasin pitoisuus ja kokonaisproteiinimäärä. Näiden kovariaattien yksittäisiä efektikokoja ei pidetty kliinisesti merkittävinä. Aiempi tupakointi, alkoholin käyttö (rajalliset tiedot) ja maksametastaasit eivät vaikuttaneet merkitsevästi afatinibin farmakokinetiikkaan.
Pediatriset potilaat
Kun 2 – < 18-vuotiaille pediatrisille potilaille annettiin afatinibia annoksena 18 mg/m2, altistus vakaassa tilassa (AUC ja Cmax) oli samankaltainen kuin aikuisilla, joille annettiin afatinibia annoksena 40–50 mg (katso myös kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Muuta tietoa lääkeaineyhteisvaikutuksista
Yhteisvaikutukset lääkeaineiden soluunottojärjestelmien kanssa
In vitro ‑tiedot viittaavat siihen, että afatinibilla ei todennäköisesti ole OATP1B1-, OATP1B3-, OATP2B1-, OAT1-, OAT3-, OCT1-, OCT2- eikä OCT3‑kuljettajaproteiinien toiminnan estosta johtuvia lääkeaineyhteisvaikutuksia.
Yhteisvaikutukset sytokromi P450 -entsyymien (CYP) kanssa
Ihmisellä todettiin, että entsyymien katalysoimilla metaboliareaktioilla on vain hyvin vähäinen merkitys afatinibin metaboliassa. Noin 2 % afatinibiannoksesta metaboloitui FMO3‑välitteisesti, ja CYP3A4‑välitteinen N‑demetylaatio oli niin vähäistä, että se alitti kvantifikaatiorajan. Afatinibi ei estä eikä indusoi CYP‑entsyymitoimintaa. Tällä lääkevalmisteella ei siis todennäköisesti ole yhteisvaikutuksia muiden CYP‑välitteisesti metaboloituvien tai CYP‑toimintaan vaikuttavien lääkkeiden kanssa.
UDP‑glukuronosyylitransferaasi 1A1:n (UGT1A1) eston vaikutus afatinibiin
In vitro ‑tiedot viittaavat siihen, että UGT1A1:n esto ei todennäköisesti aiheuta afatinibiin vaikuttavia lääkeaineyhteisvaikutuksia.
Prekliiniset tiedot turvallisuudesta
Kun hiirille ja rotille annettiin kerta‑annoksia suun kautta, afatinibin akuutti toksisuus oli vähäistä. Peroraalisissa toistuvaisannostutkimuksissa rotalla (enintään 26 viikkoa) ja minisialla (enintään 52 viikkoa) tärkeimmät vaikutukset kohdistuivat ihoon (ihomuutokset, epiteelin atrofia ja follikuliitti rotalla), ruoansulatuskanavaan (ripuli, mahan eroosiot, epiteelin atrofia rotalla ja minisialla) ja munuaisiin (papillanekroosi rotalla). Näitä muutoksia esiintyi löydöksestä riippuen kliinisesti relevantilla altistustasolla tai sitä pienemmillä tai suuremmilla altistuksilla. Molemmilla lajeilla havaittiin myös eri elinten epiteelin farmakodynaamisesti välittyvää atrofiaa.
Lisääntymistoksisuus
Vaikutusmekanismin vuoksi kaikki EGFR‑toimintaan vaikuttavat täsmälääkkeet, myös GIOTRIF, voivat aiheuttaa haittaa sikiölle. Afatinibilla tehdyissä alkion- ja sikiönkehitystutkimuksissa ei havaittu viitteitä teratogeenisuudesta. Systeeminen kokonaisaltistus (AUC) oli rotalla hieman suurempi (2,2‑kertainen) ja kanilla pienempi (0,3‑kertainen) kuin potilailla.
Kun rotalle annettiin suun kautta radioaktiivisesti leimattua afatinibia imetyspäivänä 11, se erittyi emojen maitoon.
Uros- ja naarasrotilla tehdyssä hedelmällisyystutkimuksessa, jossa eläimet saivat enintään suurimpia siedettyjä annoksia, ei havaittu merkitsevää vaikutusta hedelmällisyyteen. Systeeminen kokonaisaltistus (AUC0‑24) oli suunnilleen potilaalla havaittua vastaava tai sitä pienempi (urosrotilla 1,3‑kertainen ja naarasrotilla 0,51‑kertainen).
Rottatutkimuksessa, jossa eläimet saivat enintään suurimpia siedettyjä annoksia, ei havaittu merkitsevää vaikutusta pre-/postnataaliseen kehitykseen. Suurin naarasrottien systeeminen kokonaisaltistus (AUC0‑24) oli potilaalla havaittua altistusta pienempi (0,23‑kertainen).
Valotoksisuus
In vitro tehdyssä 3T3‑kokeessa todettiin, että afatinibi saattaa olla valotoksinen.
Karsinogeenisuus
GIOTRIF‑valmisteella ei ole tehty karsinogeenisuustutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Laktoosimonohydraatti
Mikrokiteinen selluloosa (E460)
Vedetön kolloidinen piidioksidi (E551)
Krospovidoni (tyyppi A)
Magnesiumstearaatti (E470b)
Kalvopäällyste
GIOTRIF 20 mg kalvopäällysteiset tabletit
Hypromelloosi (E464)
Makrogoli 400
Titaanidioksidi (E171)
Talkki (E553b)
Polysorbaatti 80 (E433)
GIOTRIF 30 mg, 40 mg ja 50 mg kalvopäällysteiset tabletit
Hypromelloosi (E464)
Makrogoli 400
Titaanidioksidi (E171)
Talkki (E553b)
Polysorbaatti 80 (E433)
Indigokarmiini alumiinilakka (E132)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Läpipainopakkaus ja purkki
3 vuotta
Purkin avaamisen jälkeen lääkevalmiste on käytettävä 2 kuukauden kuluessa.
Säilytys
Läpipainopakkaus
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle. Herkkä valolle.
Purkki
Pidä purkki tiiviisti suljettuna. Herkkä kosteudelle. Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
GIOTRIF tabletti, kalvopäällysteinen
20 mg (L:kyllä) 28 x 1 fol (1934,06 €)
30 mg (L:kyllä) 28 x 1 fol (1934,06 €)
40 mg (L:kyllä) 28 x 1 fol (1934,06 €)
50 mg (L:kyllä) 28 x 1 fol (1934,06 €)
PF-selosteen tieto
Yksittäispakattu PVC/PVDC‑läpipainopakkaus. Kukin läpipainopakkaus on pakattu laminoituun alumiiniseen pussiin, jossa on myös kuivatusainepussi, ja sisältää 7 x 1 kalvopäällysteistä tablettia.
Pakkauskoot: 7 x 1, 14 x 1 ja 28 x 1 kalvopäällysteistä tablettia.
Polypropyleenistä (PP) valmistettu purkki, jossa kierrettävä HDPE/PP-suljin ja kuiva-aine korkissa. Pakkauskoko 30 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
GIOTRIF 20 mg kalvopäällysteiset tabletit
Valkea tai kellertävä, pyöreä, kaksoiskupera, viistoreunainen kalvopäällysteinen tabletti, jonka toisella puolella kaiverrettu koodi ”T20” ja toisella puolella kaiverrettu Boehringer Ingelheimin logo.
GIOTRIF 30 mg kalvopäällysteiset tabletit
Tummansininen, pyöreä, kaksoiskupera ja viistoreunainen kalvopäällysteinen tabletti, jonka toisella puolella kaiverrettu koodi ”T30” ja toisella puolella kaiverrettu Boehringer Ingelheimin logo.
GIOTRIF 40 mg kalvopäällysteiset tabletit
Vaaleansininen, pyöreä, kaksoiskupera ja viistoreunainen kalvopäällysteinen tabletti, jonka toisella puolella kaiverrettu koodi ”T40” ja toisella puolella kaiverrettu Boehringer Ingelheimin logo.
GIOTRIF 50 mg kalvopäällysteiset tabletit
Tummansininen, soikea, kaksoiskupera kalvopäällysteinen tabletti, jonka toisella puolella kaiverrettu koodi ”T50” ja toisella puolella kaiverrettu Boehringer Ingelheimin logo.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
GIOTRIF tabletti, kalvopäällysteinen
20 mg 28 x 1 fol
30 mg 28 x 1 fol
40 mg 28 x 1 fol
50 mg 28 x 1 fol
- Ylempi erityiskorvaus (100 %). Afatinibi ja gefitinibi: Paikallisesti edenneen tai etäpesäkkeitä lähettäneen ei-pienisoluisen keuhkosyövän hoito erityisin edellytyksin (155).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Afatinibi ja gefitinibi: Paikallisesti edenneen tai etäpesäkkeitä lähettäneen ei-pienisoluisen keuhkosyövän hoito erityisin edellytyksin (341).
ATC-koodi
L01EB03
Valmisteyhteenvedon muuttamispäivämäärä
08.05.2026
Yhteystiedot
 BOEHRINGER INGELHEIM FINLAND KY
BOEHRINGER INGELHEIM FINLAND KY Tammasaarenkatu 5
00180 Helsinki
010 310 2800
www.boehringer-ingelheim.fi
medinfo.finland@boehringer-ingelheim.com