
TECFIDERA enterokapseli, kova 120 mg, 240 mg
Vaikuttavat aineet ja niiden määrät
Tecfidera 120 mg kovat enterokapselit
Yksi kova enterokapseli sisältää 120 mg dimetyylifumaraattia.
Tecfidera 240 mg kovat enterokapselit
Yksi kova enterokapseli sisältää 240 mg dimetyylifumaraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Enterokapseli, kova
Kliiniset tiedot
Käyttöaiheet
Tecfidera on tarkoitettu aikuispotilaiden ja vähintään 13‑vuotiaiden pediatristen potilaiden aaltomaisen MS-taudin (relapsoiva-remittoiva multippeliskleroosi [RRMS]) hoitoon.
Ehto
Hoito tulee aloittaa MS-taudin hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito tulee aloittaa MS-taudin hoitoon perehtyneen lääkärin valvonnassa.
Annostus
Aloitusannos on 120 mg kaksi kertaa vuorokaudessa. Annos suurennetaan seitsemän päivän hoidon jälkeen suositeltuun annokseen 240 mg kaksi kertaa vuorokaudessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos potilas unohtaa ottaa annoksen, hänen ei pidä ottaa kaksinkertaista annosta. Potilas saa ottaa unohtuneen annoksen vain, jos annosten väliin jää neljä tuntia aikaa. Muussa tapauksessa potilaan tulee odottaa ja ottaa seuraava annos aikataulun mukaisesti.
Annoksen pienentäminen väliaikaisesti 120 mg:aan kaksi kertaa vuorokaudessa voi vähentää haittavaikutuksina esiintyvien punastumisen ja maha-suolikanavan oireiden esiintyvyyttä. Suositeltua ylläpitoannosta 240 mg kaksi kertaa vuorokaudessa tulisi jatkaa kuukauden kuluessa.
Tecfidera otetaan ruoan kanssa (ks. kohta Farmakokinetiikka). Tecfidera-valmisteen ottaminen ruoan kanssa saattaa parantaa siedettävyyttä, jos potilaalla esiintyy punastumista tai maha-suolikanavan haittavaikutuksia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Haittavaikutukset).
Erityispotilasryhmät
Iäkkäät potilaat
Tecfidera-valmistetta sai kliinisissä tutkimuksissa vain pieni joukko 55‑vuotiaita tai sitä vanhempia potilaita eikä niissä ollut mukana riittävästi 65‑vuotiaita tai sitä vanhempia potilaita, jotta olisi voitu selvittää, reagoivatko he valmisteeseen eri tavoin kuin nuoremmat potilaat (ks. kohta Farmakokinetiikka). Vaikuttavan aineen vaikutustavan perusteella annoksen muuttamiseen iäkkäillä ei ole teoreettisia perusteita.
Munuaisten ja maksan vajaatoiminta
Tecfidera-valmistetta ei ole tutkittu munuaisten tai maksan vajaatoimintaa sairastavien potilaiden hoidossa. Kliinisten farmakologisten tutkimusten perusteella annosta ei ole tarpeen muuttaa (ks. kohta Farmakokinetiikka). Vaikeaa munuaisten tai maksan vajaatoimintaa sairastavia potilaita hoidettaessa on syytä noudattaa varovaisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Annostus vähintään 13‑vuotiaille pediatrisille potilaille on sama kuin aikuisille.
Tietoja 10–12‑vuotiaista lapsista on vähän. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset ja Farmakodynamiikka, ei voida antaa suosituksia annostuksesta.
Tecfidera-valmisteen turvallisuutta ja tehoa alle 10 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Suun kautta.
Kapselit tulee niellä kokonaisina. Kapselia tai sen sisältöä ei saa murskata, jakaa, liuottaa, imeskellä eikä pureskella, sillä mikrotablettien enteropäällyste estää maha-suolikanavan ärsytystä.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Epäilty tai vahvistettu progressiivinen multifokaalinen leukoenkefalopatia (PML)
Varoitukset ja käyttöön liittyvät varotoimet
Verikokeet/laboratoriotutkimukset
Munuaisten toiminta
Dimetyylifumaraattihoitoa kliinisissä tutkimuksissa saaneiden potilaiden munuaiskokeiden tuloksissa on havaittu muutoksia (ks. kohta Haittavaikutukset). Näiden muutosten kliinistä merkitystä ei tiedetä. Munuaisten (esim. kreatiniini, veren ureatyppi ja virtsatutkimus) toiminnan tutkimista suositellaan ennen hoidon aloittamista, 3 ja 6 kuukauden hoidon jälkeen, ja sen jälkeen 6‑12 kuukauden välein sekä kliinisen tarpeen mukaan.
Maksan toiminta
Dimetyylifumaraattihoidosta saattaa aiheutua maksavaurio, mukaan lukien maksaentsyymin pitoisuuden nousu (≥ 3 kertaa normaalin yläraja (ULN)) ja kokonaisbilirubiinitasojen nousu (≥ 2 × ULN). Maksavaurio saattaa ilmetä päivien, viikkojen tai pidemmän ajan kuluttua Tecfidera‑hoidon aloituksesta. Haittavaikutusten on havaittu hävinneen, kun hoito on lopetettu. Seerumin aminotransferaasien (esim. alaniiniaminotransferaasi (ALAT), aspartaattiaminotransferaasi (ASAT)) ja kokonaisbilirubiinitasojen määrittämistä suositellaan ennen hoidon aloittamista ja hoidon aikana kliinisen tarpeen mukaan.
Lymfosyytit
Tecfidera-hoitoa saaneille potilaille voi kehittyä lymfopenia (ks. kohta Haittavaikutukset). Ennen Tecfidera-hoidon aloittamista on määritettävä senhetkinen täydellinen verenkuva, lymfosyytit mukaan lukien.
Jos lymfosyyttimäärä on normaalialueen rajan alapuolella, mahdolliset syyt on arvioitava huolellisesti ennen hoidon aloittamista. Dimetyylifumaraattia ei ole tutkittu potilailla, joiden lymfosyyttimäärä on ennestään pieni, joten tämän potilasryhmän hoidossa on noudatettava varovaisuutta. Hoitoa ei pidä aloittaa potilaille, jolla on vaikea lymfopenia (lymfosyyttimäärä < 0,5 × 109/l).
Hoidon aloittamisen jälkeen täydellinen verenkuva, lymfosyytit mukaan lukien, on määritettävä 3 kuukauden välein.
Lymfopeenisten potilaiden tehostettua valvontaa suositellaan progressiivisen multifokaalisen leukoenkefalopatian (PML) lisääntyneen riskin vuoksi seuraavalla tavalla:
- Jos potilaalla on pitkittynyt vaikea lymfopenia (lymfosyyttimäärä on < 0,5 × 109/l) yli 6 kuukauden ajan, hoito pitää keskeyttää.
- Jos potilaan absoluuttisen lymfosyyttimäärän kohtalainen väheneminen ≥ 0,5 × 109/l – < 0,8 × 109/l pitkittyy yli 6 kuukauden ajan, Tecfidera-hoidon hyöty–riskitasapaino on arvioitava uudelleen.
- Jos potilaan lymfosyyttimäärä on paikallisen laboratorion viitealueen mukaisten normaaliarvojen alarajan (LLN) alle, absoluuttisen lymfosyyttimäärän säännöllistä tarkkailua suositellaan. Lisätekijät, jotka saattavat entisestään lisätä yksilöllistä PML:n riskiä, on otettava huomioon (ks. PML-alakohta alla).
Lymfosyyttimääriä on seurattava kunnes ne ovat palautuneet (ks. kohta Farmakodynamiikka). Lymfosyyttimäärien palauduttua ja muiden hoitovaihtoehtojen puuttuessa on hoidon keskeyttämisen jälkeen käytettävä kliinistä harkintaa päätettäessä siitä, aloitetaanko Tecfidera-hoito uudelleen vai ei.
Magneettikuvaus (MRI)
Ennen Tecfidera-hoidon aloittamista lähtötilanteen magneettikuva (yleensä 3 kuukauden sisällä otettu) pitää olla käytettävissä vertailukuvana. Magneettikuvauksen uusimisen tarvetta on arvioitava kansallisten ja paikallisten hoitosuositusten mukaan. Magneettikuvausta voidaan pitää osana tehostettua valvontaa hoidettaessa potilaita, joilla on suurentunut PML-riski. Jos PML:ää epäillään, magneettikuvaus on tehtävä välittömästi osana diagnostiikkaa.
Progressiivinen multifokaalinen leukoenkefalopatia (PML)
Tecfidera-valmisteella hoidetuilla potilailla on raportoitu PML:ää (ks. kohta Haittavaikutukset). PML on John-Cunningham-viruksen (JCV) aiheuttama opportunistinen infektio, joka voi johtaa kuolemaan tai vaikeaan toimintakyvyn heikkenemiseen.
PML-tapauksia on ilmennyt dimetyylifumaraatin ja muiden fumaraatteja sisältävien lääkevalmisteiden käytön yhteydessä, potilaan lymphopenian yhteydessä (lymfosyyttimäärä alle normaaliarvojen alarajan). Pitkittynyt kohtalainen tai vaikea lymfopenia näyttää lisäävän PML:n riskiä Tecfidera-hoidon yhteydessä, mutta riskiä ei kuitenkaan voi poissulkea lievää lymfopeniaa sairastavilla potilailla.
Lisätekijöitä, jotka saattavat vaikuttaa lisääntyneeseen PML:n riskiin lymfopenian yhteydessä, ovat seuraavat:
- Tecfidera-hoidon kesto. PML-tapaukset ovat esiintyneet noin 1–5 vuotta kestäneen hoidon jälkeen, vaikka tarkka suhde hoidon kestoon on tuntematon.
- voimakas immuunipuolustuksen kannalta tärkeiden CD4+- ja erityisesti CD8+-T-solumäärien väheneminen (ks. kohta Haittavaikutukset) ja
- aiempi immunosuppressiivinen tai immunomoduloiva hoito (ks. alla).
Lääkäreiden pitää tutkia potilaansa selvittääkseen, ovatko oireet merkki neurologisesta toimintahäiriöstä, ja jos ovat, ovatko ne MS-taudille tyypillisiä vai viittaavatko ne mahdollisesti PML:ään.
PML:ään viittaavan ensilöydöksen tai ‑oireen ilmaantuessa on lopetettava Tecfidera-hoito ja tehtävä asianmukaiset diagnostiset tutkimukset, mukaan lukien JCV:n DNA-määritys aivo-selkäydinnesteestä kvantitatiivisella polymeraasiketjureaktiomenetelmällä (PCR). PML:n oireet voivat muistuttaa MS-taudin pahenemisvaihetta. Tyypilliset PML:ään liittyvät oireet ovat moninaisia, ne etenevät vuorokausien tai viikkojen ajan, ja niihin kuuluu progressiivinen kehon toispuoleinen heikkous tai raajojen kömpelyys, näköhäiriöt sekä ajattelukyvyn, muistin ja orientaation muutokset, jotka johtavat sekavuuteen ja persoonallisuuden muutoksiin. Lääkärien pitää kiinnittää erityistä huomiota PML:ään viittaaviin oireisiin, joita potilas ei välttämättä huomaa. Potilasta on myös neuvottava kertomaan hoidosta kumppanilleen tai häntä hoitaville henkilöille, sillä he saattavat huomata oireita, joita potilas ei huomaa.
PML voi ilmetä ainoastaan, jos potilaalla on JCV-infektio. On otettava huomioon, että lymfopenian vaikutusta seerumista tehtävän JCV-vasta-ainemäärityksen tarkkuuteen ei ole tutkittu dimetyylifumaraattihoitoa saaneilla potilailla. Myös se on huomioitava, että JCV-vasta-ainemäärityksen negatiivinen tulos (lymfosyyttimäärän ollessa normaali) ei sulje pois myöhemmän JCV-infektion mahdollisuutta.
Jos potilaalle kehittyy PML, Tecfidera-hoito on lopetettava pysyvästi.
Hoitoa edeltävät immunosuppressio- tai immunomodulaatiohoidot
Tecfidera-hoidon tehoa ja turvallisuutta ei ole tutkittu silloin, kun potilaat ovat siirtyneet toisista taudin etenemiseen vaikuttavista hoidoista Tecfideraan. On mahdollista, että aiempi immunosuppressiohoito vaikuttaa PML:n kehittymiseen dimetyylifumaraattihoitoa saavilla potilailla.
PML-tapauksia on raportoitu potilailla, joita on aiemmin hoidettu natalitsumabilla, jonka käytössä PML on varmistettu riski. Lääkäreiden on huomattava, että natalitsumabihoidon äskettäisen lopettamisen jälkeen ilmenneisiin PML-tapauksiin ei välttämättä liity lymfopeniaa.
Lisäksi suurin osa Tecfidera-valmisteen käytön yhteydessä varmistetuista PML-tapauksista oli potilailla, jotka olivat aiemmin saaneet immunomoduloivaa hoitoa.
Kun potilas siirtyy toisesta taudin etenemiseen vaikuttavasta hoidosta Tecfideraan, toisen hoidon puoliintumisaika ja vaikutustapa on otettava huomioon, jotta vältetään additiivinen immuunivaikutus samalla kun pienennetään MS-taudin uudelleen aktivoitumisen riskiä. Täydellisen verenkuvan määrittämistä suositellaan ennen Tecfidera-hoidon aloittamista ja säännöllisesti hoidon aikana (ks. edellä oleva kohta Verikokeet/laboratoriotutkimukset).
Vaikea munuaisten tai maksan vajaatoiminta
Tecfidera-valmistetta ei ole tutkittu vaikeaa munuaisten tai maksan vajaatoimintaa sairastavien potilaiden hoidossa, joten näiden potilasryhmien hoidossa on syytä noudattaa varovaisuutta (ks. kohta Annostus ja antotapa).
Vaikea aktiivinen ruoansulatuselimistön sairaus
Tecfidera-valmistetta ei ole tutkittu vaikea-asteista aktiivista ruoansulatuselimistön sairautta sairastavien potilaiden hoidossa, joten tämän potilasryhmän hoidossa on syytä noudattaa varovaisuutta.
Punastuminen
Tecfidera-hoitoa kliinisissä tutkimuksissa saaneista potilaista 34 %:lla esiintyi punastumista. Suurimmalla osalla potilaista, joilla punastumista esiintyi, punastuminen oli voimakkuudeltaan lievää tai keskivaikeaa. Terveille vapaaehtoisille tehtyjen tutkimusten perusteella dimetyylifumaraatin käyttöön liittyvä punastuminen on todennäköisesti prostaglandiini-välitteistä. Lyhyestä hoitojaksosta enteropäällysteettömällä 75 mg:n asetyylisalisyylihappoannoksella saattaa olla hyötyä potilaille, jotka kokevat punastumisen sietämättömäksi (ks. kohta Yhteisvaikutukset). Punastumisen esiintyvyys ja voimakkuus vähenivät annostelujakson aikana kahdessa terveillä vapaaehtoisilla tehdyssä tutkimuksessa.
Kliinisissä tutkimuksissa kolmella yhteensä 2 560 dimetyylifumaraattihoitoa saaneesta potilaasta esiintyi vakavia punastumisoireita, jotka olivat todennäköisesti yliherkkyys- tai anafylaktoidisia reaktioita. Nämä haittavaikutukset eivät olleet hengenvaarallisia, mutta ne johtivat sairaalahoitoon. Lääkkeen määrääjien ja potilaiden on oltava tarkkana tämän riskin suhteen vakavien punastumisoireiden esiintyessä (ks. kohdat Annostus ja antotapa, Yhteisvaikutukset ja Haittavaikutukset).
Anafylaktiset reaktiot
Tecfidera-valmisteen annon jälkeisiä anafylaksia-/anafylaktoidisia reaktiotapauksia on raportoitu markkinoille tulon jälkeen (ks. kohta Haittavaikutukset). Oireita voivat olla hengenahdistus, hypoksia, hypotensio, angioedeema, ihottuma tai urtikaria. Dimetyylifumaraatin aiheuttaman anafylaksian mekanismia ei tunneta. Nämä reaktiot ilmenevät yleensä ensimmäisen annoksen jälkeen, mutta niitä saattaa esiintyä myös milloin tahansa hoidon aikana, ja ne saattavat olla vakavia ja henkeä uhkaavia. Potilaita on neuvottava lopettamaan Tecfidera-valmisteen ottaminen ja hakeutumaan välittömästi lääkärinhoitoon, jos heillä esiintyy anafylaksian oireita tai löydöksiä. Hoitoa ei pidä aloittaa uudelleen (ks. kohta Haittavaikutukset).
Infektiot
Infektioiden (60 % vs 58 %) ja vakavien infektioiden (2 % vs 2 %) esiintyvyys oli vaiheen 3 lumekontrolloiduissa tutkimuksissa samankaltaista Tecfidera-hoitoa tai lumelääkettä saaneilla potilailla. Kuitenkin jos potilaalle kehittyy vakava infektio, Tecfidera-hoidon keskeyttämistä on harkittava ja hyödyt ja riskit on arvioitava uudelleen ennen hoidon jatkamista Tecfidera-valmisteen immuunivastetta muuntavien vaikutusten vuoksi (ks. kohta Farmakodynamiikka). Tecfidera-valmistetta saavia potilaita on neuvottava ilmoittamaan infektio-oireista lääkärille. Potilaat, joilla on vakavia infektioita, eivät saa aloittaa Tecfidera-hoitoa ennen kuin infektio/infektiot on hoidettu.
Vakavien infektioiden esiintyvyyden lisääntymistä ei havaittu potilailla, joiden lymfosyyttimäärä oli alle 0,8 × 109/l tai alle 0,5 × 109/l (ks. kohta Haittavaikutukset). Jos hoitoa jatketaan, kun potilaalla on kohtalainen tai vaikea, pitkittynyt lymfopenia, opportunistisen infektion, PML mukaan lukien, mahdollisuutta ei voida sulkea pois (ks. kohdan Varoitukset ja käyttöön liittyvät varotoimet alakohta PML).
Vyöruusu (herpes zoster ‑infektiot)
Tecfidera-valmisteen käytön yhteydessä on raportoitu vyöruusutapauksia (ks. kohta Haittavaikutukset). Suurin osa tapauksista ei ollut vakavia, mutta myös vakavia tapauksia on raportoitu, mukaan lukien disseminoitunut vyöruusu, silmänseudun vyöruusu, korvan vyöruusu, neurologinen herpes zoster ‑infektio, herpes zoster ‑infektion aiheuttama meningoenkefaliitti ja herpes zoster ‑infektion aiheuttama meningomyeliitti. Näitä haittavaikutuksia saattaa ilmetä missä tahansa vaiheessa hoidon aikana. Potilaita on seurattava vyöruusun oireiden ja löydösten havaitsemiseksi etenkin, jos potilaalla on raportoitu samanaikaisesti lymfosytopenia. Jos vyöruusu todetaan, potilaalle pitää antaa siihen asianmukaista hoitoa. Jos potilaalla on vakava infektio, on harkittava hoidon keskeyttämistä siihen saakka, kunnes infektio on hävinnyt (ks. kohta Haittavaikutukset).
Hoidon aloittaminen
Hoito tulee aloittaa asteittaisesti, jotta vältettäisiin punastumisoireet ja ruoansulatuskanavaan kohdistuvat haittavaikutukset (ks. kohta Annostus ja antotapa).
Fanconin oireyhtymä
Dimetyylifumaraattia sisältävän lääkevalmisteen ja muiden fumaarihapon estereiden samanaikaisen käytön yhteydessä on raportoitu Fanconin oireyhtymätapauksia. Fanconin oireyhtymän varhainen diagnoosi ja dimetyylifumaraattihoidon lopettaminen on tärkeää, jotta munuaisten vajaatoiminta ja osteomalasia voidaan estää, sillä oireyhtymä on yleensä korjaantuva. Tärkeimmät merkit ovat proteinuria, glukosuria (jossa verensokeriarvot ovat normaalit), aminoasiduria ja fosfaturia (mahdollisesti samanaikaisesti hypofosfatemian kanssa). Oireyhtymän edetessä saattaa ilmetä muita oireita, kuten polyuriaa, polydipsiaa ja proksimaalista lihasheikkoutta. Harvoissa tapauksissa saattaa ilmetä hypofosfateemista osteomalasiaa (jonka yhteydessä on paikantamatonta luukipua), kohonnut seerumin alkalisen fosfataasin pitoisuus ja rasitusmurtumia. On tärkeää huomata, että Fanconin oireyhtymä voi ilmetä, vaikka kreatiniinipitoisuus ei olisi kohonnut eikä glomerulusten suodatusnopeus olisi pieni. Jos epäselviä oireita ilmenee, Fanconin oireyhtymä pitää ottaa huomioon ja tehdä asianmukaiset tutkimukset.
Apuaineet
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per kapseli eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Syöpälääkkeet, immunosuppressiiviset tai kortikosteroidihoidot
Tecfidera-valmisteen käyttöä yhdessä syöpälääkkeiden tai immunosuppressiivisten hoitojen kanssa ei ole tutkittu, joten niitä samanaikaisesti käytettäessä on oltava varovainen. MS-potilailla tehdyissä kliinisissä tutkimuksissa pahenemisvaiheiden samanaikaiseen hoitoon lyhytaikaisella laskimoon annettavalla kortikosteroidikuurilla ei liittynyt kliinisesti oleellista infektioiden lisääntymistä.
Rokotteet
Rokotteiden, jotka eivät sisällä eläviä taudinaiheuttajia, antoa kansallisen rokotusohjelman mukaisesti voidaan harkita Tecfidera-hoidon aikana. Kliinisessä tutkimuksessa, johon osallistui yhteensä 71 relapsoivaa-remittoivaa MS-tautia sairastavaa potilasta, Tecfidera-valmistetta 240 mg kahdesti vuorokaudessa vähintään 6 kuukauden ajan (n = 38) tai pegyloimatonta interferonia vähintään 3 kuukauden ajan (n = 33) saaneilla potilailla saavutettiin samankaltainen immuunivaste (määritettiin ≥ 2-kertaisena nousuna rokotusta edeltävästä titteristä rokotuksen jälkeiseen titteriin) tetanustoksoidille (muistiantigeeni) ja konjugoituneelle meningokokki C -polysakkaridirokotteelle (neoantigeeni). Immuunivaste konjugoimattoman 23-valenttisen pneumokokkipolysakkaridirokotteen eri serotyypeille (T-solusta riippumaton antigeeni) sen sijaan vaihteli kummassakin hoitoryhmässä. Positiivinen immuunivaste, joka määritettiin vasta-ainetitterin ≥ 4-kertaisena nousuna näille kolmelle rokotteelle, saavutettiin harvemmalla potilaalla kummassakin hoitoryhmässä. Pieniä numeerisia eroja vasteessa tetanustoksoidille ja serotyypin 3 pneumokokkipolysakkaridille havaittiin pegyloimattoman interferonin hyväksi.
Kliinisiä tietoja ei ole saatavissa eläviä heikennettyjä taudinaiheuttajia sisältävien rokotteiden annon tehosta ja turvallisuudesta Tecfidera-hoitoa saaville potilaille. Eläviin rokotteisiin saattaa liittyä suurempi kliinisen infektion riski eikä niitä saa antaa Tecfidera-hoitoa saaneille potilaille, ellei tämän riskin poikkeustapauksissa katsota olevan pienempi kuin potilaalle koituva riski, jos rokotusta ei anneta.
Muut fumaarihapon johdannaiset
Muiden (paikallisesti tai systeemisesti käytettävien) fumaarihapon johdannaisten samanaikaista käyttöä Tecfidera-hoidon aikana on vältettävä.
Dimetyylifumaraatti metaboloituu ihmisellä valtaosin esteraasien toimesta ennen kuin se pääsee systeemiseen verenkiertoon. Metaboloituminen jatkuu edelleen sitruunahappokierrossa ilman sytokromi P450 (CYP) -järjestelmän osallisuutta. CYP-entsyymien toiminnan estymistä ja induktiota selvittäneissä tutkimuksissa, P-glykoproteiinitutkimuksessa tai dimetyylifumaraatin ja monometyylifumaraatin (dimetyylifumaraatin ensisijaisen metaboliitin) proteiineihin sitoutumista koskevissa tutkimuksissa in vitro ei havaittu mahdollisia yhteisvaikutusriskejä.
Muiden aineiden vaikutukset dimetyylifumaraattiin
MS-potilaiden hoidossa yleisesti käytetyillä lääkevalmisteilla, lihakseen annettavalla interferonibeeta‑1a:lla ja glatirameeriasetaatilla on tehty kliinisiä yhteisvaikutustutkimuksia dimetyylifumaraatin kanssa. Näiden ei havaittu vaikuttavan dimetyylifumaraatin farmakokineettiseen profiiliin.
Terveille vapaaehtoisille tehtyjen tutkimusten perusteella Tecfidera-valmisteen käyttöön liittyvä punastuminen on todennäköisesti prostaglandiini-välitteistä. Kahdessa terveille vapaaehtoisille tehdyssä tutkimuksessa enteropäällysteettömän 325 mg:n (tai vastaavan) asetyylisalisyylihappoannoksen anto 30 minuuttia ennen Tecfidera-valmisteen antoa 4 vuorokauden ja 4 viikon hoitojakson ajan ei muuttanut Tecfidera-valmisteen farmakokineettistä profiilia. Asetyylisalisyylihappohoitoon liittyvät mahdolliset riskit on otettava huomioon ennen samanaikaista käyttöä Tecfidera-valmisteen kanssa potilailla, joilla on aaltomainen MS-tauti. Asetyylisalisyylihapon pitkäaikaisesta (>4 viikkoa) jatkuvasta käytöstä ei ole tehty tutkimuksia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Tecfidera-hoitoa saavien potilaiden samanaikainen hoito munuaistoksisilla lääkevalmisteilla (esim. aminoglykosideilla, diureeteilla, ei-steroidaalisilla tulehduskipulääkkeillä tai litiumilla) saattaa suurentaa munuaisiin kohdistuvien haittavaikutusten (esim. proteinurian, ks. Kohta Haittavaikutukset) riskiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet Verikokeet/laboratoriotutkimukset).
Alkoholin kohtuukäyttö ei muuttanut altistusta dimetyylifumaraatille eikä siihen liittynyt haittavaikutusten lisääntymistä. Vahvojen (alkoholia yli 30 tilavuusprosenttia sisältävien) alkoholijuomien käyttöä suurina määrinä tulisi välttää tunnin ajan Tecfidera-valmisteen ottamisesta, sillä alkoholi saattaa lisätä maha-suolikanavan haittavaikutusten esiintyvyyttä.
Dimetyylifumaraatin vaikutukset muihin aineisiin
CYP-induktiotutkimukset in vitro eivät osoittaneet yhteisvaikutusta Tecfidera-valmisteen ja suun kautta annettavien ehkäisyvalmisteiden välillä. In vivo -tutkimuksessa Tecfidera-valmisteen samanaikainen anto suun kautta annettavan yhdistelmäehkäisyvalmisteen (norgestimaatti ja etinyyliestradioli) kanssa ei vaikuttanut oleellisesti suun kautta annettavalle ehkäisyvalmisteelle altistukseen. Yhteisvaikutustutkimuksia ei ole tehty muita progestogeenejä sisältävien suun kautta annettavien ehkäisyvalmisteiden kanssa, mutta Tecfidera-valmisteen ei odoteta vaikuttavan näille valmisteille altistukseen.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Raskaana olevista naisista on saatavana kohtalaisen laajat tiedot (300–1 000 raskaudesta), jotka pohjautuvat raskausrekisteriin ja markkinoille tulon jälkeisiin spontaaneihin raportteihin. Tecfidera-raskausrekisteriin oli dokumentoitu prospektiivisesti kerätyt 289 raskauden lopputulokset MS-potilailta, jotka olivat altistuneet dimetyylifumaraatille. Dimetyylifumaraattialtistuksen keston mediaani oli 4,6 raskausviikkoa, ja kuudennen raskausviikon jälkeinen altistus oli vähäistä (44 raskautta). Näin varhaisessa vaiheessa raskautta tapahtuvalla dimetyylifumaraattialtistuksella ei havaittu epämuodostumia aiheuttavaa tai fetaalista/neonataalista toksisuutta yleisväestöön verrattuna. Pidempikestoisen tai myöhemmin raskauden aikana tapahtuvan dimetyylifumaraattialtistuksen riskejä ei tunneta.
Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi Tecfidera-valmisteen käyttöä on suositeltavaa välttää raskauden aikana. Tecfidera-valmistetta saa käyttää raskauden aikana vain, jos se on selvästi välttämätöntä ja jos mahdollinen hyöty oikeuttaa sikiölle mahdollisesti aiheutuvan riskin.
Imetys
Ei tiedetä, erittyykö dimetyylifumaraatti tai sen metaboliitit ihmisen rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä lopetetaanko rintaruokinta vai lopetetaanko Tecfidera-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Dimetyylifumaraatin vaikutuksista ihmisten hedelmällisyyteen ei ole tietoja. Prekliinisistä tutkimuksista saadut tiedot eivät viittaa siihen, että dimetyylifumaraatti lisäisi hedelmällisyyden heikentymisen riskiä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tecfidera-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin esiintyviä haittavaikutuksia ovat punastuminen (35 %) ja maha-suolikanavan oireet (ripuli [14 %], pahoinvointi [12 %], vatsakipu [10 %], ylävatsakipu [10 %]). Punastuminen ja maha-suolikanavan oireet ilmaantuvat yleensä hoidon alkuvaiheessa (lähinnä ensimmäisen kuukauden aikana). Potilailla, joilla punastumista ja maha-suolikanavan oireita esiintyy, näitä oireita saattaa esiintyä ajoittain koko Tecfidera-hoidon ajan. Hoidon keskeyttämiseen johtavia yleisimmin raportoituja haittavaikutuksia ovat punastuminen (3 %) ja maha‑suolikanavan oireet (4 %).
Tecfidera-valmistetta on saanut vaiheen 2 ja 3 lumekontrolloiduissa ja kontrolloimattomissa kliinisissä tutkimuksissa yhteensä 2 513 potilasta enimmillään 12 vuoden ajan. Kokonaisaltistus vastaa 11 318:aa potilasvuotta. Yhteensä 1 169 potilasta on saanut Tecfidera-hoitoa vähintään 5 vuoden ajan, ja 426 potilasta vähintään 10 vuoden ajan. Kontrolloimattomista kliinisistä tutkimuksista saadut tiedot ovat yhdenmukaisia lumekontrolloiduista kliinisistä tutkimuksista saatujen tietojen kanssa.
Haittavaikutustaulukko
Seuraavassa taulukossa mainitaan haittavaikutukset, joita on ilmoitettu kliinisissä tutkimuksissa, markkinoille tulon jälkeisissä turvallisuustutkimuksissa ja spontaaneissa raporteissa.
Haittavaikutukset on esitetty MedDRA-elinjärjestelmäluokituksen suositeltujen termien mukaisesti. Haittavaikutusten esiintymistiheydet on luokiteltu seuraavasti:
- hyvin yleinen (≥ 1/10)
- yleinen (≥ 1/100, < 1/10)
- melko harvinainen (≥ 1/1 000, < 1/100)
- harvinainen (≥ 1/10 000, < 1/1 000)
- hyvin harvinainen (< 1/10 000)
- tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
| MedDRA-elinjärjestelmä | Haittavaikutus | Yleisyysluokka |
| Infektiot | Gastroenteriitti | Yleinen |
| Progressiivinen multifokaalinen leukoenkefalopatia (PML) | Tuntematon | |
| Vyöruusu | Tuntematon | |
| Veri ja imukudos | Lymfopenia | Yleinen |
| Leukopenia | Yleinen | |
| Trombosytopenia | Melko harvinainen | |
| Immuunijärjestelmä | Yliherkkyys | Melko harvinainen |
| Anafylaksia | Tuntematon | |
| Hengenahdistus | Tuntematon | |
| Hypoksia | Tuntematon | |
| Hypotensio | Tuntematon | |
| Angioedeema | Tuntematon | |
| Hermosto | Poltteen tunne | Yleinen |
| Verisuonisto | Punastuminen | Hyvin yleinen |
| Kuumat aallot | Yleinen | |
| Hengityselimet, rintakehä ja välikarsina | Rinorrea | Tuntematon |
| Ruoansulatuselimistö | Ripuli | Hyvin yleinen |
| Pahoinvointi | Hyvin yleinen | |
| Ylävatsakipu | Hyvin yleinen | |
| Vatsakipu | Hyvin yleinen | |
| Oksentelu | Yleinen | |
| Dyspepsia | Yleinen | |
| Gastriitti | Yleinen | |
| Ruoansulatuskanavan oireet | Yleinen | |
| Maksa ja sappi | ASAT-arvon nousu | Yleinen |
| ALAT-arvon nousu | Yleinen | |
| Lääkkeen aiheuttama maksavaurio | Harvinainen | |
| Iho ja ihonalainen kudos | Kutina | Yleinen |
| Ihottuma | Yleinen | |
| Eryteema | Yleinen | |
| Alopesia | Yleinen | |
| Munuaiset ja virtsatiet | Proteinuria | Yleinen |
| Yleisoireet ja antopaikassa todettavat haitat | Kuumoitus | Yleinen |
| Tutkimukset | Ketoaineita virtsassa | Hyvin yleinen |
| Albumiinia virtsassa | Yleinen | |
| Veren valkosolujen määrän väheneminen | Yleinen |
Valikoitujen haittavaikutusten kuvaus
Punastuminen
Lumekontrolloiduissa tutkimuksissa punastumisen (34 % vs. 4 %) ja kuumien aaltojen (7 % vs. 2 %) esiintyvyys oli suurempi Tecfidera-hoitoa saaneilla kuin lumehoitoa saaneilla potilailla. Punastumista kuvataan yleensä kasvojen tai kaulan punoituksena tai kuumina aaltoina, mutta se voi käsittää muitakin oireita (esim. lämmön tunne, punaisuus, kutina tai poltteen tunne). Punastumisoireet ilmaantuvat yleensä hoidon alkuvaiheessa (lähinnä ensimmäisen kuukauden aikana). Potilailla, joilla punastumista esiintyy, sitä saattaa esiintyä ajoittain koko Tecfidera-hoidon ajan. Suurimmalla osalla potilaista punastumisoireet olivat voimakkuudeltaan lieviä tai kohtalaisia. Tecfidera-hoitoa saaneista potilaista kaikkiaan 3 % keskeytti hoidon punastumisen takia. Vakavia punastumisoireita, joille voi olla ominaista yleistynyt eryteema, ihottuma ja/tai kutina, havaittiin alle 1 %:lla Tecfidera-hoitoa saaneista potilaista (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Ruoansulatuselimistöön kohdistuvat haittavaikutukset
Maha-suolikanavan oireiden esiintyvyys (esim. ripuli [14 % vs. 10 %], pahoinvointi [12 % vs. 9 %], ylävatsakipu [10 % vs. 6 %], vatsakipu [9 % vs. 4 %], oksentelu [8 % vs. 5 %] ja dyspepsia [5 % vs. 3 %]) oli suurempi Tecfidera-hoitoa saaneilla potilailla verrattuna lumehoitoa saaneisiin. Maha‑suolikanavaan kohdistuvat haittavaikutukset ilmaantuvat yleensä hoidon alkuvaiheessa (lähinnä ensimmäisen kuukauden aikana). Potilailla, joilla maha-suolikanavan oireita esiintyy, nämä saattavat jatkua ajoittain koko Tecfidera-hoidon ajan. Suurimmalla osalla potilaista, joilla maha-suolikanavan oireita esiintyi, ne olivat voimakkuudeltaan lieviä tai keskivaikeita. Neljä prosenttia (4 %) Tecfidera-hoitoa saaneista potilaista keskeytti hoidon maha-suolikanavaan kohdistuvien haittavaikutusten takia. Vakavia maha-suolikanavan oireita, kuten gastroenteriittiä ja gastriittia, havaittiin 1 %:lla Tecfidera-hoitoa saaneista potilaista (ks. kohta Annostus ja antotapa).
Maksan toiminta
Lumekontrolloiduista tutkimuksista saatujen tietojen perusteella suurimmalla osalla potilaista, joiden maksan transaminaasiarvot olivat suurentuneet, arvot olivat alle 3‑kertaiset viitearvojen ylärajaan (ULN) nähden. Tecfidera-hoitoa saaneilla potilailla havaittiin suurentuneita maksan transaminaasiarvoja lumehoitoa saaneisiin verrattuna ensisijaisesti ensimmäisten kuuden hoitokuukauden aikana. ALAT- ja ASAT-arvojen suurentumista vähintään 3‑kertaisiksi ULN:ään verrattuna havaittiin vastaavasti 5 % ja 2 %:lla lumehoitoa saaneista sekä 6 % ja 2 %:lla Tecfidera-hoitoa saaneista potilaista. Hoidon keskeytti suurentuneiden maksan transaminaasiarvojen takia alle 1 % potilaista. Keskeyttämisiä tapahtui yhtä usein Tecfidera‑hoitoa saaneilla ja lumehoitoa saaneilla potilailla. Transaminaasiarvojen suurentumista vähintään 3‑kertaisiksi ULN:ään verrattuna ja samanaikaista kokonaisbilirubiinipitoisuuden suurentumista yli 2‑kertaiseksi ULN:ään verrattuna ei havaittu lumelääkekontrolloiduissa tutkimuksissa.
Markkinoille tulon jälkeisessä seurannassaTecfidera-valmisteen annon seurauksena on raportoitu maksaentsyymien pitoisuuden suurentumista ja lääkkeen aiheuttamia maksavaurioita (transaminaasiarvojen suurentumista vähintään 3‑kertaisiksi ULN:ään verrattuna ja samanaikaista kokonaisbilirubiinipitoisuuden suurentumista yli 2‑kertaiseksi ULN:ään verrattuna). Raportoidut haittavaikutukset ovat hävinneet hoidon lopettamisen myötä.
Lymfopenia
Lumekontrolloiduissa tutkimuksissa useimpien potilaiden (yli 98 %) lymfosyyttimäärät olivat normaalit ennen hoidon aloittamista. Keskimääräinen lymfosyyttimäärä pieneni ensimmäisen Tecfidera‑hoitovuoden aikana ja tasaantui sen jälkeen. Lymfosyyttimäärät pienenivät keskimäärin noin 30 % lähtötilanteen arvoista. Lymfosyyttimäärien keskiarvot ja mediaanit pysyivät normaaliarvojen rajoissa. Lymfosyyttimääriä alle 0,5 × 109/l havaittiin alle 1 %:lla lumehoitoa saaneista ja 6 %:lla Tecfidera-hoitoa saaneista potilaista. Lymfosyyttimäärä oli alle 0,2 × 109/l yhdellä Tecfidera-hoitoa saaneella potilaalla, mutta ei yhdelläkään lumehoitoa saaneista.
Kliinisissä tutkimuksissa (sekä kontrolloiduissa että kontrolloimattomissa) 41 %:lla Tecfidera-valmisteella hoidetuista potilaista oli lymfopenia (määritelty näissä tutkimuksissa arvoksi < 0,91 × 109/l). Lievää lymfopeniaa (määrät ≥ 0,8 × 109/l – < 0,91 × 109/l) havaittiin 28 %:lla potilaista; kohtalaista lymfopeniaa (määrät ≥ 0,5 × 109/l – < 0,8 × 109/l), joka jatkui vähintään kuuden kuukauden ajan, havaittiin 11 %:lla potilaista; vaikeaa lymfopeniaa (määrät < 0,5 × 109/l), joka jatkui vähintään kuuden kuukauden ajan, havaittiin 2 %:lla potilaista. Vaikean lymfopenian ryhmässä lymfosyyttimäärä pysyi suurimmalla osalla tasolla < 0,5 × 109/l hoidon jatkuessa.
Lisäksi kontrolloimattomassa, prospektiivisessa markkinoille tulon jälkeisessä tutkimuksessa Tecfidera-hoidon viikon 48 kohdalla (n = 185) CD4+-T-solut olivat vähentyneet kohtalaisesti (määrä ≥ 0,2 × 109/l – < 0,4 × 109/l) enintään 37 %:lla tai vaikeasti (< 0,2 × 109/l) 6 %:lla potilaista, kun taas CD8+-T-solut olivat vähentyneet yleisemmin eli enintään 59 %:lla potilaista määrään < 0,2 × 109/l ja 25 %:lla potilaista määrään < 0,1 × 109/l. Kontrolloiduissa ja kontrolloimattomissa kliinisissä tutkimuksissa Tecfidera-hoidon lopettaneilla potilailla, joiden lymfosyyttimäärä oli alle normaaliarvojen alarajan (lower limit of normal, LLN), seurattiin lymfosyyttimäärän palautumista normaaliarvojen alarajaan (ks. kohta Farmakodynamiikka).
Progressiivinen multifokaalinen leukoenkefalopatia (PML)
Tecfidera-valmisteen käytön yhteydessä on raportoitu John Cunningham ‑viruksen (JCV) aiheuttamia infektioita, joista on aiheutunut PML:ää (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). PML saattaa johtaa kuolemaan tai toimintakyvyn vaikeaan heikkenemiseen. Yhdessä kliinisistä tutkimuksista Tecfidera-valmistetta saaneelle 1 potilaalle kehittyi PML pitkittyneen vaikean lymfopenian yhteydessä (lymfosyyttimäärä 3,5 vuoden ajan pääasiallisesti < 0,5 × 109/l), mikä johti kuolemaan. Markkinoille tulon jälkeen PML:ää on ilmennyt myös kohtalaisen ja lievän lymfopenian yhteydessä (> 0,5 × 109/l – < LLN, paikallisen laboratorion viitealueen mukaan).
Useissa PML-tapauksissa T-solualajoukkojen määrityksessä PML-diagnoosin aikaan CD8+-T-solumäärän havaittiin vähentyneen arvoon < 0,1 × 109/l, kun taas CD4+-T-solujen määrä vaihteli (< 0,05 – 0,5 × 109/l) ja korreloi enemmän lymfopenian kokonaisvaikeuden kanssa (< 0,5 × 109/l – < LLN). Tämän seurauksena CD4+/CD8+-suhde oli kasvanut näillä potilailla.
Pitkittynyt kohtalainen tai vaikea lymfopenia näyttää lisäävän PML:n riskiä Tecfidera-hoidon yhteydessä, mutta PML:ää esiintyi myös potilailla, joilla oli lievä lymfopenia. Lisäksi suurin osa markkinoille tulon jälkeisistä PML-tapauksista esiintyi > 50-vuotiailla potilailla.
Vyöruusu (herpes zoster -infektiot)
Tecfidera-valmisteen yhteydessä on raportoitu vyöruusua (herpes zoster ‑infektioita). Pitkäaikaisessa jatkotutkimuksessa, jossa hoitoa sai 1 736 MS-potilasta, noin 5 %:lla potilaista ilmeni vähintään yksi vyöruusutapahtuma. Niistä 42 % oli lieviä, 55 % keskivaikeita ja 3 % oli vaikea-asteisia. Ensimmäisestä Tecfidera-annoksesta vyöruusun ilmaantumiseen kulunut aika oli noin 3 kuukaudesta 10 vuoteen. Neljällä potilaalla oli vakavia tapahtumia, jotka kaikki paranivat. Useimmilla tutkittavista lymfosyyttien määrä oli normaaliarvojen alarajan yläpuolella, myös niillä tutkittavilla, joilla oli vakava vyöruusu. Suurimmalla osalla tutkittavista, joiden samanaikainen lymfosyyttimäärä oli alle normaaliarvojen alarajan, lymfopenia määriteltiin kohtalaiseksi tai vaikeaksi. Markkinoille tulon jälkeiset vyöruusutapaukset eivät useimmiten olleet vakavia ja hävisivät hoidon myötä. Markkinoille tulon jälkeen vyöruusun saaneiden potilaiden absoluuttisista lymfosyyttimääristä (ALC) on vähän tietoja saatavilla. Raportoiduissa tapauksissa suurimmalla osalla potilaista oli kohtalainen (≥ 0,5 × 109/l – < 0,8 × 109/l) tai vaikea (< 0,5 × 109/l – 0,2 × 109/l) lymfopenia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Laboratoriotulosten poikkeavuudet
Lumekontrolloiduissa tutkimuksissa virtsan ketoaineiden määrät (1+ tai sitä suurempi) olivat suurempia Tecfidera-hoitoa saaneilla (45 %) kuin lumehoitoa saaneilla (10 %). Kliinisissä tutkimuksissa ei havaittu odottamattomia kliinisiä seurauksia.
Tecfidera-hoitoa saaneiden potilaiden 1,25-(OH)2-D-vitamiinipitoisuudet pienenivät suhteessa lumehoitoa saaneisiin (mediaanin prosentuaalinen lasku kahden vuoden kohdalla lähtötilanteeseen verrattuna 25 % vs.15 %) ja lisäkilpirauhashormonipitoisuudet suurenivat Tecfidera‑hoitoa saaneilla suhteessa lumehoitoa saaneisiin (mediaanin prosentuaalinen kasvu kahden vuoden kohdalla lähtötilanteesta vastaavasti 29 % vs. 15 %). Kummankin parametrin keskimääräiset arvot pysyivät viitearvojen puitteissa.
Ohimenevää keskimääräisen eosinofiilimäärän suurenemista havaittiin ensimmäisen kahden hoitokuukauden aikana.
Pediatriset potilaat
Avoimeen, 96 viikon pituiseen, satunnaistettuun, aktiivikontrolloituun tutkimukseen osallistui 10 – < 13‑vuotiaita (n = 7) ja 13 – < 18-vuotiaita (n = 71) aaltomaista MS-tautia (relapsoivaa-remittoivaa multippeliskleroosia [RRMS]) sairastavia pediatrisia potilaita, joita hoidettiin 120 mg:n annoksella kaksi kertaa vuorokaudessa 7 päivän ajan, ja sen jälkeen 240 mg:n annoksella kaksi kertaa vuorokaudessa hoidon päättymiseen asti. Pediatristen potilaiden turvallisuusprofiili vaikutti samankaltaiselta kuin aikuisilla potilailla oli aiemmin todettu.
Pediatrisen kliinisen tutkimuksen tutkimusasetelma poikkesi aikuisten lumekontrolloiduista kliinisistä tutkimuksista. Siksi kliinisen tutkimusasetelman vaikutusta numeerisiin eroihin haittatapahtumissa pediatristen ja aikuisten potilaiden välillä ei voida poissulkea. Maha-suolikanavan häiriöitä samoin kuin hengityselinten, rintakehän ja välikarsinan häiriöitä sekä päänsärkyä ja kuukautiskipuja raportoitiin haittatapahtumina yleisemmin pediatrisilla potilailla (≥ 10 %) kuin aikuisilla potilailla.
Näitä haittatapahtumia raportoitiin pediatrisilla potilailla seuraavina prosenttimäärinä:
- Päänsärkyä raportoitiin 28 %:lla Tecfidera-hoitoa saaneista potilaista vs. 36 %:lla interferonibeeta‑1a-hoitoa saaneista potilaista.
- Ruoansulatuselimistön häiriöitä raportoitiin 74 %:lla Tecfidera-hoitoa saaneista potilaista vs. 31 %:lla interferonibeeta‑1a-hoitoa saaneista potilaista. Tecfidera-hoitoa saaneilla yleisimmin raportoituja niistä olivat vatsakipu ja oksentelu.
- Hengityselinten, rintakehän ja välikarsinan häiriöitä raportoitiin 32 %:lla Tecfidera-hoitoa saaneista potilaista vs. 11 %:lla interferonibeeta‑1a-hoitoa saaneista potilaista. Tecfidera-hoitoa saaneilla yleisimmin raportoituja niistä olivat suunielun kipu ja yskä.
- Dysmenorreaa raportoitiin 17 %:lla Tecfidera-hoitoa saaneista potilaista vs. 7 %:lla interferonibeeta‑1a-hoitoa saaneista potilaista.
Suppeassa, avoimessa, 24 viikon pituisessa kontrolloimattomassa tutkimuksessa, johon osallistui 13−17‑vuotiaita pediatrisia RRMS-potilaita (120 mg kaksi kertaa vuorokaudessa 7 vuorokauden ajan, minkä jälkeen 240 mg kaksi kertaa vuorokaudessa hoidon päättymiseen asti; n = 22) ja jota seurasi 96 viikon jatkotutkimus (240 mg kaksi kertaa vuorokaudessa; n = 20), turvallisuusprofiili vaikutti samankaltaiselta kuin aikuisilla potilailla oli todettu.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tecfidera-valmisteen yliannostustapauksia on raportoitu. Näissä tapauksissa kuvatut oireet ovat olleet yhteneviä Tecfidera-valmisteen tunnetun turvallisuusprofiilin kanssa. Tecfidera-valmisteen eliminaation tehostamiseen ei ole tunnettuja hoitokeinoja tai vastalääkettä. Yliannostustapauksissa suositellaan oireenmukaisen hoidon aloittamista kliinisen tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, muut immunosuppressantit, ATC-koodi:
L04AX07
Vaikutusmekanismi
Dimetyylifumaraatin terapeuttisen vaikutuksen mekanismia MS-taudissa ei täysin tunneta. Prekliiniset tutkimukset osoittavat, että dimetyylifumaraatin farmakodynaamiset vaikutukset välittyvät ensisijaisesti transkriptiotekijä ”Nuclear factor (erythroid-derived 2)-like 2:n” (Nrf2) säätelemän signaalireitin kautta. Dimetyylifumaraatin on osoitettu aktivoivan Nrf2-riippuvaisten antioksidanttigeenien (kuten NAD(P)H dehydrogenaasi, kinoni 1; [NQO1]) ilmentymistä.
Farmakodynaamiset vaikutukset
Vaikutukset immuunijärjestelmään
Dimetyylifumaraatilla osoitettiin prekliinisissä ja kliinisissä tutkimuksissa olevan anti-inflammatorisia ja immuunivastetta muuntavia vaikutuksia. Prekliinisissä malleissa dimetyylifumaraatti ja monometyylifumaraatti, joka on dimetyylifumaraatin ensisijainen metaboliitti, vähensivät merkitsevästi tulehdusreaktiossa tapahtuvaa immuunisolujen aktivaatiota ja proinflammatoristen sytokiinien vapautumista. Kliinisissä tutkimuksissa psoriaasipotilailla dimetyylifumaraatti vaikutti lymfosyytteihin proinflammatorisia sytokiiniprofiileja (TH1, TH17) vaimentaen ja siirtäen tasapainoa kohti anti‑inflammatorista (TH2) vastetta. Dimetyylifumaraatilla todettiin terapeuttista aktiivisuutta useissa inflammatorisissa ja neuroinflammatorisissa vauriomalleissa. Vaiheen 3 tutkimuksissa MS‑potilailla (DEFINE, CONFIRM ja ENDORSE) keskimääräinen lymfosyyttimäärä pieneni ensimmäisen Tecfidera-hoitovuoden aikana keskimäärin noin 30 % lähtötilanteen arvosta ja tasaantui sen jälkeen. Näissä tutkimuksissa hoidon lopettaneilla potilailla, joiden lymfosyyttimäärä oli alle normaaliarvojen alarajan (LLN, 0,9 × 109/l), seurattiin lymfosyyttimäärän palautumista normaaliarvojen alarajaan.
Kuvassa 1 on esitetty niiden potilaiden osuudet, joiden arvioitiin Kaplan-Meierin menetelmällä saavuttavan normaaliarvojen alarajan ilman pitkittynyttä vaikea-asteista lymfopeniaa. Palautumisen lähtötilanteeksi määriteltiin viimeinen hoidon aikana mitattu absoluuttinen lymfosyyttimäärä (ALC) ennen hoidon lopettamista. Taulukossa 1, taulukossa 2 ja taulukossa 3 on esitetty niiden potilaiden arvioidut osuudet, joiden arvot olivat palautuneet normaaliarvojen alarajaan (absoluuttinen lymfosyyttimäärä ≥ 0,9 × 109/l) viikolla 12 ja viikolla 24 ja joilla oli palautumisen lähtötilanteessa lievä, keskivaikea tai vaikea lymfopenia, sekä pisteittäiset 95 %:n luottamusvälit. Elossaolofunktion Kaplan-Meier-estimaattorin keskivirhe on laskettu Greenwoodin kaavalla.
Kuva 1: Kaplan-Meierin menetelmällä arvioidut niiden potilaiden osuudet, joiden absoluuttinen lymfosyyttimäärä (ALC) palautui palautumisen lähtötilanteesta (RBL) normaaliarvojen alarajaan (LLN) (≥ 910 solua/mm3) (0,9 × 109/l)
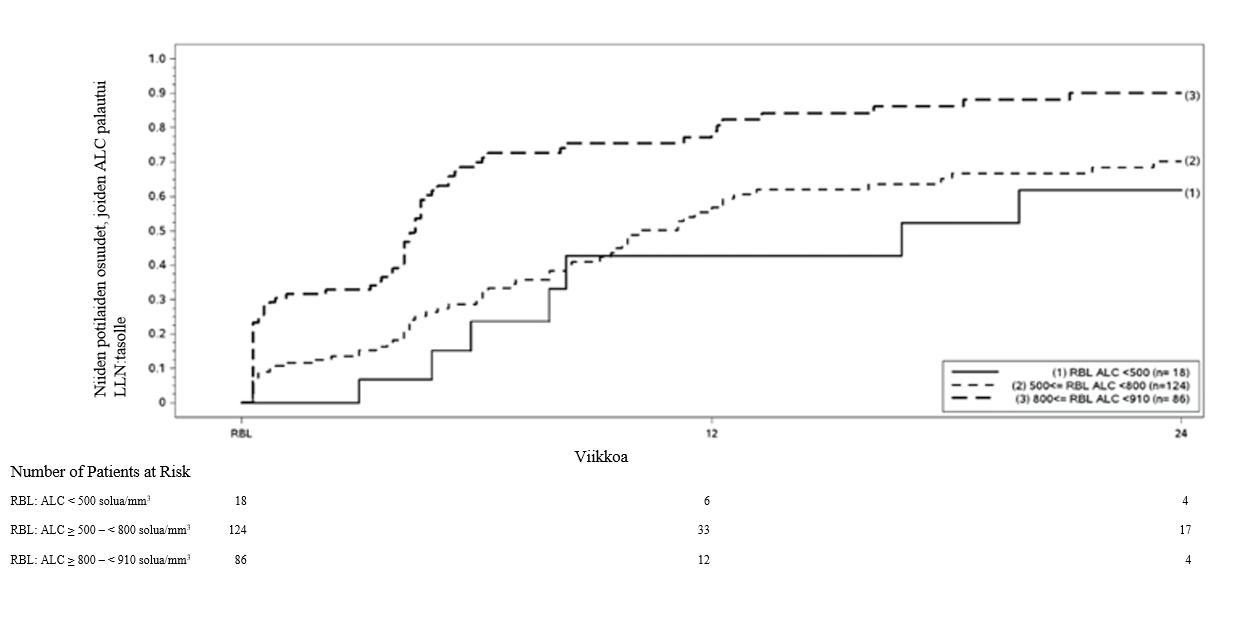
Huom.: 500 solua/mm3 = 0,5 × 109/l, 800 solua/mm3 = 0,8 × 109/l ja 910 solua/mm3 = 0,9 × 109/l.
Taulukko 1.Kaplan-Meierin menetelmällä arvioitu niiden potilaiden osuus, joiden arvot palautuivat normaaliarvojen alarajaan; potilaat, joilla oli palautumisen lähtötilanteessa lievä lymfopenia, pois lukien potilaat, joilla oli pitkittynyt vaikea-asteinen lymfopenia
Riskinalaiset potilaat, joilla lievä lymfopenia, lkma | Lähtötilanne N = 86 | Viikko 12 N = 12 | Viikko 24 N = 4 |
Niiden potilaiden osuus, joiden arvot palautuivat normaaliarvojen alarajaan (95 %:n luottamusväli) | 0,81 (0,71, 0,89) | 0,90 (0,81, 0,96) |
a Potilaat, joiden absoluuttinen lymfosyyttimäärä < 0,9 × 109/l ja ≥ 0,8 × 109/l palautumisen lähtötilanteessa, pois lukien potilaat, joilla oli pitkittynyt vaikea-asteinen lymfopenia.
Taulukko 2. Kaplan-Meierin menetelmällä arvioitu niiden potilaiden osuus, joiden arvot palautuivat normaaliarvojen alarajaan; potilaat, joilla oli palautumisen lähtötilanteessa keskivaikea lymfopenia, pois lukien potilaat, joilla oli pitkittynyt vaikea-asteinen lymfopenia
Riskinalaiset potilaat, joilla keskivaikea lymfopenia, lkma | Lähtötilanne N = 124 | Viikko 12 N = 33 | Viikko 24 N = 17 |
Niiden potilaiden osuus, joiden arvot palautuivat normaaliarvojen alarajaan (95 %:n luottamusväli) | 0,57 (0,46, 0,67) | 0,70 (0,60, 0,80) |
a Potilaat, joiden absoluuttinen lymfosyyttimäärä < 0,8 × 109/l ja ≥ 0,5 × 109/l palautumisen lähtötilanteessa, pois lukien potilaat, joilla oli pitkittynyt vaikea-asteinen lymfopenia.
Taulukko 3.Kaplan-Meierin menetelmällä arvioitu niiden potilaiden osuus, joiden arvot palautuivat normaaliarvojen alarajaan; potilaat, joilla oli palautumisen lähtötilanteessa vaikea-asteinen lymfopenia, pois lukien potilaat, joilla oli pitkittynyt vaikea-asteinen lymfopenia
Riskinalaiset potilaat, joilla vaikea lymfopenia, lkma | Lähtötilanne N = 18 | Viikko 12 N = 6 | Viikko 24 N = 4 |
Niiden potilaiden osuus, joiden arvot palautuivat normaaliarvojen alarajaan (95 %:n luottamusväli) | 0,43 (0,20, 0,75) | 0,62 (0,35, 0,88) |
a Potilaat, joiden absoluuttinen lymfosyyttimäärä < 0,5 × 109/l palautumisen lähtötilanteessa, pois lukien potilaat, joilla oli pitkittynyt vaikea-asteinen lymfopenia.
Kliininen teho ja turvallisuus
Aaltomaista (relapsoivaa-remittoivaa) MS-tautia (RRMS) sairastavilla potilailla tehtiin kaksi kahden vuoden kestoista, satunnaistettua, kaksoissokkoutettua, lumekontrolloitua tutkimusta (DEFINE, johon osallistui 1 234 potilasta ja CONFIRM, johon osallistui 1 417 potilasta). Näihin tutkimuksiin ei otettu mukaan MS-taudin progressiivisia muotoja sairastavia potilaita.
Teho (ks. taulukko 4) ja turvallisuus osoitettiin potilailla, joiden EDSS-pisteet (expanded disability status scale) olivat 0–5, joilla oli ilmennyt vähintään yksi pahenemisvaihe satunnaistamista edeltäneen vuoden aikana tai joilla kuuden viikon sisällä ennen satunnaistamista tehty aivojen magneettikuvaus osoitti vähintään yhden gadoliniumilla tehostuvan (Gd+) leesion. CONFIRM-tutkimus sisälsi arvioitsijan osalta sokkoutetun glatirameeriasetaattivertailuvalmisteryhmän (tutkimushoidon vastetta arvioiva tutkimuslääkäri/tutkija oli sokkoutettu).
DEFINE-tutkimuksessa potilasjoukkoa lähtötilanteessa kuvaavat mediaaniarvot olivat: 39 vuoden ikä, sairauden kesto 7,0 vuotta, EDSS-pistemäärä 2,0. Lisäksi 16 %:lla potilaista EDSS-pistemäärä oli yli 3,5, 28 %:lla oli ollut vähintään 2 pahenemisvaihetta edellisenä vuonna ja 42 % potilaista oli aikaisemmin saanut hoitoa jollakin hyväksytyistä MS‑taudin lääkehoidoista. MRI-ryhmässä 36 %:lla tutkimukseen mukaan otetuista potilaista oli lähtötilanteessa Gd+-leesioita (Gd+-leesioiden keskimääräinen lukumäärä 1,4).
CONFIRM-tutkimuksessa potilasjoukkoa lähtötilanteessa kuvaavat mediaaniarvot olivat: 37 vuoden ikä, sairauden kesto 6,0 vuotta, EDSS-pistemäärä 2,5. Lisäksi 17 %:lla potilaista EDSS-pistemäärä oli yli 3,5, 32 %:lla oli ollut vähintään 2 pahenemisvaihetta edellisenä vuonna ja 30 % potilaista oli aikaisemmin saanut hoitoa jollakin hyväksytyistä MS‑taudin lääkehoidoista. MRI-ryhmässä 45 %:lla tutkimukseen mukaan otetuista potilaista oli lähtötilanteessa Gd+-leesioita (Gd+-leesioiden keskimääräinen lukumäärä 2,4).
Tecfidera-hoitoa saaneiden potilaiden seuraavat päätetapahtumat pienenivät kliinisesti ja tilastollisesti merkitsevästi lumehoitoon verrattuna: DEFINE-tutkimuksen ensisijainen päätetapahtuma, pahenemisvaiheen saaneiden potilaiden osuus kahden vuoden hoidon aikana; CONFIRM-tutkimuksen ensisijainen päätetapahtuma, pahenemisvaiheiden vuosittainen määrä (ARR) kahden vuoden hoidon aikana.
Taulukko 4. DEFINE- ja CONFIRM-tutkimusten kliiniset ja MRI-päätetapahtumat
DEFINE | CONFIRM | ||||
Lume | Tecfidera 240 mg kahdesti vuoro-kaudessa | Lume | Tecfidera 240 mg kahdesti vuoro-kaudessa | Glatira-meeri-asetaatti | |
Kliiniset päätetapahtumata | |||||
Potilaiden lukumäärä | 408 | 410 | 363 | 359 | 350 |
Vuosittainen pahenemisvaihemäärä (ARR) | 0,364 | 0,172*** | 0,401 | 0,224*** | 0,286* |
Esiintyvyyssuhde (95 %:n luottamusväli) | 0,47 (0,37, 0,61) | 0,56 (0,42, 0,74) | 0,71 (0,55, 0,93) | ||
Pahenemisvaiheen saaneiden osuus | 0,461 | 0,270*** | 0,410 | 0,291** | 0,321** |
Riskitiheyssuhde (HR) (95 %:n luottamusväli) | 0,51 (0,40, 0,66) | 0,66 (0,51, 0,86) | 0,71 (0,55, 0,92) | ||
Niiden potilaiden osuus, joilla toimintakyky heikkeni (varmistettu 12 viikon ajalta) | 0,271 | 0,164** | 0,169 | 0,128# | 0,156# |
Riskitiheyssuhde (Hazard Ratio) (95 %:n luottamusväli) | 0,62 (0,44, 0,87) | 0,79 (0,52, 1,19) | 0,93 (0,63, 1,37) | ||
Niiden potilaiden osuus, joilla toimintakyky heikkeni (varmistettu 24 viikon ajalta) | 0,169 | 0,128# | 0,125 | 0,078# | 0,108# |
Riskitiheyssuhde (Hazard Ratio) (95 %:n luottamusväli) | 0,77 (0,52, 1,14) | 0,62 (0,37, 1,03) | 0,87 (0,55, 1,38) | ||
MRI-päätetapahtumatb | |||||
Potilaiden lukumäärä | 165 | 152 | 144 | 147 | 161 |
Uusien tai äskettäin laajentuneiden T2-leesioiden keskimääräinen (mediaani) lukumäärä 2 vuoden aikana | 16,5 (7,0) | 3,2 (1,0)*** | 19,9 (11,0) | 5,7 (2,0)*** | 9,6 (3,0)*** |
Leesioiden keskisuhde (95 %:n luottamusväli) | 0,15 (0,10, 0,23) | 0,29 (0,21, 0,41) | 0,46 (0,33, 0,63) | ||
Gd-leesioiden keskimääräinen (mediaani) lukumäärä 2 vuoden kuluttua | 1,8 (0) | 0,1 (0)*** | 2,0 (0,0) | 0,5 (0,0)*** | 0,7 (0,0)** |
Kerroinsuhde (Odds Ratio) (95 %:n luottamusväli) | 0,10 (0,05, 0,22) | 0,26 (0,15, 0,46) | 0,39 (0,24, 0,65) | ||
Uusien T1-hypointensiivisten leesioiden keskimääräinen (mediaani) lukumäärä 2 vuoden aikana | 5,7 (2,0) | 2,0 (1,0)*** | 8,1 (4,0) | 3,8 (1,0)*** | 4,5 (2,0)** |
Leesioiden keskisuhde (95 %:n luottamusväli) | 0,28 (0,20, 0,39) | 0,43 (0,30, 0,61) | 0,59 (0,42, 0,82) | ||
aKaikki kliinisten päätetapahtumien analyysit olivat hoitoaikeen (intent-to-treat, ITT) mukaisia; bMRI-analyysissa käytettiin MRI-kohorttia
*p-arvo alle 0,05; **p-arvo alle 0,01; ***p-arvo alle 0,0001; #ei tilastollisesti merkitsevä
Avoimeen, kontrolloimattomaan 8 vuoden pituiseen jatkotutkimukseen (ENDORSE) otettiin pivotaalitutkimuksista (DEFINE ja CONFIRM) mukaan 1 736 kriteerit täyttävää aaltomaista (relapsoivaa–remittoivaa) MS-tautia (RRMS) sairastavaa potilasta. Tutkimuksen ensisijainen tavoite oli arvioida Tecfidera-valmisteen pitkäaikaista turvallisuutta aaltomaista MS-tautia sairastavilla potilailla. Mukaan otetuista 1 736 potilaasta noin puolet (909, 52 %) sai hoitoa vähintään 6 vuoden ajan. Potilaista 501 sai kaikissa kolmessa tutkimuksessa yhtäjaksoisesti Tecfidera-hoitoa annoksella 240 mg kaksi kertaa vuorokaudessa. Potilaista 249 oli saanut DEFINE- ja CONFIRM-tutkimuksissa lumelääkettä ja sai ENDORSE-tutkimuksessa Tecfidera-hoitoa annoksella 240 mg kaksi kertaa vuorokaudessa. Yhtäjaksoista Tecfidera-hoitoa kahdesti vuorokaudessa saaneet potilaat saivat sitä enimmillään 12 vuoden ajan.
ENDORSE-tutkimuksessa yli puolet potilaista, jotka saivat Tecfidera-valmistetta 240 mg kaksi kertaa vuorokaudessa, ei saanut relapsia. Korjattu vuosittainen relapsimäärä (ARR) potilailla, jotka saivat kaikissa kolmessa tutkimuksessa yhtäjaksoisesti hoitoa kahdesti vuorokaudessa, oli DEFINE- ja CONFIRM-tutkimuksissa 0,187 (95 %:n luottamusväli: 0,156, 0,224) ja ENDORSE-tutkimuksessa 0,141 (95 %:n luottamusväli: 0,119, 0,167). Potilailla, jotka olivat saaneet aiemmin lumelääkettä, korjattu vuosittainen relapsimäärä pieneni DEFINE- ja CONFIRM-tutkimusten 0,330:sta (95 %:n luottamusväli: 0,266, 0,408) ENDORSE-tutkimuksen 0,149:ään (95 %:n luottamusväli: 0,116, 0,190).
ENDORSE-tutkimuksen potilaista suurimmalla osalla (> 75 %) ei havaittu varmistettua etenevää toimintakyvyn heikkenemistä (määriteltiin 6 kuukauden ajan etenevänä toimintakyvyn heikentymisenä). Näiden kolmen tutkimuksen yhdistetyt tulokset osoittivat, että varmistetun etenevän toimintakyvyn heikentymisen esiintyvyys oli Tecfidera-hoitoa saaneilla potilailla yhdenmukaista ja vähäistä ja että keskimääräinen EDSS-pistemäärä suureni hieman ENDORSE-tutkimuksen kuluessa. Magneettikuvausten (joita tehtiin enimmillään vuoteen 6 asti 752 potilaalle, jotka olivat aiemmin olleet DEFINE- ja CONFIRM-tutkimusten magneettikuvauskohortissa) tulokset osoittivat, että suurimmalla osalla potilaista (noin 90 %:lla) ei ollut Gd-tehosteisia leesioita. Näiden kuuden vuoden aikana uusien tai äskettäin laajentuneiden T2-leesioiden ja uusien T1-leesioiden vuotuinen korjattu keskimääräinen lukumäärä pysyi pienenä.
Teho potilailla, joiden tauti on erittäin aktiivinen:
DEFINE- ja CONFIRM-tutkimuksissa hoitovaikutuksen pahenemisvaiheisiin havaittiin olevan yhdenmukainen niiden potilaiden alaryhmässä, joilla tauti oli erittäin aktiivinen, kun taas hoitovaikutusta toimintakyvyn jatkuvaan heikkenemiseen (varmistettu 3 kuukauden ajalta) ei ollut selvästi osoitettavissa. Erittäin aktiivinen tauti määriteltiin tutkimusasetelmassa seuraavasti:
- Potilaat, joilla oli vähintään 2 pahenemisvaihetta vuoden aikana ja vähintään yksi aivojen MRI‑kuvauksessa todettu Gd-tehosteinen leesio (n = 42 DEFINE-tutkimuksessa; n = 51 CONFIRM-tutkimuksessa) tai
- Potilaat, jotka eivät olleet saaneet vastetta asianmukaisesti toteutettuun interferonibeetahoitoon (vähintään yksi hoitovuosi), kun heillä oli ollut vähintään yksi hoidon aikainen pahenemisvaihe edeltävänä vuonna ja vähintään yhdeksän aivojen MRI-tutkimuksessa todettua T2‑hyperintensiivistä leesiota tai vähintään yksi Gd-tehosteinen leesio, tai potilaat, joilla pahenemisvaiheiden määrä ei ollut muuttunut tai se oli lisääntynyt vuotta aikaisemmin verrattuna sitä edeltäviin 2 vuoteen (n = 177 DEFINE-tutkimuksessa; n = 141 CONFIRM-tutkimuksessa).
Pediatriset potilaat
Tecfidera-valmisteen turvallisuutta ja tehoa aaltomaista MS-tautia (relapsoivaa-remittoivaa multippeliskleroosia [RRMS]) sairastavilla pediatrisilla potilailla arvioitiin satunnaistetussa, avoimessa, aktiivikontrolloidussa (interferonibeeta‑1a) rinnakkaisryhmätutkimuksessa, johon osallistui 10 – < 18‑vuotiaita RRMS‑potilaita. Sataviisikymmentä potilasta satunnaistettiin saamaan dimetyylifumaraattia (240 mg suun kautta kaksi kertaa vuorokaudessa) tai interferonibeeta‑1a:ta (30 mikrog lihakseen kerran viikossa) 96 viikon ajan. Ensisijainen päätetapahtuma oli niiden potilaiden osuus, joilla ei havaittu viikolla 96 aivojen magneettikuvauksessa uusia tai äskettäin laajentuneita T2‑hyperintensiivisiä leesioita. Pääasiallinen toissijainen päätetapahtuma oli uusien tai äskettäin laajentuneiden T2‑hyperintensiivisten leesioiden lukumäärä aivojen magneettikuvauksessa viikolla 96. Esitetyt tilastotiedot ovat kuvailevia, koska ensisijaiselle päätetapahtumalle ei ollut määritetty etukäteen konfirmatorista hypoteesia.
Niiden potilaiden osuus hoitoaikeen mukaisessa (ITT) populaatiossa, joilla ei ollut uusia tai äskettäin laajentuneita T2‑leesioita magneettikuvauksessa viikolla 96 suhteessa lähtötilanteeseen, oli dimetyylifumaraattiryhmässä 12,8 % vs. interferonibeeta‑1a-ryhmässä 2,8 %. Uusien tai äskettäin laajentuneiden T2‑leesioiden keskimääräinen lukumäärä viikolla 96 suhteessa lähtötilanteeseen, lähtötilanteen T2‑leesioiden lukumäärän ja iän mukaan korjattuna (ITT-populaatio pois lukien potilaat, joilla ei ollut magneettikuvaustuloksia), oli dimetyylifumaraattiryhmässä 12,4 ja interferonibeeta‑1a‑ryhmässä 32,6.
Kliinisen pahenemisvaiheen todennäköisyys 96 viikon pituisen avoimen tutkimusjakson aikana oli dimetyylifumaraattiryhmässä 34 % ja interferonibeeta‑1a-ryhmässä 48 %.
Turvallisuusprofiili Tecfidera-hoitoa saavilla (13 – < 18‑vuotiailla) pediatrisilla potilailla oli kvalitatiivisesti yhdenmukainen aikuisilla potilailla aiemmin todetun turvallisuusprofiilin kanssa (ks. kohta Haittavaikutukset).
Farmakokinetiikka
Suun kautta annettu dimetyylifumaraatti käy esteraasien välityksellä läpi nopean presysteemisen hydrolyysin ja muuntuu pääasialliseksi metaboliitiksi, monometyylifumaraatiksi, joka on myös aktiivinen. Tecfidera-valmisteen suun kautta tapahtuneen annon jälkeen dimetyylifumaraattia ei ole plasmassa mitattavia määriä. Siksi kaikki dimetyylifumaraattiin liittyvät farmakokineettiset analyysit tehtiin plasman monometyylifumaraattipitoisuuksista. Farmakokineettiset tiedot saatiin MS‑potilaista ja terveistä vapaaehtoisista.
Imeytyminen
Monometyylifumaraatin Tmax on 2–2,5 tuntia. Koska Tecfidera- kovat enterokapselit sisältävät mikrotabletteja, joita suojaa enteropäällyste, imeytyminen alkaa vasta, kun tabletit ovat poistuneet mahalaukusta (yleensä alle 1 tunti). Ruoan kanssa annetun annoksen (240 mg kahdesti vuorokaudessa) jälkeen MS-potilaiden huippupitoisuuden (Cmax) mediaani oli 1,72 mg/l ja kokonaisaltistus (käyrän alla oleva pinta-ala (AUC)) 8,02 h.mg/l. Kaiken kaikkiaan Cmax- ja AUC-arvot suurenivat tutkituilla annoksilla (120–360 mg) likimäärin suhteessa annokseen. MS-potilailla tehdyissä tutkimuksissa annettiin kaksi 240 mg:n annosta neljän tunnin välein kolme kertaa päivässä toteutettavan hoito-ohjelman mukaisesti. Tämä johti lääkeaineen hyvin vähäiseen kertymiseen elimistöön, mikä suurensi Cmax-arvon mediaania 12 % verrattuna kahdesti vuorokaudessa annettavaan annostukseen (1,72 mg/l kahdesti vuorokaudessa verrattuna 1,93 mg/l kolme kertaa vuorokaudessa) eikä tällä ollut turvallisuuteen liittyviä vaikutuksia.
Ruoka ei vaikuttanut dimetyylifumaraattialtistukseen kliinisesti merkittävästi. Tecfidera on kuitenkin otettava ruoan kanssa, sillä se parantaa siedettävyyttä punastumisen tai maha-suolikanavan haittatapahtumien kannalta (ks. kohta Annostus ja antotapa).
Jakautuminen
Näennäinen jakautumistilavuus suun kautta annetun 240 mg dimetyylifumaraattiannoksen jälkeen on 60−90 l. Monometyylifumaraatista sitoutuu ihmisen plasman proteiineihin yleensä 27–40 %.
Biotransformaatio
Dimetyylifumaraatti metaboloituu ihmisissä valtaosin ja alle 0,1 % annoksesta poistuu muuttumattomana dimetyylifumaraattina virtsaan. Dimetyylifumaraatti metaboloituu aluksi kaikkialla ruoansulatuskanavassa, veressä ja kudoksissa olevien esteraasien välityksellä, ennen kuin se pääsee systeemiseen verenkiertoon. Metaboloituminen jatkuu edelleen sitruunahappokierrossa ilman sytokromi P450 (CYP) -järjestelmän osallistumista metaboliaan. Kerta-annostutkimuksessa, jossa annettiin 240 mg 14C-dimetyylifumaraattia, ihmisen plasmassa esiintyvän pääasiallisen metaboliitin havaittiin olevan glukoosi. Muut verenkierrossa olevat metaboliitit olivat fumaarihappo, sitruunahappo ja monometyylifumaraatti. Fumaarihapon loppupään metabolia tapahtuu sitruunahappokierrossa, jossa hiilidioksidin (CO2) uloshengitys toimii ensisijaisena poistumisreittinä.
Eliminaatio
Hiilidioksidin (CO2) uloshengitys on dimetyylifumaraatin ensisijainen poistumisreitti, ja se kattaa 60 % annoksesta. Toissijaisia poistumisreittejä ovat munuaiset (15,5 % annoksesta) ja uloste (0,9 % annoksesta).
Monometyylifumaraatin terminaalinen puoliintumisaika on lyhyt (noin 1 tunti) eikä useimmilla potilailla ole monometyylifumaraattia enää verenkierrossa 24 tunnin kuluttua. Dimetyylifumaraattia tai monometyylifumaraattia ei kerry elimistöön, kun dimetyylifumaraattia annetaan useita annoksia hoito‑ohjelman mukaisesti.
Lineaarisuus
Dimetyylifumaraattialtistus suurenee suunnilleen suhteessa annokseen, kun annetaan yksi tai useampi tutkitun suuruinen annos (120–360 mg).
Farmakokinetiikka erityispotilasryhmillä
Varianssianalyysin (ANOVA) tulosten perusteella ruumiinpaino on RRMS-potilaiden altistuksen tärkein kovariaatti (Cmax- ja AUC-arvoilla mitattuna), mutta se ei vaikuttanut kliinisissä tutkimuksissa käytettyihin turvallisuus- ja tehomittareihin.
Sukupuoli ja ikä eivät vaikuttaneet kliinisesti merkittävästi dimetyylifumaraatin farmakokinetiikkaan. Farmakokinetiikkaa ei ole tutkittu 65‑vuotiailla tai sitä vanhemmilla potilailla.
Munuaisten vajaatoiminta
Koska munuaiset ovat dimetyylifumaraatin toissijainen poistumisreitti, joka kattaa alle 16 % annetusta annoksesta, farmakokinetiikkaa ei tutkittu munuaisten vajaatoimintaa sairastavilla henkilöillä.
Maksan vajaatoiminta
Koska dimetyylifumaraatti ja monometyylifumaraatti metaboloituvat esteraasien välityksellä ilman sytokromi P450 (CYP) -järjestelmän osallistumista metaboliaan, farmakokinetiikkaa ei tutkittu maksan vajaatoimintaa sairastavilla potilailla.
Pediatriset potilaat
Dimetyylifumaraatin farmakokineettistä profiilia, kun valmistetta annettiin 240 mg kaksi kertaa vuorokaudessa, arvioitiin pienessä, avoimessa, kontrolloimattomassa tutkimuksessa aaltomaista (relapsoivaa-remittoivaa) MS-tautia (RRMS) sairastavilla 13–17-vuotiailla potilailla (n = 21). Tecfidera-valmisteen farmakokinetiikka näillä nuorilla potilailla oli yhdenmukainen aikuisilla potilailla aiemmin todetun kanssa (Cmax: 2,00 ± 1,29 mg/l; AUC0-12h: 3,62 ± 1,16 h.mg/l, mikä vastaa 24 tunnin kokonais-AUC-arvoa 7,24 h.mg/l).
Prekliiniset tiedot turvallisuudesta
Jäljempänä olevissa kohdissa Toksikologia ja Lisääntymistoksisuus kuvattuja haittavaikutuksia ei havaittu kliinisissä tutkimuksissa, mutta niitä todettiin eläimillä kliinistä altistusta vastaavilla altistustasoilla.
Genotoksisuus
Dimetyylifumaraatti ja monometyylifumaraatti olivat negatiivisia in vitro -testisarjassa (Amesin testi, nisäkässolujen kromosomipoikkeavuudet). Dimetyylifumaraatti oli negatiivinen rotalla tehdyssä in vivo -mikrotumamäärityksessä.
Karsinogeenisuus
Dimetyylifumaraattia tutkittiin hiirillä ja rotilla enimmillään kaksi vuotta kestäneissä karsinogeenisuustutkimuksissa. Dimetyylifumaraattia annettiin suun kautta hiirille annoksina 25, 75, 200 ja 400 mg/kg/vrk ja rotille annoksina 25, 50, 100 ja 150 mg/kg/vrk.
Hiirillä munuaistiehyen karsinooman esiintyvyys suureni annoksella 75 mg/kg/vrk, joka vastaa ihmiselle suositellun annoksen aiheuttamaa altistusta (AUC). Rotilla munuaistiehyen karsinooman ja kiveksen välisolujen (Leydigin solujen) adenoomien esiintyvyys suureni annoksella 100 mg/kg/vrk, josta aiheutuva altistus on noin kaksinkertainen ihmiselle suositellun annoksen aiheuttamaan altistukseen nähden. Näiden löydösten merkitystä ihmisille aiheutuvan riskin kannalta ei tunneta.
Levyepiteelipapillooman ja ‑karsinooman esiintyvyys ei-rauhasmahassa (etumahassa) suureni hiirillä ihmiselle suositeltua annosta vastaavalla altistuksella ja rotilla tätä pienemmällä altistuksella (AUC:n perusteella). Ihmisillä ei ole vastinetta jyrsijöiden etumahalle.
Toksikologia
Jyrsijöille, kaniineille ja apinoille annettiin nonkliinisissä tutkimuksissa dimetyylifumaraattisuspensiota (dimetyylifumaraattia 0,8‑prosenttisessa hydroksipropyylimetyyliselluloosassa) letkuruokintana suun kautta. Kroonisessa toksisuustutkimuksessa koirille annettiin dimetyylifumaraattikapseleita suun kautta.
Suun kautta useita dimetyylifumaraattiannoksia saaneilla hiirillä, rotilla, koirilla ja apinoilla havaittiin munuaismuutoksia. Kaikilla lajeilla havaittiin vaurioon viittaavaa munuaistiehyen epiteelin regeneraatiota. Rotilla havaittiin elinikäisen annon yhteydessä (2‑vuotinen tutkimus) munuaistiehyen hyperplasiaa. Koirilla, joille annettiin dimetyylifumaraattiannoksia suun kautta päivittäin 11 kuukauden ajan, munuaiskuoren atrofialle laskettu marginaali havaittiin annoksella, joka oli AUC:n perusteella kolminkertainen suositeltuun annokseen nähden. Apinoilla, joille annettiin dimetyylifumaraattiannoksia suun kautta päivittäin 12 kuukauden ajan, havaittiin yksittäisten solujen nekroosia annoksella, joka oli AUC:n perusteella kaksinkertainen suositeltuun annokseen nähden. Interstitiaalista fibroosia ja munuaiskuoren atrofiaa havaittiin annoksella, joka oli AUC:n perusteella kuusinkertainen suositeltuun annokseen nähden. Näiden löydösten merkitystä ihmisille ei tunneta.
Rotilla ja koirilla havaittiin kivesten siementiehyiden epiteelin degeneraatiota. Rotilla havainnot tehtiin suunnilleen suositellulla annoksella ja koirilla annoksella, joka oli kolminkertainen suositeltuun annokseen nähden (AUC:n perusteella). Näiden löydösten merkitystä ihmisille ei tunneta.
Hiirien ja rottien etumahan löydöksiä olivat levyepiteelin hyperplasia ja hyperkeratoosi, tulehdukset sekä levyepiteelipapillooma ja ‑karsinooma vähintään 3 kuukauden pituisissa tutkimuksissa. Ihmisellä ei ole vastinetta hiirien ja rottien etumahalle.
Lisääntymis- ja kehitystoksisuus
Urosrotille suun kautta ennen parittelua ja sen aikana annettu dimetyylifumaraatti (75, 250 ja 375 mg/kg/vrk) ei vaikuttanut urosten hedelmällisyyteen suurimmillakaan tutkituilla annoksilla (AUC:n perusteella vähintään kaksi kertaa suurempi kuin suositeltu annos). Naarasrotille suun kautta ennen parittelua ja sen aikana annettu dimetyylifumaraatti (25, 100 ja 250 mg/kg/vrk) ja annon jatkaminen 7. tiineyspäivään asti vähensi kiimavaiheiden määrää 14 vuorokautta kohden ja suurensi suurimmalla tutkitulla annoksella (AUC:n perusteella 11 kertaa suurempi kuin suositeltu annos) niiden eläinten lukumäärää, joiden kiimojen välinen aika oli pitkittynyt. Nämä muutokset eivät kuitenkaan vaikuttaneet hedelmällisyyteen tai elinkykyisten sikiöiden määrään.
Dimetyylifumaraatin osoitettiin läpäisevän rottien ja kaniinien istukkakalvon ja pääsevän sikiön vereen; pitoisuuksien suhde sikiön ja emon plasmassa oli rotilla 0,48–0,64 ja kaniineilla 0,1. Rotilla ja kaniineilla ei havaittu epämuodostumia millään dimetyylifumaraattiannostuksella. Dimetyylifumaraatin antaminen suun kautta (annoksina 25, 100 ja 250 mg/kg/vrk) tiineille rotille organogeneesin aikana AUC:n perusteella neljä kertaa suositeltua annosta suurempina annoksina aiheutti emolle haittavaikutuksia ja 11 kertaa suositeltua annosta suurempina annoksina sikiön pienipainoisuutta ja luutumisen viivästyneisyyttä (jalkapöydät ja takajalan varvasluut). Pienen sikiöpainon ja viivästyneen luutumisen katsottiin johtuvan emoon kohdistuneesta toksisuudesta (alentunut paino ja vähentynyt ravinnon nauttiminen).
Dimetyylifumaraatin antaminen suun kautta (25, 75 ja 150 mg/kg/vrk) tiineille kaniineille organogeneesin aikana ei vaikuttanut alkion ja sikiön kehitykseen, mutta aiheutti AUC:n perusteella seitsemän kertaa suositeltua annosta suurempina annoksina emon painon alenemista ja lisäsi 16 kertaa suositeltua annosta suurempina annoksina tiineyden keskeytymisiä.
Dimetyylifumaraatin antaminen suun kautta (25, 100 ja 250 mg/kg/vrk) rotille tiineyden ja laktaation aikana AUC:n perusteella 11 kertaa suositeltua annosta suurempina annoksina alensi F1-jälkeläisten painoa ja viivästytti F1-urosten sukukypsyyttä. Vaikutuksia F1-jälkeläisten hedelmällisyyteen ei esiintynyt. Jälkeläisten alentuneen painon katsottiin johtuvan emoon kohdistuneesta toksisuudesta.
Toksisuus nuorilla eläimillä
Kahdessa nuorilla rotilla tehdyssä toksisuustutkimuksessa, joissa dimetyylifumaraattia annettiin päivittäin suun kautta syntymänjälkeisestä päivästä 28 syntymänjälkeiseen päivään 90–93 (vastaa noin 3 vuoden ikää ihmisellä), havaittiin samankaltaista kohde-elinten toksisuutta munuaisissa ja etumahassa kuin aikuisilla eläimillä. Ensimmäisessä tutkimuksessa dimetyylifumaraatti ei suurimmallakaan annoksella 140 mg/kg/vrk (noin 4,6‑kertainen ihmiselle suositeltuun annokseen nähden pediatrisia potilaita koskevien suppeiden AUC-tietojen perusteella) vaikuttanut kehitykseen, neurologiseen käyttäytymiseen eikä urosten tai naaraiden hedelmällisyyteen. Toisessa, nuorilla urosrotilla tehdyssä tutkimuksessa suurimmallakaan dimetyylifumaraattiannoksella 375 mg/kg/vrk (noin 15‑kertainen pediatrisille potilaille suositellusta annoksesta aiheutuvaan oletettuun AUC-arvoon nähden) ei niin ikään havaittu vaikutuksia urosten sukupuolielimiin tai apuelimiin. Nuorilla urosrotilla havaittiin kuitenkin reisiluun ja lannenikamien mineraalimäärän ja -tiheyden pienentymistä. Muutoksia nuorten rottien luuston densitometriassa havaittiin myös diroksimeelifumaraatin suun kautta annon jälkeen. Diroksimeelifumaraatti on toinen fumaarihapon esteri, joka metaboloituu samaksi aktiiviseksi metaboliitiksi, monometyylifumaraatiksi, in vivo. Annos, jolla ei ole havaittavia haittavaikutuksia (No Observed Adverse Effect Level, NOAEL) nuorten rottien luuston densitometriaan, vastaa noin 1,5‑kertaista pediatrisille potilaille suositellusta annoksesta aiheutuvaa oletettua AUC-arvoa. Luustovaikutukset saattavat liittyä alentuneeseen painoon, mutta suoran vaikutuksen osuutta ei voida poissulkea. Luustolöydösten merkitys on aikuisten potilaiden kannalta vähäinen. Merkitystä pediatrisille potilaille ei tunneta.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö (enteropäällysteiset mikrotabletit)
Mikrokiteinen selluloosa
Kroskarmelloosinatrium
Talkki
Vedetön kolloidinen piidioksidi
Magnesiumstearaatti
Trietyylisitraatti
Metakryylihappo – metyylimetakrylaattikopolymeeri (1:1)
Metakryylihappo – etyyliakrylaattikopolymeeri (1:1) 30‑prosenttinen dispersio
Simetikoni
Natriumlauryylisulfaatti
Polysorbaatti 80
Kapselikuori
Liivate
Titaanidioksidi (E 171)
Briljanttisininen FCF (E 133)
Keltainen rautaoksidi (E 172)
Kapselin painatus (musta muste)
Shellakka
Kaliumhydroksidi
Musta rautaoksidi (E 172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta
Säilytys
Säilytä alle 30 °C.
Pidä läpipainopakkaukset ulkopakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TECFIDERA enterokapseli, kova
120 mg (L:ei) 14 fol (166,09 €)
240 mg (L:ei) 56 fol (998,55 €)
PF-selosteen tieto
120 mg kovat enterokapselit
14 kovaa enterokapselia PVC/PE/PVDC-PVC-alumiiniläpipainopakkauksissa.
240 mg kovat enterokapselit
56 tai 168 kovaa enterokapselia PVC/PE/PVDC-PVC-alumiiniläpipainopakkauksissa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Tecfidera 120 mg kovat enterokapselit
Kokoa 0 olevat valkovihreät kovat enterokapselit, joissa on merkintä ˮBG-12 120 mgˮ ja jotka sisältävät mikrotabletteja.
Tecfidera 240 mg kovat enterokapselit
Kokoa 0 olevat vihreät kovat enterokapselit, joissa on merkintä ˮBG-12 240 mgˮ ja jotka sisältävät mikrotabletteja.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TECFIDERA enterokapseli, kova
120 mg 14 fol
240 mg 56 fol
- Ei korvausta.
ATC-koodi
L04AX07
Valmisteyhteenvedon muuttamispäivämäärä
18.12.2024
Yhteystiedot
Bertel Jungin aukio 5 C
02600 Espoo
0207 401 200
www.biogen.fi
www.MS-nyt.fi