
COFACT injektiokuiva-aine ja liuotin, liuosta varten 250 IU, 500 IU
Vaikuttavat aineet ja niiden määrät
Cofact (neljän hyytymistekijän konsentraatti) on injektiokuiva-aine ja liuotin, liuosta varten, joka sisältää ihmisen protrombiinikompleksia. Valmiste sisältää nimellisesti seuraavassa taulukossa esitetyt määrät ihmisen hyytymistekijöitä:
| Cofact 250 IU (tekijä IX) | Cofact 500 IU (tekijä IX) | Käyttövalmiiksi liuottamisen jälkeen* (IU/ml) | |
| Vaikuttavat aineet | |||
| Hyytymistekijä II | 140 - 350 | 280 - 700 | 14 - 35 |
| Hyytymistekijä VII | 70 - 200 | 140 - 400 | 7 - 20 |
Hyytymistekijä IX | 250 | 500 | 25 |
| Hyytymistekijä X | 140 - 350 | 280 - 700 | 14 - 35 |
| Muut vaikuttavat aineet | |||
| Proteiini C | 111–390 | 222-780 | 11-39 |
| Proteiini S | 10-80 | 20-160 | 1-8 |
*10 ml:lla (Cofact 250 IU) tai 20 ml:lla (Cofact 500 IU) injektionesteisiin käytettävää vettä käyttökuntoon saatettuna
Kokonaisproteiinimäärä injektiopulloa kohti on 130 - 350 mg (Cofact 250 IU) tai 260 - 700 mg (Cofact 500 IU). Valmisteen spesifinen aktiivisuus on ≥ 0,6 IU/mg tekijä IX -aktiivisuutena ilmaistuna.
Kaikkien hyytymistekijöiden sekä proteiini C:n ja S:n (antigeeni) aktiivisuus on testattu WHO:n tai Euroopan farmakopean voimassa olevien standardien mukaisesti.
Apuaine(et), joiden vaikutus tunnetaan
Käyttökuntoon saatettuna tämä lääkevalmiste sisältää 125–195 mmol/l natriumia, enintään 44,8 mg natriumia per 10 ml.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
- Verenvuotojen hoito ja leikkauksiin liittyvä verenvuotojen ennaltaehkäisy hankinnaisessa protrombiinikompleksihyytymistekijöiden puutoksessa, kuten K-vitamiiniantagonistihoidon aiheuttama puutos, tai K-vitamiiniantagonistien yliannostustapauksessa, kun tarvitaan puutoksen nopeaa korjaamista.
- Verenvuotojen hoito ja leikkauksiin liittyvä verenvuotojen ennaltaehkäisy synnynnäisessä, jonkin K-vitamiinista riippuvan hyytymistekijän puutoksessa, kun puhdistettua, spesifistä hyytymistekijävalmistetta ei ole saatavilla.
Ehto
Hoito tulee aloittaa hyytymishäiriöihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Annostus
Seuraavassa on esitetty ainoastaan yleiset annostusohjeet. Hoito tulee aloittaa hyytymishäiriöiden hoitoon erikoistuneen lääkärin valvonnassa. Annostus ja korvaushoidon kesto riippuvat häiriön vakavuudesta, vuodon sijainnista ja laajuudesta sekä potilaan kliinisestä tilasta.
Annettava määrä ja antotiheys on laskettava kunkin potilaan kohdalla yksilöllisesti. Annosvälit tulee sovittaa protrombiinikompleksiin kuuluvien hyytymistekijöiden eripituisten verenkierron puoliintumisaikojen mukaisesti (katso kohta Farmakokinetiikka). Yksilölliset annostarpeet on mahdollista tunnistaa ainoastaan määrittämällä kyseessä olevien hyytymistekijöiden plasmapitoisuudet säännöllisesti tai määrittämällä protrombiinikompleksipitoisuudet (protrombiiniaika, INR) yleistestien avulla ja seuraamalla tiiviisti potilaan kliinistä tilaa.
Suurissa kirurgisissa toimenpiteissä on välttämätöntä seurata korvaushoitoa tarkkaan hyytymistekijäpitoisuusmääritysten avulla (spesifiset hyytymistekijäpitoisuusmääritykset ja/tai protrombiinikompleksipitoisuuksien yleistestit).
Verenvuodot ja leikkauksiin liittyvä verenvuotojen ennaltaehkäisy K-vitamiiniantagonistihoidon aikana:
Annos riippuu hoitoa edeltävästä INR-arvosta, tavoitteena olevasta INR-arvosta ja potilaan painosta. Seuraavissa taulukoissa on annettu suuntaa-antavat annokset INR-arvon korjaamiseksi INR:n eri lähtöarvoilla.
Annostaulukot edustavat ainoastaan yleisiä annostusohjeita. Ne eivät korvaa yksilöllisen annoksen arviointia kunkin potilaan kohdalla eivätkä hoidon aikaista INR-arvon ja muiden hyytymisparametrien huolellista seurantaa.
Cofactin annossuositukset (ml) tavoite-INR-arvon ≤ 2,1 saavuttamiseksi
| Lähtö-INR | 7,5 | 5,9 | 4,8 | 4,2 | 3,6 | 3,3 | 3,0 | 2,8 | 2,6 | 2,5 | 2,3 | 2,2 |
| Potilaan paino | ||||||||||||
| 50 kg | 40 | 40 | 40 | 30 | 30 | 30 | 20 | 20 | x | x | x | x |
| 60 kg | 50 | 50 | 40 | 40 | 30 | 30 | 30 | 20 | x | x | x | x |
| 70 kg | 60 | 50 | 50 | 50 | 40 | 40 | 30 | 30 | x | x | x | x |
| 80 kg | 60 | 60 | 60 | 50 | 50 | 40 | 40 | 30 | x | x | x | x |
| 90 kg | 60 | 60 | 60 | 60 | 50 | 50 | 40 | 30 | x | x | x | x |
| 100 kg | 60 | 60 | 60 | 60 | 60 | 50 | 40 | 40 | x | x | x | x |
Cofactin annossuositukset (ml) tavoite-INR-arvon ≤ 1,5 saavuttamiseksi
| Lähtö-INR | 7,5 | 5,9 | 4,8 | 4,2 | 3,6 | 3,3 | 3,0 | 2,8 | 2,6 | 2,5 | 2,3 | 2,2 |
| Potilaan paino | ||||||||||||
| 50 kg | 60 | 60 | 60 | 50 | 50 | 50 | 40 | 40 | 30 | 30 | 30 | 30 |
| 60 kg | 80 | 70 | 70 | 60 | 60 | 60 | 50 | 50 | 40 | 40 | 40 | 30 |
| 70 kg | 90 | 80 | 80 | 70 | 70 | 70 | 60 | 60 | 50 | 40 | 40 | 40 |
| 80 kg | 100 | 100 | 90 | 90 | 90 | 80 | 80 | 70 | 60 | 50 | 50 | 40 |
| 90 kg | 100 | 100 | 100 | 90 | 90 | 90 | 80 | 80 | 70 | 60 | 50 | 40 |
| 100 kg | 100 | 100 | 100 | 100 | 100 | 90 | 90 | 80 | 70 | 70 | 60 | 50 |
Annokset on laskettu Cofactin tekijä IX -pitoisuuden perusteella, koska tällä on suhteellisen lyhyt puoliintumisaika ja alhainen infuusion jälkeinen saanto verrattuna muiden protrombiinikompleksin hyytymistekijöiden vastaaviin ominaisuuksiin. Oletuksena on, että tekijä IX:n keskimääräinen plasmapitoisuus ≥ 30 % on riittävä INR-arvon ≤ 2,1 saavuttamiseksi ja ≥ 60 % on riittävä INR-arvon ≤ 1,5 saavuttamiseksi. Laskennalliset määrät on pyöristetty alaspäin 10 ml:n kerrannaisiksi, ja ylärajaksi on asetettu 60 ml tai 100 ml (katso edellä olevat taulukot). Tavoite-INR-arvot ovat Federation of Dutch Thrombosis Services -organisaation suosittelemat ja samankaltaiset kuin englantilaiset ja saksalaiset suositukset.
K-vitamiiniantagonistin aiheuttaman heikentyneen hemostaasin korjaaminen kestää noin 6 - 8 tuntia. Samaan aikaan annetun K-vitamiinin vaikutukset saavutetaan yleensä kuitenkin 4 - 6 tunnin kuluessa. Näin ollen ihmisen protrombiinikompleksia ei yleensä tarvitse antaa toistuvasti silloin, kun annetaan K-vitamiinia.
Koska mainitut suositukset ovat kokemusperäisiä ja potilaan tilan paraneminen ja vaikutuksen kesto saattavat vaihdella, INR-arvoa on seurattava hoidon aikana.
Verenvuodot ja leikkauksiin liittyvä verenvuotojen ennaltaehkäisy synnynnäisessä, jonkin K-vitamiinista riippuvan hyytymistekijän puutoksessa, kun spesifistä hyytymistekijää ei ole saatavilla:
Tarvittava laskennallinen hoitoannos perustuu kokemusperäiseen havaintoon. Sen mukaan noin 1 IU tekijää VII tai tekijää IX potilaan painokiloa kohti nostaa plasman tekijä VII -aktiivisuutta tai tekijä IX -aktiivisuutta vastaavasti 0,01 IU/ml. Edelleen 1 IU tekijää II tai tekijää X potilaan painokiloa kohti nostaa plasman tekijä II -aktiivisuutta 0,02 IU/ml tai tekijä X -aktiivisuutta 0,017 IU/ml.
Kunkin hyytymistekijän annos ilmaistaan kansainvälisinä yksikköinä (International Unit, IU), jotka perustuvat kyseisen tekijän voimassa olevaan WHO-standardiin. Yksittäisen hyytymistekijän aktiivisuus plasmassa ilmaistaan joko prosentteina (suhteessa normaaliplasmaan) tai kansainvälisinä yksikköinä (suhteessa kyseisen hyytymistekijän kansainväliseen standardiin).
Yksi kansainvälinen yksikkö (IU) hyytymistekijäaktiivisuutta vastaa kyseisen tekijän määrää 1 ml:ssa normaalia ihmisplasmaa.
Esimerkiksi tarvittavan tekijä X:n annoksen laskeminen perustuu kokemusperäiseen havaintoon, jonka mukaan 1 IU tekijää X potilaan painokiloa kohti nostaa plasman tekijä X -aktiivisuutta 0,017 IU/ml. Vaadittava annos määritetään seuraavan kaavan avulla:
Tarvittava yksikkömäärä = potilaan paino (kg) x haluttu tekijä X -pitoisuuden nousu (IU/ml) x 60
jossa 60 (ml/kg) on arvioidun saannon käänteisluku.
Jos yksilöllinen saanto on tiedossa, laskennassa pitää käyttää kyseistä arvoa.
Pediatriset potilaat
Cofact-valmisteen käytön turvallisuutta ja tehoa pediatristen potilaiden hoidossa ei ole varmistettu.
Antotapa
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Cofact annetaan laskimoon. Käyttövalmiin tuotteen suositeltu antonopeus on noin 2 ml/min.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Hoidon tulee tapahtua hyytymishäiriöiden hoitoon erikoistuneen asiantuntijan ohjauksessa.
Niillä potilailla, joilla on hankinnainen K-vitamiinista riippuvien hyytymistekijöiden puutos (esim. K-vitamiiniantagonistihoidon aiheuttama), Cofactia tulee käyttää ainoastaan, jos protrombiinikompleksipitoisuuksien nopea korjaaminen on välttämätöntä. Tällaisia tilanteita ovat mm. suuret verenvuodot ja hätäleikkaukset. Muissa tilanteissa riittää yleensä K-vitamiiniantagonistin annoksen pienentäminen ja/tai K-vitamiinin anto.
K-vitamiiniantagonisteja saavilla potilailla saattaa olla taustalla ylikorostunut hyytymistaipumus, ja ihmisen protrombiinikompleksin anto voi pahentaa tätä.
Jonkin K-vitamiinista riippuvan hyytymistekijän synnynnäisessä puutoksessa tulee käyttää spesifistä hyytymistekijävalmistetta, mikäli sellainen on saatavilla.
Jos allergisia tai anafylaktistyyppisiä reaktioita ilmenee, injektio/infuusio tulee keskeyttää välittömästi. Sokin hoidossa tulee noudattaa vakiintunutta sokin hoitokäytäntöä.
Ihmisen verestä tai plasmasta valmistettujen lääkkeiden välittämien infektioiden estämiseksi käytetään vakiintuneita toimenpiteitä. Näitä ovat luovuttajien valinta, yksittäisten luovutusten ja plasmapoolien tutkiminen tiettyjen infektiomarkkereiden suhteen sekä viruksia tehokkaasti inaktivoivat ja poistavat valmistusvaiheet. Tästä huolimatta taudinaiheuttajien siirtymismahdollisuutta ei voida täysin sulkea pois, kun annetaan ihmisen verestä tai plasmasta valmistettuja lääkkeitä. Tämä koskee myös tuntemattomia tai uusia viruksia ja muita patogeeneja.
Cofactin valmistuksessa käytettyjen toimenpiteiden katsotaan olevan tehokkaita vaipallisiin viruksiin, kuten ihmisen immuunikatovirukseen (HIV), hepatiitti B -virukseen (HBV) ja hepatiitti C -virukseen (HCV) sekä vaipattomaan hepatiitti A -virukseen. Tehokkuus muihin vaipattomiin viruksiin, kuten parvovirus B19, saattaa olla rajallinen. Parvovirustartunta saattaa olla vakava raskaana oleville naisille (sikiön saama tartunta) ja henkilöille, joilla on immuunipuutos tai lyhentynyt punasolujen elinikä (esim. hemolyyttinen anemia).
Asiankuuluvaa rokotussuojaa (hepatiitti A ja B) tulisi harkita potilaille, jotka käyttävät säännöllisesti tai toistuvasti ihmisplasmaperäisiä protrombiinikompleksivalmisteita.
Kun synnynnäistä tai hankinnaista puutosta sairastavia potilaita hoidetaan ihmisen protrombiinikompleksivalmisteella, on olemassa etenkin toistuvia annoksia käytettäessä tromboosin tai disseminoituneen intravaskulaarisen koagulaation riski. Riski saattaa olla suurempi hoidettaessa erillistä tekijä VII -puutosta. Tämä johtuu siitä, että muut K-vitamiinista riippuvat hyytymistekijät, joilla on pidemmät puoliintumisajat, saattavat kumuloitua huomattavasti normaalia korkeampina pitoisuuksina.
Ihmisen protrombiinikompleksia saavia potilaita tulee seurata tarkkaan intravaskulaarisen koagulaation tai tromboosin merkkien ja oireiden varalta. Tromboembolisten komplikaatioiden riskin vuoksi potilaita tulee seurata huolellisesti, jos ihmisen protrombiinikompleksivalmistetta annetaan:
- potilaille, joilla on joskus todettu sepelvaltimotauti
- potilaille, joilla on maksasairaus
- leikkauspotilaille
- vastasyntyneille
- potilaille, jotka kuuluvat tromboembolisten tapahtumien tai disseminoituneen intravaskulaarisen koagulaation suhteen riskiryhmään.
Kaikissa edellä mainituissa tilanteissa tulee punnita hoidosta mahdollisesti saatavat hyödyt ja näiden komplikaatioiden riskit.
Cofactin käytöstä K-vitamiinin puutoksesta johtuvassa perinataaliverenvuodossa vastasyntyneillä ei ole saatavilla tietoja.
Apuaineet
Cofact sisältää enintään 448 mg natriumia per 100 ml, mikä vastaa 22 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille. Potilaiden, joilla on ruokavalion natriumrajoitus, tulee ottaa tämä huomioon.
Pediatriset potilaat
Tiedot ovat riittämättömiä Cofact-valmisteen suosittelemiseksi lapsille ja nuorille.
Yhteisvaikutukset
Ihmisen protrombiinikompleksivalmisteet neutraloivat K-vitamiiniantagonistihoidon vaikutuksen. Yhteisvaikutuksia muiden lääkevalmisteiden kanssa ei tunneta.
Raskaus ja imetys
Ihmisen protrombiinikompleksin turvallisuutta raskauden ja imetyksen aikana ei ole osoitettu.
Eläinkokeet eivät ole soveltuvia, kun arvioidaan valmisteen käytön turvallisuutta raskauden, alkion ja sikiön kehityksen ja synnytyksen aikana sekä synnytyksen jälkeen. Tämän vuoksi ihmisen protrombiinikompleksivalmistetta tulisi käyttää raskauden ja imetyksen aikana ainoastaan, jos se on ehdottoman tarpeellista. Ks. kohdasta Varoitukset ja käyttöön liittyvät varotoimet tietoja parvovirus B19 ‑infektion riskistä raskaana oleville naisille.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia valmisteen vaikutuksesta ajokykyyn tai koneiden käyttökykyyn ei ole tehty.
Haittavaikutukset
Cofact-valmisteen haittavaikutustaulukko
Mainitut haittavaikutukset on raportoitu kliinisissä tutkimuksissa ja Cofactin markkinoille tulon jälkeisen käytön aikana. Jäljempänä olevan taulukon tiedot esitetään MedDRA-elinjärjestelmäluokituksen (SOC ja Preferred Term Level) mukaisesti. Haittavaikutusten esiintymistiheys määritellään seuraavan esitystavan mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Haittavaikutusten esiintyvyys
MedDRA-elinjärjestelmäluokka (SOC) | Haittavaikutus | Esiintyvyys |
Immuunijärjestelmä | Anafylaktinen reaktio, yliherkkyys | Tuntematon |
Hermosto | Aivohaveri, heitehuimaus | Tuntematon |
Sydän | Akuutti sydäninfarkti | Tuntematon |
Verisuonisto
| Tromboemboliset tapahtumat (embolia, syvä laskimotukos), ks. kohta Varoitukset ja käyttöön liittyvät varotoimet | Yleinen |
Hypotensio | Melko harvinainen | |
Hengityselimet, rintakehä ja välikarsina | Keuhkoembolia, hengitysvajaus | Tuntematon |
Ruoansulatuselimistö | Pahoinvointi, oksentelu | Tuntematon |
Iho ja ihonalainen kudos | Liikahikoilu, kutina, urtikaria | Tuntematon |
Yleisoireet ja antopaikassa todettavat haitat | Infuusiokohdan punoitus, infuusiokohdan ärsytys, infuusiokohdan turpoaminen
Huonovointisuus | Tuntematon |
Tutkimukset | Poikkeava maksan toiminta | Tuntematon |
Korvaushoito saattaa johtaa verenkierrossa vasta-aineiden muodostumiseen, jotka estävät yhtä tai useampaa ihmisen protrombiinikompleksin hyytymistekijää. Tämä ilmenee huonona kliinisenä vasteena, esim. jatkuvana verenvuotona.
Virusturvallisuutta koskevat tiedot, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Ihmisen protrombiinikompleksivalmisteiden käyttö suurina annoksina on yhdistetty sydäninfarktiin, disseminoituneeseen intravaskulaariseen koagulaatioon, laskimotromboosiin ja kehkoveritulppaan. Näin ollen yliannostustapauksissa tromboembolisten komplikaatioiden ja disseminoituneen intravaskulaarisen koagulaation riski on kohonnut.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: hemostaatit, hyytymistekijät IX, II, VII ja X yhdistelmävalmisteina, ATC-koodi: B02BD01.
Maksassa K-vitamiinin avulla syntetisoituvia hyytymistekijöitä II, VII, IX ja X kutsutaan yleisesti protrombiinikompleksiksi. Cofact sisältää hyytymistekijöiden lisäksi K-vitamiinista riippuvaisia hyytymisen estäjiä proteiinia C ja proteiinia S.
Tekijä VII on aktiivisen seriiniproteaasi tekijä VIIa:n esiaste, joka käynnistää veren ulkoisen hyytymisjärjestelmän. Kudostekijä-tekijä VIIa -kompleksi aktivoi hyytymistekijä X:n ja IX:n, jolloin muodostuvat tekijät IXa ja Xa. Hyytymisketjun aktivoitumisen jatkuessa protrombiini (tekijä II) aktivoituu ja muuttuu trombiiniksi. Trombiinin vaikutuksesta fibrinogeeni muuttuu fibriiniksi, mikä johtaa hyytymän muodostumiseen. Trombiinin normaali muodostuminen on ensiarvoisen tärkeää myös verihiutaleiden toiminnan kannalta osana primaaria hemostaasia.
Erillinen tekijä VII:n vaikea puutos johtaa vähentyneeseen trombiinin muodostukseen ja heikentyneestä fibriinin muodostuksesta sekä heikentyneestä primaarista hemostaasista johtuvaan vuotoherkkyyteen. Erillinen tekijä IX:n puutos on yksi klassisen hemofilian muoto (hemofilia B). Erilliset tekijä II:n tai tekijä X:n puutokset ovat erittäin harvinaisia, mutta vaikeassa muodossa ne aiheuttavat samankaltaisen vuototaipumuksen kuin klassisessa hemofiliassa.
Myös muut aineosat eli hyytymisen estäjät proteiini C ja proteiini S muodostuvat maksassa. Kofaktori proteiini S voimistaa proteiinin C biologista aktiivisuutta.
Aktivoitu proteiini C estää hyytymisen inaktivoimalla hyytymistekijöitä Va ja VIIIa. Proteiinin C kofaktori proteiini S tukee hyytymisen inaktivaatiota. Proteiinin C puutokseen liittyy lisääntynyt tromboosiriski.
K-vitamiiniantagonistihoidon aikana esiintyy K-vitamiinista riippuvien hyytymistekijöiden hankinnaista puutosta. Jos puutos kehittyy vakavaksi, seuraa vakava vuototaipumus, joka ilmenee vatsakalvontakaisina (retroperitoneaalisina) vuotoina tai aivoverenvuotoina, ei niinkään lihaksen tai nivelen verenvuotoina. Myös vaikea maksan vajaatoiminta johtaa merkittävään K-vitamiinista riippuvien hyytymistekijöiden pitoisuuksien laskuun ja kliiniseen verenvuototaipumukseen, joka on kuitenkin usein kompleksinen tila johtuen samanaikaisesta heikosta intravaskulaarisesta koagulaatiosta, alhaisista verihiutalearvoista, hyytymisinhibiittoreiden puutoksesta ja häiriintyneestä fibrinolyysistä.
Ihmisen protrombiinikompleksin anto nostaa K-vitamiinista riippuvien hyytymistekijöiden pitoisuuksia plasmassa ja saattaa tilapäisesti korjata hyytymisvajausta potilailla, joilla on yhden tai useamman tällaisen tekijän puutos.
Farmakokinetiikka
Cofactin sisältämien neljän hyytymistekijän puoliintumisajoista on annettu kirjallisuudessa seuraavat tiedot:
| Hyytymistekijä | Puoliintumisaika |
| Tekijä II | 40 - 60 tuntia |
| Tekijä VII | 4 - 6 tuntia |
| Tekijä IX | 18 - 25 tuntia |
| Tekijä X | 30 - 60 tuntia |
Prekliiniset tiedot turvallisuudesta
Cofactin vaikutuksia eläimillä ei ole tutkittu lukuun ottamatta yhtä rotilla tehtyä tutkimusta, jossa selvitettiin valmisteen mahdollista hypotensiivista vaikutusta (jota ei tutkimuksessa todettu).
Koe-eläimillä on tehty toksikologisia tutkimuksia TNBP:n (tri-n-butyylifosfaatin) ja Tween 80:n osalta. Cofact sisältää TNBP:tä enintään 0,4 µg/1 IU tekijää IX ja enintään 4 µg Tween 80:tä/1 IU tekijää IX. Käytettäessä Cofactia annossuositusten mukaan potilaan saamat TNBP- ja Tween 80 -määrät jäävät selvästi alle eläinkokeissa todettujen haitallisten määrien.
Farmaseuttiset tiedot
Apuaineet
Injektiokuiva-aine: natriumsitraatti, natriumkloridi, antitrombiini ≤ 0,6 IU/ml.
Liuotin: injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Cofact on yhteensopiva polypropyleenimateriaalin kanssa. Hyytymistekijän adsorptio joidenkin injektio- ja infuusiovälineiden sisäpintaan saattaa aiheuttaa hoidon epäonnistumisen.
Kestoaika
3 vuotta.
Käyttökuntoon saatetun valmisteen kemialliseksi ja fysikaaliseksi käytönaikaiseksi säilyvyydeksi on osoitettu 3 tuntia 15–25 °C:n lämpötilassa. Mikrobiologiselta kannalta valmiste pitää käyttää heti käyttökuntoon saattamisen jälkeen. Jos sitä ei käytetä heti, käytönaikaiset säilytysajat ja ‑olosuhteet ennen käyttöä ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2°C - 8°C). Ei saa jäätyä.
Pidä injektiopullo ulkokotelossa. Herkkä valolle.
Valmistetta voidaan säilyttää ennen käyttöä kestoajan puitteissa enintään 25 °C:ssa enintään 6 kuukauden ajan. Jos valmistetta ei käytetä tänä aikana, se on hävitettävä. Kun valmiste on otettu pois jääkaappisäilytyksestä, sitä ei saa enää palauttaa sinne takaisin. Päivämäärä, jolloin valmiste on otettu huoneenlämpöön, pitää merkitä pakkaukseen.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
COFACT injektiokuiva-aine ja liuotin, liuosta varten
250 IU (L:ei) 250 IU + 10 ml (237,27 €)
500 IU (L:ei) 500 IU + 20 ml (458,57 €)
PF-selosteen tieto
250 IU kuiva-ainetta injektiopullossa (lasia, tyyppi I), jossa tulppa (bromobutyyliä) + 10 ml liuotinta injektiopullossa (lasia, tyyppi I), jossa tulppa (bromobutyyliä) + siirtokanyyli - yhden kappaleen pakkaus.
500 IU kuiva-ainetta injektiopullossa (lasia, tyyppi II), jossa tulppa (bromobutyyliä) + 20 ml liuotinta injektiopullossa (lasia, tyyppi I), jossa tulppa (FluroTec-pinnoitettua klorobutyyliä) + siirtokanyyli - yhden kappaleen pakkaus.
Valmisteen kuvaus:
Kuiva-aine on väriltään sinertävä. Liuotin on kirkas, väritön liuos, jossa ei ole näkyviä hiukkasia.
Käyttö- ja käsittelyohjeet
Liuottaminen
Kuivattu proteiinifraktio tulee liuottaa annettuun tilavuuteen injektionesteisiin käytettävää vettä. Jos Cofact-injektiopullo ja liuotinpullo on säilytetty 2 °C - 8 °C:ssa, ne on hyvä saattaa huoneenlämpöisiksi (15 °C - 25 °C) ennen liuottamista.
Siirtokanyylin käyttö
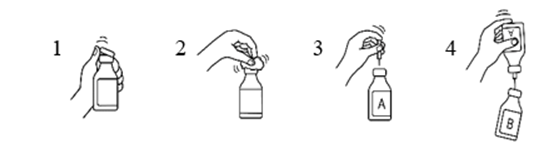
- Poista vesipullon ja kuiva-ainepullon muovisuojukset.
- Desinfioi pullojen kumitulpat 70‑prosenttiseen alkoholiin kostutetulla sideharsolapulla.
- Poista siirtokanyylin toisen pään suojus. Työnnä neula vesipulloon (A).
- Poista sitten siirtokanyylin toisen pään suojus, käännä siirtokanyylissa kiinni oleva pullo ylösalaisin ja työnnä välittömästi vapaana oleva neula kuiva-ainepulloon (B).
Kuiva-ainepullossa oleva alipaine imee veden pulloon. Suositus: veden valuessa kuiva-ainepulloa tulee pitää kallistettuna, jolloin vesi valuu pullon seinää pitkin. Tämä nopeuttaa tuotteen liukenemista. Heti kun koko vesimäärä on valunut pulloon, tyhjä pullo ja siirtokanyyli tulee poistaa samanaikaisesti.
Liukenemistapahtumaa voidaan nopeuttaa pyörittämällä pulloa varovasti ja, mikäli tarpeen, lämmittämällä se 30 °C:een. Pulloa ei koskaan saa ravistaa eikä lämmittää yli 37 °C:seen. Jos pulloa lämmitetään vesihauteessa, tulee varmistaa, että vesi ei pääse kosketuksiin suojuksen ja/tai kumitulpan kanssa.
Pääsääntöisesti kuiva-aineen tulisi olla täysin liuennut 10 minuutin kuluessa muodostaen sinisen liuoksen; sininen väri johtuu plasmaproteiini keruloplasmiinista.
Liuoksen tulee olla kirkas tai hieman opalisoiva. Älä käytä sameaa tai sakkaista liuosta. Käyttövalmiiksi liuotettu tuote tulee tarkastaa visuaalisesti hiukkasten ja värimuutosten osalta ennen antamista.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
COFACT injektiokuiva-aine ja liuotin, liuosta varten
250 IU 250 IU + 10 ml
500 IU 500 IU + 20 ml
- Ei korvausta.
ATC-koodi
B02BD01
Valmisteyhteenvedon muuttamispäivämäärä
22.10.2022
Yhteystiedot
 PROTHYA BIOSOLUTIONS FINLAND OY
PROTHYA BIOSOLUTIONS FINLAND OY Lars Sonckin kaari 14
02600 Espoo
09 6120 9122
www.prothya.com
info@prothya.fi