
CLUVOT injektio-/infuusiokuiva-aine ja liuotin, liuosta varten 250 IU, 1250 IU
Vaikuttavat aineet ja niiden määrät
Vaikuttava aine: Cluvot on puhdistettu konsentraatti ihmisen plasman hyytymistekijää XIII (FXIII). Se on valkoinen kuiva-aine.
Jokainen injektiopullo sisältää 250 tai 1250 IU ihmisen veren hyytymistekijää XIII. Kun valmiste on saatettu käyttökuntoon sekoittamalla 250 IU:n injektiopullo 4 ml:aan tai 1250 IU:n injektiopullo 20 ml:aan injektionesteisiin käytettävää vettä, liuos sisältää noin 62,5 IU/ml (250 IU / 4 ml ja 1250 IU / 20 ml) ihmisen plasman hyytymistekijää XIII.
Cluvotin spesifinen aktiivisuus on noin 6–10 IU/mg proteiinia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektio-/infuusiokuiva-aine ja liuotin, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Cluvot on tarkoitettu aikuisille ja pediatrisille potilaille
- hyytymistekijä XIII:n (FXIII:n) synnynnäisen puutoksen estohoitoon
- leikkauksen aikaisen verenvuodon hoitoon synnynnäisen FXIII:n puutoksen yhteydessä.
Annostus ja antotapa
Annostus
1 ml vastaa noin 62,5 IU:ta, ja 100 IU vastaa 1,6 ml:aa.
Tärkeää:
Annettava määrä ja antotiheys on aina sovitettava kullekin potilaalle yksilöllisen kliinisen tehon mukaan.
Annostus
Annostus on yksilöllinen potilaan painon, laboratorioarvojen ja kliinisen tilan mukaan.
Annostus rutiiniluonteisessa estohoidossa
Aloitusannos
- 40 kansainvälistä yksikköä (IU) per painokilo (IU/kg)
- Injektionopeus ei saa ylittää 4 ml:aa minuutissa.
Seuraavat annokset
- Annos määritellään viimeisimmän pienimmän FXIII-aktiivisuuden perusteella, ja antotiheys on kerran 28 vuorokaudessa (4 viikossa), jotta pienimpänä FXIII-aktiivisuutena säilyy noin 5−20 %.
- Annosmuutossuosituksen ± 5 IU/kg pitää perustua taulukossa 1 esitettyyn pienimpään FXIII-aktiivisuuteen ja potilaan kliiniseen tilaan.
- Annosmuutokset on toteutettava FXIII-pitoisuuden spesifisen herkän määrityksen perusteella. Taulukossa 1 esitetään esimerkki annosmuutoksesta standardin Berichrom®-aktiivisuusmäärityksen perusteella.
Taulukko 1. Annosmuutos Berichrom®-aktiivisuusmäärityksen perusteella
| Hyytymistekijän XIII pienin aktiivisuus (%) | Annosmuutos |
| Pienin pitoisuusmääritystulos kerran < 5 % | Suurenna 5 yksikköä/kg |
| Pienin pitoisuusmääritystulos 5–20 % | Ei muutosta |
| Pienin pitoisuusmääritystulos kaksi kertaa > 20 % | Pienennä 5 yksikköä/kg |
| Pienin pitoisuusmääritystulos kerran > 25 % | Pienennä 5 yksikköä/kg |
Yksikköinä ilmoitettu teho on määritetty Berichrom®-aktiivisuusmäärityksellä, johon viitataan voimassa olevassa veren hyytymistekijän XIII kansainvälisessä standardissa (International Standard for Blood Coagulation Factor XIII, Plasma). Yksikkö vastaa siten kansainvälistä yksikköä (International Unit).
Estohoito ennen leikkausta
Jos potilaalle suunnitellaan leikkausta ja hän on saanut viimeisen tavanomaisen estohoitoannoksen:
- 21–28 vuorokauden kuluttua: potilaalle annetaan juuri ennen leikkausta koko estohoitoannos, ja seuraava estohoitoannos annetaan 28 vuorokauden kuluttua.
- 8–21 vuorokauden kuluttua: ennen leikkausta voidaan antaa lisäannos (koko annos tai osa-annos). Annos pitää määritellä potilaan FXIII-aktiivisuuden ja kliinisen tilan mukaan ja sovitettava Cluvot-valmisteen puoliintumisajan mukaan.
- 7 vuorokauden kuluessa viimeisimmästä annoksesta: lisäannosta ei välttämättä tarvita.
Annosmuutokset saattavat poiketa näistä suosituksista ja niiden pitää olla yksilöllisiä FXIII-aktiivisuuden ja potilaan kliinisen tilan mukaan. Kaikkia potilaita on seurattava tarkoin leikkauksen aikana ja sen jälkeen.
Näin ollen FXIII:n aktiivisuuden lisääntymistä suositellaan seuraamaan FXIII:n määritysten avulla. Jos potilaalle tehdään suuri leikkaus ja verenvuodot ovat oletettavasti runsaita, tavoitteena on saavuttaa lähes normaaliarvot (terveet yksilöt: 70–140 %).
Pediatriset potilaat
Lapsille ja nuorille käytettävä annos ja antotapa perustuvat painoon, joten heihin voi yleensä soveltaa samoja ohjeita kuin aikuisiin. Annos ja/tai antotiheys on kussakin tapauksessa määritettävä kliinisen tehon ja FXIII:n aktiivisuuden perusteella. (Ks. myös kohdat Farmakodynamiikka ja Farmakokinetiikka).
Iäkkäät potilaat
Iäkkäiden potilaiden (> 65-vuotiaiden) annostusta ja antotapaa ei ole dokumentoitu kliinisissä tutkimuksissa.
Antotapa
Käyttökuntoon saatettu liuos on kirkas tai hieman opaalinhohtoinen. Valmiste on lämmitettävä ennen antoa huoneen- tai kehonlämpöiseksi. Anna hitaana injektiona tai infuusiona laskimoon erillisellä injektio‑/infuusiolaitteella (mukana pakkauksessa) nopeudella, joka tuntuu potilaasta miellyttävältä. Injektio- tai infuusionopeus ei saa ylittää noin 4 ml:aa minuutissa.
Tarkkaile potilasta välittömien reaktioiden havaitsemiseksi. Jos Cluvot-valmisteen antoon mahdollisesti liittyvä reaktio ilmaantuu, infuusionopeutta on hidastettava tai infuusion anto on lopetettava sen mukaan, miten potilaan kliininen tila edellyttää.
Ks. kohdasta Käyttö- ja käsittelyohjeet; ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jos potilaan tiedetään olevan allerginen valmisteelle (oireita ovat mm. yleistynyt urtikaria, ihottuma, verenpaineen lasku, hengenahdistus), estohoitona voidaan antaa antihistamiinia ja kortikosteroideja.
Allergiatyyppiset yliherkkyysreaktiot ovat mahdollisia Cluvotin käytön yhteydessä. Jos yliherkkyysreaktioiden merkkejä (kuten urtikaria, yleistynyt urtikaria, puristava tunne rinnassa, hengityksen vinkuminen, hypotensio ja anafylaksia) ilmenee, valmisteen käyttö on lopetettava välittömästi. Jos potilas joutuu sokkiin, on noudatettava voimassa olevia sokin hoitotoimenpiteitä.
Jos potilaalla on äskettäin ollut tromboosi, hoidossa pitää olla varovainen, koska hyytymistekijä XIII vaikuttaa fibriiniä stabiloivasti.
Immunogeenisuus
Cluvot-hoitoa saaneilla potilailla on todettu neutraloivien vasta-aineiden kehittymistä hyytymistekijälle XIII. Potilaita on siksi seurattava neutraloivien vasta-aineiden kehittymisen havaitsemiseksi. Neutraloivat vasta-aineet voivat ilmetä riittämättömänä hoitovasteena. Jos on oletettavissa, ettei FXIII-aktiivisuutta saavuteta plasmassa tai jos estohoidon aikana esiintyy verenvuotoja, FXIII:aa neutraloivat vasta-ainepitoisuudet on mitattava.
Huomautus potilaille, joilla on ruokavalion natriumrajoitus
Cluvot sisältää natriumia 124,4–195,4 mg (5,41–8,50 mmol) per annos (40 IU/keskimäärin 70 kg:n painoinen potilas), jos annetaan suositeltu annos (2800 IU = 44,8 ml). Potilaiden, joilla on ruokavalion natriumrajoitus, tulee ottaa tämä huomioon.
Virusturvallisuus
Vakiintuneita toimenpiteitä ihmisen verestä tai plasmasta valmistetuista lääkevalmisteista aiheutuvien infektioiden estämiseksi ovat verenluovuttajien valinta, erityisten infektiomerkkiaineiden seulominen luovutetusta verestä ja plasmapooleista sekä valmistuksenaikaiset tehokkaat toimenpiteet virusten inaktivoimiseksi/poistamiseksi. Tästä huolimatta taudinaiheuttajien siirtymisen mahdollisuutta ei voida täysin sulkea pois, kun käytetään ihmisen verestä tai plasmasta valmistettuja lääkevalmisteita. Tämä koskee myös tuntemattomia tai uusia viruksia ja muita taudinaiheuttajia.
Käytössä olevien toimenpiteiden katsotaan tehoavan vaipallisiin viruksiin, kuten ihmisen immuunikatovirukseen (HIV)-, hepatiitti B (HBV)- ja hepatiitti C (HCV) -viruksiin sekä vaipattomiin viruksiin kuten hepatiitti A -virus ja parvovirus B19.
Valmisteen nimi ja eränumero kehotetaan kirjaamaan Cluvot-valmisteen jokaisen antokerran yhteydessä, jotta potilas ja valmiste-erä voidaan yhdistää.
Ihmisen plasmasta valmistettuja valmisteita säännöllisesti/toistuvasti saavien potilaiden on tavallisesti harkittava asianmukaisten rokotusten (hepatiitti A:ta ja B:tä vastaan) ottamista.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Raskaus
Cluvot-valmisteen kliinisestä käytöstä raskauden aikana on vähän tietoa, mutta tiedot eivät viittaa raskauden kulkuun tai peri- ja postnataaliseen kehitykseen kohdistuviin haitallisiin vaikutuksiin.
Cluvot-valmisteen käyttöä voidaan harkita raskauden aikana, jos tarpeen.
Imetys
Ei ole tietoa FXIII:n erittymisestä ihmisen rintamaitoon. Suuren molekyylikoon perusteella erittymisen ihmisen rintamaitoon on kuitenkin epätodennäköistä, ja valmisteen proteiiniluonteen vuoksi ei ole todennäköistä, että molekyylejä siirtyisi muuttumattomina imeväiseen. Tästä syystä valmistetta voi käyttää rintaruokinnan aikana.
Hedelmällisyys
Valmisteen vaikutuksista hedelmällisyyteen ei ole tietoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia valmisteen vaikutuksesta ajokykyyn tai koneiden käyttökykyyn ei ole tehty.
Haittavaikutukset
Seuraavat haittavaikutukset perustuvat valmisteen myyntiluvan saamisen jälkeiseen käyttökokemukseen.
Haittavaikutustaulukko
Haittavaikutukset esitetään oheisessa taulukossa MedDRA-elinjärjestelmäluokituksen mukaisesti. Esiintymistiheydet on määritetty seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, <1/10), melko harvinaiset (≥1/1 000, <1/100), harvinaiset (≥1/10 000, <1/1 000), hyvin harvinaiset (<1/10 000).
| MedDRA elinluokka | Haittavaikutus | Esiintymistiheys |
| Immuunijärjestelmä | Allergis-anafylaktoidiset reaktiot (kuten yleistynyt urtikaria, ihottuma, verenpaineen lasku, hengenahdistus) | Harvinainen |
| Vasta-aineiden kehittyminen FXIII:lle | Hyvin harvinainen | |
| Yleisoireet ja antopaikassa todettavat haitat | Lämmön nousu | Harvinainen |
Jos allergis-anafylaktisia reaktioita ilmaantuu, Cluvot-valmisteen anto on keskeytettävä heti ja aloitettava asianmukainen hoito. Sokin voimassa olevat hoitosuositukset on huomioitava.
Pediatriset potilaat
Pediatristen potilaiden ja aikuispotilaiden turvallisuusprofiilissa ei ollut eroa kliinisissä tutkimuksissa.
Turvallisuus tarttuvien taudinaiheuttajien suhteen, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksia ei ole raportoitu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: hemostaatit.
ATC-koodi: B02BD07.
Hyytymistekijä XIII liittää entsymaattisen vaikutuksensa (transamidaasiaktiivisuuden) avulla aminoryhmän lysiinin glutamiiniin, mikä johtaa fibriinimolekyylien ristisidosten muodostumiseen. Fibriinin ristisidos ja stabilisaatio edistävät fibroblastien penetraatiota ja edistävät siten haavan paranemista.
Pediatriset potilaat
Kliinisissä tutkimuksissa, joissa Cluvotia annettiin estohoitona 28 vuorokauden välein alle 18-vuotiaille tutkimushenkilöille, joilla oli synnynnäinen FXIII:n puutos, pienin FXIII-aktiivisuus pysyi noin 5–20 %:ssa.
Farmakokinetiikka
Jakautuminen
Valmiste annetaan laskimoon, joten sen hyötyosuus on heti käytettävissä. Pitoisuus plasmassa vastaa tällöin annettua annosta.
Eliminaatio
Cluvot-valmisteen biologiseksi puoliintumisajaksi potilailla, joilla on synnynnäinen FXIII:n puutos, määritettiin 6,6 ± 2,29 vuorokautta (keskiarvo± SD). Cluvot metaboloituu samalla tavoin kuin endogeeninen FXIII.
Seuraavassa taulukossa on yleiskuvaus farmakokineettisistä muuttujista (aikuiset / 18-vuotiaat ja sitä vanhemmat):
| Muuttujat | Mediaani (min-max) |
| AUC ss, 0-inf (yksikköä•h/ml) | 182,9 (133,5–300,2) |
| Css, max (yksikköä /ml)* | 0,9 (0,6–1,2) |
| Css, min (yksikköä /ml)* | 0,07 (0,0–0,16) |
| Tmax (h) | 1,2 (0,7–4,2) |
| Puoliintumisaika [vrk] | 7,8 (3,1–11,02) |
| CL [ml/h/kg] | 0,22 (0,13–0,30) |
| Vss [ml/kg] | 49,4 (31,65–62,91) |
| MRT [vrk] | 11,7 (5,7–17,02) |
AUC ss, (0-inf) = Vakaan tilan plasmapitoisuuskäyrän
alle jäävä pinta-ala nollahetkestä äärettömään
* 100 %:n aktiivisuus vastaa 1 yksikköä/ml
Css, max: Vakaan tilan huippupitoisuus
Css, min: Vakaan tilan pienin pitoisuus
Tmax: Aika huippupitoisuuden saavuttamiseen
CL: Puhdistuma
Vss: Vakaan tilaan jakaantumistilavuus
MRT = Keskimääräinen viipymä
Pediatriset potilaat
Kliinisissä tutkimuksissa FXIII -konsentraattia annettiin 188 yksilöidylle tutkittavalle, joista 117 oli tutkimukseen liittyessään alle 18-vuotiaita (1 kk –<2 vuotta, n= 17; 2–<12 vuotta, n=62; 12–< 16 vuotta, n= 30; 17–18 vuotta, n=8). Farmakokineettisessä tutkimuksessa (PK 2002) 14 tutkittavasta 5 oli iältään 2–< 18-vuotiaita (2–11 vuotta, n= 3; 12–16 vuotta, n=2; 17–18 vuotta, n=0). Alle 16-vuotiailla puoliintumisaika oli lyhyempi ja puhdistuma nopeampi (puoliintumisaika: 5,7±1,00 vrk; puhdistuma: 0,291±0,12 ml/h/kg) kuin aikuisilla (puoliintumisaika: 7,1 ± 2,74 vrk, puhdistuma: 0,22 ± 0,07 ml/h/kg).
Lapsilla valmisteen puoliintumisaika on lyhyempi ja puhdistuma nopeampi kuin aikuisilla. Koska annostus kaikissa ikäryhmissä määritetään kuitenkin yksilöllisesti potilaan painon ja pienimmän FXIII:n aktiivisuuden mukaan, erityisiä ikään perustuvia annostusohjeita ei tarvita.
Prekliiniset tiedot turvallisuudesta
Cluvot-valmisteen sisältämät proteiinit ovat peräisin ihmisen plasmasta ja toimivat samalla tavoin kuin ihmisen plasman proteiinit.
Cluvot-valmisteen kerta-annoksen ja toistuvan altistuksen aiheuttamaa toksisuutta selvittäneissä eläinkokeissa ei havaittu toksisuutta.
Lisääntymistoksisuutta ja sikiön kehitystä selvittäviä tutkimuksia ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine: Ihmisen albumiini, glukoosimonohydraatti, natriumkloridi, natriumhydroksidi (pH:n säätöön).
Liuotin: Injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Cluvot-valmistetta ei saa sekoittaa muiden lääkevalmisteiden, laimentimien ja liuottimien kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet, joten se on annettava eri infuusiolaitteen kautta.
Kestoaika
3 vuotta.
Älä käytä pakkauksessa mainitun viimeisen käyttöpäivämäärän jälkeen.
Valmisteen on osoitettu säilyvän kemiallisesti ja fysikaalisesti stabiilina 24 tunnin ajan ≤ 25 °C:ssa. Mikrobiologiselta kannalta katsoen valmiste on käytettävä välittömästi. Jos sitä ei anneta välittömästi, säilytysaika voi olla enintään neljä tuntia huoneenlämmössä. Älä säilytä käyttökuntoon saatettua liuosta kylmässä. Ei saa jäätyä.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
CLUVOT injektio-/infuusiokuiva-aine ja liuotin, liuosta varten
250 IU (L:ei) 1 kpl (250 IU + 4 ml, siirtolaite Mix2Vial) (528,93 €)
1250 IU (L:ei) 1 kpl (1250 IU + 20 ml, siirtolaite Mix2Vial) (2412,01 €)
PF-selosteen tieto
Injektiopullot:
250 IU
Kuiva-aine: väritön, lasinen injektiopullo, joka on suljettu kumitulpalla (bromobutyylikumia), alumiinikorkilla ja muovikiekolla.
Liuotin (injektionesteisiin käytettävä vesi): väritön, lasinen injektiopullo.
1250 IU
Kuiva-aine: väritön, lasinen injektiopullo, joka on suljettu kumitulpalla (bromobutyylikumia), alumiinikorkilla ja muovikiekolla.
Liuotin (injektionesteisiin käytettävä vesi): väritön, lasinen injektiopullo.
Pakkaukset:
250 IU:n pakkaus
1 injektiopullo kuiva-ainetta
1 injektiopullo (4 ml) injektionesteisiin käytettävää vettä
1 suodattimella varustettu siirtolaite 20/20 (Mix2Vial)
Annostelutarvikkeet (sisälaatikko):
1 kertakäyttöinen 5 ml:n ruisku
1 laskimopunktiosetti
2 desinfektiopyyhettä
1 ei-steriili laastari
1250 IU:n pakkaus
1 injektiopullo kuiva-ainetta
1 injektiopullo (20 ml) injektionesteisiin käytettävää vettä
1 suodattimella varustettu siirtolaite 20/20 (Mix2Vial)
Annostelutarvikkeet (sisälaatikko):
1 kertakäyttöinen 20 ml:n ruisku
1 laskimopunktiosetti
2 desinfektiopyyhettä
1 ei-steriili laastari
Valmisteen kuvaus:
Valkoinen jauhe ja kirkas väritön liuotin.
Käyttö- ja käsittelyohjeet
Yleiset ohjeet
Liuoksen on oltava kirkasta tai hieman opaalinhohtoista. Kun käyttökuntoon saatettu valmiste on suodatettu/vedetty ruiskuun (ks. jäljempänä), se on tarkistettava ennen antoa silmämääräisesti, ettei liuoksessa ole hiukkasia tai värimuutoksia. Älä käytä liuosta, jos se on sameaa tai siinä on hiutaleita tai hiukkasia.
Valmisteen käyttökuntoon saattaminen ja vetäminen ruiskuun on tehtävä aseptisissa olosuhteissa.
Käyttökuntoon saattaminen
Anna liuottimen lämmetä huoneenlämpöiseksi. Varmista, että kuiva-aineen ja liuottimen sisältävien injektiopullojen irti napsautettavat flip-off-sulkimet on poistettu ja tulpat on käsitelty aseptisella liuoksella. Tulpan on sen jälkeen annettava kuivua ennen Mix2Vial-pakkauksen avaamista.
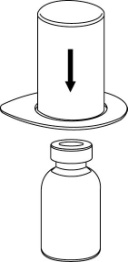
| 1. Avaa Mix2Vial-pakkaus vetämällä suojakansi pois. Älä ota Mix2Vial-laitetta pois pakkauksesta! |
| 2. Aseta liuottimen sisältävä injektiopullo tasaiselle, puhtaalle alustalle ja ota injektiopullosta tukeva ote. Ota Mix2Vial sekä pakkaus ja paina sinisen sovittimen piikki suoraan liuotinpullon tulpan läpi. |
| 3. Poista pakkaus varovasti Mix2Vial-laitteesta siten, että pidät pakkauksen reunasta kiinni ja vedät kohtisuoraan ylöspäin. Varmista, että vedät pois vain pakkauksen etkä Mix2Vial-laitetta. |
| 4. Aseta injektiopullo tasaiselle ja tukevalle alustalle. Käännä liuotinpullo ja siihen kiinnitetty Mix2Vial ylösalaisin, ja paina läpinäkyvän sovittimen piikki suoraan kuiva-aineinjektiopullon tulpan läpi. Liuotin siirtyy automaattisesti kuiva-aineen sisältävään injektiopulloon. |
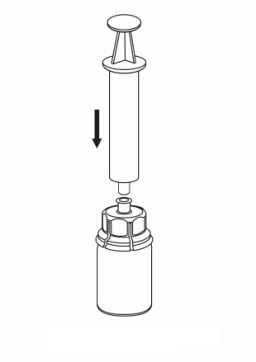
| 5. Ota toisella kädellä kiinni Mix2Vial-laitteen kuiva-aineen sisältävän injektiopullon puolelta ja toisella kädellä liuottimen sisältävän injektiopullon puolelta ja kierrä laite varovasti kahteen osaan. Hävitä liuotinpullo ja siihen kiinnitetty sininen Mix2Vial-sovitin. |
| 6. Pyörittele kuiva-aineinjektiopulloa ja siihen kiinnitettyä läpinäkyvää sovitinta, kunnes kuiva-aine on liuennut täysin. Ei saa ravistaa. |
| 7. Vedä tyhjään, steriiliin ruiskuun ilmaa. Kun kuiva-aineen sisältävä injektiopullo on oikeinpäin, kiinnitä ruisku Mix2Vial-sovittimen Luer Lock ‑liittimeen. Ruiskuta ilma kuiva-aineen sisältävään injektiopulloon. |
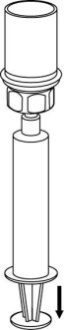
Valmisteen vetäminen ruiskuun ja anto
| 8. Kun ruiskun mäntä on alas painettuna, käännä laite ja injektiopullo ylösalaisin ja vedä liuos ruiskuun vetämällä mäntää hitaasti ulospäin. |
| 9. Kun liuos on nyt siirretty ruiskuun, ota tukeva ote ruiskun kammiosta (pitäen ruiskun mäntää samalla alaspäin) ja irrota ruiskusta läpinäkyvä Mix2Vial-sovitin.
|
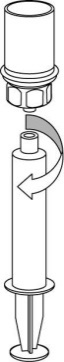
Veren pääsyä valmisteen sisältävään ruiskuun on vältettävä, koska tällöin on riski, että veri hyytyy ruiskuun ja potilas saattaa saada injektion mukana fibriinihyytymän.
Käyttökuntoon saatettu liuos on annettava hitaana injektiona laskimoon erillisellä injektio-/infuusiolaitteella (mukana pakkauksessa). Antonopeus ei saa ylittää 4 ml:aa minuutissa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
CLUVOT injektio-/infuusiokuiva-aine ja liuotin, liuosta varten
250 IU 1 kpl
1250 IU 1 kpl
- Ylempi erityiskorvaus (100 %). Krooniset hyytymishäiriöt (126).
- Peruskorvaus (40 %).
ATC-koodi
B02BD07
Valmisteyhteenvedon muuttamispäivämäärä
24.09.2021
Yhteystiedot
 CSL BEHRING AB
CSL BEHRING AB Box 712
182 17 Danderyd
Ruotsi
+46 (0) 8 544 966 70
www.cslbehring.fi
info@cslbehring.se