
BENLYSTA infuusiokuiva-aine konsentraattiliuosta varten 120 mg, 400 mg
Huomioitavaa
▼ Tähän lääkkeeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti uutta turvallisuutta koskevaa tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Benlysta 120 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo sisältää 120 mg belimumabia. Valmistamisen jälkeen liuos sisältää 80 mg belimumabia millilitrassa.
Benlysta 400 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo sisältää 400 mg belimumabia. Valmistamisen jälkeen liuos sisältää 80 mg belimumabia millilitrassa.
Belimumabi on humaani IgG1λ monoklonaalinen vasta-aine, joka valmistetaan nisäkässolulinjassa (NS0) käyttäen rekombinantti-DNA-teknologiaa.
Apuaine, jonka vaikutus tunnetaan
Benlysta 120 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo sisältää 0,6 mg polysorbaatti 80:tä.
Benlysta 400 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo sisältää 2,0 mg polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos.
Kliiniset tiedot
Käyttöaiheet
Benlysta on tarkoitettu lisälääkkeeksi vähintään 5-vuotiaille potilaille, joilla on hyvin aktiivinen autovasta-ainepositiivinen systeeminen lupus erytematosus (SLE) (esim. positiivinen anti-dsDNA ja alhainen komplementti) huolimatta tavanomaisesta hoidosta (ks. kohta Farmakodynamiikka).
Benlysta on tarkoitettu aktiivisen SLE-nefriitin hoitoon aikuispotilaille, yhdessä immunosuppressiivisten taustahoitojen kanssa (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Ehto
Hoidon aloittavan ja sitä valvovan lääkärin tulee olla perehtynyt systeemisen lupus erythematosuksen (SLE) diagnosointiin ja hoitoon.
Annostus ja antotapa
SLE:n diagnoosiin ja hoitoon perehtyneen lääkärin pitää aloittaa ja valvoa Benlysta-hoito. Benlysta-infuusion antajan on oltava infuusiohoidon antamiseen koulutettu terveydenhuollon ammattilainen.
Benlystan antaminen voi aiheuttaa vaikeita tai hengenvaarallisia yliherkkyysreaktioita ja infuusioreaktioita. Potilaiden on raportoitu saaneen akuutin yliherkkyysreaktion oireita useita tunteja infuusion antamisen jälkeen. Oireiden asianmukaisen ensihoidon jälkeen on myös havaittu kliinisesti merkitsevien reaktioiden uusimista (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Sen vuoksi Benlysta on annettava olosuhteissa, joissa tällaisten reaktioiden hoitoon tarvittavat välineet ovat välittömästi saatavilla. Potilaiden on suositeltavaa pysyä kliinisessä valvonnassa pidemmän aikaa (joitakin tunteja) ainakin kahden ensimmäisen infuusion jälkeen mahdollisen viivästyneen yliherkkyysreaktion varalta.
Benlysta-hoitoa saaville potilaille on kerrottava vaikean tai hengenvaarallisen yliherkkyysreaktion mahdollisuudesta sekä siitä, että yliherkkyys voi alkaa viivästyneesti tai oireet voivat uusia. Potilaalle on annettava pakkausseloste jokaisella Benlysta-antokerralla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Ennen Benlysta-infuusiota voidaan antaa esilääkityksenä antihistamiinia, joko antipyreetin kanssa tai ilman (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilailla, joilla on SLE tai aktiivinen SLE-nefriitti, suositeltu Benlysta-annos on 10 mg/kg päivinä 0, 14 ja 28 ja tämän jälkeen neljän viikon välein. Potilaan tilaa tulee arvioida jatkuvasti.
Benlysta-hoidon lopettamista on harkittava SLE-potilailla, jos tautitilassa ei ole tapahtunut paranemista kuuden kuukauden hoidon jälkeen.
Benlystaa on käytettävä potilaille, joilla on aktiivinen SLE-nefriitti, yhdessä kortikosteroidien ja mykofenolaatin tai syklofosfamidin kanssa induktioon tai mykofenolaatin tai atsatiopriinin kanssa ylläpitohoitoon.
Siirtyminen laskimoon annettavasta hoidosta ihonalaiseen antoon
SLE
Jos SLE-potilas siirtyy laskimoon annettavasta Benlysta-hoidosta ihonalaiseen antoon, ensimmäinen ihonalainen injektio on annettava 1–4 viikkoa viimeisen laskimoannoksen jälkeen (ks. kohta Farmakokinetiikka).
SLE‑nefriitti
Jos SLE-nefriittipotilas siirtyy laskimoon annettavasta Benlysta-hoidosta ihonalaiseen antoon, ensimmäinen ihonalainen injektio (200 mg annos) on suositeltavaa antaa 1–2 viikkoa viimeisen laskimoannoksen jälkeen. Siirtyminen voidaan toteuttaa milloin tahansa sen jälkeen, kun potilas on saanut ensimmäiset 2 laskimoannosta (ks. kohta Farmakokinetiikka).
Erityisryhmät
Iäkkäät
Tutkimustietoa Benlystan käytöstä ≥ 65-vuotiailla potilailla on rajallisesti (ks. kohta Farmakodynamiikka.). Benlystaa on käytettävä varoen iäkkäillä potilailla. Annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Belimumabia on tutkittu vain pienellä määrällä munuaisten vajaatoimintaa sairastavia SLE-potilaita. Olemassa olevan tiedon perusteella annosta ei tarvitse muuttaa potilailla, joilla on lievä, kohtalainen tai vaikea munuaisten vajaatoiminta. Tietoa ei ole vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden hoidosta tällä lääkkellä, joten varovaisuutta on noudatettava (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Benlystaa ei ole tutkittu erikseen potilailla, joilla on maksan vajaatoiminta. Maksan vajaatoiminta potilailla annosta ei todennäköisesti tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
SLE
Suositeltu Benlysta-annostus vähintään 5-vuotiaille lapsille on 10 mg/kg päivinä 0, 14 ja 28 ja sen jälkeen 4 viikon välein.
Laskimoon annettavan Benlystan turvallisuutta ja tehoa alle 5 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
SLE‑nefriitti
Laskimoon annettavan Benlystan turvallisuutta ja tehoa ei ole varmistettu alle 18 vuoden ikäisten lasten ja nuorten hoidossa. Tietoja ei ole saatavilla.
Antotapa
Benlysta annetaan infuusiona laskimoon ja se on liuotettava ja laimennettava ennen antamista. Ks. kohdasta Käyttö- ja käsittelyohjeet miten Benlysta liuotetaan, laimennetaan ja säilytetään ennen sen antoa.
Benlysta on annettava tunnin kestävänä infuusiona.
Benlystaa ei saa antaa laskimonsisäisenä boluksena.
Infuusionopeutta voidaan hidastaa tai infuusio keskeyttää, jos potilalle kehittyy infuusiosta reaktio. Infuusio on lopetettava heti, jos potilas saa haittavaikutuksen, joka voi olla hengenvaarallinen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset)
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Benlystaa ei ole tutkittu seuraavilla potilasryhmillä aikuisissa ja lapsissa, eikä sitä suositella käytettäväksi potilaille, joilla on:
- vaikea aktiivinen keskushermostolupus (ks. kohta Farmakodynamiikka)
- HIV
- hepatiitti B tai C, joko nyt tai aikaisemmin sairastettu
- hypogammaglobulinemia (IgG < 400 mg/dl) tai IgA puutos (IgA < 10 mg/dl)
- aikaisemmin tehty suuri elinsiirto tai hematopoeettinen kantasolu-/luuydinsiirto tai munuaissiirto
Samanaikainen käyttö B-soluihin kohdistetun hoidon kanssa
Saatavilla oleva tieto ei tue Benlystan käyttöä yhdistelmänä rituksimabin kanssa SLE-potilailla (ks. kohta Farmakodynamiikka). On noudatettava varovaisuutta, jos Benlystaa annetaan samanaikaisesti muiden B-soluihin kohdistettujen hoitojen kanssa.
Infuusioreaktiot ja yliherkkyys
Benlystan antaminen voi johtaa yliherkkyysreaktioihin ja infuusioreaktioihin, jotka voivat olla vaikeita ja johtaa kuolemaan. Vaikean reaktion sattuessa Benlystan antaminen on keskeytettävä ja annettava asianmukaista lääkehoitoa (ks. kohta Annostus ja antotapa). Yliherkkyysreaktion riski on suurin kahden ensimmäisen infuusion yhteydessä; riski on kuitenkin huomioitava jokaisen annettavan infuusion yhteydessä. Potilaat, joilla on ollut aikaisemmin useita lääkeallergioita tai merkittäviä yliherkkyyksiä, voivat olla muita alttiimpia saamaan tämän reaktion.
Ennen Benlysta-infuusiota voidaan antaa esilääkityksenä antihistamiinia, joko antipyreetin kanssa tai ilman. Ei ole riittävästi tietoa määrittämään, pienentääkö esilääkitys infuusioreaktioiden yleisyyttä tai vakavuutta.
Kliinisissä tutkimuksissa vakavia infuusio- ja yliherkkyysreaktioita oli noin 0,9 %:lla aikuisista potilaista. Näitä olivat anafylaktinen reaktio, bradykardia, hypotensio, angioedeema ja dyspnea. Infuusioreaktioita oli useammin kahden ensimmäisen infuusion yhteydessä ja ne vähenivät seuraavien infuusioiden yhteydessä (ks. kohta Haittavaikutukset). Potilaiden on raportoitu saaneen akuutin yliherkkyysreaktion oireita useita tunteja infuusion antamisen jälkeen. Oireiden asianmukaisen ensihoidon jälkeen on myös havaittu kliinisesti merkitsevien reaktioiden uusimista (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Sen vuoksi Benlysta on annettava olosuhteissa, joissa tällaisten reaktioiden hoitoon tarvittavat välineet ovat välittömästi saatavilla. Potilaiden on suositeltavaa pysyä kliinisessä valvonnassa pidemmän aikaa (joitakin tunteja) ainakin kahden ensimmäisen infuusion jälkeen mahdollisen viivästyneen yliherkkyysreaktion varalta. Potilaille on kerrottava, että yliherkkyysreaktiot ovat mahdollisia infuusiopäivänä ja useina sitä seuraavina päivinä. Heille on kerrottava yliherkkyysreaktioon viittaavat merkit ja oireet ja siitä, että oireet voivat uusiutua. Potilaita on neuvottava hakeutumaan välittömästi lääkärin hoitoon, jos he kokevat näitä oireita. Potilaalle on annettava pakkausseloste jokaisella Benlysta-antokerralla (ks. kohta Annostus ja antotapa).
Viivästyneitä, ei-akuutteja yliherkkyysreaktioita on havaittu ja oireina oli esim. ihottuma, pahoinvointi, väsymys, lihaskipu, päänsärky ja kasvojen turvotus.
Infektiot
Belimumabin vaikutusmekanismi saattaa suurentaa infektioiden, myös opportunisti-infektioiden kehittymisen riskiä SLE:tä sairastavilla aikuisilla ja lapsilla, ja nuoremmilla lapsilla riski voi olla suurentunut. Kontrolloiduissa kliinisissä tutkimuksissa vakavien infektioiden ilmaantuvuus oli vastaava Benlysta- ja lumelääkeryhmissä; kuolemaan johtaneita infektioita (kuten pneumonia ja sepsis) esiintyi kuitenkin useammin Benlystaa kuin lumelääketta saaneilla potilailla (ks. kohta Haittavaikutukset). Pneumokokkirokotusta tulisi harkita ennen Benlysta-hoidon aloittamista. Benlystaa ei saa aloittaa potilailla, joilla on aktiivisia vakavia infektioita (mukaan lukien vakavat krooniset infektiot). Lääkäreiden on noudatettava varovaisuutta ja arvioitava huolellisesti ovatko hyödyt riskejä suuremmat harkitessaan Benlystan käyttöä potilailla, joilla on aikaisemmin ollut toistuvia infektioita. Lääkäreiden on kehotettava potilaita olemaan yhteydessä terveydenhuollonyksikköön, jos heille kehittyy infektion oireita. Potilaita, jotka saavat infektion Benlysta-hoidon aikana on seurattava tarkoin. Lisäksi on harkittava tarkoin immunosuppressiivisen hoidon, mukaanlukien Benlystan, keskeyttämistä, kunnes infektio on parantunut. Benlystan käyttöön liittyvistä riskeistä potilailla, joilla on aktiivinen tai latentti tuberkuloosi, ei ole tietoa.
Masennus ja itsetuhoisuus
Laskimonsisäistä ja ihonalaista annostelua käyttäneissä kontrolloiduissa kliinisissä tutkimuksissa psyykkisiä häiriöitä (masennusta, itsemurha-ajatuksia ja itsemurhakäyttäytymistä, mukaan lukien itsemurhat) ilmoitettiin useammin Benlysta-valmistetta saaneilla potilailla (ks. kohta Haittavaikutukset). Ennen Benlysta-hoidon aloittamista, lääkärien tulisi arvioida masennuksen ja itsemurhan riski ottaen huomioon potilaan anamneesi ja psyykkinen nykytila, ja potilaiden seurantaa tulisi jatkaa hoidon aikana. Lääkärien on kehotettava potilaita (ja tarvittaessa heitä hoitavia henkilöitä) ottamaan yhteyttä hoitavaan terveydenhuollon ammattilaiseen, jos potilaalla ilmenee uusia psyykkisiä oireita tai aiemmat oireet pahenevat. Hoidon lopettamista on harkittava potilailla, joilla esiintyy tällaisia oireita.
Vaikeat ihon haittavaikutukset
Benlysta-hoidon yhteydessä on raportoitu Stevens-Johnsonin oireyhtymää (SJS) ja toksista epidermaalista nekrolyysiä (TEN), jotka voivat olla hengenvaarallisia tai kuolemaan johtavia. Potilaille on kerrottava SJS:n ja TEN:n merkeistä ja oireista, ja heitä tulee seurata tarkasti ihoreaktioiden varalta. Benlysta-hoito on lopetettava välittömästi ja harkittava vaihtoehtoista hoitoa, jos näihin reaktioihin viittaavia merkkejä ja oireita ilmaantuu. Benlysta-hoitoa ei saa missään vaiheessa aloittaa uudelleen, jos potilaalle on kehittynyt Benlystan käytön yhteydessä SJS tai TEN.
Progressiivinen multifokaalinen leukoenkelofalopatia
Progressiivista multifokaalista leukoenkelofalopatiaa (PML) on raportoitu potilailla, jotka saavat Benlysta-hoitoa SLE-tautiin. Lääkäreiden on oltava tietoisia erityisesti niistä PML:aan viittaavista oireista, joita potilaat eivät välttämättä huomaa (kuten kognitiiviset, neurologiset tai psykiatriset oireet tai löydökset). Potilaita on tarkkailtava tällaisten uusien tai pahenevien oireiden tai löydösten varalta ja jos näitä ilmenee, potilaan lähettämistä neurologille tai sopivia toimenpiteitä PML:n diagnosoimiseksi on harkittava, kuten kliinisesti indikoitu. Jos PML:aa epäillään, immunosuppressiivinen hoito, mukaan lukien Benlysta, on keskeytettävä kunnes PML on suljettu pois. Jos PML on vahvistettu, immunosuppressiivinen hoito, mukaan lukien Benlysta, on lopetettava.
Immunisaatio
Eläviä rokotteita ei pidä antaa 30 vuorokauden aikana ennen Benlysta-hoidon aloittamista eikä Benlysta-hoidon aikana, koska kliinistä turvallisuutta ei ole vahvistettu. Ei ole tietoa välillisestä infektion tarttumisesta elävää rokotetta saaneesta henkilöstä Benlysta-hoitoa saavalle potilaalle.
Vaikutusmekanisminsa vuoksi belimumabi voi häiritä immunisaatiovasteita. Pienessä tutkimuksessa arvioitiin immuunivastetta 23-valenttiselle pneumokokkirokotteelle. Tutkimuksessa immuunivaste eri serotyypeille oli kuitenkin rokotuksen antohetkellä samankaltainen Benlysta-valmistetta saaneilla SLE-potilailla ja SLE-potilailla, jotka saivat tavanomaista immunosupressiivista hoitoa. Ei ole riittävästi tietoa, jotta voidaan tehdä muita rokotteita koskevia johtopäätöksiä.
Rajallisen tiedon perusteella näyttää siltä, että Benlysta ei vaikuta merkittävästi kykyyn säilyttää ennen Benlysta-hoitoa saatujen immunisaatioiden suojaava immuunivaste. Alatutkimuksessa pienellä joukolla potilaita, jotka olivat aikaisemmin saaneet joko tetanus-, pneumokokki- tai influenssarokotuksen, titterit olivat suojaavalla tasolla Benlysta-hoidon jälkeenkin.
Pahanlaatuiset taudit ja lymfoproliferatiiviset häiriöt
Immuunivastetta muuntavat lääkevalmisteet, myös Benlysta, voivat lisätä pahanlaatuisten tautien riskiä. Varovaisuutta suositellaan, kun harkitaan Benlysta-hoitoa potilaille, joilla on ollut pahanlaatuisia sairauksia, tai hoidon jatkamista potilailla, joille kehittyy pahanlaatuinen tauti. Potilaita, joilla on ollut pahanlaatuinen kasvain viimeisten viiden vuoden aikana, ei ole tutkittu. Poikkeuksena potilaat, joilla on ihon tyvisolu- tai levyepiteelisolusyöpä tai kohdunkaulansyöpä, joka on saatu joko kokonaan poistettua tai muuten hoidettua asianmukaisesti.
Polysorbaatti 80 ‑pitoisuus
Tämä lääkevalmiste sisältää polysorbaatti 80:tä (ks. kohta Vaikuttavat aineet ja niiden määrät). Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Natriumpitoisuus
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevannatriumiton. Benlysta-kuiva-aine kuitenkin laimennetaan natriumia sisältävään infuusionesteeseen, ja tämä on huomioitava potilailla, joilla on ruokavalion natriumrajoitus (ks. kohta Käyttö- ja käsittelyohjeet).
Yhteisvaikutukset
In vivo yhteisvaikutustutkimuksia ei ole tehty. Joidenkin CYP450-entsyymien muodostuminen on estynyt tiettyjen sytokiinien määrän lisääntymisen takia kroonisen tulehduksen aikana. Ei tiedetä, voisiko belimumabi olla epäsuora sytokiinimodulaattori. Riskiä belimumabin aiheuttamalle epäsuoralle CYP-aktiivisuuden vähentymiselle ei voida sulkea pois. Terapeuttista seurantaa on harkittava, kun belimumabi-lääkitys aloitetaan tai lopetetaan potilailla, joita on hoidettu CYP-substraateilla, joilla on kapea terapeuttinen indeksi ja jolloin annos on yksilöllisesti säädetty (esim. varfariini).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehille ja naisille
Naisten, jotka voivat tulla raskaaksi on käytettävä tehokasta ehkäisyä Benlysta-hoidon aikana ja vähintään neljän kuukauden ajan hoidon päättymisen jälkeen.
Raskaus
Benlystan käytöstä raskaana oleville naisille on vain vähän tietoa. Lukuun ottamatta odotettua farmakologista vaikutusta, ts. B-solujen määrän alenemista, apinoilla tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).Benlystaa ei pidä käyttää raskauden aikana, ellei lääkkeestä mahdollisesti saatava hyöty ole suurempi kuin mahdollinen sikiölle koituva riski.
Imetys
Ei tiedetä, erittyykö Benlysta ihmisillä äidinmaitoon tai imeytyykö se systeemisesti suun kautta otettuna. Belimumabia kuitenkin havaittiin naarasapinoiden maidosta, kun ne olivat saaneet belimumabia 150 mg/painokg kahden viikon välein.
Koska äidin vasta-aineet (IgG) erittyvät rintamaitoon, suositellaan, että päätetään, lopetetaanko imetys vai pidättäydytäänkö Benlysta-hoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Belimumabin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Vaikutusta koiraiden ja naaraiden hedelmällisyyteen ei ole tutkittu järjestelmällisesti eläimillä tehtävissä tutkimuksissa (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Vaikutuksia ajokykyyn ja koneiden käyttökykyyn ei ole tutkittu. Belimumabin farmakologian perusteella valmisteen ei oleteta heikentävän näitä kykyjä. Potilaan kliininen tila ja Benlystan haittavaikutusprofiili on suositeltavaa pitää mielessä harkittaessa potilaan kykyä suorittaa tehtäviä, jotka vaativat harkintakykyä, motorisia tai kognitiivisia taitoja.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto aikuisilla
Belimumabin turvallisuutta SLE-potilailla on arvioitu kolmessa ennen myyntiluvan myöntämistä toteutetussa plasebokontrolloidussa tutkimuksessa, joissa valmistetta annettiin laskimoon, yhdessä myöhemmässä alueellisessa lumekontrolloidussa tutkimuksessa, jossa valmistetta annettiin laskimoon, yhdessä plasebokontrolloidussa tutkimuksessa, jossa valmistetta annettiin ihon alle, ja kahdessa markkinoilletulon jälkeen toteutetussa plasebokontrolloidussa tutkimuksessa, jossa valmistetta annettiin laskimoon. Turvallisuutta aktiivista SLE-nefriittiä sairastavilla potilailla on arvioitu yhdessä lumekontrolloidussa tutkimuksessa, jossa valmistetta annettiin laskimoon.
Seuraavassa taulukossa esitetyt tiedot kuvaavat altistusta 674 SLE-potilaalla kolmessa, ennen myyntiluvan myöntämistä toteutetuissa kliinisissä tutkimuksissa ja 470 potilaalla myöhemmin tehdyssä lumekontrolloidussa tutkimuksessa, jossa Benlystaa annettiin laskimoon (10 mg/painokg 1 tunnin ajan päivinä 0, 14, 28 ja sen jälkeen 28 vrk välein 52 viikkoon asti) ja 556 SLE-potilaan altistusta ihon alle annetulle Benlystalle (200 mg) kerran viikossa, enimmillään 52 viikon ajan. Kuvatut turvallisuustiedot sisältävät joidenkin SLE-potilaiden osalta tietoa 52 viikon jälkeiseltäkin ajalta. Tiedot kuvaavat lisäaltistusta 224 potilaalle, joilla oli aktiivinen SLE-nefriitti ja jotka saivat Benlystaa laskimoon (10 mg/painokg enimmillään 104 viikon ajan). Tiedot markkinoille tulon jälkeen saaduista raporteista on otettu mukaan.
Suurin osa potilaista sai samanaikaisesti myös yhtä tai useampaa seuraavista SLE-hoidoista: kortikosteroidit, immuunivasteen muuntajat, malarialääkkeet, ei-steroidiset anti-inflammatoriset lääkkeet.
Haittavaikutuksia raportoitiin 84 %:lla Benlystaa saaneista potilaista ja 87 %:lla plaseboa saaneista potilaista. Yleisimmin raportoitu haittavaikutus oli (≥ 5 %:lla Benlystaa ja tavanomaista hoitoa saaneista SLE-potilaista ja ≥ 1 % useammin kuin plaseboa saaneilla potilailla) nasofaryngiitti. 7 % potilaista lopetti hoidon haittavaikutusten vuoksi Benlysta-ryhmässä ja 8 % plaseboa saaneiden ryhmässä.
Yleisimmin raportoituja haittavaikutuksia (> 5 % potilaista, joilla on aktiivinen SLE-nefriitti, ja jotka saivat Benlystaa + tavanomaista hoitoa) olivat ylähengitystieinfektio, virtsatieinfektio ja vyöruusu. Haittavaikutusten vuoksi hoidon lopetti 12,9 % potilaista Benlysta-ryhmässä ja 12,9 % plaseboryhmässä.
Vaikeat ihon haittavaikutukset: Benlysta-hoidon yhteydessä on raportoitu Stevens-Johnsonin oireyhtymää (SJS) ja toksista epidermaalista nekrolyysiä (TEN) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Haittavaikutukset luokitellaan alla MedDRA-elinjärjestelmäluokituksen ja yleisyyden mukaan. Haittavaikutukset on luokiteltu yleisyyden perusteella seuraavasti:
Hyvin yleinen ≥ 1/10
Yleinen ≥ 1/100, < 1/10
Melko harvinainen ≥ 1/1 000, < 1/100
Harvinainen ≥ 1/10 000, < 1/1 000
tuntematon saatavissa oleva tieto ei riitä esiintyvyyden arviointiin
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä. Yleisyysluokka on ilmoitettu sen mukaan, kummalla lääkemuodolla se on suurempi.
| Elinjärjestelmä | Yleisyys | Haittavaikutus |
| Infektiot1 | Hyvin yleinen | Bakteeri-infektiot, esim. keuhkoputkitulehdus, virtsatieinfektio |
| Yleinen | Virusgastroentriitti, faryngiitti, nasofaryngiitti, virusperäinen ylähengitystieinfektio | |
| Veri ja imukudos | Yleinen | Leukopenia |
| Immuunijärjestelmä | Yleinen | Yliherkkyysreaktiot2 |
| Melko harvinainen | Anafylaktinen reaktio | |
| Harvinainen | Viivästyneet, ei-akuutit yliherkkyysreaktiot | |
| Psyykkiset häiriöt | Yleinen | Masennus |
| Melko harvinainen | Itsemurhakäyttäytyminen, itsemurha-ajatukset | |
| Hermosto | Yleinen | Migreeni |
| Ruoansulatuselimistö | Yleinen | Ripuli, pahoinvointi |
| Iho ja ihonalainen kudos | Yleinen | Injektiokohdan reaktiot3, nokkosihottuma, ihottuma |
| Melko harvinainen | Angioedeema | |
| tuntematon | Stevens-Johnsonin oireyhtymä, toksinen epidermaalinen nekrolyysi | |
| Luusto, lihakset ja sidekudos | Yleinen | Raajakipu |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Infuusioon tai injektioon liittyvät systeemiset reaktiot2, kuume |
1 Katso lisätietoja kohdista ‘Valikoitujen haittavaikutusten kuvaus’ ja 4.4 ‘Infektiot’.
2’ Yliherkkyysreaktiot’ kattaa joukon termejä, mm. anafylaksia, ja se voi ilmetä monina erilaisina oireina mukaan lukien hypotensio, angioedeema, urtikaria tai muut ihottumat, pruritus ja dyspnea. ’Infuusioon tai injektioon liittyvät systeemiset reaktiot’ kattaa joukon termejä ja voi ilmetä monina erilaisina oireina, kuten bradykardiana, myalgiana, päänsärkynä, ihottumana, urtikariana, kuumeena, hypotensiona, hypertensiona, huimauksena ja artralgiana. Koska merkit ja oireet ovat samanlaisia, ei ole aina mahdollista erottaa toisistaan yliherkkyysreaktioita ja infuusion tai injektion antoon liittyviä systeemisiä reaktioita.
3 Koskee vain ihon alle annettavaa lääkemuotoa.
Valikoitujen haittavaikutusten kuvaus
Seuraavat tiedot on yhdistetty kolmesta ennen myyntiluvan myöntämistä toteutetuista kliinisistä tutkimuksista, joissa käytettiin laskimoon annettavaa valmistetta (ainoastaan laskimoon annettu annos 10 mg/painokg), sekä tutkimuksesta, jossa käytettiin ihon alle annettavaa valmistetta. ’Infektioiden’ ja ’Psyykkisten häiriöiden’ kohdalla esitetään tietoja myös markkinoilletulon jälkeen toteutetusta tutkimuksesta.
Infuusioon tai injektioon liittyvät systeemiset reaktiot ja yliherkkyysreaktiot: Infuusioon tai injektioon liittyviä systeemisiä reaktioita ja yliherkkyyttä havaittiin yleensä antopäivänä, mutta akuutteja yliherkkyysreaktioita voi ilmaantua myös useiden lääkkeenantoa seuraavien päivien aikana. Potilaat, joilla on ollut aikaisemmin useita lääkeallergioita tai merkittäviä yliherkkyysreaktioita, voivat olla muita alttiimpia saamaan tämän reaktion.
Kun valmistetta annettiin laskimoon infuusioreaktioiden ja yliherkkyysreaktioiden ilmaantuvuus 3 vrk kuluessa infuusion jälkeen oli Benlysta-ryhmässä 12 % ja plaseboryhmässä 10 %. Benlystaa saaneista 1,2 % ja plaseboa saaneista 0,3 % joutui lopettamaan hoidon lopullisesti.
Infektiot: Infektioiden kokonaisilmaantuvuus rekisteröintiä edeltävissä SLE-tutkimuksissa, joissa valmistetta annettiin laskimoon tai ihon alle, oli sekä Benlysta- että plaseboryhmässä 63 %. Infektioita, joita oli vähintään 3 %:lla Benlysta-potilaista ja vähintään 1 % useammin kuin plaseboa saaneilla potilailla, olivat virusperäiset ylähengitystieinfektiot, keuhkoputkitulehdus ja bakteeriperäiset virtsatieinfektiot. Vakavia infektioita ilmeni 5 %:lla potilaista sekä Benlysta- että plaseboryhmässä; näistä vakavia opportunisti-infektioita oli Benlysta-ryhmässä 0,4 % ja plaseboryhmässä 0 %. Hoidon keskeyttämiseen johtaneita infektioita oli 0,7 %:lla Benlysta-potilaista ja 1,5 %:lla plasebopotilaista. Jotkut infektiot olivat vaikeita tai kuolemaan johtaneita.
Tietoja pediatrisilla SLE-potilailla havaituista infektioista löydät alla olevasta kohdasta ”Pediatriset potilaat”.
SLE-nefriittitutkimuksessa potilaat saivat ennestään tavanomaista hoitoa (ks. kohta Farmakodynamiikka). Infektioiden yleinen ilmaantuvuus oli 82 % Benlystaa saaneilla potilailla ja 76 % lumelääkettä saaneilla potilailla. Vakavia infektioita esiintyi 13,8 %:lla Benlystaa saaneista potilaista ja 17,0 %:lla plaseboa saaneista potilaista. Kuolemaan johtaneita infektioita esiintyi 0,9 %:lla Benlystaa saaneista (2/224) ja 0,9 %:lla plaseboa saaneista (2/224) potilaista.
Satunnaistetussa, kaksoissokkoutetussa, 52 viikkoa kestäneessä, markkinoilletulon jälkeisessä SLE-turvallisuustutkimuksessa (BEL115467) arvioitiin kuolleisuutta ja tiettyjä haittavaikutuksia aikuisilla. Vakavia infektioita esiintyi 3,7 %:lla potilaista, jotka saivat Benlystaa (10 mg/painokg laskimoon) verrattuna 4,1 %:iin lumelääkettä saaneista potilaista. Kuolemaan johtaneita infektioita (kuten pneumonia ja sepsis) kuitenkin esiintyi 0,45 %:lla (9/2002) Benlystalla hoidetuista potilaista verrattuna 0,15 %:iin (3/2001) lumelääkettä saaneista potilaista, kun taas kokonaiskuolleisuus-ilmaantuvuus oli 0,50 % (10/2002) verrattuna 0,40 %:iin (8/2001). Suurin osa kuolemaan johtaneista infektioista ilmeni Benlysta-hoidon ensimmäisten 20 viikon aikana.
Psyykkiset häiriöt: Ennen myyntiluvan myöntämistä toteutetuissa kliinisissä SLE-tutkimuksissa, joissa Benlysta-valmistetta annettiin laskimoon, vakavia psykiatrisia tapahtumia ilmoitettiin 1,2 %:lla (8/674) 10 mg/painokg Benlysta-valmistetta saaneista ja 0,4 %:lla (3/675) plaseboa saaneista potilaista. Vakavaa masennusta ilmoitettiin 0,6 %:lla (4/674) 10 mg/painokg Benlysta-valmistetta saaneista ja 0,3 %:lla (2/675) plaseboa saaneista potilaista. Benlysta-hoitoa saaneilla potilailla ilmoitettiin kaksi itsemurhaa (joista yksi 1 mg/painokg Benlysta-valmistetta saaneella potilaalla).
Markkinoilletulon jälkeisessä SLE-tutkimuksessa ilmoitettiin vakavia psykiatrisia tapahtumia 1,0 %:lla (20/2002) Benlysta-valmistetta saaneista ja 0,3 %:lla (6/2001) plaseboa saaneista potilaista. Vakavaa masennusta ilmoitettiin 0,3 %:lla (7/2002) Benlysta-valmistetta saaneista ja < 0,1 %:lla (1/2001) plaseboa saaneista potilaista. Vakavien itsemurha-ajatusten, vakavan itsemurhakäyttäytymisen ja ei-itsemurhahakuisen itsensä vahingoittamisen kokonaisilmaantuvuus oli 0,7 % (15/2002) Benlysta-valmistetta saaneilla ja 0,2 % (5/2001) plaseboryhmässä. Kummassakaan ryhmässä ei ilmoitettu yhtään itsemurhaa.
Yllä mainituista SLE-tutkimuksista, joissa valmistetta annettiin laskimoon, ei suljettu pois potilaita, joilla oli anamneesissa psyykkisiä häiriöitä.
Kliinisestä SLE-tutkimuksesta, jossa valmistetta annettiin ihon alle, suljettiin pois potilaat, joilla oli anamneesissa psyykkisiä häiriöitä. Tässä tutkimuksessa vakavia psykiatrisia tapahtumia ilmoitettiin 0,2 %:lla (1/556) Benlysta-valmistetta saaneista eikä yhdelläkään plaseboa saaneella. Kummassakaan ryhmässä ei ilmoitettu vakavia masennukseen liittyviä tapahtumia eikä itsemurhia.
Leukopenia: Leukopeniaa raportoitiin haittatapahtumana SLE-potilailla Benlysta-ryhmässä 3 %:lla potilaista ja plaseboryhmässä 2 %:lla.
Ruoansulatuselimistö: Ylipainoisilla (painoindeksi (BMI) > 30 kg/m2) Benlystaa laskimoon saaneilla SLE-potilailla oli useammin pahoinvointia, oksentelua ja ripulia kuin plaseboa saaneilla tai normaalipainoisilla potilailla (BMI ≥ 18,5–≤ 30 kg/m2). Mitkään ylipainoisten potilaiden ruoansulatuselimistön haittavaikutukset eivät olleet vakavia.
Pediatriset potilaat
Pediatrisia potilaita koskeva haittavaikutusprofiili perustuu yhteen tutkimukseen, jossa lääke annettiin ihon alle ja yhteen tutkimukseen, jossa lääke annettiin laskimoon.
52 viikkoa kestäneessä avoimessa tutkimuksessa 25 pediatrista SLE-potilasta (10–17‑vuotiaita) sai Benlysta-valmistetta ihon alle annoksina, jotka vastasivat aikuisten altistusta (200 mg painon mukaan määritetyin väliajoin taustalla olevien muiden lääkitysten lisänä). Tutkimuksessa todettiin turvallisuusprofiilin olevan Benlysta-valmistetta ihon alle saavilla pediatrisilla potilailla yhdenmukainen belimumabin tunnetun turvallisuusprofiilin kanssa.
52 viikkoa kestäneessä lumelääkekontrolloidussa tutkimuksessa 53 SLE-potilasta (6-17-vuotiaita) sai Benlysta-valmistetta (10 mg/painokg laskimoon päivinä 0, 14, 28 ja sen jälkeen 28 päivän välein, taustalla olevien muiden lääkitysten lisänä). Uusia turvallisuussignaaleja ei havaittu 12-vuotiaiden tai sitä vanhempien pediatrisessa populaatiossa (n = 43). Turvallisuustietoja alle 12-vuotiaista lapsista (n = 10) on niukasti.
Infektiot
5–11-vuotiaiden ryhmä: infektioita ilmoitettiin kahdeksalla 10:stä Benlysta-valmistetta laskimoon saaneesta potilaasta, ja kaikilla kolmella lumelääkettä saaneella potilaalla. Vakavia infektioita ilmoitettiin yhdellä 10:stä Benlysta valmistetta laskimoon saaneesta potilaasta, ja kahdella kolmesta lumelääkettä saaneesta potilaasta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
12–17-vuotiaiden ryhmä: infektioita ilmoitettiin 22:lla 43:sta Benlysta-valmistetta laskimoon saaneesta potilaasta, ja 25:llä 37:stä lumelääkettä saaneesta potilaasta. Vakavia infektioita ilmoitettiin kolmella 43:sta Benlysta-valmistetta laskimoon saaneesta potilaasta, ja kolmella 37:stä lumelääkettä saaneesta potilaasta. Avoimessa jatkovaiheessa oli yksi kuolemaan johtava infektio Benlysta-valmistetta laskimoon saaneella potilaalla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Benlystan yliannostuksesta on vain vähän kliinisiä kokemuksia. Yliannostuksen yhteydessä raportoidut haittavaikutukset ovat olleet yhdenmukaisia belimumabin odotettujen haittavaikutuksien kanssa.
Ihmisille on annettu kaksi enintään 20 mg/painokg annosta 21 vuorokauden välein laskimonsisäisenä infuusiona ilman, että haittavaikutuksia olisi ilmaantunut useammin tai että ne olisivat olleet vakavampia kuin annoksilla 1, 4 tai 10 mg/painokg.
Tahattoman yliannostuksen tapahtuessa potilasta on seurattava tarkoin ja asianmukaista tukihoitoa on annettava tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, monoklonaaliset vasta-aineet, ATC-koodi: L04AG04
Vaikutusmekanismi
Belimumabi on humaani IgG1λ monoklonaalinen vasta-aine, joka on spesifinen liukoiselle ihmisen B-lymfosyyttiä stimuloivalle proteiinille (BLyS, käytetään myös nimityksiä BAFF ja TNFSF13B). Belimumabi estää liukoisen BLySin, joka on välttämätön tekijä B-solun eloonjäämiselle, sitoutumisen sen reseptoreihin B-soluissa. Belimumabi ei sitoudu suoraan B-soluihin vaan BLySiin. Näin belimumabi estää B-solujen, myös autoreaktiivisten B-solujen eloonjäämisen ja vähentää B-solujen erilaistumista immunoglobuliinia tuottaviksi plasmasoluiksi.
SLE-potilailla ja muilla autoimmuunitauteja sairastavilla BLyS-tasot ovat koholla. Plasman BLyS-tason ja SLE-taudin aktiivisuuden välillä on yhteys. BLyS-tasojen suhteellista merkitystä SLE:n patofysiologialle ei ymmärretä täysin.
Farmakodynaamiset vaikutukset
Kliinisissä tutkimuksissa, joissa Benlystaa annettiin laskimoon, havaittiin muutoksia biomerkkiaineissa. Aikuisilla SLE-potilailla, joilla oli hypergammaglobulinemia, IgG-arvojen havaittiin normalisoituvan viikkoon 52 menessä 49 %:lla potilaista, jotka saivat Benlystaa ja 20 %:lla plasebopotilaista.
SLE-potilaista, joilla oli anti-dsDNA-vasta-aineita, 16 % Benlysta-potilaista muuttui anti-dsDNA-negatiivisiksi viikkoon 52 mennessä ja vastaavasti 7 % plasebopotilaista.
SLE-potilailla, joiden komplementtitasot olivat alhaisia, havaittiin C3:n normalisoitumista 38 %:lla ja C4:n normalisoitumista ja 44 %:lla Benlysta-potilaista viikkoon 52 mennessä. Plasebopotilailla vastaavat luvut olivat 17 % ja 18 %.
Anti-fosfolipidivasta-aineista mitattiin vain anti-kardiolipiinivasta-aineet. Anti-kardiolipiini IgA-vasta-aineet vähenivät 37 % viikkoon 52 mennessä (p = 0,0003), anti-kardiolipiini IgG-vasta-aineet vähenivät 26 % viikkoon 52 mennessä (p = 0,0324) ja anti-kardiolipiini IgM-vasta-aineet vähenivät 25 % (p = ei merkitsevä, 0,46).
Laskimonsisäisen belimumabihoidon aikana esiintyneitä muutoksia SLE-potilaiden B-solu- (mukaan lukien naiivit, muisti- ja aktivoidut B-solut sekä plasmasolut) ja IgG-arvoissa seurattiin pitkäaikaisessa kontrolloimattomassa jatkotutkimuksessa. Seitsemän ja puolen vuoden hoidon jälkeen (mukaan lukien 72 viikon päätutkimus), B-solujen alaryhmissä havaittiin huomattavaa ja pitkäkestoista vähenemistä, jolloin naiivien B-solujen vähenemisen mediaani oli 87 %, muisti-B-solujen 67 %, aktivoituneiden B-solujen 99 % ja plasmasolujen 92 % yli seitsemän vuoden hoidon jälkeen. Noin seitsemän vuoden jälkeen, IgG-arvojen havaittiin pienenevän 28 % (mediaani) ja 1,6:lla %:lla tutkittavista IgG-arvot laskivat alle 400 mg/dl. Tutkimuksen aikana raportoitujen haittatapahtumien ilmaantuvuus pysyi yleensä vakaana tai vähentyi.
Potilailla, joilla on aktiivinen SLE-nefriitti, esiintyi Benlysta‑hoidon (10 mg/painokg laskimoon) tai plasebohoidon jälkeen seerumin IgG‑pitoisuuksien suurenemista, johon liittyi proteinurian väheneminen. Benlysta‑ryhmässä havaittiin plaseboon verrattuna vähäisempää seerumin IgG‑pitoisuuksien suurenemista, mikä oli odotettavissa belimumabin tunnetun mekanismin perusteella. Viikon 104 kohdalla IgG‑arvon prosentuaalinen mediaanisuurenema lähtötilanteesta oli Benlysta‑ryhmässä 17 % ja plaseboryhmässä 37 %. Autovasta‑aineiden väheneminen, komplementtitasojen suureneminen ja verenkierron B‑solujen kokonaismäärän ja B‑solualaryhmien pieneneminen vastasi SLE‑tutkimuksissa tehtyjä havaintoja.
Yhdessä tutkimuksessa, jossa lääkettä annettiin laskimoon 6–17‑vuotiaille pediatrisille SLE-potilaille, farmakodynaaminen vaste oli yhdenmukainen aikuisia koskevien tietojen kanssa.
Immunogeenisuus
Kerätyissä näytteissä oleva aktiivi lääkeaine heikentää neutraloivia vasta-aineita ja ei-spesifisiä lääkkeen vasta-aineita (ADA) mittaavien määritysten herkkyyttä. Tämän vuoksi ei tiedetä, minkä verran tutkimuspotilailla todella on neutraloivia vasta-aineita ja ei-spesifisiä lääkkeen vasta-aineita. Antibelimumabivasta-aineiden testitulos oli jatkuvasti positiivinen kahdessa faasin III SLE-tutkimuksessa neljällä aikuisella potilaalla 563:sta (0,7 %) 10 mg/painokg-ryhmässä ja 27 potilaalla 559:stä (4,8 %) 1 mg/painokg-ryhmässä.
Faasi III:n SLE-tutkimuksissa olleista potilaista, joilla antibelimumabivasta-aineiden testitulos oli jatkuvasti positiivinen, 1/10 (10 %) plaseboryhmän, 2/27 (7 %)1 mg/kg-ryhmän ja 1/4 (25 %) 10 mg/kg-ryhmän potilaista koki infuusioreaktioita lääkkeen antopäivänä; infuusioreaktiot olivat kaikki ei-vakavia ja voimakkuudeltaan lieviä – kohtalaisia. Vain harvat ADA-potilaat kokivat vakavia/voimakkaita haittavaikutuksia. Infuusioreaktioiden määrä oli samanlainen potilailla, joilla antibelimumabivasta-aineiden testitulos oli jatkuvasti positiivinen kuin ADA-negatiivisilla potilailla: 75/552 (14 %) plaseboryhmässä, 78/523 (15 %) 1 mg/painokg-ryhmässä ja 83/559 (15 %) 10 mg/painokg-ryhmässä.
Belimumabivasta‑aineita ei havaittu SLE-nefriittitutkimuksessa, jossa 224 aikuispotilasta sai Benlystaa 10 mg/painokg laskimoon.
Yhdessä tutkimuksessa, jossa 6-17-vuotiaille pediatrisille SLE-potilaille (n = 53) annettiin lääkettä laskimoon, yksikään potilaista ei kehittänyt belimumabille vasta-aineita.
Kliininen teho ja turvallisuus
SLE
Laskimoinfuusio aikuisilla
Laskimoon annetun Benlystan tehoa arvioitiin kahdessa randomoidussa, kaksoissokkoutetussa plasebokontrolloidussa tutkimuksessa, joihin osallistui 1684 potilasta, joilla oli kliininen SLE-diagnoosi American College of Rheumatology (ACR) -luokituksen kriteerien mukaan. Potilailla oli aktiivinen SLE-tauti, määritelmänä seulontavaiheen SELENA-SLEDAI (SELENA=Safety of Estrogens in Systemic Lupus Erythematosus National Assessment; SLEDAI=Systemic Lupus Erythematosus Disease Activity Index) -pisteet ≥ 6 ja tumavasta-aine (ANA) positiivinen (ANA-titteri ≥ 1:80 ja/tai positiivinen anti-dsDNA [≥ 30 yksikköä/ml]). Potilailla oli vakiintunut SLE-lääkehoito, johon kuului (yksin tai yhdistelmänä): kortikosteroideja, malarialääkkeitä, NSAID-lääkkeitä tai muita immunosuppressantteja. Tutkimukset olivat rakenteeltaan samanlaisia, paitsi että BLISS-76-tutkimus kesti 76 viikkoa ja BLISS-52-tutkimus 52 viikkoa. Molemmissa tutkimuksissa ensisijaista tehoa mittaavat päätetapahtumat arvioitiin viikon 52 kohdalla.
Potilaat, joilla oli vaikea aktiivinen SLE-nefriitti ja potilaat, joilla oli vaikea aktiivinen keskushermosto (CNS) lupus, suljettiin tutkimusten ulkopuolelle.
BLISS-76 tehtiin pääasiassa Pohjois-Amerikassa ja Länsi-Euroopassa. Peruslääkityksenä oli kortikosteroideja (76 %; > 7,5 mg/vrk 46 %), immunosuppressantteja (56 %) ja malarialääkkeitä (63 %).
BLISS-52 tehtiin Etelä-Amerikassa, Itä-Euroopassa, Aasiassa ja Australiassa. Peruslääkityksenä oli kortikosteroideja (96 %; > 7,5 mg/vrk 69 %), immunosuppressiiveja (42 %) ja malarialääkkeitä (67 %).
Alkutilanteessa 52 %:lla potilaista tauti oli erittäin aktiivinen (SELENA SLEDAI -asteikko ≥ 10), 59 %:lla potilaista oli tautia limakalvoissa, 60 %:lla luustossa, lihaksissa ja sidekudoksessa, 16 %:lla veressä, 11 %:lla munuaisissa ja 9 %:lla verisuonistossa (BILAG A tai B lähtötilanteessa).
Ensisijainen tehoa mittaava päätetapahtuma oli yhdistelmämuuttuja (SLE Responder Index; SLE-vasteindeksi), jossa vasteen määritelmänä oli kaikkien seuraavien kriteerien täyttyminen viikon 52 kohdalla verrattuna lähtötilanteeseen:
- ≥ 4 pisteen aleneminen SELENA-SLEDAI-asteikolla, ja
- ei uusia pisteitä BILAG A -kohde-elinasteikolla (British Isles Lupus Assessment Group) tai kaksi uutta pistettä BILAG B -kohde-elinasteikolla, ja
- ei huononemista (< 0,30 pisteen kasvua) PGA (Physician’s Global Assessment) -asteikolla
SLE-vasteindeksi mittaa miten SLE-taudin aktiivisuudessa tapahtuu paranemista ilman että minkään kehon elinjärjestelmän toiminta tai potilaan yleinen vointi huononee.
Taulukko 1. Vaste viikon 52 kohdalla
| Vaste | BLISS-76 | BLISS-52 | BLISS-76 ja BLISS-52 yhdistettynä | |||||
Plasebo1 (n = 275) | Benlysta 10 mg/kg1 (n = 273) | Plasebo1 (n = 287) | Benlysta 10 mg/kg1 (n = 290) | Plasebo1 (n = 562) | Benlysta 10 mg/kg1 (n = 563) | |||
| SLE-vasteindeksi | 33,8 % | 43,2 % (p = 0,021) | 43,6 % | 57,6 % (p= 0,0006) | 38,8 % | 50,6 % (p <0,0001) | ||
| Ero plaseboon verrattuna | 9,4 % | 14,0 % | 11,8 % | |||||
| Vetosuhde (OR; 95 % CI) vs. placebo | 1,52 (1,07, 2,15) | 1,83 (1,30, 2,59) | 1,68 (1,32, 2,15) | |||||
| SLE-vasteindeksin osatekijät | ||||||||
Niiden potilaiden osuus, joilla SELENA‑ SLEDAI laski ≥4 | 35,6 % | 46,9 % (p = 0,006 | 46,0 % | 58,3 % (p = 0,0024) | 40,9 % | 52,8 % (p < 0,0001) | ||
| Niiden potilaiden osuus, joiden BILAG-indeksi ei huonontunut | 65,1 % | 69,2 % (p = 0,32) | 73,2 % | 81,4 % (p = 0,018) | 69,2 % | 75,5 % (p = 0,019) | ||
| Niiden potilaiden osuus, joiden PGA-indeksi ei huonontunut | 62,9 % | 69,2 % (p = 0,13) | 69,3 % | 79,7 % (p = 0,0048) | 66,2 % | 74,6 % (p = 0,0017) | ||
1Kaikki potilaat saivat tavanomaista hoitoa.
Näiden kahden tutkimuksen yhdistettyjen tietojen analyysissä potilaista, jotka saivat lähtötilanteessa > 7,5 mg/vrk prednisonia (tai vastaavaa) ja joiden keskimääräistä kortikosteroidiannosta pienennettiin vähintään 25 %:lla lähtötilanteesta siten, että annos vastasi ≤ 7,5 mg/vrk prednisonia viikkoina 40–52, oli Benlysta-ryhmässä 17,9 % ja plaseboryhmässä 12,3 % (p = 0,0451).
SLE:n aktivoitumista mitattiin muunnetulla SELENA SLEDAI -aktivoitumisindeksillä (SLE Flare Index). Keskimääräinen aika ensimmäiseen aktivoitumiseen oli pidempi yhdistetyssä Benlysta-ryhmässä verrattuna plaseboryhmään (110 vrk vs 84 vrk; riskitiheyssuhde 0,84; p = 0,012). Vaikeita aktivoitumisia havaittiin 52 seurantaviikon aikana 15,6 %:lla Benlysta-potilaista ja 23,7 %:lla plaseboryhmässä (havaittu hoitoero = -8,1%; riskisuhde 0,64; p = 0,0011).
Yhdistetyissä tiedoissa FACIT-väsymysasteikolla (FACIT Fatique scale) mitattuna Benlysta lievitti väsymystä verrattuna plaseboon. Keskimääräinen muutos lähtötilanteesta viikolle 52 on merkitsevästi parempi Benlystalla kuin plasebolla (4,70 vs 2,46; p = 0,0006).
Ensisijaisen päätetapahtuman yhden muuttujan ja useiden muuttujien analyysi etukäteen määritetyissä alaryhmissä osoitti, että hyöty oli suurin potilailla, joiden tauti oli aktiivisempi, mukaan lukien potilaat, joiden SELENA SLEDAI -pisteet olivat ≥ 10 tai potilaat, jotka tarvitsivat steroideja taudin hallintaan tai potilaat, joilla komplementtitasot olivat alhaisia.
Post-hoc-analyysissä on havaittu alaryhmiä, joilla vaste on voimakas, esim. potilaat, joilla on lähtötilanteessa alhainen komplementtitaso ja positiivinen anti-dsDNA. Tulokset tässä esimerkkiryhmässä, jossa tautiaktiivisuus on suurempi, ks. taulukko 2. Näistä potilaista 64,5 %:lla SELENA SLEDAI -pisteet olivat lähtötilanteessa ≥ 10.
Taulukko 2.Potilaat, joilla lähtötilanteessa alhainen komplementtitaso ja positiivinen anti-dsDNA
| Alaryhmät | Positiivinen anti-dsDNA JA alhainen komplementtitaso | |
| BLISS-76 ja BLISS-52 yhdistettynä | Plasebo (n = 287) | Benlysta 10 mg/kg (n = 305) |
SRI vaste viikon 52 kohdalla (%) Havaittu hoitoero verrattuna plaseboon (%) | 31,7 | 51,5 (p < 0,0001) 19,8 |
SRI vaste (pois lukien komplementti- ja anti-dsDNA-muutokset) viikon 52 kohdalla (%) Havaittu hoitoero verrattuna plaseboon (%) | 28,9 | 46,2 (p < 0,0001) 17,3 |
| Vaikeita taudin aktivoitumisia 52 viikon aikana | ||
| Niiden potilaiden osuus, joilla oli vakava aktivoituminen (%) | 29,6 | 19,0 |
| Havaittu hoitoero verrattuna plaseboon (%) | 10,6 | |
| Vaikeaan aktivoitumiseen kulunut aika [riskitiheyssuhde (95 % CI)] | 0,61 (0,44, 0,85) (p = 0,0038) | |
Prednisoniannoksen pienentäminen ≥ 25 % lähtötilanteesta ≤ 7.5 mg/vrk viikkoina 40–521 (%) Havaittu hoitoero verrattuna plaseboon (%) | (n = 173) 12,1 | (n = 195) 18,5 (p = 0,0964) 6,3 |
FACIT-väsymysasteikon pisteiden paraneminen lähtötilanteesta viikon 52 kohdalla (keskiarvo) Havaittu hoitoero verrattuna plaseboon (%) | 1,99 | 4,21 (p = 0,0048) 2,21 |
| BLISS-76 tutkimus yksinään | Plasebo (n = 131) | Benlysta 10 mg/kg (n = 134) |
SRI-vasteen saaneiden osuus viikolla 76 (%) Havaittu hoitoero verrattuna plaseboon (%) | 27,5 | 39,6 (p = 0,0160) 12,1 |
1Potilailla, joiden prednisoniannos >7.5 mg/vrk lähtötilanteessa
Benlysta -valmisteen tehoa ja turvallisuutta yhdistettynä yksisyklisesti rituksimabiin arvioitiin faasi III, satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa 104 viikon tutkimuksessa (BLISS/BELIEVE) 292 potilaalla. Ensisijainen päätetapahtuma oli koehenkilöiden osuus, joiden tautitila oli parantunut, minkä määritelmänä oli SLEDAI-2K pistemäärä ≤ 2 ilman immuunisalpaajan käyttöä ja kortikosteroidin käyttö prednisonia vastaavalla annoksella ≤ 5 mg/vrk viikolla 52. Tämä saavutettiin 19,4 % (n = 28/144) Benlystan ja rituksimabin yhdistelmällä hoidetuista potilaista ja 16,7 % (n = 12/72) Benlystan ja lumelääkkeen yhdistelmällä (vetosuhde 1,27; 95 % CI: 0,60, 2,71; p = 0,5342). Enemmän haittavaikutuksia (91,7 % vs. 87,5 %), vakavia haittavaikutuksia (22,2 % vs. 13,9 %) ja vakavia infektioita (9,0 % vs. 2,8 %) havaittiin Benlystan ja rituksimabin yhdistelmällä hoidetuilla potilailla verrattu Benlystan ja lumelääkkeen yhdistelmällä hoidettuihin potilaisiin.
SLE‑nefriitti
Edellä kuvatuista laskimonsisäisistä SLE-tutkimuksista suljettiin pois potilaat, joilla oli vaikea aktiivinen SLE-nefriitti. 11 %:lla potilaista oli kuitenkin lähtötilanteessa munuaiselinten alueen vaikutuksia (BILAG A- tai B-arvioinnin perusteella). Seuraava aktiivisen SLE-nefriitin tutkimus on tehty.
Laskimoon annettavan Benlystan (10 mg/painokg 1 tunnin ajan päivinä 0, 14 ja 28 ja sen jälkeen 28 vrk välein) tehoa ja turvallisuutta arvioitiin 104 viikon pituisessa, satunnaistetussa (1:1), kaksoissokkoutetussa, plasebokontrolloidussa vaiheen III tutkimuksessa (BEL114054) 448 potilaalla, joilla oli aktiivinen SLE-nefriitti. Potilailla oli kliininen SLE-diagnoosi ACR‑luokituksen (American College of Rheumatology) kriteerien mukaan, biopsialla vahvistettu luokan III, IV ja/tai V SLE-nefriitti ja seulontavaiheessa aktiivinen munuaistauti, joka vaati tavanomaista hoitoa. Tavanomainen hoito sisälsi kortikosteroideja, 0–3 laskimonsisäistä metyyliprednisoloni-annosta (500–1000 mg/annos) ja sen jälkeen oraalinen prednisoni 0,5–1 mg/kg /vrk, jolloin päivittäinen kokonaisannos on ≤ 60 mg/vrk, joka kaventuu arvoon ≤ 10 mg/vrk viikkoon 24 mennessä:
- mykofenolaattimofetiili 1-3 g/vrk suun kautta tai mykofenolaattinatrium 720–2160 mg/vrk suun kautta induktio- ja ylläpitohoitona tai
- 500 mg syklofosfamidia laskimoon 2 viikon välein 6 infuusiona induktiohoitona ja sen jälkeen atsatiopriini suun kautta tavoiteannoksella 2 mg/kg/vrk ylläpitohoitona.
Tutkimus toteutettiin Aasiassa, Pohjois-Amerikassa, Etelä-Amerikassa ja Euroopassa. Potilaiden mediaani-ikä oli 31 vuotta (vaihteluväli: 18–77 vuotta), ja valtaosa (88 %) oli naisia.
Ensisijainen tehon päätetapahtuma oli primaarinen tehoa mittaava munuaisvaste (PERR) viikon 104 kohdalla. PERR‑vasteeksi määriteltiin viikon 100 kohdalla todettu vaste, joka vahvistettiin viikon 104 kohdalla seuraavien parametrien toistomittauksella: virtsan proteiinin ja kreatiniinin suhde (uPCR) ≤ 700 mg/g (79,5 mg/mmol) ja glomerulusten laskennallinen suodatusnopeus (eGFR) ≥ 60 ml/min/1,73m2 tai ei eGFR‑arvon pienenemistä > 20 %:lla aktivoitumista edeltävästä arvosta.
Merkittäviä toissijaisia päätetapahtumia olivat:
- Täydellinen munuaisvaste (CRR), jollaiseksi määriteltiin viikon 100 kohdalla todettu vaste, joka vahvistettiin viikon 104 kohdalla seuraavien parametrien toistomittauksella: uPCR < 500 mg/g (56,8 mg/mmol) ja eGFR‑arvo ≥ 90 ml/min/1,73m2 tai ei eGFR‑arvon pienenemistä > 10 %:lla aktivoitumista edeltävästä arvosta.
- PERR viikon 52 kohdalla.
- Aika munuaistapahtumaan tai kuolemaan (munuaistapahtumaksi määriteltiin ensimmäistä kertaa esiintyvä loppuvaiheen munuaissairaus, seerumin kreatiniiniarvon kaksinkertaistuminen, munuaistoiminnan heikentyminen [proteinurian lisääntyminen ja/tai munuaisten vajaatoiminta] tai munuaissairauteen liittyvän kielletyn hoidon saaminen).
PERR- ja CRR-päätetapahtumien yhteydessä vasteeksi määritettiin steroidihodon pienentäminen arvoon ≤ 10 mg/vrk viikosta 24 alkaen. Näiden päätetapahtumien osalta hoidon katsottiin epäonnistuneen potilailla, jotka lopettivat hoidon ennenaikaisesti, saivat kiellettyjä lääkkeitä tai vetäytyivät tutkimuksesta ennenaikaisesti.
PERR:n saavutti viikon 104 kohdalla merkitsevästi suurempi osuus Benlystaa saaneista potilaista kuin plaseboa saaneista. Myös merkittävien toissijaisten päätetapahtumien osalta Benlysta‑ryhmässä todettiin merkitsevää paranemista plaseboryhmään verrattuna (taulukko 3).
Taulukko 3. Tehotulokset aikuisilla SLE-nefriittipotilailla
| Tehon päätetapahtuma | Plasebo n = 223 | Benlysta 10 mg/kg n = 223 | Ero plaseboon verrattuna | Vetosuhde (OR) / riskitiheyssuhde (HR) plaseboon verrattuna (95 % CI) | P-arvo |
PERR viikon 104 kohdalla1 Vasteen saavuttaneita | 32,3 % | 43,0 % | 10,8 % | OR 1,55 (1,04; 2,32) | 0,0311 |
| PERR:n osatekijät | |||||
| Virtsan proteiinin ja kreatiniinin suhde ≤ 700 mg/g (79,5 mg/mmol) | 33,6 % | 44,4 % | 10,8 % | OR 1,54 (1,04; 2,29) | 0,0320 |
| eGFR ≥ 60 ml/min/1,73m2 tai ei eGFR‑arvon pienenemistä > 20 %:lla aktivoitumista edeltävästä arvosta | 50,2 % | 57,4 % | 7,2 % | OR 1,32 (0,90; 1,94) | 0,1599 |
| Ei hoidon epäonnistumista³ | 74,4 % | 83,0 % | 8,5 % | OR 1,65 (1,03; 2,63) | 0,0364 |
CRR viikon 104 kohdalla1 Vasteen saavuttaneita | 19,7 % | 30,0 % | 10,3 % | OR 1,74 (1,11; 2,74) | 0,0167 |
| CRR:n osatekijät | |||||
| Virtsan proteiinin ja kreatiniinin suhde < 500 mg/g (56,8 mg/mmol) | 28,7 % | 39,5 % | 10,8 % | OR 1,58 (1,05; 2,38) | 0,0268 |
| eGFR ≥ 90 ml/min/1,73m2 tai ei eGFR‑arvon pienenemistä > 10 %:lla aktivoitumista edeltävästä arvosta | 39,9 % | 46,6 % | 6,7 % | OR 1,33 (0,90; 1,96) | 0,1539 |
| Ei hoidon epäonnistumista³ | 74,4 % | 83,0 % | 8,5 % | OR 1,65 (1,03; 2,63) | 0,0364 |
PERR viikon 52 kohdalla1 Vasteen saavuttaneita | 35,4 % | 46,6 % | 11,2 % | OR 1,59 (1,06; 2,38) | 0,0245 |
Aika munuaistapahtumaan tai kuolemaan1 Tapahtuman kokeneiden potilaiden prosenttiosuus2 Tapahtumaan kulunut aika (riskitiheyssuhde [95 % CI]) | 28,3 % | 15,7 % | - - | HR 0,51 (0,34; 0,77) | 0,0014 |
1PERR viikon 104 kohdalla oli ensisijainen tehoanalyysi; CRR viikon 104 kohdalla, PERR viikon 52 kohdalla ja munuaistapahtumaan tai kuolemaan kulunut aika sisältyivät ennalta määritettyyn testaushierarkiaan. 2Kun analyysistä jätettiin pois kuolemat (1 Benlysta‑ryhmässä; 2 plaseboryhmässä), munuaistapahtuman kokeneiden potilaiden prosenttiosuus oli Benlysta‑ryhmässä 15,2 % ja plaseboryhmässä 27,4 % (HR = 0,51; 95 % CI: 0,34, 0,78). ³Hoidon epäonnistuminen: Potilaat, jotka ottivat protokollan vastaista lääkitystä. | |||||
Benlystaa saaneista potilaista numeerisesti suurempi prosenttiosuus saavutti PERR:n plaseboa saaneisiin verrattuna viikolta 24 alkaen, ja hoitojen välinen ero säilyi viikolle 104 asti. Viikolta 12 alkaen Benlystaa saaneista potilaista numeerisesti suurempi prosenttiosuus saavutti CRR:n plaseboa saaneisiin verrattuna, ja numeerinen ero säilyi viikolle 104 asti (kuva 1).
Kuva 1. SLE-nefriittiä sairastavien aikuisten vasteprosentit käynneittäin
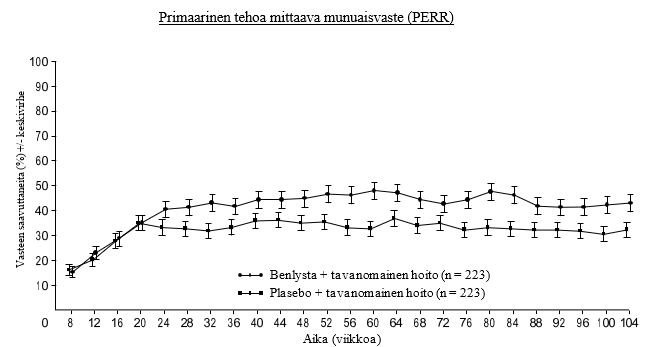
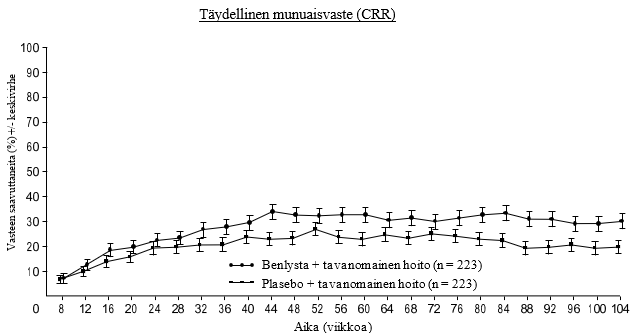
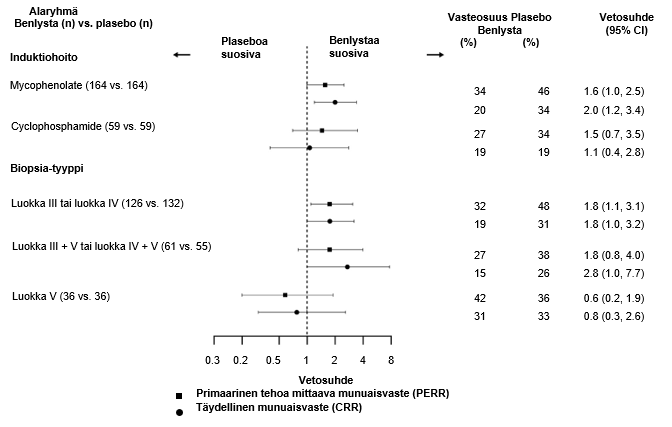
Kuvailevissa alaryhmäanalyyseissä tehon keskeisiä päätetapahtumia (PERR ja CRR) tutkittiin induktio-ohjelman (mykofenolaatti tai syklofosfamidi) ja biopsialuokan (luokka III tai IV, luokka III + V tai luokka IV + V tai luokka V) mukaan (kuva 2).
Kuva 2. PERR: n ja CRR: n kerroinsuhde viikolla 104 alaryhmissä
Ikä ja etninen tausta
Ikä
Laskimoon tai ihon alle annettavan Benlystan tehossa tai turvallisuudessa ei havaittu eroja ≥ 65-vuotiailla SLE-potilailla verrattuna lumelääkekontrolloitujen tutkimusten tutkimuspopulaatioon; ≥ 65- vuotiaiden potilaiden lukumäärä (62 potilasta (teho) ja 219 potilasta (turvallisuus)) ei kuitenkaan ole riittävän suuri, jotta voitaisiin määrittää onko vaste iäkkäillä erilainen nuoriin potilaisiin verrattuna.
Tummaihoiset potilaat
Benlysta-valmiste annettiin laskimoon tummaihoisille SLE-potilaille satunnaistetussa (2: 1), kaksoissokkoutetussa, lumelääkekontrolloidussa 52 viikon III / IV-vaiheen tutkimuksessa (EMBRACE). Tehoa arvioitiin 448 potilaalla. SRI-S2K-vastetta saavien tummaihoisten potilaiden osuus oli suurempi Benlysta-valmistetta saaneilla potilailla, mutta ero ei ollut tilastollisesti merkitsevä verrattuna plaseboon. Muiden tutkimusten tulosten mukaisesti tummaihoisilla potilailla, joilla oli korkea sairausaktiivisuus (alhainen komplementti ja positiivinen anti-dsDNA lähtötilanteessa, n = 141), SRI-S2K-vaste oli 45,1 % Benlysta 10 mg/painokg valmisteella verrattuna 24,0 % plaseboon (kerroinsuhde 3,00; 95 % CI: 1,35-6,68).
Pediatriset potilaat
SLE
Benlysta-valmisteen turvallisuutta ja tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa 52 viikon tutkimuksessa (PLUTO) 93 pediatrisella potilaalla, joilla oli kliininen SLE-diagnoosi ACR-luokitusperusteiden mukaisesti. Potilailla oli aktiivinen SLE-tauti, jonka määritelmänä oli SELENA-SLEDAI-pisteet ≥ 6 ja positiiviset autovasta-aineet seulontavaiheessa, kuten aikuistutkimuksissa on kuvattu. Potilailla oli vakaa SLE-hoito (hoitosuositusten mukaisesti), ja samanlaiset sisäänotto- ja poissulkukriteerit kuin aikuistutkimuksissa. Potilaita, joilla oli vaikea aktiivinen lupusnefriitti, vaikea aktiivinen keskushermoston lupus, primaarinen immuunipuutos, IgA-puutos tai hoitoa vaativia akuutteja tai kroonisia infektioita, ei sisällytetty tutkimukseen. Tutkimus tehtiin Yhdysvalloissa, Etelä-Amerikassa, Euroopassa ja Aasiassa. Potilaiden iän mediaani oli 15 vuotta (vaihteluväli 6–17 vuotta). 5–11-vuotiaiden ryhmässä (n = 13) SELENA-SLEDAI-pistemäärä vaihteli välillä 4–13 ja 12–17-vuotiaiden ryhmässä (n = 79) SELENA-SLEDAI-pistemäärä vaihteli välillä 4 – 20. Suurin osa (94,6 %) potilaista oli naisia. Tutkimus ei ollut voimaltaan soveltuva tilastollisiin vertailuihin ja kaikki tulokset ovat kuvailevia.
Ensisijainen tehon päätetapahtuma oli SLE-vasteindeksi (SRI) viikolla 52, kuten aikuisilla tehdyissä suonensisäisissä tutkimuksissa on kuvattu. Pediatrisista potilaista suurempi osuus Benlysta-hoitoa saaneista saavutti SRI-vasteen verrattuna plaseboa saaneisiin. Vasteet päätetapahtuman yksittäisten osatekijöiden osalta, olivat yhdenmukaisia SRI-vasteen kanssa (taulukko 4).
Taulukko 4. – Pediatrinen vaste viikolla 52
| Vaste1 | Plasebo (n = 40) | Benlysta 10 mg/kg (n = 53) |
SLE-vasteindeksi (%) Vetosuhde (OR; 95 % CI) vs. plasebo | 43,6 (17/39) | 52,8 (28/53) 1,49 (0,64, 3,46) |
| SLE-vasteindeksin osatekijät | ||
Niiden potilaiden osuus, joilla SELENA‑ SLEDAI laski ≥ 4 (%) Vetosuhde (OR; 95 % CI) vs. plasebo | 43,6 (17/39) | 54,7 (29/53) 1,62 (0,69, 3,78) |
Niiden potilaiden osuus, joiden BILAG-indeksi ei huonontunut (%) Vetosuhde (OR; 95 % CI) vs. plasebo | 61,5 (24/39) | 73,6 (39/53) 1,96 (0,77, 4,97) |
Niiden potilaiden osuus, joiden PGA-indeksi ei huonontunut (%) Vetosuhde (OR; 95 % CI) vs. plasebo | 66,7 (26/39) | 75,5 (40/53) 1,70 (0,66, 4,39) |
1Analyyseistä poissuljettiin kaikki tapaukset joista puuttui minkä tahansa osatekijän arviointi lähtötilanteessa (1 plasebo).
Potilailla, joilla oli vaikea aktivoituminen/pahenemisvaihe, mediaaniaika ensimmäiseen vaikeaan aktivoitumiseen oli tutkimuksen päivä 150 Benlysta-ryhmässä ja päivä 113 lumelääkeryhmässä. Vaikeita aktivoitumisia havaittiin 17,0 %: lla Benlysta-ryhmässä verrattuna 35,0 %: iin lumelääkeryhmässä 52 havaintoviikon aikana (havaittu ero hoitojen välillä = 18,0 %; riskisuhde = 0,36; 95 % CI: 0,15, 0,86). Tämä oli linjassa suonensisäisistä aikuistutkimuksista tehtyjen havaintojen kanssa.
Kansainvälisen pediatristen reumatutkimusten yhdistyksen/American College of Rheumatologyn (PRINTO/ACR) asettamia nuorten SLE-vasteen arviointikriteereitä käytettäessä suurempi osuus Benlysta-valmistetta saaneista pediatrisista potilaista osoitti paranemista verrattuna plaseboon (taulukko 5).
Taulukko 5. – PRINTO/ACR vaste viikolla 52
| Niiden potilaiden osuus, joilla vähintään 50 %:n paranema kahdessa viidestä osatekijästä1 ja yli 30 %:n huononema enintään yhdessä jäljellä olevista | Niiden potilaiden osuus, joilla vähintään 30 %:n paranema kolmessa viidestä osatekijästä1 ja yli 30 %:n huononema enintään yhdessä jäljellä olevista | |||
Plasebo n = 40 | Benlysta 10 mg/kg n = 53 | Plasebo n = 40 | Benlysta 10 mg/kg n = 53 | |
| Vaste, n (%) | 14/40 (35,0) | 32/53 (60,4) | 11/40 (27,5) | 28/53 (52,8) |
| Havaittu ero vs. plasebo | 25,38 | 25,33 | ||
| Vetosuhde (95 % CI) vs. plasebo | 2,74 (1,15, 6,54) | 2,92 (1,19, 7,17) | ||
1Viisi PRINTO/ACR-osatekijää olivat prosentuaalinen muutos seuraavissa muuttujissa viikolla 52: vanhempien yleinen arviointi (vanhemman GA), PGA, SELENA SLEDAI-pisteet, vuorokausivirtsan proteinurea, ja lasten elämänlaatua mittaavan kyselyn (PedsQL GC) fyysisen toiminnan pisteet.
Farmakokinetiikka
Alla mainitut laskimoon annetun valmisteen farmakokineettiset parametrit perustuvat niiden 563 SLE-potilaan parametrien arvioihin, jotka saivat Benlystaa 10 mg/painokg kahdessa faasin III tutkimuksessa.
Imeytyminen
Benlysta annetaan laskimonsisäisellä infuusiolla. Belimumabin maksimipitoisuudet seerumissa havaittiin yleensä, kun infuusio lopetettiin tai pian infuusion lopettamisen jälkeen. Korkein pitoisuus seerumissa oli 313 mikrog/ml (vaihteluväli: 173–573 mikrog/ml); tämä on laskettu simuloimalla pitoisuuden aikaprofiilia käyttäen populaatiofarmakokineettisen mallin parametrien tyypillisiä arvoja.
Jakautuminen
Belimumabi jakautui kudoksiin siten, että sen vakaan tilan jakautumistilavuus (Vss) oli noin 5 litraa.
Biotransformaatio
Belimumabi on proteiini, jonka oletettu metaboliareitti on hajoaminen pieniksi peptideiksi ja yksittäisiksi aminohapoiksi laajalti jakautuneiden proteolyyttisten entsyymien avulla. Klassisia biotransformaatiotutkimuksia ei ole tehty.
Eliminaatio
Seerumin belimumabipitoisuudet alenivat bi-eksponentiaalisesti, jakautumisen puoliintumisaika oli 1,75 vuorokautta ja terminaalinen puoliintumisaika 19,4 vuorokautta. Systeeminen puhdistuma oli 215 ml/vrk (vaihteluväli: 69–622 ml/vrk).
SLE‑nefriittitutkimus
Populaatiofarmakokinetiikan analyysi toteutettiin 224 aikuisella SLE-nefriittipotilaalla. Potilaat saivat 10 mg/painokg Benlystaa laskimoon (päivinä 0, 14 ja 28 ja sen jälkeen 28 vrk välein enimmillään 104 viikon ajan). Belimumabin puhdistuma oli SLE‑nefriittipotilailla munuaistaudin aktiivisuuden takia aluksi suurempi kuin SLE‑tutkimuksissa, mutta 24 hoitoviikon jälkeen ja koko loppututkimuksen ajan belimumabin puhdistuma ja altistus olivat samaa luokkaa kuin aikuisilla SLE‑potilailla, jotka saivat 10 mg/painokg Benlystaa laskimoon.
Erityisryhmät
Pediatriset potilaat: Farmakokineettiset parametrit perustuvat 53 potilaan populaatiofarmakokineettisen analyysin yksittäisiin parametriarvioihin pediatrisilla SLE-potilailla tehdyssä tutkimuksessa. Kun lääkettä annettiin 10 mg/painokg päivinä 0, 14 ja 28 ja sen jälkeen 4 viikon välein, belimumabialtistus oli samanlainen lapsilla ja aikuisilla SLE-potilailla. Vakaan tilan geometriset keskiarvot Cmax, Cmin ja AUC-arvoille olivat 305 μg/ml, 42 μg/ml ja 2569 päivää • μg/ml 5-11-vuotiaiden ryhmässä ja 317 μg/ml, 52 μg/ml ja 3126 päivää • μg/ml 12 - 17-vuotiaiden ryhmässä.
Iäkkäät: Benlysta-valmistetta on tutkittu vain pienellä määrällä iäkkäitä potilaita. Ikä ei vaikuttanut belimumabialtistukseen infuusiota saaneiden potilaiden populaatiofarmakokinetiikan analyyseissä. Iän vaikutusta ei kuitenkin voida täysin sulkea pois ottaen huomioon ≥ 65-vuotiaiden potilaiden pieni määrä.
Munuaisten vajaatoiminta: Erillisiä tutkimuksia ei ole tehty selvittämään munuaisten toiminnanvajauksen merkitystä belimumabin farmakokinetiikkaan. Benlystan kliinisen kehitysohjelman aikana Benlystaa tutkittiin potilailla, joilla oli SLE ja munuaisten vajaatoiminta (261 henkilöä, joilla oli kohtalainen munuaisten vajaatoiminta, kreatiniinipuhdistuma ≥ 30 ja < 60 ml/min; 14 henkilöä, joilla oli vakava munuaisten vajaatoiminta, kreatiniinipuhdistuma ≥ 15 ja < 30/min). Populaatiofarmakokinetiikkamallin mukaan arvioituna systeemisen puhdistuman heikentyminen munuaisten vajaatoimintaryhmien keskiarvojen kohdalla verrattuna potilaisiin, joiden kreatiniinipuhdistuma oli populaatiofarmakokinetiikan mediaanin mukainen (79,9 ml/min), oli 1,4 % lievissä tapauksissa (75 ml/min), 11,7 % keskivaikeissa (45 ml/min) ja 24,0 % (22,5 ml/min) vaikeissa munuaisten vajaatoimintatapauksissa. Vaikka proteinurea (≥ 2g/vrk) lisäsi belimumabin puhdistumaa ja kreatiniinipuhdistuman pieneneminen pienensi belimumabin puhdistumaa, nämä vaikutukset olivat odotetun vaihteluvälin sisällä. Sen vuoksi potilaille, joilla on munuaisten vajaatoiminta, ei suositella annosten muuttamista.
Maksan vajaatoiminta: Erillisiä tutkimuksia ei ole tehty selvittämään maksan vajaatoiminnan merkitystä belimumabin farmakokinetiikkaan. IgG1-molekyylit, kuten belimumabi, kataboloituvat laajalti levinneiden proteolyyttisten entsyymien avulla. Nämä entsyymit eivät rajoitu maksakudokseen, ja maksan toiminnan muutokset eivät todennäköisesti vaikuta belimumabin eliminoitumiseen.
Paino/painoindeksi (BMI)
Painon mukaan määritetty belimumabiannostus johtaa alipainoisten henkilöiden (BMI < 18.5) alempaan altistukseen ja ylipainoisten henkilöiden (BMI ≥ 30) suurempaan altistukseen. BMI:stä johtuvat erot altistuksessa eivät johtaneet vastaaviin eroihin tehossa. Ylipainoisten, jotka saivat 10 mg/painokg belimumabia, lisääntynyt altistus ei johtanut yleensä ottaen haittavaikutusten tai vakavien haittavaikutusten määrän lisääntymiseen verrattuna plaseboa saaneisiin. Ylipainoisilla havaittiin kuitenkin enemmän pahoinvointia, oksentelua ja ripulia. Mitkään näistä ruuansulatuskanavan tapahtumista ylipainoisilla eivät olleet vakavia. Annoksen muuttamista ei suositella alipainoisille tai ylipainoisille potilaille.
Siirtyminen laskimoon annettavasta hoidosta ihonalaiseen antoon
SLE
Kun SLE-potilaat siirtyivät laskimoon 4 viikon välein annetuista 10 mg/painokg belimumabiannoksista saamaan 200 mg annoksia ihon alle, ja käytettiin 1–4 viikon vaihtoväliä, annosta edeltävä seerumin belimumabipitoisuus oli ensimmäisen ihon alle annettavan annoksen kohdalla lähellä myöhempää ihonalaisella hoidolla saavutettua vakaan tilan minimipitoisuutta (ks. kohta Annostus ja antotapa).
Populaatiofarmakokinetiikan parametrien simulaatioissa vakaan tilan keskimääräiset belimumabipitoisuudet olivat samanlaiset annettaessa 200 mg ihon alle kerran viikossa (aikuispotilaille ja 5 – < 18‑vuotiaille, ≥ 50 kg painaville pediatrisille potilaille), 10 vrk:n välein (5 – < 18‑vuotiaille, 30 – < 50 kg painaville pediatrisille potilaille) tai 2 viikon välein (5 – < 18‑vuotiaille, 15 – < 30 kg painaville pediatrisille potilaille) kuin annettaessa 10 mg/painokg laskimoon 4 viikon välein.
SLE‑nefriitti
Kun SLE‑nefriittipotilaat siirtyvät laskimoon annettavasta belimumabista (10 mg/painokg) ihon alle annettavaan belimumabiin (200 mg viikossa) 1–2 viikon kuluttua ensimmäisistä kahdesta laskimoon annetusta annoksesta, seerumin keskimääräisten belimumabipitoisuuksien ennustetaan olevan populaatiofarmakokinetiikan simulaatioiden perusteella samaa luokkaa kuin potilailla, jotka saavat 10 mg/painokg laskimoon 4 viikon välein (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta ja reproduktiotoksisuutta koskevien tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Apinoille laskimonsisäisesti ja ihon alle annosteltu belimumabi sai odotetusti aikaan perifeeristen ja imusolukudosten B-solujen määrän vähenemistä, mihin ei liittynyt toksikologisia löydöksiä.
Lisääntymistutkimuksia on tehty kantavilla cynomolgus-apinoilla, jotka saivat belimumabia 150 mg/painokg laskimonsisäisenä infuusiona (noin 9-kertainen altistus verrattuna ihmisen oletettavaan maksimiannokseen) joka toinen viikko aina viikkoon 21 saakka. Belimumabihoidolla ei ollut suoraa tai epäsuoraa haitallista vaikutusta emoon kohdistuvaan toksisuuteen, kehitystoksisuuteen tai teratogeenisuuteen.
Hoitoon liittyviä löydöksiä olivat vain B-solujen odotettavissa oleva, korjautuva väheneminen sekä emoilla että poikasilla ja korjautuva IgM:n väheneminen poikasilla. B-solujen määrä korjaantui belimumabihoidon lopettamisen jälkeen noin 1 vuodessa synnytyksen jälkeen aikuisilla apinoilla ja kolmessa kuukaudessa poikasilla; IgM-tasot poikasilla, jotka olivat altistuneet belimumabille in utero, korjaantuivat kuuden kuukauden ikään mennessä.
Vaikutuksia koiraan ja naaraan hedelmällisyyteen arvioitiin apinoilla kuusi kuukautta kestäneissä toistuvan annoksen toksikologisissa tutkimuksissa, jossa belimumabia annettiin enintään 50 mg/painokg. Hoidosta aiheutuvia muutoksia ei havaittu koiraiden eikä naaraiden lisääntymiselimissä sukukypsillä eläimillä. Epävirallinen kuukautiskierron arviointi ei osoittanut belimumabiin liittyviä muutoksia.
Koska belimumabi on monoklonaalinen vasta-aine, genotoksisuus tutkimuksia ei ole tehty. Karsinogeenisuustutkimuksia tai hedelmällisyystutkimuksia (koirailla tai naarailla) ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Sitruunahappomonohydraatti (E330)
Natriumsitraatti (E331)
Sakkaroosi
Polysorbaatti 80 (E433)
Yhteensopimattomuudet
Benlysta ei ole yhteensopiva 5 %:n glukoosin kanssa.
Lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamattomat injektiopullot
5 vuotta
Liuotettu liuos
Jos liuosta, joka on saatu liuottamalla kuiva-aine injektionesteisiin käytettävään veteen, ei käytetä välittömästi, se on suojattava suoralta auringonvalolta ja säilytettävä jääkaapissa 2 °C–8 °C.
Liuotettu ja laimennettu infuusioneste
Liuosta, jossa Benlysta on laimennettu 9 mg/ml (0,9 %) natriumkloridi-infuusionesteeseen, 4,5 mg/ml (0,45 %) natriumkloridi-infuusionesteeseen tai laktaattipitoiseen Ringerin infuusionesteeseen, voidaan säilyttää 2 °C–8 °C lämpötilassa tai huoneenlämmössä (15 °C–25 °C).
Kokonaisaika Benlystan liuottamisesta infuusion päättymiseen ei saa ylittää kahdeksaa tuntia.
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BENLYSTA infuusiokuiva-aine konsentraattiliuosta varten
120 mg (L:ei) 1 kpl (270,65 €)
400 mg (L:ei) 1 kpl (855,63 €)
PF-selosteen tieto
Benlysta 120 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
120 mg kuiva-ainetta sisältävä injektiopullo (5 ml, tyypin 1 lasia), jossa silikonoitu kumikorkki (klorobutyyli). Pullo on suljettu repäistävällä alumiinisuojuksella.
Pakkauskoko: 1 injektiopullo
Benlysta 400 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
400 mg kuiva-ainetta sisältävä injektiopullo (20 ml, tyypin 1 lasia), jossa silikonoitu kumikorkki (klorobutyyli). Pullo on suljettu repäistävällä alumiinisuojuksella.
Pakkauskoko: 1 injektiopullo
Valmisteen kuvaus:
Valkoinen tai melkein valkoinen jauhe.
Käyttö- ja käsittelyohjeet
120 mg:n infuusionesteen valmistaminen
Liuottaminen
Liuottaminen ja laimentaminen on tehtävä aseptisissa olosuhteissa.
Anna injektiopullon lämmetä 10–15 minuuttia huoneenlämpöiseksi (15 °C–25 °C).
On suositeltavaa käyttää 21-25 gaugen neulaa, kun kumikorkki lävistetään liuottamista ja laimentamista varten.
120 mg belimumabia sisältävän kertakäyttöön tarkoitetun injektiopullon sisältö liuotetaan 1,5 ml:aan injektionesteisiin käytettävää vettä. Tämän liuoksen belimumabipitoisuus on 80 mg/ml.
Vaahtoamisen estämiseksi injektionesteisiin käytettävä vesi on ruiskutettava injektiopullon reunaa kohti. Pyörittele injektiopulloa kevyesti 60 sekuntia. Anna injektiopullon olla huoneenlämmössä (15 °C–25 °C) liuottamisen ajan, pyöritellen sitä kevyesti 60 sekunnin ajan joka viides minuutti, kunnes kuiva-aine on liuennut. Älä ravista. Yleensä kuiva-aine liukenee kokonaan 10–15 minuutissa veden lisäämisestä, mutta joskus se voi viedä jopa 30 minuuttia.
Suojaa liuotettu liuos auringolta.
Jos Benlystan liuottamiseen käytetään mekaanista liuotuslaitetta, vauhti ei saa ylittää 500 kierrosta minuutissa eikä injektiopulloa saa pyörittää pitempään kuin 30 minuuttia.
Benlystan liuottamisen jälkeen liuoksen pitäisi olla opalisoivaa ja väriltään väritöntä tai vaaleankeltaista. Liuoksessa ei pitäisi olla partikkeleita. On kuitenkin todennäköistä ja hyväksyttävää, että liuoksessa on pieniä ilmakuplia.
Liuottamisen jälkeen yhdestä injektiopullosta voidaan vetää 1,5 ml liuosta (vastaten 120 mg belimumabia).
Laimentaminen
Liuotettu lääkevalmiste laimennetaan 250 ml:aan 9 mg/ml (0,9 %) natriumkloridi-infuusionestettä, 4,5 mg/ml (0,45 %) natriumkloridi-infuusionestettä tai laktaattipitoista Ringerin infuusionestettä. Potilaille, joiden paino on 40 kg tai alle, voidaan harkita 100 ml näitä laimennusaineita sisältäviä infuusiopusseja edellyttäen, että tuloksena oleva belimumabin pitoisuus infuusiopussissa ei ylitä 4 mg/ml.
5 % glukoosi-injektioneste ei ole yhteensopiva Benlystan kanssa ja sitä ei saa käyttää.
Ota 250 ml:n (tai 100 ml:n) infuusionestepussista tai -pullosta 9 mg/ml (0,9 %) tai 4,5 mg/ml (0,45 %) natriumkloridi-infuusionestettä tai laktaattipitoista Ringerin infuusionestettä potilaan annokseen tarvittavan liuotetun Benlysta-liuoksen määrää vastaava tilavuus ja hävitä se. Lisää sitten tarvittava määrä liuotettua Benlysta-liuosta infuusiopussiin tai -pulloon. Sekoita liuos kääntämällä pussi tai pullo kevyesti toisin päin. Injektiopulloihin käyttämättä jäävä liuos on hävitettävä.
Ennen Benlystan antamista potilaalle tarkista liuoksen ulkonäkö mahdollisten partikkeleiden ja värivirheiden havaitsemiseksi. Hävitä liuos, jos havaitset siinä partikkeleita tai värivirheen.
Kokonaisaika Benlystan liuottamisesta infuusion päättymiseen ei saa ylittää kahdeksaa tuntia.
400 mg:n infuusionesteen valmistaminen
Liuottaminen
Liuottaminen ja laimentaminen on tehtävä aseptisissa olosuhteissa.
Anna injektiopullon lämmetä 10–15 minuuttia huoneenlämpöiseksi (15 °C–25 °C).
On suositeltavaa käyttää 21-25 gaugen neulaa, kun kumikorkki lävistetään liuottamista ja laimentamista varten.
400 mg belimumabia sisältävän kertakäyttöön tarkoitetun injektiopullon sisältö liuotetaan 4,8 ml:aan injektionesteisiin käytettävää vettä. Tämän liuoksen belimumabipitoisuus on 80 mg/ml.
Vaahtoamisen estämiseksi injektionesteisiin käytettävä vesi on ruiskutettava injektiopullon reunaa kohti. Pyörittele injektiopulloa kevyesti 60 sekuntia. Anna injektiopullon olla huoneenlämmössä (15 °C–25 °C) liuottamisen ajan, pyöritellen sitä kevyesti 60 sekunnin ajan joka viides minuutti, kunnes kuiva-aine on liuennut. Älä ravista. Yleensä kuiva-aine liukenee kokonaan 10–15 minuutissa veden lisäämisestä, mutta joskus se voi viedä jopa 30 minuuttia.
Suojaa liuotettu liuos auringolta.
Jos Benlystan liuottamiseen käytetään mekaanista liuotuslaitetta, vauhti ei saa ylittää 500 kierrosta minuutissa eikä injektiopulloa saa pyörittää pitempään kuin 30 minuuttia.
Benlystan liuottamisen jälkeen liuoksen pitäisi olla opalisoivaa ja väriltään väritöntä tai vaaleankeltaista. Liuoksessa ei pitäisi olla partikkeleita. On kuitenkin todennäköistä ja hyväksyttävää, että liuoksessa on pieniä ilmakuplia.
Liuottamisen jälkeen yhdestä injektiopullosta voidaan vetää 5 ml liuosta (vastaten 400 mg belimumabia).
Laimentaminen
Liuotettu lääkevalmiste laimennetaan 250 ml:aan 9 mg/ml (0,9 %) natriumkloridi-infuusionestettä, 4,5 mg/ml (0,45 %) natriumkloridi-infuusionestettä tai laktaattipitoista Ringerin infuusionestettä.
5 % glukoosi-injektioliuos ei ole yhteensopiva Benlystan kanssa ja sitä ei saa käyttää.
Ota 250 ml:n infuusionestepussista tai -pullosta 9 mg/ml (0,9 %) tai 4,5 mg/ml (0,45 %) natriumkloridi-infuusionestettä tai laktaattipitoista Ringerin infuusionestettä potilaan annokseen tarvittavan liuotetun Benlysta-liuoksen määrää vastaava tilavuus ja hävitä se. Lisää sitten tarvittava määrä liuotettua Benlysta-liuosta infuusiopussiin tai -pulloon. Sekoita liuos kääntämällä pussi tai pullo kevyesti toisin päin. Injektiopulloihin käyttämättä jäävä liuos on hävitettävä.
Ennen Benlystan antamista potilaalle tarkista liuoksen ulkonäkö mahdollisten partikkeleiden ja värivirheiden havaitsemiseksi. Hävitä liuos, jos havaitset siinä partikkeleita tai värivirheen.
Kokonaisaika Benlystan liuottamisesta infuusion päättymiseen ei saa ylittää kahdeksaa tuntia.
Antotapa
Benlysta annetaan tunnin kestävänä infuusiona.
Benlystaa ei saa antaa infuusiona samanaikaisesti muiden lääkevalmisteiden kanssa saman infuusioletkun kautta. Benlystan antamista samanaikaisesti muiden lääkevalmisteiden kanssa ei ole selvitetty fysikaalisilla tai biokemiallisilla yhteensopivuustutkimuksilla.
Benlystan ja polyvinyylikloridi- tai polyolefiinipussien välillä ei ole havaittu yhteensopimattomuutta.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BENLYSTA infuusiokuiva-aine konsentraattiliuosta varten
120 mg 1 kpl
400 mg 1 kpl
- Ei korvausta.
ATC-koodi
L04AG04
Valmisteyhteenvedon muuttamispäivämäärä
25.09.2025
Yhteystiedot
 GLAXOSMITHKLINE OY
GLAXOSMITHKLINE OY Porkkalankatu 20 A
00180 Helsinki
010 303 030
www.glaxosmithkline.fi