
FIBRYGA injektio-/infuusiokuiva-aine ja liuotin, liuosta varten 1 g
Vaikuttavat aineet ja niiden määrät
Ihmisen fibrinogeeni
Yksi FIBRYGA-pullo sisältää 1 g ihmisen fibrinogeeniä. Kun FIBRYGA on saatettu käyttökuntoon 50 ml:lla vettä injektiota varten, se sisältää noin 20 mg/ml ihmisen fibrinogeeniä.
Hyytyvän proteiinin pitoisuus on määritetty Euroopan farmakopean ihmisen fibrinogeeniä koskevan ohjeen mukaisesti.
Tuotettu ihmisluovuttajien plasmasta.
Apuaineet, joiden vaikutus tunnetaan: natriumia enintään 132 mg (5,8 mmol) pulloa kohti.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektio- / infuusiokuiva-aine ja liuotin liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Synnynnäistä hypofibrinogenemiaa tai afibrinogenemiaa sairastavien potilaiden verenvuototilanteiden hoito ja perioperatiivinen profylaksia, kun sairauteen liittyy verenvuototaipumus.
Täydentävänä hoitona hankitusta hypofibrinogenemiasta kärsivien potilaiden hallitsemattoman vakavan verenvuodon hoitamiseksi leikkaustoimenpiteiden aikana.
Ehto
Hoito tulee aloittaa hyytymishäiriöihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito on aloitettava hyytymishäiriöiden hoitoon perehtyneen lääkärin valvonnassa.
Annostus
Korvaushoidon annostus ja kesto riippuvat häiriön vakavuudesta, verenvuodon sijainnista ja määrästä ja potilaan kliinisestä tilasta.
(Toiminnallinen) fibrinogeenipitoisuus on määritettävä yksilöllisen annoksen laskemiseksi, ja annosmäärä ja -väli on määritettävä potilaskohtaisesti mittaamalla säännöllisesti fibrinogeenin pitoisuutta plasmassa ja seuraamalla jatkuvasti potilaan kliinistä tilaa ja muuta käytettyä korvaushoitoa.
Suuren leikkaustoimenpiteen yhteydessä korvaushoidon täsmällinen seuranta hyytymiskokeilla on oleellista.
1. Synnynnäistä hypofibrinogenemiaa tai afibrinogenemiaa sairastavien ja vuoto-oireisten potilaiden verenvuodon profylaksia.
Profylaktinen hoito liiallisen verenvuodon estämiseksi kirurgisten toimenpiteiden aikana on suositeltavaa, jotta fibrinogeenin pitoisuus saadaan nostettua 1 g:aan/l ja pidettyä tällä tasolla, kunnes hemostaasi on turvattu, ja yli 0,5 g/l, kunnes haavan paraneminen on täydellistä.
Leikkaustoimenpiteen tai verenvuototilanteen hoidon yhteydessä annos on laskettava seuraavalla tavalla:
Annos (mg/kg kehon painoa) = [kohdepitoisuus (g/l) - mitattu pitoisuus (g/l)]
0,018 (g/l mg/kg kehon painoa kohti)
Tätä seuraava annostus (annokset ja injektioväli) on sovitettava potilaan kliinisen tilan ja laboratoriotulosten perusteella.
Fibrinogeenin biologinen puoliintumisaika on 3‑4 päivää. Näin ollen jos kulutusta ei ole, toistuva hoito ihmisen fibrinogeenillä ei yleensä ole tarpeen. Ehkäisevässä käytössä toistuvassa annostelussa tapahtuva kertyminen huomioon ottaen annos ja annosväli on määritettävä lääkärin tietylle potilaalle asettamien hoitotavoitteiden mukaisesti.
Pediatriset potilaat
Kirurgisen toimenpiteen yhteydessä tai hoidettaessa verenvuototilannetta nuorten annos lasketaan yllä kuvatun aikuisten kaavan mukaan, ja < 12-vuotiaiden lasten annos lasketaan seuraavalla tavalla:
Annos (mg/kg kehon painoa) = [kohdepitoisuus (g/l) - mitattu pitoisuus (g/l)]
0,014 (g/l mg/kg kehon painoa kohti)
Tätä seuraava annostus on sovitettava potilaan kliinisen tilan ja laboratoriotulosten perusteella.
Iäkkäät potilaat
FIBRYGA:n kliinisissä tutkimuksissa ei ollut mukana 65‑vuotiaita ja tätä vanhempia potilaita. Näin ollen varmaa näyttöä siitä, onko heillä erilainen vaste kuin nuoremmilla potilailla ei ole.
2. Verenvuodon hoito
Verenvuoto synnynnäistä hypofibrinogenemiaa tai afibrinogenemiaa sairastaville potilaille
Verenvuototilanteita on hoidettava yllä olevien aikuisia/nuoria ja lapsia koskevien kaavojen mukaan, jotta saavutetaan suositeltu 1 g/l fibrinogeenipitoisuus plasmassa. Tämä pitoisuus on säilytettävä, kunnes hemostaasi on turvattu.
Hankittua fibrinogeenipuutosta sairastavien potilaiden verenvuoto
Aikuiset
Yleensä aluksi annetaan 1-2 g infuusio ja sen jälkeen tarvittaessa lisäinfuusioita. Vakavan verenvuodon yhteydessä, -esim. suuri leikkaustoimenpide, saatetaan tarvita suurempia annoksia (4-8 g) fibrinogeeniä.
Pediatriset potilaat
Annostus määritetään kehon painon ja kliinisen tarpeen perusteella, mutta on tavallisesti 20-30 mg/kg.
Antotapa
Infuusio tai injektio laskimoon.
FIBRYGA on annettava laskimoon hitaasti suositellulla enimmäisnopeudella 5 ml/min potilaille, joilla on synnynnäinen hypo- tai afibrinogenemia ja suositellulla enimmäisnopeudella 10 ml/min potilaille, joilla on hankittu fibrinogeenipuutos.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Tromboembolia
Tromboosin riski on olemassa, kun joko synnynnäisestä tai hankitusta puutoksesta kärsiviä potilaita hoidetaan ihmisen fibrinogeenilllä erityisesti suurella annoksella tai toistuvalla annostelulla. Potilaita, joille on annettu ihmisen fibrinogeeniä, on seurattava tarkasti tromboosin merkkien ja oireiden varalta.
Potilailla, joilla on aiemmin ollut sepelvaltimotauti tai sydäninfarkti, potilailla, joilla on maksasairaus, peri- ja post-operatiivisilla potilailla, vastasyntyneillä tai potilailla, joilla on tromboemboliatapahtumien tai DIC-oireyhtymän riski, ihmisen plasman fibrinogeenillä toteutettavan hoidon mahdollista hyötyä on punnittava tromboembolisten komplikaatioiden riskiä vasten. Varovaisuutta ja tarkkaa seurantaa on noudatettava.
Hankittuun hypofibrinogenemiaan liittyvät kaikkien hyytymistekijöiden (ei ainoastaan fibrinogeenin) ja inhibiittorien matalat plasmapitoisuudet, ja sen vuoksi tulisi harkita hoitoa hyytymistekijöitä sisältävillä verivalmisteilla. Hyytymisjärjestelmän huolellinen seuranta on tarpeen.
Allergiset tai anafylaksin kaltaiset reaktiot
Jos allergisia tai anafylaktian kaltaisia reaktioita esiintyy, injektio/infuusio on keskeytettävä välittömästi. Anafylaktisessa sokkitilanteessa noudatetaan tavanomaisia sokin hoito-ohjeita.
Natriumpitoisuus
Tämä lääkevalmiste sisältää enintään 132 mg natriumia pulloa kohti, mikä vastaa 6,6 prosenttia WHO:n suosittelemasta natriumin päivittäisestä 2 g:n enimmäissaannista aikuisella. Potilaiden, joiden natriuminsaantia on rajoitettu, on otettava tämä huomioon.
Virusturvallisuus
Tavanomaisia keinoja ehkäistä ihmisen verestä tai plasmasta valmistettujen lääkkeiden aiheuttamia infektioita ovat luovuttajien valinta, yksittäisten luovutuserien ja plasmapoolien seulonta spesifisiä infektiomarkkereita silmällä pitäen ja tehokkaiden tuotantovaiheiden käyttöönotto virusten inaktivoimiseksi/poistamiseksi. Tästä huolimatta ihmisen verestä tai plasmasta valmistettuja lääkkeitä annettaessa ei tartuntavaarallisten tekijöiden siirtymisen mahdollisuutta voida kokonaan sulkea pois. Tämä koskee myös tuntemattomia tai uusia viruksia ja muita taudinaiheuttajia.
Käytössä olevia varotoimenpiteitä pidetään tehokkaina vaipallisia viruksia, kuten HIV, HBV ja HCV vastaan, sekä vaipatonta virusta HAV vastaan. Käytössä olevien varotoimenpiteiden hyöty vaipattomia viruksia, kuten parvovirus B19:ää, vastaan voi olla rajallista. Parvovirus B19 -infektio voi olla raskaana olevilla naisilla (sikiön infektio) ja immuunipuutteesta tai lisääntyneestä erytropoieesista kärsivillä henkilöillä (esim. hemolyyttinen anemia) vakava.
Asianmukaista rokotusta (A- ja B-hepatiitti) on harkittava potilailla, jotka saavat säännöllisesti/toistuvasti ihmisen plasmasta peräisin olevia tuotteita.
Immunogeenisuus
Kun hyytymistekijöiden korvaushoitoa on annettu muissa synnynnäisissä puutoksissa, on havaittu vasta-ainereaktioita, mutta tietoja fibrinogeenikonsentraatin immunogeenisyydestä ei tällä hetkellä ole.
Yhteisvaikutukset
Ihmisen fibrinogeenituotteilla ei tiedetä olevan yhteisvaikutuksia muiden lääkkeiden kanssa.
Raskaus ja imetys
Raskaus
FIBRYGA-valmisteen turvallisuutta ihmisillä raskauden aikana ei ole tutkittu kontrolloiduissa kliinisissä tutkimuksissa. Fibrinogeenivalmisteilla synnytyskomplikaatioiden hoidossa saadun kliinisen kokemuksen perusteella haitallisia vaikutuksia raskauden kulkuun tai sikiön tai vastasyntyneen terveyteen ei ole odotettavissa. FIBRYGA-valmisteella ei ole tehty eläinkokeita lisääntymistoksisuuden selvittämiseksi (ks. kohta Prekliiniset tiedot turvallisuudesta). Koska vaikuttava aine on peräisin ihmisestä, se kataboloituu samalla tavalla kuin potilaan oma proteiini. Näiden ihmisveren fysiologisten rakenneosien ei odoteta aiheuttavan lisääntymiseen tai sikiöön kohdistuvia haittavaikutuksia.
FIBRYGA-valmisteen hyöty raskauden aikana on arvioitava ottaen huomioon, että kliinistä kokemusta fibrinogeenikonsentraateista on saatavilla, mutta tietoja kontrolloiduista kliinisistä tutkimuksista puuttuu.
Imetys
Ei tiedetä, erittyykö FIBRYGA ihmisen rintamaitoon. Vaikuttavan aineen luonteen vuoksi, ei ole odotettavissa vaikutuksia vastasyntyneisiin/imeväisiin.
Näin ollen on päätettävä, käytetäänkö FIBRYGA-hoitoa imetyksen aikana, ottaen huomioon imetyksen hyödyt lapselle ja hoidon hyödyt naiselle.
Hedelmällisyys
Hedelmällisyyttä koskevia tietoja ei ole saatavilla.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
FIBRYGA-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Turvallisuustietojen yhteenveto
Haittavaikutusten esiintymistiheydestä ei ole vankkaa tietoa tämän valmisteen kliinisistä tutkimuksista.
Kliinisissä tutkimuksissa raportoitiin seuraavia haittavaikutuksia: kuume, lääkeihottuma, laskimotulehdus ja tromboosi.
FIBRYGA-valmisteella ja muilla fibrinogeenitiivisteillä on raportoitu seuraavia haittavaikutuksia:
MedDRA-elinjärjestelmäluokka | Haittavaikutukset | Esiintymistiheys* |
Immuunijärjestelmä: | Allergiset tai anafylaksian kaltaiset reaktiot Ihoreaktiot | Tuntematon |
Verisuonisto: | Tromboemboliatilanteet (mukaan lukien sydäninfarkti ja keuhkoembolia) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) Tromboflebiitti | Tuntematon |
Yleisoireet ja antopaikassa todettavat haitat: | Ruumiinlämmön nousu (kuume) | Tuntematon |
* Esiintymistiheyttä ei tunneta, koska sitä ei voida laskea käytettävissä olevien tietojen perusteella. Lievä kuume ja ihoreaktiot olivat yksittäisiä tapauksia kliinisissä tutkimuksissa. Allergiset tai anafylaksian kaltaiset reaktiot, tromboemboliatilanteet (mukaan lukien sydäninfarkti ja keuhkoembolia) ja tromboflebiitti ovat luokkavaikutuksia. | ||
Tartuntavaarallisia tekijöitä koskeva turvallisuus, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Pediatriset potilaat
Kaksikymmentäkuusi 1–< 18-vuotiasta potilasta otettiin mukaan synnynnäistä fibrinogeenin puutosta koskevaan turvallisuusanalyysiin, ja heistä 12 nuorta oli 12–< 18-vuotiaita, 8 lasta 6–< 12-vuotiaita ja 6 lasta 1–<6-vuotiaita.
Turvallisuusprofiilissa ei kaiken kaikkiaan ole eroa aikuisten, nuorten ja lasten välillä.
Tietoa FIBRYGA-valmisteen käytöstä pediatrisille potilaille, joilla on hankittu fibrinogeenipuutos, ei ole.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostuksen välttämiseksi on aiheellista seurata säännöllisesti fibrinogeenin pitoisuutta plasmassa (ks. kohta Annostus ja antotapa).
Yliannostustapauksessa tromboembolisten komplikaatioiden kehittymisen riski on kohonnut.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: verenvuotoa estävät lääkkeet, fibrinogeeni, ATC-koodi: B02BB01
Ihmisen fibrinogeeni (hyytymistekijä I) muuttuu trombiinin, aktivoidun hyytymistekijän XIII (FXIIIa) ja kalsiumionien läsnä ollessa vakaaksi ja elastiseksi, kolmiulotteiseksi verenvuotoa tyrehdyttäväksi fibriinihyytymäksi.
Ihmisen fibrinogeenin antaminen nostaa fibrinogeenin pitoisuutta plasmassa ja voi ohimenevästi korjata fibrinogeenin puutoksesta kärsivien potilaiden hyytymisvajetta.
Avoin, prospektiivinen, satunnaistettu, kontrolloitu kahden tutkimusryhmän kerta-annoksen vaihtovuoroinen vaiheen 2 farmakokineettinen tutkimus kahdellakymmenelläkahdella potilaalla, jotka kärsivät synnynnäisestä fibrinogeenin puutoksesta (afibrinogenemia) (ks. kohta Farmakokinetiikka), arvioi myös hyytymän enimmäislujuutta (maximum clot firmness, MCF) biologisena merkkinä verenvuodon tyrehdyttämisen tehokkuudesta (FORMA-01). MCF määritettiin tromboelastometrisellä testillä (ROTEM). MCF määritettiin jokaiselle potilaalle ennen FIBRYGA-valmisteen kerta-annoksen antoa (lähtötaso) ja tunti sen jälkeen. MCF-arvot olivat merkitsevästi korkeampia FIBRYGA-valmisteen annon jälkeen kuin lähtötasolla (ks. alla oleva taulukko).
Taulukko 1: Hyytymän enimmäislujuus MCF [mm] (ITT-populaatio) n=22
Aikapiste | Keskiarvo ± keskihajonta | Mediaani (alue) |
Ennen infuusiota | 0 ± 0 | 0 (0-0) |
1 tunti infuusion jälkeen | 9,7± 3,0 | 10,0 (4,0-16,0) |
Keskimääräinen muutos (ensisijainen analyysi)* | 9,7± 3,0 | 10,0 (4,0-16,0) |
MCF = hyytymän enimmäislujuus; ITT = kaikki satunnaistetut potilaat. | ||
Prospektiivinen, avoin, kontrolloimaton, vaiheen 3 monikeskustutkimus (FORMA-02) tehtiin 25 potilaalla, jotka olivat 12–54-vuotiaita (6 nuorta, 19 aikuista) ja jotka kärsivät synnynnäisestä fibrinogeenin puutoksesta (afibrinogenemia ja hypofibrinogenemia). Tähän sisältyi 89 verenvuototilanteen hoito ja 12 kirurgista toimenpidettä. Hyytymän enimmäislujuudessa oli ROTEM:lla ja fibrinogeenin plasmapitoisuudella mitattuna merkitsevä ero lähtötasoon nähden. Verenvuototilanteiden hoidossa käytetty FIBRYGA-mediaaniannos infuusiota kohden oli 57,5 mg/kg ja kokonaisannoksen mediaani oli 59,4 mg/kg. FIBRYGA-kokonaisannoksen mediaani kirurgista toimenpidettä kohden oli 85,8 mg/kg. Riippumaton arviointikomitea arvioi objektiivista pisteytysjärjestelmää käyttäen hemostaattisen tehon kokonaisuudessaan onnistuneeksi (teho arvioitiin hyväksi tai erinomaiseksi) 98,9 %:ssa hoidetuista verenvuototapahtumista ja 100 %:ssa kirurgisista toimenpiteistä.
Toisessa prospektiivisessa, avoimessa, kontrolloimattomassa vaiheen 3 monikeskustutkimuksessa (FORMA-04) tutkittiin 14 lasta, joilla oli synnynnäinen fibrinogeenin puutos (afibrinogenemia ja hypofibrinogenemia) ja jotka olivat iältään 1–10-vuotiaita (kuusi < 6-vuotiasta ja kahdeksan 6–< 12-vuotiasta). Tähän sisältyi 10 verenvuototilanteen hoito ja 3 kirurgista toimenpidettä sekä kerta-annoksen farmakokinetiikan tarkastelu. Hyytymän enimmäislujuudessa oli ROTEM:lla ja fibrinogeenin pitoisuudella plasmassa mitattuna merkitsevä ero lähtötasoon nähden. Verenvuototilanteiden hoidossa käytetty FIBRYGA-mediaaniannos infuusiota kohden oli 70,2 mg/kg ja kokonaisannoksen mediaani oli 73,9 mg/kg. FIBRYGA-kokonaisannoksen mediaani kirurgista toimenpidettä kohden oli 108 mg/kg. Riippumaton arviointikomitea arvioi objektiivista pisteytysjärjestelmää käyttäen hemostaattisen tehon kokonaisuudessaan onnistuneeksi (teho arvioitiin hyväksi tai erinomaiseksi) 100 %:ssa hoidetuista verenvuototapahtumista ja kirurgisista toimenpiteistä.
Prospektiivisessa, satunnaistetussa, kontrolloidussa FORMA-05-tutkimuksessa tutkittiin FIBRYGA-valmisteen hemostaattista tehoa ja turvallisuutta vertaamalla kryopresipitaattiin fibrinogeenin täydennyslähteinä potilailla, joille kehittyi hankittu fibrinogeenipuutos sytoreduktiivisessa leikkauksessa levinneen vatsan alueen pahalaatuisen pseudomyxoma peritonein hoitamiseksi. Tutkimukseen sisältyi 43 aikuista potilasta koko hoidon läpikäyneiden (PP-) analyysisarjassa, 21 potilasta sai FIBRYGA-valmistetta ja 22 potilasta kryopresipitaattia. Intraoperatiivinen fibrinogeenin lisäys suoritettiin ennaltaehkäisevästi (ts. kun leikkausta oli jatkunut 60-90 minuuttia, kun havaittiin runsasta verenhukkaa, mutta ennen kuin 2 litraa verta oli menetetty) 4 g FIBRYGA-annoksin tai kahtena kryopresipitaatin viiden yksikön annoksena, toistettuna tarvittaessa. Leikkauksen 7,8 ± 1,7 tunnin aikana käytettiin 6,5 ± 3 g FIBRYGA-valmistetta (89 ± 39 mg/painokilo) tai vastaavasti 4,1 ± 2,2 kappaletta kryopresipitaatin viiden yksikön annosta. Mediaaniarvoltaan 1 yksikkö ja 0,5 yksikköä RBC:tä annettiin intraoperatiivisesti potilaille, joita hoidettiin vastaavasti FIBRYGA-valmisteella ja kryopresipitaatilla, ja mediaaniarvoltaan 0 yksikköä RBC:tä leikkauksen jälkeisten ensimmäisten 24 tunnin aikana molemmissa ryhmissä (ks. alla oleva taulukko). Tuoretta jäädytettyä plasmaa tai verihiutalekonsentraatteja ei siirretty tutkimuksen aikana. Riippumaton arviointikomitea arvioi objektiivista pisteytysjärjestelmää käyttäen fibrinogeenin täydennykseen perustuvan hemostaattisen hoidon onnistuneeksi 100 %:ssa leikkauksista molemmissa ryhmissä.
Taulukko 2: RBC*-transfuusio [yksiköitä] intraoperatiivisesti ja ensimmäisten 24 tunnin aikana postoperatiivisesti (PP-populaatio)
| Aikajakso | FIBRYGA-ryhmä (n=21) | Kryopresipitaattiryhmä (n=22) |
|---|---|---|
| Intraoperatiivisesti | 1 (0-4) | 0,5 (0-5) |
| Ensimmäiset 24 tuntia postoperatiivisesti | 0 (0-2) | 0 (0-2) |
| RBC = punasolukonsentraatit; PP = koko hoidon läpikäyneet. *muiden allogeenisten verituotteiden, kuten tuoreen jäädytetyn plasman tai verihiutalekonsentraattien transfuusiota ei tapahtunut | ||
Pediatriset potilaat
Synnynnäisen fibrinogeenin puutoksen yhteydessä FIBRYGA-valmistetta annettiin kahdessa kliinisessä tutkimuksessa (FORMA-02 ja FORMA-04) kahdellekymmenelle 1–< 18-vuotiaalle potilaalle, joista kuusi oli 12–< 18-vuotiaita nuoria, kahdeksan 6–< 12-vuotiaita lapsia ja kuusi 1–< 6-vuotiaita lapsia. Riippumaton arviointikomitea arvioi hemostaattisen tehon onnistuneeksi kaikissa hoidetuissa verenvuototilanteissa (kymmenen verenvuototilannetta nuorilla, viisi 6–< 12-vuotiailla lapsilla ja viisi 1–< 6-vuotiailla lapsilla); myös profylaksia arvioitiin yhtä onnistuneeksi neljässä näille potilaille tehdyissä kirurgisissa toimenpiteissä (yksi nuorelle ja kolme 1–< 6-vuotiaille lapsille).
Farmakokinetiikka
Ihmisen fibrinogeeni on ihmisen plasman normaali rakenneosa ja toimii endogeenisen fibrinogeenin tavoin. Fibrinogeenin biologinen puoliintumisaika plasmassa on 3‑4 päivää. FIBRYGA-valmiste annetaan laskimoon, ja se on välittömästi saatavilla annettua annosta vastaavassa pitoisuudessa plasmassa.
Avoin, prospektiivinen, satunnaistettu, kontrolloitu kahden tutkimusryhmän vaihtovuoroinen vaiheen 2 tutkimus kahdellakymmenelläkahdella potilaalla, jotka kärsivät synnynnäisestä fibrinogeenin puutoksesta (afibrinogenemia) ja olivat 12‑53-vuotiaita (6 nuorta, 16 aikuista), vertasi FIBRYGA-valmisteen kerta-annoksen farmakokineettisiä ominaisuuksia toisen kaupallisesti saatavilla olevan fibrinogeenitiivisteen vastaaviin samoilla potilailla (FORMA-01). Jokainen potilas sai laskimoonkerta-annoksen 70 mg/kg FIBRYGA-valmistetta ja vertailutuotetta. Fibrinogeenin aktiivisuuden määrittämiseksi otettiin verinäytteitä lähtötasolla ja aina 14. päivään infuusiosta. FIBRYGA-valmisteen farmakokineettiset parametrit koko hoidon läpikäyneiden (PP-) analyysissä (n=21) on esitetty yhteenvetona alla olevassa taulukossa.
Taulukko 3: Fibrinogeenin aktiivisuuden farmakokineettiset parametrit (n=21) (PP-populaatio*)
Parametri | Keskiarvo ± keskihajonta | Alue |
Puoliintumisaika [h] | 75,9 ± 23,8 | 40,0–157,0 |
Cmax [mg/dl] | 139,0 ± 36,9 | 83,0–216,0 |
AUCnorm annoksella 70 mg/kg [mg*h/ml] | 113,7 ± 31,5 | 59,7–175,5 |
Puhdistuma [ml/h/kg] | 0,67 ± 0,2 | 0,4–1,2 |
Keskimääräinen jäämäaika [h] | 106,3 ± 30,9 | 58,7–205,5 |
Jakautumistilavuus tasapainotilassa [ml/kg] | 70,2 ± 29,9 | 36,9–149,1 |
*Yksi potilas suljettiin pois PP-populaatiosta, koska tämä sai <90 % suunnitellusta FIBRYGA- ja vertailutuotteen annoksesta. | ||
Saanto (ns. in vivo recovery, IVR) määritettiin neljä tuntia infuusion jälkeen saakka saaduista pitoisuuksista. Saannon mediaani oli 1,8 mg/dl (alue 1,08‑2,62 mg/dl) nousua mg/kg kohti. Saannon mediaani viittaa siihen, että 70 mg/kg annos nostaa potilaan plasman fibrinogeenipitoisuutta noin 125 mg/dl.
Farmakokinetiikka erityisryhmissä
Tutkimukseen osallistuneiden miesten ja naisten välillä ei havaittu tilastollisesti merkittävää eroa fibrinogeenin aktiivisuudessa.
Pediatriset potilaat
FORMA-02-tutkimuksesta saatiin farmakokineettistä tietoa nuorista, jotka olivat iältään 12-vuotiaista alle 18-vuotiaisiin. PP-analyysissä havaittiin pieni ero nuorten (n = 5) ja aikuisten (n = 16) puoliintumisajassa, jotka olivat nuorilla 72,8 ± 16,5 tuntia ja aikuisilla 76,9 ± 26,1 tuntia. Puhdistuma oli lähes identtistä kummassakin ikäryhmässä, eli 0,68 ± 0,18 ml/h/kg nuorilla ja 0,66 ± 0,21 ml/h/kg aikuisilla.
FIBRYGA-valmisteen farmakokinetiikkaa tutkittiin edelleen FORMA-04-tutkimuksessa 13 lapsella, jotka olivat iältään alle 12-vuotiaita ja joilla oli synnynnäinen fibrinogeenin puutos (afibrinogenemia). Kaikki potilaat saivat kerta-annoksen 70 mg/kg FIBRYGA-valmistetta laskimoon. FIBRYGA-valmisteen farmakokineettiset parametrit esitetään yhteenvetona alla olevassa taulukossa. Saannon (IVR) mediaani oli 1,4 mg/dl (vaihteluväli 1,3–2,1 mg/dl) nousua mg/kg kohti.
Taulukko 4. Fibrinogeenin aktiivisuuden farmakokineettiset parametrit (n = 13)
Parametri | Keskiarvo + keskihajonta | Vaihteluväli |
Puoliintumisaika [h]* | 63,3 ± 12,0 | 45,6–91,6 |
Cmax [mg/dl] | 107,2 ± 16,8 | 93,0–154,0 |
AUCnorm annoksella 70 mg/kg [mg*h/ml]* | 92,0 ± 20,0 | 69,7–134,2 |
Puhdistuma [ml/h/kg]* | 0,8 ± 0,2 | 0,5–1,0 |
Keskimääräinen jäämäaika [h]* | 88,0 ± 16,8 | 63,6–126,7 |
Jakautumistilavuus tasapainotilassa [ml/kg]* | 67,6 ± 7,1 | 52,8–76,8 |
* Laskettu 10 potilaalla 13:sta, koska 3 potilaalla määrällisten arvojen lukumäärä oli riittämätön. | ||
Prekliiniset tiedot turvallisuudesta
FIBRYGA-valmisteen turvallisuus on osoitettu useissa nonkliinisissä turvallisuusfarmakologisissa (sydämeen ja verisuoniin liittyvät vaikutukset, trombogeeninen potentiaali) ja toksisuustutkimuksissa (akuutti toksisuus, paikallinen siedettävyys). Näiden tutkimusten perusteella nonkliiniset tiedot eivät paljasta erityistä vaaraa ihmisille. Laskimostaasitestissä (Wesslerin testi) FIBRYGA osoittautui ei-trombogeeniseksi aina 400 mg/kg kehon painoa annokseen asti.
Farmaseuttiset tiedot
Apuaineet
Jauhe
L-arginiinihydrokloridi
Glysiini
Natriumkloridi
Natriumsitraattidihydraatti
Liuotin
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta.
Käyttökuntoon saatetun liuoksen kemiallinen ja fysikaalinen käyttöstabiilisuus on osoitetusti 24 tuntia huoneenlämmössä (enintään 25 °C). Mikrobiologisesta näkökulmasta katsottuna tuote on käytettävä välittömästi käyttökuntoon saattamisen jälkeen. Jos sitä ei käytetä välittömästi, käytön aikaiset säilytysajat ja -olosuhteet ovat käyttäjän vastuulla. Käyttökuntoon saatettua liuosta ei saa pakastaa tai säilyttää jääkaapissa. Osittain käytetyt pullot on hävitettävä.
Säilytys
Säilytä alle 25 °C. Ei saa jäätyä. Säilytä pullo ulkopakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
FIBRYGA injektio-/infuusiokuiva-aine ja liuotin, liuosta varten
1 g (L:ei) 1 g (Octajet siirtolaite) (687,95 €)
PF-selosteen tieto
Jokainen pakkaus sisältää:
- 1 g ihmisen fibrinogeeniä 100 ml:n värittömässä tyypin II Ph. Eur. lasipullossa, joka on suljettu bromobutyylikumitulpalla ja alumiinikorkilla.
- 50 ml liuotinta (injektionesteisiin käytettävää vettä) 50 ml:n värittömässä tyypin II Ph. Eur. lasisessa injektiopullossa, joka on suljettu halobutyylikumitulpalla ja alumiinikorkilla.
- 1 nextaro-siirtolaite.
Valmisteen kuvaus:
Jauhe on valkoinen tai vaaleankeltainen ja hygroskooppinen, se voi myös näyttää hauraalta kiinteältä massalta.
Liuos on kirkas ja väritön neste.
Käyttö- ja käsittelyohjeet
Yleiset ohjeet
- Käyttökuntoon saatetun liuoksen on oltava lähes väritöntä tai hieman opalisoivaa. Älä käytä sameita liuoksia tai liuoksia, joissa on sakkaa.
- FIBRYGA on tarkoitettu vain kertakäyttöön. Älä käytä mitään osaa uudelleen.
- Mikrobiologisen turvallisuuden takia liuos on annettava välittömästi käyttökuntoon saattamisen jälkeen. Käyttökuntoon saatetun liuoksen kemiallinen ja fysikaalinen säilyvyys on osoitettu 24 tuntiin asti huoneenlämmössä (enintään 25° C) säilytettynä. Älä laita FIBRYGA-liuosta jääkaappiin tai pakasta sitä käyttökuntoon saattamisen jälkeen.
Käyttökuntoon saattaminen
| 1. Varmista, että jauhetta (FIBRYGA) sisältävä pullo ja liuotinta sisältävä injektiopullo ovat huoneenlämpöisiä. Ylläpidä tämä lämpötila käyttökuntoon saattamisen ajan. Jos lämmittämiseen käytetään vesihaudetta, on oltava varuillaan, jotta vältetään veden pääseminen kosketuksiin pullojen kumitulppien tai korkkien kanssa. Veden lämpötila ei saa ylittää 37 °C:ta. | |||
| 2. Poista korkki jauhepullosta (FIBRYGA) ja liuotinpullosta niin, että tulpan keskiosa tulee näkyviin. Puhdista kumitulpat alkoholiin kastetulla vanutupolla ja anna kumitulppien kuivua. | |||
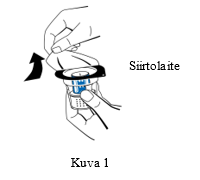
| 3. Avaa siirtolaitteen (nextaro) pakkaus vetämällä kansi pois (Kuva 1). Jätä siirtolaite puhtaaseen ulkopakkaukseen pitääksesi sen steriilinä. Älä koske piikkiin. |
| ||
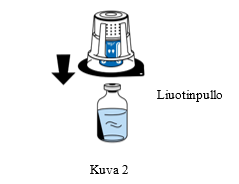
4. Aseta liuotinpullo tasaiselle, puhtaalle pinnalle ja ota siitä lujasti kiinni. Aseta siirtolaitteen sininen osa liuotinpullon päälle poistamatta siirtolaitteen ulkopakkausta. Paina siirtolaitetta suoraan ja lujasti alaspäin, kunnes se napsahtaa paikalleen (Kuva 2). Älä kierrä laitetta kiinnittämisen aikana. Huomautus: Siirtolaite on liitettävä ensin liuotinta sisältävään injektiopulloon ja sitten kylmäkuivattua jauhetta sisältävään pulloon. Muuten laitteen tyhjiö häviää eikä liuos siirry. |
| ||
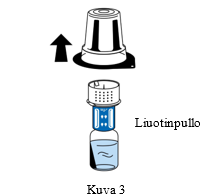
| 5. Pidä kiinni liuotinpullosta ja poista siirtolaitteen (nextaro) ulkopakkaus varovasti vetämällä sitä ylöspäin. Varmista, että siirtolaite jää lujasti kiinni liuotinpulloon (Kuva 3). |
| ||
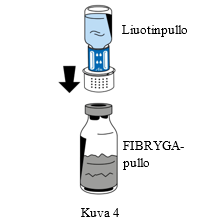
| 6. Aseta jauhetta (FIBRYGA) sisältävä pullo tasaiselle, puhtaalle alustalle ja pidä siitä tiukasti kiinni. Tartu liuotinpulloon, johon on kiinnitetty siirtolaite, ja käännä se ylösalaisin. Aseta siirtolaitteen liittimen valkoinen osa jauhetta (FIBRYGA) sisältävän pullon päälle ja paina siirtolaitetta lujasti alas, kunnes se napsahtaa paikoilleen (Kuva 4). Älä kierrä osia kiinnittämisen aikana. Liuotin virtaa automaattisesti jauhetta (FIBRYGA) sisältävään pulloon. |
| ||
| 7. Pidä liuotinpullo liitettynä ja sekoita FIBRYGA-pulloa varovasti, kunnes jauhe on liuennut kokonaan. Älä ravista pulloa, jotta vaahtoa ei muodostu. Jauheen pitäisi liueta täysin noin 5 minuutin kuluessa. Liukenemisen ei pitäisi kestää yli 20 minuuttia. Jos jauhe ei liukene 20 minuutin aikana, tuote pitää hävittää. | |||
| 8. Joskus harvoin liuoksessa voi kellua liukenematonta jauhetta injektionesteisiin käytettävän veden siirtämisen aikana tai käyttökuntoon saattamiseen kuluva aika voi pitkittyä odottamattomasti. Liukenemista voidaan nopeuttaa liikuttamalla injektiopulloa voimakkaammin vaakatasossa | |||
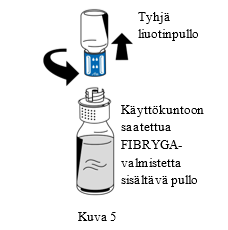
| 9. Kun käyttökuntoon saattaminen on valmis, kierrä siirtolaitetta (sininen osa) vastapäivään niin, että se irtoaa kahdeksi osaksi (Kuva 5). Älä koske siirtolaitteen Luer lock -liittimen valkoiseen osaan. |
| ||
| 10. Hävitä tyhjä liuotinpullo ja siirtolaitteen sininen osa yhdessä. | |||
Anto
| 1. Liitä ruisku varovasti siirtolaitteen valkoiseen osan Luer lock -liittimeen (Kuva 6). | 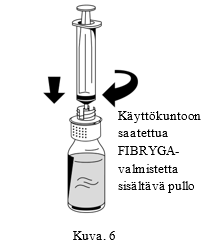 |
| 2. Käännä FIBRYGA-pullo ylösalaisin ja vedä lius ruiskuun (Kuva 7). | 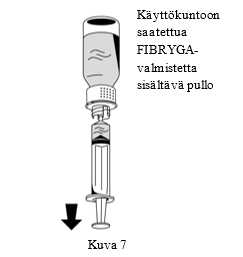 |
| 3. Kun liuos on siirretty, pidä lujasti kiinni ruiskun sylinteristä (pidä ruiskun mäntä alaspäin) ja poista ruisku siirtolaitteesta (Kuva 8). | 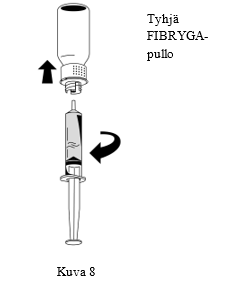 |
| 4. Hävitä siirtolaitteen valkoinen osa ja tyhjä FIBRYGA-pullo yhdessä. |
Käyttökuntoon saatetun liuoksen laskimoon antoon huoneenlämmössä suositellaan vakiomuotoista infuusiovälineistöä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
FIBRYGA injektio-/infuusiokuiva-aine ja liuotin, liuosta varten
1 g 1 g
- Ei korvausta.
ATC-koodi
B02BB01
Valmisteyhteenvedon muuttamispäivämäärä
17.11.2023
Yhteystiedot
Rajatorpantie 41 C
01640 Vantaa
09 8520 2710
www.octapharma.fi
info@octapharma.fi