
COMETRIQ kapseli, kova 20 mg + 80 mg
Vaikuttavat aineet ja niiden määrät
Yksi kova kapseli sisältää kabotsantinibi (S)-malaattia, joka vastaa 20 mg tai 80 mg kabotsantinibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, kova.
Kliiniset tiedot
Käyttöaiheet
COMETRIQ on tarkoitettu progressiivisen, medullaarisen kilpirauhaskarsinooman hoitoon aikuispotilaille, joiden paikallisesti edennyttä tai metastaattista syöpää ei voida poistaa leikkauksella.
Niiden potilaiden kohdalla, joilla RET-mutaation (rearranged during transfection) statusta ei tunneta tai se on negatiivinen, on otettava huomioon mahdollinen pienempi hyöty ennen yksilöllistä hoitopäätöstä (katso tärkeää tietoa kohdasta Farmakodynamiikka).
Ehto
Hoito tulee aloittaa syövän hoitoon tarkoitettujen lääkevalmisteiden antamiseen perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
COMETRIQ-hoito tulee aloittaa syövän hoitoon tarkoitettujen lääkevalmisteiden antamiseen perehtyneen lääkärin valvonnassa.
Annostus
COMETRIQ (kabotsantinibi) ‑kapselit ja CABOMETYX (kabotsantinibi) ‑tabletit eivät ole keskenään biologisesti samanarvoisia eivätkä siten keskenään vaihtokelpoisia (ks. kohta Farmakokinetiikka).
Suositeltu COMETRIQ-annos on 140 mg kerran vuorokaudessa, jolloin otetaan yksi 80 mg oranssi kapseli ja kolme 20 mg harmaata kapselia. Hoitoa pitää jatkaa siihen saakka, kunnes potilas ei enää kliinisesti hyödy hoidosta tai kunnes ilmenee toksisuutta, jota ei voida hyväksyä.
On odotettavissa, että huomattava osa COMETRIQ-valmisteella hoidetuista potilaista tarvitsee yhden tai useamman annoksen säätämisen (pienentämisen ja/tai keskeytyksen) toksisuuden takia. Tästä syystä potilaita pitää seurata huolellisesti ensimmäisen kahdeksan hoitoviikon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Lääkkeen epäiltyjen haittavaikutusten hoito saattaa vaatia COMETRIQ-hoidon tilapäistä keskeyttämistä ja/tai annoksen pienentämistä. Kun annoksen pienentäminen on tarpeellista, suositellaan sen pienentämistä 100 mg:aan vuorokaudessa, joka otetaan yhtenä 80 mg:n oranssina kapselina ja yhtenä 20 mg:n harmaana kapselina, ja sen jälkeen 60 mg:aan vuorokaudessa, joka otetaan kolmena 20 mg:n harmaana kapselina.
Annoksen keskeyttämisiä suositellaan CTCAE-kriteerien mukaisen asteen 3 tai sitä korkeampien toksisuuksien hoitoon taikka sietämättömien asteen 2 toksisuuksien hoitoon.
Annoksen pienentämisiä suositellaan tapauksissa, jotka jatkuessaan voivat kehittyä vakaviksi tai sietämättömiksi.
Koska useimmat tapahtumat voivat esiintyä hoitokuurin aikaisessa vaiheessa, pitää lääkärin arvioida potilas huolellisesti ensimmäisen kahdeksan hoitoviikon aikana määrittääkseen, ovatko annoksen muuttamiset tarpeellisia. Yleensä aikaisessa vaiheessa esiintyviä tapahtumia ovat hypokalsemia, hypokalemia, thrombosytopenia, hypertensio, palmoplantaarinen erytrodysestesia (kämmenten ja jalkapohjien oireyhtymä) sekä ruoansulatuselimistön tapahtumat (vatsakipu tai suukipu, limakalvotulehdus, ummetus, ripuli, oksentelu).
Joidenkin vakavien haittareaktioiden (kuten ruoansulatuskanavan fisteli) esiintyminen saattaa olla riippuvainen kumulatiivisesta annoksesta ja ilmetä hoidon myöhemmässä vaiheessa.
Jos potilaalta jää väliin yksi annos, ei väliin jäänyttä annosta pidä ottaa, jos seuraavan annoksen ottamiseen on alle 12 tuntia.
Samanaikaiset lääkevalmisteet
Varovaisuutta pitää noudattaa käytettäessä samanaikaisesti lääkevalmisteita, jotka ovat voimakkaita CYP3A4:n estäjiä, ja sellaisten lääkevalmisteiden kroonista käyttöä tulee välttää, jotka ovat voimakkaita CYP3A4:n induktoreja (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Tulisi harkita sellaisen vaihtoehtoisen samanaikaisesti käytettävän lääkevalmisteen valintaa, jolla ei ole potentiaalia tai jolla on vain vähän potentiaalia indusoida tai estää CYP3A4:tä.
Iäkkäät potilaat
Erityistä annoksen muuttamista ei suositella käytettäessä kabotsantinibia iäkkäämmille henkilöille (≥ 65‑vuotiaat). Suuntaus vakavien haittavaikutusten ilmenemisen lisääntymiseen on kuitenkin havaittu 75‑vuotiailla ja sitä vanhemmilla potilailla.
Rotu
Kabotsantinibista on vain vähän kokemusta potilailla, jotka eivät ole valkoisia.
Munuaisten vajaatoiminta
Kabotsantinibia pitää käyttää varoen potilailla, joilla on lievä tai kohtalainen munuaisten vajaatoiminta.
Kabotsantinibia ei suositella käytettäväksi potilailla, joilla on vaikea munuaisten vajaatoiminta, koska turvallisuutta ja tehoa ei ole varmistettu tälle väestöryhmälle.
Maksan vajaatoiminta
Lievästä tai kohtalaisesta maksan vajaatoiminnasta kärsivillä suositeltu kabotsantinibiannos on 60 mg kerran vuorokaudessa. Näiden potilaiden hoidossa suositellaan kokonaisturvallisuuden tarkkaa seurantaa (ks. kohta Farmakokinetiikka), sillä annoksen säätäminen tai keskeyttäminen voi olla tarpeen. Kabotsantinibia ei suositella käytettäväksi potilailla, joilla on vaikea maksan vajaatoiminta, koska turvallisuutta ja tehoa ei ole varmistettu tälle väestöryhmälle.
Sydämen vajaatoimintaa sairastavat potilaat
Sydämen vajaatoimintaa sairastavista potilaista on rajallisesti tietoja. Erityisiä annostussuosituksia ei voida antaa.
Pediatriset potilaat
Kabotsantinibin turvallisuutta ja tehoa alle 18-vuotiaiden lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
COMETRIQ otetaan suun kautta. Kapselit tulee niellä kokonaisina eikä niitä saa avata. Potilaita pitää neuvoa olemaan syömättä ainakin 2 tuntia ennen COMETRIQ-valmisteen ottamista ja 1 tunti sen jälkeen.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Annoksen pienentämistä esiintyi 79 prosentilla ja annoksen keskeyttämistä 72 prosentilla kabotsantinibilla hoidetuista potilaista keskeisessä kliinisessä tutkimuksessa. Kaksi annoksen vähennystä tarvittiin 41 prosentilla potilaista. Keskimääräinen aika ensimmäiseen annoksen vähentämiseen oli 43 päivää ja ensimmäiseen annoksen keskeyttämiseen oli 33 päivää. Tämän vuoksi potilaiden huolellista seurantaa suositellaan ensimmäisen kahdeksan hoitoviikon aikana (ks. kohta Annostus ja antotapa).
Maksatoksisuus
Kabotsantinibihoitoa saaneilla potilailla on usein havaittu poikkeamia maksan toimintakokeissa (kohonnut alaniiniaminotransferaasi [ALAT], aspartaattiaminotransferaasi [ASAT] ja bilirubiini). On suositeltavaa suorittaa maksan toimintakokeet (ALAT, ASAT ja bilirubiini) ennen kabotsantinibihoidon aloittamista ja seurata maksan toimintaa tarkkaan hoidon aikana. Jos maksan toimintakoetulokset huononevat ja sen katsotaan johtuvan kabotsantinibihoidosta (ts. jos muuta ilmeistä syytä ei ole), annosta on vähennettävä tai hoito keskeytettävä kohdassa Annostus ja antotapa esitettyjen suositusten mukaisesti.
Perforaatiot, fistelit ja vatsan sisäiset absessit
Kabotsantinibia käytettäessä on havaittu vakavia ruoansulatuskanavan perforaatioita ja fisteleitä, jotka ovat joskus johtaneet kuolemaan, sekä vatsan sisäisiä absesseja. Ennen kabotsantinibihoidon aloittamista on arvioitava huolellisesti hiljattain sädehoitoa saaneet potilaat, potilaat, joilla on tulehduksellinen suolistosairaus (esim. Crohnin tauti, haavainen paksusuolen tulehdus, peritoniitti tai divertikuliitti) tai joilla on kasvaimen infiltraatio henkitorveen tai keuhkoputkiin tai ruokatorveen sekä potilaat, joilla on komplikaatioita aiemmasta ruoansulatuskanavan kirurgiasta (varsinkin liittyen hitaaseen tai epätäydelliseen paranemiseen) tai aiemmasta rintakehän (mukaan lukien välikarsinan) alueen sädehoidosta. Hoidon aloituksen jälkeen tulee potilaita tarkkailla huolella perforaatioiden ja fisteleiden oireiden havaitsemiseksi. Muualla kuin ruoansulatuskanavassa oleva fisteli tulee sulkea pois asianmukaisesti tapauksissa, joissa hoitoa aloitettaessa ilmenee suutulehdus. Kabotsantinibi pitää keskeyttää potilailla, joilla ilmenee ruoansulatuskanavan perforaatio tai fisteli taikka muu kuin ruoansulatuskanavan fisteli.
Tromboemboliset tapahtumat
Kabotsantinibia käytettäessä on havaittu laskimotromboemboliaa, mukaan lukien keuhkoemboliaa, ja valtimoiden tromboembolisia tapahtumia, ja ne ovat toisinaan johtaneet potilaan kuolemaan. Kabotsantinibia pitää käyttää varoen potilailla, joilla on riski saada näitä tapahtumia, tai joilla on ollut niitä aiemmin. Kabotsantinibi pitää lopettaa potilailla, joille kehittyy akuutti sydäninfarkti tai jokin muu kliinisesti merkittävä valtimotromboembolinen komplikaatio.
Verenvuoto
Kabotsantinibia käytettäessä on havaittu vaikea-asteista verenvuotoa, joka on toisinaan johtanut potilaan kuolemaan. Potilaat, joilla on ennen hoidon aloittamista todisteita kasvaimen leviämisestä henkitorveen tai keuhkoputkiin tai aikaisemmin todettu veriyskä, pitää arvioida huolellisesti ennen kabotsantinibihoidon aloittamista. Kabotsantinibia ei pidä antaa potilaille, joilla on vakava verenvuoto tai joilla on hiljattain ollut veriyskä.
Aneurysmat ja valtimon dissekaatiot
VEGF-reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava tarkoin ennen kabotsantinibihoidon aloittamista potilaille, joilla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Ruoansulatuskanavan häiriöt
Yleisimmin raportoituja ruoansulatuskanavan haittavaikutuksia olivat ripuli, pahoinvointi/oksentelu, heikentynyt ruokahalu ja stomatiitti/suukipu (ks. kohta Haittavaikutukset). Lääkehoito, mukaan lukien tukihoito antiemeeteillä, ripulilääkkeillä tai haponestolääkkeillä, on aloitettava viipymättä, jotta estetään elimistön kuivuminen, elektrolyyttitasapainon häiriöt ja laihtuminen. Jos merkittävät ruoansulatuskanavan haittavaikutukset pitkittyvät tai uusiutuvat, kabotsantinibihoidon keskeyttämistä, annoksen pienentämistä tai hoidon lopettamista kokonaan pitää harkita (ks. kohta Annostus ja antotapa).
Haavakomplikaatiot
Kabotsantinibia käytettäessä on havaittu haavakomplikaatioita. Kabotsantinibihoito tulee mahdollisuuksien mukaan lopettaa vähintään 28 vuorokautta ennen suunniteltua leikkausta, mukaan lukien hammaskirurgiset tai invasiiviset hammastoimenpiteet. Päätöksen kabotsantinibihoidon jatkamisesta tulee perustua kliiniseen arviointiin haavan riittävästä paranemisesta. Kabotsantinibi pitää keskeyttää potilailla, joiden haavan paranemiseen liittyy lääketieteellisiä toimenpiteitä vaativia komplikaatioita.
Kohonnut verenpaine
Kabotsantinibia käytettäessä on havaittu kohonnutta verenpainetta, mukaan lukien hypertensiivisiä kriisejä. Verenpaine on saatava hyvään hoitotasapainoon ennen kabotsantinibihoidon aloittamista. Kabotsantinibihoidon aloittamisen jälkeen verenpainetta on seurattava varhaisessa vaiheessa ja säännöllisesti, ja sitä on hoidettava tarpeen mukaan asianmukaisella verenpainelääkityksellä. Jos verenpaine on jatkuvasti koholla verenpainelääkityksestä huolimatta, kabotsantinibihoito on keskeytettävä, kunnes verenpaine on hallinnassa, minkä jälkeen kabotsantinibihoitoa voidaan jatkaa pienemmällä annoksella. Kabotsantinibi on lopetettava, jos hypertensio on vaikea ja jatkuu verenpainelääkityksestä ja kabotsantinibiannoksen pienentämisestä huolimatta. Hypertensiivisen kriisin ilmetessä kabotsantinibihoito on lopetettava.
Sydämen vajaatoiminta
Kabotsantinibin käyttöön on liittynyt suurentunut sydämen vajaatoiminnan riski. Kabotsantinibin yleiset haittavaikutukset (esim. kohonnut verenpaine, kilpirauhasen vajaatoiminta ja valtimoveritulppatapahtumat) voivat lisätä tätä riskiä ja siten johtaa sydämen vajaatoimintaan. Potilaita pitää seurata hoidon aikana sydämen vajaatoiminnan oireiden ja löydösten varalta. Nämä haittavaikutukset on hoidettava viipymättä. Tarvittaessa on harkittava hoidon tauottamista ja/tai annoksen muuttamista (ks. kohta Annostus ja antotapa) ja hoito tyrosiinikinaasi-inhibiittorilla (TKI) on lopetettava potilailla, joille ilmaantuu vaikea-asteinen sydämen vajaatoiminta.
Osteonekroosi
Kabotsantinibia käytettäessä on havaittu leuan osteonekroositapahtumia. Suu pitää tarkastaa ennen kabotsantinibin aloittamista ja säännöllisesti kabotsantinibihoidon aikana. Potilaita tulee neuvoa suun hygienian suhteen. Kabotsantinibihoidosta tulee pidättäytyä mahdollisuuksien mukaan vähintään 28 vuorokautta ennen suunniteltua hammaskirurgista tai invasiivista hammastoimenpidettä. Varovaisuutta pitää noudattaa potilailla, jotka saavat leuan osteonekroosiin liittyviä lääkeaineita, kuten bisfosfonaatteja. Kabotsantinibi keskeytetään potilailla, joilla esiintyy leuan osteonekroosia.
Palmoplantaarinen erytrodysestesia
Kabotsantinibia käytettäessä on havaittu palmoplantaarista erytrodysestesiaa (kämmenten ja jalkapohjien oireyhtymää). Jos palmoplantaarinen erytrodysestesia on vaikea, kabotsantinibihoidon keskeyttämistä on harkittava. Kabotsantinibi pitää aloittaa uudelleen pienemmällä annoksella, kun palmoplantaarinen erytrodysestesia on parantunut asteeseen 1.
Valkuaisvirtsaisuus
Kabotsantinibia käytettäessä on havaittu valkuaisvirtsaisuutta. Virtsan valkuaispitoisuutta pitää seurata säännöllisesti kabotsantinibihoidon aikana. Kabotsantinibi pitää keskeyttää potilailla, joille kehittyy nefroottinen oireyhtymä.
Posteriorinen reversiibeli enkefalopatiaoireyhtymä
Kabotsantinibia käytettäessä on havaittu posteriorista reversiibeliä enkefalopatiaoireyhtymää (PRES). PRES pitää ottaa huomioon, jos potilaalla on siihen viittaavia oireita, kuten kouristuskohtauksia, päänsärkyä, näköhäiriöitä, sekavuutta tai henkisen vireystilan muutoksia. Kabotsantinibihoito pitää keskeyttää potilailla, joilla on PRES-oireyhtymä.
Pidentynyt QT-aika
Kabotsantinibia pitää käyttää varoen sellaisilla potilailla, joilla on aikaisemmin esiintynyt QT-ajan pidentymistä, jotka käyttävät rytmihäiriölääkkeitä, tai joilla on olennainen aikaisemmin todettu sydänsairaus, bradykardia tai elektrolyyttitasapainon häiriöitä. Kabotsantinibia käytettäessä tulee harkita säännöllistä seurantaa hoidon aikana EKG-mittauksilla ja elektrolyyttien määrityksillä (seerumin kalsium, kalium ja magnesium). Samanaikaista hoitoa vahvojen CYP3A4:n estäjien kanssa, joka saattaa lisätä kabotsantinibin plasmapitoisuuksia, on käytettävä varoen.
CYP3A4:n induktorit ja estäjät
Kabotsantinibi on CYP3A4:n substraatti. Samanaikainen kabotsantinibin anto voimakkaan CYP3A4:n estäjän ketokonatsolin kanssa aiheutti kabotsantinibin plasma-altistuksen lisääntymistä. Varovaisuutta on noudatettava annettaessa kabotsantinibia sellaisten lääkeaineiden kanssa, jotka ovat voimakkaita CYP3A4:n estäjiä. Kabotsantinibin anto samanaikaisesti voimakkaan CYP3A4:n induktorin rifampisiinin kanssa aiheutti kabotsantinibin plasma-altistuksen vähenemistä. Tästä syystä kabotsantinibin kanssa pitää välttää sellaisten lääkeaineiden kroonista antoa, jotka ovat voimakkaita CYP3A4:n induktoreja. (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset)
P-glykoproteiinin substraatit
Kabotsantinibi oli P-glykoproteiinin (P‑gp) kuljetustoiminnan estäjä (IC50 = 7,0 μM), mutta ei substraatti kaksisuuntaisessa analyysijärjestelmässä, jossa käytettiin MDCK-MDR1-soluja. Tästä syystä kabotsantinibilla saattaa olla potentiaalia lisätä samanaikaisesti annettujen P‑gp:n substraattien plasmapitoisuuksia. Potilaita pitää varoittaa P‑gp:n substraattien (esim. feksofenadiini, aliskireeni, ambrisentaani, dabigatraani eteksilaatti, digoksiini, kolkisiini, maravirokki, posakonatsoli, ranolatsiini, saxagliptiini, sitagliptiini, talinololi, tolvaptaani) käytöstä, kun he saavat kabotsantinibihoitoa.
MRP2-estäjät
MRP2-estäjien antaminen saattaa aiheuttaa kabotsantinibin plasmapitoisuuksien kohoamista. Tästä syystä samanaikaiseen MRP2-estäjien (esim. siklosporiini, efavirentsi, emtrisitabiini) käyttöön pitää suhtautua varoen.
Apuaine
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per kapseli eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutus kabotsantinibiin
CYP3A4:n estäjät ja induktorit
Annettaessa voimakasta CYP3A4:n estäjää ketokonatsolia (400 mg vuorokaudessa 27 päivän ajan) terveille vapaaehtoisille kabotsantinibin puhdistuma väheni (29 prosentilla) ja kabotsantinibin kerta-annoksen plasma-altistus lisääntyi (AUC) 38 prosentilla. Siksi voimakkaiden CYP3A4:n estäjien (esim. ritonaviiri, itrakonatsoli, erytromysiini, klaritromysiini, greippimehu) samanaikaiseen käyttöön kabotsantinibin kanssa pitää suhtautua varoen.
Annettaessa voimakasta CYP3A4:n induktoria rifampisiinia (600 mg vuorokaudessa 31 päivän ajan) terveille vapaaehtoisille kabotsantinibin puhdistuma kasvoi (4,3-kertaiseksi) ja kabotsantinibin kerta-annoksen plasma-altistus väheni (AUC) 77 prosentilla. Siksi tulee välttää vahvojen CYP3A4:n induktorien (esim. fenytoiini, karbamatsepiini, rifampisiini, fenobarbitaali tai rohdosvalmisteet, jotka sisältävät mäkikuismaa [Hypericum perforatum]) kroonista annostusta kabotsantinibin kanssa.
Mahan pH:ta muuttavat aineet
Protonipumpun estäjän (PPI) esomepratsolin (40 mg päivässä 6 päivän ajan) samanaikainen anto yhden 100 mg kabotsantinibiannoksen kanssa terveille vapaaehtoisille ei aiheuttanut kliinisesti merkittävää vaikutusta kabotsantinibin plasma-altistukselle (AUC). Annoksen muuttaminen ei ole aiheellista annettaessa vatsan pH:ta muuttavia aineita (esim, protonipumpun estäjät, H2-reseptorisalpaajat ja antasidit) samanaikaisesti kabotsantinibin kanssa.
MRP2-estäjät
In vitro -tiedot osoittavat, että kabotsantinibi on MRP2:n substraatti. Tästä syystä MRP2-estäjien antaminen saattaa aiheuttaa kabotsantinibin plasmapitoisuuksien kohoamista.
Sappisuoloja sitovat aineet
Sappisuoloja sitovat aineet, kuten kolestyramiini ja cholestagel, saattavat olla vuorovaikutuksessa kabotsantinibin kanssa ja voivat vaikuttaa imeytymiseen (tai takaisinimeytymiseen) ja siten johtaa mahdolliseen alentuneeseen altistukseen (ks. kohta Farmakokinetiikka). Näiden mahdollisten yhteisvaikutusten kliininen merkitys on tuntematon.
Kabotsantinibin vaikutus muihin lääkevalmisteisiin
Kabotsantinibin vaikutusta ehkäisevien steroidien farmakokinetiikkaan ei ole tutkittu. Koska ehkäisevän vaikutuksen ennallaan pysymistä ei voida taata, suositellaan lisäehkäisyn, kuten estemenetelmän, käyttöä.
Koska kabotsantinibi sitoutuu plasman proteiineihin suuressa määrin (kohta Farmakokinetiikka), plasman proteiinisidoksesta syrjäytymiseen perustuva yhteisvaikutus varfariinin kanssa voi olla mahdollinen. Tällaista yhdistelmää käytettäessä on seurattava INR-arvoja.
P-glykoproteiinin substraatit
Kabotsantinibi oli P-glykoproteiinin kuljetustoiminnan estäjä (IC50 = 7,0 μM), mutta ei substraatti kaksisuuntaisessa analyysijärjestelmässä, jossa käytettiin MDCK-MDR1-soluja. Siksi kabotsantinibilla saattaa olla potentiaali lisätä samanaikaisesti annettujen P‑gp:n substraattien plasmapitoisuuksia. Potilaita pitää varoittaa P‑gp:n substraattien (esim. feksofenadiini, aliskireeni, ambrisentaani, dabigatraani eteksilaatti, digoksiini, kolkisiini, maravirokki, posakonatsoli, ranolatsiini, saxagliptiini, sitagliptiini, talinololi, tolvaptaani) käytöstä, kun he saavat kabotsantinibihoitoa.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset/ehkäisy miehillä ja naisilla
Hedelmällisessä iässä olevia naisia on neuvottava välttämään raskautta kabotsantinibihoidonaikana. Kabotsantinibihoitoa saavien miespotilaiden naispuolisoita on neuvottava välttämään raskautta. Mies- ja naispotilaiden sekä heidän puolisoidensa on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään neljä kuukautta hoidon päättymisen jälkeen. Koska ehkäisypillereitä ei ehkä voida pitää “tehokkaana ehkäisymenetelmänä”, niitä tulee käyttää yhdessä jonkun toisen menetelmän, kuten estemenetelmän kanssa (ks. kohta Yhteisvaikutukset).
Raskaus
Kabotsantinibia käyttävistä raskaana olevista naisista ei ole tutkimuksia. Eläinkokeissa on havaittu alkio‑sikiö- ja teratogeenisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmiselle ei tunneta. Kabotsantinibia ei tule käyttää raskauden aikana, ellei naisen kliininen tila vaadi kabotsantinibihoitoa.
Imetys
Ei tiedetä, erittyykö/erittyvätkö kabotsantinibi/metaboliitit ihmisen rintamaitoon. Mahdollisen vahingollisen vaikutuksen takia äidin on lopetettava imetys kabotsantinibihoidon ajaksi ja vähintään neljän kuukauden ajaksi hoidon päättymisen jälkeen.
Hedelmällisyys
Ihmisen hedelmällisyydestä ei ole olemassa tietoja. Konventionaalisten turvallisuutta koskevien löydosten perusteella miesten ja naisten hedelmällisyys saattaa kompromisoitua kabotsantinibihoidon takia (ks. kohta Prekliiniset tiedot turvallisuudesta). Sekä miehiä että naisia tulee neuvoa hakemaan ja harkitsemaan hedelmällisyysneuvontaa ennen hoidon aloittamista.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kabotsantinibilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Haittareaktioita, kuten väsymystä ja heikkoutta, on yhdistetty kabotsantinibiin. Siksi suositellaan varovaisuutta autoa ajettaessa tai koneita käytettäessä.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Kaikkein yleisimmät kabotsantinibiin liittyvät vakavat haittavaikutukset ovat keuhkokuume, limakalvotulehdus, hypokalsemia, nielemishäiriö, kuivuminen, keuhkoembolia ja korkea verenpaine. Kaikkein yleisimmin esiintyviä kaikenasteisia (esiintyivät vähintään 20 prosentilla potilaista) haittavaikutuksia olivat ripuli, palmoplantaarinen erytrodysestesia, painon lasku, vähentynyt ruokahalu, pahoinvointi, uupumus, makuaistin häiriö, hiusten värin muutokset, korkea verenpaine, suutulehdus, ummetus, oksentelu, limakalvontulehdus, voimattomuus (astenia) ja puhehäiriö (dysfonia).
Kaikkein yleisimmät laboratorioarvojen poikkeamat olivat aspartaattiaminotransferaasin (ASAT) pitoisuuden nousu, alaniiniaminotransferaasin (ALAT) pitoisuuden nousu, alkalisen fosfataasin (AFOS) pitoisuuden nousu, lymfopenia, hypokalsemia, neutropenia, trombosytopenia, hypofosfatemia, hyperbilirubinemia, hypomagnesemia ja hypokalaemia.
Luettelo haittavaikutuksista taulukkomuodossa
Haittavaikutukset on luetteloitu taulukossa 1 MedDRA-elinjärjelmän ja yleisyysluokituksen mukaan. Esiintymistiheydet perustuvat kaikkiin luokkiin ja ne määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100) ja tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutusten vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Kabotsantinibille raportoidut haittavaikutukset
| Infektiot | |
| Yleinen | absessi* (mukaan lukien sisäelimissä, ihossa, hampaassa), keuhkokuume, karvatuppitulehdus, sieni-infektio (mukaan lukien iholla, suussa, sukuelimissä) |
| Melko harvinainen | Aspergilloosi |
| Umpieritys | |
| Yleinen | kilpirauhasen vajaatoiminta |
| Aineenvaihdunta ja ravitsemus | |
| Hyvin yleinen | vähentynyt ruokahalu, hypokalsemiac, hypokalemiac, hypomagnesemiac |
| Yleinen | kuivuminen*, hypoalbuminemiac, hyperbilirubinemiad, hypofosfatemiac |
| Psyykkiset häiriöt | |
| Yleinen | ahdistuneisuus, masennus, sekavuustila |
| Melko harvinainen | epänormaalit unet, delirium |
| Hermosto | |
| Hyvin yleinen | makuhäiriö, päänsärky, huimaus |
| Yleinen | aivoverisuonitapahtuma*, perifeerinen neuropatia, tuntoaistimushäiriö, makuaistin puute, vapina |
| Melko harvinainen | ataksia, huomiokyvyn häiriö, hepaattinen enkefalopatia, tajunnan menetys, puhehäiriö, posteriorinen reversiibeli enkefalopatia -oireyhtymä* |
| Silmät | |
| Yleinen | näön hämärtyminen |
| Melko harvinainen | kaihi, sidekalvontulehdus |
| Kuulo ja tasapainoelin | |
| Yleinen | korvakipu, tinnitus |
| Melko harvinainen | Huonokuuloisuus |
| Sydän | |
| Yleinen | Eteisvärinä, sydämen vajaatoiminta |
| Melko harvinainen | angina pectoris, supraventrikulaarinen takykardia |
| Tuntematon | Sydäninfarkti |
| Verisuonisto | |
| Hyvin yleinen | kohonnut verenpaine*f |
| Yleinen | alhainen verenpaineg, syvä laskimotukos*, laskimotukos*, valtimoveritulppa*, kalpeus, ääreisosien kylmyys |
| Melko harvinainen | hypertensiivinen kriisi*h, valtimoveritulppa |
| Tuntematon | aneurysmat ja valtimon dissekaatiot |
| Hengityselimet, rintakehä ja välikarsina | |
| Hyvin yleinen | puhehäiriö, suun ja nielun kipu |
| Yleinen | fisteli muualla kuin ruoansulatuskanavassa* (mukaan lukien henkitorven fisteli, pneumomediastinum, henki- ja ruokatorven välillä), keuhkoembolia*, hengitystien verenvuoto* (mukaan lukien keuhkossa, keuhkoputkessa, henkitorvessa), aspiraatiokeuhkokuume |
| Melko harvinainen | atelektaasi, nielun turvotus, pneumoniitti, ilmarinta |
| Ruoansulatuselimistö | |
| Hyvin yleinen | ripuli*, pahoinvointi*, suutulehdus, ummetus, oksentelu*, vatsakipue, dyspepsia, nielemisvaikeus, kielikipu |
| Yleinen | ruoansulatuskanavan perforaatio*, ruoansulatuskanavan fisteli*, ruoansulatuskanavan verenvuoto*, haimatulehdus, peräpukamat, anaalifissuura, peräaukon tulehdus, suupielten tulehdus |
| Melko harvinainen | Esofagiitti |
| Maksa ja sappi | |
| Yleinen | Sappikivitauti |
| Iho ja ihonalainen kudos | |
| Hyvin yleinen | palmoplantaarinen erytrodysestesia*, hiusten värin muutokset, ihottuma, kuiva iho, hiustenlähtö, eryteema |
| Yleinen | hyperkeratoosi, akne, rakkula, epänormaali karvankasvu, ihon kesiminen, ihon hypopigmentaatio |
| Melko harvinainen | ihohaava, telangiektasia |
| Tuntematon | Ihovaskuliitti |
| Luusto, lihakset ja sidekudos | |
| Hyvin yleinen | nivelkipu, lihasspasmit, raajakipu |
| Yleinen | muskuloskeletaalinen rintakipu, leuan osteonekroosi* |
| Melko harvinainen | Rabdomyolyysi |
| Munuaiset ja virtsatiet | |
| Yleinen | valkuaisvirtsaisuus*, dysuria, hematuria |
| Melko harvinainen | akuutti munuaisten vajaatoiminta |
| Sukupuolielimet ja rinnat | |
| Melko harvinainen | kuukautisten puuttuminen, emättimen verenvuoto |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | väsymys, limakalvotulehdus, voimattomuus |
| Yleinen | heikentynyt haavan paraneminen*, vilunväristykset, kasvojen turvotus |
| Melko harvinainen | kysta, kasvojen kipu, paikallinen turvotus |
| Tutkimukset | |
| Hyvin yleinen | painon lasku, seerumin ALAT-, ASAT- ja AFOS-arvon nousu, veren LDH-arvon nousu, veren TSH-arvon nousu*d, trombosytopeniaa |
| Yleinen | veren kreatiniiniarvon nousu, lymfopeniaa, neutropeniaa, lipaasiarvojen nousu |
| Melko harvinainen | lyhentynyt aktivoitu partiaalinen tromboplastiiniaika, eosinofiilien määrän kasvub, verihiutaleiden määrän kasvub |
* Ks. tarkemmat tiedot kohdasta Haittavaikutukset Valikoitujen haittavaikutusten kuvaus.
Seuraavat termit on yhdistetty asianmukaisten esiintyvyysluokitusten määrittämiseksi:
a Pienentyneet hematologiset parametrit: lymfopenia ja vähentynyt lymfosyyttien määrä; neutropenia ja vähentynyt neutrofiilien määrä; trombosytopenia ja vähentynyt trombosyyttien määrä.
b Kohonneet hematologiset parametrit: suurentunut eosinofiilien määrä ja eosinofilia; suurentunut trombosyyttien määrä ja trombosytoosi.
c Pienentyneet biokemialliset parametrit: hypoalbuminemia ja pienentynyt veren albumiinipitoisuus; hypokalsemia ja pienentynyt veren kalsiumpitoisuus; hypokalemia ja pienentynyt veren kaliumpitoisuus; hypomagnesemia ja pienentynyt veren magnesiumpitoisuus; hypofosfatemia ja pienentynyt veren fosforipitoisuus.
d Kohonneet biokemialliset parametrit: hyperbilirubinemia ja suurentunut veren bilirubiinipitoisuus; hypotyreoosi ja suurentunut kilpirauhasta stimuloivan hormonin pitoisuus veressä.
e Vatsakipu, epämukavat tuntemukset vatsassa, ylävatsakipu ja alavatsakipu.
f Hypertensio ja verenpaineen nousu.
g Hypotensio ja verenpaineen lasku.
h Hypertensiivisiä kriisejä ei ilmoitettu Cometriq-valmisteella tehdyissä kliinisissä tutkimuksissa; yleisyys perustuu kabotsantinibia koskeviin yhdistettyihin tietoihin (Cabometyx 60 mg:n tablettia koskevat tiedot mukaan lukien).
Valikoitujen haittavaikutusten kuvaus
Kilpirauhasta stimuloivan hormonin (TSH) normaalin pitoisuuden ylittävä arvo ensimmäisen annoksen jälkeen havaittiin 57 prosentilla kabotsantinibilla hoidetuista potilaista verrattuna 19 prosenttiin lumelääkettä saaneista potilaista (perusarvoista huolimatta). Yhdeksänkymmeneltäkahdelta prosentilta potilaista kabotsantinibiryhmässä oli aikaisemmin poistettu kilpirauhanen, ja 89 prosenttia käytti kilpirauhashormoneita ennen ensimmäistä annosta.
Syöpäpotilaille tehdyssä kontrolloidussa kliinisessä tutkimuksessa havaittiin Friderician menetelmällä korjatun QT-ajan (QTcF) 10 ‑ 15 ms kasvu perusarvosta päivänä 29 (mutta ei päivänä 1) kabotsantinibihoidon (annoksella 140 mg kerran vuorokaudessa) aloituksen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Tämä vaikutus ei liittynyt sydämen aaltomuodon morfologian muutokseen tai uusiin rytmeihin. Kellään kabotsantinibilla hoidetuista potilaista ei ollut QTcF-aika > 500 ms.
Ks. kohdasta Varoitukset ja käyttöön liittyvät varotoimet suositukset seuraavien haittavaikutusten seurannasta ja hoidosta: perforaatiot, fistelit ja vatsan sisäiset absessit, tromboemboliset tapahtumat, verenvuoto, aneurysmat ja valtimon dissekaatiot, ruoansulatuskanavan häiriöt, haavakomplikaatiot, kohonnut verenpaine, osteonekroosi, palmoplantaarinen erytrodysestesia, valkuaisvirtsaisuus ja posteriorinen reversiibeli enkefalopatiaoireyhtymä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kabotsantinibin yliannostukseen ei ole spesifistä hoitoa, eikä yliannostuksen mahdollisia oireita ole määritetty.
Epäillyssä yliannostustapauksessa kabotsantinibi tulee keskeyttää ja on ryhdyttävä antamaan tukihoitoa. Aineenvaihduntaan liittyviä kliinisiä laboratorioparametreja pitää seurata vähintään viikoittain tai sopivaksi katsotulla aikavälillä mahdollisten muutosten suuntauksen arvioimiseksi. Yliannostukseen liittyviä haittavaikutuksia on hoidettava oireenmukaisesti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset aineet, proteiinikinaasin estäjät, ATC-koodi: L01EX07
Vaikutusmekanismi
Kabotsantinibi on pieni molekyyli, joka estää useita tuumorin kasvuun angiogeneesiin, patologiseen luun uudelleenmuodostumista sekä syövän metastaattiseen etenemiseen yhdistettyjä reseptorityrosiinikinaaseja (RTK). Kabotsantinibia arvioitiin sen estokyvyn suhteen useita kinaaseja vastaan, ja se tunnistettiin MET- (hepatosyyttien kasvutekijän reseptoriproteiini) ja VEGF-reseptorien (verisuonen endoteelinen kasvutekijä) estäjäksi. Lisäksi kabotsantinibi estää muita tyrosiinikinaaseja, mukaan lukien RET:ä, GAS6-reseptoria (AXL), kantasolutekijän reseptoria (KIT) ja fms-kaltaista tyrosiinikinaasi-3:a (FLT3).
Farmakodynaamiset vaikutukset
Kabotsantinibi osoitti kykyä tuumorin kasvun annoksesta riippuvaan estoon, tuumorin regressioon, ja/tai se esti metastaasointia useissa prekliinisissä kasvainmalleissa.
Kabotsantinibin teho on havaittu medullaarista kilpirauhassyöpää sairastavilla potilailla, joilla oli RET:n villi tai mutatoitunut tyyppi.
Medullaarisen kilpirauhassyövän kliiniset tiedot
Satunnaistettu, kaksoissokkoutettu monikeskustutkimus, jossa kabotsantinibia (N = 219) verrattiin lumelääkkeeseen (N = 111), suoritettiin potilaille, joiden paikallisesti edennyttä tai metastaattista medullaarista kilpirauhaskarsinoomaa ei voitu poistaa leikkauksella, ja joilla oli dokumentoitu taudin radiologinen eteneminen 14 kuukauden aikana ennen tutkimukseen osallistumista. Ensisijaisena tavoitteena oli verrata etenemisvapaan elinajan (PFS) pitenemistä kabotsantinibia saaneilla potilailla lumelääkkeeseen verrattuna. Toissijaisina tavoitteina oli verrata yleistä hoitovastetta (ORR) ja kokonaiseloonjäämistä (OS). Keskitettyä, riippumatonta, sokkoutettua kuvantamistietojen arviointia käytettiin etenemisvapaan elinajan (PFS) ja yleisen hoitovasteen (ORR) arviointiin. Potilaita hoidettiin taudin etenemiseen asti tai kunnes ilmeni sietämätöntä toksisuutta.
Keskitettyyn RECIST-kriteeristön arvioon perustuvat PFS-analyysin tulokset, osoittivat tilastollisesti merkitsevän eron etenemisvapaan elinajan kestossa kabotsantinibilla lumelääkkeeseen verrattuna: keskimääräinen kesto oli 11,2 kuukautta kabotsantinibiryhmän potilailla verrattuna 4,0 kuukauteen lumelääkeryhmän potilailla (stratifioitu riskisuhde [HR] = 0,28; 95% luottamusväli: 0,19, 0,40; p<0.0001; Kuva 1). Etenemisvapaan elinajan tulokset olivat yhtäpitäviä kaikissa arvioiduissa perustieto- ja väestöalaryhmissä, mukaan lukien aikaisempaa hoitoa tyrosiinikinaasin estäjillä saaneet (joka on saattanut koostua lääkeaineista, joiden kohteena oli anti-angiogeneesiin liittyvät signaalireitit), RET-mutaatiostatuksen omaavat (mukaan lukien potilaat, joilla ei ollut dokumentoitu olevan RET-mutaatioita), aikaisemman syöpälääkitys- tai sädehoitostatuksen omaavat tai potilaat, joilla oli luumetastaaseja.
Yleinen hoitovaste (ORR) oli 27,9% kabotsantinibiryhmän potilailla ja 0% lumelääkeryhmän potilailla (p<0.0001; Taulukko 2). Objektiivisen vasteen keskimääräinen kesto oli 14,6 kuukautta (95% luottamusväli: 11,1, 17,5) kabotsantinibiryhmän potilailla.
Kuva 1: Kaplan Meier-käyrä etenemisvapaasta elinajasta
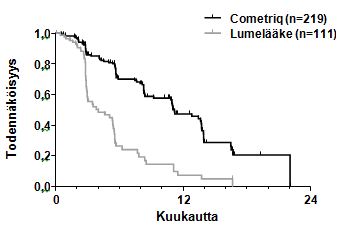
Riskipotilaiden määrä | ||||||||
Kuukausi | 0 | 3 | 6 | 9 | 12 | 15 | 18 | 21 |
Cometriq | 219 | 121 | 78 | 55 | 31 | 12 | 2 | 1 |
Lumelääke | 111 | 35 | 11 | 6 | 3 | 2 | 0 | 0 |
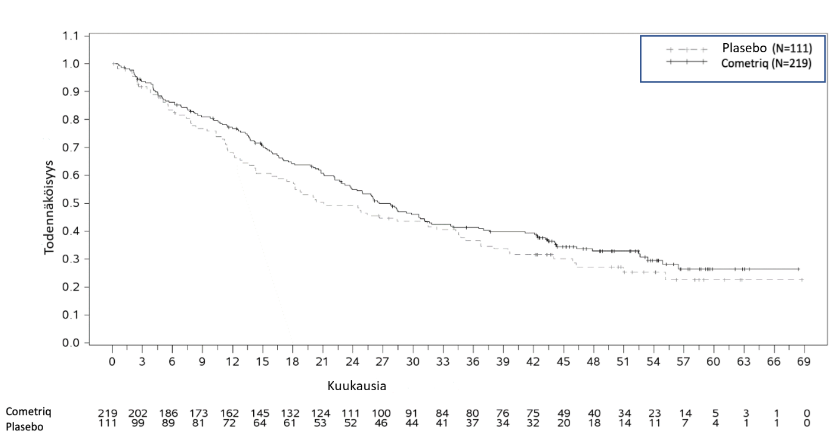
Kokonaiseloonjäämisen lopullinen analyysi suoritettiin 218 tapahtuman (kuolema) jälkeen, osoittaen lisääntyneen trendin keskimääräisessä selviytymisessä 5,5 kuukauden jälkeen kabotsantinibiryhmässä: mediaani 22,6 kuukautta kabotsantinibiryhmässä vs. 21,1 kuukautta plaseboryhmässä (HR = 0,85 [95 % CI: 0,64, 1,12], p = 0,2409).
Kuva 2: Kaplan-Meierin keskimääräinen eloonjäämiskäyrä
Taulukko 2: Yhteenveto tärkeimmistä tehoa koskevista löydöksistä
Kabotsantinibi | Lumelääke | |
Keskimääräinen etenemisvapaa elinaika | 11,2 kuukautta | 4,0 kuukautta |
HR: 0,28 (0,19, 0,40) p<0,0001 | ||
Keskimääräinen kokonaiseloonjääminen | 26,6 kuukautta | 21,1 kuukautta |
HR: 0,85 (0,64, 1,12) p = 0,2409 | ||
Yleinen hoitovastea (95%:n luottamusväli) | 27,9% (21,9%, 34,5%) | 0% |
p<0,0001 | ||
Vasteen kesto; mediaani (95%:n luottamusväli) | 14,6 kuukautta (11,1, 17,5) | N/A |
Taudin hallintab(95%:n luottamusväli) | 55,3% (48,3%, 62,2%) | 13,5% (7,6%, 21,6%) |
Kalsitoniini-vastea | 47% (49/104)c | 3% (1/40) c |
CEA-vastea | 33% (47/143) c | 2% (1/55)c |
a Hoitovaste = täydelliset + osittaiset vasteet
b Taudin hallinta = stabiili tauti + yleinen hoitovaste
c Sisältää potilaat, joiden vaste voitiin mitata
RET-mutaatiostatus
Riittävästi tietoa mutaatiostatuksen määrittämiseksi saatiin 215 potilaasta, joista 78,6% (n=169) luokiteltiin RET-mutaatiopositiivisiksi (126 jotka olivat M918T mutaatiopositiivisia) ja 21,4% (n=46) luokiteltiin RET-mutaationegatiivisiksi. Lisäksi 115 potilaalla RET-mutaatiostatusta ei voitu määrittää tai se oli epäselvä. Kaikilla kolmella ryhmällä etenemisvapaa elinaika piteni kabotsantinibiryhmässä verrattuna lumelääkeryhmään (riskisuhteet 0,23 RET-mutaatiopositiivisella, 0,53 RET-mutaationegatiivisella ja 0,30 tuntemattomalla alaryhmällä). Näissä alaryhmissä mitatut objektiiviset vasteet sopivat yleisesti yhteen etenemisvapaan elinajan tulosten kanssa, jolloin RET-mutaatiopositiivisella ryhmällä oli 32%:n, RET-mutaationegatiivisella ryhmällä 22%:n ja tuntemattomalla alaryhmällä 25%:n tuumorivaste.
Geneettinen lisäanalyysi osoitti, että pienellä osalla potilaita oli somaattisia tuumorimutaatioita HRAS-, KRAS- tai NRAS-geeneissä. Näillä potilailla (n=16) todettiin merkitsevää etenemisvapaan elinajan pitenemistä (riskisuhde 0,15) ja 31%:n objektiivinen hoitovaste. RET-mutaationegatiivisilla potilailla, joilla ei ollut todisteita RAS-mutaatiosta (n=33), todettiin etenemisvapaan elinajan pitenemistä (riskisuhde 0,87) ja pienempi 18%:n hoitovaste mutaation omaaviin alaryhmiin verrattuna.
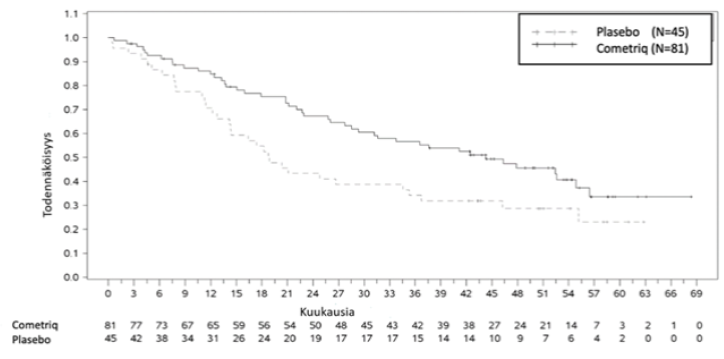
Kokonaiseloonjäämisen merkittävä paraneminen huomattiin vain RET M918T-mutaatiopositiivisten potilaiden alaryhmässä (n=81/219 kabotsantinibiryhmä). 44,3 kuukautta kabotsantinibiryhmässä vs. 18,9 kuukautta plasebo-ryhmässä (HR = 0,60, p = 0,0255). Kokonaiseloonjääminen ei parantunut muissa RET M918T –mutaation negativiisissa ja tuntemattomissa alaryhmissä.
Kuva 3: Kaplan-Meier-analyysi kokonaiseloonjäämisestä potilailla, joilla on RET M918T-mutaatio
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset kabotsantinibin käytöstä kaikkien pediatristen potilasryhmien pahanlaatuisen kiinteän kasvaimen hoidossa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Suun kautta otetun kabotsantinibin huippupitoisuus plasmassa saavutetaan 2 ‑ 5 tuntia annoksen jälkeen. Plasma-pitoisuus aikaprofiilit osoittavat toisen imeytymishuipun noin 24 tuntia annostuksen jälkeen, mikä viittaa siihen, että kabotsantinibi saattaa käydä läpi enterohepaattisen uudelleenkierron.
Toistuva kabotsantinibin annostus annoksella 140 mg 19 vuorokauden ajan sai aikaan noin 4‑kertaisesta 5‑kertaiseen kabotsantinibin keskimääräisen kertymisen (AUC-käyrään perustuen) verrattuna kerta-annokseen; vakaa tila saavutetaan suunnilleen 15. päivänä.
Runsaasti rasvaa sisältävä ateria nosti Cmax - ja AUC-arvoja (Cmax 41 % ja AUC 57 %) suhteessa paastoamiseen terveillä vapaaehtoisilla, joille annettiin yksi 140 mg:n kabotsantinibiannos. Tarkasta ruoan vaikutuksesta ei ole olemassa tietoja, kun ruoka nautitaan 1 tunti kabotsantinibin ottamisen jälkeen.
Kabotsantinibikapseleiden ja ‑tablettien ei voitu osoittaa olevan bioekvivalentteja, kun terveille tutkittaville annettiin 140 mg:n kerta-annos. Cmax-arvon havaittiin olleen tablettilääkemuodon (CABOMETYX) käytössä 19 % suurempi kuin kapselilääkemuodon (COMETRIQ) käytössä. AUC-arvon havaittiin olleen kabotsantinibitablettien (CABOMETYX) ja ‑kapselien (COMETRIQ) käytössä samankaltainen (ero < 10 %).
Jakautuminen
Kabotsantinibi sitoutuu suuressa määrin proteiiniin in vitro ihmisen plasmassa (≥ 99,7 %). Populaatiofarmakokineettisen mallin (PK-malli) perusteella jakautumistilavuus (V/F) on noin 349 l (SE: ± 2,73 %). Proteiineihin sitoutuminen ei muuttunut potilailla, joilla oli lievä tai kohtalainen munuaisten tai maksan vajaatoiminta.
Biotransformaatio
Kabotsantinibi metaboloituu in vivo. Neljä metaboliittia esiintyi plasmassa altistuksella (AUC), joka oli suurempi kuin 10% emoyhdiste: XL184‑N‑oksidi, XL184:n amidin jakautumistuote, XL184:n monohydroksisulfaatti ja 6‑desmetyyliamidin jakautumistuote sulfaatti. Kahdesta konjugoimattomasta metaboliitista (XL184-N‑oksidi ja XL184:n amidin hajoamistuote), joilla on <1% emoyhdisteen kabotsantinibin kohdistetun kinaasin estopotentiaalista, kumpikin edustaa <10% lääkkeeseen liittyvästä plasman kokonaisaltistuksesta.
Kabotsantinibi on CYP3A4:n substraatti in vitro, koska CYP3A4:n neutraloiva vasta-aine esti metaboliitin XL184 N‑oksidin muodostumista >80 prosentilla NADPH:n katalysoimassa ihmisen maksan mirosomaalien inkubaatiossa; sitä vastoin CYP1A2:n, CYP2A6:n, CYP2B6:n, CYP2C8:n, CYP2C19:n, CYP2D6:n ja CYP2E1:n neutraloivilla vasta-aineilla ei ollut vaikutusta kabotsantinibin metaboliittien muodostumiseen. CYP2C9:n neutraloivalla vasta-aineella oli vähäinen vaikutus kabotsantinibin metaboliitin muodostumiseen (eli <20% vähenemä).
Eliminaatio
Kabotsantinibin puoliintumisaika plasmassa terveille vapaaehtoisille suoritetuissa kerta-annostutkimuksissa on noin 120 tuntia. Keskimääräinen puhdistuma (CL/F) vakaassa tilassa syöpäpotilailla oli arviolta 4,4 l/h populaatiofarmakokineettisessä analyysissä. Terveille vapaaehtoisille annetun 14C-merkityn kabotsantinibin kerta-annoksen jälkeen saatiin takaisin 48 päivän keräysjakson jälkeen noin 81% annetusta radioaktiivisesta kokonaisannoksesta, jolloin 54 % saatiin takaisin ulosteesta ja 27 % virtsasta.
Farmakokinetiikka erityisryhmillä
Munuaisten vajaatoiminta
Tulokset munuaisten vajaatoiminnasta kärsiville potilaille suoritetusta tutkimuksesta osoittavat, että geometrisen pienimmän neliösumman keskiarvon suhteet plasman kabotsantinibille, Cmax- ja AUC0-inf olivat vastaavasti 19 % ja 30 % suurempia henkilöillä, joilla oli lievä munuaisten vajaatoiminta (90 % luottamusväli, jolloin Cmax 91,60 % – 155,51 %; AUC0‑inf 98,79 % – 171,26 %), ja vastaavasti 2 % ja 6-7 % suurempia (90 % luottamusväli, jolloin Cmax 78,64 % – 133,52 %; AUC0-inf 79,61 % – 140,11 %) henkilöillä, joilla oli kohtalainen munuaisten vajaatoiminta verrattuna henkilöihin, joilla oli normaali munuaisten toiminta. Vaikeasta munuaisten vajaatoiminnasta kärsiviä potilaita ei ole tutkittu.
Maksan vajaatoiminta
Tulokset maksan vajaatoiminnasta kärsiville potilaille suoritetusta tutkimuksesta osoittavat, että altistus (AUC0-inf) kasvoi 81 %:lla lievästä maksan vajaatoiminnasta kärsivillä henkilöillä ja 63 %:lla kohtalaisesta maksan vajaatoiminnasta kärsivillä henkilöillä (90 % luottamusväli, jolloin AUC0-inf: 121,44 % – 270,34 % lievässä ja 107,37 % – 246,67 % kohtalaisessa vajaatoiminnassa). Vaikeasta maksan vajaatoiminnasta kärsiviä potilaita ei ole tutkittu.
Rotu
Ei ole saatavilla tietoja, joiden perusteella eroavuutta farmakokinetiikassa rodun perusteella voitaisiin määrittää.
Prekliiniset tiedot turvallisuudesta
Seuraavia haittavaikutuksia ei ole todettu kliinisissä tutkimuksissa, mutta niitä on todettu koe-eläimillä, jotka ovat saaneet hoitoannoksia vastaavia määriä lääkeainetta. Siksi haitoilla voi olla kliinistä merkitystä:
Rotilla ja koirilla korkeintaan 6 kuukautta kestävissä toistuvan annoksen toksisuututkimuksissa toksisuuden kohde-elimet olivat ruoansulatuskanava, luuydin, imukudokset, munuaiset, lisämunuais- ja sukuelinten tiehyeiden kudokset. Näiden löydösten altistumistaso, jolla ei havaittu haittavaikutuksia (NOAEL), oli aiotulla terapeuttisella annoksella ihmisen kliinisen altistuksen alapuolella.
Kabotsantinibilla ei esiintynyt mutageenista tai klastogeenista potentiaalia normaaleissa geenitoksisuuden analyysisarjoissa. Kabotsantinibin karsinogeenisuutta on arvioitu kahdella eläinlajilla: rasH2-siirtogeenisillä hiirillä ja Sprague-Dawley-rotilla. Rotilla tehdyssä 2‑vuotisessa karsinogeenisuustutkimuksessa kabotsantinibiin liittyviin kasvainlöydöksiin kuului hyvänlaatuisen feokromosytooman ilmaantuvuuden lisääntyminen: sitä ilmeni rotilla sukupuolesta riippumatta joko sellaisenaan tai yhdessä lisämunuaisytimen pahanlaatuisen feokromosytoooman / monimuotoisen pahanlaatuisen feokromosytoooman kanssa altistuksilla, jotka olivat huomattavasti ihmiselle tarkoitetun kliinisen altistuksen alapuolella. Rotilla havaittujen kasvainleesioiden kliininen merkitys on epäselvä, mutta todennäköisesti se on vähäinen. Kabotsantinibi ei ollut karsinogeeninen rasH2-hiirimallissa hieman suuremmalla altistuksella kuin ihmiselle tarkoitettu terapeuttinen altistus.
Hedelmällisyystutkimukset rotilla ovat osoittaneet uroksen ja naaraan hedelmällisyyden heikkenemistä. Lisäksi epänormaalin niukkaa siittiöntuotantoa havaittiin uroskoirilla altistustasolla, joka oli aiotulla terapeuttisella annoksella ihmisen kliinisen altistuksen alapuolella.
Alkion-sikiön kehitystutkimuksia tehtiin rotilla ja kaneilla. Rotilla kabotsantinibi aiheutti sikiön menetyksen kiinnittymisen jälkeen, sikiön edeeman, huulihalkion/ristihuulen, ihon aplasian ja sykkyräisen tai alkeellisen hännän. Kaneilla kabotsantinibi aiheutti muutoksia sikiön pehmytkudoksissa (pienempi pernan koko, pieni tai puuttuva keuhkon keskilohko) ja lisääntyneen täydellisten epämuodostumien esiintyvyyden. Alkio-sikiötoksisuuden ja teratogeenisten löydösten altistustaso, jolla ei havaittu haittavaikutuksia (NOAEL), oli aiotulla terapeuttisella annoksella ihmisen kliinisen altistuksen alapuolella.
Nuorilla rotilla (verrattavissa yli 2-vuotiaisiin lapsipotilaisiin), joille annettiin kabotsantinibia, esiintyi valkosolujen määrän suurenemisparametreja, heikentynyttä hematopoieesia, puberteetti-ikäisen/kypsymättömän naaraan sukupuolielimiä (ilman hidastunutta emättimen aukeamista), hampaiden epämuodostumia, alentunutta luun mineraalipitoisuutta ja -tiheyttä, maksan pigmentaatiota ja sappitiehyeen hyperplasiaa. Kohdussa/munasarjoissa esiintyneet löydökset ja heikentynyt hematopoieesi näyttivät olevan ohimeneviä, kun taas luun parametrien ja maksan pigmentaation vaikutukset olivat pysyviä. Arvioita ei ole tehty nuorille rotille (verrattavissa alle 2-vuotiaisiin lapsipotilaisiin).
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Mikrokiteinen selluloosa
Kroskarmelloosinatrium
Natriumtärkkelysglykolaatti
Vedetön kolloidinen piidioksidi
Steariinihappo
Kapselin kuori
Gelatiini
Musta rautaoksidi (E172) (vain 20 mg kapselit)
Punainen rautaoksidi (E172) (vain 80 mg kapselit)
Titaanidioksidi (E171)
Painomuste
Shellakka
Musta rautaoksidi (E172)
Propyleeniglykoli
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Säilytä alle 25 °C.
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
COMETRIQ kapseli, kova
20 mg + 80 mg (L:ei) 84+28 fol (4652,50 €)
PF-selosteen tieto
PVC/PE/PCTFE-Al-läpipainopakkaus foliotaustalla, suljettu toiseen lämpösuljettuun pahvipakkaukseen.
Pahvilla varustetut läpipainopakkaukset sisältävät:
21 x 20 mg ja 7 x 80 mg kapselia (140 mg/vuorokausi -annos 7:n päivän ajaksi)
28 päivän pakkaus, joka sisältää:
112 kapselia (4 läpipainopakkausta, joissa 21 x 20 mg ja 7 x 80 mg) (140 mg/vuorokausi -annos 28:n päivän ajaksi)
Valmisteen kuvaus:
Cometriq 20 mg kovat kapselit:
Kovat kapselit ovat harmaita ja kapselin runkoon on painettu mustalla merkintä “XL184 20mg”.
Kapseli sisältää melkein valkoista tai valkoista jauhetta.
Cometriq 80 mg kovat kapselit:
Kovat kapselit ovat oransseja ja kapselin runkoon on painettu mustalla merkintä “XL184 80mg”.
Kapseli sisältää melkein valkoista tai valkoista jauhetta.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
COMETRIQ kapseli, kova
20 mg + 80 mg 84+28 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Kabotsantinibi: Munuaissyövän ja kilpirauhassyövän hoito erityisin edellytyksin (3012).
ATC-koodi
L01EX07
Valmisteyhteenvedon muuttamispäivämäärä
15.01.2026
Yhteystiedot
Kista Science Tower, Färögatan 33
SE-164 51 Kista
Sweden
+46 8 451 60 00
www.ipsen.com
info.se@ipsen.com