
YONDELIS kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 0,25 mg, 1 mg
Vaikuttavat aineet ja niiden määrät
Yondelis 0,25 mg
Yksi injektiopullollinen kuiva-ainetta sisältää 0,25 mg trabektediinia.
Yksi ml käyttökuntoon saatettua liuosta sisältää 0,05 mg trabektediinia.
Apuaineet, joiden vaikutus tunnetaan:
Yksi injektiopullollinen kuiva-ainetta sisältää 2 mg kaliumia ja 0,1 g sakkaroosia.
Täydellinen apuaineluettelo, ks. Kohta Apuaineet.
Yondelis 1 mg
Yksi injektiopullollinen kuiva-ainetta sisältää 1 mg trabektediinia.
Yksi ml käyttökuntoon saatettua liuosta sisältää 0,05 mg trabektediiinia.
Apuaineet, joiden vaikutus tunnetaan:
Yksi injektiopullollinen kuiva-ainetta sisältää 8 mg kaliumia ja 0,4 g sakkaroosia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos.
Kliiniset tiedot
Käyttöaiheet
Yondelis on tarkoitettu edenneen pehmytkudossarkooman hoitoon aikuisilla potilailla, joiden antrasykliini- ja ifosfamidihoidot ovat epäonnistuneet tai joille nämä lääkkeet eivät sovi. Tehoa koskevat tiedot perustuvat lähinnä liposarkooma- ja leiomyosarkoomapotilaisiin.
Yondelis on tarkoitettu uusiutuneen platinalle herkän munasarjasyövän hoitoon yhdessä pegyloidun liposomaalisen doksorubisiinin (PLD) kanssa.
Ehto
Valmistetta tulee käyttää vain syövän hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Yondelis tulee antaa syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa. Sitä saavat antaa vain syöpätautien erikoislääkärit tai muu nimenomaan sytotoksisten aineiden antoon perehtynyt terveydenhuollon ammattihenkilöstö.
Annostus
Pehmytkudossarkooman hoidossa suositusannos on 1,5 mg/m2 kehon pinta-alan mukaan laskettuna. Lääke annetaan 24 tuntia kestävänä infuusiona laskimoon, ja hoitojaksojen välillä pidetään 3 viikon tauko.
Munasarjasyövän hoidossa Yondelis annetaan kolmen viikon välein 3 tuntia kestävänä infuusiona annoksella 1,1 mg/m2, välittömästi 30 mg/m2 PLD-annoksen jälkeen. PLD-hoitoon liittyvien infuusioreaktioiden riskin minimoimiseksi ensimmäinen annos annetaan enintään nopeudella 1 mg/min. Jos infuusioreaktioita ei havaita, myöhemmät PLD-infuusiot voidaan antaa 1 tunnin kuluessa (tarkemmat annosteluohjeet, ks. PLD-valmisteen valmisteyhteenveto [SmPC]).
Kaikille potilaille on annettava kortikosteroideja, esim. 20 mg deksametasonia, laskimoon 30 minuuttia ennen PLD-hoitoa (yhdistelmähoito) tai Yondelisin antoa (monoterapia). Tämä esilääkitys ei ainoastaan ehkäise pahoinvointia, vaan sillä on nähtävästi myös maksaa suojaavaa vaikutusta. Muita pahoinvointilääkkeitä voidaan antaa tarpeen mukaan.
Yondelis-hoito on sallittua vain, jos potilas täyttää seuraavat kriteerit:
-
Absoluuttinen neutrofiiliarvo (ANC) ≥ 1 500/mm3
-
Trombosyyttiarvo ≥ 100 000/mm3
-
Bilirubiinipitoisuus ≤ normaaliarvojen yläraja (ULN)
-
Alkalinen fosfataasi ≤ 2,5 x ULN (jos arvojen suurentuminen voi olla luustoperäistä, harkitaan maksan isoentsyymi 5-nukleotidaasin tai gammaglutamyyli-transpeptidaasin (GGT) tutkimista)
-
Albumiini ≥ 25 g/l
-
Alaniiniaminotransferaasi (ALAT) ja aspartaattiaminotransferaasi (ASAT) ≤ 2,5 x ULN
-
Kreatiniinipuhdistuma ≥ 30 ml/min (monoterapia), seerumin kreatiniini ≤ 1,5 mg/dl (≤ 132,6 µmol/l) tai kreatiniinipuhdistuma ≥ 60 ml/min (yhdistelmähoito)
-
Kreatiinifosfokinaasi (CK) ≤ 2,5 x ULN
-
Hemoglobiini ≥ 9 g/dl
Potilaan on täytettävä samat kriteerit myös ennen jokaista uutta hoitojaksoa. Muussa tapauksessa hoitoa tulee siirtää enintään 3 viikon ajan, kunnes kriteerit täyttyvät.
Lisäksi tiettyjä veriarvoja (bilirubiini, alkalinen fosfataasi, aminotransferaasiarvot ja kreatiniinifosfokinaasi) on seurattava viikoittain ensimmäisten kahden hoitojakson ajan ja tämän jälkeen vähintään kertaalleen aina hoitojaksojen välillä.
Kaikkien hoitojaksojen aikana käytetään samaa annosta, ellei potilaalla havaita asteen 3–4 toksisuutta ja jos hoidon uusimiskriteerit täyttyvät.
Annosmuutokset hoidon aikana
Potilaiden on täytettävä edellä kuvatut lähtötilannetta koskevat kriteerit ennen kutakin uutta hoitojaksoa. Jos mitään seuraavista ilmenee missään vaiheessa hoitojaksojen välillä, myöhempien hoitojaksojen annosta on pienennettävä yhden annostason verran taulukon 1 mukaisesti:
-
Neutropenia < 500/mm3, joka kestää yli 5 vrk tai johon liittyy kuumetta tai jokin infektio
-
Trombosytopenia < 25 000/mm3
-
Bilirubiiniarvot suurenevat normaaliarvojen ylärajaa suuremmiksi ja/tai alkalisen fosfataasin arvo tasolle > 2,5 x ULN
-
Aminotransferaasiarvot (ASAT tai ALAT) suurenevat tasolle > 2,5 x ULN (monoterapia) tai > 5 x ULN (yhdistelmähoito) eivätkä normalisoidu päivään 21 mennessä
-
Potilaalle kehittyy jokin muu asteen 3 tai 4 haittavaikutus (esim. pahoinvointi, oksentelu, väsymys)
Jos annosta on kertaalleen pienennetty toksisuuden takia, annoksen suurentaminen myöhempien hoitojaksojen yhteydessä ei ole suositeltavaa. Jos tällaista toksisuutta ilmenee uudelleen myöhempien hoitojaksojen aikana, mutta potilas hyötyy kliinisesti hoidosta, annosta voidaan pienentää edelleen (ks. alla). Hematologisen toksisuuden hoitamiseksi voidaan antaa verisolujen kasvutekijöitä paikallisten vakiokäytäntöjen mukaisesti.
Taulukko 1: Yondelisin ja PLD:n annosmuutostaulukko (kun Yondelisia käytetään monoterapiana pehmytkudossarkooman (STS) hoitoon tai yhdessä PLD:n kanssa munasarjasyövän hoitoon)
Pehmytkudossarkooma | Munasarjasyöpä | ||
Yondelis | Yondelis | PLD | |
Aloitusannos | 1,5 mg/m2 | 1,1 mg/m2 | 30 mg/m2 |
Ensimmäinen annosmuutos | 1,2 mg/m2 | 0,9 mg/m2 | 25 mg/m2 |
Toinen annosmuutos | 1 mg/m2 | 0,75 mg/m2 | 20 mg/m2 |
Tarkemmat tiedot PLD-annoksen muuttamisesta, ks. PLD-valmisteen valmisteyhteenveto.
Jos annosta on tarpeen pienentää edelleen, on harkittava hoidon lopettamista.
Hoidon kesto
Kliinisissä tutkimuksissa annettavien hoitojaksojen määrää ei rajoitettu etukäteen. Hoitoa jatkettiin niin pitkään kuin siitä oli kliinistä hyötyä. Yondelisia annettiin vähintään 6 hoitojakson ajan 29,5 %:lle monoterapiaa saaneista potilaista ja 52 %:lle yhdistelmähoitoa saaneista. Kaikkia potilaita hoidettiin tässä ehdotetun annoksen ja aikataulun mukaisesti. Monoterapiaa käytettiin enimmillään 38 hoitojakson ajan ja yhdistelmähoitoa enimmillään 21 hoitojakson ajan. Potilailla, jotka ovat saaneet useita hoitojaksoja, ei ole havaittu kumuloituvaa toksisuutta.
Pediatriset potilaat
Yondelis-valmistetta ei pidä käyttää lapsuusiän sarkoomia sairastavien alle 18 vuoden ikäisten lasten hoitoon sen tehoon liittyvien huolenaiheiden vuoksi (ks. kohdasta Farmakodynamiikka lapsille tehdyn sarkoomatutkimuksen tulokset).
Iäkkäät potilaat
Erityisesti iäkkäitä potilaita koskevia tutkimuksia ei ole tehty. Yhteensä 20 % kliinisten monoterapiatutkimusten integroituun turvallisuusanalyysiin otetusta 1 164 potilaasta oli yli 65-vuotiaita. Munasarjasyövän tutkimuksissa 333 potilasta sai trabektediinia yhdessä PLD:n kanssa. Heistä 24 % oli vähintään 65-vuotiaita ja 6 % yli 75-vuotiaita. Tässä potilasryhmässä ei havaittu mitään merkitseviä eroja lääkkeen turvallisuuden suhteen. Ikä ei nähtävästi vaikuta trabektediinin puhdistumaan plasmasta eikä sen jakautumistilavuuteen. Tästä syystä annoksen rutiininomaista muuttamista vain iän perusteella ei suositella.
Maksan vajaatoiminta
Erityinen varovaisuus on suotavaa, ja annoksen muuttaminen voi olla tarpeen potilaille, joilla on maksan vajaatoiminta, sillä systeeminen trabektediinialtistus on tavallista suurempi ja myös maksatoksisuuden riski voi suurentua. Jos potilaan seerumin bilirubiiniarvot ovat koholla lähtötilanteessa, hänelle ei saa antaa Yondelis-hoitoa. Maksantoimintakokeiden tuloksia on seurattava Yondelis-hoidon aikana, koska annoksen muuttaminen voi olla aiheellista (ks. taulukko 1 ja kohta Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Valmisteella ei ole tehty tutkimuksia, joihin olisi otettu munuaisten vajaatoimintaa sairastavia potilaita (kreatiniinipuhdistuma < 30 ml/min monoterapiaa käyttävillä tai < 60 ml/min yhdistelmähoitoa saavilla). Näin ollen Yondelis-hoitoa ei saa käyttää tässä potilasryhmässä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Trabektediinin farmakokineettisten ominaisuuksien vuoksi (ks. kohta Farmakokinetiikka) annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta.
Antotapa
Laskimoon antaminen keskuslaskimoyhteyden kautta on hyvin suositeltavaa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Käyttö- ja käsittelyohjeet).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
- Yliherkkyys trabektediinille tai kohdassa Apuaineet mainituille apuaineille
- Samanaikainen vakava tai hallitsematon infektio
- Imetys (ks. kohta Raskaus ja imetys)
- Samanaikainen keltakuumerokotus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Varoitukset ja käyttöön liittyvät varotoimet
Maksan vajaatoiminta
Potilaiden maksatoiminnan on täytettävä tietyt kriteerit ennen Yondelis-hoidon aloittamista. Maksan vajaatoiminta suurentaa systeemisen trabektediinialtistuksen keskimäärin noin kaksinkertaiseksi (ks. kohta Farmakokinetiikka), ja toksisuuksien riski saattaa näin ollen suurentua. Jos potilaalla on kliinisesti huomattava maksasairaus, esim. aktiivinen krooninen hepatiitti, hänen tilaansa on seurattava tarkoin ja annosta on muutettava tarvittaessa. Jos potilaan seerumin bilirubiiniarvot ovat koholla, hänelle ei saa antaa trabektediinihoitoa (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Kreatiniinipuhdistumaa on seurattava ennen hoitoa ja sen jälkeen. Yondelis-hoitoa ei tule käyttää, jos potilaan kreatiniinipuhdistuma on < 30 ml/min (monoterapia) tai < 60 ml/min (yhdistelmähoito) (ks. kohta Annostus ja antotapa).
Neutropenia ja trombosytopenia
Yondelis-hoidon yhteydessä on ilmoitettu hyvin yleisesti asteen 3 ja 4 neutropeniaa ja trombosytopeniaa. Potilaalta on otettava täydellinen verenkuva (johon sisältyy valkosolujen erittelylaskenta ja trombosyyttiarvon mittaus) lähtötilanteessa, kerran viikossa ensimmäisten kahden hoitojakson aikana ja sen jälkeen kertaalleen aina hoitojaksojen välillä (ks. kohta Annostus ja antotapa). Jos potilaalle nousee kuume, hänen on hakeuduttava ripeästi lääkärin hoitoon. Tässä tapauksessa aktiivinen tukihoito on aloitettava välittömästi.
Yondelis-hoitoa ei pidä antaa, jos potilaan lähtötilanteen neutrofiiliarvo on alle 1 500/mm3 tai trombosyyttiarvo alle 100 000/mm3. Annoksen pienentäminen (ks. kohta Annostus ja antotapa) on suositeltavaa, jos potilaalle kehittyy vaikea neutropenia (ANC-arvo < 500/mm3), joka kestää yli 5 vrk tai johon liittyy kuumetta tai infektioita.
Pahoinvointi ja oksentelu
Kaikille potilaille on annettava pahoinvointia estävä kortikosteroidiesilääkitys, esim. deksametasoniesilääkitys (ks. Kohta Annostus ja antotapa).
Rabdomyolyysi ja CK-arvojen vaikea kohoaminen (> 5 x ULN)
Trabektediinia ei tule käyttää, jos potilaan CK-arvot ovat > 2,5 x ULN (ks. kohta Annostus ja antotapa). Rabdomyolyysia on ilmoitettu melko harvoin, ja sen yhteydessä on yleensä esiintynyt luuydintoksisuutta, vaikeita maksa-arvojen poikkeavuuksia ja/tai munuaisten vajaatoimintaa tai monielinvauriota. Tästä syystä CK-arvoja tulee seurata tarkoin, jos potilaalla saattaa olla tämäntyyppistä toksisuutta tai lihasheikkoutta tai lihaskipua. Jos potilaalle kehittyy rabdomyolyysi, on ryhdyttävä ripeästi kussakin tapauksessa aiheellisiin tukitoimiin (esim. parenteraalinen nesteytys, virtsan alkalisointi ja dialyysi). Yondelis-hoito tulee keskeyttää, kunnes potilas on toipunut täysin.
Varovaisuutta on noudatettava, jos trabektediinia käytetään samanaikaisesti sellaisten lääkevalmisteiden kanssa, joihin liittyy rabdomyolyysiä (esim. statiinit), sillä rabdomyolyysin riski voi olla tavallista suurempi.
Maksan toimintakokeiden poikkeavuudet
Useimmilla potilailla on ilmoitettu ASAT- ja ALAT-arvojen korjautuvaa akuuttia kohoamista. Yondelis-hoitoa ei saa käyttää, jos potilaan bilirubiiniarvot ovat koholla. Jos potilaan ASAT-, ALAT- ja alkalisen fosfataasin arvot ovat koholla hoitojaksojen välillä, annoksen muuttaminen voi olla tarpeen (ks. Kohta Annostus ja antotapa).
Pistoskohdan reaktiot
Valmisteen antaminen keskuslaskimoyhteyden kautta on hyvin suositeltavaa (ks. kohta Annostus ja antotapa). Potilaalle saattaa kehittyä mahdollisesti vaikeakin pistoskohdan reaktio, jos trabektediini annetaan perifeerisen laskimoyhteyden kautta.
Trabektediinin ekstravasaatio voi aiheuttaa kirurgista puhdistusta (débridement) vaativaa kudosnekroosia. Trabektediinin ekstravasaation hoitoon ei ole spesifistä vastalääkettä. Ekstravasaatio tulee hoitaa paikallisten vakiokäytäntöjen mukaisesti.
Allergiset reaktiot
Valmisteen markkinoille tulon jälkeen on ilmoitettu yliherkkyysreaktioita potilailla, jotka saivat trabektediinia joko monoterapiana tai PLD:n kanssa yhdistelmähoitona (ks. Kohdat Vasta-aiheet ja Haittavaikutukset). Ne ovat hyvin harvoissa tapauksissa johtaneet kuolemaan.
Sydämen vajaatoiminta
Potilaita on seurattava sydämeen liittyvien haittatapahtumien tai sepelvaltimoiden toimintahäiriöiden varalta.
Perusteellinen sydäntutkimus, muun muassa vasemman kammion ejektiofraktio (LVEF) on suoritettava kaikukuvauksella, tai on suoritettava moniporttinen kuvantaminen (MUGA) ennen trabektediinihoidon aloittamista ja 2–3 kuukauden välein, kunnes trabektediinin käyttö keskeytetään.
Potilailla, joiden LVEF on normaalin alarajan alapuolella (LVEF < LLN), jotka ovat saaneet aiemman kumulatiivisen >300 mg/m2:n antrasykliiniannoksen, joiden ikä on >65 vuotta tai joiden terveydelliseen taustaan sisältyy sydän- ja verisuonisairauksia (ja etenkin sydänlääkitystä saavilla potilailla), saattaa olla lisääntynyt sydämen vajaatoiminnan riski, mikäli he saavat trabektediinia monoterapiana tai yhdessä doksorubisiinin kanssa.
Potilailla, joilla on sydämeen liittyviä luokan 3 tai 4 kardiomyopatiaan viittaavia haittatapahtumia tai potilailla, joiden LVEF laskee LLN:n alapuolelle (arvioituna joko LVEF:n absoluuttisena laskuna, joka on >15 %, tai tasona 5 %), trabektediinin käyttö on keskeytettävä.
Hiussuonivuoto-oireyhtymä
Trabektediinin käytön yhteydessä havaituista hiussuonivuoto-oireyhtymätapauksista (kuolemaan johtavat tapaukset mukaan lukien) on tehty ilmoituksia. Jos käyttäjälle kehittyy oireyhtymän mahdollisia oireita, kuten selittämätön turvotus johon saattaa liittyä myös verenpaineen aleneminen, hoitavan lääkärin on mitattava seerumin albumiinipitoisuus uudelleen. Nopea seerumin albumiinitason lasku saattaa olla merkki hiussuonivuoto-oireyhtymästä. Jos hiussuonivuoto-oireyhtymän diagnoosi varmistuu muiden syiden pois sulkemisen jälkeen, hoitavan lääkärin on lopetettava trabektediinihoito ja aloitettava hiussuonivuoto-oireyhtymän hoito laitoksen ohjeistuksen mukaisesti (ks. Kohdat Annostus ja antotapa ja Haittavaikutukset).
Muuta
Yondelisin käyttöä samanaikaisesti voimakkaiden CYP3A4-estäjien kanssa tulee välttää (ks. Kohta Yhteisvaikutukset). Jos tämä ei ole mahdollista, potilasta tulee seurata tarkoin myrkytysoireiden varalta ja trabektediiniannoksen pienentämistä tulee harkita.
Varovaisuutta on noudatettava, jos trabektediinia käytetään samanaikaisesti sellaisten lääkevalmisteiden kanssa, joihin liittyy maksatoksisuutta, sillä maksatoksisuuden riski voi olla tavallista suurempi.
Trabektediinin ja fenytoiinin samanaikainen käyttö voi heikentää fenytoiinin imeytymistä ja johtaa kouristusten pahenemiseen. Trabektediinin käyttöä samanaikaisesti fenytoiinin tai elävien, heikennettyjen rokotteiden kanssa ei suositella, ja sen käyttö samanaikaisesti keltakuumerokotteen kanssa on nimenomaisesti vasta-aiheista (ks. Kohta Vasta-aiheet).
Trabektediinin ja alkoholin samanaikaista käyttöä tulee välttää (ks. Kohta Yhteisvaikutukset).
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä hoidon aikana ja 8 kuukauden ajan sen jälkeen sekä ilmoitettava välittömästi hoitavalle lääkärille mahdollisesta raskaudesta (ks. Kohta Prekliiniset tiedot turvallisuudesta).
Hedelmällisessä iässä olevien miesten on käytettävä erittäin tehokasta ehkäisyä hoidon aikana ja 5 kuukauden ajan sen jälkeen (ks. Kohta Raskaus ja imetys).
Tämä lääkevalmiste sisältää kaliumia alle 1 mmol (39 mg) per injektiopullo, eli sen voidaan sanoa olevan ”kaliumiton”.
Tarkemmat tiedot varoituksista ja varotoimista, ks. myös PLD-valmisteen valmisteyhteenveto.
Yhteisvaikutukset
Muiden aineiden vaikutukset trabektediiniin
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Trabektediini metaboloituu lähinnä CYP3A4:n välityksellä, joten trabektediinipitoisuudet plasmassa todennäköisesti suurenevat potilailla, joille annetaan samanaikaisesti tämän isoentsyymin toimintaa voimakkaasti estäviä lääkkeitä. Samoin trabektediinin samanaikainen anto voimakkaiden CYP3A4-indusorien kanssa saattaa suurentaa trabektediinin metabolista puhdistumaa. Kahdessa vaiheen 1 in vivo -yhteisvaikutustutkimuksessa on vahvistettu trabektediinialtistuksen suureneminen ketokonatsolin samanaikaisen annon aikana ja trabektediinialtistuksen pieneminen rifampisiinin samanaikaisen annon aikana.
Kun ketokonatsolia annettiin samanaikaisesti trabektediinin kanssa, trabektediinin plasma-altistus suureni noin 21 % (Cmax) ja 66 % (AUC), mutta uusia turvallisuuteen liittyviä huolenaiheita ei havaittu. Toksisuutta on seurattava huolellisesti, jos potilas saa trabektediinia yhdessä voimakkaiden CYP3A4-indusorien (esim. suun kautta annettavan ketokonatsolin, flukonatsolin, ritonaviirin, klaritromysiinin tai aprepitantin) kanssa, ja tällaisia yhdistelmähoitoja on vältettävä, jos se on mahdollista. Jos tällaiset yhdistelmähoidot ovat välttämättömiä, toksisuustapauksissa annosta on muutettava vastaavasti (ks. Kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kun rifampisiinia annettiin samanaikaisesti trabektediinin kanssa, trabektediinin plasma-altistus pieneni noin 22 % (Cmax) ja 31 % (AUC). Tästä syystä trabektediinin samanaikaista käyttöä voimakkaiden CYP3A4-indusorien (esim. rifampisiinin, fenobarbitaalin, mäkikuisman) kanssa on vältettävä, jos se on mahdollista (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Alkoholinkäyttöä on vältettävä trabektediinihoidon aikana lääkevalmisteen maksatoksisuuden vuoksi (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Prekliiniset tiedot ovat osoittaneet, että trabektediini on P-gp:n substraatti. P-gp:n estäjien kuten siklosporiinin tai verapamiilin samanaikainen anto voi vaikuttaa trabektediinin jakautumiseen elimistössä ja/tai sen eliminaatioon. Tämän yhteisvaikutuksen merkitystä esimerkiksi keskushermostotoksisuuden (CNS) suhteen ei ole selvitetty täysin. Tällaisessa tilanteessa on noudatettava varovaisuutta.
Raskaus ja imetys
Raskaus
Raskaudenaikaisesta altistuksesta ei ole riittävästi kliinistä tietoa. Trabektediinin tiedetyn vaikutusmekanismin vuoksi lääke voi kuitenkin aiheuttaa vakavia synnynnäisiä epämuodostumia, jos sitä käytetään raskauden aikana. Trabektediini läpäisi istukan, kun sitä annettiin tiineille rotille. Trabektediinia ei pidä käyttää raskauden aikana. Jos potilas tulee raskaaksi hoidon aikana, hänelle on kerrottava sikiöön mahdollisesti kohdistuvista riskeistä (ks. kohta Prekliiniset tiedot turvallisuudesta) ja tilannetta on seurattava huolellisesti. Jos trabektediinia käytetään loppuraskauden aikana, vastasyntynyttä tulee seurata tarkoin mahdollisten haittavaikutusten varalta.
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä hoidon aikana ja 8 kuukauden ajan sen jälkeen sekä ilmoitettava välittömästi hoitavalle lääkärille mahdollisesta raskaudesta (ks. Kohta Prekliiniset tiedot turvallisuudesta).
Jos raskaus alkaa hoidon aikana, on perinnöllisyysneuvontaa harkittava.
Imetys
Ei tiedetä, erittyykö trabektediini ihmisen rintamaitoon. Trabektediinin erittymistä maitoon ei ole tutkittu eläimillä. Imetys on vasta-aiheista hoidon aikana ja 3 kuukauden ajan sen jälkeen (ks. Kohta Vasta-aiheet).
Hedelmällisyys
Hedelmällisessä iässä olevien miesten on käytettävä erittäin tehokasta ehkäisyä hoidon aikana ja 5 kuukauden ajan sen jälkeen (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Trabektediinilla voi olla genotoksinen vaikutus. On syytä antaa munasolujen tai siittiöiden varastointia koskevaa neuvontaa ennen hoitoa, sillä Yondelis-hoito saattaa aiheuttaa korjautumatonta hedelmättömyyttä.
Perinnöllisyysneuvontaa suositellaan potilaille, jotka toivovat lapsia hoidon jälkeen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia valmisteen vaikutuksesta ajokykyyn tai koneidenkäyttökykyyn ei ole tehty. Trabektediinihoitoa saavilla potilailla on kuitenkin ilmoitettu väsymystä ja/tai voimattomuutta. Jos potilaalla ilmenee tällaisia oireita hoidon aikana, hän ei saa ajaa eikä käyttää koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Eriasteisia haittavaikutuksia esiintyy todennäköisesti useimmilla Yondelis-hoitoa saavista (91 %:lla monoterapiaryhmässä ja 99,4 %:lla yhdistelmähoitoryhmässä). Vakavia haittavaikutuksia, joiden vaikeusaste on 3 tai 4, esiintyy todennäköisesti alle kolmanneksella potilaista (10 % monoterapiaryhmässä ja 25 % yhdistelmähoitoryhmässä). Yleisimpiä haittavaikutuksia (kaikki vaikeusasteet) olivat neutropenia, pahoinvointi, oksentelu, ASAT-/ALAT-arvojen suureneminen, anemia, väsymys, trombosytopenia, ruokahaluttomuus ja ripuli.
1,9 %:lla monoterapiaryhmän potilaista ja 0,6 %:lla yhdistelmähoitoa saaneista esiintyi kuolemaan johtaneita reaktioita. Kyseessä oli usein monien eri tapahtumien yhdistelmä, johon kuului esimerkiksi pansytopeniaa, kuumeista neutropeniaa (johon liittyi joissakin tapauksissa sepsistä), maksa-affisiota, munuaisten vajaatoimintaa tai monielinvauriota ja rabdomyolyysiä.
Yhteenveto haittavaikutuksista taulukkomuodossa
Yondelis-valmisteen seuraavat turvallisuustiedot perustuvat kliinisissä tutkimuksissa raportoituihin haittavaikutuksiin, myyntiluvan myöntämisen jälkeisiin turvallisuustutkimuksiin sekä spontaanisti annettuihin ilmoituksiin.
Seuraavassa taulukossa esitetään pehmytkudossarkooman ja munasarjasyövän Yondelis-valmisteen suositushoitoa kussakin käyttöaiheessa saaneilla potilailla esiintyneet haittavaikutukset. Esiintymistiheydet on laskettu sekä haittavaikutusten että laboratorioarvojen mukaan.
Haittavaikutukset on lueteltu elinjärjestelmäluokan ja yleisyyden mukaan. Yleisyydet luokiteltu seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100) ja harvinaiset (≥ 1/10 000, < 1/1000).
Elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen |
Infektiot | Neutropeeninen infektio | Verenmyrkytys | Septinen sokki | |
Veri ja imukudos | Neutropenia, trombosytopenia, anemia, leukopenia | Kuumeinen neutropenia | ||
Immuunijärjestelmä | Yliherkkyys | |||
Aineenvaihdunta ja ravitsemus | Ruokahalun heikkeneminen | Nestehukka Hypokalemia | ||
Psyykkiset häiriöt | Unettomuus | |||
Hermosto | Päänsärky | Huimaus Makuaistin muutokset Perifeerinen sensorinen neuropatia Pyörtyily* | ||
Sydän | Sydämentykytys* Vasemman kammion vajaatoiminta* | |||
Verisuonisto | Hypotensio Punastelu | Hiussuonivuotooireyhtymä | ||
Hengityselimet, rintakehä ja välikarsina | Hengenahdistus Yskä | Keuhkoembolia* | Keuhkoedeema | |
Ruoansulatuselimistö | Vatsakipu Pahoinvointi Oksentelu Ummetus Ripuli Suutulehdus | Dyspepsia | ||
Maksa ja sappi | Kohonneet ALAT-arvot Kohonneet ASAT-arvot Kohonneet veren AFOSarvot Kohonneet veren bilirubiiniarvot | Kohonneet GGTarvot | Maksan vajaatoiminta | |
Iho ja ihonalainen kudos | Palmoplantaarinen erytrodysestesia* | Ihottuma Hiustenlähtö Ihon hyperpigmentaatio* | ||
Luusto, lihakset ja sidekudos | Selkäkipu Kohonneet veren kreatiinifosfokinaasiarvot | Nivelkipu Lihaskipu | Rabdomyolyysi | |
Munuaiset ja virtsatiet | Akuutti munuaisvaurio | |||
Yleisoireet ja antopaikassa todettavat haitat | Väsymys Kuume Turvotus Limakalvotulehdus* | Pistoskohdan reaktiot | Ekstravasaatio Pehmytkudosnekroosi | |
Tutkimukset | Kohonneet veren kreatiniiniarvot Laskeneet veren albumiiniarvot | Painonlasku |
* Haittavaikutus on havaittu vain munasarjasyöpää sairastavilla potilailla, sisältäen tiedot ET743‑OVA‑301, satunnaistetusta vaiheen 3 tutkimuksesta 672 potilaalla, jotka saivat joko trabektediiniä (1,1 mg/m2) ja PLD:tä (30 mg/m2) 3 viikon välein tai PLD:tä (50 mg/m2) 4 viikon välein, sekä tutkimuksesta ET743-OVC-3006, johon osallistui 576 potilasta, jotka saivat joko PLD:tä (30 mg/m2) ja sen jälkeen trabektediiniä (1,1 mg/m2) 3 viikon välein tai pelkkää PLD:tä (50 mg/m2) 4 viikon välein.
Yondelisin ja PLD:n yhdistelmätutkimuksessa ET743‑OVA‑301 muilla kuin valkoihoisilla potilailla (lähinnä aasialaisilla) esiintyi valkoihoisia potilaita enemmän asteen 3 ja 4 haittavaikutuksia (96 % muilla ja 87 % valkoihoisilla) ja vakavia haittavaikutuksia (kaikki asteet: 44 % muilla ja 23 % valkoihoisilla). Eroja havaittiin erityisesti neutropenian (93 % muilla ja 66 % valkoihoisilla), anemian (37 % muilla ja 14 % valkoihoisilla) ja trombosytopenian (41 % muilla ja 19 % valkoihoisilla) kohdalla. Hematologiseen toksisuuteen liittyvien kliinisten komplikaatioiden kuten vaikeiden infektioiden ja verenvuodon ilmaantuvuudessa ei kuitenkaan ollut eroja alaryhmien välillä. Myöskään kuolemaan tai hoidon lopettamiseen johtaneiden haittavaikutusten ilmaantuvuudessa ei ollut eroja.
Kuvaus valituista haittavaikutuksista
Yleisimmät haittavaikutukset
Veri ja imukudos
Neutropenia:
Yleisin hematologinen haittavaikutus on neutropenia. Se noudatti ennustettavaa kaavaa eli alkoi ja korjautui nopeasti, ja siihen liittyi vain harvoin kuumetta tai infektioita. Neutrofiiliarvojen nadiirin saavuttamiseen kulunut mediaaniaika oli 15 vrk, ja arvot korjautuivat viikossa. Monoterapiahoitoa saaneiden potilaiden tietojen analysointi kutakin hoitojaksoa kohti osoitti, että asteen 3 neutropeniaa esiintyi noin 19 %:ssa hoitojaksoista ja asteen 4 neutropeniaa noin 8 %:ssa. Tässä populaatiossa kuumeista neutropeniaa esiintyi 2 %:lla potilaista ja < 1 %:ssa hoitojaksoista.
Trombosytopenia:
Trombosytopeniaan liittyviä vuototapahtumia esiintyi < 1 %:lla monoterapiaa saaneista potilaista. Tämän potilaspopulaation tietojen analysointi kutakin hoitojaksoa kohti osoitti, että asteen 3 trombosytopeniaa esiintyi noin 3 %:ssa hoitojaksoista ja asteen 4 trombosytopeniaa alle 1 %:ssa.
Anemia:
Anemiaa esiintyi 93 %:lla monoterapiaa saaneista potilaista ja 94 %:lla yhdistelmähoitoa saaneista. Lähtötilanteessa anemiaa oli esiintynyt 46 %:lla monoterapiaryhmän potilaista ja 35 %:lla yhdistelmähoitoa saaneista. Monoterapiaa saaneiden potilaiden tietojen analysointi kutakin hoitojaksoa kohti osoitti, että asteen 3 anemiaa esiintyi noin 3 %:ssa hoitojaksoista ja asteen 4 anemiaa noin 1 %:ssa.
Maksa ja sappi
ASAT-/ALAT-arvojen suureneminen:
Korkeimpien ASAT- ja ALAT-arvojen saavuttamiseen kulunut mediaaniaika oli 5 päivää. Useimmiten arvojen nousu lievittyi asteen 1 tasoiseksi tai korjautui kokonaan päivään 14–15 mennessä (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet). Monoterapiaa saaneiden potilaiden tietojen analysointi kutakin hoitojaksoa kohden osoitti, että ASAT-arvojen nousua (aste 3) esiintyi 12 %:ssa hoitojaksoista ja ALAT-arvojen nousua (aste 3) 20 %:ssa. ASAT-arvojen nousua (aste 4) esiintyi 1 %:ssa hoitojaksoista ja ALAT-arvojen nousua (aste 4) 2 %:ssa. Kohonneet transaminaasiarvot korjautuivat useimmiten tasolle 1 tai hoitoa edeltäneelle tasolle 15 vrk kuluessa. Arvojen korjautumiseen kului yli 25 vrk vain alle 2 %:ssa hoitojaksoista. ALAT- ja ASAT-arvojen nousu ei ollut kumulatiivista, ja sillä oli taipumusta muuttua ajan mittaan vaikeusasteeltaan lievemmäksi.
Hyperbilirubinemia:
Bilirubiinipitoisuudet olivat suurimmillaan noin viikon kuluttua kohoamisprosessin alusta ja korjautuivat noin kahden viikon kuluttua kohoamisprosessin alusta.
Vaikeaa toksisuutta ennustavia (Hyn lain mukaisia) maksan toimintakokeiden muutoksia ja vaikeiden maksavaurioiden kliinisiä oireita esiintyi melko harvoin, ja yksittäisten merkkien ja oireiden kuten ikteruksen, hepatomegalian tai maksakivun ilmaantuvuus oli alle 1 %. Maksavaurion yhteydessä esiintyi kuolemantapauksia alle 1 %:lla potilaista.
Muut haittavaikutukset
Maksan vajaatoiminta: Harvoissa tapauksissa on ilmoitettu maksan vajaatoiminnasta (joka joissakin tapauksissa johti kuolemaan) vakavaa perussairautta sairastavilla potilailla, jotka saivat trabektediinia. Ilmoituksia on sekä kliinisistä tutkimuksista että valmisteen markkinoille tulon jälkeiseltä ajalta. Joitakin mahdollisia, mainituissa tapauksissa havaittuja riskitekijöitä, jotka ovat saattaneet lisätä trabektediinin toksisuutta, ovat annosteluohjeiden noudattamisen laiminlyöminen, mahdollinen yhteisvaikutus CYP3A4:n kanssa useiden kilpailevien CYP3A4-substraattien tai CYP3A4-estäjien takia tai deksametasoniesilääkityksen puuttuminen.
Hiussuonivuoto-oireyhtymä: Hiussuonivuoto-oireyhtymätapauksista (kuolemaan johtavat tapaukset mukaan lukien) on tehty ilmoituksia trabektediinin käytön yhteydessä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Trabektediiniyliannostuksen vaikutuksista on rajallisesti tietoa. Tärkeimmät odotettavissa olevat toksisuudet ovat ruoansulatuskanavaan kohdistuva toksisuus, luuydinsuppressio ja maksatoksisuus. Trabektediinille ei tällä hetkellä ole saatavilla spesifistä vastalääkettä. Yliannostustapauksessa potilasta tulee seurata tarkoin, ja oireenmukaisiin tukitoimiin tulee ryhtyä tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Solunsalpaajat, ATC-koodi: L01CX01.
Vaikutusmekanismi
Trabektediini sitoutuu deoksiribonukleiinihapon (DNA) matalaan uurteeseen ja saa kaksoiskierteen taipumaan syvän uurteen suuntaan. Lääkkeen sitoutuminen DNA:han käynnistää tapahtumasarjan, joka vaikuttaa useisiin eri transkriptiotekijöihin, DNA:han sitoutuviin proteiineihin ja DNA:n korjausmekanismeihin. Tämä johtaa solusyklin häiriintymiseen.
Farmakodynaamiset vaikutukset
Trabektediinilla on osoitettu olevan antiproliferatiivista vaikutusta in vitro ja in vivo useisiin eri ihmisen kasvainsolulinjoihin ja kokeellisiin kasvaimiin kuten sarkoomaan, rintasyöpään, ei-pienisoluiseen keuhkosyöpään, munasarjasyöpään ja melanoomaan.
Sydänfilmitutkimukset (EKG)
Plasebokontrolloidussa QT-/QTc-tutkimuksessa trabektediini ei pidentänyt QTc-väliä potilailla, joilla oli pitkälle edenneitä kiinteitä pahanlaatuisia kasvaimia.
Kliininen teho ja turvallisuus
Trabektediinin teho- ja turvallisuustiedot pehmytkudossarkooman hoidossa perustuvat satunnaistettuun tutkimukseen, johon otetuilla potilailla oli paikallisesti levinnyt tai etäpesäkkeinen liposarkooma tai leiomyosarkooma ja joiden tauti oli edennyt tai relapsoinut vähintään antrasykliinejä ja ifosfamidia sisältäneen hoidon jälkeen. Tässä tutkimuksessa trabektediinia annettiin joko 1,5 mg/m2 annoksena 24 tuntia kestävänä laskimoinfuusiona 3 viikon välein tai 0,58 mg/m2 annoksena 3 tuntia kestävänä laskimoinfuusiona, joka annettiin viikoittain 3 viikon ajan kutakin 4 viikon hoitojaksoa kohden. Tutkimussuunnitelmassa määritellyn taudin etenemiseen kuluneen kokonaisajan analyysi osoitti, että taudin etenemisen suhteellinen riski pieneni 26,6 % niillä potilailla, jotka saivat 24 tunnin infuusioita 3 viikon välein [riskisuhde (HR) = 0,734, luottamusväli (CI) 0,554–0,974]. Taudin etenemiseen kulunut mediaaniaika oli 24 h infuusioita 3 viikon välein saaneessa ryhmässä 3,7 kk (luottamusväli 2,1–5,4 kk) ja 3 h infuusioita viikon välein saaneessa ryhmässä 2,3 kk (luottamusväli 2,0–3,5 kk, p = 0,0302). Kokonaiselossaoloajassa ei havaittu merkitseviä eroja. 24 h infuusioita 3 viikon välein saaneessa ryhmässä kokonaiselossaoloajan mediaani oli 13,9 kk (luottamusväli 12,5–18,6 kk), ja 60,2 % potilaista oli elossa 1 vuoden kohdalla (luottamusväli 52,0–68,5 %).
Tehotietoja on saatu myös kolmesta yhden hoitoryhmän käsittäneestä vaiheen II tutkimuksesta, joissa samankaltaiset potilaspopulaatiot saivat samaa hoitoa. Näissä tutkimuksissa hoitoa arvioitiin yhteensä 100 lipo- tai leiomyosarkoomapotilaalla ja 83 potilaalla, joilla oli jokin muu sarkoomatyyppi.
Tulokset laajennetun saatavuuden ohjelmasta pehmytkudossarkoomaa (STS) sairastavilla potilailla (tutkimus ET743-SAR- 3002) osoittavat, että 903 potilaasta, joiden kokonaiselinaika (OS) arvioitiin, kokonaiselinajan mediaani oli 11,9 kuukautta (95 % CI: 11,2–13,8). Histologisen kasvaintyypityksen mukaan kokonaiselinajan mediaani oli 16,2 kuukautta (95 % CI: 14,1–19,5) potilailla, joilla oli leiomyosarkooma ja liposarkooma ja 8,4 kuukautta (95 % CI: 7,1–10,7) potilailla, joilla oli muun tyyppisiä sarkoomia. Kokonaiselinajan mediaani liposarkoomapotilailla oli 18,1 kuukautta (95 % CI: 15,0–26,4) ja leiomyosarkoomapotilailla 16,2 kuukautta (95 % CI: 11,7–24,3).
Tehotietoja on saatu myös satunnaistetusta aktiivisesti kontrolloidusta vaiheen III tutkimuksesta, jossa tutkittiin trabektediinia verrattuna dakarbatsiiniin (tutkimus ET743-SAR-3007). Tutkittavat potilaat saivat hoitoa ei-resekoitavissa olevaan tai metastaattiseen lipo- tai leimyosarkoomaan, ja heitä oli aikaisemmin hoidettu vähintään yhdellä antrasykliiniä ja ifosfamidia sisältävällä hoidolla tai yhdellä antrasykliiniä sisältävällä hoidolla ja yhdellä ylimääräisellä solunsalpaajahoidolla. Trabektediinihoitoryhmän potilaiden piti ottaa deksametasonia 20 mg laskimonsisäisenä injektiona ennen jokaista trabektediini-infuusiota. Kaikkiaan 384 potilasta satunnaistettiin trabektediiniryhmään (1,5 mg/m2 3 viikon välein 24 tunnin ajan) ja 193 potilasta dakarbatsiiniryhmään (1 g/m2 3 viikon välein). Potilaiden iän mediaani oli 56 vuotta (vaihteluväli 17–81 vuotta), ja heistä 30 % oli miehiä, 77 % kaukasialaisia, 12 % afroamerikkalaisia ja 4 % aasialaisia. Hoitokertojen määrän mediaani oli trabektediiniryhmässä 4 ja dakarbatsiiniryhmässä 2. Tutkimuksen tehon ensisijainen päätetapahtuma oli kokonaiselinaika, johon sisältyi 381 kuolematapahtumaa (66 % kaikista satunnaistetuista potilaista): 258 kuolemaa (67,2 %) trabektediiniryhmässä ja 123 kuolemaa (63,7 %) dakarbatsiiniryhmässä (HR 0,927 [95 % lv: 0,748; 1,150; p = 0,4920]). Lopullisessa analyysissa ei havaittu merkitsevää eroa elinajan mediaanissa 21,2 kuukautta kestäneessä seurannassa: trabektediiniryhmän mediaani oli 13,7 kuukautta (95 % lv: 12,2; 16,0) ja dakarbatsiiniryhmän 13,1 kuukautta (95 % lv: 9,1; 16,2]. Pääasiallisten toissijaisten päätetapahtumien yhteenveto on alla olevassa taulukossa:
Tutkimuksen ET743‑SAR‑3007 tehotulokset
Päätetapahtuma/ tutkimuspopulaatio | Trabektediini | Dakarbatsiini | HR / ristitulosuhde (OR) | p-arvo |
Ensisijainen päätapahtuma | n = 384 | n = 193 |
|
|
Kokonaiselinaika, n (%) | 258 (67,2 %) | 123 (63,7 %) | 0,927 (0,748–1,150) | 0,4920 |
Toissijaiset päätetapahtumat | n = 345 | n =173 |
|
|
Etenemisvapaa elinaika (kuukautta; 95 % lv) | 4,2 | 1,5 | 0,55 (0,44; 0,70) | < 0,0001 |
Kokonaisvasteprosentti, n (%); ristitulosuhde (95 % lv) | 34 (9,9 %) | 12 (6,9 %) | 1,47 (0,72; 3,2) | 0,33 |
Vasteen kesto (DOR) (kuukautta; 95 % lv) | 6,5 | 4,2 | 0,47 (0,17; 1,32) | 0,14 |
Kliininen hyötyprosentti (CBR), n (%); ristitulosuhde (95 % lv) | 34,2 % | 18,5 % | 2,3 (1,45; 3,7) | < 0,0002 |
Tehotietoja on saatu myös satunnaistetusta, avoimesta, vaiheen III monikeskustutkimuksesta (JapicCTI-121850). Tutkittavat olivat japanilaisia potilaita, joilla oli translokaatioon liittyvä sarkooma (TRS), joista yleisimpiä olivat myksoidinen pyörösoluinen liposarkooma (n = 24), synoviaalisarkooma (n = 18), mesenkymaalinen kondrosarkooma (n = 6) ja ekstraskeletaalinen Ewingin sarkooma / PNET, alveolaarinen pehmytosasarkooma, alveolaarinen rabdomyosarkooma ja kirkassolusarkooma (kullakin n = 5). Tutkimuksessa arvioitiin trabektediinin tehoa ja turvallisuutta verrattuna parhaimpaan tukihoitoon toisena hoitolinjana tai myöhempänä hoitona edennyttä translokaatioon liittyvää sarkoomaa sairastavilla potilailla, joilla ei saatu vastetta tavanomaisella kemoterapialla tai jotka eivät sietäneet sitä. Potilaat saivat japanilaisille potilaille suositellun trabektediiniannoksen 1,2 mg/m2 (1,2 mg/m2 3 viikon välein 24 tunnin ajan). Tutkimukseen otettiin yhteensä 76 japanilaista potilasta, joista 73 potilasta oli lopullisessa analyysijoukossa. Tutkimuksen ensisijainen päätetapahtuma oli etenemisvapaa elinaika, jonka paraneminen oli tilastollisesti merkitsevää trabektediiniryhmässä parhaimpaan tukihoitoon verrattuna [HR = 0,07; 95 % CI: 0,03–0,16; p < 0,0001]. Trabektediiniryhmässä etenemisvapaan elinajan mediaani oli 5,6 kuukautta [95 % CI: 4,1–7,5] ja parasta tukihoitoa saaneessa ryhmässä 0,9 kuukautta [95 % CI: 0,7–1,0]. Toissijaisiin päätetapahtumiin sisältyi objektiivinen vaste, jota analysoitiin RECIST-ja Choi-kriteereiden mukaan. RECIST-kriteerien mukaan kokonaisvasteluku trabektediinilla hoidetuilla potilailla oli 3 (8,1 %; 95 % CI: 1,7–21,9 %) ja kliininen hyötyluku 24 (64,9 %, 95 % CI: 47,5–79,9 %), kun taas parasta tukihoitoa saaneilla potilailla kokonaisvasteluku oli 0 (0 %, 95 % CI: 0,0–9,7 %) ja kliininen hyötyluku 0 (0 %, 95 % CI: 0,0–9,7 %). Choi-kriteerien mukaan kokonaisvasteluku trabektediinilla hoidetuilla potilailla oli 4 (10,8 %; 95 % CI: 3,0–25,4 %) ja kliininen hyötyluku 7 (18,9 %, 95 % CI: 8,0–35,2 %), kun taas parasta tukihoitoa saaneilla kokonaisvasteluku oli 0 (0 %, 95 % CI: 0,0–9,7 %) ja kliininen hyötyluku 0 (0 %, 95 % CI: 0,0–9,7 %).
Tiedot Yondelisin ja PLD:n yhdistelmähoidon tehosta uusiutuneen munasarjasyövän hoidossa perustuvat tutkimukseen ET743-OVA-301. Tässä satunnaistetussa vaiheen 3 tutkimuksessa 672 potilasta sai joko trabektediinia (1,1 mg/m2) ja PLD.tä (30 mg/m2) 3 viikon välein tai pelkkää PLD:tä (50 mg/m2) 4 viikon välein. Ensisijaiseen, etenemisvapaata elinaikaa koskeneeseen analyysiin otettiin 645 potilasta, joiden tauti oli mitattavissa. Näille potilaille tehtiin riippumaton radiologinen arviointi. Yhdistelmähoitoryhmässä taudin etenemisen riski oli 21 % pienempi kuin pelkkää PLD-hoitoa saaneilla (HR = 0,79, luottamusväli [lv] 0,65–0,96, p = 0,0190). Myös toissijaiset etenemisvapaan elossaolon ja vasteprosentin analyysit osoittivat, että yhdistelmähoidolla päästiin parempiin tuloksiin. Tärkeimpien tehon analyysien tulokset esitetään yhteenvedon muodossa alla.
Tutkimuksen ET743-OVA-301 tehokkuusanalyysit
| Yondelis+PLD | PLD | HR/ristitulosuhde (OR) | P |
Etenemisvapaa elossaolo | ||||
Riippumaton radiologinen arviointi, tauti mitattavissa * | n = 328 | n = 317 |
|
|
Etenemisvapaan elinajan mediaani (95 % lv) (kk) | 7,3 (5,9–7,9) | 5,8 (5,5–7,1) | 0,79 (0,65–0,96) | 0,0190 a |
Etenemisvapaa elossaoloprosentti 12 kk kohdalla (95 % lv) (%) | 25,8 (19,7–32,3) | 18,5 (12,9–24,9) |
|
|
Riippumaton onkologinen arviointi, kaikki satunnaistetut | n = 336 | n = 335 |
|
|
Etenemisvapaan elinajan mediaani (95 % lv) (kk) | 7,4 (6,4–9,2) | 5,6 (4,2–6,8) | 0,72 (0,60–0,88) | 0,0008 a |
Kokonaiselinaika (loppuanalyysi – n = 522 tapahtumaa, 38 % suljettiin pois) | ||||
Kaikki satunnaistetut | n = 337 | n = 335 |
|
|
Kokonaiselinajan mediaani (95 % lv) (kk) | 22,2 (19,3–25,0) | 18,9 (17,1–21,5) | 0,86 (0,72–1,02) | 0,0835 a |
Kokonaiselinaika platinalle herkässä ryhmässä (loppuanalyysi n=316 tapahtumaa) | ||||
| n = 218 | n = 212 |
|
|
Kokonaiselinajan mediaani (95 % lv) (kk) | 27,0 (24,1–31,4) | 24,1 (20,9–25,9) | 0,83 (0,67–1,04) | 0,1056 a |
Kokonaisvasteprosentti (ORR) | ||||
Riippumaton radiologinen arviointi, kaikki satunnaistetut | n = 337 | n = 335 |
|
|
Kokonaisvasteprosentti (95 % lv) (%) | 27,6 (22,9–32,7) | 18,8 (14,8–23,4) | 1,65 (1,14–2,37) | 0,0080 b |
* Ensisijainen tehokkuusanalyysi
a Log rank ‑testi
b Fisherin testi
Riippumattoman onkologisen arvioinnin perusteella potilaiden, joiden platinahoidoton aika oli < 6 kk (35 % Yondelis- ja PLD-ryhmässä ja 37 % PLD-ryhmässä), etenemisvapaa elinaika oli molemmissa ryhmissä samaa luokkaa (mediaaniaika 3,7 kk, HR = 0,89, lv 0,67–1,20). Potilailla, joiden platinahoidoton aika oli ≥ 6 kk (65 % Yondelis- ja PLD-ryhmässä ja 63 % PLD-ryhmässä), etenemisvapaan elinajan mediaani oli Yondelis- ja PLD-ryhmässä 9,7 kk ja PLD-monoterapiaryhmässä 7,2 kk (HR = 0,66, lv 0,52–0,85).
Loppuanalyysin perusteella Yondelis+PLD-hoidon vs. PLD-hoidon vaikutus kokonaiselossaoloon oli suurempi potilailla, joiden platinahoidoton aika oli ≥ 6 kk (platinalle herkkä ryhmä: 27,0 kk Yondelis- ja PLD-ryhmässä, 24,1 kk PLD-ryhmässä, HR = 0,83, lv 0,67–1,04), kuin niillä, joiden platinahoidoton aika oli < 6 kk (platinalle resistentti ryhmä: 14,2 kk Yondelis- ja PLD-ryhmässä ja 12,4 kk PLD-ryhmässä, HR = 0,92, lv 0,70–1,21).
Yondelis+PLD-hoidolla saavutettu kokonaiselossaolohyöty ei johtunut myöhemmistä hoidoista, mikä oli hyvin tasapainossa kahden hoitohaaran välillä.
Monimuuttuja-analyysissä, jossa otettiin huomioon platinahoidoton aika, hoidon vaikutus kokonaiselossaoloon oli Yondelis+PLD-ryhmässä tilastollisesti merkitsevässä määrin parempi (kaikki satunnaistetut p = 0,0285, platinalle herkkä ryhmä p = 0,0319).
Hoitoryhmien välillä ei todettu tilastollisesti merkitseviä eroja yleisluontoisissa elämänlaatumittareissa.
Yondelis+PLD-yhdistelmää relapsoituneessa munasarjasyövässä arvioitiin myös tutkimuksessa ET743-OVC-3006, vaiheen 3 tutkimuksessa, jossa munasarjasyöpää sairastavat naiset satunnaistettiin toisen platinaa sisältävän hoito-ohjelman epäonnistumisen jälkeen saamaan Yondelista (1,1 mg/m2) ja PLD:tä (30 mg/m2) 3 viikon välein tai PLD:tä (50 mg/m2) 4 viikon välein. Tutkimukseen osallistuvien tuli olla platinaherkkiä (PFI ≥ 6 kuukautta) ensimmäisen platinaa sisältävän hoito-ohjelmansa jälkeen ja heillä tuli olla täydellinen tai osittainen vaste toisen linjan platinapohjaiseen kemotarapiaan (ilman PFI-rajoitteita). Tämä tarkoittaa, että nämä potilaat voivat olla joko platinaherkkiä (PFI ≥ 6 kuukautta) tai -resistenttejä (PFI <6 kuukautta) toisen platinaa sisältävän hoito-ohjelmansa jälkeen. Post hoc -analyysi määritteli, että 42 % rekrytoiduista tutkittavista olivat platinaresistenttejä (PFI < 6 kuukautta) viimeisen platinaa sisältävän hoito-ohjelmansa jälkeen.
Tutkimuksen ET743-OVC-3006 ensisijainen päätepiste oli OS, ja toissijaiset päätepisteet sisälsivät PFS:n ja ORR:n. Tutkimuksen koko määrättiin siten, että noin 670 potilasta rekrytoitiin, jotta voitiin havaita 514 kuolemaa ja HR 0,78 OS:lle 80 %:n teholla, kun kaksipuoleinen 0.05:n merkitystaso jakautui kahden OS-analyysin poikki, väliaikainen (60 % tai 308/514 kuolemasta) ja lopullisen analyysin poikki (514 kuolemaa). Kaksi varhaista futiliteettianalyysia tehtiin Riippumattoman tietojenseurantakomitean (Independent Data Monitoring Committee IDMC) pyynnöstä. Toisen futiliteettianalyysin, joka tehtiin 45 % suunniteltujen tapahtumien kohdalla (232/514 kuolemaa), jälkeen IDMC suositteli tutkimuksen keskeyttämistä, koska (1) ensisijaisen OS-analyysin futiliteetti ja (2) haittatapahtumien epätasapainoon perustuva suuri riski eivät suosineet Yondelis+PLD-yhdistelmää. Tutkimuksen varhaisessa keskeytyksessä 9 % (52/572 hoidettua) tutkittavista lopetti hoidon, 45 % (260/576 satunnaistettua) lopetti seurannan ja 54 % (310/576 satunnaistettua) sensuroitiin OS-arvioinnista, poissulkien luotettavat PFS-arviot ja OS-päätepisteet.
Yondelis+PLD-hoidon vertailusta platinapohjaiseen hoitoon platinapohjaisille hoidoille herkillä potilailla ei ole tietoa.
Pediatriset potilaat
SAR-2005-tutkimuksen I–II-vaiheeseen osallistui 50 pediatrista potilasta, jotka sairastivat rabdomyosarkoomaa, Ewingin sarkoomaa tai muuta pehmytkudossarkoomaa kuin rabdomyosarkooma. Kahdeksan potilaan annos oli 1,3 mg/m2 ja 42 potilaan 1,5 mg/m2. Trabektediini annettiin 24 tuntia kestävänä laskimoinfuusiona kolmen viikon välein. Vaste arvioitiin täydellisesti 40 potilaalla. Yksi osittainen keskitetysti vahvistettu vaste (PR, partial response) havaittiin: kokonaisvaste ORR: 2,5 % (CI 95 % 0,1–13,2 %). PR vastasi alveolaarista rabdomyosarkoomaa sairastavan vastetta. Vasteen kesto oli 6,5 kuukautta. Vastetta ei havaittu Ewingin sarkoomalle tai NRSTS:lle, RR: 0 % (CI 95 % (0 %–30,9 %)). Kolme potilasta saavutti stabiilin tautitilan (yksi rabdomyosarkoomaa sairastava 15 hoitojakson jälkeen, yksi sukkulasolusarkoomaa sairastava kahden hoitojakson jälkeen ja yksi Ewingin sarkoomaa sairastava neljän hoitojakson jälkeen).
Haittavaikutuksiin sisältyivät palautuva maksaentsyymien nousu ja hematologiset haittatapahtumat; lisäksi ilmoitettiin myös kuumeesta, infektiosta, nestehukasta ja tromboosista/embolismista.
Farmakokinetiikka
Jakautuminen
Kun valmiste annetaan tasaisella nopeudella laskimoinfuusiona, systeeminen altistus kasvaa suhteessa annokseen, kun annos on enintään 1,8 mg/m2. Trabektediinin farmakokineettisen profiilin perusteella vaikuttaa siltä, että lääkkeen jakautuminen on monitilamallin mukaista.
Laskimoon annetun trabektediinin näennäinen jakautumistilavuus on suuri, sillä lääke sitoutuu voimakkaasti kudoksiin ja plasman proteiineihin (94–98 % plasman trabektediinista on proteiineihin sitoutunutta). Vakaassa tilassa trabektediinin jakautumistilavuus ihmisellä on yli 5 000 l.
Biotransformaatio
CYP3A4 on tärkein trabektediinin oksidatiivisesta metaboliasta vastaava sytokromi P450:n isoentsyymi kliinisesti relevantteja pitoisuuksia käytettäessä. Muut CYP450-entsyymit saattavat osallistua metaboliaan. Trabektediini ei indusoi eikä estä tärkeimpien CYP450-entsyymien toimintaa.
Eliminaatio
Muuttumaton trabektediini eliminoituu vain vähäisessä määrin (alle 1 %) ihmisen munuaisten kautta. Terminaalinen puoliintumisaika on pitkä (terminaalisen eliminaatiovaiheen pituus tutkitussa populaatiossa: 180 tunnista ylöspäin). Kun syöpäpotilaille annettiin radioaktiivisesti leimattu trabektediiniannos, keskimäärin 58 % kokonaisradioaktiivisuudesta erittyi ulosteeseen (keskihajonta 17 %) ja keskimäärin 5,8 % virtsaan (keskihajonta 1,73 %). Populaatiosta arvioidun trabektediinin plasmapuhdistuman (30,9 l/h) ja veri/plasmasuhteen (0,89) perusteella trabektediinin puhdistuma kokoverestä on noin 35 l/h. Tämä on noin puolet ihmisen maksan verenkiertonopeudesta. Trabektediinin ekstraktiosuhdetta voidaan näin ollen pitää kohtalaisena. Trabektediinin plasmapuhdistuman arviointiin käytetyssä populaatiossa potilaiden välinen vaihtelu oli 49 % ja saman potilaan eri arviointikertojen välinen vaihtelu 28 %.
Populaatiofarmakokineettinen analyysi osoitti, että trabektediinin puhdistuma plasmasta pieneni 31 %, kun se annosteltiin yhdessä PLD:n kanssa. Trabektediinin samanaikainen annostelu ei vaikuttanut PLD:n farmakokinetiikkaan.
Erityisryhmät
Populaatiofarmakokineettisen analyysin tulokset viittasivat siihen, että ikä (vaihteluväli 19–83 v), sukupuoli, paino (vaihteluväli 36–148 kg) ja elimistön pinta-ala (vaihteluväli 0,9–2,8 m2) eivät vaikuta trabektediinin puhdistumaan plasmasta. Populaatiofarmakokineettinen analyysi osoitti, että japanilaisten populaatiossa havaitut trabektediinipitoisuudet plasmassa annostuksella 1,2 mg/m² vastasivat pitoisuuksia, jotka saatiin ei-japanilaisten länsimaalaisten populaatiossa annostuksella 1,5 mg/m².
Munuaisten vajaatoiminta
Kreatiniinipuhdistuman perusteella mitatulla munuaistoiminnalla ei ollut merkittävää vaikutusta trabektediinin farmakokinetiikkaan kliinisiin tutkimuksiin osallistuneiden potilaiden puhdistuma-arvojen rajoissa (≥ 30,3 ml/min). Potilaista, joiden kreatiniinipuhdistuma on alle 30,3 ml/min, ei ole tietoja. Vain pieni osuus potilaiden saaman 14C-merkityn trabektediinikerta-annoksen kokonaisradioaktiivisuudesta erittyi virtsaan (< 9 % kaikilla tutkituilla potilailla), mikä viittaa siihen, että munuaisten vajaatoiminnalla on vain vähäinen vaikutus trabektediinin tai sen metaboliittien eliminaatioon.
Maksan vajaatoiminta
Maksan vajaatoiminnan vaikutusta trabektediinin farmakokinetiikkaan arvioitiin 15 syöpäpotilaalla, joiden annos oli 0,58–1,3 mg/ m2 3 tuntia kestävänä infuusiona. Geometrinen keskimääräinen annosnormalisoitu trabektediinialtistus (AUC) suureni 97 % (90 %:n luottamusväli: 20–222 %) 6 potilaalla, joilla oli kohtalainen maksan vajaatoiminta (kohonneet seerumin bilirubiiniarvot 1,5‑3 x ULN ja kohonneet aminotransferaasiarvot [ASAT tai ALAT] < 8 x ULN), trabektediinin kerta-annoksen 0,58 mg/m2 (n = 3) tai 0,9 mg/m2 (n = 3) annon jälkeen verrattuna 9 potilaaseen, joiden maksan toiminta oli normaali trabektediinin kerta-annoksen 1,3 mg/m2 annon jälkeen (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Prekliiniset tiedot turvallisuudesta
Prekliiniset tiedot viittaavat siihen, että trabektediini vaikuttaa rajallisessa määrin sydämeen, verisuonistoon, hengityselimistöön ja keskushermostoon, kun altistuksen AUC-arvo on kliinistä terapeuttista altistusta pienempi.
Trabektediinin vaikutusta sydän- ja verisuonitoimintaan ja hengitystoimintaan on tutkittu in vivo nukutetuilla makakeilla. Infuusioajaksi valittiin 1 h, jotta valmisteen maksimipitoisuudet plasmassa (Cmax-arvot) olisivat samaa luokkaa kuin kliinisesti todettavat arvot. Näin saavutettu plasman trabektediinipitoisuus (Cmax) oli 10,6 ± 5,4 ng/ml eli suurempi kuin 1 500 mikrog/m2 infuusion 24 tunnin aikana saaneilla potilailla (joiden Cmax oli 1,8 ± 1,1 ng/ml) ja samankaltainen kuin niillä potilailla, jotka saivat saman annoksen 3 tuntia kestävänä infuusiona (jolloin Cmax oli 10,8 ± 3,7 ng/ml).
Trabektediinin tärkeimmiksi toksisiksi vaikutuksiksi todettiin myelosuppressio ja maksatoksisuus. Muita löydöksiä olivat hematopoieesiin kohdistuva toksisuus (vaikea leukopenia, anemia ja imukudos- ja luuydintuho) sekä maksan toimintakoetulosten kohoaminen, maksasolujen degeneraatio, suoliepiteelin nekroosi ja vaikeat paikallisreaktiot injektiokohdassa. Apinoilla tehdyissä useiden hoitojaksojen pituisissa toksisuustutkimuksissa havaittiin munuaisiin kohdistuvaa toksisuutta. Näitä löydöksiä edelsivät antopaikan vaikeat paikallisreaktiot, joten on epäselvää, oliko syynä trabektediini. Näiden munuaislöydösten tulkinnassa on kuitenkin noudatettava varovaisuutta, eikä hoidon aiheuttaman toksisuuden mahdollisuutta voida sulkea pois.
Trabektediini on genotoksinen sekä in vitro että in vivo. Pitkäaikaisia karsinogeenisuustutkimuksia ei ole tehty.
Trabektediinilla ei tehty hedelmällisyystutkimuksia, mutta toistuvaisannoksia koskeneiden toksisuustutkimusten yhteydessä havaittiin sukurauhasten rajallisia histopatologisia muutoksia. Kun yhdisteen sytotoksinen ja mutageeninen luonne otetaan huomioon, on todennäköistä, että se vaikuttaa lisääntymiskykyyn.
Trabektediinin siirtymistä istukkaan ja sikiön altistumista trabektediinille havaittiin tutkimuksessa, jossa tiineet rotat saivat yhden laskimonsisäisen annoksen 14C-trabektediinia annostuksella 0,061 mg/kg. Sikiökudoksen enimmäisradioaktiivisuuspitoisuus oli sama kuin äidin plasmassa tai veressä.
Farmaseuttiset tiedot
Apuaineet
Sakkaroosi
Kaliumdivetyfosfaatti
Fosforihappo (pH:n säätöön)
Kaliumhydroksidi (pH:n säätöön)
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet. Sama koskee valmisteen laimentamista.
Kestoaika
Avaamaton injektiopullo
60 kuukautta.
Käyttökuntoon saatettu valmiste
Osoitettu säilyvän kemiallisesti ja fysikaalisesti stabiilina 30 tunnin ajan enintään 25 °C lämpötilassa.
Mikrobiologiselta kannalta käyttökuntoon saatettu liuos tulee laimentaa ja käyttää välittömästi. Jos käyttökuntoon saatettua valmistetta ei käytetä välittömästi, sen käytönaikainen säilytysaika ja käyttöä edeltävät säilytysolosuhteet ovat käyttäjän vastuulla. Ne ovat kuitenkin yleensä enintään 24 tuntia 2–8 °C:n lämpötilassa, ellei käyttökuntoon saattaminen tapahdu kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Laimennettu valmiste
Osoitettu säilyvän kemiallisesti ja fysikaalisesti stabiilina 30 tunnin ajan enintään 25 °C lämpötilassa.
Säilytys
Säilytä jääkaapissa (2 °C ‑ 8 °C).
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. Kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
YONDELIS kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
0,25 mg (L:ei) 0,25 mg (712,46 €)
1 mg (L:ei) 1 mg (2507,35 €)
PF-selosteen tieto
Yondelis 0,25 mg
Tyypin I lasista valmistettu väritön injektiopullo, jossa on butyylikumitulppa ja alumiininen repäisysinetti, sisältää 0,25 mg trabektediinia.
Yksi pakkaus sisältää yhden injektiopullon.
Yondelis 1 mg
Tyypin I lasista valmistettu väritön injektiopullo, jossa on butyylikumitulppa ja alumiininen repäisysinetti, sisältää 1 mg trabektediinia.
Yksi pakkaus sisältää yhden injektiopullon.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen kuiva-aine.
Käyttö- ja käsittelyohjeet
Valmistelu laskimoinfuusiota varten
Yondelis saatetaan käyttökuntoon ja jatkolaimennetaan ennen infuusiota. Infuusionesteen valmistuksessa on käytettävä asianmukaista aseptista tekniikkaa (ks. kohdat Käyttökuntoon saattamisen ohjeet ja Laimennusohjeet).
PLD:n kanssa yhdistelmähoitona käytettäessä infuusioletku tulee huuhdella perusteellisesti 50 mg/ml (5 %) glukoosi-infuusionesteellä PLD-annoksen annostelun jälkeen ja ennen Yondelisin antoa. Muiden laimentimien kuin 50 mg/ml (5 %) glukoosi-infuusionesteen käyttö letkun huuhtelussa voi aiheuttaa PLD:n saostumista (tarkat käsittelyohjeet, ks. PLD-valmisteen valmisteyhteenveto).
Käyttökuntoon saattamisen ohjeet
Yondelis 0,25 mg
Yksi injektiopullo, jossa on 0,25 mg trabektediinia, saatetaan käyttökuntoon lisäämällä siihen 5 ml steriiliä injektionesteisiin käytettävää vettä. Näin saadun liuoksen trabektediinipitoisuus on 0,05 mg/ml, ja se on tarkoitettu vain yhtä käyttökertaa varten.
Injektiopulloon injisoidaan ruiskulla 5 ml steriiliä injektionesteisiin käytettävää vettä. Injektiopulloa ravistellaan, kunnes valmiste on liuennut täysin. Käyttökuntoon saatettu liuos on kirkas, väritön tai hieman kellertävä liuos, jossa ei ole käytännössä lainkaan näkyviä hiukkasia.
Tämä käyttökuntoon saatettu liuos sisältää 0,05 mg/ml trabektediinia. Se tulee jatkolaimentaa, ja sitä tulee käyttää vain yhtä käyttökertaa varten.
Yondelis 1 mg
Yksi injektiopullo, jossa on 1 mg trabektediinia, saatetaan käyttökuntoon lisäämällä siihen 20 ml steriiliä injektionesteisiin käytettävää vettä. Näin saadun liuoksen trabektediinipitoisuus on 0,05 mg/ml, ja se on tarkoitettu vain yhtä käyttökertaa varten.
Injektiopulloon injisoidaan ruiskulla 20 ml steriiliä injektionesteisiin käytettävää vettä. Injektiopulloa ravistellaan, kunnes valmiste on liuennut täysin. Käyttökuntoon saatettu liuos on kirkas, väritön tai hieman kellertävä liuos, jossa ei ole käytännössä lainkaan näkyviä hiukkasia.
Tämä käyttökuntoon saatettu liuos sisältää 0,05 mg/ml trabektediinia. Se tulee jatkolaimentaa, ja sitä tulee käyttää vain yhtä käyttökertaa varten.
Laimennusohjeet
Käyttökuntoon saatettu liuos tulee laimentaa 9 mg/ml (0,9 %) NaCl-infuusionesteellä tai 50 mg/ml (5 %) glukoosi-infuusionesteellä. Tarvittava määrä lasketaan seuraavasti:
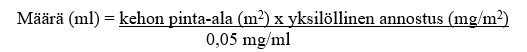
Jos valmiste aiotaan antaa keskuslaskimokatetrin kautta, asianmukainen määrä käyttökuntoon saatettua liuosta vedetään injektiopullosta ja lisätään infuusiopussiin, joka sisältää ≥ 50 ml laimenninta (9 mg/ml [0,9 %] NaCl-infuusionestettä tai 50 mg/ml [5 %] glukoosi-infuusionestettä), jolloin infuusionesteen trabektediinipitoisuus on ≤ 0,030 mg/ml.
Jos valmistetta ei voida antaa keskuslaskimoon, vaan on käytettävä perifeeristä laskimoyhteyttä, käyttökuntoon saatettua liuos tulee lisätä infuusiopussiin, joka sisältää ≥ 1 000 ml laimenninta (9 mg/ml [0,9 %] NaCl-infuusionestettä tai 50 mg/ml [5 %] glukoosi-infuusionestettä).
Ennen annostelua parenteraalinen liuos tarkistetaan silmämääräisesti näkyvien hiukkasten varalta. Kun infuusioneste on valmisteltu, se tulee antaa välittömästi.
Käsittely- ja hävittämisohjeet
Yondelis on sytotoksinen syöpälääke. Siksi sen kuten muidenkin mahdollisesti toksisten valmisteiden käsittelyssä tulee noudattaa varovaisuutta. Sytotoksisten lääkevalmisteiden asianmukaista käsittelyä ja hävittämistä koskevia menettelytapoja on noudatettava. Yondelis-valmistetta käsittelevän henkilöstön tulee saada etukäteen valmisteen asianmukaista käyttökuntoon valmistus- ja laimennustekniikkaa koskevaa koulutusta. Käyttökuntoon ja laimentamisen aikana on käytettävä suojavaatetusta, johon kuuluu kasvomaski, suojalasit ja käsineet. Raskaana olevat henkilöstön jäsenet eivät saa käsitellä tätä lääkevalmistetta.
Jos valmistetta joutuu vahingossa iholle, silmään tai limakalvolle, alue tulee huuhdella välittömästi runsaalla vedellä.
Yondelis-valmisteen ei ole havaittu olevan yhteensopimaton tyypin I lasipullojen, PVC- tai PE-pussien eikä ‑letkujen, polyisopreenisäiliöiden eikä implantoitavien titaanisten laskimoporttien kanssa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä sytotoksisia lääkevalmisteita koskevien paikallisten vaatimusten mukaisesti.
Korvattavuus
YONDELIS kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
0,25 mg 0,25 mg
1 mg 1 mg
- Ei korvausta.
ATC-koodi
L01CX01
Valmisteyhteenvedon muuttamispäivämäärä
21.05.2026
Yhteystiedot
Solnavägen 3H
113 63 Stockholm
Sweden
+46 (0)8 5333 95 00
www.immedica.com
info@immedica.com