
LYNPARZA tabletti, kalvopäällysteinen 100 mg, 150 mg
Vaikuttavat aineet ja niiden määrät
Lynparza 100 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 100 mg olaparibia.
Lynparza 150 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 150 mg olaparibia.
Apuaine, jonka vaikutus tunnetaan:
Tämä lääkevalmiste sisältää 0,24 mg natriumia per 100 mg:n tabletti ja 0,35 mg natriumia per 150 mg:n tabletti.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti).
Kliiniset tiedot
Käyttöaiheet
Munasarjasyöpä
Lynparza on tarkoitettu monoterapiana:
- pitkälle edennyttä (FIGO-levinneisyysasteet III ja IV) korkean pahanlaatuisuusasteen epiteelistä munasarja-, munanjohdin‑ tai primaaria peritoneaalista syöpää sairastavien aikuispotilaiden ylläpitohoitoon, kun potilailla on BRCA1/2-mutaatio (ituradan ja/tai somaattinen) ja potilaiden hoitovaste (täydellinen tai osittainen) on säilynyt ensilinjan platinapohjaisen solunsalpaajahoidon päättymisen jälkeen.
- uusiutunutta platinaherkkää korkean pahanlaatuisuusasteen epiteelistä munasarja-, munanjohdin‑ tai primaaria peritoneaalista syöpää sairastavien aikuispotilaiden ylläpitohoitoon, kun potilailla on hoitovaste (täydellinen tai osittainen) platinapohjaiselle solunsalpaajahoidolle.
Lynparza on tarkoitettu käytettäväksi yhdessä bevasitsumabin kanssa:
- pitkälle edennyttä (FIGO-levinneisyysasteet III ja IV) korkean pahanlaatuisuusasteen epiteelistä munasarja-, munanjohdin‑ tai primaaria peritoneaalista syöpää sairastavien aikuispotilaiden ylläpitohoitoon, kun potilaiden hoitovaste (täydellinen tai osittainen) on säilynyt platinapohjaisen solunsalpaajahoidon ja bevasitsumabin yhdistelmällä annetun ensilinjan hoidon jälkeen ja potilaiden syöpään liittyy positiivinen HRD-status (homologous recombination deficiency) määriteltynä joko BRCA1/2-mutaation ja/tai genomin epävakauden perusteella (ks. kohta Farmakodynamiikka).
Rintasyöpä
Lynparza on tarkoitettu:
- monoterapiana tai hormonaaliseen hoitoon yhdistettynä liitännäishoitona aikuispotilaille, joilla on ituradan BRCA1/2-mutaatioita ja HER2-negatiivinen korkean riskin varhaisvaiheen rintasyöpä, jota on aiemmin hoidettu esiliitännäis- tai liitännäissolunsalpaajahoidolla (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka)
- monoterapiana aikuispotilaille, joilla on ituradan BRCA1/2-mutaatioita ja HER2-negatiivinen paikallisesti edennyt tai metastasoitunut rintasyöpä. Edellytyksenä on, että potilaat ovat aiemmin saaneet antrasykliinia ja taksaania joko (esi)liitännäishoitona tai metastasoituneen taudin hoitoon, paitsi jos nämä hoidot eivät ole soveltuneet potilaille (ks. kohta Farmakodynamiikka). Jos potilaalla on hormonireseptoripositiivinen (HR-positiivinen) rintasyöpä, edellytetään myös, että tauti on edennyt aiemman hormonihoidon aikana tai sen jälkeen tai että potilas ei sovellu saamaan hormonihoitoa.
Haiman adenokarsinooma
Lynparza on tarkoitettu monoterapiana ylläpitohoitoon aikuispotilaille, joilla on ituradan BRCA1/2-mutaatioita ja metastasoitunut haiman adenokarsinooma ja joilla tauti ei ole edennyt ensilinjan solunsalpaajahoitona annetun vähintään 16 viikon pituisen platinahoidon jälkeen.
Eturauhassyöpä
Lynparza on tarkoitettu:
- monoterapiana aikuispotilaille, joilla on metastasoitunut kastraatioresistentti eturauhassyöpä (mCRPC) ja BRCA1/2-mutaatioita (ituradan ja/tai somaattisia) ja joilla tauti on edennyt aiemman uutta hormonaalista lääkeainetta sisältäneen hoidon jälkeen.
- yhdessä abirateronin ja prednisonin tai prednisolonin kanssa metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastaville aikuispotilaille, kun solunsalpaajahoito ei ole kliinisesti aiheellista (ks. kohta Farmakodynamiikka).
Kohdun limakalvon syöpä
Lynparza yhdessä durvalumabin kanssa on tarkoitettu ylläpitohoitoon aikuispotilaille, joilla on primaari pitkälle edennyt tai uusiutunut kohdun limakalvon syöpä, johon liittyy toimiva DNA:n kahdentumisvirheiden korjausmekanismi eli MMR-mekanismi (pMMR; proficient mismatch repair), ja joiden tauti ei ole edennyt durvalumabin, karboplatiinin ja paklitakselin yhdistelmällä annetun ensilinjan hoidon aikana.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Lynparza‑hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Potilasvalinta
Pitkälle edenneen munasarjasyövän ensilinjan ylläpitohoito, kun potilailla on BRCA-mutaatio:
Ennen Lynparza-hoidon aloittamista korkean pahanlaatuisuusasteen epiteelisen munasarja-, munanjohdin‑ tai primaarin peritoneaalisen syövän ensilinjan ylläpitohoitoon täytyy validoidulla testausmenetelmällä osoittaa, että potilaalla on haitallinen tai epäilty haitallinen ituradan mutaatio ja/tai somaattisia mutaatioita rintasyövän alttiusgeeneissä (BRCA) 1 tai 2.
Platinaherkän uusiutuneen munasarjasyövän ylläpitohoito:
BRCA1/2-testausta ei vaadita ennen kuin Lynparza-valmistetta käytetään monoterapiana uusiutuneen epiteelisen munasarja-, munanjohdin- tai primaarin peritoneaalisen syövän ylläpitohoitoon, kun potilaalla on täydellinen tai osittainen hoitovaste platinapohjaiselle solunsalpaajahoidolle.
HRD-positiivisen pitkälle edenneen munasarjasyövän ensilinjan ylläpitohoito yhdistelmänä bevasitsumabin kanssa:
Ennen Lynparza-valmisteen ja bevasitsumabin yhdistelmähoidon aloittamista epiteelisen munasarja-, munanjohdin- tai primaarin peritoneaalisen syövän ensilinjan ylläpitohoitoon täytyy varmistaa, että potilaalla on validoidulla testausmenetelmällä osoitettu haitallinen tai epäilty haitallinen BRCA1/2-mutaatio ja/tai genomin epävakaus (ks. kohta Farmakodynamiikka).
Ituradan BRCA-mutaation omaavan korkean riskin varhaisvaiheen rintasyövän liitännäishoito
Ennen Lynparza-hoidon aloittamista HER2-negatiivisen korkean riskin varhaisvaiheen rintasyövän liitännäishoitona täytyy varmistaa, että potilaalla on validoidulla testausmenetelmällä osoitettu haitallinen tai epäilty haitallinen gBRCA1/2-mutaatio (ks. kohta Farmakodynamiikka).
gBRCA1/2-mutaation omaavan HER2-negatiivisen metastasoituneen rintasyövän hoito monoterapiana:
Jos potilaalla on epidermaalisen kasvutekijän reseptori 2:n (HER2) suhteen negatiivinen paikallisesti edennyt tai metastasoitunut rintasyöpä ja rintasyövän alttiusgeenien ituradan mutaatioita (gBRCA1/2), haitallinen tai epäilty haitallinen gBRCA1/2-mutaatio täytyy varmistaa ennen Lynparza-hoidon aloittamista. gBRCA1/2-mutaatio on osoitettava validoidulla testausmenetelmällä tällaisiin määrityksiin perehtyneessä laboratoriossa. Rintasyöpään liittyvän kasvaimen BRCA1/2-määritysten kliinisen validoinnin osoittavia tietoja ei tällä hetkellä ole saatavilla.
Haiman metastasoituneen adenokarsinooman ensilinjan ylläpitohoito, kun potilailla on gBRCA-mutaatio:
Kun on kyseessä haiman metastasoituneen adenokarsinooman ensilinjan ylläpitohoito ja potilaalla on ituradan BRCA1/2-mutaatioita, haitallinen tai epäilty haitallinen gBRCA1/2-mutaatio täytyy varmistaa ennen Lynparza-hoidon aloittamista. gBRCA1/2-mutaatio on osoitettava validoidulla testausmenetelmällä tällaisiin määrityksiin perehtyneessä laboratoriossa. Haiman adenokarsinoomaan liittyvän kasvaimen BRCA1/2-määritysten kliinisen validoinnin osoittavia tietoja ei tällä hetkellä ole saatavilla.
BRCA1/2-mutaation omaavan metastasoituneen kastraatioresistentin eturauhassyövän hoito monoterapiana:
BRCA1/2-mutaation omaavaa metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavilla potilailla pitää olla vahvistettu haitallinen tai epäilty haitallinen BRCA1/2-mutaatio (vahvistetaan joko kasvain- tai verinäytteestä) ennen Lynparza-hoidon aloittamista (ks. kohta Farmakodynamiikka). BRCA1/2-mutaatio on osoitettava validoidulla testausmenetelmällä tällaisiin määrityksiin perehtyneessä laboratoriossa.
Metastasoituneen kastraatioresistentin eturauhassyövän hoito yhdessä abirateronin ja prednisonin tai prednisolonin kanssa:
Genomitutkimusta ei tarvita ennen Lynparza-valmisteen käyttöä yhdessä abirateronin ja prednisonin tai prednisolonin kanssa hoidettaessa metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavia potilaita.
Toimivan MMR-mekanismin omaavan (pMMR), pitkälle edenneen tai uusiutuneen kohdun limakalvon syövän ensilinjan ylläpitohoito yhdessä durvalumabin kanssa:
Ennen hoidon aloittamista täytyy varmistaa validoidulla testausmenetelmällä, että kasvaimessa on toimiva MMR-mekanismi (pMMR) (ks. kohta Farmakodynamiikka).
Potilaille, joilta tutkitaan BRCA1/2-geenien mutaatioita, on annettava perinnöllisyysneuvontaa paikallisten säädösten mukaisesti.
Annostus
Lynparza-valmistetta on saatavilla 100 mg:n ja 150 mg:n tabletteina.
Lynparza‑valmisteen suositeltu annos monoterapiana tai yhdessä muiden lääkeaineiden kanssa on 300 mg (kaksi 150 mg:n tablettia) kaksi kertaa vuorokaudessa, mikä vastaa 600 mg:n kokonaisvuorokausiannosta. Annoksen pienentämistä varten on saatavilla 100 mg:n tabletti.
Lynparza-monoterapia
Potilaiden, joilla on uusiutunut platinaherkkä (PSR) korkean pahanlaatuisuusasteen epiteelinen munasarjasyöpä, munanjohdinsyöpä tai primaari peritoneaalinen syöpä ja joilla on hoitovaste (täydellinen tai osittainen) platinapohjaiselle solunsalpaajahoidolle, on aloitettava Lynparza‑hoito viimeistään 8 viikon kuluttua viimeisen platinaa sisältävän hoitoannoksen jälkeen.
Lynparza yhdessä bevasitsumabin kanssa
Kun Lynparza-valmistetta käytetään yhdessä bevasitsumabin kanssa korkean pahanlaatuisuusasteen epiteelisen munasarja-, munanjohdin‑ tai primaarin peritoneaalisen syövän ensilinjan ylläpitohoitoon sen jälkeen, kun potilas on saanut päätökseen platinapohjaisen solunsalpaajahoidon ja bevasitsumabin yhdistelmällä annetun ensilinjan hoidon, bevasitsumabin annos on 15 mg/kg kerta-annoksena 3 viikon välein. Katso lisätiedot bevasitsumabin valmisteyhteenvedosta (ks. kohta Farmakodynamiikka).
Lynparza yhdessä hormonaalisen hoidon kanssa
Katso suositeltua annostusta koskevat tiedot yhdistelmähoidossa käytettävien hormonaalisten hoitojen (aromataasin estäjä tai antiestrogeeni ja/tai LHRH) valmisteyhteenvedoista.
Lynparza yhdessä abirateronin ja prednisonin tai prednisolonin kanssa
Kun Lynparza-valmistetta käytetään yhdessä abirateronin kanssa metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavien potilaiden hoitoon, abirateronin annos on 1 000 mg suun kautta kerran vuorokaudessa (ks. kohta Farmakodynamiikka). Abirateronihoidon yhteydessä on annettava 5 mg prednisonia tai prednisolonia suun kautta kaksi kertaa vuorokaudessa. Katso lisätiedot abirateronin täydellisestä valmisteyhteenvedosta.
Lynparza yhdessä durvalumabin kanssa
Kun Lynparza-valmistetta käytetään yhdessä durvalumabin kanssa sellaisten potilaiden ylläpitohoitoon, joiden primaariin pitkälle edenneeseen tai uusiutuneeseen kohdun limakalvon syöpään liittyy toimiva MMR-mekanismi (pMMR) ja joiden tauti ei ole edennyt durvalumabin, karboplatiinin ja paklitakselin yhdistelmällä annetun ensilinjan hoidon aikana, durvalumabin annos on 1 500 mg 4 viikon välein (ks. kohta Farmakodynamiikka). Katso lisätiedot durvalumabin täydellisestä valmisteyhteenvedosta.
Hoidon kesto
Vasta diagnosoidun pitkälle edenneen munasarjasyövän ylläpitohoito, kun potilailla on BRCA-mutaatio:
Potilaiden hoitoa voidaan jatkaa, kunnes perussairaus etenee radiologisesti todennetusti, ilmenee haittavaikutuksia, joita ei voida hyväksyä, tai enintään 2 vuoden ajan, jos radiologista näyttöä sairaudesta ei ole, kun hoitoa on jatkettu 2 vuoden ajan. Potilaat, joiden kohdalla on näyttöä sairaudesta 2 vuoden kohdalla ja jotka hoitavan lääkärin näkemyksen mukaan voivat edelleen hyötyä hoidon jatkamisesta, voivat saada hoitoa 2 vuoden jälkeenkin.
Platinaherkän uusiutuneen munasarjasyövän ylläpitohoito:
Hoitoa suositellaan jatkamaan taudin etenemiseen asti tai kunnes ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, jos potilaalla on uusiutunut platinaherkkä korkean pahanlaatuisuusasteen epiteelinen munasarjasyöpä, munanjohdinsyöpä tai primaari peritoneaalinen syöpä.
HRD-positiivisen pitkälle edenneen munasarjasyövän ensilinjan ylläpitohoito yhdessä bevasitsumabin kanssa:
Potilaiden Lynparza-hoitoa voidaan jatkaa, kunnes perussairaus etenee radiologisesti todennetusti, ilmenee haittavaikutuksia, joita ei voida hyväksyä, tai enintään 2 vuoden ajan, jos radiologista näyttöä sairaudesta ei ole, kun hoitoa on jatkettu 2 vuoden ajan. Potilaat, joiden kohdalla on näyttöä sairaudesta 2 vuoden kohdalla ja jotka hoitavan lääkärin näkemyksen mukaan voivat edelleen hyötyä Lynparza-hoidon jatkamisesta, voivat saada hoitoa 2 vuoden jälkeenkin. Bevasitsumabin valmisteyhteenvedon mukaan bevasitsumabihoidon suositeltu enimmäiskesto on 15 kuukautta mukaan lukien jaksot, joiden aikana bevasitsumabi annetaan yhdessä solunsalpaajahoidon kanssa ja ylläpitohoitona (ks. kohta Farmakodynamiikka).
Ituradan BRCA-mutaation omaavan korkean riskin varhaisvaiheen rintasyövän liitännäishoito
Suosituksena on, että potilaita hoidetaan enintään 1 vuoden ajan tai taudin uusiutumiseen asti tai kunnes ilmenee haittavaikutuksia, joita ei voida hyväksyä, sen mukaan, mikä näistä tapahtuu ensin.
gBRCA1/2-mutaation omaavan HER2-negatiivisen metastasoituneen rintasyövän hoito monoterapiana:
Hoitoa suositellaan jatkamaan taudin etenemiseen asti tai kunnes ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä.
Tehoa tai turvallisuutta koskevia tietoja Lynparza-ylläpitohoidon toteuttamisesta uudelleen sairauden ensimmäisen tai myöhemmän uusiutumisen jälkeen munasarjasyöpää sairastavilla potilailla ei ole vahvistettu. Tehoa tai turvallisuutta koskevia tietoja rintasyöpäpotilaiden hoitamisesta uudelleen Lynparza-valmisteella ei ole (ks. kohta Farmakodynamiikka).
Haiman metastasoituneen adenokarsinooman ensilinjan ylläpitohoito, kun potilailla on gBRCA-mutaatio:
Hoitoa suositellaan jatkamaan taudin etenemiseen asti tai kunnes ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä.
BRCA1/2-mutaation omaavan metastasoituneen kastraatioresistentin eturauhassyövän hoito monoterapiana:
Hoitoa suositellaan jatkamaan taudin etenemiseen asti tai kunnes ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä. Lääkkeellistä kastraatiota luteinisoivaa hormonia vapauttavalla hormonilla (LHRH) on jatkettava hoidon aikana, jos potilasta ei ole kastroitu kirurgisesti.
Metastasoituneen kastraatioresistentin eturauhassyövän hoito yhdessä abirateronin ja prednisonin tai prednisolonin kanssa:
Kun Lynparza-valmistetta käytetään yhdessä abirateronin ja prednisonin tai prednisolonin kanssa, hoitoa suositellaan jatkamaan taudin etenemiseen asti tai kunnes ilmaantuu haittavaikutuksia, joita ei voida hyväksyä. Hoitoa gonadotropiinia vapauttavan hormonin (GnRH) analogilla on syytä jatkaa hoidon aikana kaikilla potilailla, tai vaihtoehtoisesti potilaille tulee olla tehty molemminpuolinen orkiektomia. Katso lisätiedot abirateronin valmisteyhteenvedosta.
Eturauhassyöpää sairastavien potilaiden uudelleenhoidosta Lynparza-valmisteella ei ole tehoa tai turvallisuutta koskevia tietoja (ks. kohta Farmakodynamiikka).
Toimivan MMR-mekanismin omaavan (pMMR), pitkälle edenneen tai uusiutuneen kohdun limakalvon syövän ensilinjan ylläpitohoito yhdessä durvalumabin kanssa:
Hoitoa suositellaan jatkamaan taudin etenemiseen asti tai kunnes ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä. Katso lisätiedot durvalumabin valmisteyhteenvedosta.
Unohtunut annos
Jos potilas unohtaa ottaa Lynparza‑annoksen, hänen on otettava seuraava tavanomainen annos normaaliin ajankohtaan.
Annoksen muuttaminen haittavaikutusten vuoksi
Hoito voidaan keskeyttää, jotta haittavaikutukset, kuten pahoinvointi, oksentelu, ripuli ja anemia, saadaan hallintaan, ja annoksen pienentämistä voidaan harkita (ks. kohta Haittavaikutukset).
Annosta suositellaan pienentämään 250 mg:aan (yksi 150 mg:n tabletti ja yksi 100 mg:n tabletti) kaksi kertaa vuorokaudessa (mikä vastaa 500 mg:n kokonaisannosta vuorokaudessa).
Jos annosta on tarpeen edelleen pienentää, suositellaan sen pienentämistä 200 mg:aan (kaksi 100 mg:n tablettia) kaksi kertaa vuorokaudessa (mikä vastaa 400 mg:n kokonaisannosta vuorokaudessa).
Annoksen muuttaminen CYP3A‑estäjien samanaikaisen käytön vuoksi
Samanaikaista voimakkaiden tai keskivahvojen CYP3A‑estäjien käyttöä ei suositella ja vaihtoehtoisia lääkeaineita on harkittava. Jos voimakkaita CYP3A‑estäjiä on käytettävä samanaikaisesti, suositellaan Lynparza-annoksen pienentämistä 100 mg:aan (yksi 100 mg:n tabletti) kaksi kertaa vuorokaudessa (mikä vastaa 200 mg:n kokonaisannosta vuorokaudessa). Jos keskivahvoja CYP3A‑estäjiä on käytettävä samanaikaisesti, suositellaan Lynparza-annoksen pienentämistä 150 mg:aan (yksi 150 mg:n tabletti) kaksi kertaa vuorokaudessa (mikä vastaa 300 mg:n kokonaisannosta vuorokaudessa) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Erityisryhmät
Iäkkäät
Aloitusannosta ei tarvitse muuttaa iäkkäille potilaille.
Munuaisten vajaatoiminta
Suositeltu annos keskivaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma 31–50 ml/min) sairastaville potilaille on 200 mg (kaksi 100 mg:n tablettia) Lynparza‑valmistetta kaksi kertaa vuorokaudessa (mikä vastaa 400 mg:n kokonaisannosta vuorokaudessa) (ks. kohta Farmakokinetiikka).
Lynparza‑valmistetta voidaan antaa annosta muuttamatta potilaille, joilla on lievä munuaisten vajaatoiminta (kreatiniinipuhdistuma 51–80 ml/min).
Lynparza‑valmistetta ei suositella käytettäväksi potilaille, joilla on vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaissairaus (kreatiniinipuhdistuma ≤ 30 ml/min), koska turvallisuutta ja farmakokinetiikkaa ei ole tutkittu näillä potilailla. Lynparza‑valmistetta voidaan käyttää potilaille, joilla on vaikea munuaisten vajaatoiminta, ainoastaan jos lääkkeestä saatava hyöty on suurempi kuin siitä koituva mahdollinen haitta. Lisäksi on potilaiden munuaisten toimintaa ja haittavaikutuksia seurattava tarkasti.
Maksan vajaatoiminta
Lynparza-valmistetta voidaan antaa annosta muuttamatta potilaille, joilla on lievä tai keskivaikea maksan vajaatoiminta (Child-Pughin luokitus A tai B) (ks. kohta Farmakokinetiikka). Lynparza-valmistetta ei suositella käytettäväksi potilaille, joilla on vaikea maksan vajaatoiminta (Child-Pughin luokitus C), koska turvallisuutta ja farmakokinetiikkaa ei ole tutkittu näillä potilailla.
Muut kuin kaukaasialaiset potilaat
Muista kuin kaukaasialaisista potilaista on vain vähän kliinistä tietoa. Annoksen muuttaminen etnisen alkuperän vuoksi ei kuitenkaan ole tarpeen (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Lynparza‑valmisteen turvallisuutta ja tehoa lasten ja nuorten (alle 18-vuotiaiden) hoidossa ei ole varmistettu. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Antotapa
Lynparza otetaan suun kautta.
Lynparza-tabletit niellään kokonaisina eikä niitä saa pureskella, murskata, liuottaa tai jakaa. Lynparza-tabletit voidaan ottaa ateria-ajoista riippumatta.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Imetys hoidon aikana ja 1 kuukauden ajan viimeisestä annoksesta (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Hematologinen toksisuus
Lynparza-valmistetta saaneilla potilailla on ilmoitettu hematologista toksisuutta mukaan lukien kliinisiä diagnooseja ja/tai laboratoriolöydöksiä; yleensä lievää tai keskivaikeaa (CTCAE‑asteet 1 tai 2) anemiaa, neutropeniaa, trombosytopeniaa ja lymfosytopeniaa. Puhdasta punasoluaplasiaa (PRCA) (ks. kohta Haittavaikutukset) ja/tai autoimmuunihemolyyttistä anemiaa on ilmoitettu, kun Lynparza-valmistetta on käytetty yhdessä durvalumabin kanssa.
Potilaiden Lynparza‑hoitoa ei saa aloittaa, ennen kuin potilaat ovat toipuneet aiemman syöpähoidon aiheuttamasta hematologisesta toksisuudesta (hemoglobiini‑, verihiutale‑ ja neutrofiiliarvojen on oltava ≤ CTCAE‑astetta 1). Täydellisen verenkuvan tutkimista suositellaan hoidon aloitusvaiheessa ja jatkossa kerran kuukaudessa hoidon ensimmäisten 12 kuukauden aikana sekä tämän jälkeen määräajoin kaikkien parametrien kliinisesti merkittävien muutosten seuraamiseksi (ks. kohta Haittavaikutukset).
Jos potilaalle kehittyy vaikea hematologinen toksisuus tai riippuvuutta verensiirroista, on Lynparza‑hoito keskeytettävä ja aloitettava asianmukaiset hematologiset tutkimukset. Jos veriarvot ovat edelleen kliinisesti poikkeavia 4 viikon kuluttua Lynparza‑hoidon keskeyttämisen jälkeen, suositellaan luuydintutkimusta ja/tai veren sytogeneettistä analyysiä. Jos potilaalla vahvistetaan olevan punasoluaplasia tai autoimmuunihemolyyttinen anemia, Lynparza- ja durvalumabihoito on lopetettava.
Myelodysplastinen oireyhtymä / Akuutti myelooinen leukemia
Myelodysplastista oireyhtymää (MDS) / akuuttia myelooista leukemiaa (AML) on ilmennyt Lynparza-valmistetta saaneilla potilailla (ks. kohta Haittavaikutukset). Suurin osa näistä tapauksista johti kuolemaan. MDS:n/AML:n kehittymisen riski oli suurempi uusiutunutta platinaherkkää BRCA-mutatoitunutta munasarjasyöpää sairastavilla potilailla, jotka olivat saaneet vähintään kahta aiemman linjan platinapohjaista solunsalpaajahoitoa. Olaparibihoidon kesto vaihteli alle 6 kuukaudesta yli 4 vuoteen potilailla, joille kehittyi MDS/AML.
Epäiltäessä MDS:ää/AML:ää, on potilas ohjattava hematologille jatkotutkimuksiin, joissa tehdään luuydintutkimus ja verinäytteestä sytogeneettinen analyysi. Jos pitkittyneen hematologisen toksisuuden vuoksi tehtävien tutkimusten jälkeen MDS/AML-diagnoosi varmistuu, Lynparza-hoito on lopetettava ja potilasta on hoidettava asianmukaisesti.
Laskimotromboemboliset tapahtumat
Lynparza-hoitoa saaneilla potilailla on ilmennyt laskimotromboembolisia tapahtumia, lähinnä keuhkoemboliaa, mutta tapahtumilla ei ole ollut johdonmukaista kliinistä kaavaa. Ilmaantuvuuden havaittiin olevan suurempi metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavilla potilailla, jotka saivat myös androgeenideprivaatiohoitoa, verrattuna muihin hyväksyttyihin käyttöaiheisiin (ks. kohta Haittavaikutukset). Potilaita on seurattava laskimotromboosin ja keuhkoembolian kliinisten merkkien ja oireiden varalta ja hoidettava lääketieteellisesti asianmukaisesti. Potilailla, joilla on anamneesissa laskimotromboembolisia tapahtumia, saattaa olla suurentunut uusiutumisen riski, ja heitä on seurattava asianmukaisesti.
Pneumoniitti
Potilailla, jotka ovat saaneet kliinisissä tutkimuksissa Lynparza-valmistetta, on ilmoitettu pneumoniittia, kuolemaan johtaneet tapaukset mukaan lukien (ks. kohta Haittavaikutukset). Lynparza-hoito on keskeytettävä ja tutkimukset on aloitettava välittömästi, jos potilaalle ilmaantuu uusia tai pahenevia hengitysoireita, kuten hengenahdistusta, yskää ja kuumetta, tai todetaan poikkeava löydös rintakehän radiologisessa tutkimuksessa. Lynparza-hoito on keskeytettävä ja potilasta on hoidettava asianmukaisesti, jos pneumoniittidiagnoosi varmistuu.
Maksatoksisuus
Olaparibilla hoidetuilla potilailla on raportoitu maksatoksisuutta (ks. kohta Haittavaikutukset). Jos potilaalle kehittyy maksatoksisuuteen viittaavia kliinisiä oireita tai löydöksiä, potilaan kliininen arviointi ja maksan toimintaa mittaavat kokeet on tehtävä viipymättä. Hoito on keskeytettävä, jos epäillään lääkkeen aiheuttamaa maksavauriota. Jos ilmenee lääkkeen aiheuttama vaikea maksavaurio, on harkittava hoidon lopetusta, mikäli se on kliinisesti tarkoituksenmukaista.
Alkio‑ ja sikiötoksisuus
Vaikutusmekanisminsa (PARP:n [poly‑ADP‑riboosipolymeraasin] esto) perusteella Lynparza voi vahingoittaa sikiötä, jos sitä annetaan raskaana olevalle naiselle. Ei‑kliiniset tutkimukset rotilla ovat osoittaneet, että olaparibilla on haitallisia vaikutuksia alkion ja sikiön eloonjäämiseen ja se aiheuttaa sikiölle vakavia epämuodostumia ihmisen suositusannosta (300 mg kaksi kertaa vuorokaudessa) pienemmillä annoksilla.
Raskaus/ehkäisy
Lynparza‑valmistetta ei pidä käyttää raskauden aikana. Naisten, jotka voivat tulla raskaaksi, on käytettävä kahta luotettavaa ehkäisymenetelmää ennen Lynparza-hoidon aloittamista, hoidon aikana ja kuuden kuukauden ajan viimeisen Lynparza-annoksen ottamisen jälkeen. On suositeltavaa käyttää kahta erittäin tehokasta ja toisiaan täydentävää ehkäisymenetelmää. Miespotilaiden ja heidän naispuolisten kumppaniensa, jotka voivat tulla raskaaksi, on käytettävä luotettavaa ehkäisymenetelmää hoidon aikana ja kolmen kuukauden ajan viimeisen Lynparza-annoksen ottamisen jälkeen (ks. kohta Raskaus ja imetys).
Yhteisvaikutukset
Lynparza‑valmisteen antamista samanaikaisesti voimakkaiden tai keskivahvojen CYP3A‑estäjien kanssa ei suositella (ks. kohta Yhteisvaikutukset). Lynparza‑annosta on pienennettävä, jos voimakkaita tai keskivahvoja CYP3A‑estäjiä on annettava samanaikaisesti (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Lynparza‑valmisteen antamista samanaikaisesti voimakkaiden tai keskivahvojen CYP3A‑induktorien kanssa ei suositella. Tapauksissa, joissa Lynparza‑valmistetta jo käyttävä potilas tarvitsee hoitoa voimakkaalla tai keskivahvalla CYP3A‑induktorilla, lääkärin on huomioitava, että Lynparza‑valmisteen teho saattaa heikentyä huomattavasti (ks. kohta Yhteisvaikutukset).
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol (23 mg) natriumia per 100 mg:n tai 150 mg:n tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Farmakodynaamiset yhteisvaikutukset
Myelosuppressiivisen toksisuuden tehostumista ja pitkittymistä on osoitettu kliinisissä tutkimuksissa olaparibilla yhdistelmänä muiden syöpälääkevalmisteiden kanssa, mukaan lukien DNA:ta vaurioittavat aineet. Monoterapiana käytetyn Lynparza‑valmisteen suositeltu annos ei sovi käytettäväksi yhdistelmähoitona myelosuppressiivisten syöpälääkevalmisteiden kanssa.
Olaparibin yhdistelmää rokotteiden tai immuunivastetta ehkäisevien aineiden kanssa ei ole tutkittu. Siksi on noudatettava varovaisuutta, jos näitä lääkevalmisteita annetaan yhdessä Lynparza‑valmisteen kanssa, ja potilaita on seurattava tarkoin.
Farmakokineettiset yhteisvaikutukset
Muiden lääkevalmisteiden vaikutus olaparibiin
CYP3A4 ja CYP3A5 ovat isoentsyymejä, jotka pääasiassa vastaavat olaparibin metabolisesta poistumasta.
Kliininen tutkimus, jossa arvioitiin tunnetun CYP3A‑estäjän itrakonatsolin vaikutusta, osoitti, että itrakonatsolin samanaikainen käyttö olaparibin kanssa suurensi olaparibin keskimääräistä Cmax-arvoa 42 %:lla (90 % luottamusväli: 33−52 %) ja keskimääräistä AUC-arvoa 170 %:lla (90 % luottamusväli: 114−197 %). Siksi tämän isoentsyymin tunnettujen voimakkaiden estäjien (esim. itrakonatsoli, telitromysiini, klaritromysiini, ritonaviirilla tai kobisistaatilla tehostetut proteaasin estäjät, bosepreviiri ja telapreviiri) tai keskivahvojen estäjien (esim. erytromysiini, diltiatseemi, flukonatsoli ja verapamiili) käyttöä samanaikaisesti Lynparza-valmisteen kanssa ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos voimakkaita tai keskivahvoja CYP3A-estäjiä täytyy käyttää samanaikaisesti Lynparza-valmisteen kanssa, Lynparza-annosta on pienennettävä. Lynparza-annos suositellaan pienentämään 100 mg:aan kaksi kertaa vuorokaudessa (mikä vastaa 200 mg:n kokonaisannosta vuorokaudessa) voimakkaan CYP3A-estäjän käytön yhteydessä ja 150 mg:aan kaksi kertaa vuorokaudessa (mikä vastaa 300 mg:n kokonaisannosta vuorokaudessa) keskivahvan CYP3A-estäjän käytön yhteydessä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Lynparza-hoidon aikana on myös suositeltavaa välttää juomasta greippimehua, koska se on CYP3A-estäjä.
Kliininen tutkimus, jossa arvioitiin tunnetun CYP3A‑induktorin rifampisiinin vaikutusta, osoitti, että rifampisiinin samanaikainen käyttö olaparibin kanssa pienensi olaparibin keskimääräistä Cmax-arvoa 71 %:lla (90 % luottamusväli: 76−67 %) ja keskimääräistä AUC-arvoa 87 %:lla (90 % luottamusväli: 89−84 %). Tämän isoentsyymin tunnettujen voimakkaiden induktorien (esim. fenytoiini, rifampisiini, rifapentiini, karbamatsepiini, nevirapiini, fenobarbitaali ja mäkikuisma) käyttöä samanaikaisesti Lynparza-valmisteen kanssa ei suositella, sillä on mahdollista, että Lynparza-valmisteen teho heikkenee huomattavasti. Lynparza-valmisteen antamista keskivahvojen ja vahvojen induktorien (esim. efavirentsi, rifabutiini) kanssa ei myöskään suositella, sillä niiden vaikutuksen suuruutta olaparibialtistukseen ei tiedetä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Olaparibin vaikutus muihin lääkevalmisteisiin
Olaparibi estää CYP3A4:ää in vitro ja sen oletetaan olevan heikko CYP3A:n estäjä in vivo. Siksi on noudatettava varovaisuutta, kun sensitiivisiä CYP3A‑substraatteja tai substraatteja, joilla on kapea terapeuttinen marginaali (esim. simvastatiini, sisapridi, siklosporiini, torajyväalkaloidit, fentanyyli, pimotsidi, sirolimuusi, takrolimuusi ja ketiapiini) käytetään yhdessä olaparibin kanssa. Asiaankuuluvaa kliinistä seurantaa suositellaan potilaille, jotka saavat samanaikaisesti olaparibia ja CYP3A-substraatteja, joilla on kapea terapeuttinen marginaali.
CYP1A2:n, ‑2B6:n ja -3A4:n induktio on osoitettu in vitro; CYP2B6:n indusoituminen on todennäköisimmin kliinisesti merkittävää. Olaparibin kykyä indusoida CYP2C9:ää, CYP2C19:ää ja P‑gp:tä ei myöskään voida poissulkea. Näin ollen olaparibi saattaa samanaikaisesti annettuna vähentää altistusta näiden aineenvaihduntaentsyymien ja kuljettajaproteiinin substraateille. Joidenkin hormonaalisten ehkäisyvalmisteiden teho saattaa heikentyä, jos niitä käytetään samanaikaisesti olaparibin kanssa (ks. myös kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys).
Olaparibi estää effluksiproteiini P-gp:tä in vitro (IC50 = 76 µM), siksi ei voida poissulkea mahdollisuutta, että olaparibilla saattaa olla kliinisesti merkittäviä yhteisvaikutuksia P-gp:n substraattien (esim. simvastatiini, pravastatiini, dabigatraani, digoksiini ja kolkisiini) kanssa. Asiaankuuluvaa kliinistä seurantaa suositellaan potilaille, jotka saavat samanaikaisesti tämän tyyppisiä lääkevalmisteita.
In vitro olaparibin on osoitettu olevan BCRP:n, OATP1B1:n, OCT1:n, OCT2:n, OAT3:n, MATE1:n ja MATE2K:n estäjä. Ei voida poissulkea mahdollisuutta, että olaparibi saattaa lisätä altistusta BCRP:n substraateille (esim. metotreksaatti, rosuvastatiini), OATP1B1:n substraateille (esim. bosentaani, glibenklamidi, repaglinidi, statiinit ja valsartaani), OCT1:n substraateille (esim. metformiini), OCT2:n substraateille (esim. seerumin kreatiniini), OAT3:n substraateille (esim. furosemidi ja metotreksaatti), MATE1:n substraateille (esim. metformiini) ja MATE2K:n substraateille (esim. metformiini). Varovaisuutta on noudatettava erityisesti, jos olaparibia annetaan yhdistelmänä minkä tahansa statiinin kanssa.
Samanaikainen käyttö anastrotsolin, letrotsolin ja tamoksifeenin kanssa
Kliinisessä tutkimuksessa arvioitiin olaparibin ja anastrotsolin, letrotsolin tai tamoksifeenin yhdistelmää. Kliinisesti merkittäviä yhteisvaikutuksia ei havaittu.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy naisilla
Naiset eivät saa olla raskaana aloitettaessa Lynparza‑hoitoa eivätkä he saa tulla raskaaksi Lynparza‑hoidon aikana. Kaikille naisille, jotka voivat tulla raskaaksi, on tehtävä raskaustesti ennen hoidon aloittamista, ja raskaustestien tekemistä säännöllisesti on harkittava koko hoidon ajan.
Naisten, jotka voivat tulla raskaaksi, on käytettävä kahta luotettavaa ehkäisymenetelmää ennen Lynparza-hoidon aloittamista, hoidon aikana ja kuuden kuukauden ajan viimeisen Lynparza‑annoksen ottamisen jälkeen, ellei potilas ole valinnut ehkäisymenetelmäksi yhdynnästä pidättäytymistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). On suositeltavaa käyttää kahta erittäin tehokasta ja toisiaan täydentävää ehkäisymenetelmää.
Joidenkin hormonaalisten ehkäisyvalmisteiden teho voi heikentyä, kun niitä käytetään samanaikaisesti olaparibin kanssa, sillä ei voida poissulkea mahdollisuutta, että olaparibi saattaa pienentää altistusta CYP2C9‑substraateille entsyymi-induktion kautta. Tästä syystä on hoidon aikana harkittava myös hormoneista riippumattoman ehkäisymenetelmän käyttöä (ks. kohta Yhteisvaikutukset). Naisilla, joilla on hormoniriippuvainen syöpä, on harkittava kahden hormoneista riippumattoman ehkäisymenetelmän käyttöä.
Raskauden ehkäisy miehillä
Ei tiedetä, erittyvätkö olaparibi tai sen metaboliitit siemennesteeseen. Miespotilaiden on käytettävä kondomia hoidon aikana ja kolmen kuukauden ajan viimeisen Lynparza-annoksen saamisen jälkeen, jos he ovat yhdynnässä sellaisen naisen kanssa, joka on raskaana tai voi tulla raskaaksi. Miespotilaiden naispuolisten kumppanien on myös käytettävä erittäin tehokasta ehkäisyä, jos he voivat tulla raskaaksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Miespotilaiden ei pidä luovuttaa spermaa hoidon aikana eikä kolmeen kuukauteen viimeisen Lynparza-annoksen saamisen jälkeen.
Raskaus
Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta, kuten vakavia teratogeenisiä vaikutuksia sekä vaikutuksia alkion ja sikiön eloonjäämiseen rotilla maternaalisilla, systeemisillä altistuksilla, jotka ovat alhaisempia kuin altistukset ihmisillä terapeuttisilla annoksilla (ks. kohta Prekliiniset tiedot turvallisuudesta). Olaparibin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Olaparibin vaikutustavan perusteella Lynparza-valmistetta ei pidä käyttää raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä luotettavaa ehkäisyä hoidon aikana eikä 6 kuukauteen viimeisen Lynparza-annoksen jälkeen (katso edellisestä kohdasta ”Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy naisilla” lisätietoja ehkäisystä ja raskaustesteistä).
Imetys
Eläimillä tehtyjä tutkimuksia olaparibin erittymisestä rintamaitoon ei ole. Ei tiedetä, erittyvätkö olaparibi tai sen metaboliitit ihmisillä äidinmaitoon. Lynparza-valmisteen farmakologisten ominaisuuksien vuoksi Lynparza-valmisteen käyttö imetyksen aikana on vasta-aiheista eikä imettää saa kuukauteen viimeisen Lynparza-annoksen ottamisen jälkeen (ks. kohta Vasta-aiheet).
Hedelmällisyys
Kliinistä tietoa vaikutuksesta hedelmällisyyteen ei ole. Eläimillä tehdyissä tutkimuksissa ei todettu vaikutuksia hedelmöittymiseen, mutta alkion/sikiön eloonjäämiseen liittyviä haittavaikutuksia todettiin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Lynparza-valmisteella on kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Lynparza-valmistetta käyttävillä potilailla saattaa ilmetä väsymystä, asteniaa tai heitehuimausta. Jos potilaalla on näitä oireita, hänen on noudatettava varovaisuutta ajaessaan tai koneita käyttäessään.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Lynparza-hoidon yhteydessä on esiintynyt haittavaikutuksia, jotka ovat yleensä olleet vaikeusasteeltaan lieviä tai keskivaikeita (CTCAE‑astetta 1 tai 2) ja jotka eivät yleensä ole vaatineet hoidon keskeyttämistä. Kliinisissä tutkimuksissa Lynparza-valmistetta monoterapiana saaneiden potilaiden yleisimmin (≥ 10 %) todettuja haittavaikutuksia olivat pahoinvointi, väsymys/voimattomuus, anemia, oksentelu, ripuli, heikentynyt ruokahalu, päänsärky, neutropenia, makuaistin muutokset, yskä, leukopenia, huimaus, hengenahdistus, ja dyspepsia.
Vähintään vaikeusasteen 3 haittavaikutuksia, joita ilmeni yli 2 %:lla potilaista, olivat anemia (14 %), neutropenia (5 %), väsymys/voimattomuus (4 %), leukopenia (2 %) ja trombosytopenia (2 %).
Haittavaikutuksia, jotka useimmiten johtivat monoterapiana annetun hoidon keskeyttämiseen ja/tai annoksen pienentämiseen, olivat anemia (16 %), pahoinvointi (7 %), väsymys/voimattomuus (6 %), neutropenia (6 %) ja oksentelu (6 %). Haittavaikutuksia, jotka useimmiten johtivat hoidon pysyvään lopettamiseen, olivat anemia (1,7 %), pahoinvointi (0,9 %), väsymys/voimattomuus (0,8 %), trombosytopenia (0,7 %), neutropenia (0,6 %) ja oksentelu (0,5 %).
Kun Lynparza-valmistetta käytetään yhdessä bevasitsumabin kanssa munasarjasyövän hoitoon, yhdessä abirateronin ja prednisonin tai prednisolonin kanssa eturauhassyövän hoitoon tai yhdessä durvalumabin kanssa kohdun limakalvon syövän hoitoon, kun potilaat ovat ensin saaneet durvalumabin ja platinapohjaisen solunsalpaajahoidon yhdistelmää, turvallisuusprofiili on yleisesti ottaen yhdenmukainen yksittäin annettujen hoitojen turvallisuusprofiilien kanssa.
Kun olaparibia käytettiin yhdessä bevasitsumabin kanssa, haittavaikutukset johtivat olaparibihoidon keskeyttämiseen ja/tai annoksen pienentämiseen 57 %:lla potilaista ja hoidon pysyvään lopettamiseen 21 %:lla olaparibia saaneista potilaista ja 6 %:lla lumelääkettä saaneista potilaista. Haittavaikutukset, jotka useimmiten johtivat olaparibihoidon keskeyttämiseen ja/tai annoksen pienentämiseen, olivat anemia (21,7 %), pahoinvointi (9,5 %), väsymys/voimattomuus (5,4 %), oksentelu (3,7 %), neutropenia (3,6 %), trombosytopenia (3,0 %) ja ripuli (2,6 %). Haittavaikutukset, jotka useimmiten johtivat hoidon pysyvään lopettamiseen, olivat anemia (3,7 %), pahoinvointi (3,6 %) ja väsymys/voimattomuus (1,5 %).
Kun olaparibia käytettiin yhdessä abirateronin kanssa, haittavaikutukset johtivat olaparibihoidon keskeyttämiseen ja/tai annoksen pienentämiseen 50,7 %:lla potilaista ja hoidon pysyvään lopettamiseen 19,0 %:lla olaparibia saaneista potilaista ja 8,8 %:lla lumelääkettä saaneista potilaista. Haittavaikutukset, jotka useimmin johtivat olaparibihoidon keskeyttämiseen ja/tai annoksen pienentämiseen, olivat anemia (17,1 %), väsymys/voimattomuus (5,5 %), pahoinvointi (4,1 %), neutropenia (3,4 %), oksentelu (2,3 %), ripuli (2,1 %) ja laskimotromboemboliset tapahtumat (2,1 %). Haittavaikutukset, jotka useimmin johtivat hoidon pysyvään lopettamiseen, olivat anemia (4,5 %) ja väsymys/voimattomuus (1,3 %).
Kun olaparibia käytettiin yhdessä durvalumabin kanssa sen jälkeen, kun potilaat olivat saaneet durvalumabia yhdessä platinapohjaisen solunsalpaajahoidon kanssa, haittavaikutukset johtivat olaparibihoidon keskeyttämiseen ja/tai annoksen pienentämiseen 59,9 %:lla potilaista ja olaparibihoidon pysyvään lopettamiseen 10,9 %:lla potilaista. Haittavaikutukset, jotka useimmin johtivat olaparibihoidon keskeyttämiseen ja/tai annoksen pienentämiseen, olivat anemia (20,8 %), pahoinvointi (8,3 %), neutropenia (7,3 %), väsymys/voimattomuus (5,7 %), trombosytopenia (4,2 %), oksentelu (4,2 %), veren kreatiniiniarvon suureneminen (3,1 %), leukopenia (3,1 %), ruokahalun heikkeneminen (2,6 %) ja ripuli (2,1 %). Haittavaikutukset, joka useimmin johtivat olaparibihoidon pysyvään lopettamiseen, olivat anemia (3,6 %) ja neutropenia (1 %).
Haittavaikutustaulukko
Turvallisuusprofiili perustuu yhdistettyihin tietoihin, jotka saatiin 4 499 potilaasta, joilla oli kiinteitä kasvaimia ja jotka saivat Lynparza-valmistetta monoterapiana kliinisissä tutkimuksissa suositellulla annoksella.
Seuraavat haittavaikutukset todettiin kliinisissä tutkimuksissa potilailla, jotka saivat Lynparza‑valmistetta monoterapiana, ja potilaiden altistus tunnettiin. Haittavaikutukset on lueteltu taulukossa 1 MedDRA:n elinjärjestelmäluokituksen mukaan käyttäen MedDRA:n suositeltuja termejä. Suositellut termit on esitetty kussakin elinjärjestelmässä ensin yleisyyden mukaan alenevassa ja sitten vakavuuden mukaan alenevassa järjestyksessä. Haittavaikutusten esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1 Yhteenveto haittavaikutuksista
| Haittavaikutukset | ||
| MedDRA‑elinjärjestelmä | Kaikkien CTCAE‑vaikeusasteiden esiintymistiheys | Esiintymistiheys CTCAE‑astetta 3 tai suurempi |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Melko harvinainen Myelodysplastinen oireyhtymä / akuutti myelooinen leukemiaa | Melko harvinainen Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia |
| Veri ja imukudosb | Hyvin yleinen trombosytopeniaa | Hyvin yleinen Anemiaa Yleinen Neutropeniaa, trombosytopeniaa, leukopeniaa, lymfosytopeniaa |
| Immuunijärjestelmä | Melko harvinainen Harvinainen | Harvinainen Yliherkkyysa |
| Maksa ja sappi | Yleinen Tuntematon | |
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen Heikentynyt ruokahalu | Melko harvinainen Heikentynyt ruokahalu |
| Hermosto | Hyvin yleinen Heitehuimaus, päänsärky, makuaistin muutokseta | Melko harvinainen Heitehuimaus, päänsärky |
| Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen Yskäa, hengenahdistusa Melko harvinainen Pneumoniittia | Yleinen Melko harvinainen |
| Ruoansulatuselimistö | Hyvin yleinen Oksentelu, ripuli, pahoinvointi, dyspepsia Yleinen Suutulehdusa, ylävatsakipu | Yleinen Oksentelu, pahoinvointi Melko harvinainen Suutulehdusa, ripuli Harvinainen Dyspepsia, ylävatsakipu |
| Iho ja ihonalainen kudos | Yleinen Ihottumaa Melko harvinainen Dermatiittia Harvinainen Kyhmyruusu | Melko harvinainen Harvinainen |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen Väsymys (astenia mukaan lukien) | Yleinen Väsymys (astenia mukaan lukien) |
| Tutkimuksetb | Yleinen Suurentunut veren kreatiniiniarvo Melko harvinainen Suurentunut solujen keskimääräinen tilavuus | Harvinainen Suurentunut veren kreatiniiniarvo |
| Verisuonisto | Yleinen Laskimotromboemboliaa | Yleinen Laskimotromboemboliaa |
a MDS/AML-termiin sisältyvät suositellut termit akuutti myelooinen leukemia, myelodysplastinen oireyhtymä ja myelooinen leukemia.
Anemia-termiin sisältyvät suositellut termit anemia, makrosyyttinen anemia, erytrosytopenia, pienentynyt hematokriitti, pienentynyt hemoglobiiniarvo, normosyyttinen anemia ja punasoluniukkuus.
Neutropenia-termiin sisältyvät suositellut termit kuumeinen neutropenia, neutropenia, neutropeeninen infektio, neutropeeninen sepsis ja neutrofiilien niukkuus.
Trombosytopenia-termiin sisältyvät suositellut termit verihiutaleniukkuus ja trombosytopenia.
Leukopenia-termiin sisältyvät suositellut termit leukopenia ja veren valkosolujen niukkuus.
Lymfosytopenia-termiin sisältyvät suositellut termit lymfosyyttimäärän pieneneminen ja lymfosytopenia.
Yliherkkyys-termiin sisältyvät suositellut termit lääkeaineyliherkkyys ja yliherkkyys.
Transaminaasiarvojen suureneminen -termiin sisältyvät suositellut termit alaniiniaminotransferaasiarvon suureneminen, aspartaattiaminotransferaasiarvon suureneminen, maksaentsyymiarvojen suureneminen ja hypertransaminasemia.
Makuaistin muutokset -termiin sisältyvät suositellut termit makuaistin muutos ja makuaistin häiriö.
Yskä-termiin sisältyvät suositellut termit yskä ja limaa tuottava yskä.
Hengenahdistus-termiin sisältyvät suositellut termit hengenahdistus ja rasitushengenahdistus.
Pneumoniitti-termiin sisältyvät suositellut termit pneumoniitti, interstitiaalinen keuhkosairaus, akuutti interstitiaalinen pneumoniitti, eosinofiilinen pneumonia, akuutti eosinofiilinen pneumonia ja allerginen alveoliitti.
Suutulehdus-termiin sisältyvät suositellut termit aftahaavauma, suun haavauma ja suutulehdus.
Ihottuma-termiin sisältyvät suositellut termit eryteema, kesivä ihottuma, ihottuma, punoittava ihottuma, makulaarinen ihottuma, makulopapulaarinen ihottuma, papulaarinen ihottuma ja kutiava ihottuma.
Dermatiitti-termiin sisältyvät suositellut termit dermatiitti ja allerginen dermatiitti.
Laskimotromboembolia sisältää seuraavat suositellut termit: embolia, keuhkoveritulppa, tromboosi, syvä laskimotukos, onttolaskimon tukos ja laskimotromboosi.
b Kirjatut laboratoriotutkimustiedot on esitetty jäljempänä kohdissa Hematologinen toksisuus ja Muut laboratoriolöydökset.
* Havaittu myyntiluvan saamisen jälkeen.
Potilailla, jotka saivat Lynparza-valmistetta yhdessä durvalumabin kanssa saatuaan aiemmin durvalumabia yhdessä platinapohjaisen solunsalpaajahoidon kanssa, useimpia haittavaikutuksia (kakkien vaikeusasteiden haittavaikutukset ja vähintään CTCAE-asteen 3 haittavaikutukset) esiintyi yhtä yleisesti tai harvemmin kuin edellä olevassa Lynparza-monoterapian haittavaikutuksia koskevassa taulukossa on esitetty. Lynparza-valmistetta yhdessä durvalumabin kanssa saaneilla potilailla ilmoitettiin useammin trombosytopeniaa ja ihottumaa (hyvin yleisiä) ja yliherkkyyttä (yleinen). Lisäksi todettiin seuraava haittavaikutus:
Taulukko 2 Muu haittavaikutus, joka ilmoitettiin Lynparza-valmisteen ja durvalumabin yhdistelmää koskeneessa kliinisessä tutkimuksessa
| MedDRA-elinjärjestelmä | MedDRA-termi | CIOMS/ kokonaisesiintymistiheys (kaikki CTCAE-asteet) | Esiintymistiheys CTCAE-astetta 3 tai suurempi |
|---|---|---|---|
| Veri ja imukudos | Punasoluaplasia
| Yleinen | Yleinen |
Valikoitujen haittavaikutusten kuvaus
Hematologinen toksisuus
Anemia ja muut hematologiset toksisuudet olivat yleensä vaikeusasteeltaan lieviä (CTCAE-astetta 1 tai 2), mutta CTCAE-asteen 3 ja sitäkin vaikeampia tapahtumia ilmoitettiin. Anemia oli yleisin kliinisissä tutkimuksissa ilmoitettu vähintään CTCAE-asteen 3 haittavaikutus. Mediaaniaika anemian ilmaantumiseen ensimmäistä kertaa oli noin 4 viikkoa (vähintään CTCAE-asteen 3 tapahtumien kohdalla noin 7 viikkoa). Anemia hoidettiin keskeyttämällä hoito, pienentämällä annosta (ks. kohta Annostus ja antotapa) ja tarvittaessa käyttämällä verensiirtoja. Tablettilääkemuodolla tehdyissä kliinisissä tutkimuksissa anemiaan liittyvien haittavaikutusten ilmaantuvuus oli 35,2 % (vähintään CTCAE-asteen 3 haittavaikutusten ilmaantuvuus 14,8 %), ja hoito keskeytettiin 16,4 %:lla tutkittavista, annosta pienennettiin 11,1 %:lla tutkittavista ja hoito lopetettiin 2,1 %:lla tutkittavista anemian vuoksi. 15,6 % olaparibia saaneista potilaista tarvitsi ainakin yhden verensiirron. Olaparibin ja hemoglobiiniarvon laskun välillä on osoitettu olevan altistus-vasteyhteys. Lynparza-valmisteella tehdyissä kliinisissä tutkimuksissa lähtötilanteeseen verrattuna vähintään kahden CTCAE-asteen muutoksen (pienenemisen) ilmaantuvuus oli hemoglobiinin osalta 21 %, absoluuttisten neutrofiiliarvojen osalta 17 %, verihiutaleiden osalta 5 %, lymfosyyttien osalta 26 % ja leukosyyttien osalta 19 % (kaikki prosenttiluvut ovat likiarvoja).
Punasolujen keskimääräisen tilavuuden suurenemisen ilmaantuvuus lähtötilanteessa todetusta pienestä tai normaalista arvosta viitearvon ylärajaa (ULN) suuremmaksi oli noin 51 %. Arvot näyttivät palautuvan normaaleiksi hoidon keskeyttämisen jälkeen eikä kliinisiä seurauksia näyttänyt olevan.
Täydellisen verenkuvan tutkimista suositellaan hoidon aloitusvaiheessa ja jatkossa kerran kuukaudessa hoidon ensimmäisten 12 kuukauden aikana sekä tämän jälkeen määräajoin kaikkien parametrien kliinisesti merkittävien muutosten seuraamiseksi. Muutokset saattavat edellyttää hoidon keskeyttämistä tai annoksen pienentämistä ja/tai lisähoitoa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia
Myelodysplastinen oireyhtymä ja akuutti myelooinen leukemia (MDS/AML) ovat vakavia haittavaikutuksia, joita ilmeni kliinisissä tutkimuksissa melko harvoin (0,9 %) kaikissa käyttöaiheissa, kun hoito annettiin monoterapiana terapeuttisilla annoksilla. Ilmaantuvuus oli 0,5 % mukaan lukien tapahtumat, joita raportoitiin pitkän aikavälin turvallisuusseurannan aikana (määrä on laskettu kokonaisturvallisuuspopulaatiosta, jossa oli 18576 potilasta, jotka saivat kliinisissä tutkimuksissa vähintään yhden annoksen olaparibia suun kautta). Kaikilla potilailla oli mahdollisia MDS:n/AML:n kehittymistä edistäviä tekijöitä, koska he olivat saaneet aiemmin platinapohjaista solunsalpaajahoitoa. Monet potilaista olivat saaneet myös muita DNA:ta vaurioittavia lääkeaineita ja sädehoitoa. Suurin osa tapauksista raportoitiin potilailla, joilla oli ituradan rintasyövän alttiusgeenin 1 tai 2 (gBRCA1/2-geenin) mutaatio. MDS/AML-tapausten ilmaantuvuus oli gBRCA1m-potilailla (1,6 %) samankaltainen kuin gBRCA2m-potilailla (1,2 %). Joillakin potilailla oli aiemmin ollut syöpä tai todettu luuytimen dysplasia.
Viisi vuotta kestäneessä seurannassa MDS:n/AML:n ilmaantuvuus oli 8 % olaparibia saaneilla potilailla ja 4 % lumelääkettä saaneilla potilailla (SOLO2-tutkimus, jossa 45 % potilaista sai olaparibihoitoa vähintään 2 vuoden ajan). Näillä potilailla oli uusiutunut platinaherkkä BRCA-mutatoitunut munasarjasyöpä, he olivat saaneet vähintään kahta aiemman linjan platinapohjaista solunsalpaajahoitoa ja jatkaneet tutkimushoitoa taudin etenemiseen saakka. Olaparibihaarassa yhdeksän 16:sta MDS/AML-tapauksesta ilmeni olaparibihoidon lopettamisen jälkeen elossaoloseurannan aikana. MDS/AML-tapauksia havaittiin olaparibihaarassa pidentyneen kokonaiselinajan yhteydessä ja myöhään alkaneina MDS/AML-tapauksina. MDS:n/AML:n riski ensilinjan hoidossa pysyy pienenä, kun olaparibia annetaan ylläpitohoitona yhden platinapohjaisen solunsalpaajahoitolinjan jälkeen 2 vuoden ajan (1,5 % SOLO1-tutkimuksessa seurannan kestettyä 7 vuotta ja 1,1 % PAOLA-1-tutkimuksessa seurannan kestettyä 5 vuotta). Riskin pienentäminen ja hallinta, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Punasoluaplasia
Punasoluaplasiaa on ilmoitettu, kun Lynparza-valmistetta on käytetty yhdessä durvalumabin kanssa. Kliinisessä tutkimuksessa, jossa kohdun limakalvon syöpää sairastaville potilaille annettiin Lynparza-valmistetta yhdessä durvalumabin kanssa, punasoluaplasian ilmaantuvuus oli 1,6 %. Kaikki tapahtumat olivat CTCAE-astetta 3 tai 4. Tapahtumat olivat hoidettavissa, kun sekä Lynparza-hoito että durvalumabihoito lopetettiin. Suurin osa tapahtumista hoidettiin verensiirrolla ja immunosuppressiolla, ja nämä potilaat toipuivat. Yksikään tapahtumista ei johtanut kuolemaan. Riskin pienentäminen ja hallinta, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Laskimotromboemboliset tapahtumat
Miehillä, jotka saivat olaparibia ja abirateronia metastasoituneen kastraatioresistentin eturauhassyövän ensilinjan hoitona (PROpel-tutkimus), laskimotromboembolisten tapahtumien ilmaantuvuus oli 8 % olaparibi + abirateroni -haarassa ja 3,3 % lumelääke + abirateroni -haarassa. Mediaaniaika tapahtuman ilmaantumiseen tässä tutkimuksessa oli 170 päivää (vaihteluväli: 12–906 päivää). Suurin osa potilaista toipui tapahtumasta ja pystyi jatkamaan olaparibihoitoa ja tavanomaista lääkehoitoa.
Potilaat, joilla oli jokin merkittävä sydän- ja verisuonitauti, suljettiin pois. Katso kardiovaskulaariset poissulkukriteerit abirateronin valmisteyhteenvedosta (kohta Varoitukset ja käyttöön liittyvät varotoimet).
Muut laboratoriolöydökset
Lynparza-valmisteella tehdyissä kliinisissä tutkimuksissa lähtötilanteeseen verrattuna vähintään kahden CTCAE-asteen muutoksen (suurenemisen) ilmaantuvuus oli veren kreatiniinipitoisuuden osalta noin 11 %. Kaksoissokkoutetusta lumekontrolloidusta tutkimuksesta saadut tiedot osoittivat, että lähtötilanteeseen verrattuna 23 %:iin asti suurentunut mediaani säilyi yhtä suurena ajan kuluessa ja palautui lähtötasolle, kun hoito lopetettiin, eikä selviä kliinisiä jälkiseurauksia havaittu. Kreatiniiniarvot olivat lähtötilanteessa 90 %:lla potilaista CTCAE-astetta 0, ja 10 %:lla potilaista CTCAE-astetta 1.
Gastrointestinaaliset haittavaikutukset
Pahoinvointia raportoitiin yleensä hyvin varhaisessa vaiheessa ja useimmilla potilailla sitä ilmeni ensimmäisen kerran ensimmäisen Lynparza-hoitokuukauden aikana. Oksentelua raportoitiin varhaisessa vaiheessa, ja useimmilla potilailla sitä ilmeni ensimmäisen kerran kahden ensimmäisen Lynparza-hoitokuukauden aikana. Sekä pahoinvoinnin että oksentelun ilmoitettiin olevan useimmilla potilailla ajoittaista ja sitä voitiin hoitaa keskeyttämällä lääkkeen antaminen, pienentämällä annosta ja/tai käyttämällä pahoinvointilääkitystä. Ennaltaehkäisevää pahoinvointilääkitystä ei tarvittu.
Munasarjasyövän ensilinjan ylläpitohoidossa potilailla ilmeni pahoinvointitapahtumia (77 %:lla tutkittavista olaparibiryhmässä, 38 %:lla lumeryhmässä), oksentelua (40 %:lla tutkittavista olaparibiryhmässä, 15 %:lla lumeryhmässä), ripulia (34 %:lla tutkittavista olaparibiryhmässä, 25 %:lla lumeryhmässä) ja dyspepsiaa (17 %:lla tutkittavista olaparibiryhmässä, 12 %:lla lumeryhmässä). Pahoinvointitapahtumat johtivat hoidon keskeyttämiseen 2,3 %:lla olaparibihoitoa saaneista potilaista (CTCAE-aste 2) ja 0,8 %:lla lumelääkettä saaneista (CTCAE-aste 1). Olaparibihoitoa saaneista potilaista 0,8 % lopetti hoidon vaikeusasteltaan lievän (CTCAE-aste 2) oksentelun ja 0,4 % dyspepsian vuoksi. Yksikään olaparibihoitoa tai lumelääkettä saanut potilas ei keskeyttänyt hoitoa ripulin vuoksi. Yksikään lumelääkettä saanut potilas ei lopettanut hoitoa oksentelun tai dyspepsian vuoksi. Olaparibiryhmässä pahoinvointitapahtumat johtivat hoidon keskeyttämiseen 14 %:lla potilaista ja annoksen pienentämiseen 4 %:lla potilaista. Oksentelutapahtumat johtivat hoidon keskeyttämiseen 10 %:lla olaparibihoitoa saaneista potilaista. Yhdelläkään olaparibihoitoa saaneella potilaalla oksentelutapahtuma ei johtanut annoksen pienentämiseen.
Pediatriset potilaat
Pieni määrä pediatrisia potilaita sai olaparibia tutkimuksessa D0816C00025. Tässä tutkimuspopulaatiossa ei havaittu uusia turvallisuussignaaleja verrattuna Lynparza-valmisteen tunnettuun turvallisuusprofiilin aikuisilla (ks. kohta Farmakodynamiikka).
Muut erityisryhmät
Muista kuin kaukaasialaisista potilaista on saatavilla vain vähän turvallisuustietoja.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Olaparibin yliannostuksesta on vain vähän tietoa. Odottamattomia haittavaikutuksia ei raportoitu pienellä potilasjoukolla, joka otti olaparibitabletteja enintään 900 mg:n annoksen vuorokaudessa kahden päivän ajan. Yliannostuksen oireita ei ole varmistettu eikä Lynparza‑valmisteen yliannostukseen ole erityistä hoitoa. Yliannostuksen tapahtuessa lääkärin on annettava potilaalle tavanomaista tukihoitoa sekä oireiden mukaista hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut antineoplastiset lääkkeet, ATC‑koodi: L01XK01
Vaikutusmekanismi ja farmakodynaamiset vaikutukset
Olaparibi on voimakas ihmisen poly‑ADP‑riboosipolymeraasientsyymien (PARP‑1, PARP‑2 ja PARP‑3) estäjä ja sen on osoitettu estävän tiettyjen kasvainsolulinjojen kasvua in vitro ja kasvaimen kasvua in vivo joko yksinään käytettynä tai yhdistelmänä vakiintuneiden solunsalpaajahoitojen tai uusien hormonaalisten lääkeaineiden (NHA) kanssa.
PARP:eja tarvitaan DNA:n yhden juosteen katkosten tehokkaaseen korjaukseen, ja PARP:in avulla tapahtuvan korjauksen tärkeänä edellytyksenä on, että kromatiinin muuntumisen jälkeen PARP muuntaa itse itsensä ja irtoaa DNA:sta, mikä helpottaa emäksen poistokorjausjärjestelmän (base excision repair, BER) entsyymien pääsyä DNA:han. Kun olaparibi sitoutuu DNA:han liittyneen PARP:in aktiiviseen kohtaan, se estää PARP:in irtoamisen ja vangitsee sen DNA:han estäen siten DNA:n korjauksen. Replikoituvissa soluissa tämä johtaa myös DNA:n kaksoiskierteen katkosten muodostumiseen, kun replikaatiohaarukat kohtaavat PARP‑DNA-addukteja. Normaaleissa soluissa korjaus homologisella rekombinaatiolla (homologous recombination repair, HRR) on tehokas näiden DNA:n kaksoiskierteen katkosten korjauksessa. Jos kyseessä ovat syöpäsolut, joissa tehokkaan homologisella rekombinaatiolla tapahtuvan korjauksen toiminnan kannalta keskeiset oleelliset komponentit, kuten BRCA1 tai 2, puuttuvat, DNA:n kaksoiskierteen katkokset eivät voi korjautua tarkasti tai tehokkaasti, mikä johtaa huomattavan puutteelliseen homologiseen rekombinaatioon (HRD). Sen sijaan aktivoituvat vaihtoehtoiset ja virheille alttiit reitit, kuten klassinen ei‑homologinen säikeiden päiden yhteenliittäminen (nonhomologous end joining, NHEJ), mikä tekee genomista hyvin epävakaan. Useiden replikaatiosyklien jälkeen genomin epävakaus voi saavuttaa sietämättömän tason ja aiheuttaa syöpäsolun kuoleman, koska syöpäsolujen DNA‑vauriokuorma on jo normaaleihin soluihin nähden suuri. Muut mekanismit saattavat vaarantaa HRR-reitin, mutta syynä olevaa poikkeavuutta ja sen penetranssia ei ole täysin selvitetty. Täysin toimivan HRR-reitin puuttuminen on eräs keskeisistä platinaherkkyyden määräävistä tekijöistä munasarja- ja mahdollisesti muissa syövissä.
In vivo ‑malleissa, joissa BRCA1 tai BRCA2 toimii puutteellisesti, olaparibin antaminen platinahoidon jälkeen hidasti kasvaimen etenemistä ja pidensi kokonaiselinaikaa pelkkään platinahoitoon verrattuna. Tämän vaikutuksen todettiin korreloivan olaparibiylläpitohoidon keston kanssa.
Kasvaimen kasvua ehkäisevä yhdistelmävaikutus uusien hormonaalisten lääkeaineiden kanssa
Eturauhassyöpämalleilla tehdyissä prekliinisissä tutkimuksissa havaittiin kasvaimen kasvua ehkäisevä yhdistelmävaikutus, kun PARP:n estäjiä ja uuden sukupolven hormonaalisia lääkeaineita annettiin samanaikaisesti. PARP osallistuu androgeenireseptorin (AR) signaloinnin positiiviseen säätelyyn. Kun PARP/AR-signalointi on samanaikaisesti estynyt, AR-kohdegeenin suppressio tehostuu. Muiden prekliinisten tutkimusten perusteella havaittiin, että hoito uusilla hormonaalisilla lääkeaineilla estää joidenkin HRR-geenien transkriptiota ja siten aiheuttaa puutteellista homologista rekombinaatiota ja lisää herkkyyttä PARP:n estäjille ei-geneettisten mekanismien kautta.
BRCA1/2‑mutaatioiden toteaminen
Geneettiset tutkimukset on tehtävä tällaisiin määrityksiin perehtyneessä laboratoriossa käyttämällä validoitua testiä. Eri tutkimuksissa on käytetty paikallista tai keskitetysti tehtyä ituradan ja/tai somaattisten BRCA1/2-mutaatioiden testausta veri- ja/tai kasvainnäytteistä. Kudos- tai verinäytteestä saatua DNA:ta on testattu useimmissa tutkimuksissa, ja kiertävän kasvain-DNA:n testausta on käytetty eksploratiivisiin tarkoituksiin. Käytetyn testin ja kansainvälisesti sovitun luokituksen mukaan BRCA1/2-mutaatiot on luokiteltu haitallisiksi / epäillyiksi haitallisiksi tai patogeenisiksi / todennäköisesti patogeenisiksi. Positiivinen HRD-status (puutteellinen homologinen rekombinaatio) voidaan todeta haitalliseksi / epäillyksi haitalliseksi tai patogeeniseksi /todennäköisesti patogeeniseksi luokitellun BRCA1/2-mutaation havaitsemisen perusteella. HRD-positiivinen status voi määrittyä myös yhdistämällä näiden mutaatioiden ilmenemistä HRD-pistemäärään (ks. jäljempänä).
Genomin epävakauden havaitseminen
Paola-1-tutkimuksessa tutkittuihin puutteelliseen homologiseen rekombinaatioon liittyviin genomimuutoksiin kuuluivat genomin laajuinen heterotsygotian menetys, telomeerien alleeliepätasapaino ja huomattavat kromosomisiirtymät, jotka ovat jatkuvia mittareita ja joilla on ennalta määritellyt kriteerit ja pisteytys. Yhdistetty genomin epävakautta kuvaava pistemäärä (genomic instability score, GIS, kutsutaan myös HRD-pistemääräksi) määrittyy, kun yhdistettyjä mittareita ja niiden vastaavia pistemääriä käytetään tiettyjen syöpäsoluihin kertyneiden genomin poikkeamien laajuuden arvioimiseen. Pieni pistemäärä tarkoittaa, että syöpäsolujen puutteellisen homologisen rekombinaation todennäköisyys on pienempi, ja suuri pistemäärä tarkoittaa, että syöpäsolujen puutteellisen homologisen rekombinaation todennäköisyys on suurempi näytteenottohetkellä suhteessa DNA:ta vaurioittaville lääkeaineille altistumiseen. GIS-positiivisuuden määrittelyssä on käytettävä validoituja katkaisupisteitä.
HRD-positiivisuus voidaan määritellä puutteelliseen homologiseen rekombinaatioon liittyvien genomimuutosten perusteella lasketun yhdistetyn GIS-pistemäärän pohjalta. Genomimuutokset on tutkittava tällaisiin määrityksiin perehtyneessä laboratoriossa käyttämällä validoitua testiä.
Kliininen teho ja turvallisuus
Vasta diagnosoidun pitkälle edenneen munasarjasyövän ylläpitohoito, kun potilailla on BRCA-mutaatio
SOLO1‑tutkimus
Olaparibin turvallisuutta ja tehoa ylläpitohoitona tutkittiin vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa vasta diagnosoitua pitkälle edennyttä (FIGO-levinneisyysasteet III–IV) korkean pahanlaatuisuusasteen seroosia tai endometrioidia munasarjasyöpää sairastavilla potilailla, joilla oli BRCA1/2-mutaatio (BRCA1/2m) ja jotka olivat saaneet ensilinjan platinapohjaista solunsalpaajahoitoa. Tutkimuksessa satunnaistettiin 391 potilasta suhteessa 2:1 saamaan joko Lynparza-valmistetta (300 mg [kaksi 150 mg:n tablettia] 2 kertaa vuorokaudessa) tai lumelääkettä. Potilaat stratifioitiin ensilinjan platinapohjaisella solunsalpaajahoidolla saavutetun vasteen mukaan (täydellinen vai osittainen vaste). Hoitoa jatkettiin, kunnes perussairaus eteni radiologisesti todennetusti, ilmeni haittavaikutuksia, joita ei voitu hyväksyä, tai enintään 2 vuoden ajan. Potilailla, joilla täydellinen kliininen vaste säilyi (ei radiologisesti todennettua sairautta), hoidon enimmäiskesto oli 2 vuotta. Potilaat, joiden kohdalla oli näyttöä vakaana pysyvästä sairaudesta (ei näyttöä sairauden etenemisestä), saivat kuitenkin jatkaa Lynparza-hoitoa 2 vuoden jälkeenkin.
Potilaat, joilla oli ituradan tai somaattisia BRCA1/2-mutaatioita, tunnistettiin prospektiivisesti joko verinäytteestä tehtävällä ituratatutkimuksella käyttämällä paikallista testiä (n = 208) tai keskitetysti (n = 181) tai tutkimalla kasvainnäyte käyttämällä paikallista testiä (n = 2). Keskitetysti toteutetussa ituratatutkimuksessa todettiin haitallisia mutaatioita 95,3 %:lla potilaista (365/383) ja epäiltyjä haitallisia mutaatioita 4,7 %:lla potilaista (18/383). 5,5 %:lla (21/383) satunnaistetuista potilaista todettiin BRCA1/2-geenien laajoja uudelleenjärjestymiä. Paikallisen testin perusteella tutkimukseen otettujen potilaiden gBRCA-mutaatiostatus vahvistettiin jälkikäteen keskitetysti tehdyllä testillä. Potilaat, joista oli saatavilla kasvainnäyte, testattiin jälkikäteen keskitetysti tehdyllä testillä, ja 341 potilaalle onnistuttiin saamaan tulokset. Näistä potilaista 95 %:lla oli soveltuva mutaatio (tunnettu [n = 47] tai todennäköisesti patogeeninen mutaatio [n = 277]), ja kahdella gBRCAwt-potilaalla vahvistettiin olevan vain sBRCA-mutaatio. SOLO1-tutkimuksessa 389 potilaalla oli ituradan BRCA1/2-mutaatio ja 2 potilaalla oli somaattinen BRCA1/2-mutaatio.
Demografiset tiedot ja lähtötilanteen tiedot olivat yleisesti hyvin samankaltaiset olaparibi- ja lumelääkehaaroissa. Mediaani-ikä molemmissa haaroissa oli 53 vuotta. Munasarjasyöpä oli primaarikasvain 85 %:lla potilaista. Yleisin histologinen tyyppi oli seroosi (96 %), endometrioidia histologiaa ilmoitettiin 2 %:lla potilaista. Suurimmalla osalla potilaista (78 %) ECOG-suorituskykypistemäärä oli 0, eikä ole olemassa tietoja potilaista, joiden suorituskykypistemäärä olisi ollut 2−4. Potilaista 63 %:lle oli ensin tehty sytoreduktiivinen leikkaus ja suurimmalla osalla (75 %) näistä potilaista ei ollut makroskooppista jäännöstautia. Intervallisytoreduktioleikkaus tehtiin 35 %:lle potilaista ja 82 %:lla näistä potilaista ei raportoitu makroskooppista jäännöstautia. Seitsemälle potilaalle, joilla kaikilla tauti oli levinneisyysastetta IV, ei tehty sytoreduktiivista leikkausta. Kaikki potilaat olivat saaneet ensilinjan platinapohjaista hoitoa. 73 %:lla olaparibihaaran potilaista ja 77 %:lla lumehaaran potilaista ei ollut tutkijalääkärin mukaan näyttöä sairaudesta (täydellinen vaste) heidän aloittaessaan tutkimuksessa, eli ei radiologista näyttöä sairaudesta ja syöpäantigeeni 125 (CA-125) normaalilla vaihteluvälillä. 27 %:lla olaparibihaaran potilaista ja 23 %:lla lumehaaran potilaista ilmoitettiin osittainen vaste, jonka määritelmänä oli mikä tahansa mitattavissa oleva tai ei-mitattavissa oleva leesio lähtötilanteessa tai kohonnut CA-125-arvo. 93 % potilaista satunnaistettiin 8 viikon kuluessa viimeisen platinapohjaisen solunsalpaaja-annoksen saamisesta. Tutkimuksesta suljettiin pois potilaat, jotka olivat saaneet bevasitsumabia. Tästä syystä ei ole olemassa turvallisuutta ja tehoa koskevia tietoja olaparibilla hoidetuista potilaista, jotka olivat aiemmin saaneet bevasitsumabia. Tietoja on erittäin vähän potilaista, joilla on somaattinen BRCA-mutaatio.
Ensisijainen päätemuuttuja oli etenemisvapaa elinaika (PFS), joka määriteltiin aikana satunnaistamisesta tutkijalääkärin arvioimaan taudin etenemiseen muokattujen RECIST 1.1 ‑kriteerien (Response Evaluation Criteria in Solid Tumors) perusteella tai kuolemaan. Toissijaisia tehoa mittaavia päätemuuttujia olivat aika satunnaistamisesta taudin toiseen etenemiseen tai kuolemaan (PFS2), kokonaiselinaika (OS), aika satunnaistamisesta hoidon lopettamiseen tai kuolemaan (TDT), aika satunnaistamisesta ensimmäiseen seuraavaan syöpähoitoon tai kuolemaan (TFST) ja terveyteen liittyvä elämänlaatu (HRQoL). Potilaiden kasvaimet arvioitiin lähtötilanteessa ja 12 viikon välein 3 vuoden ajan ja sen jälkeen 24 viikon välein suhteessa satunnaistamispäivään, kunnes objektiivisessa radiologisessa tutkimuksessa oli todettu taudin edenneen.
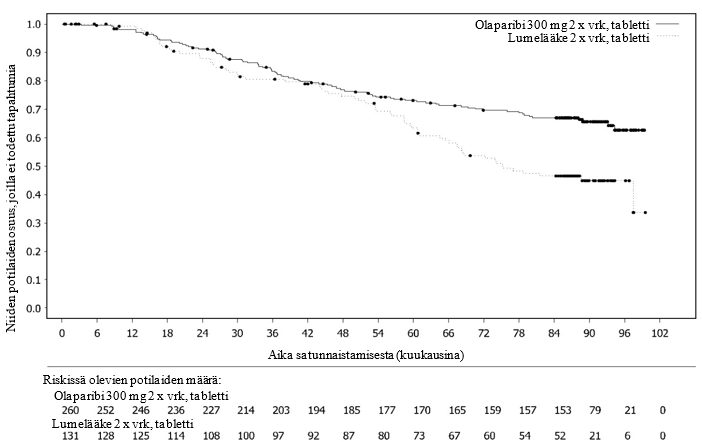
Tutkimuksessa osoitettiin kliinisesti merkittävä ja tilastollisesti merkitsevä tutkijalääkärin arvioiman PFS:n piteneminen olaparibihaarassa lumehaaraan nähden. Tutkijalääkärin tekemää etenemisvapaata elinaikaa koskevaa arviota tuki sokkoutetun riippumattoman radiologin keskitetysti tekemä etenemisvapaata elinaikaa koskeva arvio. Seitsemän vuoden kuluttua viimeisen potilaan satunnaistamisesta tehdyssä kuvailevassa analyysissä todettiin olaparibihaaraa numeerisesti suosiva, kliinisesti merkityksellinen hyöty kokonaiselossaoloajan suhteen. Tehoa koskevat tulokset on esitetty taulukossa 3 ja kuvissa 1 ja 2.
Taulukko 3 SOLO1-tutkimuksen tehoa koskevat tulokset vasta diagnosoiduista potilaista, joilla on BRCA1/2-mutaatio ja pitkälle edennyt munasarjasyöpä
| Olaparibi 300 mg kaksi kertaa vuorokaudessa | Lumelääkec | |
| PFS (maturiteetti 51 %)a | ||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 102:260 (39) | 96:131 (73) |
| Mediaaniaika (kuukausina) | Ei saavutettu | 13,8 |
| HR (95 %:n luottamusväli)b | 0,30 (0,23–0,41) | |
| p-arvo (kaksitahoinen) | p < 0,0001) | |
| PFS2 (maturiteetti 31 %) | ||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 69:260 (27) | 52:131 (40) |
| Mediaaniaika (kuukausina) | Ei saavutettu | 41,9 |
| HR (95 %:n luottamusväli)c | 0,50 (0,35–0,72) | |
| p-arvo (kaksitahoinen) | p = 0,0002 | |
| OS (maturiteetti 38 %)d | ||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 84:260 (32) | 65:131 (50) |
| Mediaaniaika (kuukausina) | Ei saavutettu | 75,2 |
| HR (95 %:n luottamusväli)b | 0,55 (0,40–0,76) | |
| TFST (maturiteetti 60 %) | ||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 135:260 (52) | 98:131 (75) |
| Mediaaniaika (kuukausina) | 64,0 | 15,1 |
| HR (95 %:n luottamusväli)c | 0,37 (0,28–0,48) | |
a Kaplan-Meier estimaattien perusteella niiden potilaiden osuus, joiden tauti ei ollut edennyt kuukauden 24 kohdalla, oli olaparibiryhmässä 74 % ja lumelääkeryhmässä 35 % ja 36 kohdalla vastaavasti 60 % ja 27 %. Olaparibi- ja lumelääkeryhmässä seuranta-ajan mediaani oli 41 kuukautta.
b Arvo, joka on < 1, suosii olaparibia. Analyysi tehtiin käyttämällä Coxin suhteellisen hasardin mallia, jossa oli kovariaattina vaste aiemmalle platinasolunsalpaajahoidolle (täydellinen tai osittainen vaste).
c Seuraavaa hoitoa saaneista 97:stä lumehaaran potilaasta 58 (60 %) sai PARP:n estäjää.
d Kaplan-Meier estimaattien perusteella niiden potilaiden osuus, jotka olivat elossa kuukauden 84 kohdalla, oli olaparibiryhmässä 67 % ja lumelääkeryhmässä 47 %.
HR = Riskisuhde; PFS: Etenemisvapaa elinaika; PFS2: Aika satunnaistamisesta taudin toiseen etenemiseen tai kuolemaan; OS: Kokonaiselinaika; TFST: Aika satunnaistamisesta ensimmäisen seuraavan hoidon alkuun tai kuolemaan.
Kuva 1 SOLO1: Kaplan-Meier-käyrä etenemisvapaalle elinajalle potilailla, joilla oli vasta diagnosoitu pitkälle edennyt BRCA1/2m-munasarjasyöpä (maturiteetti 51 % − tutkijalääkärin arvio)
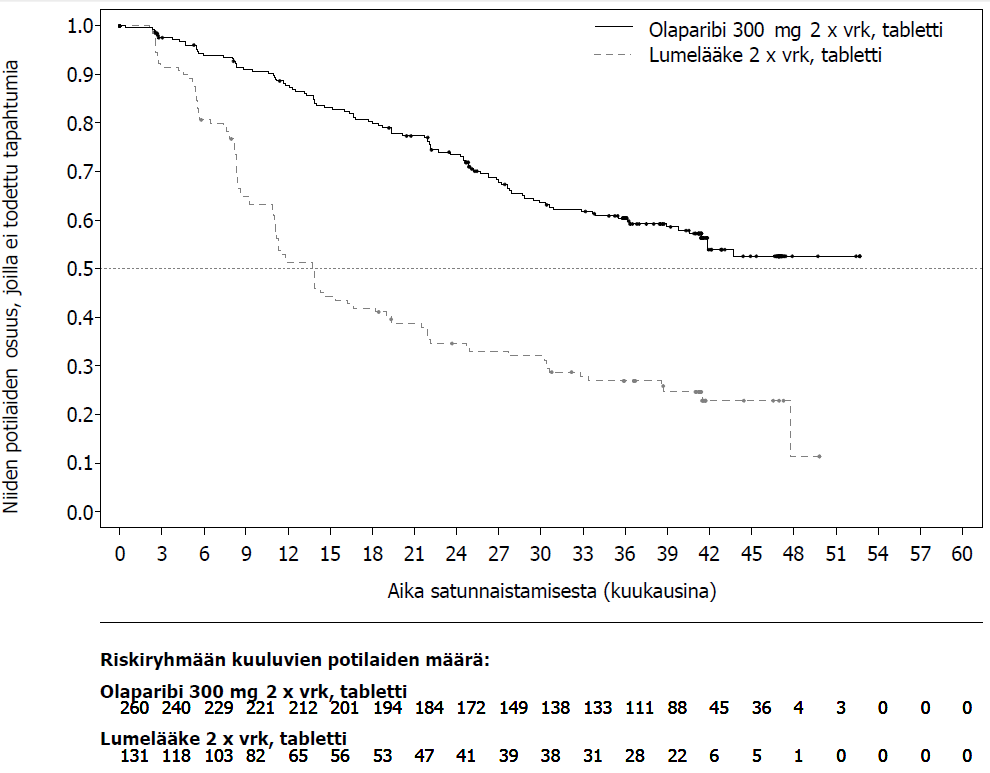
Kuva 2 SOLO1: Kaplan‑Meier-käyrä kokonaiselinajalle potilailla, joilla oli vasta diagnosoitu pitkälle edennyt BRCA1/2m-munasarjasyöpä (maturiteetti 38 %)
Yhdenmukaisia tuloksia havaittiin potilaiden alaryhmissä, jotka perustuivat näyttöön sairaudesta potilaan aloittaessa tutkimuksessa. Potilailla, joilla oli tutkijalääkärin arvion mukaan täydellinen vaste, riskisuhde oli 0,34 (95 %:n luottamusväli 0,24–0,47), etenemisvapaan elinajan mediaania ei saavutettu olaparibihaarassa ja se oli 15,3 kuukautta lumelääkehaarassa. Kuukauden 24 kohdalla 68 %:lla olaparibihaaran potilaista oli edelleen täydellinen vaste ja kuukauden 36 kohdalla vastaava osuus oli 45 %. Lumelääkehaarassa osuudet olivat 34 % ja 22 %. Potilailla, joilla oli osittainen vaste heidän aloittaessaan tutkimuksessa, etenemisvapaan elinajan riskisuhde oli 0,31 (95 %:n luottamusväli 0,18, 0,52; etenemisvapaan elinajan mediaani oli 30,9 kuukautta olaparibihaarassa ja 8,4 kuukautta lumelääkehaarassa). Potilaat, joilla oli osittainen vaste heidän aloittaessaan tutkimuksessa, joko saavuttivat täydellisen vasteen (15 % olaparibihaaran potilaista ja 4 % lumelääkehaaran potilaista kuukauden 24 kohdalla ja täydellinen vaste oli säilynyt kuukauden 36 kohdalla) tai heillä oli edelleen osittainen vaste / vakaa sairaus (43 % olaparibihaarassa ja 15 % lumelääkehaarassa kuukauden 24 kohdalla; 17 % olaparibihaarassa ja 15 % lumelääkehaarassa kuukauden 36 kohdalla). Niiden potilaiden osuus, joilla sairaus eteni 6 kuukauden kuluessa viimeisen platinapohjaisen solunsalpaajahoitoannoksen jälkeen, oli 3,5 % olaparibihaarassa ja 8,4 % lumelääkehaarassa.
Uusiutuneen platinaherkän munasarjasyövän ylläpitohoito
SOLO2-tutkimus
Olaparibin turvallisuutta ja tehoa ylläpitohoitona tutkittiin vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa tutkimuksessa potilailla, joilla oli uusiutunut platinaherkkä munasarja‑, munanjohdin- tai primaari peritoneaalinen syöpä ja ituradan BRCA1/2-mutaatio. Tutkimuksessa verrattiin taudin etenemiseen asti annetun Lynparza-ylläpitohoidon (300 mg [kaksi 150 mg:n tablettia] kaksi kertaa vuorokaudessa) tehoa lumehoitoon nähden 295 potilaalla, joilla oli korkean pahanlaatuisuusasteen seroosi tai endometrioidi uusiutunut platinaherkkä munasarjasyöpä (satunnaistamissuhde 2:1: 196 sai olaparibia ja 99 lumelääkettä) ja joiden vaste säilyi platinaa sisältävän solunsalpaajahoidon päättymisen jälkeen (täydellinen vaste [CR] tai osittainen vaste [PR]).
Tutkimukseen otettiin mukaan potilaita, jotka olivat saaneet ainakin kaksi platinaa sisältävää hoitojaksoa ja joiden tauti oli uusiutunut yli 6 kuukauden kuluttua viimeistä edellisen platinapohjaisen solunsalpaajahoidon päättymisen jälkeen. Potilaat eivät olleet voineet saada aiemmin olaparibia tai muuta PARP:n estäjää. Potilaat olivat voineet saada aiemmin bevasitsumabia, mutta ei kuitenkaan välittömästi satunnaistamista edeltävän hoitojakson aikana.
Kaikilla potilailla oli lähtötilanteessa saatu näyttöä ituradan BRCA1/2-mutaatiosta (gBRCA1/2m). Potilaat, joilla oli BRCA1/2-mutaatioita, tunnistettiin joko verinäytteestä tehtävällä ituratatutkimuksella käyttämällä paikallista testiä tai keskitetysti Myriad-yrityksen tekemällä testillä tai tutkimalla kasvainnäyte käyttämällä paikallista testiä. 4,7 %:lla (14/295) satunnaistetuista potilaista todettiin BRCA1/2-geenien laajoja uudelleenjärjestymiä.
Demografiset tiedot ja lähtötilanteen tiedot olivat yleisesti hyvin samankaltaiset olaparibi- ja lumelääkehaaroissa. Mediaani-ikä molemmissa haaroissa oli 56 vuotta. Munasarjasyöpä oli primaarikasvain yli 80 %:lla potilaista. Yleisin histologinen tyyppi oli seroosi (yli 90 %), endometrioidia histologiaa ilmoitettiin 6 %:lla potilaista. 55 % olaparibihaaran potilaista oli saanut ainoastaan kahta aiemman linjan hoitoa, ja 45 % oli saanut vähintään kolmea aiemman linjan hoitoa. 61 % lumelääkehaaran potilaista oli saanut ainoastaan kahta aiemman linjan hoitoa, ja 39 % oli saanut vähintään kolmea aiemman linjan hoitoa. Suurimmalla osalla (81 %) potilaista ECOG-suorituskykypistemäärä oli 0, eikä ole olemassa tietoja potilaista, joiden suorituskykypistemäärä oli 2−4. Aikaväli, jolloin platinapohjaista hoitoa ei annettu, oli 60 %:lla potilaista > 12 kuukautta ja 40 %:lla potilaista > 6 – 12 kuukautta. Aiempaan platinapohjaiseen solunsalpaajahoitoon saatu vaste oli 47 %:lla potilaista täydellinen ja 53 %:lla osittainen. 17 % olaparibihaaran ja 20 % lumelääkehaaran potilaista oli saanut aiemmin bevasitsumabia.
Ensisijainen päätemuuttuja oli etenemisvapaa elinaika (PFS), jonka arvioi tutkijalääkäri käyttämällä RECIST 1.1 ‑kriteerejä (Response Evaluation Criteria in Solid Tumors). Toissijaisia tehoa koskevia päätemuuttujia olivat aika satunnaistamisesta taudin toiseen etenemiseen tai kuolemaan (PFS2), kokonaiselinaika (OS), aika satunnaistamisesta hoidon lopettamiseen tai kuolemaan (TDT), aika satunnaistamisesta ensimmäiseen seuraavaan syöpähoitoon tai kuolemaan (TFST), aika satunnaistamisesta toisen seuraavan syöpähoidon alkuun tai kuolemaan (TSST) ja terveyteen liittyvä elämänlaatu (HRQoL).
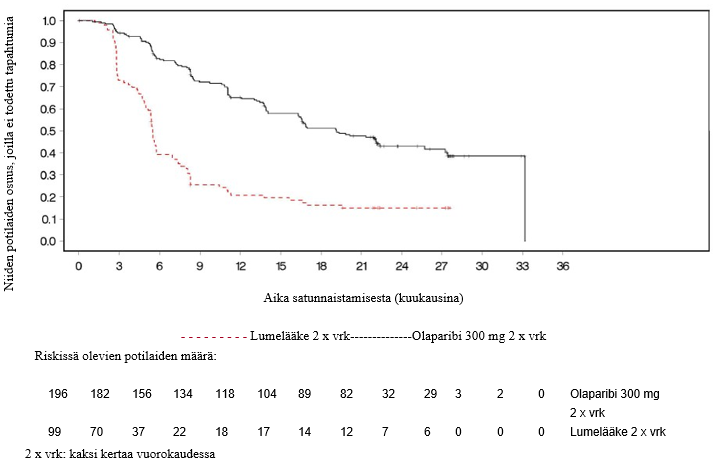
Tutkimuksen ensisijainen tavoite saavutettiin osoittamalla tutkijalääkärin arvioiman etenemisvapaan elinajan tilastollisesti merkitsevä piteneminen olaparibia saaneilla tutkittavilla lumelääkettä saaneisiin verrattuna riskisuhteella (HR) 0,30 (95 %:n luottamusväli 0,22–0,41; p < 0,0001; mediaani olaparibiryhmässä 19,1 kuukautta ja lumeryhmässä 5,5 kuukautta). Tutkijalääkärin tekemää etenemisvapaata elinaikaa koskevaa arviota tuki sokkoutetun riippumattoman radiologin keskitetysti tekemä etenemisvapaata elinaikaa koskeva arvio (riskisuhde 0,25; 95 %:n luottamusväli 0,18–0,35; p < 0,0001; mediaani olaparibia saaneilla 30,2 kuukautta ja lumelääkettä saaneilla 5,5 kuukautta). Kahden vuoden kohdalla 43 %:lla olaparibia saaneista potilaista tauti ei ollut edelleenkään edennyt, lumelääkettä saaneilla potilailla vastaava osuus oli vain 15 %.
Taulukossa 4 ja kuvassa 3 on esitetty yhteenveto tutkimuksen SOLO2 ensisijaista tavoitetta koskevasta hoitotuloksesta potilailla, joilla oli uusiutunut platinaherkkä gBRCA1/2m-munasarjasyöpä.
Taulukko 4 Yhteenveto tutkimuksen SOLO2 ensisijaista tavoitetta koskevasta hoitotuloksesta uusiutunutta platinaherkkää gBRCA1/2m-munasarjasyöpää sairastavilla potilailla
| Olaparibi 300 mg tabletti kaksi kertaa vuorokaudessa | Lumelääke | |
| PFS (maturiteetti 63 %) | ||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 107:196 (55) | 80:99 (81) |
| Mediaaniaika (kuukausina) (95 %:n luottamusväli) | 19,1 (16,3–25,7) | 5,5 (5,2–5,8) |
| HR (95 %:n luottamusväli) a | 0,30 (0,22−0,41) | |
| p-arvo (kaksitahoinen) | p < 0,0001 | |
a HR = Riskisuhde. Arvo, joka on < 1, suosii olaparibia. Analyysi tehtiin käyttämällä Coxin suhteellisten hasardien mallia, jossa oli kovariaatteina vaste aiemmalle platinasolunsalpaajahoidolle (täydellinen tai osittainen vaste) ja aika taudin etenemiseen (> 6 – 12 kuukautta ja > 12 kuukautta) viimeistä edellisen platinapohjaisen solunsalpaajahoidon yhteydessä.
PFS: Etenemisvapaa elinaika.
Kuva 3 SOLO2: Kaplan‑Meier‑käyrä etenemisvapaalle elinajalle uusiutunutta platinaherkkää gBRCA1/2m-munasarjasyöpää sairastavilla potilailla (maturiteetti 63 %, tutkijan arvio)
Kokonaiselinaikaa koskevassa lopullisessa analyysissä (maturiteetti 61 %) riskisuhde oli 0,74 (95 %:n luottamusväli 0,54–1,00; p = 0,0537; mediaani olaparibia saaneilla oli 51,7 kuukautta ja lumelääkettä saaneilla 38,8 kuukautta), mikä ei saavuttanut tilastollista merkitsevyyttä. Toissijaisille päätemuuttujille TFST ja PFS2 osoitettiin pysyvä ja tilastollisesti merkitsevä paraneminen olaparibihaarassa lumelääkkeeseen verrattuna. Kokonaiselinaikaa, TFST:tä ja PFS2:ta koskevat tulokset on esitetty taulukossa 5 ja kuvassa 4.
Taulukko 5 Yhteenveto tutkimuksen SOLO2 keskeisistä toissijaista tavoitetta koskeneista hoitotuloksista potilailla, joilla oli uusiutunut platinaherkkä gBRCA1/2m-munasarjasyöpä
| Olaparibi 300 mg tabletti kaksi kertaa vuorokaudessa | Lumelääke | |
| Kokonaiselinaika (maturiteetti 61 %) | ||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 116: 196 (59) | 65: 99 (66) |
| Mediaaniaika,kuukausina (95 %:n luottamusväli) | 51,7 (41,5–59,1) | 38,8 (31,4–48,6) |
| HR (95 %:n luottamusväli) a | 0,74 (0,54–1,00) | |
| p-arvo* (kaksitahoinen) | p = 0,0537 | |
| TFST (maturiteetti 71 %) | ||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 139:196 (71) | 86:99 (87) |
| Mediaaniaika (kuukausina) (95 %:n luottamusväli) | 27,4 (22,6–31,1) | 7,2 (6,3–8,5) |
| HR (95 %:n luottamusväli) a | 0,37 (0,28−0,48) | |
| p-arvo* (kaksitahoinen) | p < 0,0001 | |
| PFS2 (maturiteetti 40 %) | ||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 70:196 (36) | 49:99 (50) |
| Mediaaniaika (kuukausina) (95 %:n luottamusväli) | NR (24,1–NR) | 18,4 (15,4–22,8) |
| HR (95 %:n luottamusväli) a | 0,50 (0,34−0,72) | |
| p-arvo (kaksitahoinen) | p = 0,0002 | |
* Ei otettu huomioon kerrannaisuutta.
a HR = Riskisuhde. Arvo, joka on < 1, suosii olaparibia. Analyysi tehtiin käyttämällä Coxin suhteellisten hasardien mallia, jossa oli kovariaatteina vaste aiemmalle platinasolunsalpaajahoidolle (täydellinen tai osittainen vaste) ja aika taudin etenemiseen (> 6 – 12 kuukautta ja > 12 kuukautta) viimeistä edellisen platinapohjaisen solunsalpaajahoidon yhteydessä.
NR: ei saavutettu; PFS2: Aika satunnaistamisesta taudin toiseen etenemiseen tai kuolemaan; TFST: Aika satunnaistamisesta ensimmäisen seuraavan hoidon alkuun tai kuolemaan.
Kuva 4 SOLO2: Kaplan–Meier‑käyrä kokonaiselinajalle uusiutunutta platinaherkkää gBRCA1/2m-munasarjasyöpää sairastavilla potilailla (maturiteetti 61 %)
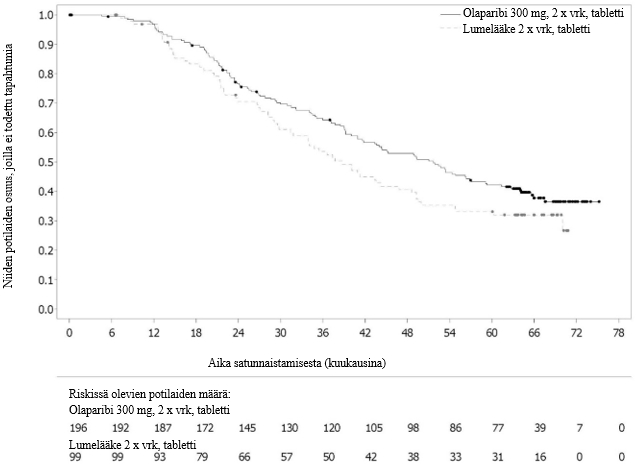
Lynparza-haarassa objektiivisen hoitovasteen saavuttaneiden osuus oli 41 % ja lumelääkehaarassa 17 % potilailla, joilla oli tutkimuksessa aloittaessaan mitattavissa oleva sairaus (lähtötilanteen valitut kohde leesiot). Lynparza-hoitoa saaneista potilaista, joilla oli tutkimuksessa aloittaessaan näyttöä sairaudesta (lähtötilanteen valitut kohde leesioit tai muut leesioit), 15,0 % saavutti täydellisen vasteen. Lumelääkettä saaneilla potilailla vastaava luku oli 9,1 %.
Kun etenemisvapaa elinaika analysoitiin, hoidon keston mediaani oli 19,4 kuukautta olaparibihaarassa ja 5,6 kuukautta lumelääkehaarassa. Suurin osa potilaista jatkoi aloitusannoksella 300 mg olaparibia kaksi kertaa vuorokaudessa. Haittatapahtuman vuoksi lääkkeen käytön keskeytti 45,1 %, annosta pienennettiin 25,1 %:lla potilaista ja lääkkeen käytön lopetti 10,8 %. Lääkkeen käyttö keskeytettiin useimmiten ensimmäisen kolmen hoitokuukauden aikana ja annosta pienennettiin useimmiten ensimmäisten 3–6 hoitokuukauden aikana. Yleisimmät käytön keskeyttämiseen tai annoksen pienentämiseen johtaneet haittavaikutukset olivat anemia, pahoinvointi ja oksentelu.
Potilaiden ilmoittamia hoitotuloksia koskevat tiedot eivät viittaa eroihin olaparibi- ja lumehoitoa saaneiden potilaiden välillä, mikä arvioitiin FACT‑O-analyysin (Functional Assessment of Cancer Therapy – Ovarian) tutkimustulosindeksin (Trial Outcome Index, TOI) muutoksena lähtötilanteeseen nähden.
Study 19 -tutkimus (D0810C00019)
Olaparibin turvallisuutta ja tehoa platinaherkän munasarjasyövän, munanjohdin ja primaari peritoneaalinen syöpä mukaan lukien, ylläpitohoitona tutkittiin laajassa vaiheen II satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa tutkimuksessa potilailla, jotka olivat saaneet vähintään kaksi platinaa sisältävää hoitojaksoa (Study 19 -tutkimus). Tutkimuksessa verrattiin taudin etenemiseen asti Lynparza-ylläpitohoidon tehoa lumehoitoon nähden 265 potilaalla (136 sai olaparibia ja 129 lumelääkettä), joilla oli platinaherkkä korkean pahanlaatuisuusasteen seroosi munasarjasyöpä ja joiden vaste säilyi platinaa sisältävän solunsalpaajahoidon päättymisen jälkeen (täydellinen tai osittainen vaste). Ensisijainen päätemuuttuja oli etenemisvapaa elinaika (PFS), jonka arvioi tutkijalääkäri käyttämällä RECIST 1.0 ‑kriteerejä. Toissijaisia tehoa koskevia päätemuuttujia olivat kokonaiselinaika (OS), taudin hallinnan aste (disease control rate, DCR), joka määriteltiin varmistettuna CR/PR + SD (stable disease, vakaa tauti), terveyteen liittyvä elämänlaatu (health related quality of life, HRQoL) ja sairauteen liittyvät oireet. TFST- ja TSST-päätemuuttujista tehtiin myös eksploratiiviset analyysit.
Tutkimukseen otettiin mukaan potilaita, joiden tauti oli uusiutunut yli 6 kuukauden kuluttua viimeistä edellisen platinapohjaisen solunsalpaajahoidon päättymisen jälkeen. Tutkimukseen mukaan ottamiseksi ei tarvittu näyttöä BRCA1/2-mutaatiosta (osalla potilaista BRCA-mutaatiostatus määritettiin jälkikäteen). Potilaat eivät olleet voineet saada aiemmin olaparibia tai muuta PARP:n estäjää. Potilaat olivat voineet saada aiemmin bevasitsumabia, mutta ei kuitenkaan välittömästi satunnaistamista edeltävän hoitojakson aikana. Uusintahoito olaparibilla ei ollut sallittu, jos tauti eteni olaparibihoidon aikana.
Potilaat, joilla oli BRCA1/2-mutaatioita, tunnistettiin joko verinäytteestä tehtävällä ituratatutkimuksella käyttämällä paikallista testiä tai keskitetysti Myriad-yrityksen tekemällä testillä tai tutkimalla kasvainnäyte käyttämällä Foundation Medicine -yrityksen suorittamaa testiä. 7,4 %:lla (10/136) satunnaistetuista potilaista todettiin BRCA1/2-geenien laajoja uudelleenjärjestymiä.
Demografiset tiedot ja lähtötilanteen tiedot olivat yleisesti hyvin samankaltaiset olaparibi- ja lumelääkehaaroissa. Mediaani-ikä molemmissa haaroissa oli 59 vuotta. Munasarjasyöpä oli primaarikasvain 86 %:lla potilaista. 44 % olaparibihaaran potilaista oli saanut ainoastaan kahta aiemman linjan hoitoa, ja 56 % oli saanut vähintään kolmea aiemman linjan hoitoa. 49 % lumelääkehaaran potilaista oli saanut ainoastaan kahta aiemman linjan hoitoa, ja 51 % oli saanut vähintään kolmea aiemman linjan hoitoa. Suurimmalla osalla (77 %) potilaista ECOG-suorituskykypistemäärä oli 0, eikä ole olemassa tietoja potilaista, joiden suorituskykypistemäärä oli 2−4. Aikaväli, jolloin platinapohjaista hoitoa ei annettu, oli 60 %:lla potilaista > 12 kuukautta ja 40 %:lla potilaista 6 – 12 kuukautta. Aiempaan platinapohjaiseen solunsalpaajahoitoon saatu vaste oli 45 %:lla potilaista täydellinen ja 55 %:lla osittainen. 6 % olaparibihaaran ja 5 % lumelääkehaaran potilaista oli saanut aiemmin bevasitsumabia.
Tutkimuksen ensisijainen tavoite saavutettiin osoittamalla etenemisvapaan elinajan tilastollisesti merkitsevä piteneminen olaparibia saaneilla tutkittavilla lumelääkettä saaneisiin verrattuna kokonaispopulaatiossa riskisuhteella 0,35 (95 %:n luottamusväli 0,25−0,49; p < 0,00001; mediaani olaparibia saaneilla 8,4 kuukautta ja lumelääkettä saaneilla 4,8 kuukautta). Kokonaiselinajan lopullisessa analyysissä (viimeinen tiedonkeruupäivä 9.5.2016), kun maturiteetti oli 79 %, olaparibin ja lumelääkkeen välisen vertailun riskisuhde oli 0,73 (95 %:n luottamusväli 0,55−0,95; p = 0,02138 [etukäteen määriteltyä merkitsevyystasoa < 0,0095 ei saavutettu]; mediaani olaparibia saaneilla oli 29,8 kuukautta ja lumelääkettä saaneilla 27,8 kuukautta). Olaparibia saaneiden ryhmässä potilaista 23,5 % (n = 32/136) jatkoi hoitoa vähintään 2 vuoden ajan verrattuna 3,9 %:iin (n = 5/128) lumelääkeryhmän potilaista. Vaikka potilasmäärät olivat pieniä, olaparibiryhmän potilaista 13,2 % (n = 18/136) jatkoi hoitoa vähintään 5 vuoden ajan verrattuna 0,8 %:iin (n = 1/128) lumelääkeryhmän potilaista.
Ennalta suunnitellussa alaryhmäanalyysissä todettiin, että potilaat, joilla oli BRCA1/2‑mutaatio ja munasarjasyöpä (n = 136, 51,3 %, sisälsi 20 potilasta, joiden kasvaimessa oli todettu somaattinen BRCA1/2-mutaatio), muodostivat alaryhmän, joka hyötyi kliinisesti eniten monoterapiana annetusta olaparibiylläpitohoidosta. Hyötyä havaittiin myös potilailla, joilla oli villityypin BRCA1/2 tai -BRCA1/2-variantti, jonka merkitys oli epäselvä (variants of uncertain significance, VUS) (BRCA1/2wt/VUS), vaikkakin voimakkuudeltaan vähäisempää. Alaryhmäanalyysien ei ollut suunniteltu sisältävän useita testejä.
Taulukossa 6 on esitetty yhteenveto Study 19 -tutkimuksen ensisijaista tavoitetta koskeneesta hoitotuloksesta potilailla, joilla oli uusiutunut platinaherkkä BRCA1/2-mutaatio- ja BRCA1/2wt/VUS-munasarjasyöpä, ja taulukossa 6 ja kuvassa 5 kaikilla potilailla.
Taulukko 6 Yhteenveto Study 19 -tutkimuksen ensisijaista tavoitetta koskeneesta hoitotuloksesta kaikilla potilailla ja uusiutunutta platinaherkkää munasarjasyöpää sairastavilla potilailla, joilla on BRCA1/2‑mutaatio ja BRCA1/2wt/VUS
| Kaikki potilaata | BRCA1/2-mutaatio | BRCA1/2wt/VUS | ||||
| Olaparibi | Lumelääke | Olaparibi | Lumelääke | Olaparibi | Lumelääke | |
| PFS – viimeinen tiedonkeruupäivä 30.6.2010 | ||||||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 60:136 (44) | 94:129 (73) | 26:74 (35) | 46:62 (74) | 32:57 (56) | 44:61 (72) |
| Mediaaniaika (kuukausina) (95 %:n luottamusväli) | 8,4 (7,4–11,5) | 4,8 (4,0–5,5) | 11,2 (8,3–NR) | 4,3 (3,0–5,4) | 7,4 (5,5–10,3) | 5,5 (3,7–5,6) |
| HR (95 %:n luottamusväli) b | 0,35 (0,25–0,49) | 0,18 (0,10–0,31) | 0,54 (0,34–0,85) | |||
| p-arvo (kaksitahoinen) | p < 0,00001 | p < 0,00001 | p = 0,00745 | |||
a Kaikki potilaat sisälsi seuraavat alaryhmät: BRCA1/2-mutaatio, BRCA1/2wt/VUS- ja BRCA1/2-status tuntematon (11 potilasta, joiden status oli tuntematon, eivät sisälly taulukkoon erillisenä alaryhmänä).
b HR = Riskisuhde. Arvo, joka on < 1, suosii olaparibia. Analyysi tehtiin käyttämällä Coxin suhteellisen hasardin mallia, jossa huomioon otettavia tekijöitä olivat hoito, etninen tausta, platinaherkkyys ja viimeiseen platinahoitoon saatu vaste.
NR: ei saavutettu; PFS: Etenemisvapaa elinaika.
Kuva 5 Study 19 -tutkimus: Kaplan‑Meier‑käyrä etenemisvapaalle elinajalle täydellisessä analyysisarjassa (maturiteetti 58 %, tutkijan arvio), viimeinen tiedonkeruupäivä 30.6.2010
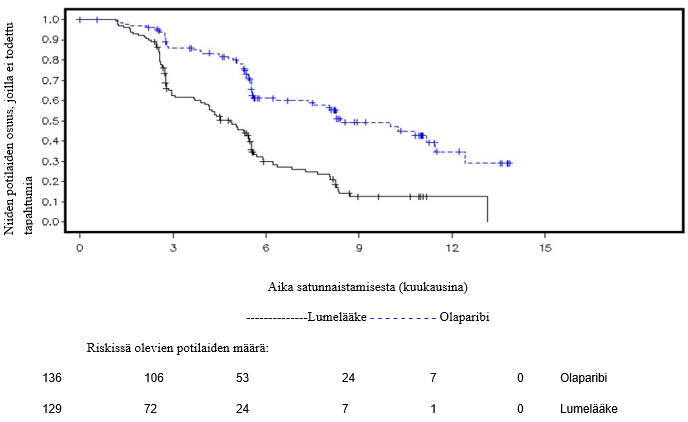
Taulukossa 7 on esitetty yhteenveto Study 19 -tutkimuksen keskeisistä toissijaista tavoitetta koskeneista hoitotuloksista potilailla, joilla oli uusiutunut platinaherkkä BRCA1/2-mutaatio- ja BRCA1/2wt/VUS-munasarjasyöpä, ja taulukossa 7 ja kuvassa 6 kaikilla Study 19 ‑tutkimuksen potilailla.
Taulukko 7 Yhteenveto Study 19 -tutkimuksen keskeisistä toissijaista tavoitetta koskeneista hoitotuloksista kaikilla potilailla ja uusiutunutta platinaherkkää munasarjasyöpää sairastavilla potilailla, joilla on BRCA1/2‑mutaatio ja BRCA1/2wt/VUS
| Kaikki potilaata | BRCA1/2-mutaatio | BRCA1/2wt/VUS | |||||||||
| Olaparibi | Lumelääke | Olaparibi | Lumelääke | Olaparibi | Lumelääke | ||||||
| OS - viimeinen tiedonkeruupäivä 9.5.2016 | |||||||||||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 98:136 (72) | 112:129 (87) | 49:74 (66) | 50:62 (81)c | 45:57 (79) | 57:61 (93) | |||||
| Mediaaniaika (kuukausina) (95 %:n luottamusväli) | 29,8 (26,9–35,7) | 27,8 (24,9–33,7) | 34,9 (29,2–54,6) | 30,2 (23,1–40,7) | 24,5 (19,8–35,0) | 26,6 (23,1–32,5) | |||||
| HR (95 %:n luottamusväli)b | 0,73 (0,55–0,95) | 0,62 (0,42–0,93) | 0,84 (0,57–1,25) | ||||||||
| p-arvo* (kaksitahoinen) | p = 0,02138 | p = 0,02140 | p = 0,39749 | ||||||||
| TFST – viimeinen tiedonkeruupäivä 9.5.2016 | |||||||||||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 106:136 (78) | 124:128 (97) | 55:74 (74) | 59:62 (95) | 47:57 (83) | 60:61 (98) | |||||
| Mediaaniaika (kuukausina) (95 %:n luottamusväli) | 13,3 (11,3–15,7) | 6,7 (5,7–8,2) | 15,6 (11,9–28,2) | 6,2 (5,3–9,2) | 12,9 (7,8–15,3) | 6,9 (5,7–9,3) | |||||
| HR (95 %:n luottamusväli)b | 0,39 (0,30–0,52) | 0,33 (0,22–0,49) | 0,45 (0,30–0,66) | ||||||||
| p-arvo* (kaksitahoinen) | p < 0,00001 | p < 0,00001 | p = 0,00006 | ||||||||
* Alaryhmäanalyysien ei ollut suunniteltu sisältävän useita testejä eikä kaikkien potilaiden TFST-päätemuuttujien arviointia ollut suunniteltu.
a Kaikki potilaat sisälsi seuraavat alaryhmät: BRCA1/2-mutaatio, BRCA1/2wt/VUS- ja BRCA1/2-status tuntematon (11 potilasta, joiden status oli tuntematon, eivät sisälly taulukkoon erillisenä alaryhmänä).
b HR = Riskisuhde. Arvo, joka on < 1, suosii olaparibia. Analyysi tehtiin käyttämällä Coxin suhteellisen hasardin mallia, jossa huomioon otettavia tekijöitä olivat hoito, etninen tausta, platinaherkkyys ja viimeiseen platinahoitoon saatu vaste.
c Noin neljäsosa lumelääkettä saaneista ja BRCA-mutaatioalaryhmään kuuluvista potilaista (14/62; 22,6 %), sai myöhemmin PARP:n estäjää.
OS: Kokonaiselinaika; TFST: Aika satunnaistamisesta ensimmäisen seuraavan hoidon alkuun tai kuolemaan.
Kuva 6 Study 19 -tutkimus: Kaplan-Meier-käyrä kokonaiselinajalle täydellisessä analyysisarjassa (maturiteetti 79 %), viimeinen tiedonkeruupäivä 9.5.2016
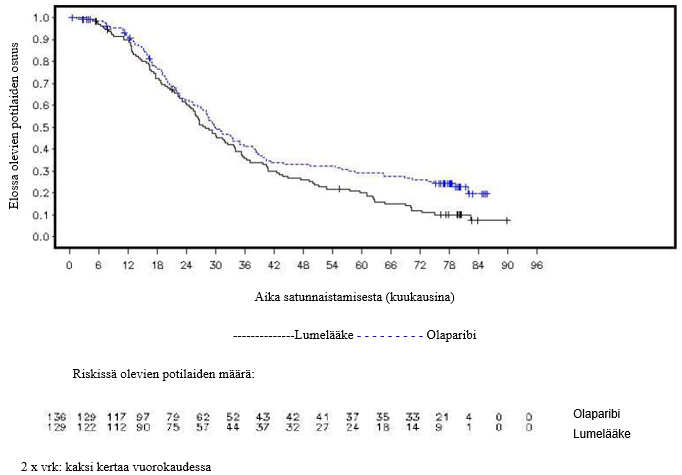
Kun etenemisvapaa elinaika analysoitiin, hoidon keston mediaani oli 8 kuukautta olaparibihaarassa ja 4 kuukautta lumelääkehaarassa. Suurin osa potilaista jatkoi aloitusannoksella olaparibia. Haittatapahtuman vuoksi lääkkeen käytön keskeytti 34,6 %, annosta pienennettiin 25,7 %:lla potilaista ja lääkkeen käytön lopetti 5,9 %. Lääkkeen käyttö keskeytettiin ja annosta pienennettiin useimmiten ensimmäisen kolmen hoitokuukauden aikana. Yleisimmät käytön keskeyttämiseen tai annoksen pienentämiseen johtaneet haittavaikutukset olivat pahoinvointi, anemia, oksentelu, neutropenia ja väsymys. Anemiaan liittyneiden haittavaikutusten ilmaantuvuus oli 22,8 % (7,4 % haittavaikutuksilla, joiden CTCAE-aste oli vähintään 3).
Potilaiden ilmoittamia hoitotuloksia koskevat tiedot eivät viittaa eroihin olaparibi- ja lumehoitoa saaneiden potilaiden välillä, mikä mitattiin sillä, miten paljon tutkimustulosindeksi (Trial Outcome Index, TOI) ja FACT-O-kokonaispistemäärä (Functional Analysis of Cancer Therapy–Ovarian total score) parani tai huononi.
OPINION-tutkimus
Olaparibin käyttöä ylläpitohoitona tutkittiin vaiheen IIIb yksihaaraisessa OPINION-monikeskustutkimuksessa uusiutunutta platinaherkkää munasarjasyöpää, munanjohdinsyöpää tai primaaria peritoneaalista syöpää sairastavilla potilailla, jotka olivat saaneet vähintään kaksi aiempaa linjaa platinapohjaista solunsalpaajahoitoa ja joilla ei ollut haitallista tai epäiltyä haitallista gBRCA-mutaatiota. Tutkimukseen otettiin mukaan potilaita, joilla oli hoitovaste (täydellinen tai osittainen) platinapohjaisen solunsalpaajahoidon päättymisen jälkeen. Yhteensä mukaan otettiin 279 potilasta, jotka saivat tässä tutkimuksessa olaparibihoitoa taudin etenemiseen asti tai kunnes ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä. Keskitetyn testauksen perusteella vahvistettiin, että 90,7 % potilaista ei kantanut gBRCA-mutaatiota ja lisäksi 9,7 %:lla potilaista tunnistettiin sBRCA-mutaatio.
Ensisijainen päätemuuttuja oli etenemisvapaa elinaika (PFS), jonka arvioi tutkijalääkäri käyttämällä muokattuja RECIST 1.1 ‑kriteerejä. Toissijaisiin päätemuuttujiin kuului kokonaiselinaika (OS).
Ylläpitohoitona käytetyllä olaparibilla osoitettiin olevan kliinistä vaikutusta potilailla, joiden uusiutuneessa platinaherkässä munasarjasyövässä ei todettu gBRCA-mutaatiota. Lopullisessa kokonaiselinajan analyysissa (viimeinen tiedonkeruupäivä 17.9.2021) kokonaiselinaikaa koskevien tulosten maturiteetti oli 52,3 %.
Yhteenveto ensisijaisen tavoitteen (etenemisvapaa elinaika, PFS) ja toissijaisen tavoitteen (kokonaiselinaika, OS) tuloksista OPINION-tutkimuksessa potilailla, joilla on uusiutunut platinaherkkä munasarjasyöpä, eikä gBRCA-mutaatiota, on esitetty taulukossa 8.
Taulukko 8 Yhteenveto keskeisten tavoitteiden tuloksista OPINION-tutkimuksessa: uusiutunutta platinaherkkää munasarjasyöpää sairastaneet potilaat, joilla ei todettu gBRCA-mutaatiota
| Olaparibitabletit, 300 mg kaksi kertaa vuorokaudessa | |
| PFS (maturiteetti 75 %) (viimeinen tiedonkeruupäivä 2.10.2020) | |
| Tapahtumien lukumäärä: potilaiden kokonaismäärä (%) | 210: 279 (75,3) |
| Mediaani PFS- (95 %:n luottamusväli) kuukausinaa | 9,2 (7,6, 10,9) |
| OS (maturiteetti 52,3 %) (viimeinen tiedonkeruupäivä 17.9.2021) | |
| Tapahtumien lukumäärä: potilaiden kokonaismäärä (%) | 146: 279 (52,3) |
| OS (95 %:n luottamusväli) kuukausina, mediaania | 32,7 (29,5, 35,3) |
aLaskettu Kaplan–Meier-menetelmällä.
PFS-ajan ja OS-ajan mediaanin luottamusvälit laskettiin Brookmeyer–Crowley-menetelmällä.
PFS = etenemisvapaa elinaika, OS = kokonaiselinaika.
HRD-positiivisen pitkälle edenneen munasarjasyövän ensilinjan ylläpitohoito
PAOLA-1-tutkimus
PAOLA-1 oli vaiheen III satunnaistettu, kaksoissokkoutettu, lumekontrolloitu monikeskustutkimus, jossa verrattiin Lynparza-valmisteen (300 mg [kaksi 150 mg :n tablettia] kaksi kertaa vuorokaudessa) ja samanaikaisesti annetun bevasitsumabihoidon (15 mg/kg kerta-annoksena 3 viikon välein infuusiona laskimoon) tehoa ja turvallisuutta lumelääkkeen ja bevasitsumabin yhdistelmään pitkälle edenneen (FIGO-levinneisyysasteet III ja IV) korkean pahanlaatuisuusasteen epiteelistä munasarja-, munanjohdin‑ tai primaaria peritoneaalista syöpää sairastavien aikuispotilaiden ylläpitohoidossa platinapohjaisen solunsalpaajahoidon ja bevasitsumabin yhdistelmällä annetun ensilinjan hoidon jälkeen. Bevasitsumabihoidon kokonaiskesto oli enintään 15 kuukautta/22 hoitojaksoa ja kattoi sekä solunsalpaajahoidon yhteydessä annetun hoidon että ylläpitohoitona annetun ajanjakson.
Tutkimukseen satunnaistettiin 806 potilasta (satunnaistamissuhde 2:1, 537 sai olaparibia ja bevasitsumabia ja 269 lumelääkettä ja bevasitsumabia), joilla ei ollut näyttöä (NED) taudista täydellisen kirurgisen poiston jälkeen tai joilla oli säilynyt täydellinen hoitovaste (CR) tai osittainen hoitovaste (PR) platinapohjaisen solunsalpaajahoidon ja bevasitsumabin yhdistelmällä annetun ensilinjan hoidon jälkeen. Potilaat olivat saaneet vähintään 4 ja enintään 9 hoitojaksoa; suurin osa (63 %) oli saanut 6 ensilinjan hoitojaksoa platina-taksaani-pohjaista solunsalpaajahoitoa ja vähintään 2 hoitojaksoa bevasitsumabia viimeisten kolmen solunsalpaajahoitojakson yhteydessä. Ennen satunnaistamista saatujen bevasitsumabihoitojaksojen määrän mediaani oli 5.
Potilaat stratifioitiin ensilinjan hoitotuloksen (sytoreduktiivisen leikkauksen ajoitus ja hoitotulos sekä vaste platinapohjaiseen solunsalpaajahoitoon) ja kasvaimen BRCA-mutaatiostatuksen (tBRCAm) mukaan, joka määritettiin prospektiivisen paikallisen testauksen perusteella. Potilaat jatkoivat bevasitsumabihoitoa ylläpitohoitona ja aloittivat Lynparza-hoidon vähintään 3 viikon ja enintään 9 viikon kuluttua viimeisimmän solunsalpaajahoitoannoksen saamisesta. Lynparza-hoitoa jatkettiin, kunnes perussairaus eteni tai ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä, tai enintään 2 vuoden ajan. Potilaat, jotka hoitavan lääkärin näkemyksen mukaan saattoivat edelleen hyötyä hoidon jatkamisesta, voivat saada hoitoa 2 vuoden jälkeenkin.
Demografiset tiedot ja lähtötilanteen tiedot olivat samankaltaiset molemmissa haaroissa hoitoaikeen mukaisessa populaatiossa (ITT-populaatiossa) ja biomarkkerien perusteella määritellyissä alaryhmissä, jotka muodostettiin tBRCA-mutaatiostatuksen (prospektiivisesti tai jälkikäteen määritetyn mutaatiostatuksen), GIS-statuksen ja HRD-statuksen (määriteltiin tässä tutkimuksessa molempien biomarkkerien yhdistelmäksi) perusteella. Potilaiden mediaani-ikä molemmissa haaroissa oli 61 vuotta, ja suurimmalla osalla (70 %:lla) ECOG-suorituskykypistemäärä oli 0. Potilaista 86 %:lla primaarikasvain oli munasarjasyöpä. Yleisin histologinen tyyppi oli seroosi (96 %), ja endometrioidia histologiaa ilmoitettiin 2 %:lla potilaista. Suurimmalla osalla potilaista (63 %:lla) oli todettu FIGO-levinneisyysaste IIIC. Kaikki potilaat olivat saaneet ensilinjan platinapohjaista hoitoa ja bevasitsumabia. Potilaita ei rajattu leikkauksen hoitotuloksen perusteella; 63 %:lla potilaista oli täydellinen sytoreduktio primaarileikkauksen tai intervallisytoreduktioleikkauksen jälkeen ja 37 %:lla oli makroskooppista jäännöstautia. Molemmissa haaroissa kolmellakymmenellä prosentilla (30 %) potilaista oli seulonnan yhteydessä todettu tBRCA-mutaatio. Biomarkkeri-alaryhmien demografiset tiedot ja lähtötilanteen tiedot olivat yhtenevät ITT-populaation demografisten tietojen ja lähtötilanteen tietojen kanssa. HRD-positiivisessa alaryhmässä 65 %:lla potilaista oli täydellinen sytoreduktio ja 35 %:lla tutkittavista oli makroskooppista jäännöstautia. Koko tutkimukseen otetussa populaatiossa 30 %:lla kummankin haaran potilaista oli seulonnan yhteydessä todettu paikallisessa testauksessa tBRCA-mutaatio (haitallinen tai patogeeninen mutaatio) ja 4 %:lla potilaista BRCA-mutaatiostatus ei ollut tiedossa. Saatavilla olevat kliiniset näytteet analysoitiin jälkikäteen 97 %:lta potilaista tBRCA-mutaatiostatuksen vahvistamiseksi ja genomin epävakautta kuvaavan pistemäärän selvittämiseksi edellä kuvatulla tavalla. Potilaista, joilla ei todettu tBRCA-mutaatiota, 29 %:lla (19 %:lla kokonaispopulaatiosta) oli positiivinen GIS-pistemäärä, joka oli tässä tutkimuksessa etukäteen määritelty siten, että yhdistetty pistemäärä oli vähintään 42. Kun tBRCA-mutaatiostatus ja positiivinen GIS-pistemäärä yhdistettiin, kokonaispopulaatiossa 48 %:lla potilaista kasvaimen HRD-status oli positiivinen, 34 %:lla negatiivinen ja 18 %:lla tuntematon.
Ensisijainen päätemuuttuja oli etenemisvapaa elinaika (PFS), joka määriteltiin aikana satunnaistamisesta tutkijalääkärin arvioimaan taudin etenemiseen muokattujen RECIST 1.1 ‑kriteerien (Response Evaluation Criteria in Solid Tumors) perusteella tai kuolemaan. Toissijaisia tehoa mittaavia päätemuuttujia olivat aika satunnaistamisesta taudin toiseen etenemiseen tai kuolemaan (PFS2), kokonaiselinaika (OS), aika satunnaistamisesta ensimmäiseen seuraavaan syöpähoitoon tai kuolemaan (TFST) ja terveyteen liittyvä elämänlaatu (HRQoL). Potilaiden kasvaimet arvioitiin RECIST 1.1 ‑kriteerien mukaan lähtötilanteessa ja 24 viikon välein (tietokonetomografia tai magneettikuvaus 12 viikon kohdalla, jos kliinisesti tai CA-125-arvojen perusteella todettu taudin eteneminen) enintään 42 kuukauden ajan tai kunnes objektiivisessa radiologisessa tutkimuksessa oli todettu taudin edenneen.
Tutkimuksessa saavutettiin sen ensisijainen päätemuuttuja ITT-populaatiossa, mikä osoitti tilastollisesti merkitsevää paranemista tutkijalääkärin arvioiman etenemisvapaan elinajan suhteen olaparibia ja bevasitsumabia saaneilla potilailla verrattuna lumelääkettä ja bevasitsumabia saaneisiin potilaisiin (HR 0,59, 95 %:n luottamusväli 0,49–0,72, p < 0,0001; yhdistetyllä olaparibi- ja bevasitsumabihoidolla mediaani 22,1 kuukautta ja yhdistetyllä lumelääke- ja bevasitsumabihoidolla 16,6 kuukautta). Tämä oli yhdenmukainen BICR:n tekemän PSF- analyysin kanssa. Suurimman osan hyödystä saivat kuitenkin biomarkkeripositiivisiksi määritellyt potilaat (tBRCA-mutaatio, GIS, positiivinen HRD-status, joka määriteltiin todetun tBRCA-mutaation ja/tai GIS-positiivisuuden perusteella).
Kokonaispopulaatiossa tehty lopullinen PFS2-analyysi (viimeinen tiedonkeruupäivä 22.3.2020, maturiteetti 53 %), oli tilastollisesti merkitsevä (HR = 0,78, 95 %:n luottamusväli 0,64–0,95, p = 0,0125, mediaani olaparibia ja bevasitsumabia saaneilla oli 36,5 kuukautta ja lumelääkettä ja bevasitsumabia saaneilla 32,6 kuukautta).
Lopullisessa kokonaiselinajan analyysissa (viimeinen tiedonkeruupäivä 22.3.2022) potilailla, joiden HRD-status oli positiivinen (tBRCA-mutaatio ja/tai GIS-positiivisuus), kokonaiselinaika oli numeerisesti pidempi olaparibia ja bevasitsumabia saaneiden haarassa verrattuna lumelääkettä ja bevasitsumabia saaneiden haaraan (ks. taulukko 9).
tBRCA-mutaatiostatuksen perusteella satunnaistettujen potilaiden alaryhmässä (241/806 potilasta) etenemisvapaan elinajan mediaani olaparibia ja bevasitsumabia saaneiden haarassa oli 37,2 kuukautta ja vastaavasti lumelääkettä ja bevasitsumabia saaneiden haarassa 22,0 kuukautta (HR = 0,34, 95 %:n luottamusväli 0,23, 0,51). Kun lopullisessa kokonaiselinajan analyysissa (viimeinen tiedonkeruupäivä 22.3.2022) tarkastellaan tBRCA-mutaatiostatuksen perusteella satunnaistettujen potilaiden alaryhmää, havaitaan kuoleman riskin olevan numeerisesti pienempi olaparibia ja bevasitsumabia saaneiden haarassa verrattuna lumelääkettä ja bevasitsumabia saaneiden haaraan (HR 0,63; 95 %:n luottamusväli 0,41, 0,97).
Tehoa koskevat tulokset muissa jälkikäteen analysoituihin kasvainnäytteisiin perustuneissa biomarkkeri-alaryhmäanalyyseissä on esitetty taulukossa 9.
Taulukko 9 Yhteenveto PAOLA-1-tutkimuksen keskeisistä tehoa koskevista löydöksistä pitkälle edennyttä munasarjasyöpää sairastavilla potilailla, joilla oli tBRCA-mutaation ja/tai GIS-pistemäärän perusteella määritelty positiivinen HRD-status (puutteellinen homologinen rekombinaatio)
tBRCA-mutaatio*, c (n = 235) | GIS-positiivinen (n = 152) | HRD-positiivinen (n = 387) | ||||
| Olaparibi ja bevasitsu-mabi | Lumelääke ja bevasitsu-mabi | Olaparibi ja bevasitsu-mabi | Lumelääke ja bevasitsu-mabi | Olaparibi ja bevasitsu-mabi | Lumelääke ja bevasitsumabi | |
| PFS, tutkijan arvio (maturiteetti 46 %), viimeinen tiedonkeruupäivä 22.3.2019a | ||||||
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 44:158 (28) | 52:77 (68) | 43:97 (44) | 40:55 (73) | 87:255 (34) | 92:132 (70) |
| Mediaaniaika (kuukausina) | 37,2 | 18,8 | 28,1 | 16,6 | 37,2 | 17,7 |
| HR (95 %:n luottamusväli)b | 0,28 (0,19, 0,42) | 0,43 (0,28, 0,66) | 0,33 (0,25, 0,45) | |||
| PFS2, tutkijan arvio (maturiteetti 40 %), viimeinen tiedonkeruupäivä 22.3.2020 | ||||||
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 44:158 (28) | 37:77 (48) | 41:97 (42) | 33:55 (60) | 85:255 (33) | 70:132 (53) |
| Mediaaniaika (kuukausina) | NR | 42,2 | 50,3 | 30,1 | 50,3 | 35,4 |
| HR (95 %:n luottamusväli)b | 0,53 (0,34, 0,82) | 0,60 (0,38, 0,96) | 0,56 (0,41, 0,77) | |||
| OS: lopullinen analyysi (maturiteetti 42 %), viimeinen tiedonkeruupäivä 22.3.2022 | ||||||
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 49:158 (31,0) | 37:77 (48,1) | 44:97 (45,4) | 32:55 (58,2) | 93:255 (36,5) | 69:132 (52,3) |
| Mediaaniaika (kuukausina) | 75,2 | 66,9 | NR | 52,0 | 75,2 | 57,3 |
| HR (95 %:n luottamusväli)b | 0,57 (0,37, 0,88) | 0,71 (0,45, 1,13) | 0,62 (0,45, 0,85) | |||
* Ennalta suunniteltu alaryhmä
a Kaplan-Meier-estimaattien perusteella niiden potilaiden osuus, joilla tauti ei ollut edennyt, oli olaparibia ja bevasitsumabia saaneiden ryhmässä 12 kuukauden kohdalla 89 % ja 24 kuukauden kohdalla 66 % ja lumelääkettä ja bevasitsumabia saaneiden ryhmässä 12 kuukauden kohdalla 71 % ja 24 kuukauden kohdalla 29 %.
b Arvo, joka on < 1, suosii olaparibia. Analyysi tehtiin käyttämällä Coxin suhteellisen hasardin mallia, joka oli stratifioitu seulonnan kohdalla todetun ensilinjan hoitovasteen ja seulonnan yhteydessä tehdyllä laboratoriotutkimuksella varmistetun tBRCA-statuksen mukaan.
c Myriad-yrityksen määrittämä tBRCA-mutaatiostatus
d HRD-positiivinen, tBRCA-mutaatio pois lukien: määritelmänä oli Myriad-yrityksen määrittämä genomin epävakautta kuvaava pistemäärä (GIS) ≥ 42 (ennakkoon määritelty katkaisupiste)
CI = Luottamusväli; HR = Riskisuhde; NR: Ei saavutettu
Kuva 7 PAOLA-1: Kaplan–Meier-käyrä etenemisvapaalle elinajalle pitkälle edennyttä munasarjasyöpää sairastavilla potilailla (jotka määriteltiin HRD-positiivisiksi PAOLA-1-tutkimuksessa) (maturiteetti 46 %, tutkijan arvio)
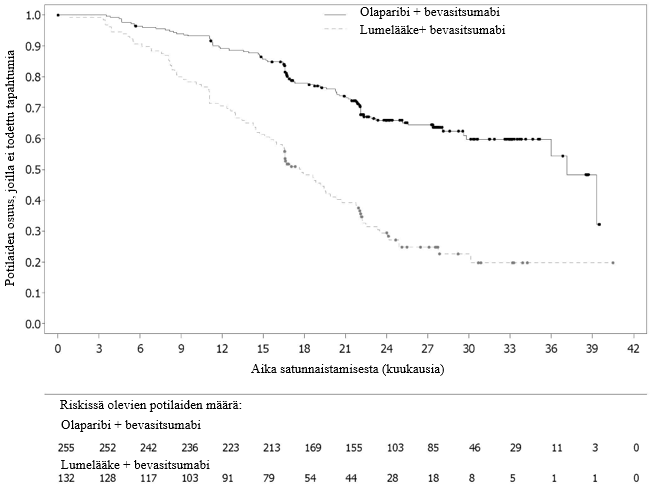
Kuva 8 PAOLA-1: Kaplan–Meier-käyrä lopulliselle kokonaiselinajalle positiivisen HRD-statuksen mukaan (tBRCA-mutaatio mukaan lukien) (viimeinen tiedonkeruupäivä 22.3.2022)
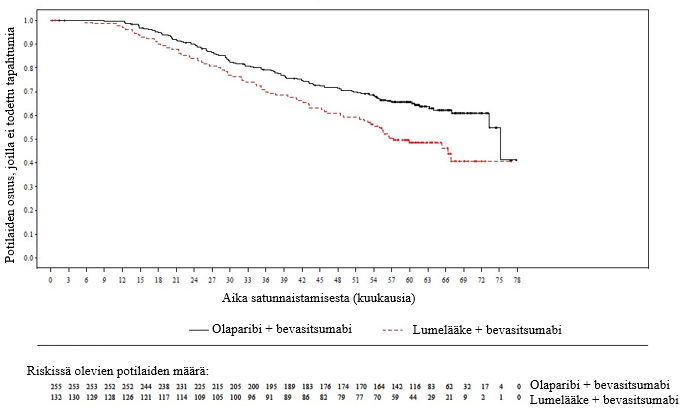
Ituradan BRCA-mutaation omaavan korkean riskin varhaisvaiheen rintasyövän liitännäishoito
OlympiA
Olaparibiliitännäishoidon turvallisuutta ja tehoa tutkittiin potilailla, joilla oli ituradan BRCA1/2-mutaatioita ja HER2-negatiivinen korkean riskin varhaisvaiheen rintasyöpä ja jotka olivat saaneet definitiivistä paikallishoitoa sekä esiliitännäis- tai liitännäissolunsalpaajahoitoa. Kyseessä oli vaiheen III satunnaistettu, kaksoissokkoutettu, rinnakkaisryhmillä toteutettu, lumekontrolloitu monikeskustutkimus (OlympiA). Potilaiden edellytettiin jatkaneen loppuun asti vähintään 6 hoitojaksoa esiliitännäis- tai liitännäissolunsalpaajahoitoa, sisältäen antrasykliiniä, taksaania tai molempia. Aikaisemmin annettu platinahoito aiemman syövän (esimerkiksi munasarjasyövän) hoitoon tai rintasyövän liitännäis- tai esiliitännäishoitoon oli sallittu. Korkean riskin varhaisvaiheen rintasyöpäpotilaat määriteltiin seuraavasti:
- esiliitännäishoitona solunsalpaajia saaneet potilaat: potilailla, joilla oli joko kolmoisnegatiivinen rintasyöpä tai hormonireseptoripositiivinen rintasyöpä, oli täytynyt olla leikkaushetkellä invasiivinen jäännöstauti rinnassa ja/tai poistetuissa imusolmukkeissa (ei täydellistä patologista vastetta). Lisäksi potilailla, joilla oli hormonireseptoripositiivinen rintasyöpä, CPS&EG-pistemäärän täytyi olla vähintään 3. Pisteytys perustui ennen hoidon aloittamista määritettyyn kliiniseen levinneisyysasteeseen ja hoidon jälkeen määritettyyn patologiseen levinneisyysasteeseen (CPS), estrogeenireseptoristatukseen (ER-statukseen) sekä histologiseen erilaistumisasteeseen taulukossa 10 esitetyllä tavalla.
Taulukko 10 Varhaisvaiheen rintasyövän levinneisyysasteen, reseptoristatuksen ja erilaistumisasteen pisteytystä koskevat vaatimukset tutkimuksen sisäänottoa varten*
| Aste/ominaisuus | Pisteet | |
|---|---|---|
Kliininen levinneisyysaste (ennen hoidon aloittamista) | I/IIA | 0 |
| IIB/IIIA | 1 | |
| IIIB/IIIC | 2 | |
| Patologinen levinneisyysaste (hoidon jälkeen) | 0/I | 0 |
| IIA/IIB/IIIA/IIIB | 1 | |
| IIIC | 2 | |
| Reseptoristatus | ER-positiivinen | 0 |
| ER-negatiivinen | 1 | |
| Tumagradus | Tumagradus 1–2 | 0 |
| Tumagradus 3 | 1 | |
* Potilaiden, joilla oli hormonireseptoripositiivinen rintasyöpä, oli saatava yhteensä vähintään 3 pistettä.
- liitännäishoitona solunsalpaajia saaneet potilaat: potilailla, joilla oli kolmoisnegatiivinen rintasyöpä, oli täytynyt olla joko imusolmukepositiivinen tauti tai imusolmukenegatiivinen tauti, jossa primaarikasvaimen koko oli vähintään 2 cm; potilailla, joilla oli HR-positiivinen, HER2-negatiivinen tauti, oli täytynyt olla vähintään 4 patologisesti positiiviseksi vahvistettua imusolmuketta.
Potilaat satunnaistettiin (1:1) saamaan joko 300 mg olaparibia (kaksi 150 mg:n tablettia) kaksi kertaa vuorokaudessa (n = 921) tai lumelääkettä (n = 915). Satunnaistaminen stratifioitiin hormonireseptoristatuksen (HR-positiivinen/HER2-negatiivinen vs. kolmoisnegatiivinen), aiemman esiliitännäis- vs. liitännäissolunsalpaahoidon ja nykyisen rintasyövän hoitoon annetun aiemman platinahoidon (annettu vs. ei annettu) mukaan. Hoitoa jatkettiin vähintään 1 vuoden ajan tai taudin uusiutumiseen asti tai kunnes ilmeni haittavaikutuksia, joita ei voitu hyväksyä. Potilaat, joilla oli HR-positiivisia kasvaimia, saivat myös hormonaalista hoitoa.
Ensisijainen päätemuuttuja oli tautivapaa elossaolo invasiivisen syövän suhteen (invasive disease-free survival, IDFS), joka määriteltiin ajaksi satunnaistamisesta päivään, jolloin tauti uusiutui ensimmäisen kerran. Uusiutumiseksi katsottiin invasiivinen lokoregionaalinen syöpä, etäpesäkkeinen uusiutuminen, kontralateraalinen invasiivinen rintasyöpä, uusi syöpä tai mistä tahansa syystä johtuva kuolema. Toissijaisia tavoitteita olivat kokonaiselinaika (OS), tautivapaa elossaolo etäpesäkkeisen syövän suhteen (distant disease-free survival, DDFS; määriteltiin ajaksi satunnaistamisesta siihen, että saatiin näyttöä rintasyövän ensimmäisestä etäpesäkkeisestä uusiutumisesta), uusien primaarien kontralateraalisten rintasyöpien (invasiiviset ja ei-invasiiviset), uuden primaarin munasarjasyövän, uuden primaarin munanjohdinsyövän ja uuden primaarin peritoneaalisyövän ilmaantuvuus sekä potilaiden ilmoittamia hoitotuloksia koskevat tiedot, jotka perustuivat FACIT-Fatigue- ja EORTC QLQ-C30-kyselyihin.
Soveltuvuus tutkimukseen määritettiin keskitetysti Myriad-yrityksen tekemällä testillä tai paikallisesti tehdyllä gBRCA-testillä, jos sellainen oli saatavilla. Potilaat, jotka otettiin tutkimukseen paikallisen gBRCA-testin tuloksen perusteella, antoivat näytteen jälkikäteen tehtävää vahvistustestiä varten. OlympiA-tutkimukseen otetuista 1 836 potilaasta 1 623:lla vahvistettiin gBRCA-mutaatio keskitetyllä testauksella joko prospektiivisesti tai retrospektiivisesti.
Demografiset tiedot ja lähtötilanteen tiedot olivat hyvin samankaltaiset molemmissa hoitohaaroissa. Tutkittavien mediaani-ikä oli 42 vuotta. 67 % potilaista oli valkoihoisia, 29 % aasialaisia ja 2,6 % mustaihoisia. Olaparibihaaran potilaista kaksi (0,2 %) ja lumehaaran potilaista neljä (0,4 %) oli miehiä. 61 % potilaista oli premenopausaalisia. 89 %:lla potilaista ECOG-suorituskykypistemäärä oli 0 ja 11 %:lla ECOG-suorituskykypistemäärä oli 1. Potilaista 82 %:lla oli kolmoisnegatiivinen rintasyöpä ja 18 %:lla HR-positiivinen tauti. 50 % potilaista oli aiemmin saanut solunsalpaajia esiliitännäishoitona ja 50 % liitännäishoitona. 94 % potilaista oli saanut antrasykliiniä ja taksaania. 26 % kaikista potilaista oli saanut aiemmin rintasyöpään platinahoitoa. Olaparibihaarassa 87 % ja lumelääkehaarassa 92 % HR-positiivista tautia sairastavista potilaista sai samanaikaisesti hormonaalista hoitoa. Kaikkiaan 89,5 % HR-positiivista tautia sairastavista potilaista sai hormonaalista hoitoa, mukaan lukien letrotsolia (23,7 %), tamoksifeenia (40,9 %), anastrotsolia (17,2 %) tai eksemestaania (14,8 %).
Tutkimuksessa saavutettiin sen ensisijainen päätemuuttuja, joka osoitti tilastollisesti merkitsevän paranemisen IDFS:n suhteen olaparibihaarassa verrattuna lumehaaraan. IDFS-tapahtumia ilmeni 284 potilaalla, mikä vastasi 12 %:a olaparibihaaran potilaista (etäpesäkkeinen 8 %:lla, paikallinen/alueellinen 1,4 %:lla, kontralateraalinen invasiivinen rintasyöpä 0,9 %:lla, toinen primaari maligniteetti muualla kuin rinnassa 1,2 %:lla ja kuolema 0,2 %:lla) ja 20 %:a lumehaaran potilaista (etäpesäkkeinen 13 %:lla, paikallinen/alueellinen 2,7 %:lla, kontralateraalinen invasiivinen rintasyöpä 1,3 %:lla, toinen primaari maligniteetti muualla kuin rinnassa 2,3 %:lla ja kuolema 0 %:lla). Tilastollisesti merkitsevä paraneminen havaittiin myös DDFS:n suhteen olaparibihaarassa verrattuna lumehaaraan. Kokonaiselinajan (OS) primaarianalyysin (maturiteetti 10 %, viimeinen tiedonkeruupäivä 12.7.2021) kohdalla havaittiin kokonaiselinajan tilastollisesti merkitsevä paraneminen olaparibihaarassa lumehaaraan verrattuna (HR = 0,68 [98,5 %:n luottamusväli 0,47, 0,97], p‑arvo = 0,0091). Noin viisi vuotta viimeisen potilaan satunnaistamisen jälkeen tehdyssä ennalta määritellyssä analyysissä, jonka kohdalla seuranta-ajan mediaani oli olaparibihaarassa 6,2 vuotta ja lumelääkehaarassa 6,1 vuotta, kokonaiselinaika oli edelleen pidempi olaparibihaarassa kuin lumelääkehaarassa. Tehoa koskevat tulokset täydellisestä analyysisarjasta on esitetty taulukossa 11 ja kuvissa 9 ja 10.
Taulukko 11 OlympiA-tutkimuksen tehoa koskevat tulokset liitännäishoitoa saaneilla varhaisvaiheen rintasyöpää sairastavilla potilailla, joilla on ituradan BRCA-mutaatio
| Olaparibi 300 mg kaksi kertaa vuorokaudessa (N = 921) | Lumelääke (N = 915) | ||
| IDFS (maturiteetti 15 %) – viimeinen tiedonkeruupäivä 27.3.2020 | |||
| Tapahtumien lukumäärä: potilaiden kokonaismäärä (%) | 106:921 (12) | 178:915 (20) | |
| HR (99,5 %:n luottamusväli)a | 0,58 (0,41, 0,82) | ||
| P-arvo (kaksitahoinen)b | 0,0000073 | ||
| Niiden potilaiden prosenttiosuus (95 %:n luottamusväli), joilla ei ollut invasiivista tautia 3 vuoden kohdallac | 86 (83, 88) | 77 (74, 80) | |
| DDFS (maturiteetti 13 %) – viimeinen tiedonkeruupäivä 27.3.2020 | |||
| Tapahtumien lukumäärä: potilaiden kokonaismäärä (%) | 89:921 (10) | 152:915 (17) | |
| HR (99,5 %:n luottamusväli)a | 0,57 (0,39, 0,83) | ||
| P-arvo (kaksitahoinen)b | 0,0000257 | ||
| Niiden potilaiden prosenttiosuus (95 %:n luottamusväli), joilla ei ollut etäpesäkkeistä tautia 3 vuoden kohdallac | 88 (85, 90) | 80 (77, 83) | |
| Kokonaiselinaika (maturiteetti 14 %) – viimeinen tiedonkeruupäivä 5.6.2024 [viiden vuoden seuranta-analyysi] | |||
| Tapahtumien lukumäärä: potilaiden kokonaismäärä (%) | 107:921 (12) | 143:915 (16) | |
| HR (95 %:n luottamusväli)a | 0,72 (0,56, 0,93) | ||
| Elossa olevien potilaiden prosenttiosuus (95 %:n luottamusväli) 3 vuoden kohdallac Elossa olevien potilaiden prosenttiosuus (95 %:n luottamusväli) 4 vuoden kohdallac Elossa olevien potilaiden prosenttiosuus (95 %:n luottamusväli) 5 vuoden kohdallac Elossa olevien potilaiden prosenttiosuus (95 %:n luottamusväli) 6 vuoden kohdallac |
|
| |
a Perustuu ositettuun Coxin suhteellisen hasardin malliin; < 1 tarkoittaa, että riski oli pienempi olaparibihaarassa kuin lumehaarassa.
b p-arvo perustuu ositettuun log rank -testiin.
c Prosenttiosuudet on laskettu Kaplan–Meier-estimaattien perusteella.
DDFS = tautivapaa elossaolo etäpesäkkeisen syövän suhteen (distant disease-free survival); IDFS = tautivapaa elossaolo invasiivisen syövän suhteen (invasive disease-free survival), HR = riskisuhde (hazard ratio)
Kuva 9 OlympiA: Kaplan–Meier-käyrä tautivapaalle elossaololle invasiivisen syövän suhteen (IDFS) liitännäishoitoa saaneilla korkean riskin varhaisvaiheen rintasyöpää sairastavilla potilailla, joilla on ituradan BRCA-mutaatio (viimeinen tiedonkeruupäivä 27.3.2020)
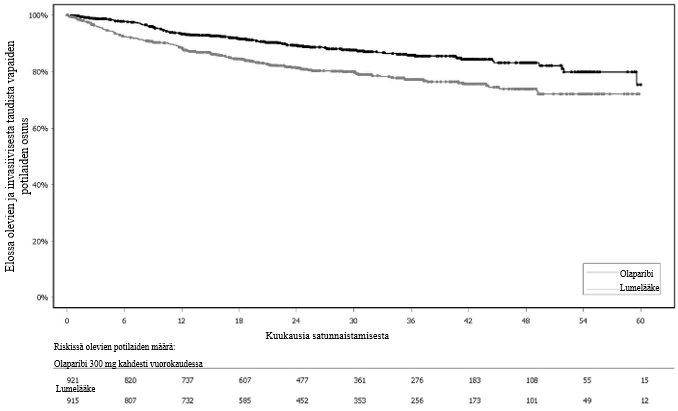
Kuva 10 OlympiA: Kaplan–Meier-käyrä kokonaiselinajalle liitännäishoitoa saaneilla korkean riskin varhaisvaiheen rintasyöpää sairastavilla potilailla, joilla on ituradan BRCA-mutaatio (viimeinen tiedonkeruupäivä 5.6.2024)
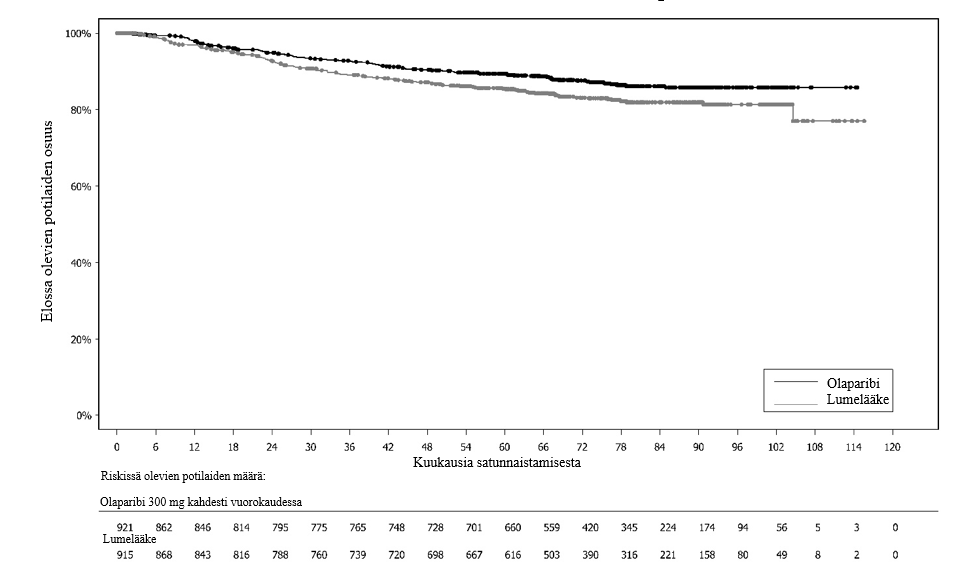
gBRCA1/2-mutaation omaava HER2-negatiivinen metastasoitunut rintasyöpä
OlympiAD (tutkimus D0819C00003)
Olaparibin turvallisuutta ja tehoa potilailla, joilla on gBRCA1/2-mutaatioita ja HER2-negatiivinen metastasoitunut rintasyöpä, tutkittiin vaiheen III satunnaistetussa, avoimessa, kontrolloidussa tutkimuksessa (OlympiAD). Tässä tutkimuksessa 302 potilasta, joilla oli todettu haitallinen tai epäilty haitallinen gBRCA-mutaatio, satunnaistettiin suhteessa 2:1 saamaan joko Lynparza-valmistetta (300 mg [kaksi 150 mg:n tablettia] 2 kertaa vuorokaudessa) tai lääkärin valitsemaa solunsalpaajahoitoa (kapesitabiinia, 42 %, eribuliinia, 35 %, tai vinorelbiinia, 17 %), kunnes tauti eteni tai ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä. Potilaat, joilla oli BRCA1/2-mutaatioita, tunnistettiin verinäytteestä tehtävällä ituratatutkimuksella käyttämällä paikallista testiä tai keskitetysti Myriad-yrityksen tekemällä testillä. Potilaat stratifioitiin seuraavien tekijöiden perusteella: aiempien solunsalpaajahoitojen saaminen metastasoituneen rintasyövän hoitoon (kyllä/ei), hormonireseptoripositiivinen (HR-positiivinen) vai kolmoisnegatiivinen, aiempi rintasyövän platinahoito (kyllä/ei). Ensisijainen päätemuuttuja oli PFS, jonka arvioi sokkoutettu riippumaton keskitetty arvioijataho (BICR) RECIST 1.1 -kriteerien mukaan. Toissijaisia päätemuuttujia olivat PFS2, kokonaiselinaika, objektiivinen hoitovaste (ORR) ja HRQoL.
Potilaiden oli täytynyt saada antrasykliinia, ellei se ollut vasta-aiheista, ja taksaania joko (esi)liitännäishoitona tai metastasoituneen syövän vuoksi. Jos potilaalla oli HR-positiivinen (ER- ja/tai PgR-positiivinen) kasvain, edellytettiin, että potilas oli saanut ainakin yhtä hormonihoitoa (liitännäishoitona tai metastasoituneen syövän hoitoon) ja tauti oli edennyt hormonihoidosta huolimatta, tai että tauti oli hoitavan lääkärin mielestä hormonihoitoon soveltumaton. Aiempi platinahoito oli sallittua metastasoituneen syövän hoidossa edellyttäen, ettei taudin etenemisestä platinahoidon aikana ollut näyttöä, ja (esi)liitännäishoitona edellyttäen, että viimeinen annos oli saatu vähintään 12 kuukautta ennen satunnaistamista. Aiempi hoito PARP:n estäjällä, mukaan lukien olaparibilla, ei ollut sallittua.
Demografiset tiedot ja lähtötilanteen tiedot olivat yleisesti hyvin samankaltaiset olaparibi- ja vertailuhaaroissa (ks. taulukko 12).
Taulukko 12 Potilaiden demografiset ja lähtötilanteen ominaispiirteet OlympiAD-tutkimuksessa
Olaparibi 300 mg kaksi kertaa vuorokaudessa n = 205 | Solunsalpaaja n = 97 | |
| Ikä vuosina (mediaani) | 44 | 45 |
| Sukupuoli (%) | ||
| Nainen | 200 (98) | 95 (98) |
| Mies | 5 (2) | 2 (2) |
| Rotu (%) | ||
| Valkoihoinen | 134 (65) | 63 (65) |
| Aasialainen | 66 (32) | 28 (29) |
| Muu | 5 (2) | 6 (6) |
| ECOG-suorituskykypistemäärä (%) | ||
| 0 | 148 (72) | 62 (64) |
| 1 | 57 (28) | 35 (36) |
| Taudin yleinen luokitus | ||
| Metastasoitunut | 205 (100) | 97 (100) |
| Paikallisesti edennyt | 0 | 0 |
| Uusi metastasoitunut rintasyöpä (%) | 26 (13) | 12 (12) |
| Hormonireseptoristatus (%) | ||
| HR+ | 103 (50) | 49 (51) |
| Kolmoisnegatiivinen rintasyöpä | 102 (50) | 48 (49) |
| gBRCA-mutaation tyyppi (%) | ||
| gBRCA1 | 117 (57) | 51 (53) |
| gBRCA2 | 84 (41) | 46 (47) |
| gBRCA1 ja gBRCA2 | 4 (2) | 0 |
| ≥ 2 etäpesäkkeiden sijaintia (%) | 159 (78) | 72 (74) |
| Etäpesäkkeen sijainti (%) | ||
| Vain luusto | 16 (8) | 6 (6) |
| Muu | 189 (92) | 91 (94) |
| BICR:n mukaan mitattavissa oleva sairaus (%) | 167 (81) | 66 (68) |
| Edennyt tauti satunnaistamishetkellä (%) | 159 (78) | 73 (75) |
| Kasvaimen erilaistumisaste diagnosointihetkellä | ||
| Hyvin erilaistunut (G1) | 5 (2) | 2 (2) |
| Kohtalaisesti erilaistunut (G2) | 52 (25) | 23 (24) |
| Heikosti erilaistunut (G3) | 108 (53) | 55 (57) |
| Erilaistumaton (G4) | 4 (2) | 0 |
| Erilaistumisastetta ei voida arvioida (GX) | 27 (13) | 15 (16) |
| Puuttuu | 9 (4) | 2 (2) |
| Metastasoituneen rintasyövän hoitoon annettujen aiempien linjojen solunsalpaajahoitojen määrä (%) | ||
| 0 | 68 (33) | 31 (32) |
| 1 | 80 (39) | 42 (43) |
| 2 | 57 (28) | 24 (25) |
| Aiempi platinapohjainen hoito (%) | 55 (27) | 21 (22) |
| vain (esi)liitännäishoitona | 12 (6) | 6 (6) |
| vain metastasoituneen taudin hoidossa | 40 (20) | 14 (14) |
| (esi)liitännäishoitona ja metastasoituneen taudin hoidossa | 3 (1) | 1 (1) |
| Aiempi antrasykliinihoito | ||
| (esi)liitännäishoitona | 169 (82) | 76 (78) |
| metastasoituneen taudin hoidossa | 41 (20) | 16 (17) |
| Aiempi taksaanihoito | ||
| (esi)liitännäishoitona | 146 (71) | 66 (68) |
| metastasoituneen taudin hoidossa | 107 (52) | 41 (42) |
| Aiempi antrasykliini- ja taksaanihoito | 204 (99,5) | 96 (99) |
Hoitohaarassa 0,5 % ja vertailuhaarassa 8 % potilaista sai seuraavaksi PARP:n estäjää, ja hoitohaarassa 29 % ja vertailuhaarassa 42 % potilaista sai seuraavaksi platinahoitoa.
Tehon ensisijaiselle päätemuuttujalle PFS:lle osoitettiin tilastollisesti merkitsevä paraneminen olaparibia saaneilla potilailla vertailuhaaran potilaisiin verrattuna (ks. taulukko 13 ja kuva 11).
Taulukko 13 Yhteenveto OlympiAD-tutkimuksen keskeisistä tehoa koskevista löydöksistä HER2-negatiivista metastasoitunutta rintasyöpää sairastavilla potilailla, joilla on gBRCA1/2-mutaatio
| Olaparibi 300 mg kaksi kertaa vuorokaudessa | Solunsalpaaja | |
| PFS (maturiteetti 77 %) – viimeinen tiedonkeruupäivä 9.12.2016 | ||
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 163:205 (80) | 71:97 (73) |
| Mediaaniaika (kuukausina) (95 %:n luottamusväli) | 7,0 (5,7–8,3) | 4,2 (2,8–4,3) |
| HR (95 %:n luottamusväli) | 0,58 (0,43–0,80) | |
| p-arvo (kaksitahoinen)a | p = 0,0009 | |
| PFS2 (maturiteetti 65 %) – viimeinen tiedonkeruupäivä 25.9.2017b | ||
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 130:205 (63) | 65:97 (67) |
| Mediaaniaika (kuukausina) (95 %:n luottamusväli) | 12,8 (10,9–14,3) | 9,4 (7,4–10,3) |
| HR (95 %:n luottamusväli) | 0,55 (0,39–0,77) | |
| p-arvo (kaksitahoinen)a | p = 0,0005 | |
| Kokonaiselinaika (maturiteetti 64 %) – viimeinen tiedonkeruupäivä 25.9.2017 | ||
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 130:205 (63) | 62:97 (64) |
| Mediaaniaika (kuukausina) (95 %:n luottamusväli) | 19,3 (17,2–21,6)c | 17,1 (13,9–21,9) |
| HR (95 %:n luottamusväli) | 0,90 (0,66–1,23) | |
| p-arvo (kaksitahoinen)a | p = 0,5131 | |
| Vahvistettu objektiivinen hoitovaste – viimeinen tiedonkeruupäivä 9.12.2016 | ||
| Objektiivisen hoitovasteen saavuttaneiden määrä: niiden potilaiden kokonaismäärä, joiden sairaus oli mitattavissa (%) | 87:167 (52)d | 15:66 (23) |
| 95 %:n luottamusväli | 44,2–59,9 | 13,3–35,7 |
| Vasteen kesto – viimeinen tiedonkeruupäivä 9.12.2016 | ||
| Mediaani, kuukausina (95 %:n luottamusväli) | 6,9 (4,2, 10,2) | 7,9 (4,5, 12,2) |
a Ositetun log-rank-testin perusteella.
b Post-hoc-analyysi.
c Pois rajattujen potilaiden seuranta-ajan mediaani oli olaparibiryhmässä 25,3 kuukautta ja vertailuryhmässä 26,3 kuukautta.
d Vahvistettu hoitovaste (BICR:n mukaan) määriteltiin kirjatuksi CR- tai PR-vasteeksi, joka on vahvistettu toistetulla kuvantamistutkimuksella vähintään 4 viikkoa sen käynnin jälkeen, jolla vaste ensin havaittiin. Olaparibihaarassa 8 % potilaista, joilla oli mitattavissa oleva sairaus, saavutti täydellisen hoitovasteen, vertailuhaarassa taas 1,5 %; 74/167 (44 %) olaparibihaaran potilaista ja 14/66 (21 %) solunsalpaajahoitohaaran potilaista saavutti osittaisen hoitovasteen. Kolmoisnegatiivista rintasyöpää sairastavien potilaiden alaryhmässä vahvistettu objektiivisen hoitovasteen saavuttaneiden osuus oli 48 % (41/86) olaparibihaarassa ja 12 % (4/33) vertailuhaarassa. HR+-potilaiden alaryhmässä vahvistettu objektiivisen hoitovasteen saavuttaneiden osuus oli 57 % (46/81) olaparibihaarassa ja 33 % (11/33) vertailuhaarassa.
HR: riskisuhde; HR+: hormonireseptoripositiivinen; PFS: etenemisvapaa elinaika; PFS2: aika toiseen etenemiseen tai kuolemaan
Kuva 11 OlympiAD: Kaplan-Meier-käyrä BICR:n arvioimalle etenemisvapaalle elinajalle HER2-negatiivista metastasoitunutta rintasyöpää sairastavilla potilailla, joilla on gBRCA1/2-mutaatio (maturiteetti 77 %), viimeinen tiedonkeruupäivä 9.12.2016
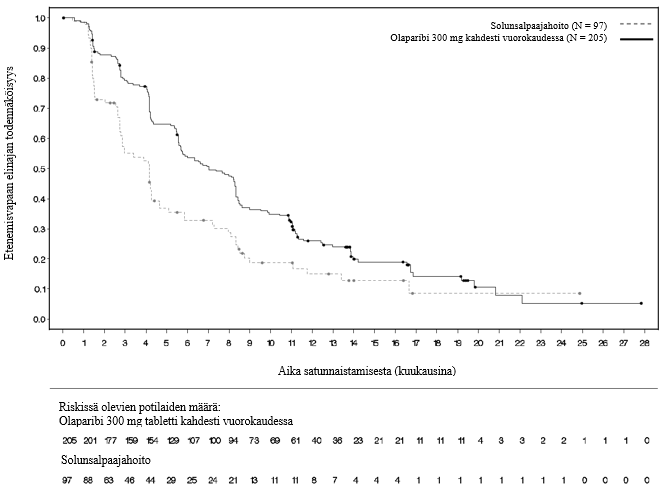
Kaikissa ennalta määritellyissä potilaiden alaryhmissä havaittiin yhdenmukaisia tuloksia (ks. kuva 12). Alaryhmäanalyysi viittasi olaparibin PFS-hyötyyn vertailuvalmisteeseen verrattuna kolmoisnegatiivisen rintasyövän (riskisuhde 0,43; 95 %:n luottamusväli 0,29–0,63, n = 152) ja hormonireseptoripositiivisen (HR+) rintasyövän (riskisuhde 0,82; 95 %:n luottamusväli 0,55–1,26, n = 150) alaryhmissä.
Kuva 12 PFS (BICR), Forest-kuvaaja ennalta määritettyjen alaryhmien mukaan
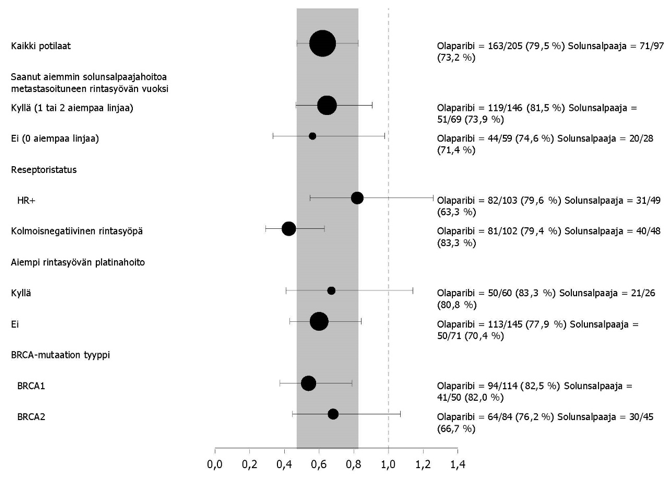
Post hoc ‑analyysissä, joka tehtiin alaryhmästä, jossa potilaiden tauti ei ollut edennyt muun solunsalpaajahoidon kuin platinahoidon aikana, etenemisvapaan elinajan mediaani oli olaparibihaarassa (n = 22) 8,3 kuukautta (95 %:n luottamusväli 3,1–16,7) ja solunsalpaajahoitohaarassa (n = 16) 2,8 kuukautta (95 %:n luottamusväli 1,4–4,2) riskisuhteella 0,54 (95 %:n luottamusväli 0,24–1,23).Potilaiden määrä on kuitenkin liian pieni, jotta tehokkuudesta voitaisiin tehdä merkityksellisiä johtopäätöksiä tässä alaryhmässä.
Seitsemän miespotilasta satunnaistettiin (5 olaparibihaaraan ja 2 vertailuhaaraan). PFS-analyysin kohdalla yhdellä olaparibihaaran potilaalla oli vahvistettu osittainen vaste, jonka kesto oli 9,7 kuukautta. Vertailuhaarassa ei ollut vahvistettuja vasteita.
Kuva 13 OlympiAD: Kaplan-Meier-käyrä kokonaiselinajalle gBRCA1/2-mutaation omaavaa HER2-negatiivista metastasoitunutta rintasyöpää sairastavilla potilailla (maturiteetti 64 %), viimeinen tiedonkeruupäivä 25.9.2017
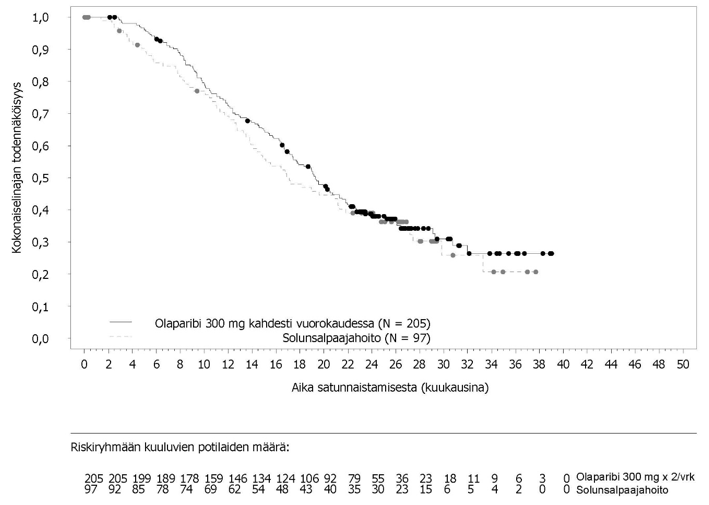
Kokonaiselinajan analyysi potilaista, jotka eivät olleet aiemmin saaneet solunsalpaajahoitoa metastasoituneen rintasyövän hoitoon, viittasi hyötyyn näillä potilailla riskisuhteella 0,45 (95 %:n luottamusväli 0,27–0,77), kun taas myöhemmissä hoitolinjoissa riskisuhde oli suurempi kuin 1.
Ylläpitohoito haiman metastasoituneen adenokarsinooman ensilinjan hoidon jälkeen, kun potilailla on ituradan BRCA-mutaatio:
POLO-tutkimus
Olaparibin turvallisuutta ja tehoa ylläpitohoitona tutkittiin satunnaistetussa (3:2), kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa 154 potilaalla, joilla oli ituradan BRCA1/2-mutaatioita ja haiman metastasoitunut adenokarsinooma. Potilaat saivat joko 300 mg (kaksi 150 mg:n tablettia) Lynparza-valmistetta kaksi kertaa vuorokaudessa (n = 92) tai lumelääkettä (n = 62), kunnes sairaus eteni radiologisesti todennetusti tai ilmeni haittavaikutuksia, joita ei voitu hyväksyä. Edellytyksenä oli, että potilaiden tauti ei ollut edennyt ensilinjan platinapohjaisen solunsalpaajahoidon aikana, ja että potilaat olivat saaneet jatkuvaa platinahoitoa vähintään 16 viikon ajan. Platinahoito voitiin keskeyttää milloin tahansa tämän jälkeen, jos potilaalla ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä, samalla kuitenkin jatkaen muita jäljelle jääneitä lääkehoitoja suunnitellun hoito-ohjelman mukaisesti tai kunnes ilmeni jonkin muun komponentin aiheuttamia toksisia vaikutuksia, joita ei voitu hyväksyä. Jos potilas sieti koko platinaa sisältävän solunsalpaajahoidon taudin etenemiseen asti, hänen ei katsottu soveltuvan tähän tutkimukseen. Ylläpitohoito aloitettiin 4–8 viikon kuluttua ensilinjan solunsalpaajahoidon komponentin/komponenttien viimeisen annoksen antamisesta, jos tauti ei ollut edennyt ja jos aiemman syöpähoidon kaikki haittavaikutukset olivat lievittyneet CTCAE-asteeseen 1 lukuun ottamatta alopesiaa, asteen 3 perifeeristä neuropatiaa ja Hb-arvoa ≥ 9 g/dl.
Potilaista, joilla oli ituradan BRCA1/2-mutaatioita, 31 % tunnistettiin aiemmilla paikallisten testien tuloksilla ja 69 % keskitetysti tehdyllä testillä. Olaparibihaarassa 32 %:lla potilaista oli ituradan BRCA1-mutaatio, 64 %:lla ituradan BRCA2-mutaatio ja 1 %:lla sekä ituradan BRCA1- että ituradan BRCA2-mutaatio. Lumelääkehaarassa 26 %:lla potilaista oli ituradan BRCA1-mutaatio ja 73 %:lla ituradan BRCA2-mutaatio, eikä yhdelläkään potilaalla ollut sekä ituradan BRCA1- että ituradan BRCA2-mutaatiota. Kaikkien aiemmilla paikallisten testien tuloksilla tunnistettujen potilaiden BRCA-mutaatiostatus vahvistettiin lähetettyjen näytteiden osalta keskitetysti tehdyllä testillä. 98 %:lla potilaista oli haitallinen mutaatio ja 2 %:lla oli epäilty haitallinen mutaatio. 5,2 %:lla (8/154) satunnaistetuista potilaista todettiin BRCA1/2-geenien laajoja uudelleenjärjestymiä.
Demografiset tiedot ja lähtötilanteen tiedot olivat yleisesti hyvin samankaltaiset olaparibi- ja lumelääkehaaroissa. Mediaani-ikä molemmissa haaroissa oli 57 vuotta. 30 % olaparibihaaran potilaista ja 20 % lumelääkehaaran potilaista oli ≥ 65‑vuotiaita. 58 % olaparibihaaran potilaista ja 50 % lumelääkehaaran potilaista oli miehiä. Olaparibihaarassa 89 % potilaista oli valkoihoisia ja 11 % muita kuin valkoihoisia. Lumelääkehaarassa 95 % potilaista oli valkoihoisia ja 5 % muita kuin valkoihoisia. Suurimmalla osalla potilaista ECOG-suorituskykypistemäärä oli 0 (71 %:lla olaparibihaarassa ja 61 %:lla lumelääkehaarassa). Kaiken kaikkiaan ennen solunsalpaajahoitoa etäpesäkkeistä 72 % sijaitsi maksassa, 10 % keuhkoissa ja 50 % muualla. Mediaaniaika alkuperäisestä diagnoosista satunnaistamiseen oli molemmissa haaroissa 6,9 kuukautta (vaihteluväli 3,6–38,4 kuukautta).
Kaiken kaikkiaan 75 % potilaista sai FOLFIRINOX-hoitoa, jossa hoitojaksojen määrän mediaani oli 9 (vaihteluväli 4–61), 8 % sai FOLFOX- tai XELOX-hoitoa, 4 % sai GEMOX-hoitoa ja 3 % sai gemsitabiinia ja sisplatiinia. Loput 10 % potilaista saivat muuta solunsalpaajahoitoa. Ensilinjan solunsalpaajahoidon kesto metastasoituneen taudin hoidossa oli 4–6 kuukautta 77 %:lla, > 6 – < 12 kuukautta 19 %:lla ja ≥ 12 kuukautta 4 %:lla olaparibihaaran potilaista sekä 4–6 kuukautta 80 %:lla, > 6 – < 12 kuukautta 17 %:lla ja ≥ 12 kuukautta 3 %:lla lumehaaran potilaista. Aika ensilinjan solunsalpaajahoidon komponentin tai komponenttien viimeisen annoksen saamisesta tutkimushoidon aloittamiseen oli molemmissa haaroissa noin 1 kuukausi. Parhaana vasteena ensilinjan solunsalpaajahoitoon 7 % olabaribia saaneista ja 5 % lumelääkettä saaneista potilaista saavutti täydellisen vasteen, 44 % olaparibia saaneista ja 44 % lumelääkettä saaneista potilaista saavutti osittaisen vasteen ja 49 %:lla olaparibia saaneista ja 50 %:lla lumelääkettä saaneista potilaista oli vakaa tauti. Satunnaistamishetkellä 85 %:lla olaparibihaaran potilaista ja 84 %:lla lumehaaran potilaista oli mitattavissa oleva sairaus. Mediaaniaika ensilinjan platinapohjaisen solunsalpaajahoidon aloittamisesta satunnaistamiseen oli 5,7 kuukautta (vaihteluväli 3,4–33,4 kuukautta).
PFS-analyysin ajankohtana 33 % olaparibihaaran potilaista ja 13 % lumehaaran potilaista jatkoi tutkimushoitoa. 49 % olaparibihaaran potilaista ja 74 % lumehaaran potilaista sai seuraavaa hoitoa. 42 % olaparibihaaran potilaista ja 55 % lumehaaran potilaista sai seuraavana hoitona platinaa. 1 % olaparibihaaran potilaista ja 15 % lumehaaran potilaista sai seuraavana hoitona PARP:n estäjää. Niistä 33:sta (36 %) olaparibihaaran potilaasta ja 28:sta (45 %) lumehaaran potilaasta, jotka saivat ensimmäistä seuraavaa platinaa sisältävää hoitoa, 8:lla olaparibihaaran potilaalla ja 6:lla lumehaaran potilaalla oli vakaa tauti, ja yhdellä olaparibihaaran potilaalla ja kahdella lumehaaran potilaalla todettiin vaste.
Ensisijainen päätemuuttuja oli etenemisvapaa elinaika (PFS), joka määriteltiin aikana satunnaistamisesta BICR:n arvioimaan taudin etenemiseen tai kuolemaan. BICR arvioi taudin etenemisen käyttämällä RECIST 1.1 ‑kriteerejä (Response Evaluation Criteria in Solid Tumors), joita oli muokattu sellaisten potilaiden arviointiin, joilla ei ollut näyttöä sairaudesta. Toissijaisia tehoa mittaavia päätemuuttujia olivat kokonaiselinaika (OS), aika satunnaistamisesta taudin toiseen etenemiseen tai kuolemaan (PFS2), aika satunnaistamisesta ensimmäiseen seuraavaan syöpähoitoon tai kuolemaan (TFST), objektiivinen hoitovaste (ORR), vasteen kesto, vasteosuus, aika vasteeseen ja terveyteen liittyvä elämänlaatu (HRQoL).
Tutkimuksessa osoitettiin tilastollisesti merkitsevä PFS:n piteneminen olaparibihaarassa lumehaaraan nähden (taulukko 14). BICR:n tekemä etenemisvapaata elinaikaa koskeva arvio vastasi tutkijalääkärin arviota.
Kokonaiselinaikaa koskevassa lopullisessa analyysissä niiden potilaiden osuus, jotka olivat elossa ja seurannassa, oli 28 % olaparibihaarassa ja 18 % lumehaarassa.
Taulukko 14: POLO-tutkimuksen tehoa koskevat tulokset haiman metastasoitunutta adenokarsinoomaa sairastavista potilaista, joilla on gBRCA-mutaatio
| Olaparibi 300 mg kaksi kertaa vuorokaudessa | Lumelääke | |
| PFS (maturiteetti 68 %)a, b (BICR, viimeinen tiedonkeruupäivä 15.1.2019) | ||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 60:92 (65) | 44:62 (71) |
| Mediaaniaika kuukausina (95 %:n luottamusväli) | 7,4 (4,14–11,01) | 3,8 (3,52–4,86) |
| HR (95 %:n luottamusväli)c, d | 0,53 (0,35–0,82) | |
| p-arvo (kaksitahoinen) | p = 0,0038 | |
| OS (maturiteetti 70 %)e (viimeinen tiedonkeruupäivä 21.7.2020) | ||
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 61:92 (66) | 47:62 (76) |
| Mediaaniaika kuukausina (95 %:n luottamusväli) | 19,0 (15,28–26,32) | 19,2 (14,32–26,12) |
| HR (95 %:n luottamusväli)d | 0,83 (0,56–1,22) | |
| p-arvo (kaksitahoinen) | p = 0,3487 | |
a Kaplan-Meier-estimaattien perusteella niiden potilaiden osuus, jotka olivat elossa ja joiden tauti ei ollut edennyt kuukauden 12 kohdalla, oli olaparibiryhmässä 34 % ja lumelääkeryhmässä 15 % ja kuukauden 24 kohdalla vastaavasti 22 % ja 10 %.
b Etenemisvapaan elinajan kohdalla pois rajattujen potilaiden seuranta-ajan mediaani oli 9,1 kuukautta olaparibihaarassa ja 3,8 kuukautta lumelääkehaarassa.
c Arvo, joka on < 1, suosii olaparibia.
d Analyysi tehtiin käyttämällä log-rank-testiä.
e Kokonaiselinajan kohdalla pois rajattujen potilaiden seuranta-ajan mediaani oli 31,3 kuukautta olaparibihaarassa ja 23,9 kuukautta lumehaarassa.
HR: riskisuhde, OS: kokonaiselinaika, PFS: etenemisvapaa elinaika.
Kuva 14 POLO: Kaplan-Meier-käyrä etenemisvapaalle elinajalle haiman metastasoitunutta adenokarsinooma sairastavilla potilailla, joilla on gBRCA-mutaatio (maturiteetti 68 % – BICR, viimeinen tiedonkeruupäivä 15.1.2019)
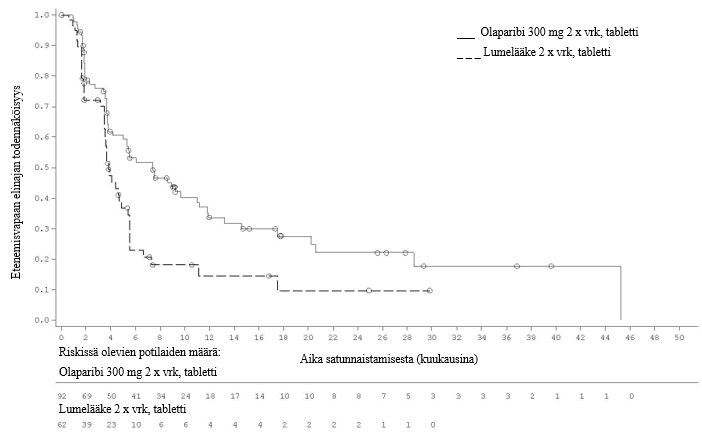
Kuva 15: POLO: Kaplan–Meier-käyrä kokonaiselinajalle haiman metastasoitunutta adenokarsinoomaa sairastavilla potilailla, joilla on gBRCA-mutaatio (maturiteetti 70 %, viimeinen tiedonkeruupäivä 21.7.2020)
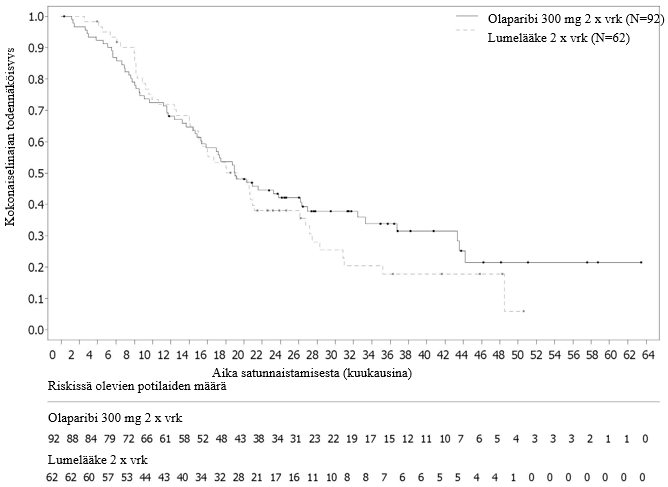
BRCA1/2-mutaation omaava metastasoitunut kastraatioresistentti eturauhassyöpä:
PROfound-tutkimus
Olaparibin turvallisuutta ja tehoa tutkittiin metastasoitunutta kastraatioresistenttiä eturauhassyöpää (mCRPC) sairastavilla miehillä vaiheen III satunnaistetussa, avoimessa monikeskustutkimuksessa, jossa arvioitiin Lynparza-valmisteen tehoa verrattuna tutkijan valitsemaan uuteen hormonaaliseen lääkeaineeseen (new hormonal agent, NHA; entsalutamidi tai abirateroniasetaatti), jota annettiin vertailuhaarassa.
Edellytyksenä oli, että potilaan tauti oli edennyt aiemman NHA-hoidon aikana, kun hormonaalista lääkeainetta oli käytetty metastasoituneen eturauhassyövän ja/tai kastraatioresistentin eturauhassyövän hoitoon. Jotta potilaat voitiin ottaa mukaan kohorttiin A, heillä piti olla haitallisia tai epäiltyjä haitallisia mutaatioita joko BRCA1- tai BRCA2-geeneissä. Potilaat, joilla oli ATM-mutaatioita, satunnaistettiin myös kohorttiin A, mutta positiivista hyöty-riskisuhdetta ei voitu osoittaa tässä alapopulaatiossa. Potilaat, joilla oli mutaatioita muissa geeneissä, satunnaistettiin kohorttiin B.
Tässä tutkimuksessa 387 potilasta satunnaistettiin suhteessa 2:1 saamaan joko olaparibia (300 mg [2 x 150 mg:n tabletti] kaksi kertaa vuorokaudessa) tai vertailuvalmistetta. Kohortissa A oli 245 potilasta (162 sai olaparibia ja 83 vertailuvalmistetta) ja kohortissa B oli 142 potilasta (94 sai olaparibia ja 48 vertailuvalmistetta). Potilaat stratifioitiin aiemman taksaanin käytön mukaan ja sen mukaan, oliko mitattavissa olevasta sairaudesta näyttöä. Hoitoa jatkettiin, kunnes tauti eteni. Potilaille, jotka oli satunnaistettu saamaan vertailuvalmistetta, annettiin mahdollisuus siirtyä käyttämään olaparibia, kun sokkoutettu riippumaton keskitetty arvioijataho (BICR) oli radiologisesti vahvistanut taudin edenneen. Potilaat, joiden kasvaimissa oli BRCA1- tai BRCA2-mutaatio, otettiin mukaan prospektiivisen, keskitetysti toteutetun testauksen perusteella, lukuun ottamatta kolmea potilasta, jotka otettiin mukaan paikallisen testauksen tuloksen perusteella. Niistä 160 potilaasta, joilla havaittiin BRCA1- tai BRCA2-mutaatio PROfound-tutkimuksessa, 114 potilasta testattiin jälkikäteen sen selvittämiseksi, olivatko tunnistetut BRCA1/2-mutaatiot ituradan vai somaattisia mutaatiota. Näistä potilaista 63:lla BRCA1/2-mutaatio todettiin ituradan verinäytteessä, joten niiden määriteltiin olevan alkuperältään ituradan mutaatioita. Jäljelle jääneillä 51 potilaalla kasvaimen BRCA1/2-mutaatiota ei havaittu ituradan verinäytteessä, joten näiden BRCA1/2-mutaatioiden määriteltiin olevan alkuperältään somaattisia. Loppujen 46 potilaan osalta ei tiedetä, oliko kyseessä alkuperältään ituradan vai somaattinen mutaatio.
Potilaiden, joilla oli BRCA1/2-mutaatioita, demografiset tiedot ja lähtötilanteen tiedot olivat yleisesti hyvin samankaltaiset olaparibi- ja vertailuhaaroissa. Mediaani-ikä oli 68 vuotta olaparibihaarassa ja 67 vuotta vertailuhaarassa. Aiempi hoito olaparibihaarassa jakautui seuraavasti: 71 % tutkittavista oli saanut taksaania, 41 % entsalutamidia, 37 % abirateroniasetaattia ja 20 % sekä entsalutamidia että abirateroniasetaattia. Aiempi hoito vertailuhaarassa jakautui seuraavasti: 60 % tutkittavista oli saanut taksaania, 50 % entsalutamidia, 36 % abirateroniasetaattia ja 14 % sekä entsalutamidia että abirateroniasetaattia. Kaikkiaan 58 %:lla potilaista olaparibihaarassa ja 55 %:lla vertailuhaarassa oli mitattavissa oleva sairaus, kun he aloittivat tutkimuksessa. Olaparibihaarassa 89 %:lla potilaista oli etäpesäkkeitä luustossa, 62 %:lla imusolmukkeissa, 23 %:lla hengityselimissä ja 12 %:lla maksassa. Vertailuhaarassa 86 %:lla potilaista oli etäpesäkkeitä luustossa, 71 %:lla imusolmukkeissa, 16 %:lla hengityselimissä ja 17 %:lla maksassa. Kummassakin hoitohaarassa useimpien potilaiden (93 %) ECOG-suorituskykypistemäärä oli 0 tai 1. Kipua kuvaavat pistemäärät olivat lähtötilanteessa BPI-SF-asteikolla (pahin kipu) 0 – < 2 (52 %), 2–3 (10 %) ja > 3 (34 %) olaparibihaarassa ja 0 – < 2 (45 %), 2–3 (7 %) ja > 3 (45 %) vertailuhaarassa. Lähtötilanteen PSA-arvon mediaani oli 57,48 µg/l olaparibihaarassa ja 103,95 µg/l vertailuhaarassa.
Tutkimuksen ensisijainen päätemuuttuja oli radiologisesti todennettu etenemisvapaa elinaika (rPFS) kohortissa A BICR:n arvioimana. BICR arvioi taudin etenemisen käyttämällä RECIST 1.1 -kriteerejä (pehmytkudos) ja PCWG3-kriteerejä (Prostate Cancer Working Group) (luusto). Keskeisiä toissijaisia päätemuuttujia olivat vahvistettu objektiivinen hoitovaste (ORR) BICR:n arvioimana, radiologisesti todennettu etenemisvapaa elinaika BICR:n arvioimana, aika kivun pahenemiseen (time to pain progression, TTPP) sekä kokonaiselinaika (OS).
Tutkimuksessa osoitettiin tilastollisesti merkitsevä radiologisesti todennetun etenemisvapaan elinajan piteneminen BICR:n arvioimana sekä lopullisen kokonaiselinajan tilastollisesti merkitsevä piteneminen olaparibihoidolla verrattuna vertailuvalmisteeseen kohortissa A.
Niitä potilaita koskevat tulokset, joilla oli BRCA1/2-mutaatio, esitetään taulukossa 15 BICR:n arvioimassa radiologisesti todennetussa etenemisvapaassa elinajassa havaittiin tilastollisesti merkitsevä piteneminen olaparibihoidolla verrattuna tutkijan valitsemaan NHA-hoitoon potilailla, joilla oli BRCA1/2-mutaatio. Kokonaiselinajan lopullisessa analyysissä havaittiin nimellisesti tilastollisesti merkitsevä kokonaiselinajan piteneminen potilailla, joilla oli BRCA1/2-mutaatio ja jotka oli satunnaistettu saamaan Lynparza-valmistetta, verrattuna potilaisiin, jotka saivat vertailuvalmistetta.
Taulukko 15 Yhteenveto keskeisistä tehoa koskevista löydöksistä PROfound-tutkimuksen potilailla, joilla oli BRCA1/2-mutaation omaava metastasoitunut kastraatioresistentti eturauhassyöpä
Olaparibi 300 mg kaksi kertaa vuorokaudessa (N = 102) | Tutkijan valitsema NHA (N = 58) | |
| rPFS BICR:n arvioimanaa,b,c viimeinen tiedonkeruupäivä 4.6.2019 | ||
| Tapahtumien lukumäärä: potilaiden kokonaismäärä (%) | 62:102 (61)c | 51:58 (88) c |
| rPFS, mediaani (95 %:n luottamusväli) [kk] | 9,8 (7,6, 11,3) | 3,0 (1,8, 3,6) |
| HR (95 %:n luottamusväli)c | 0,22 (0,15, 0,32) | |
| BICR:n vahvistama ORRa | ||
| Objektiivisen vasteen saaneiden lukumäärä: niiden potilaiden kokonaismäärä, joilla oli mitattavissa oleva sairaus lähtötilanteessa (%) | 25:57 (44) | 0:33 (0) |
| Kerroinsuhde (95 %:n luottamusväli) | Ei laskettavissa (ei laskettavissa, ei laskettavissa) | |
| Kokonaiselinaikaa viimeinen tiedonkeruupäivä 20.3.2020 c | ||
| Tapahtumien lukumäärä: potilaiden kokonaismäärä (%) | 53:102 (52) | 41:58 (71) |
| Kokonaiselinaika, mediaani (95 %:n luottamusväli [kk] | 20,1 (17,4, 26,8) | 14,4 (10,7, 18,9) |
| HR (95 %:n luottamusväli) | 0,63 (0,42, 0,95) | |
a Ei otettu huomioon kerrannaisuutta
b rPFS, maturiteetti 71 %
c Riskisuhde ja luottamusväli laskettiin käyttämällä Coxin suhteellisten riskitiheyksien mallia, jossa termeinä ovat hoito, faktori ja hoito x faktori yhdysvaikutus.
BICR = sokkoutettu riippumaton keskitetty arvioijataho, blinded independent central review; NHA = uusi hormonaalinen lääkeaine; ORR = objektiivinen hoitovaste; rPFS = radiologisesti todennettu etenemisvapaa elinaika.
Kuva 16: Potilaat, joilla on BRCA1/2-mutaatio: Kaplan-Meier-käyrä radiologisesti todennetulle etenemisvapaalle elinajalle (BICR:n arvioimana)
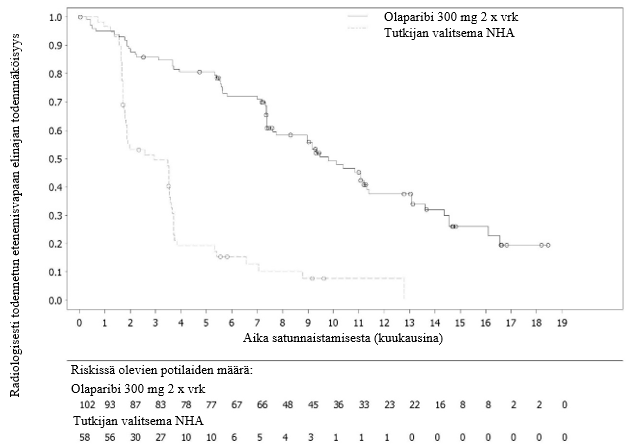
Kuva 17: Potilaat, joilla on BRCA1/2-mutaatio: KaplanMeier-käyrä kokonaiselinajalle
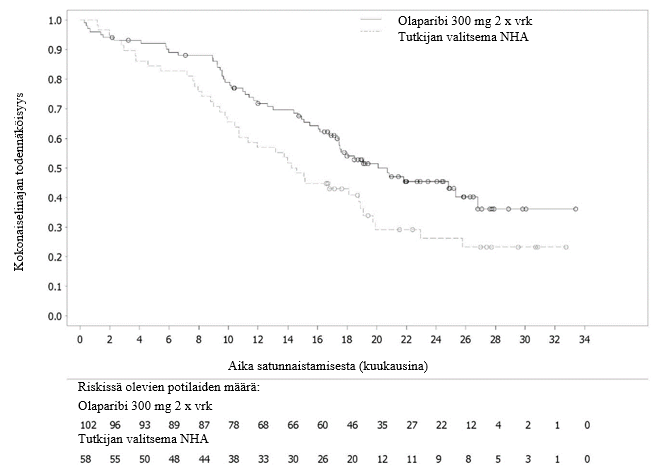
Metastasoituneen kastraatioresistentin eturauhassyövän ensilinjan hoito
PROpel
Olaparibin turvallisuutta ja tehoa tutkittiin metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavilla miehillä vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa, jossa arvioitiin Lynparza-valmisteen (300 mg [2 kpl 150 mg:n tabletteja] kahdesti vuorokaudessa) tehoa yhdistelmänä abirateronin (1 000 mg [2 kpl 500 mg:n tabletteja] kerran vuorokaudessa) kanssa verrattuna vertailuhaaraan, jossa potilaat saivat lumelääkkeen ja abirateronin yhdistelmää. Kummankin haaran potilaat saivat myös 5 mg prednisonia tai prednisolonia kahdesti vuorokaudessa.
Tutkimuksessa satunnaistettiin 796 potilasta (suhteessa 1:1; 399 potilaalle olaparibi- ja abirateronihoito ja 397 potilaalle lumelääke- ja abirateronihoito), joiden kohdalla oli näyttöä histologisesti vahvistetusta eturauhasen adenokarsinoomasta ja metastasoitumisesta. Metastasoitumisen määritelmänä oli vähintään yksi dokumentoitu metastasoitunut leesio joko luuston kuvantamistutkimuksessa, TT-kuvassa tai magneettikuvassa. Potilaat eivät olleet saaneet aiemmin solunsalpaajia tai uusia hormonaalisia lääkeaineita metastasoituneen kastraatioresistentin eturauhassyövän hoitoon. Potilaat, jotka olivat saaneet uusia hormonaalisia lääkeaineita (lukuun ottamatta abirateronihoitoa) ennen taudin kehittymistä metastasoituneeksi kastraatioresistentiksi eturauhassyöväksi ja joiden tauti ei hoidon aikana ollut edennyt PSA-pitoisuuden perusteella (kliinisesti tai radiologisesti arvioituna), soveltuivat osallistujiksi, kunhan hoito oli lopetettu vähintään 12 kuukautta ennen satunnaistamista. Ensimmäisen sukupolven antiandrogeenihoito (esim. bikalutamidi, nilutamidi, flutamidi) oli myös sallittua edellyttäen, että noudatettiin 4 viikon pituista jaksoa lääkeaineen poistumiseksi elimistöstä. Dosetakselihoito oli sallittua esiliitännäishoidon/liitännäishoidon aikana paikallisen eturauhassyövän hoidossa ja metastasoituneen hormoniherkän eturauhassyövän (mHSPC) hoidossa, kunhan tällaisen hoidon aikana tai välittömästi hoidon jälkeen ei ilmennyt taudin etenemisen merkkejä. Kaikki potilaat saivat GnRH-analogihoitoa tai heille oli aiemmin tehty molemminpuolinen orkiektomia. Potilaat ositettiin etäpesäketilanteen (vain luustoetäpesäkkeitä, viskeraalisia etäpesäkkeitä tai muita) ja mHSPC-vaiheessa saadun dosetakselihoidon (saanut tai ei saanut) perusteella. Hoitoa jatkettiin, kunnes perussairaus eteni radiologisesti todennetusti tai ilmeni haittavaikutuksia, joita ei voitu hyväksyä.
Demografiset tiedot ja lähtötilanteen tiedot olivat samankaltaiset molemmissa hoitohaaroissa. Kaikkien tutkittavien potilaiden mediaani-ikä oli 69 vuotta, ja suurin osa (71 %) potilaista kuului vähintään 65-vuotiaiden ikäryhmään. 189 potilasta (24 %) oli saanut aiemmin dosetakselihoitoa mHSPC-vaiheessa. Yhteensä 434 potilaalla (55 %:lla) oli luustoetäpesäkkeitä (metastaaseja luustossa, ei muualla), 105 potilaalla (13 %:lla) oli viskeraalisia etäpesäkkeitä (pehmytkudosmetastaaseja jossakin elimessä, esimerkiksi maksassa tai keuhkoissa) ja 257 potilaalla (32 %:lla) oli muita etäpesäkkeitä (esimerkiksi potilaat, joilla oli luustoetäpesäkkeitä ja etäpesäkkeitä imusolmukkeissa tai potilaat, joilla tauti ilmeni vain etäpesäkkeinä imusolmukkeissa). Molemmissa hoitohaaroissa useimpien potilaiden (70 %) ECOG-suorituskykyluokka oli 0. Olaparibiryhmässä oli 103 oireista potilasta (25,8 %) ja lumelääkeryhmässä 80 potilasta (20,2 %). Oireisille potilaille oli tunnusomaista pistemäärä ≥ 4 BPI-SF-lomakkeen (Brief Pain Inventory-Short Form) kohdassa 3 ja/tai opiaattien käyttö lähtötilanteessa.
Potilaiden tutkimukseenotto ei perustunut biomarkkeristatukseen. HRR-geenimutaatiostatus arvioitiin jälkikäteen kiertävän kasvain-DNA:n ja kasvainkudostutkimusten perusteella, jotta hoitovaikutuksen johdonmukaisuus täydellisessä analyysijoukossa voitiin arvioida. Homologiseen rekombinaatioon perustuvan korjausreitin geenimutaatio todettiin tutkituista potilaista 198 potilaalla kiertävän kasvain-DNA:n perusteella ja 118 potilaalla kasvainkudoksen perusteella. Niiden potilaiden jakauma, joilla oli homologiseen rekombinaatioon perustuvan korjausreitin geenimutaatio, oli hyvin tasapainoinen kahden hoitohaaran välillä.
Ensisijainen päätemuuttuja oli radiologisesti todennettu etenemisvapaa elinaika, joka määriteltiin aikana satunnaistamisesta radiologiseen taudin etenemiseen RECIST 1.1- ja PCWG-3-kriteerien (luusto) perusteella tutkijalääkärin arvioimana. Tärkeä toissijainen päätemuuttuja oli kokonaiselinaika (OS). Muita toissijaisia päätemuuttujia olivat aika satunnaistamisesta taudin toiseen etenemiseen tai kuolemaan (PFS2), aika satunnaistamisesta ensimmäiseen seuraavaan syöpähoitoon tai kuolemaan (TFST) ja terveyteen liittyvä elämänlaatu (HRQoL).
Tutkimuksessa saavutettiin sen ensisijainen päätemuuttuja, eli taudin radiologisesti todennetun etenemisen tai kuoleman riski oli tutkijalääkärin arvion mukaan olaparibi- ja abirateronihoitohaarassa tilastollisesti merkitsevästi pienempi kuin lumelääke- ja abirateronihoitohaarassa. Riskisuhde oli 0,66; 95 %:n luottamusväli 0,54, 0,81; p < 0,0001; radiologisesti todennetun etenemisvapaan elinajan mediaani oli olaparibi- ja abirateronihoitohaarassa 24,8 kuukautta ja lumelääke- ja abirateronihoitohaarassa 16,6 kuukautta. Radiologisesti todennettua etenemisvapaata elinaikaa koskevaa, tutkijalääkärin tekemää arviota tuki sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR:n) tekemä arvio. BICR:n tekemä radiologisesti todennetun etenemisvapaan elinajan herkkyysanalyysi vastasi tutkijalääkärin arvioon perustuvaa analyysiä. Riskisuhde oli 0,61; 95 %:n luottamusväli 0,49, 0,74; p < 0,0001; radiologisesti todennetun etenemisvapaan elinajan mediaani oli 27,6 kuukautta olaparibi- ja abirateronihoitohaarassa ja 16,4 kuukautta lumelääke- ja abirateronihoitohaarassa.
Alaryhmien tulokset olivat yhtenevät olaparibi- ja abirateronihoitohaaran kokonaistulosten kanssa verrattuna lumelääke- ja abirateronihoitohaaraan kaikissa ennalta määritellyissä alaryhmissä, joihin kuuluivat potilaat, jotka joko olivat tai eivät olleet saaneet taksaania mHSPC-vaiheessa, potilaat, joilla oli eri tavoin metastasoitunut tauti lähtötilanteessa (vain luustoetäpesäkkeitä vs. viskeraalisia etäpesäkkeitä vs. muita), sekä potilaat, joilla joko oli tai ei ollut HRR-mutaatiota (kuva 20).
Tehoa koskevat tulokset on esitetty taulukossa 16, taulukossa 17, kuvassa 18 ja kuvassa 19.
Taulukko 16 Yhteenveto keskeisistä tehoa koskevista löydöksistä metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavien potilaiden hoidossa PROpel-tutkimuksessa
Olaparibi ja abirateroni N = 399 | Lumelääke ja abirateroni N = 397 | ||
|---|---|---|---|
| rPFS (tutkijalääkärin arvion mukaan) (maturiteetti 50 %) (viimeinen tiedonkeruupäivä 30.7.2021) | |||
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 168:399 (42,1) | 226:397 (56,9) | |
| Ajan mediaani (95 %:n luottamusväli) (kk) | 24,8 (20,5, 27,6) | 16,6 (13,9, 19,2) | |
| Riskisuhde (95 %:n luottamusväli)a | 0,66 (0,54, 0,81) | ||
| p‑arvob | < 0,0001 | ||
| Kokonaiselinajan lopullinen analyysi (maturiteetti 48 %) (viimeinen tiedonkeruupäivä 12.10.2022) | |||
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 176:399 (44,1) | 205:397 (51,6) | |
| Ajan mediaani (95 %:n luottamusväli) (kk) | 42,1 (38,4, NC) | 34,7 (31,0, 39,3) | |
| Riskisuhde (95 %:n luottamusväli)a | 0,81 (0,67, 1,00) | ||
| p‑arvob | p = 0,0544 | ||
| Elossa 36 kuukauden kohdalla, % (95 %:n luottamusväli)c | 56,9 (51,7, 61,7) | 49,5 (44,3, 54,5) | |
a Riskisuhde ja luottamusväli laskettiin käyttämällä Coxin suhteellisten riskitiheyksien mallia, jossa tehtiin korjaukset alkuperäisessä yhdistämisstrategiassa valittujen muuttujien eli metastaasien ja mHSPC-vaiheessa annetun dosetakselihoidon suhteen. Tasahavaintojen käsittelyyn käytettiin Efron-lähestymistapaa. Riskisuhde < 1 suosii olaparibi 300 mg kahdesti vuorokaudessa + abirateroni 1 000 mg kerran vuorokaudessa -hoitoa.
b Kaksitahoinen p‑arvo laskettiin käyttämällä log-rank-testiä, jossa tehtiin ositus samojen muuttujien perusteella kuin oli valittu alkuperäisessä yhdistämisstrategiassa.
c Laskettu Kaplan–Meier-menetelmällä.
NC = ei laskettavissa
Taulukko 17 rPFS-ajan alaryhmäanalyysit tutkijalääkärin arvion mukaan – PROpel (viimeinen tiedonkeruupäivä 30.7.2021)
Olaparibi ja abirateroni
| Lumelääke ja abirateroni
| |
|---|---|---|
| Radiologisesti todennettu etenemisvapaa elinaika (rPFS) tutkijalääkärin arvion mukaan | ||
| HRR-mutaatiostatuksen aggregoidut alaryhmäanalyysit a | ||
| HRR-mutaatio | N = 111 | N = 115 |
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 43:111 (38,7) | 73:115 (63,5) |
| Mediaani (kuukausia) | NC | 13,86 |
| Riskisuhde (95 %:n luottamusväli) b | 0,50 (0,34, 0,73) | |
| Ei HRR-mutaatiota | N = 279 | N = 273 |
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 119:279 (42,7) | 149:273 (54,6) |
| Mediaani (kuukausia) | 24,11 | 18,96 |
| Riskisuhde (95 %:n luottamusväli) b | 0,76 (0,60, 0,97) | |
| BRCA-mutaatiostatuksen aggregoidut alaryhmäanalyysit a | ||
| BRCA-mutaatio | N = 47 | N = 38 |
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 14:47 (29,8) | 28:38 (73,7) |
| Mediaani (kuukausia) | NC | 8,38 |
| Riskisuhde (95 %:n luottamusväli) b | 0,23 (0,12, 0,43) | |
| Ei BRCA-mutaatiota | N = 343 | N = 350 |
| Tapahtumien määrä: potilaiden kokonaismäärä (%) | 148:343 (43,1) | 194:350 (55,4) |
| Mediaani (kuukausia) | 24,11 | 18,96 |
| Riskisuhde (95 %:n luottamusväli) b | 0,76 (0,61, 0,94) | |
a Aggregoidut alaryhmät ryhmiteltiin kiertävän kasvain-DNA:n ja kasvainkudoksen perusteella.
b Analyysissä käytettiin Coxin suhteellisten riskitiheyksien mallia, jossa termeinä olivat hoitoryhmä, alaryhmäfaktori ja hoito x alaryhmä yhdysvaikutus. Luottamusväli on laskettu profiilin todennäköisyyden perusteella. Riskisuhde < 1 suosii olaparibi 300 mg kahdesti vuorokaudessa -hoitoa.
Kuva 18 PROpel: Kaplan–Meier-kuvaaja radiologisesti todennetusta etenemisvapaasta elinajasta (tutkijan arvioimana) (maturiteetti 50 %), viimeinen tiedonkeruupäivä 30.7.2021
Kuva 19 PROpel: Kaplan–Meier-kuvaaja kokonaiselinajasta (maturiteetti 48 %), viimeinen tiedonkeruupäivä 12.10.2022
Kuva 20 PROpel: Forest-kuvaaja radiologisesti todennetun etenemisvapaan elinajan alaryhmäanalyysistä (tutkijan arvion mukaan) (maturiteetti 50 %), viimeinen tiedonkeruupäivä 30.7.2021
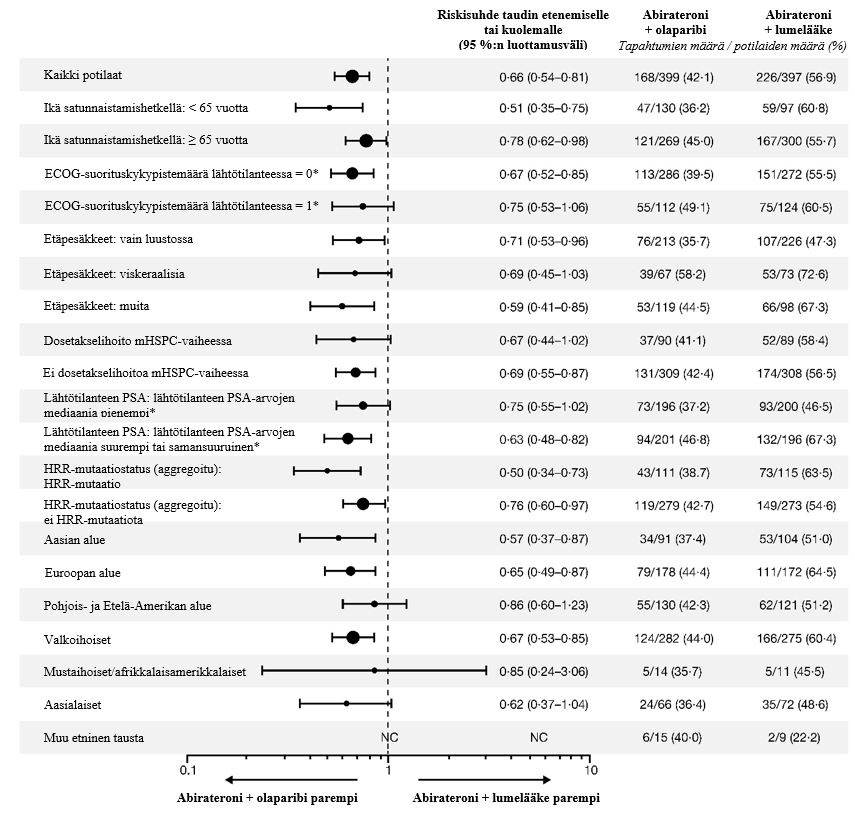
Kussakin alaryhmäanalyysissä käytettiin Coxin suhteellisten riskitiheyksien mallia. Termeinä olivat hoito, faktori ja hoito x faktori ‑yhdysvaikutus. Riskisuhde < 1 viittaa taudin etenemisen pienempään riskiin olaparibihoidon yhteydessä. Pallon koko on verrannollinen tapahtumien määrään. Kaikki tämän kuvan alaryhmät perustuvat sähköisen tietojenkeruulomakkeen (eCRF) tietoihin.
*Potilaat, joilta puuttui arvio lähtötilanteessa, on suljettu pois. ECOG: Eastern Cooperative Oncology Group; HRR-mutaatio: homologiseen rekombinaatioon perustuvan korjausreitin geenimutaatio; mHSPC: metastasoitunut hormoniherkkä eturauhassyöpä; NC: ei laskettavissa; PSA: prostataspesifinen antigeeni.
Toimivan MMR-mekanismin omaavan (pMMR), pitkälle edenneen tai uusiutuneen kohdun limakalvon syövän ensilinjan ylläpitohoito
DUO‑E-tutkimus
DUO‑E oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen III monikeskustutkimus, jossa potilaille, joilla oli pitkälle edennyt tai uusiutunut kohdun limakalvon syöpä, annettiin ensilinjan hoitona platinapohjaista solunsalpaajahoitoa yhdistelmänä durvalumabin kanssa ja tämän jälkeen durvalumabia joko yhdistelmänä olaparibin kanssa tai ilman olaparibia. Potilailla oli oltava kohdun limakalvon syöpä, joka vastasi jotakin seuraavista: äskettäin todettu levinneisyysasteen III tauti (RECIST v1.1 ‑kriteerien mukaisesti mitattavissa oleva tauti leikkaushoidon tai diagnostisen biopsian jälkeen), äskettäin todettu levinneisyysasteen IV tauti (riippumatta siitä, oliko tautia jäljellä leikkauksen tai diagnostisen biopsian jälkeen) tai taudin uusiutuminen (RECIST v1.1 ‑kriteerien mukaisesti joko mitattavissa tai ei-mitattavissa oleva tauti), jossa mahdollisuudet saavuttaa kuratiivinen tulos pelkällä leikkaushoidolla tai yhdistelmähoidolla olivat huonot. Jos potilaalla oli uusiutunut tauti, aiempi solunsalpaajahoito katsottiin hyväksyttäväksi vain, jos se oli annettu liitännäishoitona ja jos viimeisen solunsalpaaja-annoksen antopäivän ja taudin myöhemmän uusiutumisen välillä oli kulunut vähintään 12 kuukautta. Tutkimukseen otetuilla potilailla oli kohdun limakalvon epiteliaalisia karsinoomia; kaikki histologiset muodot sallittiin, mukaan lukien karsinosarkoomat. Potilaat, joilla oli kohdun limakalvon sarkooma, suljettiin pois.
Satunnaistaminen stratifioitiin kasvainkudoksen DNA:n kahdentumisvirheiden korjausmekanismin (MMR-mekanismin) statuksen (toimiva vs. puutteellinen), tautitilanteen (uusiutunut vs. äskettäin todettu) ja maantieteellisen alueen (Aasia vs. muu maailma) mukaan. Potilaat satunnaistettiin suhteessa 1:1:1 johonkin seuraavista hoitohaaroista:
- Platinapohjainen solunsalpaajahoito: Platinapohjaista solunsalpaajahoitoa (paklitakselia ja karboplatiinia) 3 viikon välein enintään 6 hoitojakson ajan ja durvalumabia muistuttavaa lumelääkettä 3 viikon välein. Solunsalpaajahoidon päätyttyä potilaat, joiden taudin ei todettu objektiivisesti edenneen, saivat durvalumabia muistuttavaa lumelääkettä 4 viikon välein ja olaparibia muistuttavia lumelääketabletteja kahdesti vuorokaudessa ylläpitohoitona taudin etenemiseen saakka.
- Platinapohjainen solunsalpaajahoito + durvalumabi: Platinapohjaista solunsalpaajahoitoa (paklitakselia ja karboplatiinia) 3 viikon välein enintään 6 hoitojakson ajan ja 1 120 mg durvalumabia 3 viikon välein. Solunsalpaajahoidon päätyttyä potilaat, joiden taudin ei todettu objektiivisesti edenneen, saivat 1 500 mg durvalumabia 4 viikon välein ja olaparibia muistuttavia lumelääketabletteja kahdesti vuorokaudessa ylläpitohoitona taudin etenemiseen saakka.
- Platinapohjainen solunsalpaajahoito + durvalumabi + olaparibi: Platinapohjaista solunsalpaajahoitoa (paklitakselia ja karboplatiinia) 3 viikon välein enintään 6 hoitojakson ajan ja 1 120 mg durvalumabia 3 viikon välein. Solunsalpaajahoidon päätyttyä potilaat, joiden taudin ei todettu objektiivisesti edenneen, saivat 1 500 mg durvalumabia 4 viikon välein ja 300 mg:n olaparibitabletteja kahdesti vuorokaudessa ylläpitohoitona taudin etenemiseen saakka.
Potilaat, jotka lopettivat jommankumman valmisteen (olaparibi/lumelääke tai durvalumabi/lumelääke) käytön muista syistä kuin taudin etenemisen vuoksi, saattoivat jatkaa toisen valmisteen käyttöä, jos tämä oli asianmukaista toksisuusnäkökohtien ja tutkijan harkinnan mukaan.
Hoitoa jatkettiin, kunnes tauti eteni RECIST v1.1 ‑kriteerien mukaisesti tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä. Kasvaimen status arvioitiin ensimmäisten 18 viikon aikana satunnaistamisen jälkeen 9 viikon välein ja sen jälkeen 12 viikon välein.
Ensisijainen päätemuuttuja oli etenemisvapaa elinaika, joka määriteltiin aikana satunnaistamisesta tutkijalääkärin arvioimaan taudin etenemiseen RECIST 1.1 ‑kriteerien perusteella tai kuolemaan. Toissijaisia tehoa koskevia päätetapahtumia olivat kokonaiselinaika, objektiivinen hoitovaste (ORR) ja vasteen kesto.
Tutkimuksessa osoitettiin etenemisvapaan elinajan tilastollisesti merkitsevä piteneminen ITT-populaatiossa potilailla, joiden hoitona oli platinapohjainen solunsalpaajahoito + durvalumabi + olaparibi, verrattuna pelkästään platinapohjaista solunsalpaajahoitoa saaneisiin (HR = 0,55; 95 %:n luottamusväli 0,43, 0,69). Etenemisvapaan elinajan analyysin ajankohtana kokonaiselinaikaa koskevien alustavien tietojen maturiteetti oli 28 % ja tapahtumia oli todettu 199:llä 718 potilaasta.
DNA:n kahdentumisvirheiden korjausmekanismin (MMR-mekanismin) status määritettiin keskitetysti käyttäen immunohistokemiallista MMR-testipaneelia. Tutkimukseen satunnaistettiin yhteensä 718 potilasta, joista 575:llä (80 %) kasvaimeen liittyi toimiva MMR-mekanismi (pMMR) ja 143:lla (20 %) kasvaimeen liittyi puutteellinen MMR-mekanismi (dMMR).
Potilailla, joiden MMR-mekanismi oli toimiva (kasvaimen status oli pMMR), demografiset tiedot ja sairauden ominaispiirteet lähtötilanteessa olivat yleisesti hyvin samankaltaiset eri hoitohaaroissa. Kaikkien kolmen hoitohaaran demografiset tiedot lähtötilanteessa olivat: iän mediaani 64 vuotta (vaihteluväli: 22–86), ikä vähintään 65 vuotta 48 %:lla, ikä vähintään 75 vuotta 8 %:lla, valkoihoisia 56 %, aasialaisia 30 % ja mustaihoisia tai afrikkalaisamerikkalaisia 6 %. Sairauden ominaispiirteet olivat: ECOG-toimintakykyluokka 0 (69 %) tai 1 (31 %), 47 % tapauksista äskettäin todettuja ja 53 % tapauksista uusiutuneita tauteja. Histologiset alatyypit olivat endometrioidi (54 %), seroosi (26 %), karsinosarkooma (8 %), sekamuotoinen epiteliaalinen (4 %), kirkassoluinen (3 %), erilaistumaton (2 %), musinoosi (< 1 %) ja muu (3 %).
Tulokset potilailla, joiden kasvaimen status oli pMMR, esitetään yhteenvetona taulukossa 18 ja kuvassa 21. Seuranta-ajan mediaani sensuroiduilla potilailla, joiden kasvaimen status oli pMMR, oli platinapohjaisen solunsalpaajahoidon, durvalumabin ja olaparibin hoitohaarassa 15,2 kuukautta ja platinapohjainen solunsalpaajahoidon hoitohaarassa 12,8 kuukautta. Etenemisvapaan elinajan analyysin ajankohtana kokonaiselinaikaa koskevien alustavien tietojen maturiteetti oli 29 % ja tapahtumia oli todettu 110:llä 383 potilaasta.
Taulukko 18 Yhteenveto tehoa koskevista tuloksista pitkälle edennyttä tai uusiutunutta kohdun limakalvon syöpää sairastavilla potilailla DUO‑E-tutkimuksessa (potilaat, joiden kasvaimen status oli pMMR)
Platinapohjainen solunsalpaajahoito + durvalumabi + olaparibi N = 191 | Platinapohjainen solunsalpaajahoito N = 192 | ||
| PFS (tutkijan arvio) (viimeinen tiedonkeruupäivä 12.4.2023) | |||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 108:191 (56,5) | 148:192 (77,1) | |
| Mediaania (95 %:n luottamusväli), kuukausina | 15,0 (12,4–18,0) | 9,7 (9,2–10,1) | |
| HR (95 %:n luottamusväli) | 0,57 (0,44–0,73) | ||
| OSb (viimeinen tiedonkeruupäivä 12.4.2023) | |||
| Tapahtumien määrä: Potilaiden kokonaismäärä (%) | 46:191 (24,1) | 64:192 (33,3) | |
| Mediaania (95 %:n luottamusväli), kuukausina | Ei saavutettu (ei saavutettu, ei saavutettu) | 25,9 (25,1, ei saavutettu) | |
| HR (95 %:n luottamusväli) | 0,69 (0,47–1,00) | ||
| Objektiivisen hoitovasteen saavuttaneiden osuusc (viimeinen tiedonkeruupäivä 12.4.2023) | |||
| Objektiivisen hoitovasteen saavuttaneiden määrä: niiden potilaiden kokonaismäärä, joiden tauti oli lähtötilanteessa mitattavissa (%) | 90:147 (61,2) | 92:156 (59,0) | |
| Vasteen kesto (viimeinen tiedonkeruupäivä 12.4.2023) | |||
| Mediaania (95 %:n luottamusväli), kuukausina | 18,7 (10,5, ei saavutettu) | 7,6 (7,1, 10,2) | |
aLaskettu Kaplan–Meier-menetelmällä.
bPerustuu ensimmäiseen välianalyysiin.
cVaste: Paras objektiivinen hoitovaste eli vahvistettu täydellinen hoitovaste tai osittainen hoitovaste.
HR: riskisuhde; OS: kokonaiselinaika; PFS: etenemisvapaa elinaika.
Kuva 21 DUO‑E: Kaplan–Meier-käyrä etenemisvapaalle elinajalle (potilaat, joiden kasvaimen status oli pMMR)
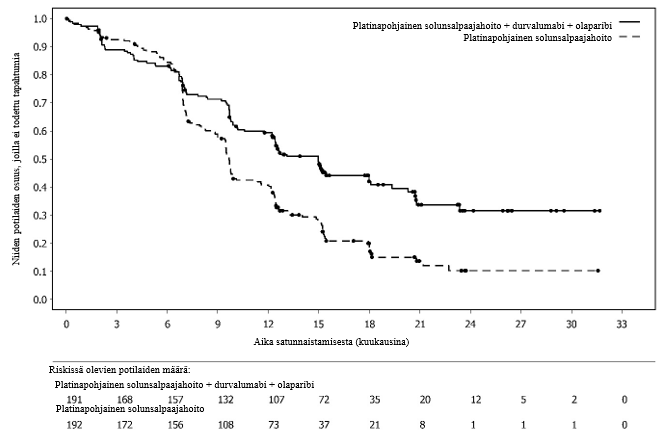
Potilailla, joiden kasvaimen status oli pMMR, etenemisvapaan elinajan riskisuhteet (HR) olivat 0,44 (95 %:n luottamusväli 0,31, 0,61) potilailla, jotka olivat positiivisia PD-L1:n ilmentymisen suhteen (236/383; 62 %), ja 0,87 (95 %:n luottamusväli 0,59, 1,28) potilailla, jotka olivat negatiivisia PD-L1:n ilmentymisen suhteen (140/383; 37 %), platinapohjainen solunsalpaajahoito + durvalumabi + olaparibi ‑hoitohaarassa verrattuna platinapohjaisen solunsalpaajahoidon hoitohaaraan. Positiivisuus PD-L1:n ilmentymisen suhteen määriteltiin tilanteeksi, jossa PD-L1-positiivisten solujen osuus kasvaimen pinta-alasta (tumour area positive, TAP) oli ≥ 1 %.
Pediatriset potilaat
Lynparza-valmisteen turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole varmistettu. Tutkimus D0816C00025 oli avoin vaiheen 1 monikeskustutkimus, jossa arvioitiin Lynparza-monoterapian turvallisuutta, siedettävyyttä, farmakokinetiikkaa, farmakodynamiikkaa ja alustavaa tehoa vähintään 6 kuukauden ja alle 18 vuoden ikäisillä pediatrisilla potilailla, joilla oli uusiutuneita tai refraktorisia kiinteitä tai primaarisia keskushermoston kasvaimia (lymfoidisia maligniteetteja lukuun ottamatta) ja joille ei ollut saatavilla tavanomaisia hoitovaihtoehtoja. Tutkimukseen otettiin mukaan 16 potilasta, jotka olivat vähintään 6- ja alle 18-vuotiaita ja joilla oli todettu puutteellinen homologiseen rekombinaatioon perustuva korjausmekanismi (HRR) tai HRR-geenimutaatio paikallisella testillä tai gBRCA-mutaatio keskitetysti tehdyllä testillä. Lynparza-valmistetta annettiin kerta-annoksena päivänä 1 ja sen jälkeen kaksi kertaa vuorokaudessa jatkuvalla aikataululla. Tutkimukseen mukaan otetuista 16 potilaasta 13 potilasta, jotka olivat vähintään 12- ja alle 18-vuotiaita, sai 300 mg olaparibia tablettimuodossa kaksi kertaa vuorokaudessa ja 3 potilasta, jotka olivat vähintään 6- ja alle 12-vuotiaita, sai 200 mg olaparibia tablettimuodossa kaksi kertaa vuorokaudessa, kunnes tauti eteni tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä. Tutkimukseen otetuilla 12 osallistujalla, joilla oli mitattavissa oleva sairaus lähtötilanteessa, ei havaittu objektiivista hoitovastetta. Tutkimustulosten perusteella ei voitu tehdä johtopäätöstä, että tällaisen käytön hyödyt olisivat suuremmat kuin sen riskit. Ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Farmakokinetiikka
300 mg:n annoksena annetun olaparibitabletin farmakokinetiikalle ovat tunnusomaisia noin 7 l/h:n näennäinen plasmapuhdistuma, noin 158 l:n näennäinen jakautumistilavuus ja 15 tunnin terminaalinen puoliintumisaika. Useita annoksia käytettäessä AUC-kumulaatiosuhteen havaittiin olevan 1,8 ja farmakokinetiikka vaikutti olevan jonkin verran ajasta riippuvaista.
Imeytyminen
Suun kautta tablettina annettu olaparibi (2 x 150 mg) imeytyy nopeasti ja mediaanihuippupitoisuudet plasmassa saavutetaan tyypillisesti 1,5 tunnin kuluttua antamisesta.
Olaparibin antaminen samanaikaisesti ruuan kanssa pienensi sen imeytymisnopeutta (tmax viivästyi 2,5 tunnilla ja Cmax pieneni noin 21 %), mutta ei merkittävästi vaikuttanut imeytyneen olaparibin määrään (AUC suureni 8 %). Tämän vuoksi Lynparza voidaan ottaa joko aterian yhteydessä tai tyhjään mahaan (ks. kohta Annostus ja antotapa).
Jakautuminen
In vitro plasma proteiineihin sitoutuminen on noin 82 % 10 µg/ml pitoisuudella, joka on suunnilleen Cmax.
Olaparibin sitoutuminen ihmisen plasmaproteiineihin in vitro oli annoksesta riippuvaista; sitoutunut fraktio oli noin 91 % 1 µg/ml pitoisuudella ja se pieneni 82 %:iin 10 µg/ml pitoisuudella ja 70 %:iin 40 µg/ml pitoisuudella. Puhdistettuja proteiineja sisältävissä liuoksissa albumiiniin sitoutunut olaparibifraktio oli noin 56 % eikä se ollut riippuvainen olaparibipitoisuuksista. Samalla analyysillä mitattuna happamaan alfa-1-glykoproteiiniin sitoutunut fraktio oli 29 % 10 µg/ml pitoisuudella, ja sitoutumisella oli taipumus vähentyä suuremmilla pitoisuuksilla.
Biotransformaatio
CYP3A4:n ja CYP3A5:n on osoitettu in vitro olevan olaparibin metaboloitumisesta ensisijaisesti vastaavia entsyymejä (ks. kohta Yhteisvaikutukset).
Annettaessa naispotilaille suun kautta 14C‑olaparibia suurin osa (70 %) verenkierron radioaktiivisuudesta plasmassa aiheutui muuttumattomasta olaparibista ja se oli pääkomponenttina sekä virtsassa (15 % annoksesta) että ulosteessa (6 % annoksesta). Olaparibi metaboloituu laajasti. Suurin osa metaboliasta johtuu hapetusreaktioista, jonka jälkeen osalle muodostuneista komponenteista tapahtuu glukuronidi‑ tai sulfaattikonjugaatio. Plasmasta todettiin 20, virtsasta 37 ja ulosteesta 20 metaboliittia. Suurin osa niistä vastasi < 1 %:a annetusta lääkkeestä. Merkittävimmät komponentit verenkierrossa olivat piperatsin-3-ol-ryhmä, jonka rengas oli avautunut, ja kaksi metaboliittia, joille oli tapahtunut mono-oksigenaatio (noin 10 % kumpaakin). Toinen näistä metaboliiteista oli myös päämetaboliitti kuonaeritteissä (6 % virtsan ja 5 % ulosteen radioaktiivisuudesta).
Olaparibi esti in vitro vain vähän tai ei lainkaan UGT1A4:a, UGT1A9:ää, UGT2B7:ää tai CYP1A2:ta, CYP2A6:ta, CYP2B6:ta, CYP2C8:aa, CYP2C9:ää, CYP2C19:ää, CYP2D6:ta tai CYP2E1:tä eikä sen odoteta olevan minkään näiden CYP‑entsyymien kliinisesti merkittävä ajasta riippuvainen estäjä. Olaparibi esti UGT1A1:tä in vitro, mutta fysiologiaan perustuvat farmakokineettiset mallinnukset viittaavat siihen, että tällä ei ole kliinistä merkitystä. In vitro olaparibi on P-glykoproteiinin (effluksikuljettajaproteiini) substraatti, mutta tällä ei todennäköisesti ole kliinistä merkitystä (ks. kohta Yhteisvaikutukset).
In vitro ‑tiedot myös osoittavat, että olaparibi ei ole OATP1B1:n, OATP1B3:n, OCT1:n, BCRP:n tai MRP2:n substraatti ja että se ei ole OATP1B3:n, OAT1:n tai MRP2:n estäjä.
Eliminaatio
14C‑olaparibin kerta‑annoksen jälkeen noin 86 % annostellusta radioaktiivisuudesta pystyttiin toteamaan 7 vuorokauden keräysjakson aikana, noin 44 % virtsasta ja noin 42 % ulosteesta. Suurin osa materiaalista erittyi metaboliitteina.
Erityisryhmät
Populaatiofarmakokineettisissä analyyseissä potilaan ikä, sukupuoli, paino, kasvaimen sijainti tai rotu (kaukaasialaiset ja japanilaiset potilaat mukaan lukien) eivät olleet merkitseviä kovariaatteja.
Munuaisten vajaatoiminta
Lievää munuaisten vajaatoimintaa (kreatiniinipuhdistuma 51–80 ml/min) sairastavien potilaiden AUC‑arvo suureni 24 % ja Cmax‑arvo 15 % verrattuna potilaisiin, joiden munuaisen toiminta oli normaali. Lynparza-annosta ei tarvitse muuttaa potilailla, joilla on lievä munuaisten vajaatoiminta.
Keskivaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma 31–50 ml/min) sairastavien potilaiden AUC‑arvo suureni 44 % ja Cmax‑arvo 26 % verrattuna potilaisiin, joiden munuaisten toiminta oli normaali. Lynparza‑annoksen muuttamista suositellaan potilailla, joilla on keskivaikea munuaisten vajaatoiminta (ks. kohta Annostus ja antotapa).
Tietoa potilaista, joilla on vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaissairaus (kreatiniinipuhdistuma < 30 ml/min) ei ole.
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa (Child-Pughin luokitus A) sairastavien potilaiden AUC‑arvo suureni 15 % ja Cmax‑arvo 13 % ja potilailla, jotka sairastavat keskivaikeaa maksan vajaatoimintaa (Child-Pughin luokitus B), AUC-arvo suureni 8 % ja Cmax-arvo pieneni 13 % verrattuna potilaisiin, joiden maksan toiminta oli normaali. Lynparza-annosta ei tarvitse muuttaa potilailla, joilla on lievä tai keskivaikea maksan vajaatoiminta (ks. kohta Annostus ja antotapa). Tietoja ei ole potilaista, joilla on vaikea maksan vajaatoiminta (Child-Pughin luokitus C).
Pediatriset potilaat
Olaparibin farmakokinetiikkaa arvioitiin D0816C00025-tutkimuksessa 14 lapsella ja nuorella, jotka olivat vähintään 6- ja alle 18-vuotiaita. Potilaat saivat olaparibia joko 300 mg kaksi kertaa vuorokaudessa (8 potilasta, vähintään 12- ja alle 18-vuotiaita) tai 200 mg kaksi kertaa vuorokaudessa (3 potilasta, 6–11 vuotiaita). Lisäksi 3 potilasta (vähintään 12- ja alle 18-vuotiaita) sai hoitoa signaalintunnistusvaiheessa, ja heille annettiin Lynparza-valmistetta 300 mg:n tablettina kaksi kertaa vuorokaudessa, kunnes tauti eteni tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä. Molemmilla ryhmillä plasmassa todetut pitoisuudet osoittivat, että altistus oli vastaavanlainen kuin aikuisilla 300 mg:n annoksella.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttama toksisuus
Kuusi kuukautta kestäneissä rotilla ja koirilla tehdyissä toistuvan altistuksen aiheuttamaa toksisuutta koskevissa tutkimuksissa olaparibin suun kautta annetut vuorokausiannokset olivat hyvin siedettyjä. Toksisuuden merkittävin ensisijainen kohde‑elin molemmissa lajeissa oli luuydin ja tällaiseen toksisuuteen liittyi muutoksia perifeerisissä hematologisissa parametreissä. Nämä muutokset korjaantuivat 4 viikon kuluessa olaparibin annon lopettamisesta. Rotilla havaittiin myös hyvin vähäisiä degeneratiivisia vaikutuksia maha-suolikanavaan. Näitä löydöksiä todettiin altistuksilla, jotka olivat pienempiä kuin kliinisesti käytetyt annokset. Ihmisen luuydinsoluilla tehdyt tutkimukset osoittivat myös, että suora altistus olaparibille voi aiheuttaa toksisuutta luuydinsoluissa ex vivo.
Genotoksisuus
Olaparibilla ei todettu mutageenista vaikutusta, mutta se oli klastogeeninen nisäkässoluissa in vitro. Rotille suun kautta annettuna olaparibi indusoi mikrotumia luuytimessä. Tämä klastogeenisuus vastaa olaparibin tunnettua farmakologiaa ja viittaa mahdolliseen genotoksiseen vaikutukseen ihmisessä.
Karsinogeenisuus
Olaparibilla ei ole tehty karsinogeenisuustutkimuksia.
Lisääntymistoksisuus
Naaraiden hedelmällisyyttä koskeneessa tutkimuksessa, jossa rotille annettiin olaparibia hedelmöityneen munasolun kiinnittymiseen asti, ei todettu vaikutuksia parittelukykyyn tai tiinehtyvyyteen, vaikka joillakin eläimillä todettiin pitkittynyt kiima. Alkioiden ja sikiöiden eloonjäänti kuitenkin hiukan väheni.
Rotilla tehdyissä alkion/sikiön kehitystä koskeneissa tutkimuksissa annoksilla, jotka eivät aiheuttaneet merkittävää toksisuutta emolle, olaparibi vähensi alkioiden/sikiöiden eloonjäämistä, pienensi sikiöiden painoa ja aiheutti sikiöille kehityshäiriöitä mukaan lukien huomattavia silmien epämuodostumia (esim. anoftalmia, mikroftalmia), nikamien/kylkiluiden epämuodostumia ja sisäelinten sekä luuston poikkeavuuksia.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Kopovidoni
Piioksidi, kolloidinen, vedetön
Mannitoli
Natriumstearyylifumaraatti
Tabletin päällyste
Hypromelloosi
Makrogoli 400
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Musta rautaoksidi (E172) (vain 150 mg:n tableteissa)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LYNPARZA tabletti, kalvopäällysteinen
100 mg (L:ei) 56 fol (7x8 fol) (2196,80 €)
150 mg (L:ei) 56 fol (7x8 fol) (2576,44 €)
PF-selosteen tieto
Alu/alu läpipainopakkaus ilman repäisyviivoja, jossa on 8 kalvopäällysteistä tablettia.
Pakkauskoot:
56 kalvopäällysteistä tablettia (7 läpipainopakkausta).
Moniannospakkaus, jossa on 112 kalvopäällysteistä tablettia (kaksi 56 tabletin pakkausta).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Lynparza 100 mg kalvopäällysteinen tabletti
Keltainen tai tummankeltainen soikea kaksoiskupera tabletti, jonka toiselle puolelle on kaiverrettu ”OP100” ja toisella puolella ei ole merkintöjä.
Lynparza 150 mg kalvopäällysteinen tabletti
Vihreä tai vihreänharmaa soikea kaksoiskupera tabletti, jonka toiselle puolelle on kaiverrettu ”OP150” ja toisella puolella ei ole merkintöjä.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
LYNPARZA tabletti, kalvopäällysteinen
100 mg 56 fol
150 mg 56 fol
- Ylempi erityiskorvaus (100 %). Olaparibi: Munasarja-, munanjohdin- tai primaarin vatsakalvon, varhaisvaiheen rintasyövän sekä etäpesäkkeisen kastraatioresistentin eturauhassyövän hoito erityisin edellytyksin (1511).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Olaparibi: Munasarja-, munanjohdin- tai primaaria vatsakalvon syöpää, rintasyöpää, eturauhassyöpää tai haimasyöpää sairastavien aikuispotilaiden hoito erityisin edellytyksin (389).
ATC-koodi
L01XK01
Valmisteyhteenvedon muuttamispäivämäärä
12.02.2026
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi