
SAXENDA injektioneste, liuos, esitäytetty kynä 6 mg/ml
Vaikuttavat aineet ja niiden määrät
1 ml liuosta sisältää 6 mg liraglutidia*. Yksi esitäytetty kynä sisältää 18 mg liraglutidia 3 ml:ssa.
* Ihmisen glukagonin kaltaisen peptidi 1:n (GLP-1) analogi, joka on tuotettu yhdistelmä-DNA-tekniikalla Saccharomyces cerevisiae -hiivassa.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
Aikuiset
Saxenda on tarkoitettu painon hallinnan avuksi vähäenergiaisen ruokavalion ja fyysisen aktiivisuuden lisäämisen rinnalla aikuisille potilaille, joiden painoindeksi (BMI) on lähtötilanteessa
- ≥ 30 kg/m² (lihavuus) tai
- ≥ 27–alle 30 kg/m² (ylipaino), kun potilaalla on lisäksi vähintään yksi painoon liittyvä sairaus, kuten dysglykemia (diabeteksen esiaste tai tyypin 2 diabetes mellitus), kohonnut verenpaine, dyslipidemia tai obstruktiivinen uniapnea.
Saxenda-hoito on lopetettava 12 viikon jälkeen 3,0 mg:n vuorokausiannoksella, jos potilaan paino ei ole laskenut vähintään 5 % lähtöpainosta.
Nuoret (≥ 12‑vuotiaat)
Saxenda-valmistetta voidaan käyttää painon hallinnan apuna terveellisen ruokavalion ja fyysisen aktiivisuuden lisäämisen rinnalla nuorille, vähintään 12‑vuotiaille potilaille, jotka täyttävät seuraavat ehdot:
- lihavuus (BMI, joka vastaa kansainvälisten raja-arvojen mukaan vähintään aikuisten painoindeksiä 30 kg/m2)* ja
- paino yli 60 kg.
Saxenda-hoito on lopetettava ja arvioitava uudelleen, jos potilaan BMI tai BMI z ‑arvo ei ole pienentynyt vähintään 4 %, kun potilas on käyttänyt 12 viikon ajan 3,0 mg:n vuorokausiannosta tai suurinta siedettyä annosta.
*IOTF:n (International Obesity Task Force) määrittelemät lihavuuden BMI-raja-arvot sukupuolen mukaan 12–18-vuotiaille (ks. taulukko 1) tutkimuksen 4180 tutkimusasetelman mukaisesti, ks. kohta Farmakodynamiikka.
Taulukko 1 IOTF:n määrittelemät lihavuuden BMI-raja-arvot sukupuolen mukaan 12–18-vuotiaille
Ikä (vuotta) | BMI, joka vastaa kansainvälisten raja-arvojen mukaan aikuisten BMI:tä 30 kg/m2. | |
| Pojat | Tytöt | |
| 12 | 26,02 | 26,67 |
| 12,5 | 26,43 | 27,24 |
| 13 | 26,84 | 27,76 |
| 13,5 | 27,25 | 28,20 |
| 14 | 27,63 | 28,57 |
| 14,5 | 27,98 | 28,87 |
| 15 | 28,30 | 29,11 |
| 15,5 | 28,60 | 29,29 |
| 16 | 28,88 | 29,43 |
| 16,5 | 29,14 | 29,56 |
| 17 | 29,41 | 29,69 |
| 17,5 | 29,70 | 29,84 |
| 18 | 30,00 | 30,00 |
Lapset (6 – < 12‑vuotiaat)
Saxenda on tarkoitettu painon hallinnan avuksi terveellisen ruokavalion ja fyysisen aktiivisuuden lisäämisen rinnalla lapsille, 6 – < 12‑vuotiaille potilaille, jotka täyttävät seuraavat ehdot:
- lihavuus (BMI ≥ 95. persentiilin tasolla)* ja
- paino ≥ 45 kg.
Saxenda-hoito on lopetettava ja arvioitava uudelleen, jos potilaan BMI tai BMI z ‑arvo ei ole pienentynyt vähintään 4 %, kun potilas on käyttänyt 12 viikon ajan 3,0 mg:n vuorokausiannosta tai suurinta siedettyä annosta.
*Yhdysvaltain tautikeskuksen (CDC) määrittelemät lihavuuden BMI-raja-arvot (≥ 95. persentiilin taso) sukupuolen mukaan 6 – < 12‑vuotiaille (ks. taulukko 2) tutkimuksen 4392 tutkimusasetelman mukaisesti, ks. kohta Farmakodynamiikka.
Taulukko 2 Lihavuuden BMI-raja-arvot [≥ 95. persentiilin taso, BMI = paino (kg) jaettuna pituuden (m) neliöllä] sukupuolen mukaan 6 – < 12‑vuotiaille lapsille
| Ikä (vuotta) | Lihavuus BMI ≥ 95. persentiilin tasolla | |
|---|---|---|
| Pojat | Tytöt | |
| 6 | 18,41 | 18,84 |
| 6,5 | 18,76 | 19,23 |
| 7 | 19,15 | 19,68 |
| 7,5 | 19,59 | 20,17 |
| 8 | 20,07 | 20,70 |
| 8,5 | 20,57 | 21,25 |
| 9 | 21,09 | 21,82 |
| 9,5 | 21,62 | 22,40 |
| 10 | 22,15 | 22,98 |
| 10,5 | 22,69 | 23,57 |
| 11 | 23,21 | 24,14 |
| 11,5 | 23,73 | 24,71 |
Annostus ja antotapa
Annostus
Aikuiset
Aloitusannos on 0,6 mg kerran vuorokaudessa. Annosta pitää nostaa 3,0 mg:aan kerran vuorokaudessa 0,6 mg:n lisäyksin vähintään yhden viikon välein gastrointestinaalisen siedettävyyden parantamiseksi (ks. taulukko 3). Jos annoksen suurentamista seuraavaan annosaskelmaan ei siedetä kahtena peräkkäisenä viikkona, tulee harkita hoidon keskeyttämistä. Yli 3,0 mg:n vuorokausiannoksia ei suositella.
Taulukko 3 Annoskoon suurentamisaikataulu
| Annos | Viikot | |
Annoskoon suurentaminen 4 viikkoa | 0,6 mg | 1 |
| 1,2 mg | 1 | |
| 1,8 mg | 1 | |
| 2,4 mg | 1 | |
| Ylläpitoannos | 3,0 mg | |
Nuoret (≥ 12‑vuotiaat)
Vähintään 12- ja alle 18-vuotiailla nuorilla on noudatettava samaa annoskoon suurentamisaikataulua kuin aikuisilla (ks. taulukko 3). Annosta suurennetaan 3,0 mg:aan (ylläpitoannos) tai suurimpaan siedettyyn annokseen asti. Yli 3,0 mg:n vuorokausiannoksia ei suositella.
Lapset (6 – < 12‑vuotiaat)
Vähintään 6- ja alle 12‑vuotiailla lapsilla on noudatettava samaa annoskoon suurentamisaikataulua kuin aikuisilla (ks. taulukko 3). Annosta suurennetaan 3,0 mg:aan (ylläpitoannos) tai suurimpaan siedettyyn annokseen asti. Yli 3,0 mg:n vuorokausiannoksia ei suositella. Liraglutidihoidon lapsille saa aloittaa vain lasten lihavuuden hoitoon perehtynyt lääkäri.
Unohtunut annos
Jos annos on jäänyt väliin ja sen tavallisesta pistoajasta on enintään 12 tuntia, potilaan pitää ottaa annos mahdollisimman pian. Jos seuraavaan annokseen on alle 12 tuntia aikaa, potilaan ei pidä ottaa väliin jäänyttä annosta, vaan jatkaa kerran vuorokaudessa otettavaa hoitoa seuraavasta suunnitellusta annoksesta. Väliin jääneen annoksen korvaamiseksi ei pidä ottaa ylimääräistä tai isompaa annosta.
Tyypin 2 diabetes mellitusta sairastavat potilaat
Saxenda-valmistetta ei pidä käyttää samanaikaisesti toisen GLP-1-reseptoriagonistin kanssa.
Kun Saxenda-hoito aloitetaan, on harkittava samanaikaisesti annettavan insuliinin tai insuliinin eritystä lisäävän lääkkeen (esimerkiksi sulfonyyliurea) annoksen pienentämistä hypoglykemiariskin pienentämiseksi. Verensokerin omaseuranta on välttämätöntä insuliinin tai insuliinin eritystä lisäävän lääkkeen annoksen säätämiseksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Erityisryhmät
Iäkkäät potilaat (≥ 65-vuotiaat)
Annosta ei tarvitse säätää iän perusteella. 75-vuotiaiden ja sitä vanhempien potilaiden hoidosta on vain vähän kokemusta, eikä käyttöä näille potilaille suositella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen säätäminen ei ole tarpeen, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma ≥ 30 ml/min). Saxenda-valmistetta ei suositella potilaille, joilla on vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma < 30 ml/min), loppuvaiheen munuaissairaus mukaan luettuna (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset ja Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen säätämistä ei suositella, jos potilaalla on lievä tai keskivaikea maksan vajaatoiminta. Saxenda-valmistetta ei suositella potilaille, joilla on vaikea maksan vajaatoiminta ja sen käytössä pitää noudattaa varovaisuutta potilailla, joilla on lievä tai keskivaikea maksan vajaatoiminta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Annoksen säätäminen ei ole tarpeen vähintään 6-vuotiailla nuorilla ja lapsilla.
Saxenda-valmisteen turvallisuutta ja tehoa alle 6 vuoden ikäisten lasten hoidossa ei ole varmistettu (ks. kohta Farmakodynamiikka).
Antotapa
Saxenda annetaan vain ihonalaisena pistoksena. Sitä ei saa pistää laskimoon tai lihakseen.
Saxenda annetaan kerran vuorokaudessa mihin vuorokaudenaikaan tahansa, aterioista riippumatta. Se pistetään vatsaan, reiteen tai olkavarteen. Pistoskohtaa ja -ajoitusta voidaan muuttaa ilman, että annosta tarvitsee säätää. On kuitenkin suositeltavaa pistää Saxenda suunnilleen samaan aikaan päivästä, kun sopivin aika on valittu. Pistoskohtia on aina vaihdeltava pistosalueen amyloidisaostumien riskin pienentämiseksi (ks. kohta Haittavaikutukset).
Lisätietoja annostuksesta on kohdassa Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Yliherkkyys liraglutidille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Aspiraatio yleisanestesian tai syvän sedaation yhteydessä
GLP-1 -reseptoriagonisteja yleisanestesiassa tai syvässä sedaatiossa saaneilla potilailla on ilmoitettu aspiraatiopneumoniatapauksista. Siksi on otettava huomioon lisääntynyt riski mahaan jääneestä sisällöstä viivästyneen tyhjentymisen takia (ks. kohta Haittavaikutukset) ennen yleisanestesian tai syvän sedaation aikana suoritettavia toimenpiteitä.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Potilaat, joilla on sydämen vajaatoiminta
New York Heart Associationin (NYHA) luokan IV sydämen vajaatoimintapotilaiden hoidosta ei ole kliinistä kokemusta, ja sen vuoksi liraglutidia ei suositella käytettäväksi näille potilaille.
Erityisryhmät
Liraglutidin turvallisuutta ja tehoa painon hallinnassa ei ole varmistettu:
- 75-vuotiaille ja sitä vanhemmille potilaille,
- potilaille, jotka käyttävät muita painonhallintavalmisteita,
- potilaille, joiden lihavuus on sekundääristä, liittyen endokrinologisiin häiriöihin tai syömishäiriöihin tai sellaiseen lääkehoitoon, joka voi aiheuttaa painon nousua,
- vaikeaa munuaisten vajaatoimintaa sairastaville potilaille,
- vaikeaa maksan vajaatoimintaa sairastaville potilaille.
Käyttöä näille potilaille ei suositella (ks. kohta Annostus ja antotapa).
Liraglutidin käyttöä painonhallinnassa ei tutkittu potilailla, joilla on lievä tai keskivaikea maksan vajaatoiminta, joten sitä pitää käyttää varoen näillä potilailla (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Valmisteen vaikutuksesta potilaisiin, joilla on tulehduksellinen suolistosairaus tai diabeettinen gastropareesi, on niukasti kokemusta. Liraglutidia ei suositella näille potilaille, koska sen käyttöön liittyy ohimeneviä ruoansulatuselimistön haittavaikutuksia, kuten pahoinvointia, oksentelua ja ripulia.
Haimatulehdus
GLP-1-reseptoriagonistien käytön yhteydessä on todettu akuutteja haimatulehduksia. Potilaille tulee kertoa akuutille haimatulehdukselle tyypillisistä oireista. Jos haimatulehdusta epäillään, liraglutidilääkitys tulee keskeyttää. Jos akuutti haimatulehdus varmistuu, liraglutidilääkitystä ei pidä aloittaa uudelleen.
Sappikivitauti ja sappirakkotulehdus
Painonhallintaan liittyvissä kliinisissä tutkimuksissa havaittiin, että sappikivitautia ja sappirakkotulehdusta esiintyi enemmän liraglutidilla hoidetuilla potilailla kuin lumelääkettä saavilla potilailla. Merkittävä painonlasku voi suurentaa sappikivitaudin ja sen myötä sappirakkotulehduksen riskiä, mutta tämä selittää liraglutidin käyttöön liittyneen suuremman sairastuneiden määrän vain osittain. Sappikivitauti ja sappirakkotulehdus voivat johtaa sairaalahoidon tarpeeseen ja sappirakon poistoon. Potilaille tulee kertoa sappikivitaudille ja sappirakkotulehdukselle tyypillisistä oireista.
Kilpirauhassairaus
Tyypin 2 diabetesta koskevissa kliinisissä tutkimuksissa on raportoitu esiintyneen kilpirauhaseen kohdistuvia haittatapahtumia kuten struumaa erityisesti potilailla, joilla on aiemmin ollut kilpirauhassairaus. Liraglutidia pitää sen vuoksi käyttää varoen potilaille, joilla on kilpirauhassairaus.
Sydämen syke
Kliinisissä tutkimuksissa havaittiin sydämen sykkeen nopeutuminen liraglutidin käytön yhteydessä (ks. kohta Farmakodynamiikka). Sykettä pitää seurata säännöllisin väliajoin tavallisten hoitokäytäntöjen mukaisesti. Potilaille tulee kertoa nopeutuneen sydämen sykkeen oireista (sydämentykytys tai nopea syke levossa). Jos potilaalla on nopeutunut sydämen leposyke ja se on kliinisesti merkitsevää ja pitkittynyttä, liraglutidihoito on keskeytettävä.
Kuivuminen
GLP-1-reseptoriagonistilla hoidetuilla potilailla on raportoitu esiintyneen merkkejä ja oireita kuivumisesta, mukaan lukien munuaisten toimintahäiriötä ja akuuttia munuaisten vajaatoimintaa. Liraglutidilla hoidettaville potilaille tulee kertoa mahdollisesta kuivumisriskistä, joka liittyy ruoansulatuselimistöön kohdistuviin haittavaikutuksiin, ja heitä pitää ohjeistaa ryhtymään varotoimiin nestevajauksen estämiseksi.
Hypoglykemia tyypin 2 diabetes mellitusta sairastavilla potilailla
Tyypin 2 diabetes mellitusta sairastavilla potilailla, jotka saavat liraglutidia yhdessä insuliinin ja/tai sulfonyyliurean kanssa, voi olla suurentunut hypoglykemian riski. Hypoglykemian riskiä voidaan pienentää insuliini- ja/tai sulfonyyliurea-annosta pienentämällä.
Pediatriset potilaat
Liraglutidihoitoa saaneilla nuorilla (≥ 12-vuotiailla) on ilmoitettu kliinisesti merkittäviä hypoglykemiatapahtumia. Potilaille on kerrottava hypoglykemian tyypillisistä oireista ja oikeista toimintatavoista.
Hyperglykemia diabetes mellitusta sairastavilla potilailla, jotka saavat insuliinihoitoa
Saxenda-valmistetta ei saa käyttää insuliinin korvikkeena diabetes mellitusta sairastaville potilaille. Diabeettista ketoasidoosia on ilmoitettu esiintyneen insuliinista riippuvaisilla potilailla insuliinin käytön nopean keskeyttämisen tai annoksen pienentämisen jälkeen (ks. kohta Annostus ja antotapa).
Apuaineet
Saxenda sisältää alle 1 mmol natriumia (23 mg) annosta kohden eli se on olennaisesti ”natriumiton”.
Yhteisvaikutukset
In vitro -tutkimusten mukaan liraglutidilla on hyvin vähäinen potentiaali aiheuttaa farmakokineettisiä yhteisvaikutuksia muiden sytokromi P450 -entsyymien (CYP) välityksellä metaboloituvien ja plasmaproteiinien sitoutumiseen vaikuttavien aineiden kanssa.
Liraglutidi hidastaa hieman mahan tyhjentymistä, joten se saattaa vaikuttaa suun kautta samanaikaisesti otettavien lääkkeiden imeytymiseen. Yhteisvaikutustutkimuksissa ei ole osoitettu kliinisesti merkitsevää imeytymisviivettä, ja sen vuoksi annoksen säätämistä ei tarvita.
Yhteisvaikutustutkimuksia on tehty 1,8 mg:lla liraglutidia. Vaikutus mahan tyhjenemisnopeuteen oli samanlainen 1,8 mg:lla ja 3,0 mg:lla liraglutidia (parasetamolin AUC0-300 min). Vain harva liraglutidilla hoidettu potilas raportoi vähintään yhdestä vaikeasta ripulijaksosta. Ripuli voi vaikuttaa samanaikaisesti suun kautta otettavien lääkkeiden imeytymiseen.
Varfariini ja muut kumariinijohdokset
Yhteisvaikutustutkimuksia ei ole tehty. Kliinisesti merkitsevää yhteisvaikutusta ei voida sulkea pois varfariinin ja muiden sellaisten vaikuttavien aineiden kanssa, jotka liukenevat heikosti tai joilla on kapea terapeuttinen indeksi. On suositeltavaa, että varfariinia tai muita kumariinijohdoksia saavien potilaiden INR-arvoa (International Normalised Ratio) seurataan useammin liraglutidihoidon alussa.
Parasetamoli (asetaminofeeni)
Liraglutidi ei muuttanut parasetamolin kokonaisaltistusta 1 000 mg:n parasetamolikerta-annoksen jälkeen. Parasetamolin Cmax pieneni 31 % ja mediaani-tmax viivästyi enintään 15 minuuttia. Samanaikaisesti käytettävän parasetamolin annosta ei tarvitse muuttaa.
Atorvastatiini
Liraglutidi ei muuttanut atorvastatiinin kokonaisaltistusta 40 mg:n atorvastatiinikerta-annoksen jälkeen. Tämän vuoksi atorvastatiinin annosta ei tarvitse muuttaa, kun sitä annetaan liraglutidin kanssa. Atorvastatiinin Cmax pieneni 38 % ja mediaani-tmax viivästyi 1 tunnista 3 tuntiin, kun atorvastatiinia annettiin yhdessä liraglutidin kanssa.
Griseofulviini
Liraglutidi ei muuttanut griseofulviinin kokonaisaltistusta 500 mg:n griseofulviinikerta-annoksen jälkeen. Griseofulviinin Cmax suureni 37 %, mediaani-tmax ei muuttunut. Griseofulviinin ja muiden heikosti liukenevien ja solukalvoja hyvin läpäisevien aineiden annoksia ei tarvitse muuttaa.
Digoksiini
Liraglutidin anto 1 mg:n digoksiinikerta-annoksen kanssa johti digoksiinin AUC-arvon pienentymiseen 16 %:lla; Cmax pieneni 31 %. Digoksiinin mediaani-tmax viivästyi 1 tunnista 1,5 tuntiin. Näiden tulosten perusteella digoksiinin annosta ei tarvitse muuttaa.
Lisinopriili
Liraglutidin anto 20 mg:n lisinopriilikerta-annoksen kanssa johti lisinopriilin AUC-arvon pienentymiseen 15 %:lla; Cmax pieneni 27 %. Liraglutidi viivästytti lisinopriilin mediaani-tmax:ia 6 tunnista 8 tuntiin. Näiden tulosten perusteella lisinopriilin annosta ei tarvitse muuttaa.
Suun kautta otettavat ehkäisyvalmisteet
Liraglutidi pienensi etinyyliestradiolin Cmax-arvoa 12 % ja levonorgestreelin Cmax-arvoa 13 % ehkäisyvalmisteen kerta-annoksen jälkeen. Liraglutidi viivästytti kummankin aineen tmax-aikaa 1,5 tuntia. Liraglutidilla ei ollut kliinisesti merkitsevää vaikutusta etinyyliestradiolin eikä myöskään levonorgestreelin kokonaisaltistukseen. Liraglutidin samanaikaisen käytön ei siis odoteta vaikuttavan ehkäisyvalmisteiden tehoon.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisilla tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Liraglutidin käytöstä raskaana oleville naisille on vain vähän tietoa. Eläinkokeet ovat osoittaneet reproduktiivista toksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmisille ei tunneta.
Liraglutidia ei pidä käyttää raskauden aikana. Jos potilas suunnittelee raskautta tai tulee raskaaksi, liraglutidihoito on lopetettava.
Imetys
Ei tiedetä, erittyykö liraglutidi ihmisen rintamaitoon. Eläinkokeissa liraglutidin ja sitä rakenteellisesti muistuttavien metaboliittien siirtyminen rintamaitoon on ollut vähäistä. Imeväisikäisillä rotanpoikasilla tehdyissä prekliinisissä tutkimuksissa on todettu hoitoon liittyvää neonataalivaiheen kasvun hidastumista (ks. kohta Prekliiniset tiedot turvallisuudesta). Kokemuksen puutteen vuoksi Saxenda-valmistetta ei tule käyttää imetyksen aikana.
Hedelmällisyys
Lukuun ottamatta hienoista vähenemistä elävien sikiöiden lukumäärässä, eläinkokeet eivät osoittaneet haitallisia vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Saxenda-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Saxenda-hoidon aikana saattaa kuitenkin ilmetä huimausta, lähinnä ensimmäisten 3 kuukauden aikana. Jos huimausta ilmenee, ajettaessa tai koneita käytettäessä on noudatettava varovaisuutta.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto:
Saxenda-valmisteen turvallisuutta arvioitiin viidessä kaksoissokkoutetussa, lumelääkekontrolloidussa tutkimuksessa, joihin osallistui 5 813 ylipainoista tai lihavaa aikuispotilasta, joilla oli vähintään yksi painoon liittyvä lisäsairaus. Kaiken kaikkiaan ruoansulatuskanavan reaktiot olivat useimmin raportoituja haittavaikutuksia Saxenda-hoidon aikana (67,9 %) (ks. kohta "Valittujen haittavaikutusten kuvaus").
Taulukoitu haittavaikutuslista
Taulukossa 4 luetellaan aikuisilla raportoidut haittavaikutukset. Haittavaikutukset on lueteltu elinjärjestelmäluettelon ja esiintymistiheyden mukaan. Esiintymistiheydet on määritetty seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, <1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Jokaisessa esiintymistiheysryhmässä haittavaikutukset on esitetty vakavuuden mukaan vähenevässä järjestyksessä.
Taulukko 4 Aikuisilla raportoidut haittavaikutukset
| MedDRA:n elinjärjestelmäluettelo | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Tuntematon |
| Immuunijärjestelmä | Anafylaktinen reaktio | ||||
| Aineenvaihdunta ja ravitsemus | Hypoglykemia* | Kuivuminen | |||
| Psyykkiset häiriöt | Unettomuus** | ||||
| Hermosto | Päänsärky | Huimaus Dysgeusia | |||
| Sydän | Takykardia | ||||
| Ruoansulatuselimistö | Pahoinvointi Oksentelu Ripuli Ummetus | Suun kuivuminen Dyspepsia Mahatulehdus Gastroesofageaalinen refluksitauti Ylävatsan kivut Ilmavaivat Röyhtäily Vatsan turvotus | Haimatulehdus*** Viivästynyt mahan tyhjeneminen**** | Suolitukos† | |
| Maksa ja sappi | Sappikivitauti*** | Sappirakkotulehdus*** | |||
| Iho ja ihonalainen kudos | Ihottuma | Urtikaria | Ihoamyloidoosi | ||
| Munuaiset ja virtsatiet | Akuutti munuaisten vajaatoiminta Munuaisten toimintahäiriö | ||||
| Yleisoireet ja antopaikassa todettavat haitat | Pistoskohdan reaktiot Heikotus Uupumus | Huonovointisuus | |||
| Tutkimukset | Suurentunut lipaasiarvo Suurentunut amylaasiarvo |
*Hypoglykemiaa (perustuen potilaiden itse raportoimiin oireisiin, joita ei ole vahvistettu verenglukoosimittauksilla) raportoitiin esiintyneen potilailla, joilla ei ollut tyypin 2 diabetes mellitusta ja joita hoidettiin Saxenda-valmisteella yhdessä ruokavalion ja liikunnan kanssa. Katso lisätietoja kohdasta ”Valittujen haittavaikutusten kuvaus”.
** Unettomuutta esiintyi lähinnä hoidon ensimmäisten 3 kuukauden aikana.
*** Ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
**** Kontrolloiduista faasin 2, 3a ja 3b kliinisistä tutkimuksista.
† Haittavaikutus markkinoilletulon jälkeisistä lähteistä
Valittujen haittavaikutusten kuvaus
Hypoglykemia potilailla, jotka eivät sairasta tyypin 2 diabetes mellitusta
Kliinisissä tutkimuksissa, joihin osallistui ylipainoisia tai lihavia potilaita, joilla ei ollut tyypin 2 diabetes mellitusta ja joita hoidettiin Saxenda-valmisteella yhdessä ruokavalion ja liikunnan kanssa, ei raportoitu vaikeita hypoglykemiatapahtumia (joissa potilas olisi tarvinnut ulkopuolista apua). 1,6 % Saxenda-valmisteella hoidetuista potilaista ja 1,1 % lumelääkettä saaneista potilaista raportoi hypoglykemian oireista. Näitä tapahtumia ei kuitenkaan varmistettu verenglukoosimittauksilla. Suurin osa tapahtumista oli lieviä.
Hypoglykemia tyypin 2 diabetes mellitusta sairastavilla potilailla
Kliinisessä tutkimuksessa, johon osallistui ylipainoisia tai lihavia potilaita, jotka sairastivat tyypin 2 diabetes mellitusta ja joita hoidettiin Saxenda-valmisteella yhdessä ruokavalion ja liikunnan kanssa, 0,7 % Saxenda-valmisteella hoidetuista potilaista raportoi vaikeaa hypoglykemiaa (jossa potilas tarvitsi ulkopuolista apua) ja sitä raportoitiin esiintyneen vain potilailla, joita hoidettiin samanaikaisesti sulfonyyliurealla. Lisäksi tästä potilasjoukosta 43,6 % Saxenda-valmisteella hoidetuista potilaista ja 27,3 % lumelääkettä saavista potilaista raportoi varmistetuista oireellisista hypoglykemioista. Potilasjoukossa, jota ei hoidettu samanaikaisesti sulfonyyliurealla, 15,7 % Saxenda-valmisteella hoidetuista potilaista ja 7,6 % lumelääkkeellä hoidetuista potilaista raportoi varmistettuja oireellisia hypoglykemiatapahtumia (määritetty plasman glukoosipitoisuus ≤3,9 mmol/l, mihin liittyi oireita).
Hypoglykemia tyypin 2 diabetes mellitusta sairastavilla potilailla, joita hoidetaan insuliinilla
Kliinisessä tutkimuksessa, johon osallistui ylipainoisia tai lihavia potilaita, jotka sairastivat tyypin 2 diabetes mellitusta ja joita hoidettiin insuliinilla ja liraglutidin 3,0 mg:n vuorokausiannoksella yhdessä ruokavalion ja liikunnan sekä enintään kahden peroraalisen diabeteslääkkeen kanssa, liraglutidin 3,0 mg:n vuorokausiannoksella hoidetuista potilaista 1,5 % raportoi vaikeaa hypoglykemiaa (jossa potilas tarvitsi ulkopuolista apua). Tutkimuksessa liraglutidin 3,0 mg:n vuorokausiannoksella hoidetuista potilaista 47,2 % ja lumelääkettä saaneista potilaista 51,8 % raportoi vahvistettuja oireellisia hypoglykemioita (määritetty plasman glukoosipitoisuus ≤ 3,9 mmol/l, mihin liittyi oireita). Potilasjoukossa, jota hoidettiin samanaikaisesti sulfonyyliurealla, liraglutidin 3,0 mg:n vuorokausiannoksella hoidetuista potilaista 60,9 % ja lumelääkettä saaneista potilaista 60,0 % raportoi vahvistettuja oireellisia hypoglykemiatapahtumia.
Ruoansulatuselimistön haittavaikutukset
Useimmat ruoansulatuselimistön tapahtumista olivat lieviä tai keskivaikeita sekä ohimeneviä ja suurin osa ei johtanut hoidon keskeyttämiseen. Reaktioita esiintyi tavallisesti hoidon ensimmäisten viikkojen aikana, ja ne vähenivät muutaman päivän tai muutaman viikon aikana hoitoa jatkettaessa.
65-vuotiailla ja sitä vanhemmilla potilailla Saxenda voi lisätä ruoansulatuselimistön häiriöitä.
Potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma ≥ 30 ml/min) voi olla enemmän ruoansulatuselimistön häiriöitä, kun heitä hoidetaan Saxenda-valmisteella.
Akuutti munuaisten vajaatoiminta
GLP-1-reseptoriagonisteilla hoidetuilla potilailla on raportoitu esiintyneen akuuttia munuaisten vajaatoimintaa. Suurin osa näistä raportoiduista tapahtumista esiintyi potilailla, joilla oli ollut pahoinvointia, oksentelua tai ripulia, joka johti vähentyneeseen nestetilavuuteen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Allergiset reaktiot
Harvoja anafylaktisia reaktioita, joissa on oireina esim. matalaa verenpainetta, sydämentykytystä, hengenahdistusta ja turvotusta, on raportoitu liraglutidin kauppaantuonnin jälkeisessä käytössä. Anafylaktiset reaktiot voivat olla mahdollisesti hengenvaarallisia. Jos epäillään anafylaktista reaktiota, liraglutidihoito pitää lopettaa eikä sitä saa aloittaa uudelleen (ks. kohta Vasta-aiheet).
Pistoskohdan reaktiot
Pistoskohdan reaktioita on raportoitu esiintyneen Saxenda-valmisteella hoidetuilla potilailla. Nämä reaktiot olivat yleensä lieviä ja ohimeneviä, ja suurin osa hävisi hoitoa jatkettaessa.
Takykardia
Kliinisissä tutkimuksissa takykardiaa raportoitiin 0,6 %:lla Saxenda-valmisteella hoidetuista potilaista ja 0,1 %:lla lumelääkkeellä hoidetuista potilaista. Suurin osa tapahtumista oli lieviä tai keskivaikeita. Tapahtumat olivat yksittäisiä ja suurin osa hävisi Saxenda-hoitoa jatkettaessa.
Ihoamyloidoosi
Pistoskohdassa voi esiintyä ihoamyloidoosia (ks. kohta Annostus ja antotapa).
Pediatriset potilaat
Kliinisessä tutkimuksessa, johon osallistui vähintään 12- ja alle 18-vuotiaita lihavia nuoria, 125 potilasta sai Saxenda-valmistetta 56 viikon ajan.
Lihavilla nuorilla havaittujen haittavaikutusten esiintymistiheys, tyyppi ja vaikeusaste olivat yleisesti ottaen samanlaisia kuin aikuisilla. Oksentelua esiintyi nuorilla kaksi kertaa suuremmalla esiintymistiheydellä kuin aikuisilla.
Vähintään yhdestä kliinisesti merkittävästä hypoglykemiatapahtumasta ilmoittaneiden potilaiden prosenttiosuus oli liraglutidiryhmässä suurempi (1,6 %) kuin lumeryhmässä (0,8 %). Tutkimuksessa ei esiintynyt vaikeita hypoglykemiatapahtumia.
Kliinisessä tutkimuksessa, johon osallistui vähintään 6- ja alle 12‑vuotiaita lihavia lapsia (tutkimus 4392), 56 potilasta sai Saxenda-valmistetta 56 viikon ajan.
Lihavilla lapsilla havaittujen haittavaikutusten esiintymistiheys, tyyppi ja vaikeusaste olivat yleisesti ottaen samanlaisia kuin nuorilla ja aikuisilla.
Lapsilla ilmoitettiin ruoansulatuselimistön tapahtumia sekä liraglutidi- että lumeryhmässä enemmän kuin nuorilla ja aikuisilla, ja lapsilla havaittiin kaksi kertaa niin paljon oksentelua kuin nuorilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa ja liraglutidin kauppaantuonnin jälkeisessä käytössä on raportoitu yliannostuksia aina 72 mg:aan saakka (24-kertainen määrä painonhallintaan suositeltuun annokseen nähden). Raportoituja tapahtumia olivat voimakas pahoinvointi, voimakas oksentelu ja vakava hypoglykemia.
Yliannostustapauksissa tulee aloittaa asianmukainen tukihoito potilaan kliinisten oireiden perusteella. Potilasta on tarkkailtava kuivumisen kliinisten merkkien varalta, ja verensokeria on seurattava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Diabeteslääkkeet, GLP-1-analogit.
ATC-koodi: A10BJ02
Vaikutusmekanismi
Liraglutidi on asyloitu ihmisen glukagoninkaltaisen peptidi 1:n (GLP-1) analogi, jonka aminohappojärjestys on 97 %:sesti homologinen ihmisen endogeenisen GLP-1:n kanssa. Liraglutidi sitoutuu GLP-1-reseptoreihin (GLP-1R) ja aktivoi niitä.
GLP-1 on fysiologinen ruokahalun ja ravinnonoton säätelijä, mutta tarkkaa vaikutusmekanismia ei tunneta täysin. Eläinkokeissa liraglutidi, perifeerisesti annettuna, kulkeutui tiettyihin aivojen alueisiin, jotka osallistuvat ruokahalun säätelyyn. Liraglutidi lisäsi GLP-1R:n spesifin aktivoinnin kautta kylläisyyttä ja vähensi nälkäsignaaleja, mikä johti painon laskuun.
GLP-1-reseptorit ilmentyvät myös tietyissä kohdissa sydämessä, verisuonistossa, immuunijärjestelmässä ja munuaisissa. Ateroskleroosia tutkivissa hiirimalleissa liraglutidi ehkäisi aorttaplakin etenemistä ja vähensi plakin tulehdusta. Lisäksi liraglutidilla oli hyödyllinen vaikutus plasman lipideihin. Liraglutidi ei vähentänyt plakin kokoa jo muodostuneissa plakeissa.
Farmakodynaamiset vaikutukset
Liraglutidi laskee ihmisen painoa pääasiassa vähentämällä rasvan määrää siten, että viskeraalinen rasva vähenee enemmän suhteessa ihonalaiseen rasvaan. Liraglutidi säätelee ruokahalua lisäämällä täysinäisyyden ja kylläisyyden tunnetta samalla, kun se vähentää näläntunnetta ja ruokahalua, mikä johtaa vähentyneeseen ravinnonottoon. Liraglutidi ei lisää energiankulutusta lumelääkkeeseen verrattuna.
Liraglutidi stimuloi insuliinin eritystä ja vähentää glukagonin eritystä glukoosista riippuvaisesti, mikä johtaa paasto- ja aterianjälkeisen glukoosin laskuun. Glukoosipitoisuutta alentava vaikutus on selvempi diabeteksen esiastetta ja diabetesta sairastavilla potilailla kuin potilailla, joiden veren glukoosipitoisuus on normaali. Kliiniset tutkimukset viittaavat siihen, että liraglutidi parantaa ja ylläpitää beetasolujen toimintaa, kun mittareina on käytetty HOMA-B-määritystä (beetasolutoiminnan homeostaasimallimääritys) ja proinsuliini-insuliini-suhdetta.
Kliininen teho ja turvallisuus
Liraglutidin tehoa ja turvallisuutta painonhallinnassa, jossa hoitokeinoina käytettiin lisäksi nautitun energiamäärän vähentämistä ja liikunnan lisäämistä, tutkittiin neljässä faasin 3 satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa tutkimuksessa, joihin osallistui yhteensä 5 358 aikuispotilasta.
- Tutkimus 1 (SCALE Obesity & Pre-Diabetes - 1839): Yhteensä 3 731 lihavaa (BMI ≥ 30 kg/m²) tai ylipainoista (BMI ≥ 27 kg/m²) potilasta, joilla oli dyslipidemia ja/tai kohonnut verenpaine, ositettiin seulontahetkellä olleen diabeteksen esiasteen tilan ja lähtötilanteen BMI:n (≥ 30 kg/m² tai < 30 kg/m²) mukaan. Kaikki 3 731 potilasta satunnaistettiin 56 viikon hoitoon ja 2 254 potilasta, joilla oli valikointihetkellä diabeteksen esiaste, satunnaistettiin 160 viikon hoitoon. Molempia hoitojaksoja seurasi 12 viikon lääkkeetön tai lumelääke-seurantajakso. Kaikilla potilailla oli taustahoitona elämäntapamuutos vähäenergisen ruokavalion ja liikuntaohjauksen muodossa. Tutkimus 1:n 56 viikon osiossa arvioitiin painonlaskua kaikilla 3 731 satunnaistetulla potilaalla (2 590 potilasta jatkoi tutkimuksen loppuun). Tutkimus 1:n 160 viikon osuudessa arvioitiin aikaa tyypin 2 diabeteksen puhkeamiseen 2 254 satunnaistetulla potilaalla, joilla oli diabeteksen esiaste (1 128 potilasta jatkoi tutkimuksen loppuun).
- Tutkimus 2 (SCALE Diabetes - 1922): 56 viikon tutkimus, jossa arvioitiin 846 satunnaistetun (628 potilasta jatkoi tutkimuksen loppuun), lihavan ja ylipainoisen potilaan painonlaskua, kun potilailla oli riittämättömästi kontrolloitu tyypin 2 diabetes mellitus (HbA1c välillä 7–10 %). Aiempi hoito tutkimuksen lähtötilanteessa oli joko pelkkä ruokavalio ja liikunta, metformiini, sulfonyyliurea tai glitatsoni ainoana vaikuttavana aineena tai minkä tahansa edellä mainittujen yhdistelmä.
- Tutkimus 3 (SCALE Sleep Apnoea - 3970): 32 viikon tutkimus, jossa arvioitiin 359 satunnaistetun (276 potilasta jatkoi tutkimuksen loppuun), lihavan potilaan uniapnean vaikeutta ja painonlaskua, kun potilailla oli keskivaikea tai vaikea obstruktiivinen uniapnea.
- Tutkimus 4 (SCALE Maintenance - 1923): 56 viikon tutkimus, jossa arvioitiin 422 satunnaistetun (305 potilasta jatkoi tutkimuksen loppuun), lihavan ja ylipainoisen potilaan painonhallintaa ja painonlaskua, kun potilailla oli kohonnut verenpaine tai dyslipidemia aiemman, vähäenergisellä ruokavaliolla saavutetun vähintään 5 %:n painonlaskun jälkeen.
Paino
Kaikissa tutkituissa ryhmissä liraglutidilla saavutettiin suurempi lihavien/ylipainoisten potilaiden painonlasku kuin lumelääkkeellä. Tutkimusten kaikissa ryhmissä suurempi osa potilaista saavutti ≥ 5 %:n ja ≥ 10 %:n painonlaskun liraglutidilla kuin lumelääkkeellä (taulukot 5–7). Tutkimus 1:n 160 viikon osuudessa painonlasku tapahtui pääosin ensimmäisenä vuotena ja pysyi koko 160 viikon ajan. Tutkimuksessa 4 suurempi osa potilaista säilytti ennen hoidon aloitusta saavutetun painonlaskun liraglutidin (81,4 %) avulla lumelääkkeeseen (48,9 %) verrattuna. Spesifit tiedot painonlaskusta, vastaajista, ajan vaikutuksesta lääkeaineen aktiivisuuteen ja painonmuutoksen kumulatiivisesta jakaumasta (%) tutkimuksissa 1–4 esitetään taulukoissa 5–9 ja kuvissa 1, 2 ja 3.
Painonlaskuvaste 12 viikon liraglutidihoidon (3,0 mg) jälkeen
Varhaisen hoitovasteen potilaiksi määriteltiin sellaiset potilaat, jotka saavuttivat ≥ 5 %:n painonlaskun, kun heitä oli hoidettu 12 viikkoa liraglutidin hoitoannoksella (4 viikkoa annoskoon suurentamista ja 12 viikkoa hoitoannosta). Tutkimus 1:n 56 viikon osuudessa 67,5 % saavutti ≥ 5 %:n painonlaskun 12 viikon jälkeen. Tutkimuksessa 2 50,4 % saavutti ≥ 5 %:n painonlaskun 12 viikon jälkeen. Kun liraglutidihoitoa jatketaan, 86,2 % näistä varhaisen hoitovasteen potilaista ennustetaan saavuttavan ≥ 5 %:n painonlaskun ja 51 %:n ennustetaan saavuttavan ≥ 10 %:n painonlaskun 1 vuoden hoidon jälkeen. Yhden vuoden hoidon jälkeen varhaisen hoitovasteen potilaiden ennustettu keskimääräinen painonlasku on 11,2 % lähtötilanteen painosta (9,7 % miehillä ja 11,6 % naisilla). Potilaista, jotka saavuttivat < 5 %:n painonlaskun, kun heitä oli hoidettu 12 viikkoa liraglutidin hoitoannoksella, 93,4 % ei saavuttanut ≥ 10 %:n painonlaskua 1 vuoden jälkeen.
Glukoositasapaino
Liraglutidihoito paransi glykeemisiä parametrejä merkittävästi alaryhmissä, joissa potilailla oli normaali veren glukoosipitoisuus, diabeteksen esiaste tai tyypin 2 diabetes mellitus. Tutkimus 1:n 56 viikon osiossa liraglutidilla hoidetut potilaat sairastuivat tyypin 2 diabetes mellitukseen harvemmin verrattuna lumelääkkeellä hoidettuihin potilaisiin (0,2 % vs. 1,1 %). Potilaista, joilla oli lähtötilanteessa diabeteksen esiaste, useammalla potilaalla se oli väistynyt verrattuna lumelääkkeellä hoidettuihin potilaisiin (69,2 % vs. 32,7 %). Tutkimus 1:n 160 viikon osuudessa ensisijainen päätetapahtuma tehon suhteen oli se osuus potilaista, joilla puhkesi tyypin 2 diabetes mellitus, arvioituna ennen puhkeamista kuluneen ajan mukaan. Viikolla 160, hoidon vielä jatkuessa, 3 %:lla Saxenda-valmisteella hoidetuista ja 11 %:lla lumelääkkeellä hoidetuista potilaista todettiin tyypin 2 diabetes mellitus. Aika ennen tyypin 2 diabetes mellituksen arvioitua puhkeamista oli liraglutidi 3,0 mg:lla hoidetuilla potilailla 2,7 kertaa pidempi (95 % luottamusväli [1.9, 3.9]), ja riskisuhde tyypin 2 diabetes mellituksen kehittymisriskille oli 0,2 liraglutidilla verrattuna lumelääkkeeseen.
Kardiometaboliset riskitekijät
Liraglutidihoito paransi systolista verenpainetta ja vyötärön ympärysmittaa merkitsevästi lumelääkkeeseen verrattuna (taulukot 5, 6 ja 7).
Apnea-hypopnea-indeksi (AHI)
Liraglutidihoito pienensi obstruktiivisen uniapnean vaikeusastetta merkitsevästi. Tämä arvioitiin AHI-indeksin muutoksena lähtötilanteesta lumelääkkeeseen verrattuna (taulukko 8).
Taulukko 5 Tutkimus 1: Painon, glykemian ja kardiometabolisten parametrien muutokset lähtötilanteesta viikkoon 56 mennessä
| Saxenda (n = 2437) | Lumelääke (n = 1 225) | Saxenda vs. lumelääke | ||||
| Paino | ||||||
| Lähtötaso, kg (SD) | 106,3 (21,2) | 106,3 (21,7) | - | |||
| Keskimääräinen muutos viikkoon 56 mennessä, % (95 % CI) | -8,0 | -2,6 | -5,4** (-5,8; -5,0) | |||
| Keskimääräinen muutos viikkoon 56 mennessä, kg (95 % CI) | -8,4 | -2,8 | -5,6** (-6,0; -5,1) | |||
| ≥5 %:n painonlaskun viikkoon 56 mennessä saavuttaneiden potilaiden osuus, % (95 % CI) | 63,5 | 26,6 | 4,8** (4,1; 5,6) | |||
| >10 %:n painonlaskun viikkoon 56 mennessä saavuttaneiden potilaiden osuus, % (95 % CI) | 32,8 | 10,1 | 4,3** (3,5; 5,3) | |||
| Glykemia ja kardiometaboliset tekijät | Lähtötaso | Muutos | Lähtötaso | Muutos | ||
| HbA1c, % | 5,6 | -0,3 | 5,6 | -0,1 | -0,23** (-0,25; -0,21) | |
| fP-Gluk, mmol/l | 5,3 | -0,4 | 5,3 | -0,01 | -0,38** (-0,42; -0,35) | |
| Systolinen verenpaine, mmHg | 123,0 | -4,3 | 123,3 | -1,5 | -2,8** (-3,6; -2,1) | |
| Diastolinen verenpaine, mmHg | 78,7 | -2,7 | 78,9 | -1,8 | -0,9* (-1,4; -0,4) | |
| Vyötärön ympärysmitta, cm | 115,0 | -8,2 | 114,5 | -4,0 | -4,2** (-4,7; -3,7) | |
Täydellinen analyysijoukko. Painon, HbA1c:n, glukoosin, verenpaineen ja vyötärön ympärysmitan osalta lähtötilanteen arvot ovat keskiarvoja, muutokset lähtötilanteesta viikkoon 56 asti ovat arvioituja keskiarvoja (pienimpiä neliösummia) ja hoitokontrastit viikkona 56 ovat arvioituja hoitoeroja. ≥5 %:n / >10 %:n painonlaskun saavuttaneiden potilaiden määristä esitetään arvioidut vetosuhteet (odds ratio). Puuttuvat lähtötilanteen jälkeiset arvot imputoitiin käyttämällä LOCF-menetelmää (Last observation carried forward, viimeisin käytetty havainto). * p<0,05. ** p<0,0001. CI = luottamusväli. fP-Gluk = paastoplasman glukoosi. SD = standardipoikkeama.
Taulukko 6 Tutkimus 1: Painon, glykemian ja kardiometabolisten parametrien muutokset lähtötilanteesta viikkoon 160 mennessä
| Saxenda (n = 1472) | Lumelääke (n = 738) | Saxenda vs. lumelääke | |||
| Paino | |||||
| Lähtötaso, kg (SD) | 107,6 (21,6) | 108,0 (21,8) | |||
| Keskimääräinen muutos viikkoon 160 mennessä, % (95% CI) | -6,2 | -1,8 | -4,3** (-4,9; -3,7) | ||
| Keskimääräinen muutos viikkoon 160 mennessä, kg (95% CI) | -6,5 | -2,0 | -4,6** (-5,3; -3,9) | ||
| ≥5 %:n painonlaskun viikkoon 160 mennessä saavuttaneiden potilaiden osuus, % (95% CI) | 49,6 | 23,4 | 3,2** (2,6; 3,9) | ||
| >10 %:n painonlaskun viikkoon 160 mennessä saavuttaneiden potilaiden osuus, % (95% CI) | 24,4 | 9,5 | 3,1** (2,3; 4,1) | ||
| Glykemia ja kardiometaboliset tekijät | Lähtötaso | Muutos | Lähtötaso | Muutos | |
| HbA1c, % | 5,8 | -0,4 | 5,7 | -0,1 | -0,21** (-0,24; -0,18) |
| fP-Gluk, mmol/l | 5,5 | -0,4 | 5,5 | 0,04 | -0,4** (-0,5; -0,4) |
| Systolinen verenpaine, mmHg | 124,8 | -3,2 | 125,0 | -0,4 | -2,8** (-3,8; -1,8) |
| Diastolinen verenpaine, mmHg | 79,4 | -2,4 | 79,8 | -1,7 | -0,6 (-1,3; 0,1) |
| Vyötärön ympärysmitta, cm | 116,6 | -6,9 | 116,7 | -3,4 | -3,5** (-4,2; -2,8) |
Täydellinen analyysijoukko. Painon, HbA1c:n, glukoosin, verenpaineen ja vyötärön ympärysmitan osalta lähtötilanteen arvot ovat keskiarvoja, muutokset lähtötilanteesta viikkoon 160 asti ovat arvioituja keskiarvoja (pienimpiä neliösummia) ja hoitokontrastit viikkona 160 ovat arvioituja hoitoeroja. ≥5 %:n / >10 %:n painonlaskun saavuttaneiden potilaiden määristä esitetään arvioidut vetosuhteet (odds ratio). Puuttuvat lähtötilanteen jälkeiset arvot imputoitiin käyttämällä LOCF-menetelmää (Last observation carried forward, viimeisin käytetty havainto). ** p<0,0001. CI = luottamusväli. fP-Gluk = paastoplasman glukoosi. SD = standardipoikkeama.
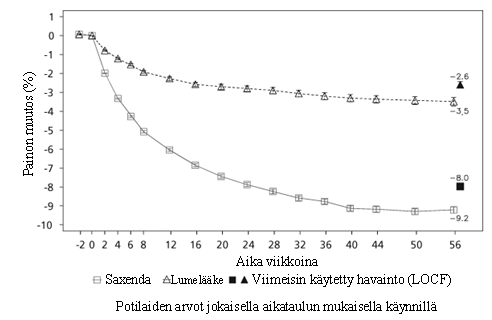
Kuva 1 Painon muutos (%) lähtötilanteesta tutkimuksen 1 aikana (0–56 viikkoa)
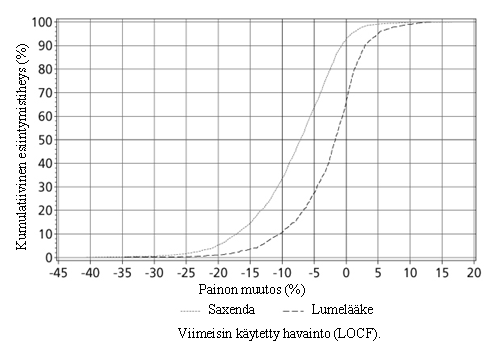
Kuva 2 Painonmuutoksen kumulatiivinen jakauma (%) 56 viikon hoidon jälkeen tutkimuksessa 1
Taulukko 7 Tutkimus 2: Painon, glykemian ja kardiometabolisten parametrien muutokset lähtötilanteesta viikkoon 56 mennessä
| Saxenda (n = 412) | Lumelääke (n = 211) | Saxenda vs. lumelääke | |||
| Paino | |||||
| Lähtötaso, kg (SD) | 105,6 (21,9) | 106,7 (21,2) | - | ||
| Keskimääräinen muutos viikkoon 56 mennessä, % (95 % CI) | -5,9 | -2,0 | -4,0** (-4,8; -3,1) | ||
| Keskimääräinen muutos viikkoon 56 mennessä, kg (95 % CI) | -6,2 | -2,2 | -4,1** (-5,0; -3,1) | ||
| ≥5 %:n painonlaskun viikkoon 56 mennessä saavuttaneiden potilaiden osuus, % (95 % CI) | 49,8 | 13,5 | 6,4** (4,1; 10,0) | ||
| >10 %:n painonlaskun viikkoon 56 mennessä saavuttaneiden potilaiden osuus, % (95 % CI) | 22,9 | 4,2 | 6,8** (3,4; 13,8) | ||
| Glykemia ja kardiometaboliset tekijät | Lähtötaso | Muutos | Lähtötaso | Muutos | |
| HbA1c, % | 7,9 | -1,3 | 7,9 | -0,4 | -0,9** (-1,1; -0,8) |
| fP-Gluk, mmol/l | 8,8 | -1,9 | 8,6 | -0,1 | -1,8** (-2,1; -1,4) |
| Systolinen verenpaine, mmHg | 128,9 | -3,0 | 129,2 | -0,4 | -2,6* (-4,6; -0,6) |
| Diastolinen verenpaine, mmHg | 79,0 | -1,0 | 79,3 | -0,6 | -0,4 (-1,7; 1,0) |
| Vyötärön ympärysmitta, cm | 118,1 | -6,0 | 117,3 | -2,8 | -3,2** (-4,2; -2,2) |
Täydellinen analyysijoukko. Painon, HbA1c:n, glukoosin, verenpaineen ja vyötärön ympärysmitan osalta lähtötilanteen arvot ovat keskiarvoja, muutokset lähtötilanteesta viikkoon 56 asti ovat arvioituja keskiarvoja (pienimpiä neliösummia) ja hoitokontrastit viikkona 56 ovat arvioituja hoitoeroja. ≥5 %:n / >10 %:n painonlaskun saavuttaneiden potilaiden määristä esitetään arvioidut vetosuhteet (odds ratio). Puuttuvat lähtötilanteen jälkeiset arvot imputoitiin käyttämällä LOCF-menetelmää (Last observation carried forward, viimeisin käytetty havainto). * p<0,05. ** p<0,0001. CI = luottamusväli. fP-Gluk = paastoplasman glukoosi. SD = standardipoikkeama.
Taulukko 8 Tutkimus 3: Painon ja apnea-hypopnea-indeksin muutokset lähtötilanteesta viikkoon 32 mennessä
| Saxenda (n = 180) | Lumelääke (n = 179) | Saxenda vs. lumelääke | ||||
| Paino | ||||||
| Lähtötaso, kg (SD) | 116,5 (23,0) | 118,7 (25,4) | - | |||
| Keskimääräinen muutos viikkoon 32 mennessä, % (95 % CI) | -5,7 | -1,6 | -4,2** (-5,2; -3,1) | |||
| Keskimääräinen muutos viikkoon 32 mennessä, kg (95 % CI) | -6,8 | -1,8 | -4,9** (-6,2; -3,7) | |||
| ≥5 %:n painonlaskun viikkoon 32 mennessä saavuttaneiden potilaiden osuus, % (95 % CI) | 46,4 | 18,1 | 3,9** (2,4; 6,4) | |||
| >10 %:n painonlaskun viikkoon 32 mennessä saavuttaneiden potilaiden osuus, % (95 % CI) | 22,4 | 1,5 | 19,0** (5,7; 63,1) | |||
| Lähtötaso | Muutos | Lähtötaso | Muutos | |||
| Apnea-hypopnea-indeksi, tapahtumia/tunti | 49,0 | -12,2 | 49,3 | -6,1 | -6,1* (-11,0; -1,2) | |
Täydellinen analyysijoukko. Lähtötilanteen arvot ovat keskiarvoja, muutokset lähtötilanteesta viikkoon 32 asti ovat arvioituja keskiarvoja (pienimpiä neliösummia) ja hoitokontrastit viikkona 32 ovat arvioituja hoitoeroja (95 % CI). ≥5 %:n / >10 %:n painonlaskun saavuttaneiden potilaiden määristä esitetään arvioidut vetosuhteet (odds ratio). Puuttuvat lähtötilanteen jälkeiset arvot imputoitiin käyttämällä LOCF-menetelmää (Last observation carried forward, viimeisin käytetty havainto). * p<0,05. ** p<0,0001. CI = luottamusväli. SD = standardipoikkeama.
Taulukko 9 Tutkimus 4: Painon muutokset lähtötilanteesta viikkoon 56 mennessä
| Saxenda (n = 207) | Lumelääke (n = 206) | Saxenda vs. lumelääke | |
| Lähtötaso, kg (SD) | 100,7 (20,8) | 98,9 (21,2) | - |
| Keskimääräinen muutos viikkoon 56 mennessä, % (95 % CI) | -6,3 | -0,2 | -6,1** (-7,5; -4,6) |
| Keskimääräinen muutos viikkoon 56 mennessä, kg (95 % CI) | -6,0 | -0,2 | -5,9** (-7,3; -4,4) |
| ≥5 %:n painonlaskun viikkoon 56 mennessä saavuttaneiden potilaiden osuus, % (95 % CI) | 50,7 | 21,3 | 3,8** (2,4; 6,0) |
| >10 %:n painonlaskun viikkoon 56 mennessä saavuttaneiden potilaiden osuus, % (95 % CI) | 27,4 | 6,8 | 5,1** (2,7; 9,7) |
Täydellinen analyysijoukko. Lähtötilanteen arvot ovat keskiarvoja, muutokset lähtötilanteesta viikkoon 56 asti ovat arvioituja keskiarvoja (pienimpiä neliösummia) ja hoitokontrastit viikkona 56 ovat arvioituja hoitoeroja. ≥5 %:n / >10 %:n painonlaskun saavuttaneiden potilaiden määristä esitetään arvioidut vetosuhteet (odds ratio). Puuttuvat lähtötilanteen jälkeiset arvot imputoitiin käyttämällä LOCF-menetelmää (Last observation carried forward, viimeisin käytetty havainto). ** p<0,0001. CI = luottamusväli. SD = standardipoikkeama.
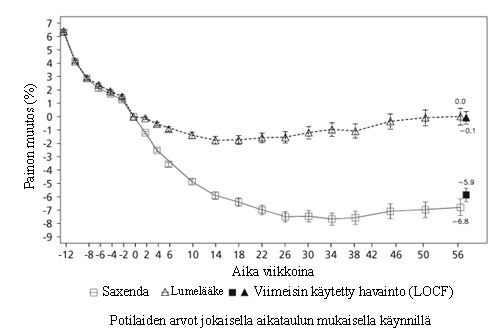
Kuva 3 Painon muutos (%) satunnaistamisesta (viikko 0) tutkimuksen 4 aikana
Ennen viikkoa 0 potilaita hoidettiin vain vähäenergisellä ruokavaliolla ja liikunnalla. Viikolla 0 potilaat satunnaistettiin ryhmiin, jotka saivat joko Saxenda-valmistetta tai lumelääkettä.
Immunogeenisuus
Proteiineja tai peptidejä sisältävillä lääkevalmisteilla saattaa olla immunogeenisiä vaikutuksia, joten potilaille voi kehittyä liraglutidivasta-aineita liraglutidihoidon aikana. Kliinisissä tutkimuksissa 2,5 %:lle liraglutidilla hoidetuista potilaista kehittyi liraglutidivasta-aineita. Vasta-aineiden muodostumisen ei ole todettu heikentävän liraglutidin tehoa.
Sydän- ja verisuonivaikutusten arviointi
Ulkopuolinen, itsenäinen asiantuntijaryhmä arvioi merkittävät sydän- ja verisuoniperäiset haittavaikutukset (major adverse cardiovascular events, MACE). Sellaisiksi katsottiin ei-fataali sydäninfarkti, ei-fataali aivohalvaus sekä sydän- ja verisuoniperäinen kuolema. Kaikkien Saxenda-valmisteen kliinisissä pitkäaikaistutkimuksissa liraglutidilla hoidettujen potilaiden joukossa oli 6 MACE-tapahtumaa ja lumelääkkeellä hoidettujen potilaiden joukossa 10 MACE-tapahtumaa. Riskisuhde ja 95 % CI on 0,33 [0,12; 0,90] verrattaessa liraglutidia lumelääkkeeseen. Liraglutidin käytön yhteydessä kliinisissä faasin 3 tutkimuksissa on havaittu keskimääräinen sydämen sykkeen nopeutuminen lähtötasosta 2,5 lyönnillä minuutissa (vaihdellen tutkimuksissa välillä 1,6–3,6 lyöntiä minuutissa). Sydämen syke saavutti huipputiheytensä noin 6 viikon jälkeen. Sydämen syketason keskimääräisen nousun pitkäkestoisia kliinisiä vaikutuksia ei ole vahvistettu. Sydämen sykkeen muutos palautui, kun liraglutidihoito lopetettiin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Liraglutidin pitkäaikaiseen sydän- ja verisuonivaikutustutkimukseen (LEADER (Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcomes Results)) osallistui 9 340 potilasta, joiden tyyppi 2 diabetes oli riittämättömässä hoitotasapainossa. Valtaosalla heistä oli todettu sydän- ja verisuonitauti. Potilaat jaettiin satunnaisesti joko joko liraglutidi-ryhmään, jossa päivittäisannos oli enintään 1,8 mg (4 668), tai lumelääkeryhmään (4 672), molemmissa taustalla vakiohoito.
Altistumisen kesto oli 3,5 vuoden ja 5 vuoden välillä. Keski-ikä oli 64 vuotta ja keskimääräinen BMI oli 32,5 kg/m2. Keskimääräinen HbA1c-lähtöarvo oli 8,7, mikä on liraglutidia saaneilla potilailla parantunut 3 vuoden jälkeen 1,2 % ja lumelääkettä saaneilla potilailla 0,8 %. Ensisijainen päätetapahtuma oli aika satunnaistamisesta ensimmäiseen mihin tahansa vakavaan sydän- ja verisuonihaittatapahtumaan (major adverse cardiovascular event, MACE): sydän- ja verisuonikuolema, ei-fataali sydäninfarkti tai ei-fataali aivohalvaus.
Liraglutidi vähensi merkitsevästi vakavien sydän- ja verisuonihaittatapahtumien määrää (ensisijaiset päätetapahtumat, MACE) verrattuna lumelääkkeeseen (liraglutidi 3,41 vs. lumelääke 3,90 sataa potilasvuotta kohden). Riski vähentyi 13 %:lla, HR 0,87 [0,78, 0,97] [95 % CI]) (p = 0,005) (ks. kuva 4).
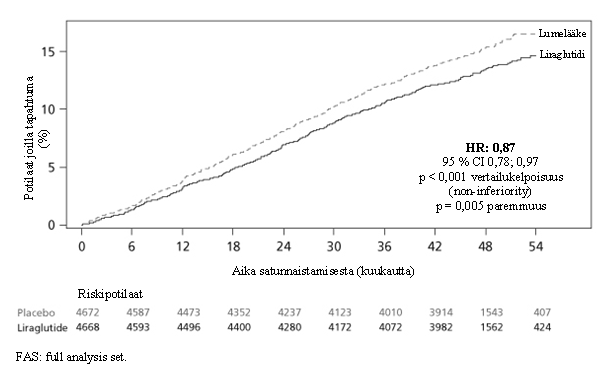
Kuva 4 Kaplan Meier -kuvaaja, aika ensimmäiseen MACE-tapahtumaan – FAS-joukko
Pediatriset potilaat
Kaksoissokkoutetussa tutkimuksessa, jossa Saxenda-valmisteen tehoa ja turvallisuutta painon pudottamisessa verrattiin lumelääkkeeseen vähintään 12-vuotiailla lihavilla nuorilla, Saxenda oli (painoindeksin keskihajontapistemäärän [BMI-SDS] perusteella arvioidun) painonlaskun suhteen parempi kuin lumelääke 56 viikon hoidon jälkeen (taulukko 10).
Niiden potilaiden osuus, joilla painoindeksi pieneni vähintään 5 % ja vähintään 10 %, oli suurempi liraglutidia saaneilla potilailla kuin lumelääkettä saaneilla, ja lisäksi keskimääräinen BMI ja paino pienenivät liraglutidia saaneilla enemmän (taulukko 10). Kun potilaat olivat olleet käyttämättä tutkimuslääkettä 26 viikon seurantajakson ajan, liraglutidia saaneilla potilailla havaittiin painon alkaneen nousta uudelleen verrattuna lumelääkettä saaneisiin potilaisiin (taulukko 10).
Taulukko 10 Tutkimus 4180: Painon ja BMI:n muutos lähtötilanteesta viikkoon 56 mennessä ja BMI-SDS-pistemäärän muutos viikosta 56 viikkoon 82
| Saxenda (N = 125) | Lumelääke (N = 126) | Saxenda vs. lumelääke | |
| BMI-SDS | |||
| Lähtötaso, BMI-SDS (SD) | 3,14 (0,65) | 3,20 (0,77) | |
| Keskimääräinen muutos viikkoon 56 mennessä (95 % CI) | -0,23 | -0,00 | -0,22* (-0,37; -0,08) |
| Viikko 56, BMI-SDS (SD) | 2,88 (0,94) | 3,14 (0,98) | |
| Keskimääräinen muutos viikosta 56 viikkoon 82, BMI-SDS (95% CI) | 0,22 | 0,07 | 0,15** (0,07; 0,23) |
| Paino | |||
| Lähtötaso, kg (SD) | 99,3 (19,7) | 102,2 (21,6) | - |
| Keskimääräinen muutos viikkoon 56 mennessä, % (95 % CI) | -2,65 | 2,37 | -5,01** (-7,63; -2,39) |
| Keskimääräinen muutos viikkoon 56 mennessä, kg (95 % CI) | -2,26 | 2,25 | -4,50** (-7,17; -1,84) |
| BMI | |||
| Lähtötaso, kg/m2 (SD) | 35,3 (5,1) | 35,8 (5,7) | - |
| Keskimääräinen muutos viikkoon 56 mennessä, kg/m2 (95 % CI) | -1,39 | 0,19 | -1,58** (-2,47; -0,69) |
| ≥ 5 %:n BMI:n pienenemisen viikkoon 56 mennessä saavuttaneiden potilaiden osuus, % (95 % CI) | 43,25 | 18,73 | 3,31** (1,78; 6,16) |
| ≥ 10 %:n BMI:n pienenemisen viikkoon 56 mennessä saavuttaneiden potilaiden osuus, % (95 % CI) | 26,08 | 8,11 | 4,00** (1,81; 8,83) |
Täydellinen analyysijoukko. BMI-SDS-pistemäärän, painon ja BMI:n osalta lähtötilanteen arvot ovat keskiarvoja, muutokset lähtötilanteesta viikkoon 56 asti ovat arvioituja keskiarvoja (pienimpiä neliösummia) ja hoitokontrastit viikkona 56 ovat arvioituja hoitoeroja. BMI-SDS-pistemäärän osalta arvot viikon 56 kohdalla ovat keskiarvoja, muutokset viikosta 56 viikkoon 82 asti ovat arvioituja keskiarvoja (pienimpiä neliösummia) ja hoitokontrastit viikkona 82 ovat arvioituja hoitoeroja. ≥ 5 %:n / > 10 %:n BMI:n pienenemisen saavuttaneiden potilaiden määristä esitetään arvioidut vetosuhteet (odds ratio). Puuttuvat havainnot imputoitiin lumelääkehaarasta käyttämällä jump to reference ‑moni-imputointimenetelmää (x 100).
*p < 0,01, **p < 0,001. CI = luottamusväli. SD = standardipoikkeama.
Siedettävyyden perusteella annosta suurennettiin 103 potilaalla (82,4 %) 3,0 mg:n ylläpitoannokseen asti, 11 potilaalla (8,8 %) 2,4 mg:n ylläpitoannokseen asti, 4 potilaalla (3,2 %) 1,8 mg:aan asti ja 4 potilaalla (3,2 %) 1,2 mg:aan asti. 3 potilasta (2,4 %) jatkoi 0,6 mg:n annoksella.
56 viikon hoidon jälkeen ei todettu vaikutuksia kasvuun tai murrosiän kehitykseen.
Tutkimuksessa, jossa oli 16 viikon pituinen kaksoissokkoutettu vaihe ja 36 viikon pituinen avoin vaihe, arvioitiin Saxenda-valmisteen tehoa ja turvallisuutta pediatrisilla potilailla, joilla oli Prader-Willin oireyhtymä ja jotka olivat lihavia. Tutkimuksessa oli mukana 32 potilasta, jotka olivat iältään 12–< 18-vuotiaita (osa A) ja 24 potilasta, jotka olivat iältään 6–< 12-vuotiaita (osa B). Potilaat satunnaistettiin suhteessa 2:1 saamaan joko Saxenda-valmistetta tai lumelääkettä. Potilaiden, joiden paino oli alle 45 kg, annoksen suurentaminen aloitettiin pienemmällä 0,3 mg:n annoksella 0,6 mg:n annoksen sijaan, ja heidän annostaan suurennettiin 2,4 mg:n enimmäisannokseen.
Keskimääräisen BMI-SDS-pistemäärän perusteella arvioitu hoitoero 16 viikon kohdalla (osa A: ‑0,20 vs ‑0,13, osa B: ‑0,50 vs ‑0,44) ja 52 viikon kohdalla (osa A: ‑0,31 vs ‑0,17, osa B: ‑0,73 vs ‑0,67) oli samanlainen Saxenda-valmistetta ja lumelääkettä saaneilla.
Tutkimuksessa ei havaittu uusia turvallisuuteen liittyviä huolenaiheita.
56 viikon pituisessa kaksoissokkoutetussa tutkimuksessa satunnaistettiin 82 lasta, jotka olivat 6 – < 12‑vuotiaita ja lihavia, suhteessa 2:1 saamaan joko liraglutidia 3,0 mg tai lumelääkettä kerran vuorokaudessa. Kaikki potilaat saivat terveellistä ruokavaliota ja fyysistä aktiivisuutta koskevaa neuvontaa koko tutkimuksen ajan.
Hoidon päättyessä (viikolla 56) BMI:n todettiin parantuneen liraglutidin käytön yhteydessä enemmän ja kliinisesti merkittävästi verrattuna lumelääkkeeseen (ks. taulukko 11). Lisäksi niiden potilaiden osuus, joilla BMI pieneni vähintään 5 %, oli suurempi liraglutidia saaneilla kuin lumelääkettä saaneilla (ks. taulukko 11).
Taulukko 11 SCALE KIDS 4392: Tulokset viikolla 56
| SAXENDA (N = 56) | Lumelääke (N = 26) | SAXENDA vs. lumelääke |
| BMI | |||
| Lähtötaso, keskimääräinen BMI, kg/m2 (SD) | 30,9 (4,7) | 31,3 (7,0) | |
| Keskimääräinen muutos lähtötasosta, % (95 % CI) | ‑5,80 | 1,60 | ‑7,40 (‑11,56; ‑3,24) |
| Lähtötasoon nähden ≥ 5 %:n BMI:n pienenemisen viikkoon 56 mennessä saavuttaneiden potilaiden osuus, OR (95 % CI) | 46,2 % | 8,7 % | 6,27 (1,36; 28,79) |
| Paino | |||
| Lähtötaso, keskimääräinen paino, kg (SD) | 69,8 (17,7) | 71,0 (23,2) | |
| Keskimääräinen muutos lähtötasosta, % (95 % CI) | 1,59 | 9,96 | ‑8,37 (‑13,39; ‑3,34) |
BMI: painoindeksi, SD: standardipoikkeama, CI: luottamusväli, OR: vetosuhde (odds ratio).
BMI:n ja painon osalta lähtötilanteen arvot ovat keskiarvoja, muutokset lähtötilanteesta viikkoon 56 asti ovat arvioituja keskiarvoja (pienimpiä neliösummia) ja hoitokontrastit viikkona 56 ovat arvioituja hoitoeroja. Lähtötilanteeseen nähden ≥ 5 %:n BMI:n pienenemisen saavuttaneiden potilaiden määristä esitetään arvioidut vetosuhteet (odds ratio).
ANCOVA: Viikon 56 vasteet analysoitiin kovarianssianalyysimallilla, jossa tekijöinä olivat satunnaistettu hoito, ositusryhmät (sukupuoli ja Tanner-luokitus lähtötilanteessa) ja ositusryhmien välinen yhdysvaikutus ja kovariaattina vastaavan päätetapahtuman lähtötaso. RD‑MI (retrieved dropouts multiple imputation): Puuttuvat havainnot imputoitiin moni-imputoinnilla (x 1 000) tutkimukseen palanneiden osallistujien tiedoista riippumatta siitä, mihin hoitohaaraan heidät oli satunnaistettu.
Farmakokinetiikka
Imeytyminen
Liraglutidi imeytyi hitaasti subkutaanisen annon jälkeen. Huippupitoisuus saavutettiin noin 11 tunnin kuluttua annoksen ottamisesta. Lihavien (BMI 30–40 kg/m2) potilaiden keskimääräinen vakaan tilan liraglutidipitoisuus (AUCτ/24) 3 mg:n liraglutidiannoksen annon jälkeen oli noin 31 nmol/l. Liraglutidille altistuminen lisääntyi suhteessa annokseen. Liraglutidin absoluuttinen biologinen hyötyosuus ihonalaisen annon jälkeen on noin 55 %.
Jakautuminen
Ihon alle annetun annoksen keskimääräinen näennäinen jakautumistilavuus on 20–25 l (noin 100 kg painavalla henkilöllä). Liraglutidi sitoutuu kattavasti plasman proteiineihin (> 98 %).
Biotransformaatio
Kun terveille koehenkilöille annettiin kerta-annoksena [3H]-liraglutidia, 24 tunnin aikana todettu pääasiallisin ainesosa plasmassa oli muuttumatonta liraglutidia. Plasmassa havaittiin kaksi vähäistä metaboliittia (≤ 9 % ja ≤ 5 % radioaktiivisesta kokonaisaltistumasta plasmassa).
Eliminaatio
Liraglutidi metaboloituu endogeenisesti samalla tavoin kuin suuret proteiinit, eikä mikään tietty elin toimi pääasiallisena eliminoitumisreittinä. [3H]-liraglutidiannoksen jälkeen muuttumatonta liraglutidia ei havaittu virtsassa tai ulosteissa. Vain vähäinen määrä annetusta radioaktiivisuudesta erittyi liraglutidin metaboliiteissa virtsaan (6 %) tai ulosteisiin (5 %). Radioaktiivisuus erittyi virtsaan ja ulosteisiin pääasiallisesti ensimmäisten 6–8 päivän aikana kolmena vähäisenä metaboliittina.
Liraglutidin keskimääräinen puhdistuma ihon alle annetun annoksen jälkeen on noin 0,9–1,4 l/h ja eliminaation puoliintumisaika on noin 13 tuntia.
Erityisryhmät
Iäkkäät potilaat
Ylipainoisista ja lihavista (18–82-vuotiaista) potilaista saatujen tietojen perusteella suoritetun populaatiofarmakokineettisen analyysin tulosten mukaan iällä ei ollut kliinisesti merkitsevää vaikutusta liraglutidin farmakokinetiikkaan. Annosta ei tarvitse säätää iän perusteella.
Sukupuoli
Populaatiofarmakokineettisistä analyyseistä saatujen tietojen perusteella naisten painon mukaan sopeutettu liraglutidin puhdistuma on 24 % vähäisempi kuin miesten. Altistus-vastetietojen perusteella annosta ei tarvitse säätää sukupuolen perusteella.
Rotu
Valkoihoisista ja mustaihoisista sekä aasialaista ja latinalaista/ei-latinalaista alkuperää olevista ylipainoisista ja lihavista potilaista tehdyn farmakokineettisen populaatioanalyysin perusteella rodulla ei ole kliinisesti merkitsevää vaikutusta liraglutidin farmakokinetiikkaan.
Paino
Liraglutidialtistus on sitä pienempi mitä suurempi on lähtötilanteen paino. 3,0 mg:n päivittäinen liraglutidiannos antoi riittävän systeemisen altistuksen 60–234 kg:n painoalueella kliinisten tutkimusten altistus-vaste-tietojen perusteella arvioituna. Liraglutidialtistusta ei tutkittu yli 234 kg painavilla potilailla.
Maksan vajaatoimintapotilaat
Kerta-annostutkimuksessa (0,75 mg) arvioitiin liraglutidin farmakokinetiikkaa potilaissa, joilla oli eriasteista maksan vajaatoimintaa. Liraglutidille altistuminen oli 13–23 % pienempi potilailla, joilla oli lievä tai keskivaikea maksan vajaatoiminta, verrattuna terveisiin koehenkilöihin. Altistuminen oli merkittävästi vähäisempää (44 %), kun potilaalla oli vaikea maksan vajaatoiminta (Child Pugh -pisteet olivat > 9).
Munuaisten vajaatoiminta
Kerta-annostutkimuksessa (0,75 mg) liraglutidille altistuminen oli vähäisempää, kun potilailla oli munuaisten vajaatoimintaa, verrattuna henkilöihin, joiden munuaistoiminta oli normaali. Altistuminen liraglutidille oli 33 % vähäisempää, kun potilaalla oli lievä munuaisten vajaatoiminta (kreatiniinipuhdistuma CrCl 50–80 ml/min), ja 14 % vähäisempää, kun potilaalla oli keskivaikea munuaisten vajaatoiminta (CrCl 30–50 ml/min), ja 27 % vähäisempää, kun potilaalla oli vaikea munuaisten vajaatoiminta (CrCl < 30 ml/min), ja 26 % vähäisempää, kun potilaalla oli dialyysiä vaativa loppuvaiheen munuaissairaus.
Pediatriset potilaat
Lihavilla nuorilla potilailla, iältään vähintään 12 ja alle 18 vuotta (134 potilasta, paino 62–178 kg) tehdyissä kliinisissä tutkimuksissa arvioitiin 3,0 mg:n liraglutidiannoksen farmakokineettisiä ominaisuuksia. Liraglutidialtistus nuorilla (ikä vähintään 12 ja alle 18 vuotta) oli samankaltainen kuin lihavilla aikuisilla.
3,0 mg:n liraglutidiannoksen farmakokineettisiä ominaisuuksia arvioitiin myös 6 – < 12‑vuotiailla lihavilla lapsilla tehdyissä kliinisissä tutkimuksissa (59 potilasta, paino 35–114 kg). Liraglutidialtistus oli lapsilla (6 – < 12‑vuotiailla) suurempi kuin aikuisilla ja nuorilla. Kun altistus korjattiin painon suhteen, se oli samankaltainen kuin aikuisilla ja nuorilla.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta tai genotoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Rotilla ja hiirillä tehdyissä 2-vuotisissa karsinogeenisuustutkimuksissa havaittiin ei-letaaleja kilpirauhasen C-solukasvaimia. Rotilla ei todettu NOAEL-arvoa (no observed adverse effect level). Näitä kasvaimia ei havaittu apinoissa 20 kuukauden hoitojakson aikana. Jyrsijöistä tehdyt havainnot johtuvat ei-genotoksisesta, erityisestä GLP-1-reseptorivälitteisestä mekanismista, jolle jyrsijät ovat erityisen herkkiä. Merkitys ihmiselle on todennäköisesti pieni, mutta sitä ei voida sulkea täysin pois. Muita hoitoon liittyviä kasvaimia ei ole havaittu.
Eläinkokeet eivät osoittaneet suoria haitallisia vaikutuksia hedelmällisyyteen, mutta suurimmilla annoksilla esiintyi hieman enemmän varhaisvaiheen sikiökuolemia. Kun liraglutidia annettiin sikiöiän keskivaiheilla, se aiheutti emon painon alenemista ja sikiöiden kasvun hidastumista, epäselviä kylkiluumuutoksia rotilla ja luuston rakennemuutoksia kaneissa. Liraglutidille altistettujen vastasyntyneiden rottien kasvu hidastui ja pysyi hitaana vieroituksen jälkeen niillä rotilla, jotka oli altistettu suurille annoksille. Ei ole tiedossa, johtuuko poikasten kasvun hidastuminen suoran GLP-1-vaikutuksen aiheuttamasta maidon saannin vähenemisestä vai vähentyneen energiamäärän aiheuttamasta emon maidontuotannon vähenemisestä.
Nuorilla rotilla liraglutidi viivästytti sukukypsyyden saavuttamista sekä uroksilla että naarailla kliinisesti merkittävillä altistuksilla. Viivästyminen ei vaikuttanut kummallakaan sukupuolella hedelmällisyyteen tai lisääntymiskykyyn eikä naarailla kykyyn ylläpitää tiineyttä.
Farmaseuttiset tiedot
Apuaineet
Dinatriumfosfaattidihydraatti
Propyleeniglykoli
Fenoli
Kloorivetyhappo (pH:n säätöön)
Natriumhydroksidi (pH:n säätöön)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Saxenda-valmisteeseen lisätyt aineet voivat aiheuttaa liraglutidin hajoamista. Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
30 kuukautta
Käyttöönoton jälkeen: 1 kuukausi
Säilytys
Säilytä jääkaapissa (2 °C - 8 °C).
Ei saa jäätyä.
Älä säilytä lähellä jääkaapin pakastelokeroa.
Käyttöönoton jälkeen: Säilytä alle 30°C tai säilytä jääkaapissa (2 °C - 8 °C).
Pidä kynän suojus paikallaan. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SAXENDA injektioneste, liuos, esitäytetty kynä
6 mg/ml (L:ei) 5 x 3 ml (178,82 €)
PF-selosteen tieto
Sylinteriampulli (tyypin 1 lasia), joka sisältää bromobutyylistä valmistetun männän ja bromobutyyli/polyisopreenista valmistetun laminoidun kumisulkimen. Sylinteriampulli on esitäytetyssä, kertakäyttöisessä moniannoskynässä, joka on valmistettu polypropeenista, polyasetaalista, polykarbonaatista ja akrylonitriilibutadieenistyreenistä.
Jokaisessa kynässä on 3 ml liuosta ja kynästä saa 0,6 mg:n, 1,2 mg:n, 1,8 mg:n, 2,4 mg:n ja 3,0 mg:n annoksia.
Pakkauskoot ovat 1, 3 ja 5 esitäytettyä kynää.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas ja väritön tai melkein väritön, isotoninen liuos, pH = 8,15.
Käyttö- ja käsittelyohjeet
Liuosta ei tule käyttää, jos se ei ole kirkasta ja väritöntä tai melkein väritöntä.
Jäätynyttä Saxenda-valmistetta ei saa käyttää.
Kynä on tarkoitettu käytettäväksi kertakäyttöisten NovoFine- tai NovoTwist-neulojen kanssa, joiden pituus on enintään 8 mm ja läpimitta ohuimmillaan 32 G.
Neulat eivät sisälly pakkaukseen.
Potilasta tulee neuvoa hävittämään injektioneula aina pistoksen jälkeen ja säilyttämään kynä ilman neulaa. Näin ehkäistään kontaminoitumista, infektioita ja liuoksen vuotamista ulos kynästä. Samalla varmistetaan, että annostus on tarkka.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SAXENDA injektioneste, liuos, esitäytetty kynä
6 mg/ml 5 x 3 ml
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Liraglutidi (lihavuus): Aikuisten ja nuorten lihavuuden hoito erityisin edellytyksin (3051).
ATC-koodi
A10BJ02
Valmisteyhteenvedon muuttamispäivämäärä
26.06.2025
Yhteystiedot
 NOVO NORDISK FARMA OY
NOVO NORDISK FARMA OY Linnoitustie 6
02600 Espoo
020 762 5300
www.novonordisk.fi