
ZEJULA kapseli, kova 100 mg
Vaikuttavat aineet ja niiden määrät
Yksi kova kapseli sisältää niraparibitosylaattimonohydraattia määrän, joka vastaa 100 mg:aa niraparibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kova kapseli sisältää 254,5 mg laktoosimonohydraattia (katso kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yhden kovan kapselin kuori sisältää myös 0,0172 mg tartratsiini-väriainetta (E 102).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kova kapseli (kapseli).
Kliiniset tiedot
Käyttöaiheet
Zejula on tarkoitettu käytettäväksi:
- monoterapiana edennyttä epiteelistä (FIGO-asteet III ja IV) korkean pahanlaatuisuusasteen (high grade) munasarja-, munanjohdin- tai primaaria peritoneaalista syöpää sairastavien aikuispotilaiden ylläpitohoitoon, kun päättyneellä platinapohjaisella ensilinjan solunsalpaajahoidolla on saavutettu edelleen jatkuva (täydellinen tai osittainen) hoitovaste.
- monoterapiana uusiutunutta platinaherkkää korkean pahanlaatuisuusasteen (high grade) seroosia epiteelistä munasarja-, munanjohdin- tai primaaria peritoneaalista syöpää sairastavien aikuispotilaiden ylläpitohoitoon, kun platinapohjaisella solunsalpaajahoidolla on saavutettu (täydellinen tai osittainen) hoitovaste.
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Zejula-hoidon tulee aloittaa lääkäri, jolla on kokemusta syöpälääkkeiden käytöstä, ja hänen tulee myös valvoa hoitoa.
Annostus
Munasarjasyövän ensilinjan ylläpitohoito
Zejula-valmisteen suositeltu aloitusannos on 200 mg (kaksi 100 mg:n kapselia) kerran vuorokaudessa. Jos potilas painaa ≥ 77 kg ja lähtötilanteen verihiutaleiden määrä on ≥ 150 000/μl, Zejula-valmisteen suositeltu aloitusannos on 300 mg (kolme 100 mg:n kapselia) kerran vuorokaudessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset.).
Uusiutuneen munasarjasyövän ylläpitohoito
Annos on kolme 100 mg:n kovaa kapselia kerran päivässä, mikä vastaa 300 mg:n kokonaisannosta päivässä.
Potilaita on kehotettava ottamaan annos suurin piirtein samaan aikaan joka päivä. Lääkkeen ottaminen nukkumaan mentäessä voi auttaa hallitsemaan pahoinvointia.
Hoitoa suositellaan jatkettavan sairauden etenemiseen tai toksisuuden esiintymiseen saakka.
Annoksen unohtaminen
Jos potilas unohtaa ottaa annoksen, hänen on otettava seuraava annos normaalisti seuraavalla lääkkeenottokerralla.
Annoksen muuttaminen haittavaikutusten vuoksi
Suositellut annosmuutokset haittavaikutusten vuoksi on lueteltu taulukoissa 1, 2 ja 3.
Yleensä hoito suositellaan keskeytettävän (kuitenkin enintään 28 peräkkäisen päivän ajaksi), jotta potilas voi toipua haittavaikutuksesta, ja jatkettavan sen jälkeen samalla annoksella. Jos haittavaikutus toistuu, on suositeltavaa keskeyttää hoito ja aloittaa se uudelleen pienemmällä annoksella. Jos haittavaikutukset pysyvät 28 päivän keskeytyksen jälkeenkin, Zejula-hoito on suositeltavaa lopettaa. Ellei haittavaikutuksia saada hallintaan tällä tavalla (annon keskeyttäminen ja annoksen pienentäminen), Zejula-hoito on suositeltavaa lopettaa.
Taulukko 1: Suositellut annoksen muutokset haittavaikutusten vuoksi | ||
Aloitusannos | 200 mg | 300 mg |
Ensimmäinen annospienennys | 100 mg/vrk | 200 mg/vrk (kaksi 100 mg:n kapselia) |
Toinen annospienennys | Lopeta Zejula-hoito. | 100 mg/vrk* (yksi 100 mg:n kapseli) |
*Jos annosta on tarpeen pienentää alle tason 100 mg/vrk, Zejula-hoito on lopetettava.
Taulukko 2: Annoksen muutokset muiden kuin hematologisten haittavaikutusten vuoksi | |
Muu kuin hematologinen CTCAE-vaikeusasteen* ≥ 3 hoitoon liittyvä haittavaikutus, kun sen ehkäisy ei ole mahdollista tai kun haittavaikutus jatkuu hoidosta huolimatta | Ensimmäinen ilmaantumiskerta: • Keskeytä Zejula-hoito enintään 28 päivän ajaksi tai kunnes haittavaikutus on hävinnyt. • Aloita Zejula-hoito uudestaan pienemmällä annoksella taulukon 1 mukaisesti. |
Toinen ilmaantumiskerta: • Keskeytä Zejula-hoito enintään 28 päivän ajaksi tai kunnes haittavaikutus on hävinnyt. • Aloita Zejula-hoito uudestaan pienemmällä annoksella tai lopeta se taulukon 1 ohjeiden mukaisesti. | |
CTCAE-vaikeusasteen ≥ 3 hoitoon liittyvä haittavaikutus, joka kestää yli 28 päivää, kun potilaalle annetaan Zejulaa 100 mg/päivä | Lopeta hoito. |
*CTCAE = Common Terminology Criteria for Adverse Events, haittatapahtumien yleiset terminologiset kriteerit
Taulukko 3: Annoksen muutokset hematologisten haittavaikutusten vuoksi | |
Zejula-hoidon aikana on havaittu hematologisia haittavaikutuksia etenkin hoidon aloitusvaiheessa. Sen vuoksi suositellaan, että potilaan täydellistä verenkuvaa seurataan viikoittain ensimmäisen hoitokuukauden aikana ja annosta muutetaan tarpeen mukaan. Ensimmäisen kuukauden jälkeen täydellistä verenkuvaa suositellaan seurattavan kuukausittain ja sen jälkeen säännöllisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Yksilöllisten laboratorioarvojen perusteella viikoittainen seuranta toisen hoitokuukauden ajan voi olla tarpeen. | |
Hematologinen haittavaikutus, joka vaatii verensiirtoa tai hematopoieettisen kasvutekijän antamista | • Potilaille, joiden verihiutaleiden määrä on ≤ 10 000/μl, on harkittava verihiutaleiden siirtoa. Jos potilaalla on muita verenvuodon riskitekijöitä, esimerkiksi hyytymisenestolääkkeiden tai verihiutaleiden estolääkkeiden samanaikainen käyttö, niiden keskeyttämistä on harkittava ja/tai potilaalle on siirrettävä enemmän verihiutaleita. • Aloita Zejula-hoito uudestaan pienemmällä annoksella. |
Verihiutaleiden määrä < 100 000/μl | Ensimmäinen ilmaantumiskerta: • Keskeytä Zejula-hoito enintään 28 päivän ajaksi ja seuraa veriarvoja viikoittain, kunnes verihiutaleiden määrä on ≥ 100 000/µl. • Aloita Zejula-hoito uudestaan samalla tai pienemmällä annoksella kliinisen arvioinnin perusteella taulukon 1 mukaisesti. • Jos verihiutalemäärä on < 75 000/μl missä tahansa hoidon vaiheessa, aloita Zejula-hoito uudestaan pienemmällä annoksella taulukon 1 mukaisesti. |
Toinen ilmaantumiskerta: • Keskeytä Zejula-hoito enintään 28 päivän ajaksi ja seuraa veriarvoja viikoittain, kunnes verihiutaleiden määrä on ≥ 100 000/µl. • Aloita Zejula-hoito uudestaan pienemmällä annoksella taulukon 1 mukaisesti. • Lopeta Zejula-hoito, ellei verihiutaleiden määrä palaudu hyväksyttävälle tasolle niiden 28 päivän aikana, kun potilas ei saa hoitoa, tai jos potilaan annosta on jo pienennetty annokseen 100 mg kerran päivässä. | |
Neutrofiilien määrä < 1 000/µl tai hemoglobiiniarvo < 8 g/dl | • Keskeytä Zejula-hoito enintään 28 päivän ajaksi ja seuraa veriarvoja viikoittain, kunnes neutrofiilien määrä on ≥ 1 500/µl tai kunnes hemoglobiiniarvo on ≥ 9 g/dl. • Aloita Zejula-hoito uudestaan pienemmällä annoksella taulukon 1 mukaisesti. • Lopeta Zejula-hoito, ellei neutrofiilien määrä ja/tai hemoglobiiniarvo palaudu hyväksyttävälle tasolle niiden 28 päivän aikana, kun potilas ei saa hoitoa, tai jos potilaan annosta on jo pienennetty annokseen 100 mg kerran päivässä. |
Myelodysplastisen oireyhtymän tai akuutin myelooisen leukemian diagnoosi vahvistettu | • Lopeta Zejula-hoito pysyvästi. |
Pienipainoiset potilaat, jotka saavat ylläpitohoitoa uusiutuneeseen munasarjasyöpään
NOVA-tutkimuksessa noin 25 prosenttia potilaista painoi alle 58 kg ja noin 25 prosenttia potilaista painoi yli 77 kg. Pienipainoisille potilaille vaikeusasteen 3 tai 4 haittavaikutuksia ilmaantui enemmän (78 %) kuin painavammille potilaille (53 %). Vain 13 prosentilla pienipainoisista potilaista säilytettiin 300 mg:n annos hoitojakson 3 jälkeen. Alle 58 kg:n painoisille potilaille aloitusannokseksi voidaan harkita 200 mg:aa.
Iäkkäät potilaat
Iäkkäiden potilaiden (≥ 65-vuotiaiden) annosta ei tarvitse muuttaa. Kliinisiä tietoja vähintään 75-vuotiaista potilaista on vähän.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa potilailla, jotka sairastavat lievää tai keskivaikeaa munuaisten vajaatoimintaa. Tietoja vaikeaa munuaisten vajaatoimintaa tai loppuvaiheen munuaissairautta sairastavista potilaista, jotka käyvät hemodialyysissa, ei ole, joten näillä potilailla lääkettä on käytettävä varoen (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa potilailla, jotka sairastavat lievää maksan vajaatoimintaa (joko aspartaattiaminotransferaasi (ASAT) > viitearvojen yläraja ja kokonaisbilirubiini (TB) ≤ viiterajan yläraja tai mikä tahansa ASAT ja TB > 1,0 – 1,5 x viiterajan yläraja). Potilalle, jotka sairastavat keskivaikeaa maksan vajaatoimintaa (mikä tahansa ASAT ja TB > 1,5 – 3,0 x viiterajan yläraja) suositeltu Zejulan aloitusannos on 200 mg kerran päivässä. Tietoja vaikeaa maksan vajaatoimintaa sairastavista potilaista ei ole (mikä tahansa ASAT ja TB > 3 x viiterajan yläraja), joten näillä potilailla lääkettä on käytettävä varoen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Potilaat, joiden ECOG-suorituskykyluokka on 2–4
Saatavilla ei ole kliinisiä tietoja potilaista, joiden ECOG-suorituskykyluokka on 2–4.
Pediatriset potilaat
Niraparibin turvallisuutta ja tehoa lasten ja alle 18-vuotiaiden nuorten hoidossa ei ole vielä määritetty. Tietoja ei ole saatavilla.
Antotapa
Zejula on tarkoitettu käytettäväksi suun kautta. Kapselit on nieltävä kokonaisina veden kanssa. Kapseleita ei saa pureskella tai murskata.
Zejula kapselit voidaan ottaa aterioista riippumatta (ks. kohta Farmakokinetiikka).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Hematologiset haittavaikutukset
Zejula-hoitoa saaneilla potilailla on ilmoitettu hematologisia haittavaikutuksia (trombosytopeniaa, anemiaa ja neutropeniaa) (ks. kohta Haittavaikutukset). Pienipainoisilla potilailla tai potilailla, joilla lähtötilanteen verihiutaleiden määrä on pienentynyt saattaa olla suurentunut riski asteen 3 tromposytopeniaan (ks. kohta Annostus ja antotapa.)
Täydellisen verenkuvan seuranta viikoittain ensimmäisen hoitokuukauden aikana ja sen jälkeen kuukausittain 10 seuraavan hoitokuukauden ajan sekä säännöllisesti sen jälkeen on suositeltavaa. Näin voidaan seurata hematologisten parametrien kliinisesti merkittäviä muutoksia hoidon aikana (ks. kohta Annostus ja antotapa).
Jos potilaalle kehittyy vaikeaa ja itsepintaista hematologista toksisuutta (mukaan lukien pansytopeniaa), joka ei häviä 28 päivän kuluessa lääkityksen keskeyttämisestä, Zejula-hoito on lopetettava.
Trombosytopenian riskin vuoksi hyytymisenestolääkkeitä ja sellaisia lääkkeitä, joiden tiedetään vähentävän trombosyyttien määrää, on käytettävä varoen (ks. kohta Haittavaikutukset).
Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia
Myelodysplastista oireyhtymää / akuuttia myelooista leukemiaa (MDS/AML), myös kuolemaan johtaneita tapauksia, on havaittu Zejula-monoterapiaa tai yhdistelmähoitoa saaneilla potilailla kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeen (ks. kohta Haittavaikutukset).
Ennen myelodysplastisen oireyhtymän / akuutin myelooisen leukemian kehittymistä näiden potilaiden Zejula-hoito oli kestänyt kliinisissä tutkimuksissa 0,5 kuukaudesta > 4,9 vuoteen. Tapaukset olivat sekundaariselle syöpähoitoon liittyvälle myelodysplastiselle oireyhtymälle / akuutille myelooiselle leukemialle tyypillisiä. Kaikki potilaat olivat saaneet platinaa sisältäviä kemoterapiahoitoja, ja monet olivat saaneet myös muita DNA:ta vaurioittavia lääkeaineita ja sädehoitoa. Jotkin potilaat olivat sairastaneet luuydinlamaa. NOVA-tutkimuksessa myelodysplastisen oireyhtymän/akuutin myelooisen leukemian ilmaantuvuus oli korkeampi gBRCAmut-kohortissa (7,4 %) kuin non-gBRCAmut-kohortissa (1,7 %).
Jos myelodysplastista oireyhtymää / akuuttia myelooista leukemiaa epäillään tai potilaalla on pitkittynyt hematologinen toksisuus, potilas on lähetettävä hematologille lisäarviointia varten. Jos myelodysplastinen oireyhtymä / akuutti myelooinen leukemia vahvistetaan, Zejula-hoito on keskeytettävä ja potilas hoidettava asianmukaisesti.
Hypertensio ja hypertensiivinen kriisi
Zejulan käyttöön liittyvästä hypertensiosta ja hypertensiivisestä kriisistä on ilmoitettu (ks. kohta Haittavaikutukset). Olemassa oleva hypertensio on saatava asianmukaisesti hallintaan ennen Zejula-hoidon aloittamista. Zejula-hoidon aikana verenpainetta on seurattava vähintään viikoittain kahden kuukauden ajan, tämän jälkeen kuukausittain ensimmäisen vuoden ajan ja sen jälkeen säännöllisesti. Verenpaineen kotiseurantaa voidaan harkita joillain potilailla, ohjeistaen heitä olemaan yhteydessä lääkäriin mikäli verenpaine kohoaa.
Hypertensio on hoidettava verenpainelääkkeillä ja tarvittaessa muuttamalla Zejulan annosta (ks. kohta Annostus ja antotapa). Kliinisessä ohjelmassa verenpainemittaukset tehtiin kunkin 28 päivän mittaisen hoitojakson ensimmäisenä päivänä niin kauan kuin potilaat jatkoivat Zejula-hoitoa. Useimmissa tapauksissa verenpaine saatiin asianmukaisesti hallintaan tavanomaisella verenpainehoidolla ja lisäksi tarvittaessa Zejulan annosta muuttamalla (ks. kohta Annostus ja antotapa). Zejula-hoito on lopetettava hypertensiivisen kriisin ilmaantuessa tai jos lääketieteellisesti merkittävää korkeaa verenpainetta ei saada verenpainelääkkeillä asianmukaisesti hallintaan.
Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES)
Zejula-hoitoa saaneilla potilailla on raportoitu PRES:ä (ks. kohta Haittavaikutukset). PRES on harvinainen, palautuva neurologinen häiriö, joka voi ilmetä nopeasti kehittyvinä oireina kuten kouristuskohtauksina, päänsärkynä, mielentilan muutoksina, näköhäiriöinä ja kortikaalisena sokeutena, ja oireisiin saattaa liittyä hypertensiota. PRES-diagnoosin varmistaminen vaatii aivojen kuvantamistutkimuksen, mieluiten magneettikuvannuksen (MRI).
Jos potilaalla diagnosoidaan PRES, suositellaan Zejula-hoidon lopettamista ja verenpaineen sekä muiden oireiden asianmukaista hoitoa. Zejula-hoidon uudelleen aloittamisen turvallisuutta ei tunneta potilailla, joilla on aikaisemmin todettu PRES.
Raskaus/ehkäisy
Zejulaa ei saa käyttää raskauden aikana eikä naisille, jotka voivat tulla raskaaksi ja jotka eivät halua käyttää erittäin tehokasta ehkäisyä hoidon aikana ja kuuden kuukauden ajan viimeisen Zejula-annoksen ottamisesta (ks. kohta Raskaus ja imetys). Kaikkien naisten, jotka voivat tulla raskaaksi, on tehtävä raskaustesti ennen hoidon aloittamista.
Maksan vajaatoiminta
Keskivaikeaa maksan vajaatoimintaa sairastavilta potilailta kerätyn datan perusteella niraparibialtistus saattaa olla suurentunut vaikeaa maksan vajaatoimintaa sairastavilla potilailla. Tästä syystä kyseisiä potilaita tulee seurata huolellisesti (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Laktoosi
Kovat Zejula-kapselit sisältävät laktoosimonohydraattia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Tartratsiini (E 102)
Tämä lääkevalmiste sisältää tartratsiinia (E 102), joka saattaa aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Farmakodynaamiset yhteisvaikutukset
Niraparibin ja rokotteiden tai immunosuppressiivisten aineiden yhdistelmää ei ole tutkittu.
Tietoja niraparibin käytöstä yhdessä sytotoksisten lääkevalmisteiden kanssa on vähän. Varovaisuutta on noudatettava, jos niraparibia käytetään yhdessä rokotteiden, immunosuppressiivisten aineiden tai muiden sytotoksisten lääkevalmisteiden kanssa.
Farmakokineettiset yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset niraparibiin
Niraparibi CYP-entsyymien (CYP1A2 ja CYP3A4) substraattina
Niraparibi on karboksyyliesteraasien ja UDP-glukuronosyylitransferaasien substraatti in vivo. Niraparibin oksidatiivinen metabolia on vähäistä in vivo. Zejulan annosta ei tarvitse muuttaa, kun sitä annetaan samanaikaisesti sellaisten lääkevalmisteiden kanssa, joiden tiedetään estävän (esimerkiksi itrakonatsoli, ritonaviiri ja klaritromysiini) tai indusoivan (esimerkiksi rifampisiini, karbamatsepiini ja fenytoiini) CYP-entsyymejä.
Niraparibi effluksikuljettajaproteiinien (Pgp, BCRP, BSEP, MRP2 ja MATE1/2) substraattina
Niraparibi on P-glykoproteiinin (Pgp:n) ja rintasyövän resistenssiproteiinin (BCRP:n) substraatti. Sen suuren läpäisevyyden ja biologisen hyötyosuuden vuoksi kliinisesti merkityksellisten yhteisvaikutusten riski näitä kuljettajaproteiineja estävien lääkevalmisteiden kanssa on kuitenkin epätodennäköinen. Näin ollen Zejulan annosta ei tarvitse muuttaa, kun sitä annetaan samanaikaisesti sellaisten lääkevalmisteiden kanssa, joiden tiedetään estävän Pgp:tä (esimerkiksi amiodaroni, verapamiili) tai BCRP:tä (esimerkiksi osimertinibi, velpatasviiri ja eltrombopagi).
Niraparibi ei ole sappisuolapumpun (BSEP:n) tai monilääkeresistenssiin liittyvän proteiini 2:n (MRP2) substraatti. Ensisijainen päämetaboliitti M1 ei ole Pgp:n, BCRP:n, BSEP:n tai MRP2:n substraatti. Niraparibi ei ole monilääke- ja toksiiniekstruusio (MATE)-1:n tai MATE-2:n substraatti, kun taas M1 on kummankin substraatti.
Niraparibi maksan soluunoton kuljettajaproteiinien (OATP1B1, OATP1B3 ja OCT1) substraattina
Niraparibi ja M1 eivät ole orgaanisten anionien kuljettajapolypeptidi 1B1:n (OATP1B1), 1B3:n (OATP1B3) tai orgaanisten kationien kuljettaja 1:n (OCT1) substraatteja. Zejulan annosta ei tarvitse muuttaa, kun sitä annetaan samanaikaisesti sellaisten lääkevalmisteiden kanssa, joiden tiedetään estävän OATP1B1- tai 1B3-kuljettajaproteiineja (esimerkiksi gemfibrotsiili, ritonaviiri) tai OCT1-kuljettajaproteiinia (esimerkiksi dolutegraviiri).
Niraparibi munuaisten soluunoton kuljettajaproteiinien (OAT1, OAT3 ja OCT2) substraattina
Niraparibi ja M1 eivät ole orgaanisten anionien kuljettaja 1:n (OAT1), 3:n (OAT3) tai orgaanisten kationien kuljettaja 2:n (OCT2) substraatteja. Zejulan annosta ei tarvitse muuttaa, kun sitä annetaan samanaikaisesti sellaisten lääkevalmisteiden kanssa, joiden tiedetään estävän OAT1-kuljettajaproteiinia (esimerkiksi probenesidi), OAT3-kuljettajaproteiinia (esimerkiksi probenesidi, diklofenaakki) tai OCT2-kuljettajaproteiinia (esimerkiksi simetidiini, kinidiini).
Niraparibin vaikutukset muihin lääkevalmisteisiin
CYP-entsyymien (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ja CYP3A4) esto
Niraparibi ja M1 eivät ole minkään vaikuttavia aineita metaboloivan CYP-entsyymin (CYP1A1/2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ja CYP3A4/5) estäjiä.
Vaikka CYP3A4-entsyymin estämistä maksassa ei oleteta tapahtuvan, mahdollisuutta estää CYP3A4:ää suolistossa ei ole vahvistettu asianmukaisilla niraparibipitoisuuksilla. Näin ollen on syytä varovaisuuteen, kun niraparibia käytetään yhdessä sellaisten vaikuttavien aineiden kanssa, jotka metaboloituvat CYP3A4-välitteisesti, ja etenkin sellaisten vaikuttavien aineiden kanssa, joiden terapeuttinen alue on kapea (kuten siklosporiini, takrolimuusi, alfentaniili, ergotamiini, pimotsidi, ketiapiini ja halofantriini).
UDP-glukuronosyylitransferaasien (UGT) esto Niraparibilla ei ollut estävää vaikutusta UGT-isomuotoihin (UGT1A1, UGT1A4, UGT1A9 ja UGT2B7) 200 μM asti in vitro. Siksi kliinisesti merkityksellisen UGT-eston mahdollisuus on niraparibilla minimaalinen.
CYP-entsyymien (CYP1A2 ja CYP3A4) indusointi
Niraparibi ja M1 eivät ole CYP3A4:n indusoijia in vitro. In vitro niraparibi indusoi CYP1A2:ta heikosti suurina pitoisuuksina, eikä tämän vaikutuksen kliinistä merkityksellisyyttä ole täysin voitu sulkea pois. M1 ei ole CYP1A2:n indusoija. Näin ollen on syytä varovaisuuteen, kun niraparibia käytetään yhdessä sellaisten vaikuttavien aineiden kanssa, jotka metaboloituvat CYP1A2-välitteisesti, ja etenkin sellaisten vaikuttavien aineiden kanssa, joiden terapeuttinen alue on kapea (kuten klotsapiini, teofylliini ja ropiniroli).
Effluksikuljettajaproteiinien (P‑gp, BCRP, BSEP, MRP2 ja MATE1/2) esto
Niraparibi ei ole BSEP:n tai MRP2:n estäjä. In vitro niraparibi estää P‑gp:tä hyvin heikosti, ja BCRP:N osalta estävä pitoisuus IC50 = 161 µM ja 5,8 µM. Näin ollen kliinisesti merkityksellistä, joskin epätodennäköistä, yhteisvaikutusta, joka liittyy näiden effluksikuljettajaproteiinien estoon, ei voida sulkea pois. On siis syytä olla varovainen, kun niraparibi yhdistetään BCRP:n substraatteihin (kuten irinotekaani, rosuvastatiini, simvastatiini, atorvastatiini ja metotreksaatti).
Niraparibi on MATE1:n ja MATE2:n estäjä (IC50-arvot ovat 0,18 µM ja ≤ 0,14 µM). Sellaisten samanaikaisesti annettujen lääkevalmisteiden, jotka ovat näiden kuljettajien substraatteja (kuten metformiini), plasmapitoisuuksien suurenemista ei voida sulkea pois.
Tärkein päämetaboliitti M1 ei vaikuta olevan Pgp:n, BCRP:n, BSEP:n, MRP2:n tai MATE1/2:n estäjä.
Maksan soluunoton kuljettajaproteiinien (OATP1B1, OATP1B3 ja OCT1) esto
Niraparibi ja M1 eivät ole orgaanisten anionien kuljettajapolypeptidi 1B1:n (OATP1B1) tai 1B3:n (OATP1B3) estäjiä.
In vitro niraparibi estää heikosti orgaanisten kationien kuljettajaproteiini 1:tä (OCT1); estävä pitoisuus IC50 = 34,4 µM. On syytä olla varovainen, kun niraparibi yhdistetään sellaisiin vaikuttaviin aineisiin, joiden soluunoton kuljettajaproteiini on OCT1 (esimerkiksi metformiini).
Munuaisten soluunoton kuljettajaproteiinien (OAT1, OAT3 ja OCT2) esto
Niraparibi ja M1 eivät ole orgaanisten anionien kuljettaja 1:n (OAT1), 3:n (OAT3) ja orgaanisten kationien kuljettaja 2:n (OCT2) estäjiä.
Kaikki kliiniset tutkimukset on tehty vain aikuispotilailla.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy naisilla
Naiset, jotka voivat tulla raskaaksi, eivät saa tulla raskaaksi hoidon aikana, eivätkä he saa olla raskaana hoidon alussa. Kaikkien naisten, jotka voivat tulla raskaaksi, on tehtävä raskaustesti ennen hoidon aloittamista. Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä hoidon aikana ja kuuden kuukauden ajan viimeisen Zejula-annoksen ottamisesta.
Raskaus
Tietoja niraparibin käytöstä raskaana olevilla naisilla ei ole tai niitä on vain vähän. Lisääntymis- ja kehitystoksisuutta koskevia eläinkokeita ei ole tehty. Niraparibi voi vaikutusmekanisminsa vuoksi aiheuttaa haittaa alkiolle tai sikiölle, mukaan lukien alkion kuoleman ja teratogeenisia vaikutuksia, jos sitä annetaan raskaana olevalle naiselle. Zejulaa ei pidä käyttää raskauden aikana.
Imetys
Ei tiedetä, erittyvätkö niraparibi tai sen metaboliitit ihmisen rintamaitoon. Imetys on vasta-aiheista Zejulan käytön aikana ja yhden kuukauden ajan viimeisen annoksen ottamisesta (ks. kohta Vasta-aiheet).
Hedelmällisyys
Kliinisiä tietoja hedelmällisyydestä ei ole. Rotilla ja koirilla havaittiin ohimenevää spermatogeneesin heikentymistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Zejulalla on kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Zejulaa käyttävillä potilailla voi esiintyä asteniaa, väsymystä, huimausta tai keskittymisvaikeuksia. Potilaiden, joilla esiintyy näitä oireita, on noudatettava varovaisuutta ajaessaan tai käyttäessään koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kaikkien vaikeusasteiden haittavaikutuksia, joita ilmaantui ≥ 10 prosentille 851:stä Zejulaa monoterapiana saaneesta potilaasta PRIMA-tutkimuksen (aloitusannos 200 mg tai 300 mg) ja NOVA-tutkimuksen yhdistetyissä tiedoissa, olivat pahoinvointi, anemia, trombosytopenia, väsymys, ummetus, oksentelu, päänsärky, unettomuus, verihiutaleiden määrän väheneminen, neutropenia, vatsakipu, ruokahalun heikentyminen, ripuli, hengenahdistus, hypertensio, astenia, huimaus, neutrofiilien määrän väheneminen, yskä, nivelkipu, selkäkipu, valkosolujen määrän väheneminen sekä kuumat aallot.
Yleisimmät vakavat haittavaikutukset, joiden hoidonaikainen yleisyys oli > 1 %, olivat trombosytopenia ja anemia.
Taulukko haittavaikutuksista
Seuraavia haittavaikutuksia on todettu kliinisten tutkimusten ja myyntiluvan myöntämisen jälkeisen seurannan perusteella potilailla, jotka saivat Zejulaa monoterapiana (ks. taulukko 4). Haittavaikutusten yleisyys perustuu PRIMA- ja NOVA-tutkimusten yhdistettyyn tietoon haittavaikutuksista (kiinteä aloitusannos 300 mg/vrk), jossa potilaan altistuminen tunnetaan ja määritellään seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000) ja erittäin harvinainen (< 1/10 000). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 4: Taulukoitu luettelo haittavaikutuksista
| Elinjärjestelmä | Kaikkien CTCAE*-vaikeusasteiden yleisyys | CTCAE*-vaikeusasteen 3 tai 4 yleisyys |
| Infektiot | Hyvin yleinen Virtsatieinfektio Yleinen Keuhkoputkitulehdus, sidekalvotulehdus | Melko harvinainen Virtsatieinfektio Keuhkoputkitulehdus |
| Hyvän‑ ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Yleinen Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia** | Yleinen Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia** |
| Veri ja imukudos | Hyvin yleinen Trombosytopenia, anemia, neutropenia, leukopenia Melko harvinainen Pansytopenia, kuumeinen neutropenia | Hyvin yleinen Trombosytopenia, anemia, neutropenia Yleinen Leukopenia Melko harvinainen Pansytopenia, kuumeinen neutropenia |
| Immuunijärjestelmä | Yleinen Yliherkkyys† | Melko harvinainen Yliherkkyys |
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen Heikentynyt ruokahalu Yleinen Hypokalemia | Yleinen Hypokalemia Melko harvinainen Heikentynyt ruokahalu |
| Psyykkiset häiriöt | Hyvin yleinen Unettomuus Yleinen Ahdistuneisuus, masennus, kognitiivinen heikentyminen†† Melko harvinainen sekavuus | Melko harvinainen Unettomuus, ahdistuneisuus, masennus, sekavuus |
| Hermosto | Hyvin yleinen Päänsärky, huimaus Yleinen Makuhäiriöt Harvinainen Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES)** | Melko harvinainen Päänsärky |
| Sydän | Hyvin yleinen Sydämentykytys Yleinen Takykardia | |
| Verisuonisto | Hyvin yleinen Hypertensio Harvinainen Hypertensiivinen kriisi | Yleinen Hypertensio |
| Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen Hengenahdistus, yskä, nenänielun tulehdukset Yleinen Nenäverenvuoto Melko harvinainen pneumoniitti | Melko harvinainen Hengenahdistus, nenäverenvuoto, pneumoniitti |
| Ruoansulatuselimistö | Hyvin yleinen Pahoinvointi, ummetus, oksentelu, vatsakipu, ripuli, ruoansulatushäiriöt Yleinen Suun kuivuminen, vatsan turvotus, limakalvojen tulehdus, stomatiitti | Yleinen Pahoinvointi, oksentelu, vatsakipu Melko harvinainen Ripuli, ummetus, limakalvojen tulehdus, stomatiitti, suun kuivuminen |
| Iho ja ihonalainen kudos | Yleinen Valoherkkyys, ihottuma | Melko harvinainen Valoherkkyys, ihottuma |
| Luusto, lihakset ja sidekudos | Hyvin yleinen Selkäkipu, nivelkipu Yleinen Lihaskipu | Melko harvinainen Selkäkipu, nivelkipu, lihaskipu |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen Väsymys, astenia Yleinen Perifeerinen turvotus | Yleinen Väsymys, astenia |
| Tutkimukset | Yleinen Kohonnut gammaglutamyylitransferaasiarvo, kohonnut ASAT-arvo, kohonnut veren kreatiniinipitoisuus, kohonnut ALAT-arvo, kohonnut veren alkalisen fosfataasin pitoisuus, painonlasku | Yleinen Kohonnut gammaglutamyylitransferaasiarvo, kohonnut ALAT-arvo Melko harvinainen Kohonnut ASAT-arvo, kohonnut veren alkalisen fosfataasin pitoisuus |
*CTCAE = Common Terminology Criteria for Adverse Events, versio 4.02
** Perustuu niraparibin kliinisten tutkimusten tietoihin. Tämä ei rajoitu pivotaaliseen ENGOT-OV16 monoterapiatutkimukseen.
† käsittää yliherkkyyden, yliherkkyyden lääkeaineille, anafylaktisen reaktion, lääkeaineihottuman, angioödeeman ja urtikarian.
†† käsittää muistin heikkenemisen, keskittymisvaikeudet.
Potilailla, jotka saivat Zejula-hoitoa aloitusannoksella 200 mg lähtötilanteen painon tai verihiutalemäärän perusteella, haittavaikutusten esiintymistiheys oli samankaltainen tai pienempi kuin potilailla, joilla oli käytössä kiinteä aloitusannos 300 mg (taulukko 4).
Jäljempänä annetaan tarkempaa tietoa trombosytopenian, anemian ja neutropenian esiintymistiheyksistä.
Tiettyjen haittavaikutusten kuvaus
Hematologisia haittavaikutuksia (trombosytopeniaa, anemiaa, neutropeniaa), joiden osalta tehtiin myös kliinisiä diagnooseja ja/tai laboratoriolöydöksiä, ilmaantui yleensä niraparibihoidon alussa, ja niiden ilmaantuvuus väheni ajan myötä.
NOVA- ja PRIMA-tutkimuksissa Zejula-hoitoon soveltuvien potilaiden lähtötilanteen hematologiset parametrit ennen hoitoa olivat seuraavat: neutrofiilien absoluuttinen määrä (ANC) ≥ 1 500 solua/µl; verihiutaleet ≥ 100 000 solua/µl ja hemoglobiini ≥ 9 g/dl (NOVA) ja ≥ 10 g/dl (PRIMA). Kliinisessä ohjelmassa hematologisia haittavaikutuksia hoidettiin laboratorioarvojen seurannalla ja annosmuutoksilla (ks. kohta Annostus ja antotapa).
PRIMA-tutkimuksessa potilailla, joiden Zejula-aloitusannos määräytyi lähtötilanteen painon tai verihiutaleiden määrän perusteella, vähintään asteen 3 trombosytopenian esiintyminen vähentyi 48 prosentista 21 prosenttiin, vähintään asteen 3 anemian 36 prosentista 23 prosenttiin ja vähintään asteen 3 neutropenian 24 prosentista 15 prosenttiin verrattuna potilasryhmään, jossa oli käytössä 300 mg:n kiinteä aloitusannos. Hoito lopetettiin trombosytopenian vuoksi 3 prosentilla, anemian vuoksi 3 prosentilla ja neutropenian vuoksi 2 prosentilla potilaista.
Trombosytopenia
PRIMA-tutkimuksessa 39 prosentilla Zejula-hoitoa saaneista potilaista esiintyi asteen 3/4 trombosytopeniaa, kun vastaava osuus lumehoitoa saaneilla oli 0,4 %. Trombosytopenian kehittymiseen kulunut mediaaniaika ensimmäisen annoksen jälkeen oli 22 päivää (vaihteluväli: 15–335 päivää) ja trombosytopenian mediaanikesto 6 päivää (vaihteluväli: 1–374 päivää). Hoito lopetettiin trombosytopenian vuoksi 4 prosentilla niraparibia saaneista potilaista.
NOVA-tutkimuksessa noin 60 prosentille Zejulaa saaneista potilaista kehittyi jonkinasteinen trombosytopenia, ja 34 prosentille potilaista kehittyi vaikeusasteen 3/4 trombosytopenia. Niistä potilaista, joiden verihiutaleiden määrä oli lähtötilanteessa alle 180 × 109/l, 76 prosentille kehittyi jonkinasteinen trombosytopenia ja 45 prosentille kehittyi vaikeusasteen 3 tai 4 trombosytopenia. Trombosytopenian kehittymiseen kulunut mediaaniaika vaikeusasteesta riippumatta ja vaikeusasteille 3 tai 4 oli 22 ja 23 päivää. Uusien trombosytopeniatapausten ilmaantuvuus sen jälkeen, kun annoksia oli muutettu voimakkaasti kahden ensimmäisen hoitokuukauden aikana jaksosta 4 alkaen, oli 1,2 %. Trombosytopenian mediaanikesto vaikeusasteesta riippumatta oli 23 päivää, ja vaikeusasteen 3/4 trombosytopenian mediaanikesto oli 10 päivää. Zejula-hoitoa saavilla potilailla, joille kehittyy trombosytopenia, voi olla suurentunut verenvuodon riski. Kliinisessä ohjelmassa trombosytopeniaa hoidettiin laboratoriokoeseurannalla, annosta muuttamalla ja tarvittaessa verihiutaleiden siirrolla (ks. kohta Annostus ja antotapa). Zejula-hoito lopetettiin trombosytopeniatapahtumien (trombosytopenian ja verihiutaleiden määrän pienentymisen) takia noin kolmella prosentilla potilaista.
NOVA-tutkimuksessa 48:lla 367 potilaasta (13 %) esiintyi verenvuotoa ja samanaikaista trombosytopeniaa. Kaikkien trombosytopenian yhteydessä esiintyneiden verenvuototapahtumien vaikeusaste oli 1 tai 2 lukuun ottamatta yhtä tapahtumaa, jossa samanaikaisen vakavan pansytopeniahaittavaikutuksen yhteydessä todettiin vaikeusasteen 3 petekioita ja hematoomia. Trombosytopeniaa esiintyi yleisemmin potilailla, joiden verihiutaleiden määrä oli lähtötilanteessa < 180 × 109/l. Noin 76 prosentille Zejula-hoitoa saaneista potilaista, joiden verihiutaleiden määrä oli lähtötilanteessa pieni (< 180 × 109/l), kehittyi jonkinasteinen trombosytopenia, ja 45 prosentille potilaista kehittyi vaikeusasteen 3/4 trombosytopenia. Pansytopeniaa on havaittu < 1 prosentilla niraparibia saaneista potilaista.
Anemia
PRIMA-tutkimuksessa 31 prosentilla Zejula-hoitoa saaneista potilaista esiintyi asteen 3/4 anemiaa, kun vastaava osuus lumehoitoa saaneilla oli 2 %. Anemian kehittymiseen kulunut mediaaniaika ensimmäisen annoksen jälkeen oli 80 päivää (vaihteluväli: 15–533 päivää) ja anemian mediaanikesto 7 päivää (vaihteluväli: 1–119 päivää). Hoito lopetettiin anemian vuoksi 2 prosentilla niraparibia saaneista potilaista.
NOVA-tutkimuksessa noin 50 prosentille potilaista kehittyi jonkinasteinen anemia, ja 25 prosentille potilaista kehittyi vaikeusasteen 3/4 anemia. Vaikeusasteesta riippumatta anemian kehittymiseen kulunut mediaaniaika oli 42 päivää, ja vaikeusasteen 3/4 tapahtumissa mediaaniaika oli 85 päivää. Jonkinasteisen anemian mediaanikesto oli 63 päivää, ja vaikeusasteen 3/4 tapahtumissa mediaanikesto oli 8 päivää. Zejula-hoidon aikana voi esiintyä jatkuvasti jonkinasteista anemiaa. Kliinisessä ohjelmassa anemiaa hoidettiin laboratoriokoeseurannalla, annosta muuttamalla (ks. kohta Annostus ja antotapa) ja tarvittaessa punasolujen siirrolla. Zejula-hoito lopetettiin anemian takia yhdellä prosentilla potilaista.
Neutropenia
PRIMA-tutkimuksessa 21 prosentilla Zejula-hoitoa saaneista potilaista esiintyi asteen 3/4 neutropeniaa, kun vastaava osuus lumehoitoa saaneilla oli 1 %. Neutropenian kehittymiseen kulunut mediaaniaika ensimmäisen annoksen jälkeen oli 29 päivää (vaihteluväli: 15–421 päivää) ja neutropenian mediaanikesto 8 päivää (vaihteluväli 1–42 päivää). Hoito lopetettiin neutropenian vuoksi 2 prosentilla niraparibia saaneista potilaista.
NOVA-tutkimuksessa noin 30 prosentille Zejulaa saaneista potilaista kehittyi jonkinasteinen neutropenia, ja 20 prosentille potilaista kehittyi vaikeusasteen 3/4 neutropenia. Jonkinasteisen neutropenian kehittymiseen kulunut mediaaniaika oli 27 päivää, ja vaikeusasteen 3/4 tapahtumissa mediaaniaika oli 29 päivää. Jonkinasteisen neutropenian mediaanikesto oli 26 päivää, ja vaikeusasteen 3/4 tapahtumissa mediaanikesto oli 13 päivää. Lisäksi noin kuudelle prosentille niraparibilla hoidetuista potilaista annettiin granulosyyttiryhmiä stimuloivaa kasvutekijää (G‑CSF) samanaikaisena neutropeniahoitona. Zejula-hoito lopetettiin neutropenian takia kahdella prosentilla potilaista.
Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia
Kliinisissä tutkimuksissa myelodysplastista oireyhtymää (MDS) / akuuttia myelooista leukemiaa (AML) esiintyi 1 %:lla Zejula-hoitoa saaneista potilaista. Tapauksista 41 % johti kuolemaan. Ilmaantuvuus 75 kuukauden elossaoloseurannan kohdalla oli suurempi uusiutunutta munasarjasyöpää sairastavilla potilailla, jotka olivat saaneet vähintään kahta aiempaa platinapohjaista solunsalpaajahoitolinjaa ja joilla oli itulinjan BRCA (gBRCA) ‑mutaatio. Kaikilla potilailla oli MDS:n/AML:n kehittymiseen myötävaikuttavia tekijöitä aiemman platinaa sisältävän solunsalpaajahoidon seurauksena. Monet olivat myös saaneet muita DNA:ta vaurioittavia lääkeaineita ja sädehoitoa. Valtaosa tapauksista raportoitiin gBRCAmut‑potilailla. Osalla potilaista oli anamneesissa aiempi syöpä tai luuydinlama.
PRIMA‑tutkimuksessa MDS:n/AML:n ilmaantuvuus oli 0,8 % Zejula-hoitoa saaneilla potilailla ja 0,4 % lumelääkettä saaneilla potilailla.
NOVA‑tutkimuksessa MDS:n/AML:n kokonaisilmaantuvuus 75 kuukauden seurannan kohdalla oli 3,8 % Zejula-hoitoa saaneilla ja 1,7 % lumelääkettä saaneilla potilailla, joilla oli uusiutunut munasarjasyöpä ja jotka olivat saaneet vähintään kahta aiempaa platinapohjaista solunsalpaajahoitolinjaa. MDS:n/AML:n ilmaantuvuus gBRCAmut‑kohortissa oli 7,4 % Zejula-hoitoa saaneilla potilailla ja 3,1 % lumelääkettä saaneilla potilailla ja non‑gBRCAmut‑kohortissa 1,7 % Zejula-hoitoa saaneilla potilailla ja 0,9 % lumelääkettä saaneilla potilailla.
Hypertensio
PRIMA-tutkimuksessa 6 prosentilla Zejula-hoitoa saaneista potilaista esiintyi asteen 3/4 hypertensiota, kun vastaava osuus lumehoitoa saaneilla oli 1 %. Hypertension kehittymiseen kulunut mediaaniaika ensimmäisen annoksen jälkeen oli 50 päivää (vaihteluväli: 1–589 päivää) ja hypertension mediaanikesto 12 päivää (vaihteluväli 1–61 päivää). Hoito lopetettiin hypertension vuoksi 0 prosentilla potilaista.
NOVA-tutkimuksessa jonkinasteinen hypertensio kehittyi 19,3 prosentille Zejulalla hoidetuista potilaista. Vaikeusasteen 3/4 hypertensio ilmaantui 8,2 prosentille potilaista. Hypertensio hoidettiin nopeasti verenpainelääkkeillä. Zejula-hoito lopetettiin hypertension takia < 1 prosentilla potilaista.
Pediatriset potilaat
Pediatrisilla potilailla ei ole tehty tutkimuksia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta.
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Zejulan yliannostukseen ei ole erityistä hoitoa, eikä yliannostuksen oireita tunneta. Yliannostustapauksessa lääkärin on annettava tavanomaista elintoimintoja tukevaa hoitoa sekä oireenmukaista hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, muut antineoplastiset lääkeaineet, ATC-koodi: L01XK02.
Vaikutusmekanismi ja farmakodynaamiset vaikutukset
Niraparibi on poly(ADP-riboosi)-polymeraasi (PARP) -entsyymien, PARP1:n ja PARP2:n, estäjä. Nämä entsyymit osallistuvat DNA:n korjaukseen. In vitro -tutkimukset ovat osoittaneet, että niraparibin indusoimaan sytotoksisuuteen saattaa liittyä PARP-entsyymien toiminnan estymistä ja PARPDNA-kompleksien muodostumisen lisääntymistä, mikä johtaa DNA:n vaurioitumiseen, apoptoosiin ja solukuolemaan. Niraparibin indusoiman sytotoksisuuden lisääntymistä havaittiin kasvainsolulinjoissa, joissa oli tai ei ollut kasvaimen BReast CAncer (BRCA) 1 ja 2 -suppressorigeenin virheitä. Potilaalta otetuissa ortotooppisissa korkean pahanlaatuisuusasteen serooseissa munasarjasyövän ksenograftikasvaimissa (PDX), joita kasvatettiin hiirissä, niraparibin on osoitettu vähentävän sellaisten kasvaimien kasvua, jotka ovat BRCA 1 ja 2 -mutantteja, BRCA-villityyppiä mutta joissa on homologisen rekombinaation (HR) virhe, ja BRCA-villityyppiä, joissa ei ole havaittavissa olevaa HR-virhettä.
Kliininen teho ja turvallisuus
Munasarjasyövän ensilinjan ylläpitohoito
PRIMA oli vaiheen 3 kaksoissokkoutettu, lumekontrolloitu tutkimus, jossa platinapohjaisella ensilinjan solunsalpaajahoidolla täydellisen tai osittaisen hoitovasteen saavuttaneet potilaat (n = 733) satunnaistettiin saamaan niraparibia tai vastaavaa lumetta suhteessa 2:1. PRIMA-tutkimuksessa aloitusannosta 300 mg x 1 annettiin 475 potilaalle (joista 317 satunnaistettiin niraparibiryhmään ja 158 lumeryhmään) jatkuvina 28 vuorokauden hoitojaksoina. PRIMA-tutkimuksen aloitusannosta muutettiin tutkimussuunnitelman muutoksessa 2. Tämän jälkeen potilaat, joiden lähtötilanteen paino oli ≥ 77 kg ja lähtötilanteen verihiutalemäärä ≥ 150 000/µl, saivat päivittäin niraparibihoitoa annoksella 300 mg (n = 34) tai lumetta (n = 21). Potilaat, joiden lähtötilanteen paino oli < 77 kg tai lähtötilanteen verihiutalemäärä < 150 000/μl, saivat päivittäin niraparibihoitoa annoksella 200 mg (n = 122) tai lumetta (n = 61).
Potilaat satunnaistettiin platinapohjaisen ensilinjan solunsalpaajahoidon päätyttyä (ja heidän saatuaan mahdollisesti leikkaushoitoa). Tutkimushenkilöt satunnaistettiin 12 viikon kuluessa viimeisen kemoterapianjakson ensimmäisestä päivästä. Tutkimushenkilöt saivat vähintään kuusi ja enintään yhdeksän platinapohjaista hoitojaksoa. Kirurgisen intervalli debulking-leikkauksen jälkeen tutkimushenkilöillä oli vähintään kaksi leikkauksen jälkeistä platinapohjaista hoitojaksoa. Tutkimuksesta ei suljettu pois potilaita, jotka olivat saaneet solunsalpaajahoitoa yhdistelmänä bevasitsumabin kanssa mutta joille bevasitsumabia ei voitu antaa ylläpitohoitona. Potilaat eivät olleet saaneet aiempaa PARP-entsyymien toimintaa estävää hoitoa, mukaan lukien niraparibi. Neoadjuvanttisolunsalpaajahoidon ja sen jälkeisen intervallileikkaushoidon saaneilla potilailla oli joko havaittavissa olevaa jäännöstautia tai ei jäännöstautia. Tutkimuksesta suljettiin pois levinneisyysasteen III tautia sairastavat potilaat, joilla oli saavutettu primaarisen sytoreduktiivisen leikkauksen jälkeen täydellinen sytoreduktiotulos (ts. ei havaittavissa olevaa jäännöstautia). Satunnaistamisessa käytettiin seuraavia ositusperusteita: paras hoitovaste ensilinjan platinahoidon aikana (täydellinen vs. osittainen vaste), neoadjuvanttisolunsalpaajahoito (kyllä vs. ei) sekä homologisen rekombinaation puutos (HRD) (positiivinen (potilaat, joilla on homologisen rekombinaation puutos) vs. negatiivinen (potilaat, joilla ei ole homologisen rekombinaation puutosta) tai ei määritelty). Homologisen rekombinaation puutos testattiin HRD-testillä kasvainkudoksesta, jota oli otettu talteen alkuperäisen diagnoosin ajankohtana. CA-125-arvojen oli oltava normaaleja (tai CA-125-arvon piti olla > 90 prosenttia pienempi kuin lähtötilanteessa) potilaan ensilinjan hoidon aikana, ja niiden piti pysyä vakaina vähintään seitsemän päivän ajan.
Hoito aloitettiin hoitojakson 1 päivänä 1 niraparibi-annoksella 200 mg tai 300 mg tai vastaavalla lumelääkeannoksella, ja sitä annettiin kerran vuorokaudessa jatkuvina 28 päivän hoitojaksoina. Potilaat kävivät tutkimuskäynneillä joka hoitojakson aikana (4 viikon ± 3 vuorokauden välein).
Ensisijainen päätetapahtuma oli etenemisvapaa elinaika (PFS), joka määritettiin RECIST-kriteerien (Response Evaluation Criteria in Solid Tumours, versio 1.1) mukaisesti sokkoutetun, riippumattoman keskitetyn arvioinnin avulla. Tärkeä toissijainen tavoite oli kokonaiselinaika (OS). PFS tutkittiin hierarkkisesti: ensin potilailla, joilla oli homologisen rekombinaation puutos, ja sitten koko populaatiossa. Mediaani-ikä oli 62 vuotta: vaihteluväli niraparibihoitoon satunnaistetuilla oli 32–85 vuotta ja lumehoitoon satunnaistetuilla 33–88 vuotta. 89 % kaikista potilaista oli valkoihoisia. 69 % niraparibihoitoon satunnaistetuista ja 71 % lumehoitoon satunnaistetuista potilaista oli ECOG-suorituskykyluokaltaan 0 tutkimuksen lähtötilanteessa. Koko populaatiosta 65 % sairasti levinneisyysasteen III tautia ja 35 % levinneisyysasteen IV tautia. Kasvaimen yleisin sijainti koko populaatiossa (≥ 80 % potilaista) oli munasarja; kasvaimen yleisin (> 90 % potilaista) histologinen tyyppi oli seroosi. 67 % potilaista sai neoadjuvanttisolunsalpaajahoitoa. 69 prosentilla potilaista platinapohjainen ensilinjan solunsalpaajahoito oli tuottanut täydellisen hoitovasteen. Kaikkiaan 6 niraparibipotilasta oli saanut munasarjasyövän aiempana hoitona bevasitsumabia.
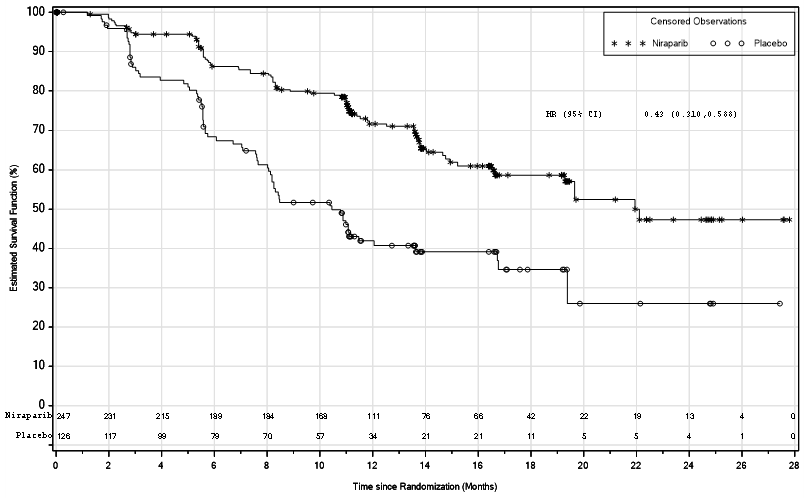
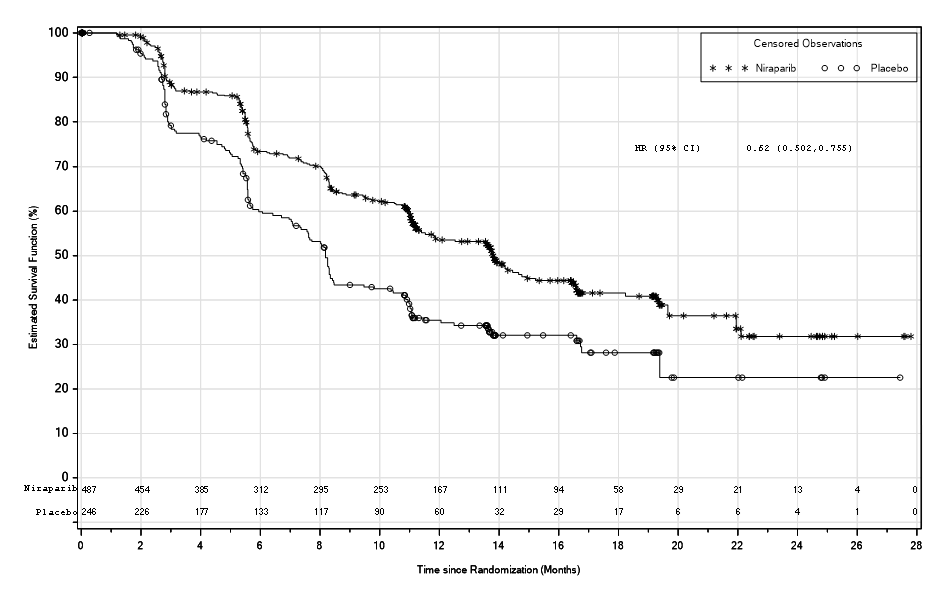
PRIMA-tutkimuksessa PFS oli tilastollisesti merkitsevästi parempi niraparibihoitoon satunnaistetuilla potilailla kuin lumehoitoon satunnaistetuilla potilailla sekä potilailla, joilla oli homologisen rekombinaation puutos, että koko populaatiossa (taulukko 5 ja kuvat 1 ja 2).
Toissijaisiin tehon päätetapahtumiin kuuluivat etenemisvapaa elinaika ensimmäisen myöhemmän hoidon jälkeen (PFS2) ja kokonaiselinaika (OS) (taulukko 5).
Taulukko 5: Tehoon liittyvät tulokset – PRIMA (sokkoutettu, riippumaton, keskitetty arviointi)
| Potilaat, joilla homologisen rekombinaation puutos | Koko populaatio | |||
Niraparibi (N = 247) | Lume (N = 126) | Niraparibi (N = 487) | Lume (N = 246) | |
| PFS-mediaani (95 %:n luottamusväli) | 21,9 (19,3 – ei arvioitavissa) | 10,4 (8,1–12,1) | 13,8 (11,5–14,9) | 8,2 (7,3–8,5) |
| Riskitiheyssuhde (95 %:n luottamusväli) | 0,43 (0,31–0,59) | 0,62 (0,50–0,76) | ||
| p-arvo | < 0,0001 | < 0,0001 | ||
PFS2 Riskitiheyssuhde (95 %:n luottamusväli) | 0,84 (0,485–1,453) | 0,81 (0,577–1,139) | ||
OS* Riskitiheyssuhde (95 %:n luottamusväli) | 0,61 (0,265–1,388) | 0,70 (0,44–1,11) | ||
PFS = etenemisvapaa elinaika; OS = kokonaiselinaika; PFS2 = etenemisvapaa elinaika ensimmäisen myöhemmän hoidon jälkeen.
*Ensisijaisen PFS-analyysin ajankohtana arvioitu elossaolo-osuus kahden vuoden kuluttua satunnaistamisesta oli Zejula-hoitoa saaneilla 84 % ja lumehoitoa saaneilla 77 % koko populaatiossa.
PFS2- ja OS-tiedot eivät vielä ole kypsät.
Kuva 1: PRIMA: etenemisvapaa elinaika (PFS) potilailla, joiden kasvaimissa oli homologisen rekombinaation puutos (ITT-populaatio, N = 373)
Kuva 2: PRIMA: etenemisvapaa elinaika (PFS) koko populaatiossa (ITT-populaatio, N = 733)
Alaryhmäanalyysit
Potilailla, joilla oli homologisen rekombinaation puutos, BRCA-mutaatiopositiivista munasarjasyöpää sairastaneiden potilaiden alaryhmässä (N = 223) havaittu riskitiheyksien suhde oli 0,40 (95 %:n luottamusväli: 0,27–0,62). Alaryhmässä, jossa potilailla oli homologisen rekombinaation puutos mutta ei BRCA-mutaatiota (N = 150), havaittu riskitiheyksien suhde oli 0,50 (95 %:n luottamusväli: 0,31–0,83). Potilailla, joilla ei ollut homologisen rekombinaation puutosta (N = 249), havaittu riskitiheyksien suhde oli 0,68 (95 %:n luottamusväli: 0,49–0,94).
Eksploratiivisissä alaryhmäanalyyseissä potilailla, jotka saivat Zejula-hoitoa annoksella 200 mg tai 300 mg lähtötilanteen painon tai verihiutalemäärän perusteella, tehotulokset (tutkijan arvioima PFS) olivat samankaltaiset potilailla, joilla oli homologisen rekombinaation puutos (riskitiheyksien suhde 0,54; 95 %:n luottamusväli: 0,33–0,91), ja koko populaatiossa (riskitiheyksien suhde 0,68; 95 %:n luottamusväli: 0,49–0,94). Potilailla, joilla ei ollut homologisen rekombinaation puutosta 200 mg:n annos vaikutti johtavan vähäisempään hoitovaikutukseen kuin 300 mg:n annos.
Uusiutuneen platinaherkän munasarjasyövän ylläpitohoito
Niraparibin turvallisuutta ja tehoa ylläpitohoitona tutkittiin vaiheen 3 satunnaistetussa kaksoissokkoutetussa lumelääkekontrolloidussa kansainvälisessä tutkimuksessa (NOVA) potilailla, joilla oli uusiutunut pääasiassa korkean pahanlaatuisuusasteen (high grade) seroosi epiteelinen munasarja-, munanjohdin- tai primaari peritoneaalinen syöpä ja joiden sairaus oli platinaherkkä. Platinaherkkyys määriteltiin täydellisenä tai osittaisena, yli kuusi kuukautta kestävänä vasteena potilaiden toiseksi viimeiseen platinapohjaiseen hoitoon. Jotta potilas soveltuisi niraparibihoitoon, tällä piti olla vaste (täydellinen tai osittainen) viimeisen platinapohjaisen kemoterapian päättymisen jälkeen. CA-125-arvojen oli oltava normaaleja (tai CA-125-arvon piti olla > 90 prosenttia pienempi kuin lähtötilanteessa) viimeisen platinahoidon jälkeen, ja niiden piti pysyä vakaina vähintään seitsemän päivän ajan. Potilaat eivät olleet voineet saada aiempaa hoitoa PARP-entsyymien estäjällä, Zejula mukaan luettuna. Soveltuvat potilaat jaoteltiin jompaankumpaan kahdesta kohortista gBRCA-mutaatiotestin tulosten perusteella. Kummassakin kohortissa potilaat satunnaistettiin suhdetta 2:1 käyttäen niraparibi- ja lumelääkeryhmiin. Potilaat sijoitettiin gBRCAmut-kohorttiin gBRCA-analyysia varten otettujen, satunnaistamista edeltävien verinäytteiden perusteella. Kasvaimen BRCA (tBRCA) ‑mutaatiota ja homologisen rekombinaation puutosta (HRD) koskevat testit tehtiin käyttäen HRD-testiä kasvainkudokseen, jota oli otettu ensimmäisen diagnoosin yhteydessä tai sairauden uusiutuessa.
Kummassakin kohortissa satunnaistaminen ositettiin seuraavilla perusteilla: sairauden etenemiseen viimeistä edellisen, ennen tutkimukseen osallistumista annetun platinahoidon jälkeen kulunut aika (6 ‑ < 12 kuukautta ja ≥ 12 kuukautta), bevasitsumabin mahdollinen käyttö viimeistä edellisen tai viimeisen platinahoidon yhteydessä ja paras vaste viimeisimmän platinahoidon aikana (täydellinen vaste ja osittainen vaste).
Potilaat aloittivat hoidon hoitojakson 1 ensimmäisenä päivänä 300 mg:n niraparibiannoksella tai vastaavalla lumelääkeannoksella, jotka annettiin kerran päivässä jatkuvina 28 päivän jaksoina. Potilaat kävivät tutkimuskäynnillä joka hoitojaksossa (4 viikkoa ± 3 päivää).
NOVA-tutkimuksessa 48 prosentilla potilaista lääkkeen anto keskeytettiin hoitojaksolla 1. Noin 47 prosenttia jatkoi lääkitystä pienemmällä annoksella hoitojaksolla 2.
NOVA-tutkimuksessa yleisin käytetty annos niraparibilla hoidetuilla potilailla oli 200 mg.
Etenemisvapaa elinaika (progression-free survival, PFS) määritettiin RECIST-kriteerien (Response Evaluation Criteria in Solid Tumours, versio 1.1) tai kliinisten löydösten ja oireiden sekä CA125-arvon kohoamisen perusteella. Etenemisvapaa elinaika mitattiin satunnaistamisesta (joka tehtiin enintään kahdeksan viikon kuluttua kemoterapian päättymisestä) sairauden etenemiseen tai kuolemaan.
Etenemisvapaata elinaikaa koskeva ensisijaisen tehoanalyysin määritti sokkoutettu riippumaton keskitetty arvioijataho, ja se määritettiin ja arvioitiin prospektiivisesti gBRCAmut-kohortin ja non‑gBRCAmut-kohortin osalta erikseen. Kokonaiselinajan (OS) analyysit olivat toissijaisia tulosmuuttujia.
Toissijaisia tehon päätetapahtumia olivat kemoterapiavapaa aika (chemotherapy‑free interval, CFI), aika ensimmäiseen seuraavaan hoitoon (time to first subsequent therapy, TFST), etenemisvapaa elinaika ensimmäistä hoitoa seuraavan hoidon jälkeen (PFS2) ja kokonaiselinaika (OS).
Demografia, sairauden piirteet lähtötilanteessa ja aiempi hoitohistoria olivat yleisesti ottaen hyvin tasapainossa gBRCAmut-kohortin (n = 203) ja non‑gBRCAmut-kohortin (n = 350) niraparibi- ja lumelääkeryhmien välillä. Mediaani-ikä hoitoryhmissä ja kohorteissa oli 57–63 vuotta. Ensisijaisen kasvaimen paikka useimmilla potilailla (> 80 %) kummassakin kohortissa oli munasarja, ja useimmilla potilailla (> 84 %) oli histologialtaan serooseja kasvaimia. Suuri osa kummankin kohortin kummankin ryhmän potilaista oli saanut vähintään kolme solunsalpaajahoitolinjaa aikaisemmin, mukaan luettuna 49 prosenttia gBRCAmut-kohortin ja 34 prosenttia non‑gBRCAmut-kohortin niraparibia saaneista potilaista. Useimmat potilaat olivat iältään 18–64-vuotiaita (78 %), valkoihoisia (86 %) ja EGOC-suorituskykyluokaltaan 0 (68 %).
Hoitojaksojen mediaanimäärä gBRCAmut-kohortissa oli suurempi niraparibiryhmässä kuin lumelääkeryhmässä (14 vs. 7 hoitojaksoa). Useammat potilaat niraparibiryhmässä kuin lumelääkeryhmässä jatkoivat hoitoa yli 12 kuukautta (54,4 % vs. 16,9 %).
Hoitojaksojen mediaanimäärä koko non-gBRCAmut-kohortissa oli suurempi niraparibiryhmässä kuin lumelääkeryhmässä (8 vs. 5 hoitojaksoa). Useammat potilaat niraparibiryhmässä kuin lumelääkeryhmässä jatkoivat hoitoa yli 12 kuukautta (34,2 % vs. 21,1 %).
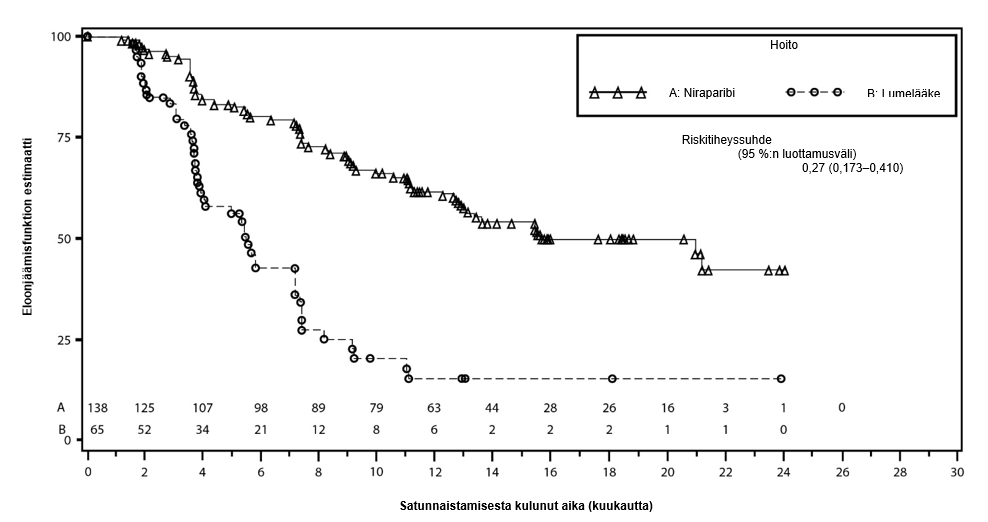
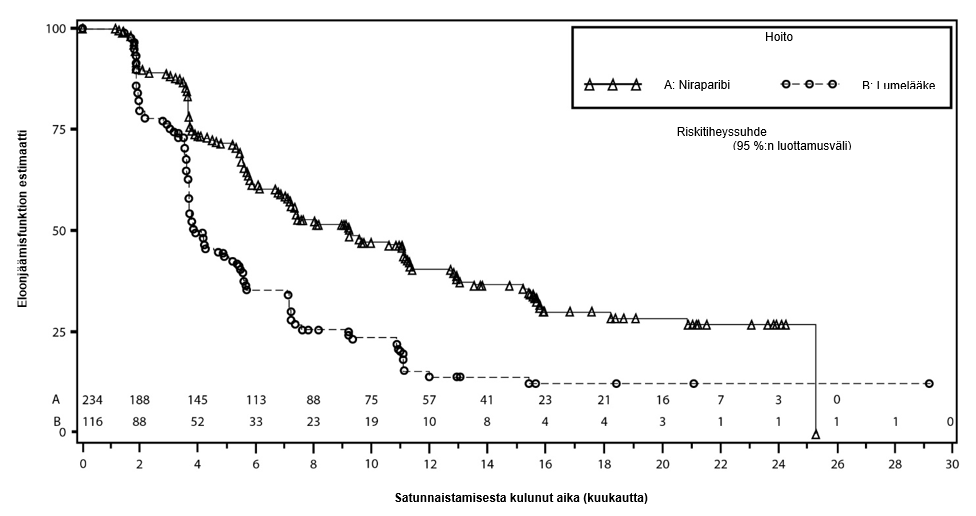
Tutkimuksen ensisijainen tavoite, tilastollisesti merkitsevästi parantunut etenemisvapaa elinaika pelkällä niraparibilla annettavassa ylläpitohoidossa suhteessa lumelääkkeeseen, saavutettiin gBRCAmut-kohortissa sekä koko non‑gBRCAmut-kohortissa. Taulukossa 6 ja kuvissa 3 ja 4 esitetään ensisijaista päätetapahtumaa eli etenemisvapaata elinaikaa koskevat tulokset tehon ensisijaisissa populaatioissa (gBRCAmut-kohortti ja koko non‑gBRCAmut-kohortti).
Taulukko 6: Yhteenveto NOVA-tutkimuksen ensisijaista tavoitetta koskevista tuloksista
| gBRCAmut-kohortti | Non‑gBRCAmut-kohortti | |||
niraparibi (N = 138) | lumelääke (N = 65) | niraparibi (N = 234) | lumelääke (N = 116) | |
| Etenemisvapaan elinajan mediaani (95 %:n luottamusväli) | 21,0 (12,9, ei arvioitavissa) | 5,5 (3,8, 7,2) | 9,3 (7,2, 11,2) | 3,9 (3,7, 5,5) |
| p-arvo | < 0,0001 | < 0,0001 | ||
Riskisuhde (niraparibi:lumel.) (95 %:n luottamusväli) | 0,27 (0,173, 0,410) | 0,45 (0,338, 0,607) | ||
Kuva 3: NOVA: Kaplan‑Meierin käyrä gBRCAmut-kohortin etenemisvapaasta elinajasta riippumattoman arviointikomitean arvion perusteella (hoitoaikeen mukainen populaatio (ITT), N = 203)
Kuva 4: NOVA: Kaplan‑Meierin käyrä koko non‑gBRCAmut-kohortin etenemisvapaasta elinajasta riippumattoman arviointikomitean arvion perusteella (hoitoaikeen mukainen populaatio, N = 350)
Toissijaiset tehon päätetapahtumat NOVA‑tutkimuksessa
Loppuanalyysissä PFS2‑mediaani gBRCAmut‑kohortissa oli 29,9 kuukautta niraparibia saaneilla potilailla ja 22,7 kuukautta lumelääkettä saaneilla potilailla (HR = 0,70; 95 %:n luottamusväli: 0,50–0,97). Non‑gBRCAmut‑kohortissa PFS2‑mediaani oli niraparibia saaneilla potilailla 19,5 kuukautta ja lumelääkettä saaneilla potilailla 16,1 kuukautta (HR = 0,80; 95 %:n luottamusväli 0,63–1,02).
Kokonaiselinajan loppuanalyysissä OS‑mediaani gBRCAmut‑kohortissa (n = 203) oli niraparibia saaneilla potilailla 40,9 kuukautta ja lumelääkettä saaneilla potilailla 38,1 kuukautta (HR = 0,85; 95 %:n luottamusväli: 0,61–1,20). gBRCAmut‑kohortin OS-tuosten kypsyys oli 76 %. Non‑gBRCAmut‑kohortissa (n = 350) OS‑mediaani oli 31,0 kuukautta niraparibia saaneilla potilailla ja 34,8 kuukautta lumelääkettä saaneilla potilailla (HR = 1,06; 95 %:n luottamusväli: 0,81–1,37). Non‑gBRCAmut‑kohortin OS-tulosten kypsyys oli 79 %.
Validoiduilla kyselytyökaluilla (FOSI ja EQ-5D) saatujen, potilaiden ilmoittamia tuloksia koskevien tietojen mukaan niraparibilla hoidetut potilaat eivät raportoineet eroja elämänlaatuun liittyvissä mittareissa lumelääkkeeseen nähden.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Zejulan käytöstä kaikkien pediatristen potilasryhmien munasarjasyövän hoidossa (pois luettuina rabdomyosarkooma ja itusolukasvaimet).
Farmakokinetiikka
Imeytyminen
Kun potilaille annettiin 300 mg:n kerta-annos niraparibia paasto-olosuhteissa, niraparibi oli mitattavissa plasmasta 30 minuutin kuluessa. Niraparibin keskimääräinen huippupitoisuus plasmassa (Cmax) saavutettiin noin kolmessa tunnissa (804 ng/ml [% variaatiokerroin: 50,2 %]). Kun niraparibia annettiin useampi 30–400 mg:n annos suun kautta kerran päivässä, niraparibin kertyminen oli noin kaksin- tai kolminkertaista.
Systeeminen altistus (Cmax ja AUC) niraparibille lisääntyi annoksesta riippuvalla tavalla, kun niraparibiannos kasvoi 30 mg:sta 400 mg:aan. Niraparibin absoluuttinen biologinen hyötyosuus on noin 73 prosenttia, mikä tarkoittaa minimaalista ensikierron vaikutusta. Niraparibin populaatiofarmakokineettisessä analyysissa yksilöiden välisten erojen biologisessa hyväksikäytettävyydessä variaatiokertoimen (CV) arvioitiin olevan 31 %.
Samaan aikaan nautittu runsasrasvainen ateria ei vaikuttanut merkittävästi niraparibin farmakokinetiikkaan 300 mg:n niraparibikapselin antamisen jälkeen.
Tabletti- ja kapselimuotojen on osoitettu olevan keskenään bioekvivalentteja. Kun 108 potilaalle, joilla oli kiinteitä kasvaimia, annettiin paasto-olosuhteissa joko yksi 300 mg:n tabletti tai kolme 100 mg:n kapselia niraparibia, geometristen keskiarvojen suhteiden (tabletti vs. kapseli) 90 %:n luottamusvälit asettuivat Cmax-, AUClast- ja AUC∞-arvojen osalta bioekvivalenssin raja-arvojen (0,80 ja 1,25) välille.
Jakautuminen
Niraparibi sitoutui kohtalaisesti proteiineihin ihmisen plasmassa (83 %), pääasiassa seerumin albumiiniin. Niraparibin populaatiofarmakokineettisessä analyysissa näennäinen jakautumistilavuus (Vd/F) oli syöpäpotilailla 1 311 l (kun potilaan paino on 70 kg) (CV 116 %), mikä viittaa siihen, että niraparibin kudosjakautuminen on laajaa.
Biotransformaatio
Niraparibi metaboloituu pääasiassa karboksyyliesteraasien välityksellä, jolloin se muuntuu tärkeäksi inaktiiviseksi metaboliitiksi, M1:ksi. Massatasetutkimuksessa M1 ja M10 (myöhemmin muodostuvat M1-glukuronidit) olivat tärkeimpiä kiertäviä metaboliitteja.
Eliminaatio
Suun kautta otetun 300 mg:n niraparibikerta-annoksen jälkeen niraparibin keskimääräinen puoliintumisaika (t½) oli 48–51 tuntia (noin 2 vuorokautta). Populaatiofarmakokineettisessä analyysissa niraparibin ilmeinen kokonaispuhdistuma (CL/F) oli syöpäpotilailla 16,5 l/h (CV 23,4 %).
Niraparibi eliminoituu enimmäkseen hepatobiliaarisen reitin ja munuaisten kautta. Suun kautta otetusta 300 mg:n [14C]‑niraparibikerta-annoksesta keskimäärin 86,2 prosenttia (vaihteluväli 71–91 %) erittyi virtsaan ja ulosteeseen 21 päivän kuluessa. Annoksen radioaktiivisuudesta 47,5 prosenttia (vaihteluväli 33,4–60,2 %) poistui virtsan mukana ja 38,8 prosenttia (vaihteluväli 28,3–47 %) ulosteen mukana. Yhdistetyissä näytteissä, jotka kerättiin kuuden päivän aikana, 40 prosenttia annoksesta oli erittynyt virtsaan pääasiassa metaboliitteina ja 31,6 prosenttia ulosteeseen pääasiassa muuttumattomana niraparibina.
Erityispotilasryhmät
Munuaisten vajaatoiminta
Potilastietojen populaatiofarmakokineettisessa analyysissa lievää (kreatiniinipuhdistuma 60-90 ml/min) ja keskivaikeaa (kreatiniinipuhdistuma 30-60 ml/min) munuaisten vajaatoimintaa sairastavilla potilailla niraparibin puhdistuma oli lievästi heikentynyt verrattuna henkilöihin, joiden munuaiset toimivat normaalisti (7-17 % suurempi altistuminen lievässä munuaisten vajaatoiminnassa ja 17-38% suurempi altistuminen keskivaikeassa munuaisten vajaatoiminnassa). Erot altistumisessa eivät edellytä annoksen muuttamista. Kliinisissä tutkimuksissa ei ollut potilaita, joilla olisi ollut vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaissairaus ja jotka olisivat käyneet hemodialyysissä (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Kliinisten tutkimusten potilastietojen populaatiofarmakokineettisessä analyysissa lievä maksan vajaatoiminta (n = 155) ei vaikuttanut niraparibin puhdistumaan. Syöpäpotilailla tehdyssä kliinisessä tutkimuksessa, jossa NCI-ODWG -kriteerejä käytettiin maksan vajaatoiminta-asteen luokitteluun, niraparibin AUCinf keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla (n = 8) oli 1,56 (90 % CI: 1,06-2,30) kertaa niraparibin AUCinf -arvo potilailla, joilla oli normaali maksan toiminta (n = 9) 300 mg kerta-annoksen antamisen jälkeen. Niraparibiannoksen muuttamista suositellaan potilaille, joilla on keskivaikea maksan vajaatoiminta (ks. kohta Annostus ja antotapa). Keskivaikea maksan vajaatoiminta ei vaikuttanut niraparibin Cmax -arvoon eikä niraparibin sitoutumiseen proteiineihin. Niraparibin farmakokinetiikkaa ei ole arvioitu potilailla, joilla on vaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Paino, ikä ja rotu
Populaatiofarmakokineettisessä analyysissa Niraparibin jakaantumistilavuus kasvoi painon lisääntyessä. Painon ei havaittu vaikuttavan puhdistumaan tai kokonaisaltistukseen. Farmakokinetiikan perusteella paino ei edellytä annosmuutoksia.
Populaatiofarmakokineettisessä analyysissä Niraparibin puhdistuma pienentyi iän myötä. Niraparibialtistuksen arvioitiin olevan 23 % suurempi 91-vuotiailla potilailla kuin 30-vuotiailla potilailla. Iän ei kuitenkaan katsota edellyttävän annosmuutoksia.
Eri etnisistä taustoista saatuja tietoja ei ole riittävästi, jotta niiden perusteella voitaisiin tehdä johtopäätöksiä etnisen taustan vaikutuksesta niraparibin farmakokinetiikkaan.
Pediatriset potilaat
Niraparibin farmakokinetiikkaa ei ole tutkittu pediatrisilla potilailla.
Prekliiniset tiedot turvallisuudesta
Turvallisuusfarmakologia
In vitro -tutkimuksissa niraparibi esti dopamiinin kuljettajaproteiinia pitoisuuksilla, jotka ovat ihmisten altistumistasoja pienempiä. Hiirillä kerta-annokset niraparibia lisäsivät dopamiinin ja sen metaboliittien solunsisäisiä pitoisuuksia korteksissa. Toisessa hiirillä tehdyistä kahdesta kerta-annostutkimuksesta havaittiin lokomotorisen aktiivisuuden vähenemistä. Näiden löydösten kliinistä merkitystä ei tiedetä. Rotilla ja koirilla tehdyissä toistuvan annoksen myrkyllisyyttä käsittelevissä tutkimuksissa ei havaittu vaikutusta käyttäytymiseen liittyviin ja/tai neurologisiin parametreihin arvioiduilla keskushermoston altistustasoilla, jotka olivat samoja tai pienempiä kuin oletetut terapeuttiset altistumistasot.
Toistuvan annoksen myrkyllisyys
Spermatogeneesin heikentymistä havaittiin rotilla ja koirilla kliinisesti käytettyjä pienemmillä altistumistasoilla, ja ne korjaantuivat yleensä neljän viikon kuluessa siitä, kun lääkkeen anto oli lopetettu.
Genotoksisuus
Bakteerimutageenisuustestissä (Amesin testi) niraparibi ei ollut mutageeninen, mutta nisäkkäiden kromosomipoikkeavuustestissä in vitro ja rottien luuytimen mikrotumatestissä in vivo se oli klastogeeninen. Tämä klastogeenisuus on johdonmukaista niraparibin primaarisesta farmakologiasta johtuvan perimän epävakauden vuoksi, ja se viittaa siihen, että niraparibi voi olla genotoksista ihmisille.
Lisääntymistoksikologia
Niraparibilla ei ole tehty lisääntymis- ja kehitystoksisuustutkimuksia.
Karsinogeenisuus
Niraparibilla ei ole tehty karsinogeenisuustutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Magnesiumstearaatti
Laktoosimonohydraatti
Kapselin kuori
Titaanidioksidi (E 171)
Liivate
Briljanttisininen FCF (E 133)
Erytrosiini (E 127)
Tartratsiini (E 102)
Painomuste
Sellakka (E 904)
Propyleeniglykoli (E 1520)
Kaliumhydroksidi (E 525)
Musta rautaoksidi (E 172)
Natriumhydroksidi (E 524)
Povidoni (E 1201)
Titaanidioksidi (E171)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Säilytä alle 30 °C.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ZEJULA kapseli, kova
100 mg 56 x 1 fol (4610,01 €), 84 x 1 fol (6813,86 €)
PF-selosteen tieto
Aclar-/PVC-/alumiinikalvosta valmistetut perforoidut yksittäispakatut läpipainopakkaukset. Pakkauskoko on 84 × 1, 56 × 1 ja 28 × 1 kova kapseli.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kova kapseli, jonka koko on noin 22 mm × 8 mm; valkoinen runko, johon on painettu mustalla musteella ”100 mg”, ja violetti kansi, johon on painettu ”Niraparib” valkoisella musteella.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ZEJULA kapseli, kova
100 mg 56 x 1 fol, 84 x 1 fol
- Ylempi erityiskorvaus (100 %). Niraparibi: Munasarjasyövän, munanjohtimen syövän tai primaarin vatsakalvon syövän hoito erityisin edellytyksin (1510).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Niraparibi: Monoterapiana uusiutunutta platinaherkkää korkean pahanlaatuisuusasteen seroosia epiteelistä munasarja-, munanjohdin- tai primaaria vatsakalvon syöpää sairastavien aikuispotilaiden hoito erityisin edellytyksin (3009).
ATC-koodi
L01XK02
Valmisteyhteenvedon muuttamispäivämäärä
03.01.2024
Yhteystiedot
 GLAXOSMITHKLINE OY
GLAXOSMITHKLINE OY Porkkalankatu 20 A
00180 Helsinki
010 303 030
www.glaxosmithkline.fi