
HEMLIBRA injektioneste, liuos 30 mg/ml, 150 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Lääkäri
Tämä opas on tarkoitettu terveydenhuollon ammattilaisille A-hemofiliapotilaan Hemlibra-hoidon turvallisuuden varmistamiseksi ja tärkeiden tunnistettujen riskien minimoimiseksi.
Potilas
Tämä potilaskortti sisältää tärkeitä turvallisuutta koskevia tietoja, joista potilaan on oltava tietoinen ennen Hemlibra-hoidon aloittamista ja hoidon aikana.
Tämä potilasopas on tarkoitettu potilaalle ja/tai potilasta hoitavalle henkilölle A-hemofiliapotilaan Hemlibra-hoidon turvallisuuden varmistamiseksi ja riskien ehkäisemiseksi tai vähentämiseksi.
Terveydenhuollon ammattilainen
Tämä opas on tarkoitettu laboratoriohenkilökunnalle A-hemofiliapotilaan Hemlibra-hoidon turvallisuuden varmistamiseksi ja tärkeiden tunnistettujen riskien minimoimiseksi.
Vaikuttavat aineet ja niiden määrät
Hemlibra 30 mg/ml injektioneste, liuos
Yksi ml injektionestettä sisältää 30 mg emisitsumabia*
Yksi 0,4 ml:n injektiopullo sisältää 12 mg emisitsumabia pitoisuutena 30 mg/ml.
Yksi 1 ml:n injektiopullo sisältää 30 mg emisitsumabia pitoisuutena 30 mg/ml.
Hemlibra 150 mg/ml injektioneste, liuos
Yksi ml injektionestettä sisältää 150 mg emisitsumabia*
Yksi 0,4 ml:n injektiopullo sisältää 60 mg emisitsumabia pitoisuutena 150 mg/ml.
Yksi 0,7 ml:n injektiopullo sisältää 105 mg emisitsumabia pitoisuutena 150 mg/ml.
Yksi 1 ml:n injektiopullo sisältää 150 mg emisitsumabia pitoisuutena 150 mg/ml.
Yksi 2 ml:n injektiopullo sisältää 300 mg emisitsumabia pitoisuutena 150 mg/ml.
* Emisitsumabi on humanisoitu, muokattu monoklonaalinen immunoglobuliini G4 (IgG4) ‑vasta-aine, joka on tuotettu yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa (nisäkässolut).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
Hemlibra on tarkoitettu verenvuotojen tavanomaiseen ennaltaehkäisyyn potilaille, jotka sairastavat A‑hemofiliaa (synnynnäistä hyytymistekijä VIII:n [FVIII:n] puutosta):
- kun potilaalla on vasta-aineita hyytymistekijä VIII:lle
-
kun potilaalla ei ole vasta-aineita hyytymistekijä VIII:lle, ja hänellä on
- vaikea-asteinen sairaus (FVIII < 1 %)
- keskivaikea sairaus (FVIII ≥ 1 % ja ≤ 5 %), johon liittyy voimakas verenvuototaipumus.
Hemlibraa voidaan käyttää kaikenikäisille potilaille.
Ehto
Hoito tulee aloittaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito pitää aloittaa hemofilian ja/tai verenvuotosairauksien hoitoon perehtyneen lääkärin valvonnassa.
Annostus
Hoito (myös tavanomainen ennaltaehkäisy) vasta-aineen ohittavilla aineilla (esim. aktivoidulla protrombiinikompleksikonsentraatilla [aPCC] ja aktivoidulla rekombinantilla ihmisen hyytymistekijä VIIa:lla [rFVIIa]) pitää lopettaa päivää ennen Hemlibra-hoidon aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Estohoitoa hyytymistekijä VIII:lla (FVIII) voidaan jatkaa Hemlibra-hoidon ensimmäisten 7 päivän ajan.
Suositeltu annos on 3 mg/kg kerran viikossa ensimmäisen 4 viikon ajan (aloitusannos), minkä jälkeen annetaan viikosta 5 alkaen ylläpitoannoksena 1,5 mg/kg kerran viikossa, 3 mg/kg kerran kahdessa viikossa tai 6 mg/kg kerran neljässä viikossa. Kaikki annokset annetaan injektiona ihon alle.
Aloitusannos on sama ylläpitohoidon annostuksesta riippumatta.
Ylläpitohoidon annostus pitää valita lääkärin ja potilaan tai potilasta hoitavan henkilön parhaaksi katsoman annostuksen perusteella, jotta tuetaan hoitoon sitoutumista.
Potilaalle annettava annos (mg) ja tilavuus (ml) lasketaan seuraavasti:
-
Aloitusannos (3 mg/kg) kerran viikossa ensimmäisen 4 viikon ajan:
Potilaan paino (kg) x annos (3 mg/kg) = annettava emisitsumabin kokonaismäärä (mg) -
Tämän jälkeen viikosta 5 alkaen ylläpitoannos joko 1,5 mg/kg kerran viikossa, 3 mg/kg kerran kahdessa viikossa tai 6 mg/kg kerran neljässä viikossa:
Potilaan paino (kg) x annos (1,5 mg/kg, 3 mg/kg tai 6 mg/kg) = annettava emisitsumabin kokonaismäärä (mg)
Ihon alle annettavan Hemlibra-injektion kokonaistilavuus lasketaan seuraavasti:
annettava emisitsumabin kokonaismäärä (mg) ÷ injektiopullon pitoisuus (mg/ml) = annettavan Hemlibra-injektion kokonaistilavuus (ml).
Eri Hemlibra-pitoisuuksia (30 mg/ml ja 150 mg/ml) ei saa yhdistää samaan ruiskuun koostettaessa annettavaa kokonaistilavuutta.
Annettava tilavuus ei saa olla suurempi kuin 2 ml/injektio.
Esimerkkejä:
Potilaan paino 16 kg, ylläpitoannos 1,5 mg/kg kerran viikossa:
- Esimerkki aloitusannoksesta (ensimmäiset 4 viikkoa): 16 kg x 3 mg/kg = aloitusannokseen tarvitaan 48 mg emisitsumabia.
- Laske annettava tilavuus jakamalla laskettu annos 48 mg pitoisuudella 150 mg/ml: 48 mg emisitsumabia ÷ 150 mg/ml = injisoitava määrä Hemlibra 150 mg/ml ‑valmistetta on 0,32 ml.
- Valitse asianmukainen annos ja tilavuus saatavana olevista vahvuuksista.
- Esimerkki ylläpitoannoksesta (viikosta 5 alkaen): 16 kg x 1,5 mg/kg = ylläpitoannokseen tarvitaan 24 mg emisitsumabia.
- Laske annettava tilavuus jakamalla laskettu annos 24 mg pitoisuudella 30 mg/ml: 24 mg emisitsumabia ÷ 30 mg/ml = kerran viikossa injisoitava määrä Hemlibra 30 mg/ml ‑valmistetta on 0,8 ml.
- Valitse asianmukainen annos ja tilavuus saatavana olevista vahvuuksista.
Potilaan paino 40 kg, ylläpitoannos 3 mg/kg kerran kahdessa viikossa:
- Esimerkki aloitusannoksesta (ensimmäiset 4 viikkoa): 40 kg x 3 mg/kg = aloitusannokseen tarvitaan 120 mg emisitsumabia.
- Laske annettava tilavuus jakamalla laskettu annos 120 mg pitoisuudella 150 mg/ml: 120 mg emisitsumabia ÷ 150 mg/ml = injisoitava määrä Hemlibra 150 mg/ml ‑valmistetta on 0,8 ml.
- Valitse asianmukainen annos ja tilavuus saatavana olevista vahvuuksista.
- Esimerkki ylläpitoannoksesta (viikosta 5 alkaen): 40 kg x 3 mg/kg = ylläpitoannokseen tarvitaan 120 mg emisitsumabia.
- Laske annettava tilavuus jakamalla annos 120 mg pitoisuudella 150 mg/ml: 120 mg emisitsumabia ÷ 150 mg/ml = kerran kahdessa viikossa injisoitava määrä Hemlibra 150 mg/ml ‑valmistetta on 0,8 ml.
- Valitse asianmukainen annos ja tilavuus saatavana olevista vahvuuksista.
Potilaan paino 60 kg, ylläpitoannos 6 mg/kg kerran neljässä viikossa:
- Esimerkki aloitusannoksesta (ensimmäiset 4 viikkoa): 60 kg x 3 mg/kg = aloitusannokseen tarvitaan 180 mg emisitsumabia.
- Laske annettava tilavuus jakamalla laskettu annos 180 mg pitoisuudella 150 mg/ml: 180 mg emisitsumabia ÷ 150 mg/ml = injisoitava määrä Hemlibra 150 mg/ml ‑valmistetta on 1,20 ml.
- Valitse asianmukainen annos ja tilavuus saatavana olevista vahvuuksista.
- Esimerkki ylläpitoannoksesta (viikosta 5 alkaen): 60 kg x 6 mg/kg = ylläpitoannokseen tarvitaan 360 mg emisitsumabia.
- Laske annettava tilavuus jakamalla laskettu annos 360 mg pitoisuudella 150 mg/ml: 360 mg emisitsumabia ÷ 150 mg/ml = kerran neljässä viikossa injisoitava määrä Hemlibra 150 mg/ml ‑valmistetta on 2,4 ml.
- Valitse asianmukainen annos ja tilavuus saatavana olevista vahvuuksista.
Hoidon kesto
Hemlibra on tarkoitettu pitkäaikaiseen verenvuotoja ennaltaehkäisevään hoitoon.
Annosmuutokset hoidon aikana
Hemlibran annosmuutoksia ei suositella.
Annosten viivästyminen tai ottamatta jääminen
Jos potilaan hoito-ohjelman mukainen Hemlibra-injektio ihon alle jää ottamatta, potilasta pitää neuvoa injisoimaan saamatta jäänyt annos mahdollisimman pian, viimeistään vuorokautta ennen seuraavaa hoito-ohjelman mukaista injektion antopäivää. Sen jälkeen potilaan pitää injisoida seuraava annos tavanomaisena injektion antopäivänä. Potilas ei saa injisoida kahta annosta samana päivänä korvatakseen unohtamansa annoksen.
Erityiset potilasryhmät
Pediatriset potilaat
Pediatrisille potilaille ei suositella annosmuutoksia (ks. kohta Farmakokinetiikka). Tietoja alle 1 vuoden ikäisistä potilaista ei ole saatavilla.
Iäkkäät potilaat
Annosmuutoksia ei suositella ≥ 65-vuotiaille potilaille (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka). Tietoja yli 77‑vuotiaista potilaista ei ole saatavilla.
Munuaisten ja maksan vajaatoiminta
Lievää munuaisten tai maksan vajaatoimintaa sairastaville potilaille ei suositella annosmuutoksia (ks. kohta Farmakokinetiikka). Hemlibra-valmisteen käytöstä keskivaikeaa munuaisten tai maksan vajaatoimintaa sairastaville potilaille on vähän tietoja. Emisitsumabia ei ole tutkittu vaikeaa munuaisten tai maksan vajaatoimintaa sairastavilla potilailla.
Hoito perioperatiivisessa tilanteessa
Emisitsumabin turvallisuutta ja tehoa leikkauksen yhteydessä ei ole tutkittu. Potilaille on kliinisissä tutkimuksissa tehty kirurgisia toimenpiteitä keskeyttämättä estohoitoa emisitsumabilla.
Jos perioperatiivisena aikana tarvitaan vasta-aineen ohittavia aineita (kuten aPCC ja rFVIIa), katso vasta-aineen ohittavien aineiden annostusohjeet kohdasta Varoitukset ja käyttöön liittyvät varotoimet. Jos perioperatiivisena aikana tarvitaan hyytymistekijää VIII (FVIII), ks. kohta Yhteisvaikutukset.
Potilaan hemostaattisen tilan seuranta, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet hyytymiskokeiden tulokset, joihin emisitsumabi ei vaikuta.
Immuunivasteen siedätys (ITI, immune tolerance induction)
Emisitsumabin turvallisuutta ja tehoa ITI-hoitoa saavilla potilailla ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Hemlibra on tarkoitettu ainoastaan ihon alle annettavaksi, ja sen annossa pitää noudattaa asianmukaista aseptista tekniikkaa (ks. kohta Käyttö- ja käsittelyohjeet).
Injektion anto pitää rajoittaa suositeltuihin injektiokohtiin: vatsaan, olkavarren ulkosivuun ja reisiin (ks. kohta Farmakokinetiikka).
Ihonalainen Hemlibra-injektio olkavarren ulkosivuun pitää antaa potilasta hoitavan henkilön tai terveydenhuollon ammattilaisen toimesta.
Injektiokohdan vaihteleminen saattaa ehkäistä injektiokohdan reaktioita tai vähentää niitä (ks. kohta Haittavaikutukset). Hemlibraa ei saa antaa ihon alle alueelle, jonka iho punoittaa tai aristaa tai jolla on mustelmia, kovettumia, luomia tai arpia.
Muut ihon alle annettavat lääkevalmisteet injisoidaan Hemlibra-hoidon aikana mieluiten eri anatomiselle alueelle.
Injektion antaminen, kun sen antaja on potilas itse ja/tai potilasta hoitava henkilö
Hemlibra on tarkoitettu käytettäväksi terveydenhuollon ammattilaisen opastuksessa. Kun potilas ja/tai häntä hoitava henkilö on saanut asianmukaisen opastuksen ihon alle annettavien injektioiden injektiotekniikkaan, hän voi injisoida Hemlibran itse tai potilasta hoitava henkilö voi injisoida sen, jos lääkäri katsoo sen asianmukaiseksi.
Lääkärin ja potilasta hoitavan henkilön pitää arvioida, voiko lapsi injisoida Hemlibra-hoidon itse. Hoidon injisoimista itse ei kuitenkaan suositella, jos lapsi on alle 7-vuotias.
Kattavat ohjeet Hemlibran antoon, ks. kohta Käyttö- ja käsittelyohjeet ja pakkausseloste.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Hemlibraan ja aktivoituun protrombiinikompleksikonsentraattiin (aPCC) liittyvä tromboottinen mikroangiopatia
Kliinisessä tutkimuksessa Hemlibraa estohoitona sekä aktivoitua protrombiinikompleksikonsentraattia (aPCC) keskimäärin > 100 U/kg/vrk suuruisina kumulatiivisina annoksina vähintään 24 tunnin ajan saaneilla potilailla on raportoitu tromboottista mikroangiopatiaa (ks. kohta Haittavaikutukset). Tromboottisen mikroangiopatian hoitoon kuului tukihoito ja mahdollisesti plasmafereesi ja hemodialyysi. Tromboottisen mikroangiopatian paranemista todettiin viikon kuluessa aPCC:n käytön lopettamisen ja Hemlibra-hoidon keskeyttämisen jälkeen. Tällainen nopea paraneminen eroaa epätyypillisen hemolyyttis-ureemisen oireyhtymän ja tromboottisen mikroangiopatian klassisten muotojen, kuten tromboottisen trombosytopeenisen purppuran, tavanomaisesta kliinisestä kulusta (ks. kohta Haittavaikutukset). Yksi potilas aloitti Hemlibra-hoidon uudelleen tromboottisen mikroangiopatian parannuttua, ja hoito jatkui turvallisesti.
Hemlibraa estohoitona saavia potilaita pitää seurata aPCC:n käytön aikana tromboottisen mikroangiopatian kehittymisen havaitsemiseksi. Jos potilaalle kehittyy tromboottiseen mikroangiopatiaan sopivia kliinisiä oireita ja/tai laboratoriolöydöksiä, lääkärin pitää välittömästi lopettaa aPCC-hoito ja keskeyttää Hemlibra-hoito sekä hoitaa oireet ja löydökset siten kuin kliinisesti on aiheellista. Kun tromboottinen mikroangiopatia on täysin parantunut, lääkärin ja potilaan / potilasta hoitavan henkilön pitää arvioida Hemlibra-estohoidon jatkamisen hyödyt ja riskit tapauskohtaisesti. Jos vasta-aineen ohittavan aineen käyttö Hemlibra-estohoitoa saavalle potilaalle on aiheellista, ks. jäljempänä vasta-aineen ohittavien aineiden annostusohjeet.
Potilaan hoidossa on oltava varovainen, jos tromboottisen mikroangiopatian riski on suuri (esim. potilaan sairaushistorian tai suvussa esiintyneen tromboottisen mikroangiopatian vuoksi) tai jos potilas käyttää samaan aikaan lääkevalmisteita, joiden tiedetään olevan tromboottisen mikroangiopatian kehittymisen riskitekijöitä (esim. siklosporiini, kiniini, takrolimuusi).
Hemlibraan ja aktivoituun protrombiinikompleksikonsentraattiin (aPCC) liittyvät tromboemboliat
Kliinisessä tutkimuksessa Hemlibraa estohoitona sekä aktivoitua protrombiinikompleksikonsentraattia keskimäärin > 100 U/kg/vrk suuruisina kumulatiivisina annoksina vähintään 24 tunnin ajan saaneilla potilailla on raportoitu vakavia tromboottisia tapahtumia (ks. kohta Haittavaikutukset). Yhdessäkään tapauksessa ei tarvittu hyytymistä estävää hoitoa. Tapahtumien todettiin lieventyneen tai parantuneen kuukauden kuluessa siitä, kun aPCC:n käyttö oli lopetettu ja Hemlibra-hoito oli keskeytetty (ks. kohta Haittavaikutukset). Yksi potilas aloitti Hemlibra-hoidon uudelleen tromboottisen tapahtuman parannuttua, ja hoito jatkui turvallisesti.
Hemlibraa estohoitona saavia potilaita pitää seurata aPCC:n käytön aikana tromboembolian kehittymisen havaitsemiseksi. Jos potilaalla on tromboottisiin tapahtumiin sopivia kliinisiä oireita, kuvantamislöydöksiä ja/tai laboratoriolöydöksiä, lääkärin pitää välittömästi lopettaa aPCC-hoito ja keskeyttää Hemlibra-hoito sekä hoitaa tällaiset oireet ja löydökset siten kuin kliinisesti on aiheellista. Kun tromboottinen tapahtuma on täysin parantunut, lääkärin ja potilaan / potilasta hoitavan henkilön pitää arvioida Hemlibra-estohoidon jatkamisen hyödyt ja riskit tapauskohtaisesti. Jos vasta-aineen ohittavan aineen käyttö Hemlibra-estohoitoa saavalle potilaalle on aiheellista, ks. jäljempänä vasta-aineen ohittavien aineiden annostusohjeet.
Ohjeet vasta-aineen ohittavien aineiden käyttöön Hemlibra-estohoitoa saaville potilaille
Hoito vasta-aineen ohittavilla aineilla pitää lopettaa päivää ennen Hemlibra-hoidon aloittamista.
Jos vasta-aineen ohittavien aineiden käyttö on tarpeen Hemlibra-estohoidon aikana, lääkärin on aina keskusteltava potilaan ja/tai potilasta hoitavan henkilön kanssa vasta-aineen ohittavan aineen tarkasta annoksesta ja annostusaikataulusta.
Hemlibra lisää potilaan veren hyytymistä. Vasta-aineen ohittavaa ainetta saatetaan siten tarvita pienempi annos kuin ilman Hemlibra-estohoitoa. Vasta-aineen ohittavien aineiden annos ja hoidon kesto riippuvat verenvuodon sijainnista ja laajuudesta sekä potilaan kliinisestä tilasta. aPCC:n käyttöä pitää välttää, ellei muita hoitovaihtoehtoja ole saatavissa. Jos aPCC:n käyttö Hemlibra-estohoitoa saavalle potilaalle on aiheellista, aloitusannos saa olla enintään 50 U/kg, ja laboratorioarvojen seuranta on suositeltavaa (mukaan lukien munuaisten toiminnan seuranta, trombosyyttien määritys ja tromboosien toteamiseksi tehtävät tutkimukset, näihin kuitenkaan rajoittumatta). Jos verenvuotoa ei saada hallintaan, kun aPCC:n aloitusannos on enintään 50 U/kg, aPCC-lisäannoksia voidaan antaa lääkärin ohjauksessa tai valvonnassa ottaen huomioon laboratorioseuranta tromboottisen mikroangiopatian tai tromboembolian toteamiseksi ja verenvuotojen toteaminen ennen toistuvaa antoa. aPCC:n kokonaisannos ei saa hoidon ensimmäisten 24 tunnin aikana ylittää annosta 100 U/kg. Hoitavan lääkärin on arvioitava tromboottisen mikroangiopatian ja tromboembolian riskiä tarkoin verenvuotojen riskiin nähden, jos ensimmäisten 24 tunnin aikana harkitaan aPCC-hoitoa enimmäisannosta 100 U/kg suuremmalla annostuksella.
Kliinisissä tutkimuksissa ei havaittu tromboottista mikroangiopatiaa eikä tromboottisia tapahtumia, kun Hemlibra-estohoitoa saavat potilaat käyttivät pelkästään aktivoitua rekombinanttia hyytymistekijä VIIa:ta (rFVIIa).
Vasta-aineen ohittavien aineiden annostusohjeita pitää noudattaa vähintään 6 kuukauden ajan Hemlibra-estohoidon lopettamisen jälkeen (ks. kohta Farmakokinetiikka).
Immunogeenisuus
Kliinisissä tutkimuksissa on melko harvoin havaittu tehon heikkenemiseen johtavaa neutraloivien emisitsumabivasta‑aineiden kehittymistä ja plasman emisitsumabipitoisuuden laskua (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). Potilaita, joilla on kliinisiä merkkejä tehon heikkenemisestä (esim. lisääntyneitä vuototapahtumia), on pikaisesti tutkittava etiologian arvioimiseksi ja muita terapeuttisia hoitovaihtoehtoja on harkittava, jos epäillään neutraloivia emisitsumabivasta-aineita.
Emisitsumabin vaikutus hyytymiskokeisiin
Emisitsumabi korvaa puuttuvan aktivoituneen hyytymistekijä VIII:n (FVIIIa) tenaasikofaktoriaktiivisuuden. Sisäiseen hyytymisreittiin perustuvat veren hyytymisen laboratoriokokeet, mukaan lukien aktivoitu hyytymisaika (ACT) ja aktivoitu partiaalinen tromboplastiiniaika (esim. aPTT), mittaavat kokonaishyytymisaikaa, mukaan lukien trombiinin aktivoiman hyytymistekijä VIII:n muuntumiseen hyytymistekijä VIIIa:ksi tarvittavan ajan. Tällaisissa sisäiseen reittiin perustuvissa kokeissa hyytymisajan tulos on emisitsumabia käytettäessä liian lyhyt, koska emisitsumabi ei vaadi trombiinin aiheuttamaa aktivaatiota. Liian lyhyt sisäisen reitin hyytymisaika puolestaan häiritsee kaikkia aktivoituun partiaaliseen tromboplastiiniaikaan perustuvia yhden hyytymistekijän määrityksiä, kuten yksivaiheista hyytymistekijä VIII:n aktiivisuuden määritystä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet, taulukko 1). Emisitsumabi ei kuitenkaan vaikuta kromogeenisilla tai immunomääritysmenetelmillä tehtyihin yhden hyytymistekijän määrityksiin, joten niitä voidaan käyttää hyytymisparametrien arvioimiseen hoidon aikana, jolloin on huomioitava erityisesti jäljempänä kuvatut kromogeeniset hyytymistekijä VIII:n aktiivisuuden määritykset.
Kromogeenisissa FVIII:n aktiivisuutta mittaavissa testeissä voidaan käyttää joko ihmisen tai naudan hyytymisproteiineja. Ihmisen hyytymistekijöitä sisältävät määritykset ovat emisitsumabille herkkiä, mutta saattavat yliarvioida emisitsumabin kliinisen hemostaattisen vaikutuksen. Naudan hyytymistekijöitä sisältävät määritykset eivät sitä vastoin ole emisitsumabille herkkiä (eivät mittaa aktiivisuutta), joten niitä voidaan käyttää endogeenisen tai infusoidun FVIII:n aktiivisuuden seuraamiseen tai hyytymistekijä VIII:n vasta-aineiden mittaamiseen.
Emisitsumabin aktiivisuus säilyy FVIII:n vasta-aineista huolimatta, minkä vuoksi hyytymiseen perustuvat, FVIII:n toiminnan estymistä mittaavat Bethesda-määritykset antavat väärän negatiivisen tuloksen. Näiden sijasta voidaan käyttää nautaperäiseen FVIII:n kromogeeniseen testiin perustuvaa kromogeenista Bethesda-määritystä, joka ei ole herkkä emisitsumabille.
Nämä kaksi farmakodynaamista markkeria eivät kuvasta emisitsumabin todellista hemostaattista vaikutusta in vivo (aktivoitu partiaalinen tromboplastiiniaika on liian lyhyt ja raportoitu FVIII:n aktiivisuus saattaa ylikorostua), mutta ne antavat suhteellisen osoituksen emisitsumabin veren hyytymistä edistävästä vaikutuksesta.
Yhteenvetona voidaan todeta, että sisäiseen hyytymisreittiin perustuvien veren hyytymistä mittaavien laboratoriokokeiden tuloksia ei pidä käyttää Hemlibra-hoitoa saaville potilaille lääkkeen aktiivisuuden seuraamiseen, hyytymistekijän korvaushoidossa tai hyytymisen estämiseen tarvittavan annoksen määrittämiseen eikä FVIII:n vasta-aineiden titterin mittaamiseen. Sisäiseen hyytymisreittiin perustuvien veren hyytymistä mittaavien laboratoriokokeiden käytössä pitää noudattaa varovaisuutta, koska jos potilaalla on verenvuotoepisodi, tulosten virhetulkinta voi johtaa potilaan alihoitamiseen ja siten vaikea-asteisiin tai hengenvaarallisiin verenvuotoihin.
Taulukossa 1 esitetään laboratoriokokeet, joihin emisitsumabi vaikuttaa ja ei vaikuta. Emisitsumabin puoliintumisaika on pitkä, joten vaikutukset hyytymismääritykseen voivat säilyä 6 kuukauteen saakka viimeisen annoksen jälkeen (ks. kohta Farmakokinetiikka).
Taulukko 1 Hyytymiskokeiden tulokset, joihin emisitsumabi vaikuttaa ja joihin se ei vaikuta
Tulokset, joihin emisitsumabi vaikuttaa | Tulokset, joihin emisitsumabi ei vaikuta |
- aktivoitu partiaalinen tromboplastiiniaika (aPTT) - FVIII:n vasta-ainetitterien Bethesda-määritykset (veren hyytymiseen perustuvat) - yksivaiheiset aktivoituun partiaaliseen tromboplastiiniaikaan perustuvat yhden hyytymistekijän määritykset - aktivoituun partiaaliseen tromboplastiiniaikaan perustuva aktivoidun proteiini C:n resistenssi (APC‑res) - aktivoitu hyytymisaika (ACT) | - FVIII:n vasta-aine-titterien Bethesda-määritykset (naudan kromogeeninen) - tromboplastiiniaika (TT) - yksivaiheiset protrombiiniaikaan perustuvat yhden hyytymistekijän määritykset - kromogeeniseen menetelmään perustuvat yhden hyytymistekijän, muun kuin FVIII:n1, määritykset - Immunoperusteiset määritykset (esim. ELISA, turbidimetriset menetelmät) - hyytymistekijöitä koskevat geenitestit (esim. hyytymistekijä V Leiden, protrombiini 20210) |
1Hyytymistekijä VIII:n aktiivisuuden kromogeenisten määritysten osalta huomioitavia tärkeitä seikkoja, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Pediatriset potilaat
Alle 1 vuoden ikäisistä lapsista ei ole tietoja. Vastasyntyneiden ja imeväisikäisten lasten hemostaattisen järjestelmän kehittyminen on dynaamista ja asteittaista, joten hyöty-riskiarviossa on huomioitava tämän potilasryhmän veren hyytymistä edistävien ja estävien proteiinien suhteelliset pitoisuudet, mukaan lukien mahdollinen tromboosiriski (esim. keskuslaskimokatetriin liittyvä tromboosi).
Koulutusmateriaali
Kaikkien lääkärien, jotka oletettavasti määräävät tai käyttävät Hemlibra-valmistetta tai valvovat Hemlibra-valmisteen käyttöä, on varmistettava, että he ovat saaneet lääkärin koulutusmateriaalin ja perehtyneet siihen. Terveydenhuollon ammattilaisten on selitettävä ja käytävä läpi Hemlibra-hoidon hyödyt ja riskit potilaan ja häntä hoitavan henkilön kanssa ja varmistettava, että heille on annettu potilaskortti ja potilaan / potilasta hoitavan henkilön opas. Potilasta / potilasta hoitavaa henkilöä on ohjeistettava pitämään potilaskortti aina mukanaan ja näyttämään se vastaanotolla kaikille muille hoitaville terveydenhuollon ammattilaisille.
Yhteisvaikutukset
Emisitsumabilla ei ole tehty riittäviä tai hyvin kontrolloituja yhteisvaikutustutkimuksia.
Kliininen kokemus viittaa emisitsumabin ja aPCC:n väliseen yhteisvaikutukseen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Prekliinisten kokeiden perusteella rekombinantin hyytymistekijä VIIa:n tai hyytymistekijä VIII:n käytössä emisitsumabin kanssa on tromboositaipumuksen mahdollisuus. Emisitsumabi lisää potilaan veren hyytymistä, joten hemostaasin aikaansaamiseen tarvittava rFVIIa- tai FVIII-annos voi olla pienempi kuin potilailla, jotka eivät ole saaneet Hemlibra-estohoitoa.
Tromboottisen komplikaation ilmetessä lääkärin pitää kliinisen tarpeen mukaan harkita rFVIIa- tai FVIII-hoidon lopettamista ja Hemlibra-estohoidon keskeyttämistä. Jatkohoito pitää räätälöidä yksilöllisen kliinisen tilanteen mukaan.
- Annosmuutoksista päätettäessä pitää ottaa huomioon lääkevalmisteiden puoliintumisajat ja etenkin se, ettei emisitsumabihoidon keskeyttämisellä välttämättä ole välitöntä vaikutusta.
- Seuranta kromogeenisella FVIII-määrityksellä voi ohjata hyytymistekijöiden antoa, ja tromboositaipumuksen kantajuuden testausta voidaan harkita.
Fibrinolyysiä estävien lääkeaineiden samanaikaisesta antamisesta aPCC:n tai rekombinantin hyytymistekijä VIIa:n kanssa potilaille, jotka käyttävät Hemlibraa estolääkityksenä, on vähän kokemusta. Kun emisitsumabihoitoa saavat potilaat käyttävät systeemisiä fibrinolyysiä estäviä lääkeaineita yhdistelmänä aPCC:n tai rekombinantin hyytymistekijä VIIa:n kanssa, tromboottisten tapahtumien mahdollisuus pitää kuitenkin ottaa huomioon.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy
Hemlibraa saavien naisten, jotka voivat tulla raskaaksi, pitää käyttää tehokasta ehkäisyä Hemlibra-hoidon aikana ja vähintään 6 kuukauden ajan hoidon päättymisen jälkeen (ks. kohta Farmakokinetiikka).
Raskaus
Kliinisiä emisitsumabitutkimuksia ei ole tehty raskaana olevilla naisilla. Hemlibralla ei ole tehty eläinten lisääntymistä koskevia tutkimuksia. Ei tiedetä, voiko raskaana olevalle naiselle annettu emisitsumabi vahingoittaa sikiötä tai voiko se vaikuttaa lisääntymiskykyyn. Hemlibraa voidaan käyttää raskauden aikana vain, jos hoidon mahdolliset hyödyt äidille ovat siitä sikiölle aiheutuvia mahdollisia riskejä suuremmat. Samalla on otettava huomioon, että tromboosin vaara on suurentunut raskauden aikana ja synnytyksen jälkeen ja että moniin raskausajan komplikaatioihin liittyy yleistyneen suonensisäisen hyytymisen (DIC-oireyhtymä) suurempi riski.
Imetys
Ei tiedetä, erittyykö emisitsumabi ihmisen rintamaitoon. Emisitsumabin vaikutusta maidon erittymiseen tai emisitsumabin erittymistä rintamaitoon ei ole tutkittu. Ihmisen IgG:n tiedetään erittyvän rintamaitoon. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Hemlibra-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Ihmisistä ei ole hedelmällisyyttä koskevia tietoja saatavissa. Emisitsumabin vaikutusta miehen ja naisen hedelmällisyyteen ei tunneta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Hemlibra-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Hemlibra-valmisteen kokonaisturvallisuusprofiili perustuu kliinisistä tutkimuksista ja valmisteen markkinoille tulon jälkeisestä seurannasta saatuihin tietoihin. Kliinisissä Hemlibra-tutkimuksissa raportoituja vakavimpia haittavaikutuksia olivat tromboottinen mikroangiopatia ja tromboottiset tapahtumat, mukaan lukien kavernoottinen sinustromboosi ja pinnallinen laskimotromboosi yhdessä samanaikaisen ihonekroosin kanssa (ks. jäljempänä ja kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vähintään yhden Hemlibra-annoksen saaneilla potilailla yleisimmin (≥ 10 %) raportoituja haittavaikutuksia olivat injektiokohdan reaktiot (19,4 %), nivelsärky (14,2 %) ja päänsärky (14,0 %).
Kliinisissä tutkimuksissa Hemlibra-estohoitoa saaneista potilaista yhteensä kolme (0,7 %) keskeytti hoidon haittavaikutusten vuoksi; näitä haittavaikutuksia olivat tromboottinen mikroangiopatia, ihonekroosi samanaikaisesti pinnallisen tromboflebiitin kanssa sekä päänsärky.
Haittavaikutustaulukko
Seuraavat haittavaikutukset perustuvat valmisteen markkinoille tulon jälkeisestä seurannasta saatuihin tietoihin sekä viiden vaiheen III kliinisen tutkimuksen yhdistettyihin tietoihin (aikuisia ja nuoria koskevat tutkimukset [BH29884 – HAVEN 1, BH30071 – HAVEN 3 ja BO39182 – HAVEN 4], kaikkia ikäryhmiä koskeva tutkimus [BO41423 – HAVEN 6] sekä pediatrisia potilaita koskeva tutkimus [BH29992 – HAVEN 2]). Tutkimuksissa oli mukana yhteensä 444 A-hemofiliaa sairastavaa potilasta, jotka saivat vähintään yhden Hemlibra-annoksen tavanomaisena estohoitona (ks. kohta Farmakodynamiikka). Kliiniseen tutkimukseen osallistuneista potilaista 307 (69,1 %) oli aikuisia (joista kaksi oli naisia), 61 (13,7 %) oli nuoria (≥ 12 – < 18-vuotiaita), 71 (16,0 %) oli lapsia (≥ 2 – < 12‑vuotiaita) ja viisi (1,1 %) oli imeväis- ja taaperoikäisiä (1 kuukaudesta < 2‑vuotiaisiin). Tutkimuksissa altistuksen kestoajan mediaani oli 32 viikkoa (vaihteluväli: 0,1–94,3 viikkoa).
Vaiheen III kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeisessä seurannassa ilmenneet haittavaikutukset on lueteltu MedDRA-elinjärjestelmäluokituksen mukaisesti (taulukko 2). Kunkin haittavaikutuksen esiintyvyysluokat perustuvat seuraavaan esitystapaan: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 2 Yhteenveto yhdistetyissä kliinisissä Hemlibra-tutkimuksissa (HAVEN) ja valmisteen markkinoille tulon jälkeisessä seurannassa havaituista haittavaikutuksista
| Elinjärjestelmäluokka | Haittavaikutus (suositeltu termi, MedDRA) | Esiintyvyys | |
| Veri ja imukudos | Tromboottinen mikroangiopatia | Melko harvinainen | |
| Hermosto | Päänsärky | Hyvin yleinen | |
| Verisuonisto | Pinnallinen tromboflebiitti | Melko harvinainen | |
| Kavernoottinen sinustromboosia | Melko harvinainen | ||
| Ruoansulatuselimistö | Ripuli | Yleinen | |
| Iho ja ihonalainen kudos | Ihonekroosi | Melko harvinainen | |
| Angioedeema | Melko harvinainen | ||
| Urtikaria | Yleinen | ||
| Ihottuma | Yleinen | ||
| Luusto, lihakset ja sidekudos | Nivelsärky | Hyvin yleinen | |
| Lihassärky | Yleinen | ||
| Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan reaktio | Hyvin yleinen | |
| Kuume | Yleinen | ||
| Heikentynyt terapeuttinen vasteb | Melko harvinainen | ||
| Immuunijärjestelmä | Yliherkkyys | Melko harvinainen | |
a Verisuonisto on kavernoottisen sinustromboosin toissijainen elinjärjestelmäluokka. b Tehon menetystä (heikentynyt terapeuttinen vaste), joka ilmenee lisääntyneinä vuototapahtumina, on raportoitu neutraloivien emisitsumabivasta‑aineiden ja plasman emisitsumabipitoisuuden laskun yhteydessä (ks. Valikoitujen haittavaikutusten kuvaus ja kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka). | |||
Valikoitujen haittavaikutusten kuvaus
Tromboottinen mikroangiopatia
Tromboottisia mikroangiopatiatapahtumia raportoitiin yhdistetyissä vaiheen III kliinisissä tutkimuksissa alle 1 %:lla (3/444) potilaista ja 9,7 %:lla (3/31) potilaista, jotka saivat emisitsumabihoidon aikana vähintään yhden aPCC-annoksen. Kaikki 3 tromboottista mikroangiopatiatapahtumaa ilmenivät annettaessa aktivoitua protrombiinikompleksikonsentraattia keskimäärin > 100 U/kg/vrk suuruisina kumulatiivisina annoksina vähintään 24 tunnin ajan hoitotapahtuman aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Potilailla oli trombosytopeniaa, mikroangiopatista hemolyyttistä anemiaa ja akuutteja munuaisvaurioita, mutta ei vaikea-asteista ADAMTS13:n aktiivisuuden puutosta. Yksi potilas aloitti Hemlibra-hoidon uudelleen tromboottisen mikroangiopatian häviämisen jälkeen eikä tromboottinen mikroangiopatia uusiutunut.
Tromboottiset tapahtumat
Vakavia tromboottisia tapahtumia raportoitiin yhdistetyissä vaiheen III kliinisissä tutkimuksissa alle 1 %:lla (2/444) potilaista ja 6,5 %:lla (2/31) potilaista, jotka saivat emisitsumabihoidon aikana vähintään yhden aPCC-annoksen. Molemmat vakavat tromboottiset tapahtumat ilmenivät annettaessa aktivoitua protrombiinikompleksikonsentraattia keskimäärin > 100 U/kg/vrk suuruisina kumulatiivisina annoksina vähintään 24 tunnin ajan hoitotapahtuman aikana. Yksi potilas aloitti Hemlibra-hoidon uudelleen tromboottisen tapahtuman häviämisen jälkeen eikä tromboottinen tapahtuma uusiutunut (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Emisitsumabin ja aPCC-hoidon väliset yhteisvaikutukset kliinisissä pivotaalitutkimuksissa
Hemlibraa estohoitona saavilla potilailla oli 82 aPCC-hoitokertaa*, joista kahdeksassa (10 %) aPCC:n keskimääräinen kumulatiivinen annos oli > 100 U/kg/vrk vähintään 24 tunnin ajan; kahteen näistä kahdeksasta hoitokerrasta liittyi tromboottisia tapahtumia ja kolmeen liittyi tromboottinen mikroangiopatia (taulukko 3). Muihin aPCC-hoitokertoihin ei liittynyt tromboottista mikroangiopatiaa eikä tromboottisia tapahtumia. 68 %:ssa kaikista aPCC-hoitokerroista potilas sai vain yhden infuusion < 100 U/kg.
Taulukko 3 aPCC-hoito* yhdistetyissä vaiheen III kliinisissä tutkimuksissa
| aPCC-hoidon kesto | aPCC:n keskimääräinen kumulatiivinen annos 24 tunnin aikana (U/kg/vrk) | ||
| < 50 | 50–100 | > 100 | |
| < 24 tuntia | 9 | 47 | 13 |
| 24–48 tuntia | 0 | 3 | 1b |
| > 48 tuntia | 1 | 1 | 7a,a,a,b |
* aPCC-hoitokerran määritelmänä on kaikki potilaan mistä tahansa syystä saamat aPCC-annokset, kunnes hoidossa oli 36 tunnin tauko. Se käsittää kaikki aPCC-hoitokerrat, lukuun ottamatta ensimmäisten 7 hoitopäivän hoitokertoja ja 30 päivää Hemlibra-hoidon lopettamisen jälkeen tapahtuneita hoitokertoja.
a Tromboottinen mikroangiopatia
b Tromboottinen tapahtuma
Injektiokohdan reaktiot
Injektiokohdan reaktioita raportoitiin yhdistetyissä vaiheen III kliinisissä tutkimuksissa hyvin yleisesti (19,4 %). Kaikki Hemlibran kliinisissä tutkimuksissa havaitut injektiokohdan reaktiot raportoitiin ei-vakavina, ne olivat vaikeusasteeltaan lieviä tai keskivaikeita, ja 94,9 % hävisi ilman hoitoa. Yleisimmin raportoituja injektiokohdan reaktioita olivat injektiokohdan punoitus (10,6 %), injektiokohdan kipu (4,1 %), injektiokohdan kutina (2,9 %) ja injektiokohdan turvotus (2,7 %).
Immunogeenisuus
Hemlibralla tehdyissä vaiheen III kliinisissä tutkimuksissa neutraloivien emisitsumabivasta-aineiden kehittyminen ja siihen liittyvä emisitsumabipitoisuuden pieneneminen oli melko harvinaista (ks. kohta Farmakodynamiikka). Yhdellä potilaalla, jolla kehittyi neutraloivia emisitsumabivasta-aineita ja emisitsumabipitoisuus pieneni, teho heikkeni (ilmeni verenvuotona) viiden viikon hoidon jälkeen, mikä johti Hemlibra-hoidon keskeyttämiseen myöhemmin (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Pediatriset potilaat
Tutkimuksiin osallistui yhteensä 137 pediatrista potilasta, joista 5 (3,6 %) oli imeväisiä ja taaperoita (1 kk – alle 2 vuoden ikäisiä), 71 (51,8 %) oli lapsia (2 – alle 12 vuoden ikäisiä) ja 61 (44,5 %) oli nuoria (12 – alle 18 vuoden ikäisiä). Yleisesti ottaen Hemlibran turvallisuusprofiili oli yhdenmukainen imeväisillä, lapsilla, nuorilla ja aikuisilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Hemlibran yliannostuksesta on vähän kokemusta.
Oireet
Tahaton yliannostus saattaa aiheuttaa veren lisääntynyttä hyytymistaipumusta.
Hoito
Yliannostuksen tahattomasti saaneen potilaan on otettava heti yhteyttä lääkäriin, ja potilaan tilaa on seurattava tarkoin.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: hemostaatit, muut systeemisesti käytettävät hemostaatit, ATC-koodi: B02BX06
Vaikutusmekanismi
Emisitsumabi on humanisoitu, muokattu monoklonaalinen immunoglobuliini G4 (IgG4) ‑vasta-aine, jossa on kaksoisspesifinen vasta-ainerakenne.
Emisitsumabi tuo aktivoituneen hyytymistekijä IX:n ja hyytymistekijä X:n yhteen ja korvaa näin puuttuvan aktivoituneen hyytymistekijä VIII:n (FVIIIa) toiminnan, jota tarvitaan tehokkaaseen hemostaasiin.
Emisitsumabilla ei ole rakenteellista yhteyttä eikä sekvenssin vastaavuutta FVIII:n kanssa, joten se ei sinänsä indusoi eikä lisää FVIII:n suorien vasta-aineiden kehittymistä.
Farmakodynaamiset vaikutukset
Hemlibra-estohoito lyhentää aktivoitua partiaalista tromboplastiiniaikaa (aPTT) ja lisää raportoitua FVIII:n aktiivisuutta (ihmisen hyytymistekijöihin perustuvassa kromogeenisessä määrityksessä). Nämä kaksi farmakodynaamista markkeria eivät kuvasta emisitsumabin todellista hemostaattista vaikutusta in vivo (aktivoitu partiaalinen tromboplastiiniaika on liian lyhyt ja raportoitu FVIII:n aktiivisuus saattaa ylikorostua), mutta ne antavat suhteellisen viitteen emisitsumabin veren hyytymistä edistävästä vaikutuksesta.
Kliininen teho ja turvallisuus
Hemlibran tehoa tavanomaisena estohoitona A-hemofiliapotilaille tutkittiin viidessä kliinisessä tutkimuksessa (kolmessa aikuisia ja nuoria A-hemofiliapotilaita, joilla oli tai ei ollut FVIII:n vasta-aineita, koskeneessa tutkimuksessa [HAVEN 1, HAVEN 3 ja HAVEN 4], yhdessä pediatrisia A‑hemofiliapotilaita, joilla oli FVIII:n vasta-aineita, koskeneessa tutkimuksessa [HAVEN 2] ja kaikenikäisistä potilaista koostuneessa tutkimusryhmässä, jossa potilailla oli lievä tai keskivaikea A‑hemofilia, mutta ei ollut FVIII:n vasta-aineita [HAVEN 6]).
Kliiniset tutkimukset aikuisilla ja nuorilla A-hemofiliapotilailla, jolla oli tai ei ollut FVIII:n vasta-aineita
A-hemofiliaa sairastavat potilaat (ikä ≥ 12 vuotta ja paino > 40 kg), joilla ei ollut vasta-aineita hyytymistekijä VIII:lle (tutkimus BH30071 – HAVEN 3)
HAVEN 3 ‑tutkimus oli satunnaistettu, avoin, vaiheen III kliininen monikeskustutkimus, jossa oli mukana 152 aikuista ja nuorta miestä (ikä ≥ 12 vuotta ja paino > 40 kg). He sairastivat vaikea-asteista A-hemofiliaa, heillä ei ollut vasta-aineita hyytymistekijä VIII:lle, ja he olivat saaneet aiemmin hyytymistekijä VIII:aa joko jaksoittaisena hoitona (tarpeen mukaan) tai estohoitona. Potilaat saivat Hemlibraa ihon alle ensimmäisten neljän viikon ajan annoksina 3 mg/kg kerran viikossa, minkä jälkeen annos oli joko 1,5 mg/kg kerran viikossa (hoitohaarat A ja D) tai 3 mg/kg kerran kahdessa viikossa (hoitohaara B) tai ei lainkaan estohoitoa (hoitohaara C). Hoitohaaran C potilailla oli mahdollisuus siirtyä Hemlibra-hoitoon (3 mg/kg kerran kahdessa viikossa) sen jälkeen, kun he olivat olleet vähintään 24 viikkoa ilman estohoitoa. Hoitohaaroissa A ja B oli sallittua suurentaa annos 24 viikon jälkeen 3 mg:aan/kg viikoittain, jos potilaalla oli kaksi tai useampia kriteerit täyttäviä verenvuotoja (eli vakaassa tilassa ilmaantuvia spontaaneja ja kliinisesti merkittäviä verenvuotoja). Hoitohaaran D potilaiden annos voitiin titrata suuremmaksi toisen kriteerit täyttävän verenvuodon jälkeen. Viiden potilaan ylläpitoannosta suurennettiin primaarianalyysin ajankohtana.
Kahdeksankymmentäyhdeksän potilasta, jotka olivat aiemmin saaneet hyytymistekijä VIII:aa jaksoittaisesti (tarpeen mukaan), satunnaistettiin suhteessa 2:2:1 saamaan Hemlibra-hoitoa joko kerran viikossa (hoitohaara A; N = 36), kerran kahdessa viikossa (hoitohaara B; N = 35) tai ei lainkaan estohoitoa (hoitohaara C; N = 18). Ositus tehtiin edeltävien 24 viikon aikana esiintyneiden verenvuotojen määrän perusteella (< 9 tai ≥ 9). Kuusikymmentäkolme potilasta, jotka olivat aiemmin saaneet hyytymistekijä VIII:aa estohoitona, otettiin mukaan Hemlibra-hoitoa (1,5 mg/kg kerran viikossa) saavaan hoitohaaraan D.
Tutkimuksen ensisijainen tavoite oli arvioida Hemlibra-estohoidon tehoa jaksoittaista hyytymistekijä VIII ‑hoitoa aiemmin saaneilla potilailla, kun viikoittain (hoitohaara A) tai kerran kahdessa viikossa (hoitohaara B) annettua estohoitoa verrattiin siihen, ettei estohoitoa annettu (hoitohaara C). Arvio perustui hyytymistekijähoitoa vaatineiden verenvuotojen lukumäärään (ks. taulukko 4). Tutkimuksen muita tavoitteita olivat hoitohaaran A tai B ja hoitohaaran C satunnaistettu vertailu Hemlibra-estohoidon kaikkia verenvuotoja, spontaaneja verenvuotoja, nivelten verenvuotoja ja kohdenivelten verenvuotoja vähentävän tehon suhteen (ks. taulukko 4) sekä potilaan mieluiten käyttämän hoidon arviointi kyselyn avulla.
Hemlibra-estohoidon tehoa verrattiin myös aiempaan hyytymistekijä VIII ‑estohoitoon (hoitohaara D) potilailla, jotka olivat ennen tutkimukseen mukaan tuloa osallistuneet ei-interventiotutkimukseen (ks. taulukko 5). Vertailuun otettiin vain ei-interventiotutkimukseen osallistuneita potilaita, koska verenvuotoja ja hoitoa koskevat tiedot oli kerätty samalla datan rakeisuudella, jota käytettiin tutkimuksessa HAVEN 3.
Ei-interventiotutkimus on havainnoiva tutkimus, jonka keskeisenä tavoitteena oli kerätä tarkkoja kliinisiä tietoja A-hemofiliaa sairastavien potilaiden verenvuotoepisodeista ja hemofilian hoitoon käyttämistä lääkevalmisteista interventiotutkimuksen ulkopuolella.
A-hemofiliaa sairastavat potilaat (iältään ≥ 12 vuotta), joilla oli FVIII:n vasta-aineita (tutkimus BH29884 – HAVEN 1)
Satunnaistetussa, avoimessa kliinisessä monikeskustutkimuksessa (HAVEN 1) oli mukana 109 A-hemofiliaa sairastavaa nuorta ja aikuista miestä (iältään ≥ 12-vuotiaita), joilla oli FVIII:n vasta-aineita ja jotka olivat aiemmin saaneet jaksoittaista hoitoa tai estohoitoa vasta-aineen ohittavilla aineilla (aPCC ja rFVIIa). Potilaat saivat tutkimuksessa Hemlibra-estohoitoa viikoittain (hoitohaarat A, C ja D) annoksina 3 mg/kg kerran viikossa neljän viikon ajan, jonka jälkeen 1,5 mg/kg kerran viikossa, tai he eivät saaneet estohoitoa (hoitohaara B). Hoitohaaraan B satunnaistettujen potilaiden oli mahdollista siirtyä Hemlibra-estohoitoon, kun he olivat olleet vähintään 24 viikkoa ilman estohoitoa. Hemlibra-estohoidossa oli sallittua suurentaa annos 3 mg:aan/kg kerran viikossa 24 viikon jälkeen, jos potilaalla oli kaksi tai useampia kriteerit täyttäviä verenvuotoja (eli vakaassa tilassa ilmenneitä spontaaneja ja varmistettuja kliinisesti merkittäviä verenvuotoja). Kahden potilaan ylläpitoannos titrattiin ensisijaisen analyysin ajankohtana annokseen 3 mg/kg kerran viikossa.
Viisikymmentäkolme potilasta, jotka olivat aiemmin saaneet jaksoittain (tarpeen mukaan) vasta-aineen ohittavia aineita, satunnaistettiin suhteessa 2:1 joko saamaan Hemlibra-estohoitoa (hoitohaara A) tai ei saamaan estohoitoa (hoitohaara B). Ositus tehtiin edeltävien 24 viikon aikana esiintyneiden verenvuotojen määrän perusteella (< 9 tai ≥ 9).
Neljäkymmentäyhdeksän estohoitoa vasta-aineen ohittavilla aineilla aiemmin saanutta potilasta otettiin mukaan hoitohaaraan C, jossa he saivat Hemlibra-estohoitoa. Seitsemän potilasta, jotka olivat saaneet aiemmin hoitoa vasta-aineen ohittavilla aineilla jaksoittaisesti (tarpeen mukaan) ja jotka olivat ennen tutkimukseen mukaan tuloa osallistuneet ei-interventiotutkimukseen, mutta eivät voineet osallistua HAVEN 1 -tutkimukseen ennen hoitohaarojen A ja B sulkemista, otettiin mukaan hoitohaaraan D, jossa he saivat Hemlibra-estohoitoa.
Tutkimuksen ensisijainen tavoite oli arvioida viikoittain annetun Hemlibra-estohoidon tehoa verrattuna siihen, ettei estohoitoa annettu (hoitohaara A vs. hoitohaara B), potilailla, jotka olivat aiemmin saaneet jaksoittaista (tarpeen mukaista) hoitoa vasta-aineen ohittavilla aineilla. Tätä arvioitiin hyytymistekijähoitoa vaativien verenvuotojen lukumäärällä ajan mittaan (vähintään 24 viikkoa tai hoidon keskeyttämispäivänä) (ks. taulukko 6). Tutkimuksen toissijaisia tavoitteita olivat hoitohaarojen A ja B satunnaistettu vertailu viikoittain annetun Hemlibra-estohoidon kaikkia verenvuotoja, spontaaneja verenvuotoja, nivelten verenvuotoja ja kohdenivelten verenvuotoja vähentävästä tehosta (ks. taulukko 6) sekä potilaiden terveyteen liittyvän elämänlaadun ja terveydentilan arviointi (ks. taulukot 10 ja 11). Kaikkien tutkimuspotilaiden keskimääräinen altistusaika (keskihajonta) oli 21,38 viikkoa (12,01). Keskimääräiset altistusajat (keskihajonta) olivat hoitohaarassa A 28,86 viikkoa (8,37), hoitohaarassa B 8,79 viikkoa (3,62), hoitohaarassa C 21,56 viikkoa (11,85) ja hoitohaarassa D 7,08 viikkoa (3,89). Yksi hoitohaaran A potilaista lopetti tutkimuksen ennen Hemlibra-hoidon aloitusta.
Tutkimuksessa arvioitiin myös viikoittain annetun Hemlibra-estohoidon tehoa verrattuna vasta-aineen ohittavien aineiden aiempaan jaksoittaiseen (tarpeen mukaiseen) käyttöön tai niiden käyttöön estohoitona (erilliset vertailut) potilailla, jotka olivat ennen tutkimukseen mukaan tuloa osallistuneet ei-interventiotutkimukseen (hoitohaarat A ja C) (ks. taulukko 7).
A-hemofiliaa sairastavat potilaat (iältään ≥ 12 vuotta), joilla oli tai ei ollut FVIII:n vasta-aineita (tutkimus BO39182 – HAVEN 4)
Hemlibraa tutkittiin yhden hoitohaaran vaiheen III kliinisessä monikeskustutkimuksessa, jossa oli mukana 41 aikuista ja nuorta miestä (ikä ≥ 12 vuotta ja paino > 40 kg). Tutkittavilla oli A-hemofilia ja vasta-aineita hyytymistekijä VIII:lle tai vaikea-asteinen A-hemofilia, mutta ei hyytymistekijä VIII:n vasta-aineita, ja he olivat aiemmin saaneet joko jaksoittaisesti hoitoa (tarpeen mukaan) tai estohoitoa vasta-aineen ohittavilla aineilla tai hyytymistekijä VIII:lla. Potilaat saivat Hemlibra-estohoitoa 3 mg/kg kerran viikossa neljän viikon ajan ja sen jälkeen 6 mg/kg kerran neljässä viikossa.
Tutkimuksen ensisijainen tavoite oli arvioida kerran neljässä viikossa annetun Hemlibra-estohoidon tehoa verenvuotojen riittävässä hallinnassa, mikä perustui hoidettuihin verenvuotoihin. Muina tavoitteina oli arvioida Hemlibra-estohoidon kliinistä tehoa kaikkiin verenvuotoihin, hoidettuihin spontaaneihin verenvuotoihin, hoidettuihin nivelten verenvuotoihin ja hoidettuihin kohdenivelten verenvuotoihin (ks. taulukko 8). Myös potilaan mieluiten käyttämää hoitoa arvioitiin kyselyn avulla.
Lievää tai keskivaikeaa A‑hemofiliaa sairastavat potilaat (kaikenikäiset), joilla ei ollut FVIII:n vasta-aineita (tutkimus BO41423 – HAVEN 6)
HAVEN 6 ‑tutkimus oli avoin, yhden hoitohaaran vaiheen III kliininen monikeskustutkimus, jossa oli mukana 71 emisitsumabihoitoa saanutta (kaikenikäistä) potilasta. Potilailla oli lievä (n = 20 [28,2 %]) tai keskivaikea (n = 51 [71,8 %]) A‑hemofilia, mutta ei FVIII:n vasta-aineita, ja tutkija arvioi estohoidon olevan näille potilaille aiheellinen. Valtaosa potilaista oli miehiä (69 potilasta [97,2 %]), ja kaksi potilasta oli naisia (2,8 %). Tutkimukseen mukaan tullessaan 34 potilasta (47,9 %) sai jaksoittaista FVIII-hoitoa ja 37 potilasta (52,1 %) sai FVIII-estohoitoa. Potilaat saivat Hemlibra-valmistetta ihon alle annoksina 3 mg/kg kerran viikossa neljän ensimmäisen viikon ajan, minkä jälkeen potilaat saivat toiveensa mukaisesti jotakin seuraavista ylläpitohoidoista viikosta 5 alkaen: 1,5 mg/kg kerran viikossa (n = 24 [33,8 %]), 3 mg/kg kahden viikon välein (n = 39 [54,9 %]) tai 6 mg/kg neljän viikon välein (n = 8 [11,3 %]). 24 viikon jälkeen annos voitiin titrata tasolle 3 mg/kg viikoittain, jos potilaalla oli vähintään kaksi vaatimukset täyttävää verenvuotoa (eli spontaania ja kliinisesti merkittävää verenvuotoa vakaassa tilassa). Välianalyysin ajankohtana yhdenkään potilaan ylläpitoannosta ei ollut titrattu suuremmaksi.
Tutkimuksen ensisijainen tehoa koskeva tavoite oli arvioida Hemlibra-estohoidon tehoa ajan mittaan ilmaantuneiden hyytymistekijähoitoa vaatineiden verenvuotojen lukumäärän (eli hoidettujen verenvuotojen lukumäärä, ks. taulukko 9) perusteella. Muina tavoitteina oli arvioida Hemlibra-estohoidon tehoa ajan mittaan ilmaantuneiden kaikkien verenvuotojen, spontaanien verenvuotojen, nivelten verenvuotojen ja kohdenivelten verenvuotojen lukumäärän perusteella sekä arvioida potilaan raportoimaa terveyteen liittyvää elämänlaatua ajan mittaan CATCH (Comprehensive Assessment Tool of Challenges in Haemophilia) ‑kyselyllä.
Tehoa koskevat tulokset
HAVEN 3
Hemlibra-estohoidon tehoa koskevia tuloksia verrattiin siihen, ettei estohoitoa käytetty. Vertailu tehtiin hoidettujen verenvuotojen, kaikkien verenvuotojen, hoidettujen spontaanien verenvuotojen, hoidettujen nivelten verenvuotojen ja hoidettujen kohdenivelten verenvuotojen määrän suhteen. Vertailu esitetään taulukossa 4.
Taulukko 4. HAVEN 3 ‑tutkimus: Verenvuotojen vuotuistettu lukumäärä Hemlibra-estohoitoa saaneessa hoitohaarassa verrattuna hoitohaaraan, joka ei saanut estohoitoa, iältään ≥ 12-vuotiailla potilailla, joilla ei ollut FVIII:n vasta-aineita
| Päätetapahtuma | Hoitohaara C: ei estohoitoa (N = 18) | Hoitohaara A: Hemlibra 1,5 mg/kg viikoittain (N = 36) | Hoitohaara B: Hemlibra 3 mg/kg kerran 2 viikossa (N = 35) |
| Hoidetut verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 38,2 (22,9; 63,8) | 1,5 (0,9; 2,5) | 1,3 (0,8; 2,3) |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | NA | 96 % (0,04), < 0,0001 | 97 % (0,03), < 0,0001 |
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 0,0 (0,0; 18,5) | 55,6 (38,1; 72,1) | 60,0 (42,1; 76,1) |
| Verenvuotojen vuotuistetun lukumäärän mediaani (kvartiiliväli) | 40,4 (25,3; 56,7) | 0 (0; 2,5) | 0 (0; 1,9) |
| Kaikki verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 47,6 (28,5; 79,6) | 2,5 (1,6; 3,9) | 2,6 (1,6; 4,3) |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | NA | 95 % (0,05), < 0,0001 | 94 % (0,06), < 0,0001 |
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 0 (0,0; 18,5) | 50 (32,9; 67,1) | 40 (23,9; 57,9) |
| Hoidetut spontaanit verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 15,6 (7,6; 31,9) | 1,0 (0,5; 1,9) | 0,3 (0,1; 0,8) |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | NA | 94 % (0,06), < 0,0001 | 98 % (0,02), < 0,0001 |
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 22,2 (6,4; 47,6) | 66,7 (49,0; 81,4) | 88,6 (73,3; 96,8) |
| Hoidetut nivelten verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 26,5 (14,67; 47,79) | 1,1 (0,59; 1,89) | 0,9 (0,44; 1,67) |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | NA | 96 % (0,04), < 0,0001 | 97 % (0,03), < 0,0001 |
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 0 (0; 18,5) | 58,3 (40,8; 74,5) | 74,3 (56,7; 87,5) |
| Hoidetut kohdenivelten verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 13,0 (5,2; 32,3) | 0,6 (0,3; 1,4) | 0,7 (0,3; 1,6) |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | NA | 95 % (0,05), < 0,0001 | 95 % (0,05), < 0,0001 |
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 27,8 (9,7; 53,5) | 69,4 (51,9; 83,7) | 77,1 (59,9; 89,6) |
Esiintyvyyssuhde ja luottamusväli on saatu negatiivisesta binomiregressiomallista (NBR) ja p-arvo ositetulla Waldin testillä vertaamalla verenvuotojen lukumäärää mainituissa hoitohaaroissa. Hoitohaara C: mukana vain hoitojakso ilman estohoitoa. Verenvuotojen määritelmät on muokattu ISTH-kriteereistä. Hoidetut verenvuodot = verenvuodot, jotka hoidettiin hyytymistekijä VIII:lla. Kaikki verenvuodot = verenvuodot, jotka hoidettiin tai joita ei hoidettu hyytymistekijä VIII:lla. Sisältää vain tiedot ennen annoksen titraamista suuremmaksi niillä potilailla, joiden annos titrattiin suuremmaksi. Emisitsumabille altistuneiden potilaiden hoito aloitettiin aloitusannoksella 3 mg/kg/viikko 4 viikon ajan. Kvartiiliväli: 25. persentiilistä 75. persentiiliin. NA = ei oleellinen (not applicable) | |||
Kliinisen HAVEN 3 ‑tutkimuksen potilaskohtaisessa analyysissa Hemlibra-estohoito vähensi (68 %) verenvuotoihin hoitoa saaneiden potilaiden verenvuotojen lukumäärää tilastollisesti merkitsevästi (p < 0,0001) verrattuna aiempaan estohoitoon hyytymistekijä VIII:lla, mikä perustui ennen tutkimukseen mukaan tuloa tehdystä ei-interventiotutkimuksesta kerättyihin tietoihin (ks. taulukko 5).
Taulukko 5. HAVEN 3 ‑tutkimus:Verenvuotojen vuotuistetun lukumäärän (hoidettuja verenvuotoja) potilaskohtainen vertailu Hemlibra-estohoidon ja aiemman hyytymistekijä VIII:lla toteutetun estohoidon välillä
| Päätetapahtuma | Hoitohaara D ei-interventiotutkimus: Aiempi estohoito hyytymistekijä VIII:lla (N = 48) | Hoitohaara D: Hemlibra 1,5 mg/kg viikoittain (N = 48) |
| Tehoa koskevan jakson mediaani (viikkoa) | 30,1 | 33,7 |
| Hoidetut verenvuodot | ||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 4,8 (3,2; 7,1) | 1,5 (1; 2,3) |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | 68 % (0,32), < 0,0001 | |
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 39,6 (25,8; 54,7) | 54,2 (39,2; 68,6) |
| Verenvuotojen vuotuistetun lukumäärän mediaani (kvartiiliväli) | 1,8 (0; 7,6) | 0 (0; 2,1) |
Esiintyvyyssuhde ja luottamusväli on saatu negatiivisesta binomiregressiomallista (NBR) ja p-arvo ositetulla Waldin testillä vertaamalla verenvuotojen vuotuistettua lukumäärää mainituissa hoitohaaroissa. Potilaskohtaiset vertailutiedot on saatu ei-interventiotutkimuksesta. Mukaan on otettu vain niiden potilaiden tiedot, jotka osallistuivat ei-interventiotutkimukseen ja HAVEN 3 ‑tutkimukseen. Sisältää vain tiedot ennen annoksen titraamista suuremmaksi niillä potilailla, joiden annos titrattiin suuremmaksi. Hoidetut verenvuodot = verenvuodot, jotka hoidettiin hyytymistekijä VIII:lla. Verenvuotojen määritelmät on muokattu ISTH-kriteereistä. Kvartiiliväli: 25. persentiilistä 75. persentiiliin. Hoitoon sitoutumisen havaittiin olleen emisitsumabiestohoidossa parempi kuin aiemman FVIII-estohoidon yhteydessä, mutta verenvuotojen vuotuistetussa lukumäärässä ei todettu eroa niiden potilaiden välillä, jotka saivat ≥ 80 % tai < 80 % FVIII-estohoitoannoksista valmisteyhteenvedossa mainittujen tavanomaisten vaatimusten mukaisesti (tietojen tulkinnassa on oltava varovainen, koska otoskoot olivat pienet). FVIII:n puoliintumisaika on lyhyt, joten sen käytön lopettamisen jälkeen ei oletettavasti esiinny jäännösvaikutusta. Vain viisi ensimmäistä emisitsumabiannosta piti antaa valvotusti, jotta varmistettiin turvallisuus sekä injektiotekniikan osaaminen. Seuraavat emisitsumabiannokset oli sallittua pistää kotona itsehoitona samoin kuin FVIII-estohoidossa. Kaikki potilaat olivat hemofilian asiantuntijoiden hoidossa. Siten varmistettiin, että potilaskohtaiseen vertailuun mukaan otetut potilaat saivat riittävän FVIII-estohoidon, jotta kaikissa tutkimuskeskuksissa kaikki potilaat saivat vastaavan tavanomaisen estohoidon. | ||
HAVEN 1
Hemlibra-estohoidon tehoa koskevia tuloksia verrattiin siihen, ettei estohoitoa käytetty. Vertailu tehtiin hoidettujen verenvuotojen, kaikkien verenvuotojen, hoidettujen spontaanien verenvuotojen, hoidettujen nivelten verenvuotojen ja hoidettujen kohdenivelten verenvuotojen määrän suhteen. Vertailu esitetään taulukossa 6.
Taulukko 6. HAVEN 1: Verenvuotojen vuotuistettu määrä Hemlibra-estohoitohaarassa verrattuna siihen, ettei estohoitoa käytetty; vertailu tehtiin ≥ 12-vuotiailla potilailla, joilla oli vasta-aineita FVIII:lle
| Päätetapahtuma | Hoitohaara B: ei estohoitoa | Hoitohaara A: 1,5 mg/kg Hemlibraa viikoittain | |
| N = 18 | N = 35 | ||
| Hoidetut verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 23,3 (12,33; 43,89) | 2,9 (1,69; 5,02) | |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | 87 % (0,13), < 0,0001 | ||
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 5,6 (0,1; 27,3) | 62,9 (44,9; 78,5) | |
| Verenvuotojen vuotuistetun lukumäärän mediaani (kvartiiliväli) | 18,8 (12,97; 35,08) | 0 (0; 3,73) | |
| Kaikki verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 28,3 (16,79; 47,76) | 5,5 (3,58; 8,60) | |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | 80 % (0,20), < 0,0001 | ||
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 5,6 (0,1; 27,3) | 37,1 (21,5; 55,1) | |
| Hoidetut spontaanit verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 16,8 (9,94; 28,30) | 1,3 (0,73; 2,19) | |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | 92 % (0,08), < 0,0001 | ||
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 11,1 (1,4; 34,7) | 68,6 (50,7; 83,1) | |
| Hoidetut nivelten verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 6,7 (1,99; 22,42) | 0,8 (0,26; 2,20) | |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | 89 % (0,11), 0,0050 | ||
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 50,0 (26,0; 74,0) | 85,7 (69,7; 95,2) | |
| Hoidetut kohdenivelten verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 3,0 (0,96; 9,13) | 0,1 (0,03; 0,58) | |
| Vähenemä (%, esiintyvyyssuhde), p‑arvo | 95 % (0,05), 0,0002 | ||
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 50,0 (26,0; 74,0) | 94,3 (80,8; 99,3) | |
Esiintyvyyssuhde ja luottamusväli on saatu negatiivisesta binomiregressiomallista (NBR) ja p-arvo ositetulla Waldin testillä vertaamalla verenvuotojen lukumäärää mainituissa hoitohaaroissa. Hoitohaara B: mukana vain hoitojakso ilman estohoitoa. Verenvuotojen määritelmät on muokattu ISTH-kriteereistä. Hoidetut verenvuodot = verenvuodot, jotka hoidettiin vasta-aineen ohittavilla aineilla. Kaikki verenvuodot = verenvuodot, jotka hoidettiin tai joita ei hoidettu vasta-aineen ohittavilla aineilla. Sisältää vain tiedot ennen annoksen titraamista suuremmaksi niillä potilailla, joiden annos titrattiin suuremmaksi. Emisitsumabille altistuneiden potilaiden hoito aloitettiin aloitusannoksella 3 mg/kg/viikko 4 viikon ajan. Kvartiiliväli: 25. persentiilistä 75. persentiiliin. | |||
HAVEN 1 ‑tutkimuksen potilaskohtaisessa analyysissä Hemlibra-estohoito vähensi hoidettuja verenvuotoja tilastollisesti merkitsevästi (p = 0,0003) ja kliinisesti merkittävästi (79 %) verrattuna aiemmasta estohoidosta vasta-aineen ohittavalla aineella ei-interventiotutkimuksessa kerättyihin tietoihin ennen tähän tutkimukseen mukaan tuloa (ks. taulukko 7).
Taulukko 7. HAVEN 1: Potilaskohtainen vertailu verenvuotojen vuotuistetusta määrästä (hoidettuja verenvuotoja) Hemlibra-estohoidon ja aiemman vasta-aineen ohittavalla aineella toteutetun estohoidon välillä (ei-interventiotutkimuksessa [NIS, non-intervention study] mukana olleet potilaat)
| Päätetapahtuma | Hoitohaara CNIS: aiempi estohoito vasta-aineen ohittavalla aineella | Hoitohaara C: Hemlibra 1,5 mg/kg viikoittain |
| N = 24 | N = 24 | |
| Hoidetut verenvuodot | ||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 15,7 (11,08; 22,29) | 3,3 (1,33; 8,08) |
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 12,5 (2,7; 32,4) | 70,8 (48,9; 87,4) |
| Verenvuotojen vuotuistetun lukumäärän mediaani (kvartiiliväli) | 12,0 (5,73; 24,22) | 0,0 (0,00; 2,23) |
| Vähenemä (%, esiintyvyyssuhde), p-arvo | 79 % (0,21), 0,0003 | |
Esiintyvyyssuhde ja luottamusväli on saatu negatiivisesta binomiregressiomallista (NBR) ja p-arvo ositetulla Waldin testillä vertaamalla verenvuotojen vuotuistettua lukumäärää mainituissa hoitohaaroissa. Potilaskohtaiset vertailutiedot on saatu ei-interventiotutkimuksesta. Mukana vain potilaat, jotka osallistuivat ei-interventiotutkimukseen ja tutkimukseen HAVEN 1. Sisältää vain tiedot ennen annoksen titraamista suuremmaksi niillä potilailla, joiden annos titrattiin suuremmaksi. Hoidetut verenvuodot = verenvuodot, jotka hoidettiin vasta-aineen ohittavilla aineilla. Verenvuotojen määritelmät on muokattu ISTH-kriteereistä. Kvartiiliväli: 25. persentiilistä 75. persentiiliin. Hoitoon sitoutumisen havaittiin olleen emisitsumabiestohoidossa parempi kuin aiemman vasta-aineen ohittavalla aineella annetun estohoidon yhteydessä, mutta verenvuotojen vuotuistetussa lukumäärässä ei todettu eroa niiden potilaiden välillä, jotka saivat ≥ 80 % tai < 80 % vasta-aineen ohittavalla aineella annetun estohoidon annoksista valmisteyhteenvedossa mainittujen tavanomaisten vaatimusten mukaisesti (tietojen tulkinnassa on oltava varovainen, koska otoskoot olivat pienet). Vasta-aineen ohittavien aineiden puoliintumisaika on lyhyt, joten sen käytön lopettamisen jälkeen ei oletettavasti esiinny jäännösvaikutusta. Vain viisi ensimmäistä emisitsumabiannosta piti antaa valvotusti, jotta varmistettiin turvallisuus sekä injektiotekniikan osaaminen. Seuraavat emisitsumabiannokset oli sallittua pistää kotona itsehoitona samoin kuin vasta-aineen ohittavalla aineella annetussa estohoidossa. | ||
HAVEN 4
Primaarianalyysi kerran neljässä viikossa annetun Hemlibra-estohoidon tehoa koskevista tuloksista hoidettujen verenvuotojen, kaikkien verenvuotojen, hoidettujen spontaanien verenvuotojen, hoidettujen nivelten verenvuotojen ja hoidettujen kohdenivelten verenvuotojen osalta esitetään taulukossa 8. Tehoa arvioitiin neljälläkymmenelläyhdellä ≥ 12‑vuotiaalla potilaalla, ja havainnointiajan mediaani oli 25,6 viikkoa (vaihteluväli 24,1–29,4).
Taulukko 8. HAVEN 4: Verenvuotojen vuotuistettu määrä Hemlibra-estohoitoa saaneilla ≥ 12‑vuotiailla potilailla, joilla oli tai ei ollut vasta-aineita FVIII:lle
| Hemlibra 6 mg/kg kerran neljässä viikossa | |||
| Päätetapahtumat | aVerenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | bVerenvuotojen vuotuistetun lukumäärän mediaani (kvartiiliväli) | Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) |
| N | 41 | 41 | 41 |
| Hoidetut verenvuodot | 2,4 (1,4; 4,3) | 0,0 (0,0; 2,1) | 56,1 (39,7; 71,5) |
| Kaikki verenvuodot | 4,5 (3,1; 6,6) | 2,1 (0,0; 5,9) | 29,3 (16,1; 45,5) |
| Hoidetut spontaanit verenvuodot | 0,6 (0,3; 1,5) | 0,0 (0,0; 0,0) | 82,9 (67,9; 92,8) |
| Hoidetut nivelten verenvuodot | 1,7 (0,8; 3,7) | 0,0 (0,0; 1,9) | 70,7 (54,5; 83,9) |
| Hoidetut kohdenivelten verenvuodot | 1,0 (0,3; 3,3) | 0,0 (0,0; 0,0) | 85,4 (70,8; 94,4) |
a Laskettu negatiivisella binomiregressiomallilla (NBR) b Laskettu verenvuotojen vuotuistettu lukumäärä Verenvuotojen määritelmät on muokattu ISTH-kriteereistä. Hoidetut verenvuodot = verenvuodot, jotka hoidettiin hyytymistekijä VIII:lla tai rekombinantilla hyytymistekijä VIIa:lla Kaikki verenvuodot = verenvuodot, jotka hoidettiin tai joita ei hoidettu hyytymistekijä VIII:lla tai rekombinantilla hyytymistekijä VIIa:lla Emisitsumabia saaneiden potilaiden hoito aloitettiin annoksella 3 mg/kg/viikko 4 viikon ajan. Kvartiiliväli: 25. persentiilistä 75. persentiiliin | |||
HAVEN 6 (välianalyysi)
Tehoa arvioitiin 51:llä keskivaikeaa A‑hemofiliaa sairastavalla potilaalla, jotka olivat iältään 2−56‑vuotiaita. Havainnointiajan mediaani oli 30,4 viikkoa (vaihteluväli: 17,4–61,7). Välianalyysi Hemlibra-estohoidon tehoa keskivaikeaa A-hemofiliaa sairastavilla potilailla (ks. kohta Käyttöaiheet) koskevista tuloksista hoidettujen verenvuotojen, kaikkien verenvuotojen, hoidettujen spontaanien verenvuotojen, hoidettujen nivelten verenvuotojen ja hoidettujen kohdenivelten verenvuotojen määrien suhteen esitetään taulukossa 9.
Taulukko 9. HAVEN 6: Verenvuotojen vuotuistettu määrä Hemlibra-estohoitoa saaneilla keskivaikeaa A‑hemofiliaa sairastavilla potilailla, joilla ei ollut vasta-aineita FVIII:lle
| cHemlibra 1,5 mg/kg kerran viikossa, 3 mg/kg kahden viikon välein tai 6 mg/kg neljän viikon välein | |||
| Päätetapahtumat | aVerenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | bVerenvuotojen vuotuistetun lukumäärän mediaani (kvartiiliväli) | Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) |
| N | 51 | 51 | 51 |
| Hoidetut verenvuodot | 0,9 (0,43; 1,89) | 0,0 (0,00; 0,00) | 78,4 (64,7; 88,7) |
| Kaikki verenvuodot | 2,6 (1,81; 3,81) | 1,7 (0,00; 3,90) | 43,1 (29,3; 57,8) |
| Hoidetut spontaanit verenvuodot | 0,1 (0,03; 0,30) | 0,0 (0,00; 0,00) | 94,1 (83,8; 98,8) |
| Hoidetut nivelten verenvuodot | 0,3 (0,10; 0,84) | 0,0 (0,00; 0,00) | 90,2 (78,6; 96,7) |
| Hoidetut kohdenivelten verenvuodot | 0,1 (0,02; 0,26) | 0,0 (0,00; 0,00) | 96,1 (86,5; 99,5) |
a Laskettu negatiivisella binomiregressiomallilla (NBR) b Laskettu verenvuotojen vuotuistettu lukumäärä Verenvuotojen määritelmät on muokattu ISTH-kriteereistä. Hoidetut verenvuodot = verenvuodot, jotka hoidettiin FVIII:lla. Kaikki verenvuodot = verenvuodot, jotka hoidettiin tai joita ei hoidettu FVIII:lla. Emisitsumabille altistuneet potilaat saivat aloitusannoksen 3 mg/kg/viikko 4 viikon ajan. Kvartiiliväli; 25. persentiilistä 75. persentiiliin c 1,5 mg/kg kerran viikossa (n = 16); 3 mg/kg kahden viikon välein (n = 30); 6 mg/kg neljän viikon välein (n = 5) | |||
Terveyteen liittyvää elämänlaatua koskevat hoitotulokset
Terveyteen liittyvää elämänlaatua ja terveydentilaa arvioitiin kliinisissä HAVEN-tutkimuksissa kliinisen hoitotuloksen arviointiin käytettävillä mittareilla. HAVEN 1 ja 2 ‑tutkimuksissa käytettiin hemofiliaspesifisen elämänlaatukyselyn aikuisille ( > 18‑vuotiaille) (Haem-A-QoL) ja nuorille (Haemo-QoL-SF, 8 – < 18-vuotiaat) tarkoitettuja versioita, joista tutkimussuunnitelmassa määriteltiin kiinnostaviksi päätetapahtumiksi fyysistä terveyttä kuvaava pisteytys (eli kivulias turvotus, nivelkipu, kipu liikuttaessa, kävelyvaikeudet pitkillä kävelymatkoilla ja pidemmän valmistautumisajan tarve) ja kokonaispisteytys (kaikkien osa-alueiden yhteispisteet). HAVEN 2 ‑tutkimuksessa käytettiin lisäksi Adapted InhibQoL ‑elämänlaatukyselyä sekä Aspects of Caregiver Burden ‑kyselyä, jotta saatiin potilaita hoitavien henkilöiden raportoimat < 12‑vuotiaiden pediatristen potilaiden terveyteen liittyvää elämänlaatua koskevat tiedot. HAVEN 6 ‑tutkimuksessa arvioitiin aikuisten ja pediatristen potilaiden sekä pediatrisia potilaita hoitavien henkilöiden terveyteen liittyvää elämänlaatua CATCH (Comprehensive Assessment Tool of Challenges in Haemophilia) ‑kyselyn avulla. Siinä tarkastellut osa-alueet olivat näkemys riskeistä ja hemofilian vaikutus päivittäisiin toimintoihin, sosiaalisiin toimintoihin, vapaa-ajan toimintoihin ja työhön/kouluun sekä sairauden jatkuva ajatteleminen ja hoidon kuormittavuus. Terveyteen liittyvän elämänlaadun muutosta mitattiin EQ-5D-5L-mittarin (EuroQoL Five-Dimension-Five Levels Questionnaire) IUS-asteikon (Index Utility Score) ja VAS-asteikon (Visual Analog Scale) perusteella.
HAVEN 1 ‑tutkimuksen terveyteen liittyvää elämänlaatua koskevat hoitotulokset (Haem-A-QoL)
Tämän tutkimuksen lähtötilanteen kokonaispisteet (keskiarvo = 41,14 Hemlibra-estohoitoa saaneilla ja 44,58 potilailla, jotka eivät saaneet estohoitoa) ja fyysistä terveyttä kuvaavan asteikon pisteet (keskiarvo = 52,41 Hemlibra-estohoitoa saaneilla ja 57,19 potilailla, jotka eivät saaneet estohoitoa) olivat samankaltaiset Hemlibra-estohoitoa saaneilla ja potilailla, jotka eivät saaneet estohoitoa. Taulukossa 10 esitetään yhteenveto Hemlibra-estohoitoa saaneen hoitohaaran (hoitohaara A) ja hoitohaaran, jonka potilaat eivät saaneet estohoitoa (hoitohaara B), Haem-A-QoL-kokonaispisteiden vertailusta ja fyysistä terveyttä kuvaavan asteikon lähtötilanteen mukaan korjatuista pisteistä 24 viikon jälkeen. Viikoittaisessa Hemlibra-estohoidossa osoitettiin ennalta määritettyjen Haem-A-QoL:n päätetapahtumien (fyysistä terveyttä kuvaavan asteikon pisteet) parantuneen viikolla 25 tehdyssä arviossa tilastollisesti merkitsevästi ja kliinisesti merkittävästi.
Taulukko 10. HAVEN 1: Fyysistä terveydentilaa koskevien Haem-A-QoL-pisteiden ja kokonaispisteiden muutos ≥ 18-vuotiailla potilailla, joilla oli FVIII:n vasta-aineita, vertailtaessa Hemlibra-estohoitoa siihen, ettei estohoitoa käytetty
| Haem-A-QoL viikolla 25 | Hoitohaara B: ei estohoitoa (N = 14) | Hoitohaara A: 1,5 mg/kg Hemlibraa viikoittain (N = 25) |
| Fyysisen terveydentilan pisteet (vaihteluväli 0–100) | ||
| Korjattu keskiarvo | 54,17 | 32,61 |
| Ero korjattuina keskiarvoina (95 %:n luottamusväli) | 21,55 (7,89; 35,22) | |
| p-arvo | 0,0029 | |
| Kokonaispisteet (vaihteluväli 0–100) | ||
| Korjattu keskiarvo | 43,21 | 29,2 |
| Ero korjattuina keskiarvoina (95 %:n luottamusväli) | 14,01 (5,56; 22,45) | |
Hoitohaara B: mukana vain hoitojakso ilman estohoitoa. Sisältää vain tiedot ennen annoksen titraamista suuremmaksi niillä potilailla, joiden annos titrattiin suuremmaksi. Emisitsumabille altistuneiden potilaiden hoito aloitettiin aloitusannoksella 3 mg/kg/viikko 4 viikon ajan. Haem-A-QoL pisteet 0–100; pienemmät pisteet kuvastavat parempaa terveyteen liittyvää elämänlaatua. Kliinisesti merkittävä ero: kokonaispisteet 7 pistettä; fyysinen terveys 10 pistettä. Analyysit perustuvat niiden henkilöiden tietoihin, joilta saatiin vastaukset sekä lähtötilanteen että viikon 25 arviointeihin. | ||
HAVEN 1 ‑tutkimuksen terveyteen liittyvää elämänlaatua koskevat hoitotulokset (EQ-5D-5L)
Taulukossa 11 esitetään yhteenveto Hemlibra-estohoitoa saaneen hoitohaaran (hoitohaara A) ja hoitohaaran, jonka potilaat eivät saaneet estohoitoa (hoitohaara B), lähtötilanteen mukaan korjatun EQ-5D-5L-utiliteetti-indeksiluvun ja VAS-asteikon (Visual Analogue Scale) pistemäärän välisestä vertailusta 24 viikon hoidon jälkeen.
Taulukko 11. HAVEN 1: ≥ 12-vuotiaiden potilaiden EQ-5D-5L-indeksiluku viikolla 25
| EQ-5D-5L-indeksiluku 24 viikon jälkeen | Hoitohaara B: ei estohoitoa (N = 16) | Hoitohaara A: 1,5 mg/kg Hemlibraa viikoittain (N = 29) |
| VAS-asteikko | ||
| Korjattu keskiarvo | 74,36 | 84,08 |
| Ero korjattuina keskiarvoina (95 %:n luottamusväli) | -9,72 (-17,62; -1,82) | |
| Utiliteetin indeksiluku | ||
| Korjattu keskiarvo | 0,65 | 0,81 |
| Ero korjattuina keskiarvoina (95 %:n luottamusväli) | -0,16 (-0,25; -0,07) | |
Hoitohaara B: mukana vain hoitojakso ilman estohoitoa. Sisältää vain tiedot ennen annoksen titraamista suuremmaksi niillä potilailla, joiden annos titrattiin suuremmaksi. Emisitsumabille altistuneiden potilaiden hoito aloitettiin aloitusannoksella 3 mg/kg/viikko 4 viikon ajan. Suurempi indeksiluku osoittaa parempaa elämänlaatua. Kliinisesti merkittävä ero: VAS-asteikko 7 pistettä; utiliteetin indeksiluku 0,07 pistettä. Analyysit perustuvat niiden henkilöiden tietoihin, joilta saatiin vastaukset sekä lähtötilanteen että viikon 25 arviointeihin. | ||
HAVEN 6 ‑tutkimuksen terveyteen liittyvät hoitotulokset
HAVEN 6 ‑tutkimuksessa arvioitiin viikolla 25 kaikenikäisten keskivaikeaa A‑hemofiliaa sairastavien potilaiden terveyteen liittyvää elämänlaatua CATCH-kyselyn perusteella. CATCH-kysely (versio 1.0) on validoitu työkalu, jolla arvioidaan hemofilian ja sen hoidon vaikutusta. Kyselystä on olemassa eri versiot aikuisille potilaille, pediatrisille potilaille ja pediatrisia potilaita hoitaville henkilöille. Hemlibra-estohoidon vaikutus terveyteen liittyvään elämänlaatuun pysyi yleisesti vakaana, ja kaikissa vastaajaryhmissä havaittiin yhdenmukaisesti paranemista CATCH-kyselyn hoidon kuormittavuutta koskevassa osiossa.
Pediatriset potilaat
Pediatriset A-hemofiliaa sairastavat potilaat (< 12-vuotiaat tai 12–17-vuotiaat, joiden paino oli < 40 kg), joilla oli FVIII:n vasta-aineita (tutkimus BH29992 – HAVEN 2)
Viikoittain annettavaa Hemlibra-estohoitoa tutkittiin yhden hoitohaaran avoimessa kliinisessä monikeskustutkimuksessa pediatrisilla A-hemofiliaa sairastavilla potilailla (< 12 vuoden ikäisiä tai 12–17 vuoden ikäisiä, joiden paino on < 40 kg), joilla oli FVIII:n vasta-aineita. Potilaat saivat Hemlibra-estohoitoa annoksina 3 mg/kg kerran viikossa ensimmäisten 4 viikon ajan ja sen jälkeen 1,5 mg/kg kerran viikossa.
Tutkimuksessa arvioitiin farmakokinetiikkaa, turvallisuutta ja tehoa sekä viikoittain annetun Hemlibra-estohoidon tehoa verrattuna aiempaan jaksoittaisesti käytettyyn ja estohoitona annettuun hoitoon vasta-aineen ohittavilla aineilla. Tutkimuksessa mukana olleet potilaat olivat ennen tutkimukseen mukaan tuloa osallistuneet ei-interventiotutkimukseen (potilaskohtainen vertailu).
Tehoa koskevat tulokset
HAVEN 2 (välianalyysi)
Tehoa arvioitiin välianalyysin ajankohtana 59 potilaalla, jotka olivat iältään < 12-vuotiaita ja jotka olivat saaneet Hemlibra-estohoitoa viikoittain vähintään 12 viikon ajan. Potilasjoukossa oli mukana neljä iältään < 2-vuotiasta potilasta, 17 iältään 2 – < 6-vuotiasta potilasta ja 38 iältään 6 – < 12-vuotiasta potilasta. Verenvuotojen vuotuistettu lukumäärä ja niiden potilaiden prosenttiosuus, joilla ei ollut verenvuotoja, laskettiin (ks. taulukko 12). Näiden potilaiden havainnointiajan mediaani oli 29,6 viikkoa (vaihteluväli: 18,4–63,0 viikkoa).
Taulukko 12. HAVEN 2: Tehoa koskevat tiedot (välianalyysi)
| Päätetapahtuma | aVerenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) bN = 59 | cVerenvuotojen vuotuistetun lukumäärän mediaani (kvartiiliväli) bN = 59 | Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) bN = 59 |
| Hoidetut verenvuodot | 0,3 (0,1; 0,5) | 0 (0; 0) | 86,4 (75; 94) |
| Kaikki verenvuodot | 3,8 (2,2; 6,5) | 0 (0; 3,4) | 55,9 (42,4; 68,8) |
| Hoidetut spontaanit verenvuodot | 0 (0; 0,2) | 0 (0; 0) | 98,3 (90,9; 100) |
| Hoidetut nivelten verenvuodot | 0,2 (0,1; 0,4) | 0 (0; 0) | 89,8 (79,2; 96,2) |
| Hoidetut kohdenivelten verenvuodot | 0,1 (0; 0,7) | 0 (0; 0) | 96,6 (88,3; 99,6) |
Kvartiiliväli: 25. persentiilistä 75. persentiiliin a Laskettu negatiivisella binomiregressiomallilla (NBR) b Tehoa koskevat tiedot perustuvat < 12-vuotiaisiin potilaisiin, jotka olivat HAVEN 2 ‑tutkimuksessa mukana vähintään 12 viikkoa (N = 59), sillä tutkimuksen pääasiallisena tavoitteena oli tutkia hoidon tehoa iän perusteella. c Laskettu verenvuotojen vuotuistettu lukumäärä Verenvuotojen määritelmät on muokattu ISTH-kriteereistä. Hoidetut verenvuodot = verenvuodot, jotka hoidettiin vasta-aineen ohittavilla aineilla. Kaikki verenvuodot = verenvuodot, jotka hoidettiin tai joita ei hoidettu vasta-aineen ohittavilla aineilla. Emisitsumabia saaneiden potilaiden hoito aloitettiin annoksella 3 mg/kg/viikko 4 viikon ajan. | |||
Potilaskohtaisessa analyysissä viikoittain annettu Hemlibra-hoito vähensi vähintään 12 viikon ajan Hemlibra-estohoitoa saaneilla 18 pediatrisella potilaalla hoidettujen verenvuotojen määrää kliinisesti merkitsevästi (98 %) verrattuna heillä ennen tutkimukseen mukaan tuloa ei-interventiotutkimuksessa todettujen verenvuotojen määrään (taulukko 13).
Taulukko 13. HAVEN 2: Potilaskohtainen vertailu verenvuotojen vuotuistetusta lukumäärästä (hoidettuja verenvuotoja) Hemlibra-estohoidossa verrattuna aiempaan estohoitoon vasta-aineen ohittavalla aineella
| Päätetapahtuma | Aiempi hoito vasta-aineen ohittavalla aineella * (N = 18) | Hemlibra-estohoito (N = 18) | |
| Hoidetut verenvuodot | |||
| Verenvuotojen vuotuistettu lukumäärä (95 %:n luottamusväli) | 19,8 (15,3; 25,7) | 0,4 (0,15; 0,88) | |
| Vähenemä (%) (esiintyvyyssuhde) | 98 % (0,02) | ||
| Potilaita (%), joilla ei verenvuotoja (95 %:n luottamusväli) | 5,6 (0,1; 27,3) | 77,8 (52,4; 93,6) | |
| Verenvuotojen vuotuistetun lukumäärän mediaani (kvartiiliväli) | 16,2 (11,49; 25,78) | 0 (0; 0) | |
* 18 potilaasta 15 potilasta oli saanut aiemmin estohoitoa ja 3 tutkittavaa oli saanut aiemmin jaksoittaista hoitoa (tarpeen mukaan). Esiintyvyyssuhde ja luottamusväli on saatu negatiivisesta binomiregressiomallista (NBR) ja p-arvo ositetulla Waldin testillä vertaamalla verenvuotojen vuotuistettua lukumäärää mainituissa hoitohaaroissa. Potilaskohtaiset vertailutiedot on saatu ei-interventiotutkimuksesta. Mukana vain potilaat, jotka osallistuivat ei-interventiotutkimukseen ja HAVEN 2 ‑tutkimukseen. Verenvuotojen määritelmät on muokattu ISTH-kriteereistä. Hoidetut verenvuodot = verenvuodot, jotka hoidettiin vasta-aineen ohittavilla aineilla. Emisitsumabia saaneiden potilaiden hoito aloitettiin annoksella 3 mg/kg/viikko 4 viikon ajan. Kvartiiliväli: 25. persentiilistä 75. persentiiliin. Hoitoon sitoutumisen havaittiin olleen emisitsumabiestohoidossa parempi kuin aiemman vasta-aineen ohittavalla aineella annetun estohoidon yhteydessä, mutta verenvuotojen vuotuistetussa lukumäärässä ei todettu eroa niiden potilaiden välillä, jotka saivat ≥ 80 % tai < 80 % vasta-aineen ohittavalla aineella annetun estohoidon annoksista valmisteyhteenvedossa mainittujen tavanomaisten vaatimusten mukaisesti (tietojen tulkinnassa on oltava varovainen, koska otoskoot olivat pienet). Vasta-aineen ohittavien aineiden puoliintumisaika on lyhyt, joten sen käytön lopettamisen jälkeen ei oletettavasti esiinny jäännösvaikutusta. Vain viisi ensimmäistä emisitsumabiannosta piti antaa valvotusti, jotta varmistettiin turvallisuus sekä injektiotekniikan osaaminen. Seuraavat emisitsumabiannokset oli sallittua pistää kotona itsehoitona samoin kuin vasta-aineen ohittavalla aineella annetussa estohoidossa. | |||
Pediatristen potilaiden terveyteen liittyvää elämänlaatua koskevat hoitotulokset
HAVEN 2 ‑tutkimuksen terveyteen liittyvää elämänlaatua koskevat hoitotulokset (Haemo-QoL-SF H)
HAVEN 2 ‑tutkimuksen viikolla 25 arvioitiin iältään ≥ 8 – < 12-vuotiaiden potilaiden terveyteen liittyvää elämänlaatua lapsille tarkoitetulla Haemo-QoL-SF-kyselyllä (ks. taulukko 14). Haemo-QoL-SF on validi ja luotettava terveyteen liittyvän elämänlaadun mittari.
Iältään < 12‑vuotiaiden potilaiden terveyteen liittyvää elämänlaatua arvioitiin viikolla 25 myös huoltajan täyttämällä kyselyllä Adapted InhibQoL, jossa selvitettiin huoltajan kuormitusta (Aspects of Caregiver Burden) (ks. taulukko 14). Adapted InhibQoL on validi ja luotettava terveyteen liittyvän elämänlaadun mittari.
Taulukko 14. HAVEN 2: Potilaiden ja huoltajien raportoima potilaan (< 12‑vuotiaiden) fyysistä terveydentilaa kuvaavan pisteytyksen muutos lähtötilanteesta viikkoon 25 potilaiden Hemlibra-estohoidon jälkeen
| Haemo-QoL-SF-kysely | |
| Fyysistä terveydentilaa kuvaava pisteytys (vaihteluväli 0–100)a | |
| Lähtötilanteen pisteiden keskiarvo (95 %:n luottamusväli) (n = 18) | 29,5 (16,4–42,7) |
| Keskimääräinen muutos lähtötilanteesta (95 %:n luottamusväli) (n = 15) | -21,7 (-37,1 – -6,3) |
| Huoltajille tarkoitettu mukautettu kysely (Adapted InhibQoL) sekä huoltajan kuormitusta selvittävä kysely (aspects of caregiver burden) | |
| Fyysistä terveydentilaa kuvaava pisteytys (vaihteluväli 0–100)a | |
| Lähtötilanteen pisteiden keskiarvo (95 %:n luottamusväli) (n = 54) | 37,2 (31,5–42,8) |
| Keskimääräinen muutos lähtötilanteesta (95 %:n luottamusväli) (n = 43) | -32,4 (-38,6 – -26,2) |
a Pienempi pistemäärä (luvun negatiivinen muutos) kuvastaa parempaa toimintakykyä. Analyysit perustuvat niiden henkilöiden tietoihin, joilta saatiin vastaukset sekä lähtötilanteen että viikon 25 arviointeihin. | |
Vasta-aineen ohittavan aineen tai hyytymistekijä VIII:n käytöstä leikkausten ja toimenpiteiden aikana on vähän kokemusta. Vasta-aineen ohittavan aineen tai hyytymistekijä VIII:n käytöstä leikkausten ja toimenpiteiden aikana päätti tutkija.
Jos verenvuotoja ilmenee hoidosta huolimatta, emisitsumabiestohoitoa saavalle potilaalle pitää antaa saatavissa olevia hoitoja. Vasta-aineen ohittavia aineita koskevat tiedot, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Immunogeenisuus
Kaikkien terapeuttisten proteiinien tavoin emisitsumabihoitoa saaville potilaille voi kehittyä immuunivaste. Yhdistetyissä kliinisissä tutkimuksissa yhteensä 739 potilaalta tutkittiin vasta-aineet emisitsumabille. Vasta-aineita emisitsumabille todettiin 36:lla potilaalla (4,9 %), ja heistä 19:llä potilaalla (2,6 %) emisitsumabivasta-aineet olivat neutraloivia in vitro. Näistä 19 potilaasta, neutraloivilla emisitsumabivasta-aineilla ei ollut kliinisesti merkittävää vaikutusta Hemlibran farmakokinetiikkaan tai tehoon 15:llä potilaalla, kun taas neljällä potilaalla (0,5 %) havaittiin plasman emisitsumabipitoisuuden pienenemistä. Yhdellä potilaalla (0,1 %), jolla kehittyi neutraloivia emisitsumabivasta-aineita ja plasman emisitsumabipitoisuus pieneni, teho heikkeni viiden viikon hoidon jälkeen ja Hemlibra-hoito keskeytettiin. Yleisesti ottaen Hemlibran turvallisuusprofiili oli yhdenmukainen potilailla, joilla oli emisitsumabivasta-aineita (mukaan lukien neutraloivat vasta-aineet) ja niillä, joilla näitä ei ollut (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Iäkkäät potilaat
HAVEN 1-, HAVEN 3-, HAVEN 4- ja HAVEN 6 ‑tutkimukset tukevat Hemlibran käyttöä 65‑vuotiaille ja vanhemmille A-hemofiliapotilaille, eikä suppeiden tietojen perusteella ole siihen viittaavaa näyttöä, että hoidon teho tai turvallisuus olisi erilainen 65-vuotiailla ja vanhemmilla potilailla.
Farmakokinetiikka
Emisitsumabin farmakokinetiikkaa tutkittiin terveiden tutkittavien tilamallittomalla analyysillä ja tekemällä populaatiofarmakokineettinen analyysi tietokannasta, joka koostui 389:n A-hemofiliaa sairastavan potilaan tiedoista.
Imeytyminen
A-hemofiliapotilaille ihon alle annetun valmisteen imeytymisen puoliintumisaika oli 1,6 vuorokautta.
Kun A-hemofiliapotilaiden ihon alle oli ensimmäisten 4 hoitoviikon aikana annettu useita annoksia 3 mg/kg kerran viikossa, plasman pienimpien emisitsumabipitoisuuksien keskiarvo (± keskihajonta) viikolla 5 oli 52,6 ± 13,6 µg/ml.
Vakaan tilan Ctrough- ja Cmax-arvojen ennakoitu keskiarvo (± keskihajonta) sekä Cmax- ja Ctrough-arvojen suhteen keskiarvo (± keskihajonta) suositeltujen ylläpitoannosten ollessa 1,5 mg/kg kerran viikossa, 3 mg/kg kerran kahdessa viikossa tai 6 mg/kg kerran neljässä viikossa esitetään taulukossa 15.
Taulukko 15. Emisitsumabipitoisuuksien keskiarvo (± keskihajonta) vakaassa tilassa
Ylläpitoannos | |||
Parametrit | 1,5 mg/kg kerran viikossa (QW) | 3 mg/kg kerran kahdessa viikossa (Q2W) | 6 mg/kg kerran neljässä viikossa (Q4W) |
Cmax, ss (µg/m) | 54,9 ± 15,9 | 58,1 ± 16,5 | 66,8 ± 17,7 |
Cavg, ss (µg/ml) | 53,5 ± 15,7 | 53,5 ± 15,7 | 53,5 ± 15,7 |
Ctrough, ss (µg/ml) | 51,1 ± 15,3 | 46,7 ± 16,9 | 38,3 ± 14,3 |
Cmax/Ctrough ratio | 1,08 ± 0,03 | 1,26 ± 0,12 | 1,85 ± 0,46 |
Cavg, ss = keskimääräinen pitoisuus vakaassa tilassa; Cmax, ss = suurin pitoisuus plasmassa vakaassa tilassa; Ctrough, ss = jäännöspitoisuus vakaassa tilassa. Farmakokineettiset parametrit saatu populaatiofarmakokineettisestä mallista. | |||
Aikuisten/nuorten (≥ 12-vuotiaiden) ja lasten (< 12-vuotiaiden) farmakokineettisten profiilien havaittiin olevan samankaltaiset annostuksen ollessa kerran viikossa (3 mg/kg/viikko 4 viikon ajan, minkä jälkeen 1,5 mg/kg/viikko) (ks. kuva 1).
Kuva 1. Plasman emisitsumabipitoisuuden keskiarvon (± 95 %:n luottamusväli) vertailu aikaprofiilien suhteen iältään ≥ 12-vuotiaiden (tutkimukset HAVEN 1 ja HAVEN 3) ja < 12‑vuotiaiden (tutkimus HAVEN 2) potilaiden välillä
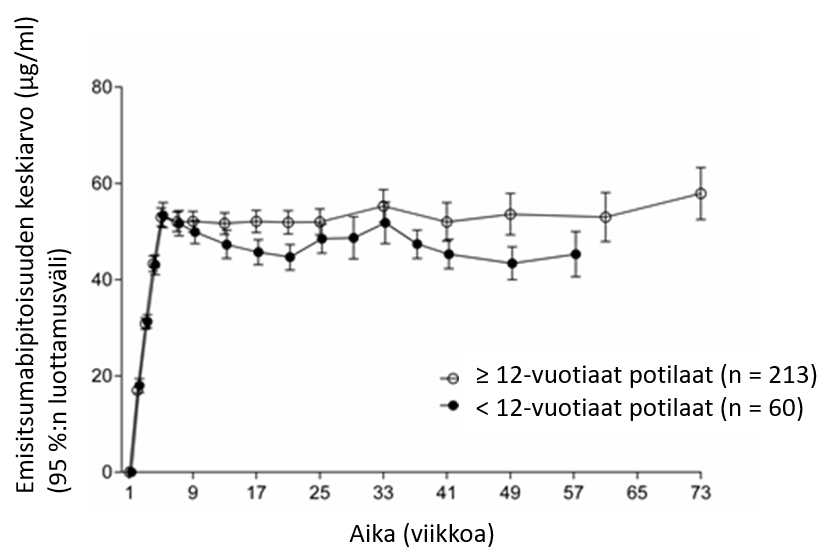
Absoluuttinen biologinen hyötyosuus oli terveille tutkittaville ihon alle annettujen annosten 1 mg/kg antamisen jälkeen 80,4–93,1 % injektiokohdasta riippuen. Farmakokineettisten profiilien havaittiin olevan samankaltaiset, jos valmiste annettiin ihon alle vatsaan, olkavarteen tai reiteen. Emisitsumabin antopaikkaa voidaan vaihdella näiden anatomisten kohtien välillä (ks. kohta Annostus ja antotapa).
Jakautuminen
Terveille tutkittaville laskimoon annetun emisitsumabikerta-annoksen 0,25 mg/kg jälkeen vakaan tilan jakautumistilavuus oli 106 ml/kg (eli 70 kg:n painoisella aikuisella 7,4 l).
Populaatiofarmakokineettisessä analyysissä arvioitu A-hemofiliapotilaan näennäinen jakautumistilavuus (V/F) useiden ihon alle annettujen emisitsumabiannosten jälkeen oli 10,4 l.
Metabolia
Emisitsumabin metaboliaa ei ole tutkittu. IgG-vasta-aineet kataboloituvat pääasiassa lysosomaalisen proteolyysin kautta ja sen jälkeen eliminoituvat tai palautuvat takaisin elimistön käyttöön.
Eliminaatio
Terveille tutkittaville laskimoon annetun annoksen 0,25 mg/kg jälkeen emisitsumabin kokonaispuhdistuma oli 3,26 ml/kg/vrk (eli 70 kg:n painoisella aikuisella 0,228 l/vrk) ja terminaalisen puoliintumisajan keskiarvo oli 26,7 vuorokautta.
Terveille tutkittaville ihon alle annetun kerta-annoksen jälkeen eliminaation puoliintumisaika oli noin 4–5 viikkoa.
A-hemofiliapotilaille ihon alle annettujen useiden injektioiden jälkeen näennäinen puhdistuma oli 0,272 l/vrk ja eliminaation näennäinen puoliintumisaika oli 26,8 vuorokautta.
Annoslineaarisuus
Emisitsumabin farmakokinetiikka oli A-hemofiliapotilailla ensimmäisen Hemlibra-annoksen jälkeen suhteessa annokseen annosvälillä 0,3−6 mg/kg. Altistus (Cavg, ss) useiden annosten jälkeen on verrannollinen annoksilla 1,5 mg/kg kerran viikossa, 3 mg/kg joka toinen viikko ja 6 mg/kg joka neljäs viikko.
Erityiset potilasryhmät
Pediatriset potilaat
Iän vaikutusta emisitsumabin farmakokinetiikkaan tutkittiin populaatiofarmakokineettisessä analyysissä, johon otettiin mukaan 5 imeväisikäistä (ikä ≥ 1 kuukaudesta < 2 vuoteen), 55 lasta (< 12‑vuotiasta) ja 50 nuorta (12 – < 18-vuotiasta), jotka sairastivat A-hemofiliaa. Ikä ei vaikuttanut pediatrisilla potilailla emisitsumabin farmakokinetiikkaan.
Iäkkäät
Iän vaikutusta emisitsumabin farmakokinetiikkaan tutkittiin populaatiofarmakokineettisessä analyysissä, johon otettiin mukaan kolmetoista iältään 65-vuotiasta tai vanhempaa tutkittavaa (yksikään tutkittava ei ollut yli 77-vuotias). Suhteellinen biologinen hyötyosuus pieneni iän lisääntyessä, mutta emisitsumabin farmakokinetiikassa ei havaittu kliinisesti tärkeitä eroja < 65-vuotiaiden ja ≥ 65-vuotiaiden tutkittavien välillä.
Etninen ryhmä
A-hemofiliapotilaiden populaatiofarmakokineettinen analyysi osoitti, että etninen tausta ei vaikuttanut emisitsumabin farmakokinetiikkaan. Tämä demografinen tekijä ei edellytä annoksen muuttamista.
Sukupuoli
Tiedot naispotilaista ovat liian suppeat johtopäätöksen tekemiseen.
Munuaisten vajaatoiminta
Munuaisten vajaatoiminnan vaikutusta emisitsumabin farmakokinetiikkaan ei ole tutkittu varsinaisissa tutkimuksissa.
Valtaosalla populaatiofarmakokineettisessä analyysissa mukana olleista A-hemofiliapotilaista munuaisten toiminta oli normaali (N = 332; kreatiniinipuhdistuma [CLcr] ≥ 90 ml/min) tai heillä oli lievää munuaisten vajaatoimintaa (N = 27; CLcr 60–89 ml/min). Lievä munuaisten vajaatoiminta ei vaikuttanut emisitsumabin farmakokinetiikkaan. Hemlibran käytöstä potilaille, joilla on keskivaikeaa munuaisten vajaatoimintaa, on vähän tietoja saatavissa (vain kahden potilaan CLcr oli 30–59 ml/min), ja Hemlibran käytöstä vaikeaa munuaisten vajaatoimintaa sairastaville potilaille ei ole tietoja saatavilla. Keskivaikean ja vaikean munuaisten vajaatoiminnnan vaikutusta emisitsumabin farmakokinetiikkaan ei voida päätellä.
Emisitsumabi on monoklonaalinen vasta-aine, ja se poistuu elimistöstä kataboloitumalla eikä erittymällä munuaisten kautta, joten annosmuutokset eivät munuaisten vajaatoimintaa sairastavilla potilailla oletettavasti ole tarpeen.
Maksan vajaatoiminta
Maksan vajaatoiminnan vaikutusta emisitsumabin farmakokinetiikkaan ei ole tutkittu varsinaisissa tutkimuksissa. Valtaosalla populaatiofarmakokineettisessä analyysissä mukana olleilla A‑hemofiliapotilailla oli normaali maksan toiminta (bilirubiini ja ASAT ≤ ULN, N = 300) tai lievä maksan vajaatoiminta (bilirubiini ≤ ULN ja ASAT > ULN tai bilirubiini 1,0–1,5 × ULN ja ASAT mikä tahansa, N = 51). Vain kuudella potilaalla oli keskivaikeaa maksan vajaatoimintaa (1,5 × ULN, bilirubiini ≤ 3 × ULN ja ASAT mikä tahansa). Lievä maksan vajaatoiminta ei vaikuttanut emisitsumabin farmakokinetiikkaan (ks. kohta Annostus ja antotapa). Emisitsumabin turvallisuutta ja tehoa maksan vajaatoimintaa sairastavilla potilailla ei ole erityisesti tutkittu. Kliinisissä tutkimuksissa oli mukana lievää ja keskivaikeaa maksan vajaatoimintaa sairastavia potilaita. Tietoja Hemlibran käytöstä vaikeaa maksan vajaatoimintaa sairastaville potilaille ei ole saatavilla.
Emisitsumabi on monoklonaalinen vasta-aine, ja se poistuu elimistöstä kataboloitumalla eikä metaboloitumalla maksassa, joten annosmuutokset eivät maksan vajaatoimintaa sairastavilla potilailla oletettavasti ole tarpeen.
Muut erityiset potilasryhmät
Mallinnus osoittaa, että antovälin pidentäminen potilaille, joilla on hypoalbuminemia ja ikään nähden pieni kehonpaino, vähentää altistusta emisitsumabille. Simulaatiot osoittavat, että nämä potilaat hyötyisivät silti kliinisesti merkityksellisestä vuotokontrollista. Kliinisiin tutkimuksiin ei otettu mukaan yhtään tällaista potilasta.
Prekliiniset tiedot turvallisuudesta
Akuuttia ja toistuvan altistuksen aiheuttamaa toksisuutta, mukaan lukien farmakologisen turvallisuuden päätetapahtumia ja lisääntymistoksisuuden päätetapahtumia koskevien tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Hedelmällisyys
Emisitsumabi ei aiheuttanut muutoksia cynomolgus-apinaurosten tai ‑naaraiden lisääntymiselimiin, kun suurin tutkittu annos oli 30 mg/kg/viikko (vastaa 11 kertaa suurinta ihmiselle tarkoitettua annosta 3 mg/kg/viikko, AUC-arvon perusteella).
Teratogeenisuus
Emisitsumabin mahdollisista haittavaikutuksista alkion ja sikiön kehitykseen ei ole tietoja saatavissa.
Pistoskohdan reaktiot
Eläimillä havaittiin korjautuvaa verenvuotoa, perivaskulaarista mononukleaarisolujen infiltraatiota, ihonalaiskudoksen degeneraatiota/nekroosia sekä ihonalaiskudoksen endoteelin turvotusta ihon alle annetun injektion jälkeen.
Farmaseuttiset tiedot
Apuaineet
L-arginiini
L-histidiini
L-asparagiinihappo
Poloksameeri 188
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Hemlibran ja polypropeeni- tai polykarbonaattiruiskujen ja ruostumattomasta teräksestä valmistettujen neulojen välillä ei ole havaittu yhteensopimattomuutta.
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo
Hemlibra 30 mg/ml injektioneste, liuos
2 vuotta.
Hemlibra 150 mg/ml injektioneste, liuos
2 vuotta.
Kun valmiste on otettu jääkaapista, avaamatonta injektiopulloa voidaan säilyttää huoneenlämmössä (alle 30 °C) enintään 7 vuorokautta.
Kun valmistetta on säilytetty huoneenlämmössä, avaamaton injektiopullo voidaan laittaa takaisin jääkaappiin. Jos valmistetta on säilytetty jääkaapin ulkopuolella ja se laitetaan takaisin jääkaappiin, yhteenlaskettu säilytysaika muussa kuin jääkaappilämpötilassa ei saa ylittää 7:ää vuorokautta. Injektiopulloja ei saa milloinkaan altistaa yli 30 °C:n lämpötilalle. Injektiopullot, joita on säilytetty huoneenlämmössä yli 7 vuorokautta tai altistettu yli 30 °C:n lämpötilalle, on hävitettävä.
Neulalla puhkaistu injektiopullo ja täytetty ruisku
Kun lääkevalmiste on siirretty injektiopullosta ruiskuun, se on mikrobiologiselta kannalta käytettävä heti. Jos lääkevalmistetta ei käytetä heti, käytönaikaiset säilytysajat ja -olosuhteet ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2 °C – 8 ºC).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Avatun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
HEMLIBRA injektioneste, liuos
30 mg/ml (L:ei) 1 ml (2091,31 €)
150 mg/ml (L:ei) 0,4 ml (4002,00 €), 0,7 ml (6868,04 €), 1 ml (9734,09 €), 2 ml (19287,59 €)
PF-selosteen tieto
Hemlibra 30 mg/ml injektioneste, liuos
Kirkkaasta tyypin I lasista valmistettu 3 ml:n injektiopullo, jossa fluorihartsikalvolla laminoitu butyylikumitulppa ja alumiinisella puristekorkilla sinetöity harmaa muovinen irti napsautettava levy. Yksi injektiopullo sisältää 12 mg emisitsumabia 0,4 ml:ssa injektionestettä. Yksi pakkaus sisältää yhden injektiopullon.
Kirkkaasta tyypin I lasista valmistettu 3 ml:n injektiopullo, jossa fluorihartsikalvolla laminoitu butyylikumitulppa ja alumiinisella puristekorkilla sinetöity taivaansininen muovinen irti napsautettava levy. Yksi injektiopullo sisältää 30 mg emisitsumabia 1 ml:ssa injektionestettä. Yksi pakkaus sisältää yhden injektiopullon.
Hemlibra 150 mg/ml injektioneste, liuos
Kirkkaasta tyypin I lasista valmistettu 3 ml:n injektiopullo, jossa fluorihartsikalvolla laminoitu butyylikumitulppa ja alumiinisella puristekorkilla sinetöity violetti muovinen irti napsautettava levy. Yksi injektiopullo sisältää 60 mg emisitsumabia 0,4 ml:ssa injektionestettä. Yksi pakkaus sisältää yhden injektiopullon.
Kirkkaasta tyypin I lasista valmistettu 3 ml:n injektiopullo, jossa fluorihartsikalvolla laminoitu butyylikumitulppa ja alumiinisella puristekorkilla sinetöity turkoosi muovinen irti napsautettava levy. Yksi injektiopullo sisältää 105 mg emisitsumabia 0,7 ml:ssa injektionestettä. Yksi pakkaus sisältää yhden injektiopullon.
Kirkkaasta tyypin I lasista valmistettu 3 ml:n injektiopullo, jossa fluorihartsikalvolla laminoitu butyylikumitulppa ja alumiinisella puristekorkilla sinetöity ruskea muovinen irti napsautettava levy. Yksi injektiopullo sisältää 150 mg emisitsumabia 1 ml:ssa injektionestettä. Yksi pakkaus sisältää yhden injektiopullon.
Kirkkaasta tyypin I lasista valmistettu 3 ml:n injektiopullo, jossa fluorihartsikalvolla laminoitu butyylikumitulppa ja alumiinisella puristekorkilla sinetöity keltainen muovinen irti napsautettava levy. Yksi injektiopullo sisältää 300 mg emisitsumabia 2 ml:ssa injektionestettä. Yksi pakkaus sisältää yhden injektiopullon.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Väritön tai kellertävä liuos.
Käyttö- ja käsittelyohjeet
Hemlibra-liuos on steriili, säilytysaineeton ja käyttövalmis liuos annettavaksi laimentamattomana ihon alle.
Hemlibra pitää tarkistaa ennen antoa silmämääräisesti sen varmistamiseksi, ettei liuoksessa ole hiukkasia eikä värimuutoksia. Hemlibra on väritön tai hieman keltainen liuos. Hemlibra-liuos pitää hävittää, jos siinä on näkyvissä hiukkasia tai jos liuos on värjäytynyttä.
Ei saa ravistaa.
Hemlibra-liuoksen sisältävät injektiopullot ovat yhtä käyttökertaa varten.
Hemlibra-liuoksen vetämiseen injektiopullosta ja injisointiin ihon alle tarvitaan ruisku, suodattimellinen siirtoneula tai suodattimellinen injektiopullon sovitin sekä injektioneula.
Ks. käyttöön liittyvät suositukset jäljempänä:
Hemlibra-liuoksen enintään 1 ml:n määrän injisointiin pitää käyttää 1 ml:n ruiskua, kun taas vähintään 1 ml:n ja enintään 2 ml:n määrien injisointiin pitää käyttää 2–3 ml:n ruiskua.
Ks. injektiopullojen yhdistämistä ruiskussa koskevat käsittelyohjeet kohdasta ”Käyttöohjeet”. Hemlibraa eri pitoisuuksina sisältävien injektiopullojen (30 mg/ml ja 150 mg/ml) sisältöä ei saa yhdistää samaan ruiskuun määrätyn annoksen antamiseksi.
1 ml:n ruisku
Valintaperusteet: Läpinäkyvä polypropeeni- tai polykarbonaattiruisku, jossa Luer‑lock-kärki, mitta-asteikko 0,01 ml:n välein.
2–3 ml:n ruisku
Valintaperusteet: Läpinäkyvä polypropeeni- tai polykarbonaattiruisku, jossa Luer‑lock-kärki, mitta-asteikko 0,1 ml:n välein.
Suodattimellinen siirtoneula
Suodattimellisen siirtoneulan valintaperusteet: Ruostumattomasta teräksestä valmistettu neula, jossa Luer‑lock-liitin, paksuus 18 G, pituus 35 mm (1½″), 5 µm suodatin ja mieluiten puolitylppä kärki.
Suodattimellinen injektiopullon sovitin
Suodattimellisen injektiopullon sovittimen valintaperusteet: Polypropeenista valmistettu sovitin, jossa Luer‑lock-liitin ja 5 µm suodatin (sopii injektiopullon kaulaan, jonka ulkoläpimitta on 15 mm).
Injektioneula
Valintaperusteet: Ruostumattomasta teräksestä valmistettu neula, jossa Luer‑lock-liitin, paksuus 26 G (hyväksyttävät paksuudet: 25–27 G), pituus mieluiten 9 mm (3/8″) tai enintään 13 mm (½″), mieluiten varustettu turvamekanismilla.
Ks. valmisteen antoa koskevia lisätietoja kohdasta Annostus ja antotapa ja pakkausselosteesta (kohta 7 Käyttöohjeet).
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
HEMLIBRA injektioneste, liuos
30 mg/ml 1 ml
150 mg/ml 0,4 ml, 0,7 ml, 1 ml, 2 ml
- Ylempi erityiskorvaus (100 %). Emisitsumabi (A-hemofilia, vasta-aineita): A-hemofiliaa sairastavien potilaiden verenvuotoja ennaltaehkäisevä hoito erityisin edellytyksin (1503), Emisitsumabi (A-hemofilia, ei vasta-aineita): Vaikea-asteista A-hemofiliaa sairastavien potilaiden verenvuotoja ennaltaehkäisevä hoito erityisin edellytyksin (1520).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Emisitsumabi (A-hemofilia, vasta-aineita): A-hemofiliaa sairastavien potilaiden verenvuotoja ennaltaehkäisevä hoito erityisin edellytyksin (397), Emisitsumabi (A-hemofilia, ei vasta-aineita): Vaikea-asteista A-hemofiliaa sairastavien potilaiden verenvuotoja ennaltaehkäisevä hoito erityisin edellytyksin (3047).
ATC-koodi
B02BX06
Valmisteyhteenvedon muuttamispäivämäärä
27.03.2025
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com