
LOKELMA jauhe oraalisuspensiota varten 5 g, 10 g
Vaikuttavat aineet ja niiden määrät
Lokelma 5 g jauhe oraalisuspensiota varten
Yksi annospussi sisältää 5 g natriumsirkoniumsyklosilikaattia
Yksi 5 g:n annospussi sisältää noin 400 mg natriumia.
Lokelma 10 g jauhe oraalisuspensiota varten
Yksi annospussi sisältää 10 g natriumsirkoniumsyklosilikaattia
Yksi 10 g:n annospussi sisältää noin 800 mg natriumia.
Lääkemuoto
Jauhe oraalisuspensiota varten
Kliiniset tiedot
Käyttöaiheet
Lokelma on tarkoitettu aikuisten hyperkalemian hoitoon (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Annostus ja antotapa
Annostus
Korjausvaihe
Suositeltu Lokelma-valmisteen aloitusannos on 10 g, joka annetaan suspensiona vedessä kolme kertaa vuorokaudessa suun kautta. Kun veren normaali kaliumpitoisuus on saavutettu, noudatetaan ylläpitoannostusta (ks. alla).
Tavallisesti normaali kaliumpitoisuus saavutetaan 24–48 tunnin kuluessa. Jos 48 tunnin kuluttua potilaalla todetaan edelleen hyperkalemiaa, samaa annostusta voidaan jatkaa vielä 24 tunnin ajan. Jos veren normaalia kaliumpitoisuutta ei ole saavutettu, kun hoitoa on jatkettu 72 tuntia, on harkittava vaihtoehtoisia hoitomenetelmiä.
Ylläpitovaihe
Kun veren normaali kaliumpitoisuus on saavutettu, on pyrittävä löytämään pienin mahdollinen tehokas annos Lokelma-valmistetta hyperkalemian uusiutumisen ehkäisemiseen. Suositeltu aloitusannos on 5 g kerran vuorokaudessa. Annosta voidaan tarvittaessa suurentaa jopa 10 g:aan kerran vuorokaudessa tai pienentää joka toinen päivä annettavaan 5 g:aan normaalin kaliumpitoisuuden ylläpitämiseksi. Ylläpitohoidossa ei saa käyttää 10 g vuorokaudessa ylittävää annostusta.
Seerumin kaliumpitoisuutta on seurattava säännöllisesti hoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Unohtunut annos
Jos potilas unohtaa ottaa annoksen, häntä on neuvottava ottamaan seuraava annos tavanomaiseen aikaan.
Erityisryhmät
Potilaat, joilla on munuaisten vajaatoiminta
Normaalia annostusta ei tarvitse muuttaa potilailla, joilla on munuaisten vajaatoiminta ja jotka eivät saa jatkuvaa hemodialyysihoitoa.
Lokelma-valmistetta annetaan dialyysipotilaille vain dialyysihoidon välipäivinä. Suositeltu aloitusannos on 5 g kerran vuorokaudessa. Veren normaalin kaliumpitoisuuden (4,0–5,0 mmol/l) saavuttamiseksi annosta voidaan suurentaa tai pienentää viikoittain siten, että annoksen muuttaminen perustuu pidemmän dialyysivälin jälkeen ennen dialyysia mitattuun seerumin kaliumpitoisuuteen. Annosta voidaan muuttaa yhden viikon välein 5 g:n lisäyksin niin, että annos on enintään 15 g kerran vuorokaudessa dialyysihoidon välipäivinä. Annoksen säätämisen aikana suositellaan seerumin kaliumpitoisuuden mittaamista kerran viikossa. Kun veren normaali kaliumpitoisuus on saavutettu, kaliumpitoisuus on tutkittava säännöllisesti (esimerkiksi kerran kuukaudessa tai tiheämmin kliinisen arvioinnin perusteella, jos esimerkiksi ruokavalion sisältämän kaliumin määrä muuttuu tai potilas käyttää seerumin kaliumpitoisuuteen vaikuttavaa lääkettä).
Potilaat, joilla on maksan vajaatoimintaa
Normaalia annostusta ei tarvitse muuttaa potilailla, joilla on maksan vajaatoiminta.
Iäkkäät henkilöt
Tälle potilasryhmälle ei ole erityisiä annossuosituksia eikä annosteluohjeita.
Pediatriset potilaat
Lokelma-valmisteen turvallisuutta ja tehoa lasten ja alle 18-vuotiaiden nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Suun kautta.
Annospussi(e)n koko sisältö tyhjennetään juomalasiin, jossa on noin 45 ml vettä, ja sekoitetaan hyvin. Mauton neste on juotava sen ollessa vielä sameaa. Jauhe ei liukene. Jos jauhe laskeutuu lasin pohjalle, neste on sekoitettava uudelleen ja juotava sen jälkeen. Tarvittaessa koko annoksen ottaminen varmistetaan huuhtelemalla lasi vedellä.
Suspensio voidaan ottaa aterian yhteydessä tai tyhjään mahaan.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle.
Varoitukset ja käyttöön liittyvät varotoimet
Seerumin kaliumpitoisuus
Seerumin kaliumpitoisuutta on seurattava, jos se on kliinisesti aiheellista, kuten potilaan lääkitykseen tehtyjen seerumin kaliumpitoisuuteen vaikuttavien muutosten jälkeen (koskee esimerkiksi reniini-angiotensiini-aldosteronijärjestelmän estäjiä ja diureetteja) tai Lokelma-valmisteen annoksen säätämisen jälkeen.
Seurantatiheys riippuu useista eri tekijöistä, kuten potilaan muusta lääkityksestä, kroonisen munuaissairauden etenemisestä ja ravinnon kautta saadun kaliumin määrästä.
Hypokalemia
Hypokalemiaa saatetaan todeta (ks. kohta Haittavaikutukset). Näissä tapauksissa annoksen säätäminen ylläpitoannostuksen yhteydessä kuvatulla tavalla saattaa olla tarpeen kohtalaisen tai vaikean hypokalemian ehkäisemiseksi. Jos potilaalla todetaan vaikea hypokalemia, Lokelma-valmisteen käyttö on keskeytettävä ja potilaan tilanne arvioitava uudelleen.
Aiemmin todetun sydämen vajaatoiminnan paheneminen
Potilaita, joilla on aiemmin todettu sydämen vajaatoiminta, on tarkkailtava sydämen vajaatoiminnan pahenemiseen viittaavien oireiden varalta. Tämä koskee etenkin potilaita, joilla lisääntynyt natriumin saanti saattaa johtaa nesteylikuormitukseen ja dekompensaatioon. Sydämen vajaatoiminnan pahenemisen oireita voivat olla lisääntynyt hengenahdistus, edeema ja nopea painonnousu, ja niitä on hoidettava tavanomaisen kliinisen käytännön mukaisesti (ks. kohta Haittavaikutukset).
QT-ajan pidentyminen
Hyperkalemian korjauksen aikana voidaan havaita QT-ajan pitenemistä, mikä on seerumin pienentyneen kaliumpitoisuuden fysiologinen seuraus.
Yhteisvaikutuksen riski röntgenkuvauksen yhteydessä
Natriumsirkoniumsyklosilikaatti saattaa näkyä samentumana röntgenkuvassa. Röntgenhoitajien on pidettävä tämä mielessä, jos potilaalle suunnitellaan vatsan alueen röntgentutkimusta.
Suolen puhkeaminen
Suolen puhkeamisen riskiä Lokelma-valmisteen käytön yhteydessä ei tällä hetkellä tunneta. Koska kaliumia sitovien aineiden, kuten Lokelma-valmisteen, käytön yhteydessä on raportoitu suolen puhkeamia, on kiinnitettävä erityistä huomioita merkkeihin ja oireisiin, jotka liittyvät suolen puhkeamiseen.
Natriumpitoisuus
Tämä lääkevalmiste sisältää noin 400 mg natriumia per 5 g:n annos, mikä vastaa 20 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Lokelma-valmisteen sisältämä natriummäärä katsotaan suureksi. Tämä on erityisesti otettava huomioon potilailla, jotka noudattavat vähäsuolaista ruokavaliota.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset natriumsirkoniumsyklosilikaattiin
Koska natriumsirkoniumsyklosilikaatti ei imeydy tai metaboloidu elimistössä, muilla lääkevalmisteilla ei odoteta olevan vaikutusta natriumsirkoniumsyklosilikaatin farmakologiseen toimintaan.
Natriumsirkoniumsyklosilikaatin vaikutukset muihin lääkevalmisteisiin
Koska natriumsirkoniumsyklosilikaatti ei imeydy tai metaboloidu elimistössä eikä se merkitsevästi sido muita lääkevalmisteita, sen vaikutus muihin lääkevalmisteisiin on vähäistä. Natriumsirkoniumsyklosilikaatti voi tilapäisesti suurentaa mahanesteen pH-arvoa absorboimalla vetyioneja. Tämä voi muuttaa samanaikaisesti annettujen lääkevalmisteiden liukoisuuteen ja imeytymiseen liittyvää kinetiikkaa, jos niiden hyötyosuus on riippuvainen ympäristön pH-arvosta. Terveillä tutkittavilla tehdyissä kliinisissä yhteisvaikutustutkimuksissa natriumsirkoniumsyklosilikaatin samanaikainen anto amlodipiinin, klopidogreelin, atorvastatiinin, furosemidin, glipitsidin, varfariinin, losartaanin tai levotyroksiinin kanssa ei aiheuttanut kliinisesti merkittäviä yhteisvaikutuksia. Samalla tavoin kuin annettaessa dabigatraania samanaikaisesti muiden mahahapon eritykseen vaikuttavien lääkeaineiden kanssa, dabigatraanin Cmax- ja AUC-arvot olivat noin 40 % pienempiä myös silloin, kun sitä annettiin samanaikaisesti natriumsirkoniumsyklosilikaatin kanssa.
Annostuksen muuttamista tai antamista eri aikoina ei edellytä millekään näistä lääkevalmisteista.
Natriumsirkoniumsyklosilikaatti on kuitenkin annettava vähintään 2 tuntia ennen suun kautta annettavia lääkevalmisteita, joiden hyötyosuus riippuu kliinisesti merkitsevästi mahanesteen pH-arvosta, tai vähintään 2 tuntia niiden jälkeen.
Esimerkkejä lääkevalmisteista, jotka kohonneeseen mahanesteen pH-arvoon liittyvien mahdollisten yhteisvaikutusten välttämiseksi on annettava 2 tuntia ennen natriumsirkoniumsyklosilikaattia tai 2 tuntia sen jälkeen, ovat atsoliryhmän sienilääkkeet (ketokonatsoli, itrakonatsoli ja posakonatsoli), HIV-lääkeaineet (atatsanaviiri, nelfinaviiri, indinaviiri, ritonaviiri, sakinaviiri, raltegraviiri, ledipasviiri ja rilpiviriini) ja tyrosiinikinaasin estäjät (erlotinibi, dasatinibi ja nilotinibi).
Natriumsirkoniumsyklosilikaattia voidaan käyttää samanaikaisesti ja se voidaan ottaa yhtä aikaa suun kautta annettavien lääkevalmisteiden kanssa, joiden hyötyosuus ei riipu pH-arvosta.
Eräässä toisessa terveillä tutkittavilla tehdyssä yhteisvaikutustutkimuksessa Lokelma-valmisteen 15 g:n annoksen samanaikainen anto takrolimuusin 5 mg:n annoksen kanssa pienensi takrolimuusin AUC-arvoa 37 % ja Cmax-arvoa 29 %. Näin ollen takrolimuusi on otettava vähintään 2 tuntia ennen Lokelma-valmistetta tai vähintään 2 tuntia sen jälkeen. Samassa tutkimuksessa Lokelma-valmisteen ja siklosporiinin samanaikaisella annolla ei todettu kliinisesti merkittäviä yhteisvaikutuksia.
Raskaus ja imetys
Raskaus
Natriumsirkoniumsyklosilikaatin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi Lokelma-valmisteen käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Synnyttäneillä rotilla tehdyissä kokeissa naaraiden altistus natriumsirkoniumsyklosilikaatille ei vaikuttanut syntyneiden poikasten kehitykseen. Fysikokemiallisten ominaisuuksiensa vuoksi natriumsirkoniumsyklosilikaatti ei imeydy systeemisesti eikä sen odoteta erittyvän ihmisillä äidinmaitoon. Vaikutuksia imetettäviin vauvoihin ei ole odotettavissa, sillä imettävän naisen systeeminen altistus natriumsirkoniumsyklosilikaatille on erittäin pieni. Lokelma-valmistetta voidaan käyttää imetyksen aikana.
Hedelmällisyys
Natriumsirkoniumsyklosilikaatin vaikutuksista ihmisen hedelmällisyyteen ei ole tietoja. Rotilla natriumsirkoniumsyklosilikaattihoito ei vaikuttanut hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Lokelma-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitetut haittavaikutukset olivat hypokalemia (4,1 %) ja edeemaan liittyvät tapahtumat (5,7 %).
Kahdessa avoimessa kliinisessä tutkimuksessa, joissa 874 tutkittavaa sai Lokelma-valmistetta enintään 1 vuoden ajan, raportoitiin seuraavia tutkijoiden mukaan Lokelma-valmisteeseen liittyviä tapahtumia: maha-suolikanavaan liittyvät tapahtumat [ummetus (2,9 %), pahoinvointi (1,6 %), ripuli (0,9 %), vatsakipu tai vatsan turvotus (0,5 %) ja oksentelu (0,5 %)] ja yliherkkyysreaktiot [ihottuma (0,3 %) ja kutina (0,1 %)]. Nämä tapahtumat olivat luonteeltaan lieviä tai kohtalaisia eikä minkään niistä raportoitu olleen vakava, ja yleensä ne hävisivät potilaan jatkaessa hoitoa. Avoimen tutkimusasetelman vuoksi syy-yhteyttä näiden tapahtumien ja Lokelma-valmisteen käytön välillä ei voida täysin varmistaa.
Kliinisissä tutkimuksissa maissa, joiden väestö on pääasiassa aasialaista, ilmeni ummetusta 8,9 %:n arvioidulla esiintymistiheydellä sellaisilla Lokelma-valmistetta saaneilla potilailla, jotka eivät saaneet dialyysihoitoa. Tämä haittavaikutus hävisi, kun annosta muutettiin tai hoito keskeytettiin.
Kolmesta Lokelma-valmistetta koskeneesta lumekontrolloidusta kliinisestä tutkimuksesta, joihin osallistuneet potilaat eivät saaneet dialyysihoitoa, tehtiin yhdistetty analyysi. Analyysin mukaan joillakin potilailla, joilla oli aiemmin todettu sydämen vajaatoiminta, todettiin sydämen vajaatoiminnan paheneminen. Tätä ilmeni hoidon aikana 13,6 %:n esiintymistiheydellä (30/220) Lokelma-valmistetta saaneilla ja 5,7 %:n esiintymistiheydellä (12/209) lumelääkettä saaneilla potilailla. Useimmissa tapauksissa tämä haittavaikutus hävisi asianmukaisella kliinisellä hoidolla ilman Lokelma-hoidon lopettamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukoitu luettelo haittavaikutuksista
Lokelma-valmisteen turvallisuusprofiilia arvioitiin kliinisissä tutkimuksissa, joihin osallistui 1 760 potilasta. Näistä potilaista 507:n altistusaika oli yksi vuosi.
Kliinisissä tutkimuksissa havaitut ja valmisteen markkinoilletulon jälkeen ilmoitetut haittavaikutukset on esitetty taulukossa 1. Tässä luetellut haittavaikutukset on luokiteltu esiintymistiheyden ja elinjärjestelmän mukaan. Haittavaikutukset on luokiteltu esiintymistiheyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1.Luettelo kliinisissä tutkimuksissa todetuista ja markkinoilletulon jälkeen ilmoitetuista haittavaikutuksista
| Elinjärjestelmä | Hyvin yleinen | Yleinen |
Aineenvaihdunta ja ravitsemus Ruoansulatuselimistö | Hypokalemia Ummetus | |
| Yleisoireet ja antopaikassa todettavat haitat | Edeemaan liittyvät tapahtumat | |
| Sydän | Aiemmin todetun sydämen vajaatoiminnan paheneminen |
Valikoitujen haittavaikutusten kuvaus
Hypokalemia
Kliinisissä tutkimuksissa 4,1 %:lle Lokelma-valmistetta saaneista potilaista kehittyi hypokalemia, jossa seerumin kaliumpitoisuus oli alle 3,5 mmol/l. Tämä hoidettiin säätämällä Lokelma-valmisteen annosta tai keskeyttämällä Lokelma-hoito.
Edeemaan liittyvät tapahtumat
Edeemaan liittyviä tapahtumia, kuten nesteretentiota, yleistä turvotusta, hypervolemiaa, paikallista turvotusta, edeemaa, perifeeristä edeemaa ja ääreisosien turvotusta, raportoitiin 5,7 %:lla Lokelma-valmistetta saaneista potilaista. Näitä tapahtumia havaittiin ainoastaan ylläpitovaiheessa, ja ne olivat tavallisempia potilailla, joita hoidettiin 15 g:n annoksella. Näistä tapahtumista jopa 53 % hoidettiin aloittamalla nesteenpoistolääkitys tai säätämällä diureetin annostusta. Muiden potilaiden tila ei edellyttänyt hoitoa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‐haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Natriumsirkoniumsyklosilikaatin yliannostus voi johtaa hypokalemiaan. Seerumin kaliumpitoisuus on tarkistettava ja potilaalle on tarvittaessa annettava kaliumlisää.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut lääkevalmisteet, hyperkalemia- ja hyperfosfatemialääkkeet
ATC-koodi: V03AE10
Vaikutusmekanismi
Natriumsirkoniumsyklosilikaatti on imeytymätön, ei-polymeerinen epäorgaaninen jauhe, jolla on yhdenmukainen mikrohuokosrakenne, joka sitoo kaliumia vaihtamalla sitä vety- ja natriumkationeihin. In vitro tehdyissä tutkimuksissa natriumsirkoniumsyklosilikaatti oli erittäin selektiivinen kaliumionien suhteen, vaikka saatavilla olisi ollut muita kationeja, kuten kalsiumia ja magnesiumia. Natriumsirkoniumsyklosilikaatti sitoo kaliumia maha-suolikanavan kaikissa osissa ja pienentää vapaan kaliumin pitoisuutta maha-suolikanavassa, jolloin seerumin kaliumpitoisuus pienenee ja kaliumin erittyminen ulosteeseen lisääntyy, mikä lievittää hyperkalemiaa.
Farmakodynaamiset vaikutukset
Natriumsirkoniumsyklosilikaatti alkaa pienentää seerumin kaliumpitoisuutta jo 1 tunnin kuluttua lääkkeen ottamisesta, ja normaali kaliumpitoisuus saavutetaan yleensä 24–48 tunnin kuluessa. Natriumsirkoniumsyklosilikaatti ei vaikuta seerumin kalsium- tai magnesiumpitoisuuksiin tai natriumin poistumiseen virtsan mukana. Vaikutuksen suuruudella ja lähtötilanteen seerumin kaliumpitoisuuden välillä on selvä korrelaatio: mitä suurempi potilaan seerumin kaliumpitoisuus on lähtötilanteessa, sitä enemmän seerumin kaliumpitoisuus pienenee. Seerumin kaliumpitoisuuden pienenemisen seurauksena kaliumin erittyminen virtsaan vähenee. Tutkimuksessa, jossa terveille tutkittaville annettiin Lokelma-valmistetta 5 g tai 10 g kerran vuorokaudessa neljän vuorokauden ajan, seerumin kaliumpitoisuuden ja virtsaan erittyneen kaliumin kokonaismäärän annosriippuvaisen pienenemisen yhteydessä todettiin ulosteeseen erittyneen kaliumin määrän keskimäärin suurentuneen. Tilastollisesti merkitseviä muutoksia natriumin erittymisessä virtsaan ei havaittu.
Ei ole tehty tutkimuksia, joissa arvioitaisiin natriumsirkoniumsyklosilikaatin farmakodynamiikkaa, kun se otetaan aterian yhteydessä tai tyhjään mahaan.
Natriumsirkoniumsyklosilikaatin on myös osoitettu sitovan ammoniumia sekä in vitro että in vivo, jolloin ammoniumia poistuu ja seerumin bikarbonaattipitoisuus suurenee. Seerumin bikarbonaattipitoisuus suureni Lokelma-valmistetta 5 g kerran vuorokaudessa saaneilla potilailla 1,1 mmol/l, 10 g kerran vuorokaudessa saaneilla 2,3 mmol/l ja 15 g kerran vuorokaudessa saaneilla 2,6 mmol/l. Lumelääkettä saaneiden potilaiden ryhmässä seerumin bikarbonaattipitoisuus suureni keskimäärin 0,6 mmol/l. Ympäristössä, jossa muita reniiniin ja aldosteroniin vaikuttavia tekijöitä ei kontrolloitu, Lokelma muutti seerumin keskimääräistä aldosteronipitoisuutta (‑30 – ‑31 %) verrattuna lumelääkeryhmään (+14 %) käytetystä annoksesta riippumatta. Tutkimuksissa ei havaittu johdonmukaisia vaikutuksia systoliseen tai diastoliseen verenpaineeseen.
Lisäksi veren keskimääräisen ureatyppipitoisuuden havaittiin pienentyneen 5 g (1,1 mg/dl) ja 10 g (2,0 mg/dl) kolme kertaa vuorokaudessa saaneiden ryhmissä; pitoisuus sen sijaan suureni hieman sekä lumelääkettä (0,8 mg/dl) että natriumsirkoniumsyklosilikaattia pienellä annoksella (0,3 mg/dl) saaneiden ryhmissä.
Kliininen teho ja turvallisuus
Lokelma-valmisteen kaliumia vähentävä vaikutus on osoitettu kolmessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa hyperkalemiapotilailla. Kaikissa kolmessa tutkimuksessa arvioitiin Lokelma-hoidon aloitusvaiheen vaikutusta hyperkalemian korjaantumiseen ensimmäisten 48 tunnin aikana, ja kahdessa tutkimuksessa arvioitiin myös saavutetun normaalin kaliumpitoisuuden ylläpitoa. Ylläpitohoitoa koskeviin tutkimuksiin osallistui potilaita, joilla oli krooninen munuaissairaus (58 %), sydämen vajaatoiminta (10 %), diabetes (62 %) tai jotka saivat lääkehoitoa reniini-angiotensiini-aldosteronijärjestelmän estäjällä (68 %). Näiden lisäksi kahdessa avoimessa ylläpitohoitoa koskeneessa tutkimuksessa arvioitiin Lokelma-valmisteen pitkän aikavälin turvallisuutta. Näihin viiteen tutkimukseen osallistuneista Lokelma-valmistetta saaneista 1 760 potilaasta 507 sai lääkitystä vähintään 360 päivän ajan. Näiden lisäksi Lokelma-valmisteen tehoa ja turvallisuutta tutkittiin kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa 196:lla jatkuvaa hemodialyysihoitoa saavalla potilaalla, joilla oli hyperkalemia ja jotka saivat Lokelma-valmistetta 8 viikon ajan. Näissä tutkimuksissa Lokelma vähensi seerumin kaliumia ja ylläpiti seerumin normaalia kaliumtasoa riippumatta hyperkalemian aiheuttajasta, potilaan iästä, sukupuolesta, etnisestä taustasta, muista samanaikaisista sairauksista tai reniini-angiotensiini-aldosteronijärjestelmän estäjän samanaikaisesta käytöstä. Potilaiden ruokavaliota ei rajoitettu, vaan heitä kehotettiin noudattamaan tavanomaista ruokavaliotaan ilman erityisiä muutoksia.
Tutkimus 1
Kaksivaiheinen lumekontrolloitu korjaus- ja ylläpitohoitoa koskeva tutkimus
Kaksiosainen, kaksoissokkoutettu, satunnaistettu, lumekontrolloitu kliininen tutkimus, johon osallistui 753 potilasta (ikä keskimäärin 66 vuotta, vaihteluväli 22–93 vuotta), joilla oli hyperkalemia (5 – ≤ 6,5 mmol/l, lähtötason kaliumarvo keskimäärin 5,3 mmol/l). Tutkimukseen otettiin potilaita, joilla oli krooninen munuaissairaus, sydämen vajaatoiminta tai diabetes tai joita hoidettiin reniini-angiotensiini-aldosteronijärjestelmän estäjällä.
Korjausvaiheessa potilaat satunnaistettiin saamaan Lokelma-valmistetta (1,25 g, 2,5 g, 5 g tai 10 g) tai lumelääkettä, joita annettiin kolme kertaa vuorokaudessa ensimmäisten 48 tunnin ajan (taulukko 2).
Taulukko 2.Korjausvaihe (tutkimus 1): normokaleemisten tutkittavien prosentuaalinen määrä 48 tunnin Lokelma-hoidon jälkeen
| Lokelma-annostus 3 kertaa vuorokaudessa | |||||
| Lumelääke | 1,25 g | 2,5 g | 5 g | 10 g | |
| N | 158 | 154 | 141 | 157 | 143 |
| Seerumin kaliumpitoisuus lähtötilanteessa, mmol/l | 5,3 | 5,4 | 5,4 | 5,3 | 5,3 |
| Normokaleemisia 48 tunnin kuluttua, % | 48 | 51 | 68 | 78 | 86 |
| p‑arvo vs. lumelääke | NS | < 0,001 | < 0,001 | < 0,001 | |
NS: ei merkitsevä
Lokelma 10 g kolme kertaa vuorokaudessa oli pienentänyt seerumin kaliumpitoisuutta 0,7 mmol/l 48 tunnin kohdalla (p < 0,001 vs. lumelääke); 1 tunnin kuluttua ensimmäisen annoksen ottamisesta kaliumin todettiin vähentyneen tilastollisesti merkitsevästi 14 %. Potilailla, joiden kaliumarvot olivat lähtötilanteessa suuremmat, todettiin parempi hoitovaste Lokelma-hoitoon. Potilailla, joiden kaliumarvot olivat ennen hoidon aloittamista yli 5,5 mmol/l (lähtötason keskiarvo 5,8 mmol/l), kaliumpitoisuus oli 48 tunnin kohdalla pienentynyt keskimäärin 1,1 mmol/l. Vastaavasti potilailla, joiden kaliumarvot olivat enintään 5,3 mmol/l, kaliumpitoisuus oli suurimmalla annostuksella pienentynyt keskimäärin 0,6 mmol/l lähtötasoon verrattuna.
Potilaat, joilla veren kaliumpitoisuus normalisoitui Lokelma-valmisteen saamisen jälkeen korjausvaiheen aikana, satunnaistettiin uudelleen saamaan kerran vuorokaudessa joko lumelääkettä tai Lokelma-valmistetta samalla annoksella, jota he olivat saaneet kolme kertaa vuorokaudessa korjausvaiheen aikana (Taulukko 3).
Taulukko 3. Ylläpitovaihe (12 päivää, tutkimus 1): normokaleemisten päivien keskimääräinen lukumäärä
| Hoito ylläpitovaiheessa (kerran vuorokaudessa) | |||||
| Lumelääke | Lokelma | p‑arvo vs. lumelääke | |||
| Lokelma-annos korjausvaiheessa | n | Päiviä | n | Päiviä | |
| Tutkittavat, jotka saivat 1,25 g kolme kertaa vuorokaudessa | 41 | 7,6 | 49 | 7,2 | NS |
| Tutkittavat, jotka saivat 2,5 g kolmekertaa vuorokaudessa | 46 | 6,2 | 54 | 8,6 | 0,008 |
| Tutkittavat, jotka saivat 5 g kolme kertaa vuorokaudessa | 68 | 6,0 | 64 | 9,0 | 0,001 |
| Tutkittavat, jotka saivat 10 g kolme kertaa vuorokaudessa | 61 | 8,2 | 63 | 10,2 | 0,005 |
NS: ei merkitsevä
Lokelma-hoidon päätyttyä ylläpitovaiheen lopussa potilaiden keskimääräiset kaliumarvot nousivat lähelle lähtötilanteen arvoja.
Tutkimus 2
Monivaiheinen lumekontrolloitu ylläpitohoitoa koskeva tutkimus ja sen avoin vaihe
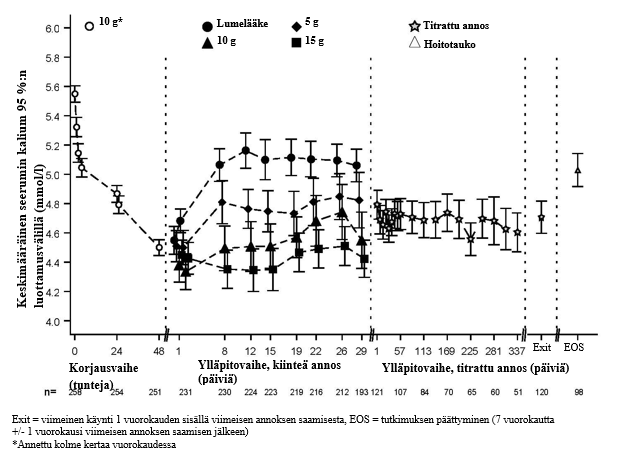
Tutkimuksen korjausvaiheessa 258 hyperkalemiapotilaalle (kaliumpitoisuus lähtötilanteessa keskimäärin 5,6, vaihteluväli 4,1–7,2 mmol/l) annettiin 10 g Lokelma-valmistetta kolme kertaa vuorokaudessa 48 tunnin ajan. Kaliumpitoisuuden havaittiin pienentyneen 1 tunnin kuluttua ensimmäisen 10 g:n Lokelma-annoksen ottamisen jälkeen. Mediaaniaika kaliumpitoisuuden normalisoitumiseen oli 2,2 tuntia. 66 % potilaista oli saavuttanut normokalemian 24 tunnin kohdalla ja 88 % potilaista 48 tunnin kohdalla. Vaikeammin hyperkaleemisilla potilailla oli parempi hoitovaste: potilailla, joiden kaliumarvo oli lähtötilanteessa < 5,5, seerumin kaliumpitoisuus pieneni 0,8 mmol/l, potilailla, joiden kaliumarvo oli lähtötilanteessa 5,5–5,9, se pieneni 1,2 mmol/l, ja potilailla, joiden kaliumarvo oli lähtötilanteessa ≥ 6 mmol/l, se pieneni 1,5 mmol/l.
Normokalemian saavuttaneet potilaat (kaliumpitoisuus 3,5–5 mmol/l) satunnaistettiin kaksoissokkoutetusti saamaan kerran vuorokaudessa jotakin kolmesta Lokelma-annoksesta [5 g (n = 45), 10 g (n = 51) tai 15 g (n = 56)] tai lumelääkettä (n = 85) 28 vuorokauden ajan (kaksoissokkoutettu satunnaistettu lääkehoidon lopettamisvaihe).
Niiden tutkittavien osuus, joilla tutkimuspäivinä 8–29 (kolmen viikon jaksolla) mitattu seerumin kaliumpitoisuus oli keskimäärin < 5,1 mmol/l, oli suurempi Lokelma-valmistetta kerran vuorokaudessa saaneilla (5 g saaneiden ryhmässä 80 %, 10 g saaneiden ryhmässä 90 % ja 15 g saaneiden ryhmässä 94 %) kuin lumelääkeryhmässä (46 %). Seerumin kaliumpitoisuus pieneni 5 g kerran vuorokaudessa Lokelma-valmistetta saaneilla keskimäärin 0,77 mmol/l, 71 % näistä potilaista pysyi normokaleemisina, 10 g kerran vuorokaudessa saaneiden seerumin kaliumpitoisuus pieneni keskimäärin 1,10 mmol/l ja 76 % pysyi normokaleemisina, 15 g kerran vuorokaudessa saaneilla seerumin kaliumpitoisuus pieneni keskimäärin 1,19 mmol/l ja 85 % pysyi normokaleemisina. Lumelääkeryhmässä vastaavat luvut olivat 0,44 mmol/l ja 48 %.
Tulokset ylläpitovaihetta arvioineesta (avoimesta) tutkimuksesta, jossa säädettiin Lokelma-annosta: avoimeen 11 kuukautta kestäneeseen vaiheeseen osallistui 123 potilasta. Potilaiden osuus, joiden seerumin keskimääräinen kaliumpitoisuus oli < 5,1 mmol/l, oli 88 % ja seerumin keskimääräinen kaliumpitoisuus oli 4,66 mmol/l. Alle 3,5 mmol/l olevia seerumin kaliumarvoja mitattiin alle 1 %:lla potilaista. 77 %:lla potilaista arvot olivat 3,5–5,1 mmol/l, ja 93 %:lla arvot olivat 3,5–5,5 mmol/l riippumatta muista mahdollisista seerumin kaliumiin vaikuttavista tekijöistä. Hoito lopetettiin tutkimuksen päättyessä (päivä 365).
Kaplan-Meierin menetelmällä arvioidut ajat relapsiin ylläpitovaiheessa osoittivat, että keskimääräinen aika relapsoitumiseen riippui Lokelma-valmisteen annostuksesta: 5 g:n annoksella ajan mediaani oli 4–21 vuorokautta, lähtötilanteen seerumin kaliumarvojen mukaan. Seerumin kaliumpitoisuutta on seurattava määräajoin ja Lokelma-valmisteen annosta säädettävä kohdan Annostus ja antotapa mukaisesti.
Kuvassa 1 esitetään keskimääräiset seerumin kaliumarvot tutkimuksen korjaus- ja ylläpitovaiheiden aikana.
Kuva 1. Korjaus- ja ylläpitovaiheet (tutkimus 2): seerumin keskimääräinen kaliumpitoisuus ajan funktiona 95 %:n luottamusvälillä
Tutkimus 3
Tutkimus hyperkalemiapotilailla, joilla oli krooninen munuaissairaus
Tämä tutkimus oli kaksoissokkoutettu lumekontrolloitu annoksen suurentamista koskeva tutkimus, johon osallistui 90 potilasta (60 potilasta Lokelma-ryhmässä ja 30 verrokkia). Tutkittavilla oli lähtötilanteessa glomerulusten laskennallinen suodatusnopeus 30–60 ml/min/1,73 m2 ja hyperkalemia (seerumin kaliumpitoisuus lähtötilanteessa 5,2 mmol/l, vaihteluväli 4,6–6,0 mmol/l). Potilaat satunnaistettiin saamaan Lokelma-valmistetta suurenevin annoksin (0,3 g, 3 g ja 10 g) tai lumelääkettä kolme kertaa vuorokaudessa aterian yhteydessä 2–4 vuorokauden ajan. Ensisijainen päätemuuttuja oli hoidon ensimmäisten kahden päivän aikana saavutettu seerumin kaliumpitoisuuden muutosnopeus lähtötasoon verrattuna. Tutkimuksessa saavutettiin tehon ensisijainen päätemuuttuja 3 g:n ja 10 g:n Lokelma-annoksilla lumelääkkeeseen verrattuna. Lokelma pienensi seerumin kaliumpitoisuutta 10 g:n annoksella keskimäärin enintään 0,92 mmol/l ja 3 g:n annoksella 0,43 mmol/l. Vuorokausivirtsan keräys osoitti, että Lokelma vähensi kaliumin erittymistä virtsaan lähtötasoon verrattuna 15,8 mmol / 24 tuntia, kun taas lumelääke lisäsi sitä 8,9 mmol / 24 tuntia (p < 0,001). Natriumin erittymisessä ei havaittu muutoksia lumelääkkeeseen verrattuna (10 g: lisäys 25,4 mmol / 24 tuntia, lumelääke: lisäys 36,9 mmol / 24 tuntia (ero ei ollut tilastollisesti merkitsevä)).
Tutkimus 4
Kaksivaiheinen avoin turvallisuutta ja tehoa koskeva moniannos- ja monikeskustutkimus
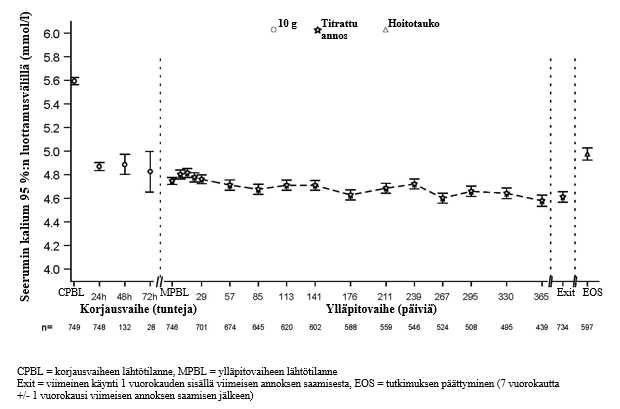
Tässä tutkimuksessa arvioitiin Lokelma-valmisteen pitkän aikavälin (enintään 12 kuukautta) vaikutuksia 751 tutkittavalla, joilla oli hyperkalemia (kaliumpitoisuus lähtötilanteessa keskimäärin 5,59 mmol/l, vaihteluväli 4,3–7,6 mmol/l). Muita samanaikaisia sairauksia olivat krooninen munuaissairaus (65 %), diabetes (64 %), sydämen vajaatoiminta (15 %) ja kohonnut verenpaine (83 %). Tutkittavista 51 % ilmoitti käyttävänsä diureetteja ja 70 % reniini-angiotensiini-aldosteronijärjestelmän estäjiä. Korjausvaiheen aikana annettiin Lokelma-valmistetta 10 g:n annoksella kolme kertaa vuorokaudessa vähintään 24 tunnin ja enintään 72 tunnin ajan. Tutkittavat, jotka saavuttivat normokalemian (3,5–5,0 mmol/l, nämä arvot mukaan lukien) 72 tunnin kuluessa, osallistuivat tutkimuksen ylläpitovaiheeseen. Kaikki ylläpitovaiheeseen osallistuneet tutkittavat saivat Lokelma-valmistetta aloitusannoksella 5 g kerran vuorokaudessa. Tätä annosta voitiin titrausohjelman mukaisesti suurentaa 5 g kerrallaan kerran vuorokaudessa (enimmäisannos 15 g kerran vuorokaudessa) tai pienentää (vähimmäisannos 5 g joka toinen päivä).
Korjausvaiheessa 24 tunnin ajan Lokelma-valmistetta saaneista tutkittavista 494/748 (66 %) saavutti normokalemian, ja 24 tunnin kohdalla seerumin kaliumpitoisuus oli pienentynyt keskimäärin 0,81 mmol/l (n = 748). Korjausvaiheessa 48 tunnin ajan Lokelma-valmistetta saaneista tutkittavista 563/748 (75 %) saavutti normokalemian, ja 48 tunnin kohdalla seerumin kaliumpitoisuus oli pienentynyt keskimäärin 1,02 mmol/l (n = 104). Korjausvaiheessa 72 tunnin ajan Lokelma-valmistetta saaneista tutkittavista 583/748 (78 %) saavutti normokalemian, ja 72 tunnin kohdalla seerumin kaliumpitoisuus oli pienentynyt 1,10 mmol/l (n = 28). Lähtötilanteen kaliumpitoisuus vaikutti normokalemian saavuttamiseen siten, että kaliumpitoisuus pieneni tutkimuslääkehoidon aloittamisen jälkeen eniten tutkittavilla, joilla oli lähtötilanteessa suurimmat seerumin kaliumpitoisuudet, mutta näillä tutkittavilla normokalemian saavuttaneiden osuus oli pienin. Seerumin kaliumpitoisuus oli 126 potilaalla lähtötilanteessa ≥ 6,0 mmol/l (lähtötason kaliumarvo keskimäärin 6,28 mmol/l). Korjausvaiheen lopussa arvot olivat pienentyneet näillä tutkittavilla keskimäärin 1,37 mmol/l.
Taulukko 4. Korjausvaihe (tutkimus 4): niiden tutkittavien osuus, joilla seerumin kaliumpitoisuudet olivat 3,5–5,0 mmol/l, nämä arvot mukaan lukien, tai 3,5–5,5 mmol/l, nämä arvot mukaan lukien, korjausvaiheen tutkimuspäivien mukaan – hoitoaiepopulaatio (ITT)
| Korjausvaihe | Lokelma 10 g kolme kertaa vuorokaudessa (N = 749) | |||||
| Seerumin kalium 3,5−5,0 mmol/l, nämä arvot mukaan lukien | Seerumin kalium 3,5−5,5 mmol/l, nämä arvot mukaan lukien | |||||
| n/N | Osuus | 95 %:n luottamus- väli | n/N | Osuus | 95 %:n luottamus- väli | |
| Korjausvaihe, 24 tunnin kohdalla | 494/748 | 0,660 | 0,625, 0,694 | 692/748 | 0,925 | 0,904, 0,943 |
| Korjausvaihe, 48 tunnin kohdalla | 563/748 | 0,753 | 0,720, 0,783 | 732/748 | 0,979 | 0,965, 0,988 |
| Korjausvaihe, 72 tunnin kohdalla / korjausvaihe, viimeinen arvo | 583/748 | 0,779 | 0,748, 0,809 | 738/748 | 0,987 | 0,976, 0,994 |
Huomattava: Yhdellä tutkittavalla annoksen saamisen jälkeinen arvo mitattiin, kun viimeisen annoksen antamisesta oli kulunut yli 1 vuorokausi. Siksi tämä tutkittava soveltui korjausvaiheen hoitoaiepopulaatioon (ITT), mutta mittausajankohta suljettiin pois analyysistä.
Normokalemia säilyi potilaiden jatkaessa lääkehoitoa, ja seerumin keskimääräinen kaliumpitoisuus suureni lääkehoidon keskeyttämisen jälkeen. Lähtötilanteessa reniini-angiotensiini-aldosteronijärjestelmän estäjiä käyttäneistä potilaista 89 % ei keskeyttänyt lääkehoitoa reniini-angiotensiini-aldosteronijärjestelmän estäjillä ja 74 % pystyi jatkamaan samalla annoksella ylläpitovaiheen aikana. Potilaista, jotka eivät käyttäneet reniini-angiotensiini-aldosteronijärjestelmän estäjiä lähtötilanteessa, 14 % pystyi aloittamaan tämän hoidon. Ylläpitovaiheen aikana 75,6 %:lla tutkittavista kaliumpitoisuus pysyi normaalina reniini-angiotensiini-aldosteronijärjestelmän estäjien käytöstä huolimatta.
Kuvassa 2 esitetään keskimääräiset seerumin kaliumarvot tutkimuksen korjaus- ja ylläpitovaiheiden aikana.
Kuva 2: Korjaus- ja ylläpitovaiheet 12 kuukauden mittaisessa avoimessa tutkimuksessa (tutkimus 4) – seerumin keskimääräinen kaliumpitoisuus ajan funktiona 95 %:n luottamusvälillä
Tutkimus 5
Satunnaistettu, kaksoissokkoutettu, lumekontrolloitu tutkimus jatkuvaa hemodialyysihoitoa saavilla potilailla
Tässä tutkimuksessa 196 potilasta (keskimääräinen ikä 58 vuotta, vaihteluväli 20–86 vuotta), joilla oli loppuvaiheen munuaissairaus, vakaana vähintään 3 kuukauden ajan jatkunut dialyysihoito ja ennen dialyysiä todettu persistentti hyperkalemia, satunnaistettiin saamaan dialyysihoidon välipäivinä kerran vuorokaudessa Lokelma-valmistetta 5 g:n annoksella tai lumelääkettä. Satunnaistamishetkellä keskimääräinen seerumin kaliumpitoisuus oli 5,8 mmol/l (vaihteluväli 4,2₋7,3 mmol/l) Lokelma-ryhmässä ja 5,9 mmol/l (vaihteluväli 4,2–7,3 mmol/l) lumelääkeryhmässä. Ennen dialyysiä mitattavan seerumin kaliumpitoisuuden 4,0–5,0 mmol/l saavuttamiseksi aikana, jolloin annosta säädettiin (neljä ensimmäistä viikkoa), annosta voitiin suurentaa viikoittain seerumin kaliumpitoisuuden perusteella 5 g:n lisäyksin niin, että annos oli enintään 15 g kerran vuorokaudessa. Kaliumpitoisuus mitattiin pidemmän dialyysivälin jälkeen ennen dialyysiä. Kun jakso, jolloin annosta säädettiin, oli päättynyt, saavutettua annosta ei muutettu seuraavan neljän viikon mittaisen arviointijakson aikana. Kun jakso, jolloin annosta säädettiin, oli päättynyt, 37 % potilaista sai Lokelma-valmistetta 5 g:n annoksella, 43 % 10 g:n annoksella ja 19 % 15 g:n annoksella. Vasteen saaneiksi katsottiin tutkittavat, joilla pidemmän dialyysivälin jälkeen ennen dialyysiä mitattu seerumin kaliumpitoisuus oli pysyvästi 4,0–5,0 mmol/l vähintään kolmen dialyysihoitokerran yhteydessä neljästä ja jotka eivät saaneet oireenmukaista hoitoa arviointijakson aikana. Vasteen saaneiden osuus oli 41 % Lokelma-ryhmässä ja 1 % lumelääkeryhmässä (p < 0,001) (ks. kuva 3).
Post hoc ‑analyyseissä niiden kertojen määrä, joilla potilaan seerumin kaliumpitoisuus oli 4,0₋5,0 mmol/l pidemmän dialyysivälin jälkeen arviointijaksolla, oli suurempi Lokelma-ryhmässä. Lokelma-ryhmässä arvot olivat kaikilla neljällä käyntikerralla näiden rajojen sisällä 24 %:lla potilaista, mutta lumelääkeryhmässä eivät yhdelläkään potilaalla. Post hoc ‑analyysit osoittivat, että arviointijaksolla pidemmän dialyysivälin jälkeen mitattu seerumin kaliumpitoisuus pysyi välillä 3,5–5,5 mmol/l Lokelma-ryhmässä 70 %:lla ja lumelääkeryhmässä 21 %:lla potilaista vähintään kolmen dialyysihoitokerran yhteydessä neljästä.
Hoidon päätyttyä seerumin keskimääräinen dialyysin jälkeen mitattu kaliumpitoisuus oli Lokelma-ryhmässä 3,6 mmol/l (vaihteluväli 2,6–5,7 mmol/l) ja lumelääkeryhmässä 3,9 mmol/l (vaihteluväli 2,2–7,3 mmol/l). Painonousussa dialyysikertojen välillä ei havaittu eroja Lokelma- ja lumelääkeryhmien välillä. Painonnousu dialyysikertojen välillä määriteltiin ennen dialyysiä mitatuksi painoksi, josta oli vähennetty edellisen dialyysikerran yhteydessä mitattu paino dialyysin jälkeen, ja se mitattiin pidemmän dialyysivälin jälkeen.
Kuva 3: Ennen dialyysiä mitatut seerumin keskimääräiset kaliumpitoisuudet ajan funktiona jatkuvaa dialyysihoitoa saavilla potilailla
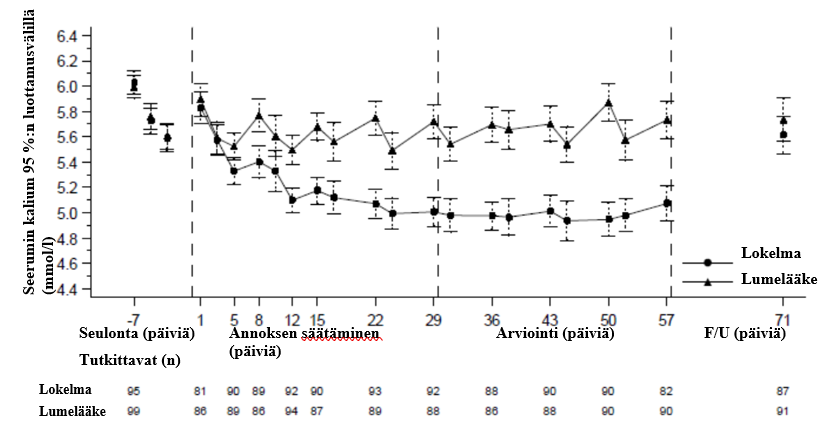
F/U = seurantajakso
Esitetyt virhepalkit vastaavat 95 %:n luottamusvälejä.
n = niiden potilaiden lukumäärä, joilta ei puutu kaliumin mittaustuloksia tietyltä käynniltä.
Tutkimus 6 – PRIORITIZE‑HF
Kyseessä oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu tutkimus, jonka tavoitteena oli arvioida lumelääkkeeseen verrattuna, salliiko Lokelma-valmistetta sisältävä hoito-ohjelma reniini-angiotensiini-aldosteronijärjestelmän (RAA-järjestelmän) estäjien annosten suurentamisen tavoitetasolle 3 kuukauden kohdalla potilailla, joilla on sydämen vajaatoiminta ja hyperkalemia tai joilla on suuri hyperkalemian kehittymisen riski. Tutkimuksen ensisijaisena päätemuuttujana olivat tutkittavien osuudet seuraavissa neljässä luokassa 3 kuukauden kohdalla: ei angiotensiinikonvertaasin (ACE:n) estäjiä / angiotensiinireseptorin (ATR:n) salpaajia / ATR:n salpaajan ja neprilysiinin estäjien yhdistelmiä (ARNI-lääkkeitä) tai näiden käyttö tavoiteannosta pienemmällä annoksella, ei mineralokortikoidireseptoriantagonistia (MRA-lääkettä); ACE:n estäjä / ATR:n salpaaja / ARNI-lääke tavoiteannoksella, ei MRA-lääkettä; MRA-lääke tavoiteannosta pienemmällä annoksella; MRA-lääke tavoiteannoksella.
Sydämen vajaatoimintaa sairastavat potilaat, joiden NYHA (New York Heart Association) ‑luokka oli II–IV, vasemman kammion ejektiofraktio (LVEF) ≤ 40 %, glomerulusten laskennallinen suodatusnopeus ([eGFR]) 20–59 ml/min/1,73 m2ja seerumin kaliumpitoisuus 4,0–5,5 mmol/l, satunnaistettiin (1:1) saamaan Lokelma-valmistetta tai lumelääkettä 3 kuukauden ajan. RAA-järjestelmän estäjien annokset pyrittiin suurentamaan hoitosuositusten mukaisiksi, mutta tämä ei ollut pakollista, ja Lokelma-valmisteen tai lumelääkkeen annoksia säädettiin samanaikaisesti hyperkalemian ehkäisemiseksi.
Tutkimus lopetettiin ennenaikaisesti COVID‑19-pandemian aikana, koska rekrytointiin liittyi haasteita ja riittävää turvallisuusseurantaa oli vaikeaa varmistaa, kun potilaat eivät pystyneet osallistumaan tutkimuskäynneille tai tulemaan laboratoriokokeisiin. Satunnaistettuja potilaita oli tästä johtuen 182 eikä suunnitelman mukaisesti 280. Tutkimuksen ennenaikaisen lopettamisen vuoksi ensisijaisista ja muista tehon mittareista ei voida tehdä varmoja johtopäätöksiä.
Tutkimus 7 – REALIZE‑K
Kyseessä oli vaiheen 4 prospektiivinen, kaksoissokkoutettu tutkimus, jossa tutkimushoito lopetettiin satunnaistetusti osalta tutkittavista ja jonka tavoitteena oli määrittää Lokelma-valmisteen teho ja turvallisuus MRA-hoitoa optimoitaessa potilailla, joilla oli sydämen vajaatoiminta ja pienentynyt ejektiofraktio. Ensisijainen päätemuuttuja oli optimaalisen vasteen esiintyvyys, ja optimaalisen vasteen määritelmänä oli yhdistelmämuuttuja, joka käsitti normaalin kaliumpitoisuuden seerumissa (3,5–5,0 mmol/l), kun spironolaktoniannos oli ≥ 25 mg vuorokaudessa, ilman hyperkalemiasta johtuvaa muuta oireenmukaisen hoidon tarvetta.
Tähän tutkimukseen otettiin mukaan aikuisia, joilla oli todettu sydämen vajaatoiminta (kesto ≥ 3 kuukautta, LVEF ≤ 40 %, oireiden NYHA-luokka II–IV ja ≥ 4 viikkoa kestänyt hoito vakailla annoksilla ACE:n estäjää / ATR:n salpaajaa / ARNI-lääkettä ja beeta-adrenergisten reseptorien salpaajaa [ellei tämä ollut vasta-aiheinen]). Osallistuminen sallittiin potilaille, jotka eivät saaneet MRA-lääkettä, ja potilaille, jotka saivat spironolaktonia tai eplerenonia < 25 mg kerran vuorokaudessa.
Seulonnan jälkeen potilaat osallistuivat avoimeen sisäänajovaiheeseen, jossa oli kaksi kohorttia. Kohortissa 1 oli potilaita, joilla oli jo näyttöä hyperkalemiasta (määritelmänä seerumin kalium 5,1–5,9 mmol/l) ja eGFR ≥ 30 ml/min/1,73 m2. Tämän kohortin potilaat saivat Lokelma-valmistetta kaliumarvojen korjaamiseksi normaalialueelle, minkä jälkeen aloitettiin spironolaktonihoito tai suurennettiin spironolaktoniannosta tutkimussuunnitelman mukaisesti. Kohortissa 2 oli potilaita, joilla oli suuri hyperkalemiariski (määritelmänä joko seerumin kaliumpitoisuus > 5,0 mmol/l, joka oli todettu edeltävien 36 kuukauden aikana, ja eGFR ≥ 30 ml/min/1,73 m2TAI seerumin kaliumpitoisuus 4,5–5,0 mmol/l ja eGFR 30–60 ml/min/1,73 m2TAI seerumin kaliumpitoisuus 4,5–5,0 mmol/l ja ikä > 75 vuotta). Näillä potilailla aloitettiin spironolaktonihoito tai spironolaktoniannosta suurennettiin kohti tavoiteannosta. Potilaat, joille kehittyi hyperkalemia, saivat Lokelma-valmistetta kaliumarvojen korjaamiseksi normaalialueelle. Jos taas potilaalle ei kehittynyt hyperkalemiaa 4 viikon kuluessa, hänen osallistumisensa tutkimukseen keskeytettiin.
Tässä tutkimuksessa Lokelma-valmisteen käyttö suurensi optimaalisen vasteen esiintyvyyttä (ensisijainen päätemuuttuja) verrattuna lumelääkkeeseen (kerroinsuhde 4,45 [95 %:n luottamusväli 2,89–6,86], p < 0,001, arvioidut osuudet 71 % [Lokelma] ja 36 % [lumelääke]). Tulokset olivat vastaavat, kun analyysistä suljettiin pois potilaat, jotka saivat Lokelma-valmistetta 15 g:n annoksella satunnaistamisajankohtana. Lumelääkkeeseen verrattuna Lokelma paransi myös toissijaisia päätemuuttujia, joita olivat normokalemian esiintyvyys käytettäessä samaa spironolaktoniannosta kuin satunnaistamisajankohtana ja ilman hyperkalemian vuoksi tarvittavaa oireenmukaista muuta hoitoa (kerroinsuhde 4,58 [95 %:n luottamusväli 2,78–7,55], p < 0,001; arvioidut osuudet 58 % [Lokelma] ja 23 % [lumelääke]); spironolaktoniannoksen ≥ 25 mg/vrk käytön esiintyvyys (kerroinsuhde 4,33 [95 %:n luottamusväli 2,50–7,52], p < 0,001; arvioidut osuudet 81 % [Lokelma] ja 50 % [lumelääke]); aika hyperkalemian ensimmäiseen ilmaantumiseen (seerumin K+ > 5,0 mmol/l) (riskitiheyksien suhde [HR] 0,51 [95 %:n luottamusväli 0,37–0,71], p < 0,001); ja aika hyperkalemiasta johtuvaan spironolaktoniannoksen pienentämiseen ensimmäistä kertaa tai spironolaktonihoidon lopettamiseen (riskitiheyksien suhde 0,37 [95 %:n luottamusväli 0,17–0,73], p = 0,006).
Tutkimus 8 – STABILIZE-CKD
Kyseessä oli vaiheen 3 kaksoissokkoutettu, rinnakkaisryhmillä toteutettu, lumekontrolloitu tutkimus, jossa tutkimushoito lopetettiin satunnaistetusti osalta tutkittavista. Tutkimuksen tavoitteena oli arvioida, onko Lokelma ACE:n estäjien / ATR:n salpaajien lisänä parempi kuin lumelääke kroonisen munuaistaudin etenemisen hidastamisessa ajan myötä potilailla, joilla on hyperkalemia tai joiden hyperkalemiariski on suurentunut. Samanaikaisia ensisijaisia päätemuuttujia olivat koko eGFR-kuvaajan kulmakerroin (satunnaistamisesta hoidon päättymiseen) ja pitkän aikavälin eGFR-kuvaajan kulmakerroin (alkaen 12 viikon kuluttua satunnaistamisesta, hoidon päättymiseen asti).
Tutkimukseen otettiin mukaan potilaita, joiden eGFR oli 25–59 ml/min/1,73 m2 ja albumiinin ja kreatiniinin suhde virtsassa 200 – 5 000 mg/g ja joilla oli hyperkalemia (seerumin kaliumpitoisuus [sK+] > 5,0 – ≤ 6,5 mmol/l), kun hoito ACE:n estäjillä / ATR:n salpaajilla oli riittävää tai rajoitettua, tai normokalemia, kun hoito ACE:n estäjillä / ATR:n salpaajilla oli rajoitettua. Tutkimuksesta suljettiin pois potilaat, joilla oli NYHA-luokan III–IV kongestiivinen sydämen vajaatoiminta seulonta-ajankohtana tai joilla oli aiemmin todettu vaikea tai oireinen sydämen vajaatoiminta.
Tutkimus sisälsi seulontajakson, aloitusvaiheen (jonka aikana osallistujat saivat Lokelma-valmistetta avoimesti enintään 72 tunnin ajan normokalemian ylläpitämiseksi tai saavuttamiseksi), 3 kuukauden mittaisen sisäänajovaiheen (jossa oli tavoitteena suurentaa lisinopriilin tai valsartaanin annos suurimmalle siedetylle tasolle siten, että kaliumpitoisuuksien hallintaan annettiin avoimessa asetelmassa Lokelma-valmistetta), alun perin 24 kuukauden mittaiseksi suunnitellun satunnaistetun sokkoutetun ylläpitovaiheen (potilaat satunnaistettiin suhteessa 1:1 saamaan sokkoutetusti Lokelma-valmistetta tai vastaavaa lumelääkettä, ja sekä lisinopriilin tai valsartaanin että Lokelma-valmisteen/lumelääkkeen annoksia säädettiin ja seurattiin tehon ja turvallisuuden arviointeja varten) ja seurantakäynnin.
Tutkimus lopetettiin ennenaikaisesti rekrytointiin liittyvien haasteiden vuoksi, jolloin otoskooksi jäi suunnitellun 1 360 potilaan sijaan 760 satunnaistettua potilasta ja satunnaistamisen jälkeinen seuranta-aika lyheni (mediaani noin 8–9 kuukautta suunnitellun 24 kuukauden sijaan). Tämän vuoksi eGFR-kuvaajan kulmakertoimesta ja vakavista munuaisperäisistä tapahtumista ei voida tehdä johtopäätöksiä.
Lokelma-valmistetta koskeneista lumekontrolloiduista kliinisistä tutkimuksista (PRIORITIZE‑HF, REALIZE‑K ja STABILIZE‑CKD), joihin osallistuneet potilaat eivät saaneet dialyysihoitoa, tehtiin yhdistetty analyysi. Analyysin mukaan potilailla, joilla oli aiemmin todettu sydämen vajaatoiminta, sydämen vajaatoiminta paheni useammalla Lokelma-valmistetta saaneista potilaista kuin lumelääkettä saaneista (ks. kohta Haittavaikutukset).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Lokelma-valmisteen käytöstä hyperkalemian hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Natriumsirkoniumsyklosilikaatti on epäorgaaninen, liukenematon yhdiste, joka ei metaboloidu entsymaattisesti. Lisäksi kliinisissä tutkimuksissa on osoitettu, ettei Lokelma imeydy systeemisesti. Eräässä rotilla in vivo tehdyssä massatasapainotutkimuksessa natriumsirkoniumsyklosilikaatin todettiin erittyvän ulosteeseen eikä systeemisestä imeytymisestä saatu näyttöä. Näiden tekijöiden ja Lokelma-valmisteen liukenemattomuuden vuoksi ei ole tehty in vivo tai in vitro tutkimuksia, joissa arvioitaisiin valmisteen vaikutusta sytokromi P450 (CYP450) -entsyymeihin tai kuljettajaproteiinien toimintaan.
Eliminaatio
Natriumsirkoniumsyklosilikaatti poistuu ulosteen mukana.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, karsinogeenisuutta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
Ei ole.
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LOKELMA jauhe oraalisuspensiota varten
5 g (L:ei) 30 x 5 g (248,39 €)
10 g (L:ei) 30 x 10 g (316,20 €)
PF-selosteen tieto
5 g tai 10 g jauhetta PET/alu/LLDPE- tai PET/LDPE/alu/EAA/LLDPE-laminaatista valmistetuissa annospusseissa
Pakkauskoot: 3 tai 30 annospussia
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai harmaa jauhe.
Käyttö- ja käsittelyohjeet
Ei erityisvaatimuksia hävittämisen suhteen.
Korvattavuus
LOKELMA jauhe oraalisuspensiota varten
5 g 30 x 5 g
10 g 30 x 10 g
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Natriumsirkoniumsyklosilikaatti ja patiromeeri: Kroonista munuaissairautta sairastavien aikuispotilaiden hyperkalemian hoito erityisin edellytyksin / Natriumsirkoniumsyklosilikaatti: Sydämen vajaatoimintaa sairastavien aikuispotilaiden hyperkalemian hoito erityisin edellytyksin (3020).
ATC-koodi
V03AE10
Valmisteyhteenvedon muuttamispäivämäärä
24.07.2025
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi