
JEVTANA infuusiokonsentraatti ja liuotin, liuosta varten 60 mg
Vaikuttavat aineet ja niiden määrät
Yksi ml konsentraattia sisältää 40 mg kabatsitakselia.
Yhdessä injektiopullossa on 1,5 ml (nimellistilavuus) konsentraattia sisältäen 60 mg kabatsitakselia.
Kun konsentraatti on laimennettu koko liuotinmäärällä, yksi ml liuosta sisältää 10 mg kabatsitakselia.
Huomaa: Sekä JEVTANA 60 mg/1,5 ml konsentraatti-injektiopullo (täyttötilavuus: 73,2 mg kabatsitakselia/1,83 ml) että liuotininjektiopullo (täyttötilavuus: 5,67 ml) sisältävät ylitäytön kompensoimaan liuottamisessa syntyvää nestehävikkiä. Tällä ylitäytöllä varmistetaan, että kun mukana toimitettu KOKO liuotinmäärä on käytetty laimennukseen, liuos sisältää 10 mg/ml kabatsitakselia.
Apuaine, jonka vaikutus tunnetaan:
Polysorbaatti 80 (E 433) 1,56 g jokaisessa 60 mg:n konsentraattipullossa, mikä vastaa 1,04 g/ml (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yksi injektiopullo liuotinta sisältää 573,3 mg etanolia (96 %).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti ja liuotin, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
JEVTANA yhdistelmänä prednisonin tai prednisolonin kanssa on tarkoitettu metastaattista, kastraatioresistenttiä eturauhassyöpää sairastavien aikuisten potilaiden hoitoon, joita on aiemmin hoidettu dosetakselilla (ks. kohta Farmakodynamiikka).
Ehto
Valmistetta tulee antaa solunsalpaajalääkityksen antoon erikoistuneissa yksiköissä ja syöpälääkitykseen perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
JEVTANA-valmisteen käyttö on rajattava solunsalpaajien antoon erikoistuneisiin yksiköihin ja se tulee annostella syöpälääkkeiden antoon perehtyneen lääkärin valvonnassa. Vakavien yliherkkyysreaktioiden, kuten hypotension ja bronkospasmin hoitoon tarkoitettujen välineiden ja laitteiden täytyy olla saatavilla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Esilääkitys
Suositeltu esilääkitys on annettava vähintään 30 minuuttia ennen jokaista JEVTANA-infuusiota yliherkkyysriskin ja sen vaikeusasteen lieventämiseksi. Esilääkitykseksi suositellaan seuraavia laskimonsisäisiä lääkevalmisteita:
- antihistamiini (dekskloorifeniramiini 5 mg tai difenhydramiini 25 mg tai vastaava),
- kortikosteroidi (deksametasoni 8 mg tai vastaava) ja
- H2-antagonisti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Profylaktista antiemeettistä lääkitystä suun kautta tai laskimoon voidaan antaa tarpeen mukaan.
Koko hoidon ajan on varmistettava potilaan riittävä nesteytys komplikaatioiden, kuten munuaisten vajaatoiminnan estämiseksi.
Annostus
Suositeltu JEVTANA-annos on 25 mg/m² 1 tunnin infuusiona laskimoon joka 3. viikko yhdistettynä prednisoniin tai prednisoloniin annoksella 10 mg suun kautta päivittäin hoidon ajan.
Annosmuutokset
Annosta on muutettava, jos potilaalla esiintyy seuraavia haittavaikutuksia (gradus-arvot viittaavat CTCAE 4.0-luokitukseen, Common Terminology Criteria for Adverse Events):
Taulukko 1 – Suositellut annosmuutokset haittavaikutusten takia kabatsitakselihoitoa saaville potilaille
| Haittavaikutukset | Annosmuutokset |
| Pitkittynyt gradus ≥ 3 neutropenia (yli 1 viikon) asianmukaisesta hoidosta huolimatta mukaan lukien G-CSF | Hoidon keskeytys kunnes neutrofiiliarvo on >1 500 solua/mm3, sitten laske kabatsitakseliannosta 25 mg/m2:stä 20 mg/m2:een |
| Kuumeinen neutropenia tai neutropeninen infektio | Hoidon keskeytys kunnes tila paranee tai korjautuu ja kunnes neutrofiiliarvo on >1 500 solua/mm3, sitten laske kabatsitakseli-annosta 25 mg/m2:stä 20 mg/m2:een |
| Gradus ≥ 3 ripuli tai jatkuva ripuli huolimatta oikeasta hoidosta, kuten nesteytyksestä ja elektrolyyttitiputuksesta | Hoidon keskeytys kunnes tila paranee tai ripuli loppuu, sitten laske kabatsitakseliannosta 25 mg/m2:stä 20 mg/m2:een |
| Gradus > 2 perifeerinen neuropatia | Hoidon keskeytys kunnes tila paranee, sitten laske kabatsitakseliannosta 25 mg/m2:stä 20 mg/m2:een |
Jos potilas saa edelleen jonkin näistä reaktioista annoksella 20 mg/m2, voidaan vielä harkita annoksen laskemista 15 mg/m2 asti tai JEVTANA-hoidon lopettamista. Tietoa potilaista, jotka ovat saaneet pienempää annosta kuin 20 mg/m2, on vain vähän.
Erityisryhmät
Potilaat, joilla on maksan vajaatoiminta
Kabatsitakseli metaboloituu suuressa määrin maksassa. Potilaille, joilla on lievä maksan vajaatoiminta (kokonaisbilirubiini > 1, ≤ 1,5 x normaaliarvon yläraja (ULN) tai aspartaattiaminotransferaasiarvo (ASAT) > 1,5 x ULN, on annettava pienempi kabatsitakseliannos 20 mg/m2. Kabatsitakseli on annettava varoen ja tarkassa turvallisuusseurannassa, kun potilaalla on lievä maksan vajaatoiminta.
Potilailla, joilla on kohtalainen maksan vajaatoiminta (kokonaisbilirubiini > 1,5, ≤ 3,0 x ULN), suurin siedetty annos (MTD) oli 15 mg/m2. Jos kohtalaista maksan vajaatoimintaa sairastavalle potilaalle päätetään antaa hoito, kabatsitakseliannosta 15 mg/m2 ei saa ylittää. Tämän annoksen tehokkuudesta on kuitenkin vain vähän tietoa.
Kabatsitakselia ei saa antaa potilaille, jotka sairastavat vaikeaa maksan vajaatoimintaa (kokonaisbilirubiini > 3 x normaaliarvon yläraja (ULN)) (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Potilaat, joilla on munuaisten vajaatoiminta
Kabatsitakseli poistuu elimistöstä munuaisten kautta vain vähän. Annoksen muuttaminen ei ole tarpeen potilaille, joilla on munuaisten vajaatoiminta, joka ei edellytä hemodialyysihoitoa. Potilaita, joilla on loppuvaiheen munuaissairaus (CLCR< 15 ml/min/1,73 m2), pitäisi hoitaa varoen potilaan kunnon ja vähäisen saatavilla olevan tiedon mukaan ja seurata tarkasti hoidon aikana (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Iäkkäät
Iäkkäille potilaille ei suositella erityistä kabatsitakseliannoksen muuttamista (ks. myös kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset ja Farmakokinetiikka).
Muiden lääkevalmisteiden samanaikainen käyttö
Vältä samanaikaista käyttöä lääkevalmisteilla, jotka ovat CYP3A-entsyymin voimakkaita induktoreita tai voimakkaita inhibiittoreita. Jos potilas kuitenkin tarvitsee samanaikaista voimakasta CYP3A:n estäjää, on harkittava kabatsitakseliannoksen pienentämistä 25 %:lla (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Pediatriset potilaat
Ei ole asianmukaista käyttää JEVTANA-valmistetta pediatristen potilaiden hoitoon.
JEVTANA-valmisteen turvallisuutta ja tehokkuutta lapsille ja alle 18-vuotiaille nuorille ei ole varmistettu (ks. kohta Farmakodynamiikka).
Antotapa
JEVTANA annetaan laskimoon.
Lääkevalmisteen valmistus- ja annostusohjeet, ks. kohta Käyttö- ja käsittelyohjeet.
Älä käytä PVC-infuusionestepakkauksia tai polyuretaani-infuusiolaitteita.
JEVTANA-valmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Vasta-aiheet
- Yliherkkyys kabatsitakselille, muille taksaaneille tai polysorbaatti 80:lle tai kohdassa Apuaineet mainituille apuaineille.
- Neutrofiiliarvo on alle 1 500/mm3.
- Vaikea maksan vajaatoiminta (kokonaisbilirubiini > 3 x ULN).
- Keltakuumerokotteen samanaikainen anto (ks. kohta Yhteisvaikutukset)
Varoitukset ja käyttöön liittyvät varotoimet
Yliherkkyysreaktiot
Kaikille potilaille pitäisi antaa esilääkitys ennen kabatsitakseli-infuusion antamista (ks. kohta Annostus ja antotapa).
Potilaita on seurattava tarkasti yliherkkyysreaktioiden varalta varsinkin ensimmäisen ja toisen infuusion aikana. Yliherkkyysreaktioita voi esiintyä muutaman minuutin kuluessa kabatsitakseli-infuusion aloittamisesta, joten laitteet ja välineet hypotension ja bronkospasmin hoitoon on oltava saatavilla. Vaikeita reaktioita mukaan lukien yleistynyttä ihottumaa/eryteemaa, hypotensiota ja bronkospasmeja voi esiintyä. Vaikeat yliherkkyysreaktiot vaativat kabatsitakselihoidon välitöntä keskeyttämistä ja asiaankuuluvaa hoitoa. JEVTANA-hoito on lopetettava yliherkkyysreaktion saaneilta potilailta (ks. kohta Vasta-aiheet).
Luuydinlama
Luuydinlama, jonka merkkinä ilmenee neutropeniaa, anemiaa, trombosytopeniaa tai pansytopeniaa, voi ilmaantua (ks. ”Neutropeniariski” ja ”Anemia” kohdassa Varoitukset ja käyttöön liittyvät varotoimet jäljempänä).
Neutropeniariski
Potilaille, joita hoidetaan kabatsitakselilla, voidaan antaa profylaktista G-CSF:ää ASCOn (American Society of Clinical Oncology) ohjeiden ja/tai paikallisen hoitokäytännön mukaan neutropeniariskin pienentämiseksi tai neutropeenisten komplikaatioiden hoitamiseksi (kuumeinen neutropenia, pitkittynyt neutropenia tai neutropeninen infektio). Primaarista estolääkitystä G-CSF:llä pitäisi harkita potilaille, joilla on korkea kliininen riskiprofiili (yli 65-vuotias, huono yleiskunto, aikaisempia kuumeisen neutropenian jaksoja, laajoja aikaisempia sädehoitojaksoja, huono ravitsemustila tai muu vaikea yleissairaus) altistaen heidät suurentuneelle komplikaatioriskille pitkittyneen neutropenian johdosta. G-CSF:n käytön on osoitettu pienentävän neutropenian esiintymistä ja lieventävän sen vaikeusastetta.
Neutropenia on kabatsitakselihoidon yleisin haittavaikutus (ks. kohta Haittavaikutukset). Täydellisen verenkuvan seuraaminen on tärkeää viikoittain 1. syklin ajan ja sen jälkeen ennen jokaista hoitosykliä, jotta annosta voidaan muuttaa tarvittaessa.
Annosta pitää laskea kuumeisen neutropenian tai pitkittyneen neutropenian ilmaantuessa oikeasta hoidosta huolimatta (ks. kohta Annostus ja antotapa).
Jatka hoitoa vasta kun neutrofiiliarvo on tasolla ≥ 1500/mm3 (ks. kohta Vasta-aiheet).
Ruoansulatuselimistö
Oireet, kuten mahakipu tai mahan arkuus, kuume, jatkuva ummetus ja ripuli, joihin voi liittyä neutropeniaa, voivat olla ruoansulatuskanavaan kohdistuvan toksisuuden ensimmäisiä ilmenemismuotoja, ja ne on tutkittava ja hoidettava pian. Kabatsitakselihoidon viivästyttäminen tai lopettaminen voi olla tarpeen.
Pahoinvoinnin, oksennuksen, ripulin ja nestehukan riski
Jos potilailla esiintyy ripulia kabatsitakselihoidon jälkeen, heitä voidaan hoitaa tavanomaisilla ripulilääkevalmisteilla. Potilaiden asianmukaisesta nesteyttämisestä on huolehdittava. Ripulia voi esiintyä yleisimmin potilailla, jotka ovat saaneet aikaisemmin vatsan alueen tai lantion sädehoitoa. Kuivuminen on yleisempää 65-vuotiailla tai sitä vanhemmilla potilailla. Potilaiden nestetasapaino on palautettava ja seerumin elektrolyyttitasoa, erityisesti kaliumia, on seurattava ja korjattava asianmukaisilla menetelmillä. Hoidon keskeyttäminen tai annoksen vähentäminen voi olla tarpeen gradus ≥3 ripulissa (ks. kohta Annostus ja antotapa). Jos potilailla esiintyy pahoinvointia tai oksentamista, heitä voidaan hoitaa yleisesti käytetyillä pahoinvointilääkkeillä.
Vakavien ruoansulatuselimistön reaktioiden riski
Kabatsitakselihoitoa saaneilla potilailla on ilmoitettu ruoansulatuskanavan verenvuotoa ja perforaatiota, ileusta sekä koliittia, joskus jopa kuolemaan johtaneina (ks. kohta Haittavaikutukset). Varovaisuutta on noudatettava hoidettaessa potilaita, jotka ovat erityisen suuressa vaarassa saada ruoansulatuskanavan komplikaatioita. Näitä ovat esimerkiksi neutropeniapotilaat, iäkkäät potilaat, potilaat, jotka käyttävät samanaikaisesti NSAID-valmisteita (tulehduskipulääkkeitä), veren hyytymistä estäviä lääkkeitä tai antikoagulantteja, sekä potilaat, jotka ovat saaneet sädehoitoa lantion alueelle tai joilla on ollut ruoansulatuskanavan sairaus, kuten haavauma tai ruoansulatuskanavan verenvuoto.
Perifeerinen neuropatia
Perifeeristä neuropatiaa, perifeeristä sensorista neuropatiaa (esim. parestesiaa, dysestesiaa) ja perifeeristä motorista neuropatiaa on havaittu kabatsitakselia saaneilla potilailla. Kabatsitakselihoitoa saavia potilaita on neuvottava kertomaan lääkärille ennen hoidon jatkamista, jos heille on kehittynyt neuropatian oireita, kuten kipua, polttelua, pistelyä, tunnottomuutta tai heikkoutta. Lääkärin on arvioitava ilmennyt neuropatia tai sen paheneminen ennen jokaista hoitokertaa. Hoidon antoa on siirrettävä, kunnes oireet paranevat. Kabatsitakseliannosta on pienennettävä 25 mg/m2:sta 20 mg/m2:een perifeerisessä neuropatiassa, jossa gradus > 2 (ks. kohta Annostus ja antotapa).
Anemia
Anemiaa on havaittu kabatsitakselia saaneilla potilailla (ks. kohta Haittavaikutukset). Hemoglobiini ja hematokriitti on tarkistettava ennen kabatsitakselihoitoa ja jos potilaalla on anemian tai verenhukan oireita. Varovaisuutta suositellaan, jos potilaan hemoglobiini < 10 g/dl ja on ryhdyttävä asianmukaisiin, kliinisen vasteen mukaisiin toimiin.
Munuaisten vajaatoiminnan riski
Munuaisten toimintahäiriöitä on raportoitu sepsiksen, ripulista johtuvan vaikean kuivumisen, oksentelun ja obstruktiivisen virtsatietaudin yhteydessä. Munuaisten vajaatoimintaa, myös kuolemaan johtaneita tapauksia on raportoitu. Munuaisten vajaatoiminnan ilmetessä syy täytyy selvittää ja potilasta on hoidettava tehokkaasti.
Riittävä nesteytys on varmistettava kabatsitakselihoidon aikana. Potilasta on neuvottava ilmoittamaan välittömästi, jos päivittäinen virtsamäärä muuttuu merkittävästi. Seerumin kreatiniini on mitattava hoidon alussa, jokaisen verikokeen yhteydessä ja aina, kun potilas ilmoittaa virtsamäärän muuttuneen. Kabatsitakselihoito on lopetettava, jos on viitteitä munuaisten toiminnan heikkenemisestä munuaisten vajaatoimintaan, joka on luokkaa gradus ≥3 (CTCAE 4,0).
Hengityselimet, rintakehä ja välikarsina
Interstitiaalista pneumoniaa/pneumoniittia ja interstitiaalista keuhkosairautta on ilmoitettu ja ne ovat voineet johtaa kuolemaan (ks. kohta Haittavaikutukset).
Jos kehittyy uusia keuhko-oireita tai olemassa olevat pahenevat, potilaita on seurattava tarkkaan, tutkittava nopeasti ja hoidettava asianmukaisesti. Kabatsitakselihoito suositellaan keskeytettäväksi, kunnes diagnoosi on selvillä. Aikainen tukihoito voi auttaa parantamaan potilaan tilaa. Kabatsitakselihoidon uudelleen aloittamisen hyödyt on arvioitava huolellisesti.
Sydämen rytmihäiriöiden riski
Sydämen rytmihäiriöitä, yleisimmin takykardiaa ja eteisvärinää, on raportoitu (ks. kohta Haittavaikutukset).
Iäkkäät henkilöt
Iäkkäät henkilöt (≥65-vuotiaat) saavat yleensä enemmän tiettyjä haittavaikutuksia, kuten neutropeniaa ja kuumeista neutropeniaa (ks. kohta Haittavaikutukset).
Potilaat, joilla on maksan vajaatoiminta
JEVTANA-hoito on vasta-aiheinen vaikeaa maksan vajaatoimintaa sairastaville potilaille (kokonaisbilirubiini > 3 x ULN) (ks. kohdat Vasta-aiheet ja Farmakokinetiikka).
Lievää (kokonaisbilirubiini > 1, ≤ 1,5 x normaaliarvon yläraja (ULN) tai ASAT > 1,5 x ULN) maksan vajaatoimintaa sairastaville potilaille annettavaa annosta on pienennettävä (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Yhteisvaikutukset
Voimakkaiden CYP3A:n estäjien samanaikaista käyttöä on vältettävä, sillä ne voivat lisätä kabatsitakselin pitoisuutta plasmassa (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset). Jos voimakkaan CYP3A:n estäjän samanaikaista antoa ei voida välttää, toksisuuden merkkejä on seurattava tarkkaan ja kabatsitakseliannoksen pienentämistä on harkittava (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Voimakkaiden CYP3A-entsyymi-induktorien samanaikaista käyttöä on vältettävä, sillä ne voivat vähentää kabatsitakselin pitoisuutta plasmassa (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Apuaineet
Tämä lääkevalmiste sisältää 573 mg alkoholia (etanolia) per liuotinpullo. Alkoholimäärä yhdessä annoksessa tätä lääkevalmistetta vastaa alle 11 ml:aa olutta tai 5 ml:aa viiniä. Tämän lääkevalmisteen sisältämä pieni määrä alkoholia ei aiheuta havaittavia vaikutuksia. Erityisiä varotoimia on kuitenkin noudatettava riskiryhmillä, kuten potilailla, joilla on maksasairaus tai epilepsia tai joilla on ollut alkoholiriippuvuus.
Polysorbaatti 80 (E 433)
Tämä lääke sisältää 1,56 g polysorbaatti 80:tä jokaisessa 60 mg:n konsentraattipullossa, mikä vastaa 1,04 g/ml. Polysorbaatit voivat aiheuttaa allergisia reaktioita. Polysorbaateilla voi olla sydän- ja verisuonivaikutuksia (hypotensio/sydämen vajaatoiminta). Sydän- ja verisuonivaikutusten riskin minimoimiseksi harkitse infuusionopeuden hidastamista.
Polysorbaatin mahdollinen vaikutus QT-ajan pidentymiseen ja kääntyvien kärkien takykardiaan on otettava huomioon, kun sitä käytetään samanaikaisesti QT/QTc-aikaa pidentävien lääkkeiden kanssa tai potilailla, joilla on synnynnäinen oireyhtymä.
Raskauden ehkäisy
Miesten on käytettävä ehkäisyä kabatsitakselihoidon aikana ja 4 kuukautta hoidon päättymisen jälkeen (ks. kohta Raskaus ja imetys).
Yhteisvaikutukset
In vitro -tutkimukset ovat osoittaneet, että kabatsitakseli metaboloituu lähinnä CYP3A:n kautta (80-90 %) (ks. kohta Farmakokinetiikka).
CYP3A:n estäjät
Ketokonatsolin, voimakkaan CYP3A:n estäjän, toistetut annokset (400 mg kerran vuorokaudessa) pienensivät kabatsitakselin puhdistumaa 20 %, josta seurasi AUC-arvon suureneminen 25 %:lla. Tästä syystä yhteiskäyttöä voimakkaiden CYP3A-estäjien kanssa (esim. ketokonatsoli, itrakonatsoli, klaritromysiini, indinaviiri, nefatsodoni, nelfinaviiri, ritonaviiri, sakinaviiri, telitromysiini, vorikonatsoli) pitäisi välttää, sillä kabatsitakselin pitoisuus plasmassa voi suurentua (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Aprepitantin, kohtalaisen CYP3A:n estäjän, samanaikainen antaminen ei vaikuttanut kabatsitakselin puhdistumaan.
CYP3A-entsyymi-induktorit
Rifampisiinin, voimakkaan CYP3A-entsyymi-induktorin, toistetut annokset (600 mg kerran vuorokaudessa) suurensivat kabatsitakselin puhdistumaa 21 %, josta seurasi AUC-arvon pieneneminen 17 %:lla. Tästä syystä yhteiskäyttöä voimakkaiden CYP3A-induktorien kanssa (esim. fenytoiini, karbamatsepiini, rifampiini, rifabutiini, rifapentiini, fenobarbitaali) pitäisi välttää, sillä kabatsitakselin pitoisuus plasmassa voi pienentyä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Lisäksi potilaiden pitäisi välttää mäkikuisman käyttöä.
OATP1B1
In vitro kabatsitakselin on myös osoitettu estävän orgaanisten anionien kuljettajaproteiinipolypeptidejä (Organic Anion Transport Polypeptides) OATP1B1. Yhteisvaikutuksen riski OATP1B1:n substraattien (esim. statiinit, valsartaani, repaglinidi) kanssa on mahdollista, varsinkin infuusion aikana (1 tunti) ja enintään 20 minuuttia infuusion päättymisen jälkeen. OATP1B1:n substraattien annostelu on suositeltavaa 12 tuntia ennen infuusiota ja vähintään 3 tuntia infuusion päättymisen jälkeen.
Rokotukset
Elävien tai elävien heikennettyjen rokotteiden antaminen potilaille, joiden immuunivastetta on heikennetty kemoterapeuttisilla aineilla, voi aiheuttaa vakavia tai hengenvaarallisia infektioita. JEVTANA-hoitoa saavien potilaiden pitäisi välttää eläviä heikennettyjä rokotteita. Kuolleita tai inaktivoituja taudinaiheuttajia sisältäviä rokotteita voidaan käyttää, mutta vaste näihin rokotteisiin voi olla pienentynyt.
Raskaus ja imetys
Raskauden ehkäisy
Kabatsitakselin käyttöön liittyvän genotoksisuuden riskin (ks. kohta Prekliiniset tiedot turvallisuudesta) vuoksi miesten on käytettävä tehokasta ehkäisymenetelmää kabatsitakselihoidon aikana ja 4 kuukautta hoidon päättymisen jälkeen.
Raskaus
Ei ole olemassa tietoja kabatsitakselin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta äidille haitallisilla annoksilla (ks. kohta Prekliiniset tiedot turvallisuudesta) ja että kabatsitakseli läpäisee istukan (ks. kohta Prekliiniset tiedot turvallisuudesta). Kuten muut sytotoksiset lääkeaineet, kabatsitakseli voi aiheuttaa sikiövaurioita, jos sitä annetaan raskaana oleville naisille.
Kabatsitakselia ei ole tarkoitettu käytettäväksi naisille.
Imetys
Olemassa olevat farmakokineettiset tiedot koe-eläimistä ovat osoittaneet kabatsitakselin ja sen metaboliittien erittyvän rintamaitoon (ks. kohta Prekliiniset tiedot turvallisuudesta).
Hedelmällisyys
Eläinkokeiden perusteella on havaittu, että kabatsitakseli vaikutti urosrottien ja -koirien lisääntymisjärjestelmään ilman toiminnallista vaikutusta hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta). Kuitenkin, kun otetaan huomioon taksaanien farmakologinen aktiivisuus, niiden aneugeeniseen mekanismiin perustuva genotoksinen vaikutus ja useiden tämän luokan yhdisteiden vaikutus hedelmällisyyteen eläinkokeissa, vaikutusta miesten hedelmällisyyteen ei voida sulkea pois.
Kabatsitakselilla hoidettavia miehiä neuvotaan hakemaan ohjeita siemennesteen säilyttämisestä ennen hoidon aloittamista.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kabatsitakseli-valmisteella on kohtalainen vaikutus ajokykyyn ja koneiden käyttökykyyn, sillä se voi aiheuttaa väsymystä ja huimausta. Potilaita pitäisi kehottaa olemaan ajamatta tai käyttämättä koneita, jos heillä esiintyy näitä haittavaikutuksia hoidon aikana.
Haittavaikutukset
Yhteenveto haittavaikutusprofiilista
JEVTANA-valmisteen turvallisuus yhdistelmänä prednisonin tai prednisolonin kanssa arvioitiin kolmessa satunnaistetussa, avoimessa, kontrolloidussa tutkimuksessa (TROPIC, PROSELICA ja CARD). Tutkimuksiin osallistui yhteensä 1 092 potilasta, joilla oli levinnyt, kastraatioresistentti eturauhassyöpä ja joita hoidettiin kabatsitakseliannoksella 25 mg/m2 3 viikon välein. Potilaille annettiin keskimäärin 6–7 sykliä kabatsitakselia.
Näiden kolmen tutkimuksen yhdistettyjen tulosten analyyseihin perustuvat ilmaantuvuudet on esitetty seuraavassa luettelossa sekä jäljempänä taulukossa.
Kun otetaan huomioon kaikkiin gradus-luokkiin kuuluvat haitat, yleisimmät haittavaikutukset olivat anemia (99,0 %), leukopenia (93,0 %), neutropenia (87,9 %), trombosytopenia (41,1 %), ripuli (42,1 %), väsymys (25,0 %) ja heikkous (15,4 %). Yleisimmät gradus ≥ 3-luokan haittavaikutukset, joita ilmeni vähintään 5 %:lla potilaista, olivat neutropenia (73,1 %), leukopenia (59,5 %), anemia (12,0 %), kuumeinen neutropenia (8,0 %) ja ripuli (4,7 %).
Haittavaikutusten vuoksi hoidon keskeyttäneiden potilaiden osuudet kabatsitakselihoitoa saaneista potilaista olivat samankaltaiset kaikissa kolmessa tutkimuksessa (18,3 % TROPIC-, 19,5 % PROSELICA- ja 19,8 % CARD-tutkimuksessa). Yleisimmät haittavaikutukset (> 1,0 %), jotka johtivat kabatsitakselihoidon keskeyttämiseen, olivat hematuria, väsymys ja neutropenia.
Yhteenveto haittavaikutuksista taulukossa
Haittavaikutukset on lueteltu taulukossa 2 MedDRA:n elinjärjestelmä- ja yleisyysluokituksen mukaan. Jokaisen yleisyysryhmän sisällä haittavaikutukset on listattu vakavuuden mukaan alenevassa järjestyksessä. Haittavaikutusten voimakkuus on luokiteltu CTCAE 4.0-luokituksen mukaan (gradus ≥3 = G≥3). Yleisyydessä on otettu huomioon kaikki haitat niiden voimakkuudesta riippumatta ja ne on määritelty seuraavasti: Hyvin yleinen (≥1/10); yleinen (≥1/100, <1/10); melko harvinainen (≥1/1 000, <1/100); harvinainen (≥1/10 000, <1/1 000); hyvin harvinainen (<1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
Taulukko 2: Raportoidut haittavaikutukset ja veriarvojen poikkeamat kabatsitakselille yhdistelmänä prednisonin tai prednisolonin kanssa yhdistetyssä analyysissä (n = 1 092)
| Elinluokka | Haittavaikutus | Kaikkia gradus-luokkia n (%) | Gradus >3 n (%) | ||
|---|---|---|---|---|---|
| Hyvin yleinen | Yleinen | Melko harvinainen | |||
| Infektiot | Neutropeeninen infektio/sepsis* | 48 (4.4) | 42 (3,8) | ||
| Septinen sokki | 10 (0,9) | 10 (0,9) | |||
| Verenmyrkytys | 13 (1,2) | 13 (1,2) | |||
| Selluliitti | 8 (0,7) | 3 (0,3) | |||
| Virtsatietulehdus | 103 (9,4) | 19 (1,7) | |||
| Influenssa | 22 (2,0) | 0 | |||
| Virtsarakontulehdus | 22 (2,0) | 2 (0,2) | |||
| Ylempien hengitysteiden infektio | 23 (2,1) | 0 | |||
| Vyöruusu | 14 (1,3) | 0 | |||
| Kandidiaasi | 11 (1,0) | 1 (< 0,1) | |||
| Veri ja imukudos | Neutropeniaa* | 950 (87,9) | 790 (73,1) | ||
| Anemia a | 1 073 (99,0) | 130 (12,0) | |||
| Leukopeniaa | 1 008 (93,0) | 645 (59,5) | |||
| Trombosytopeniaa | 478 (44.1) | 44 (4.1) | |||
| Kuumeinen neutropenia | 87 (8,0) | 87 (8,0) | |||
| Immuunijärjestelmä | Yliherkkyys | 7 (0,6) | 0 | ||
| Aineenvaihdunta ja ravitsemus | Vähentynyt ruokahalu | 192 (17.6) | 11 (1,0) | ||
| Kuivuminen | 27 (2,5) | 11 (1,0) | |||
| Hyperglykemia | 11 (1,0) | 7 (0,6) | |||
| Hypokalemia | 8 (0,7) | 2 (0,2) | |||
| Psyykkiset häiriöt | Unettomuus | 45 (4,1) | 0 | ||
| Ahdistus | 13 (1,2) | 0 | |||
| Sekava olo | 12 (1,1) | 2 (0,2) | |||
| Hermosto | Dysgeusia | 64 (5,9) | 0 | ||
| Makuhäiriöt | 56 (5,1) | 0 | |||
| Perifeerinen neuropatia | 40 (3,7) | 2 (0,2) | |||
| Perifeerinen sensorinen neuropatia | 89 (8.2) | 6 (0,5) | |||
| Polyneuropatia | 9 (0,8) | 2 (0,2) | |||
| Tuntoharhat | 46 (4,2) | 0 | |||
| Heikentynyt tunto | 18 (1,6) | 1 (< 0,1) | |||
| Heitehuimaus | 63 (5,8) | 0 | |||
| Päänsärky | 56 (5,1) | 1 (< 0,1) | |||
| Letargia | 15 (1,4) | 1 (< 0,1) | |||
| Iskias | 9 (0,8) | 1 (< 0,1) | |||
| Silmät | Sidekalvotulehdus | 11 (1,0) | 0 | ||
| Kyynelnesteen erityksen lisääntyminen | 22 (2,0) | 0 | |||
| Kuulo ja tasapainoelin | Tinnitus | 7 (0,6) | 0 | ||
| Huimaus | 15 (1,4) | 1 (< 0,1) | |||
| Sydän* | Eteisvärinä | 14 (1,3) | 5 (0,5) | ||
| Takykardia | 11 (1,0) | 1 (< 0,1) | |||
| Verisuonisto | Hypotensio | 38 (3,5) | 5 (0,5) | ||
| Syvä laskimotromboosi | 12 (1,1) | 9 (0,8) | |||
| Hypertensio | 29 (2,7) | 12 (1,1) | |||
| Ortostaattinen hypotension | 6 (0,5) | 1 (< 0,1) | |||
| Kuumat aallot | 23 (2,1) | 1 (< 0,1) | |||
| Punastelu | 9 (0,8) | 0 | |||
| Hengityselimet, rintakehä ja välikarsina | Hengenahdistus | 97 (8,9) | 9 (0,8) | ||
| Yskä | 79 (7,2) | 0 | |||
| Suunielun kipu | 26 (2,4) | 1 (< 0,1) | |||
| Keuhkokuume | 26 (2,4) | 16 (1.5) | |||
| Keuhkoembolia | 30 (2,7) | 23 (2,1) | |||
| Ruoansulatuselimistö | Ripuli | 460 (42,1) | 51 (4,7) | ||
| Pahoinvointi | 347 (31,8) | 14 (1,3) | |||
| Oksentelu | 207 (19,0) | 14 (1,3) | |||
| Ummetus | 202 (18,5) | 8 (0,7) | |||
| Vatsakipu | 105 (9,6) | 15 (1,4) | |||
| Ruuansulatushäiriö | 53 (4,9) | 0 | |||
| Ylävatsakipu | 46 (4,2) | 1 (< 0,1) | |||
| Peräpukamat | 22 (2,0) | 0 | |||
| Refluksitauti | 26 (2,4) | 1 (< 0,1) | |||
| Peräsuolen verenvuoto | 14 (1,3) | 4 (0,4) | |||
| Suun kuivuminen | 19 (1,7) | 2 (0,2) | |||
| Vatsan turvotus | 14 (1,3) | 1 (< 0,1) | |||
| Suutulehdus | 46 (4,2) | 2 (0,2) | |||
| Ileus* | 7 (0,6) | 5 (0,5) | |||
| Gastriitti | 10 (0,9) | 0 | |||
| Koliitti* | 10 (0,9) | 5 (0,5) | |||
| Ruoansulatuskanavan perforaatio | 3 (0,3) | 1 (< 0,1) | |||
| Ruoansulatuskanavan verenvuoto | 2 (0,2) | 1 (< 0,1) | |||
| Iho ja ihonalainen kudos | Hiusten lähtö | 80 (7,3) | 0 | ||
| Ihon kuivuminen | 23 (2,1) | 0 | |||
| Eryteema | 8 (0,7) | 0 | |||
| Kynsisairaus | 18 (1,6) | 0 | |||
| Luusto, lihakset ja sidekudos | Selkäkipu | 166 (15,2) | 24 (2,2) | ||
| Nivelkipu | 88 (8,1) | 9 (0,8) | |||
| Raajojen kipu | 76 (7,0) | 9 (0,8) | |||
| Lihaskouristukset | 51 (4,7) | 0 | |||
| Lihaskipu | 40 (3,7) | 2 (0,2) | |||
| Rinnan lihasten ja luuston kipu | 34 (3,1) | 3 (0,3) | |||
| Lihasheikkous | 31 (2,8) | 1 (0,2) | |||
| Kylkikipu | 17 (1,6) | 5 (0,5) | |||
| Munuaiset ja virtsatiet | Akuutti munuaisten vajaatoiminta | 21 (1,9) | 14 (1,3) | ||
| Munuaisten vajaatoiminta | 8 (0,7) | 6 (0,5) | |||
| Kivulias virtsaaminen | 52 (4,8) | 0 | |||
| Munuaiskoliikki | 14 (1,3) | 2 (0,2) | |||
| Verivirtsaisuus | 205 (18,8) | 33 (3,0) | |||
| Tiheävirtsaisuus | 26 (2,4) | 2 (0,2) | |||
| Hydronefroosi | 25 (2,3) | 13 (1,2) | |||
| Virtsaumpi | 36 (3,3) | 4 (0,4) | |||
| Inkontinenssi | 22 (2,0) | 0 | |||
| Virtsateiden obstruktio | 8 (0,7) | 6 (0,5) | |||
| Sukupuolielimet ja rinnat | Lantion kipu | 20 (1,8) | (5 (0,5) | ||
| Yleisoireet ja antopaikassa todettavat haitat | Väsymys | 333 (30,5) | 42 (3,8) | ||
| Heikkous | 227 (20,8) | 32 (2,9) | |||
| Kuume | 90 (8,2) | 5 (0,5) | |||
| Perifeerinen edeema | 96 (8,8) | 2 (0,2) | |||
| Limakalvojen tulehdus | 23 (2,1) | 1 (< 0,1) | |||
| Kipu | 36 (3,3) | 7 (0,6) | |||
| Rintakipu | 11 (1,0) | 2 (0,2) | |||
| Edeema | 8 (0,7) | 1 (< 0,1) | |||
| Vilunväreet | 12 (1,1) | 0 | |||
| Huonovointisuus | 21 (1,9) | 0 | |||
| Tutkimukset | Painonlasku | 81 (7,4) | 0 | ||
| Kohonnut aspartaattiamino-transferaasi | 13 (1,2) | 1 (< 0,1) | |||
| Kohonneet transaminaasit | 7 (0,6) | 1 (< 0,1) | |||
a perustuen laboratorioarvoihin
* ks. lisäselvitys alla
Valikoituja haittavaikutuksia
Neutropenia ja siihen liittyvät kliiniset tapahtumat
G-CSF:n käytön on osoitettu vähentävän neutropenian esiintymistä ja vaikeusastetta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Gradus ≥ 3 neutropenian esiintyvyys laboratoriotietojen perusteella vaihteli G-CSF:n käytön mukaan 44,7 %:sta 76,7 %:iin siten, että esiintyvyys oli pienin, kun G-CSF:ää käytettiin estolääkityksenä. Samoin gradus ≥ 3 kuumeisen neutropenian esiintyvyys vaihteli 3,2 %:sta 8,6 %:iin.
Neutropeenisia komplikaatioita (kuumeista neutropeniaa, neutropeenisia infektioita, neutropeenista sepsistä ja neutropeenista koliittia), jotka joissakin tapauksissa johtivat kuolemaan, ilmoitettiin 4,0 %:lla primaarista estolääkitystä G-CSF:llä saaneista potilaista ja 12,8 %:lla muista potilaista.
Sydämen toiminta- ja rytmihäiriöt
Yhdistettyjen tulosten analyysissä sydäntapahtumia ilmoitettiin 5,5 %:lla potilaista, ja heistä 1,1 % sai gradus ≥ 3-luokan sydämen rytmihäiriön. Takykardian yleisyys kabatsitakselia saaneilla oli 1,0 %, ja alle 0,1 % tapauksista oli luokkaa gradus ≥ 3. Eteisvärinän yleisyys oli 1,3 %. Sydämen vajaatoimintaa raportoitiin 2 potilaalla (0,2 %), ja yksi tapauksista johti kuolemaan. Yhdellä potilaalla raportoitiin kuolemaan johtanut kammiovärinä (0,3 %) ja 3 potilaalla sydänpysähdys (0,5 %). Tutkijan arvion mukaan yksikään tapauksista ei liittynyt hoitoon.
Verivirtsaisuus
Yhdistettyjen tietojen analyysissä verivirtsaisuutta (kaikkia vaikeusasteita) havaittiin 18,8 %:lla potilaista annoksella 25 mg/m2 (ks. kohta Farmakodynamiikka). Silloin kun sekoittavat syyt, kuten taudin eteneminen, mittaustekniset syyt, infektiot tai antikoagulantti-/tulehduskipu-/aspiriini-lääkitys, oli kirjattu, niitä todettiin melkein puolessa näistä tapauksista.
Muut laboratorioarvojen poikkeamat
Yhdistettyjen tietojen analyysissä gradus ≥ 3 anemian sekä lisääntyneen ASAT:n, ALAT:n ja bilirubiinin esiintyvyydet poikkeavien laboratorioarvojen perusteella olivat 12,0 %, 1,3 %, 1,0 % ja 0,5 %.
Ruoansulatuselimistö
Koliittia (mukaan lukien enterokoliitti ja neutropeeninen enterokoliitti) ja gastriittia on havaittu. Ruoansulatuskanavan verenvuotoa, ruoansulatuskanavan perforaatiota ja ileusta (suolitukosta) on myös ilmoitettu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hengityselimet, rintakehä ja välikarsina
Interstitiaalisia pneumonia-/pneumoniittitapauksia ja interstitiaalisia keuhkosairaustapauksia, joskus kuolemaan johtaneita, on ilmoitettu. Tapausten yleisyys on tuntematon (koska saatavissa oleva tieto ei riitä arviointiin) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Munuaiset ja virtsatiet
Sädehoidon myöhäisreaktiona ilmenevää kystiittiä, mukaan lukien hemorragista kystiittiä, on ilmoitettu melko harvoin.
Pediatriset potilaat (ks. kohta Annostus ja antotapa)
Muut erityisryhmät
Ikääntyneet potilaat
Kabatsitakselia eturauhassyöpätutkimuksissa annoksella 25 mg/m2 saaneista 1 092 potilaasta 755 oli 65-vuotiaita tai sitä vanhempia, joista 238 potilasta oli yli 75-vuotiaita. Seuraavia ei-hematologisia haittavaikutuksia raportoitiin ≥ 5 % yleisemmin potilailla, jotka olivat 65-vuotiaita tai sitä vanhempia, verrattuna nuorempiin: väsymys (33,5 % vs. 23,7 %), astenia (23,7 % vs. 14,2 %), ummetus (20,4 % vs. 14,2 %) ja hengenahdistus (10,3 % vs. 5,6 %). Neutropenia (90,9 % vs. 81,2 %) ja trombosytopenia (48,8 % vs. 36,1 %) olivat 5 % yleisempiä 65-vuotiailla tai vanhemmilla verrattuna nuorempiin potilaisiin. Suurimmat ikäryhmien väliset erot ilmoitetussa ilmaantuvuudessa olivat gradus ≥ 3 neutropeniassa (ilmaantuvuus 14 % suurempi ≥ 65-vuotiailla potilailla kuin < 65-vuotiailla) ja kuumeisessa neutropeniassa (ilmaantuvuus 4 % suurempi ≥ 65-vuotiailla potilailla kuin < 65-vuotiailla) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kabatsitakselille ei ole tunnettua vastalääkettä. Yliannostuksen odotettavissa olevat komplikaatiot ovat haittavaikutusten paheneminen kuten luuydinsuppressio ja ruuansulatuselimistön ongelmat.
Yliannostustapauksessa potilas pitäisi pitää erikoistuneessa yksikössä ja tarkan valvonnan alaisena. Potilaille pitäisi antaa terapeuttista G-CSF:ää mahdollisimman pian yliannostuksen huomaamisen jälkeen. Muuten on hoidettava oireiden mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Syöpälääkkeet, taksaanit, ATC-koodi: L01CD04
Vaikutusmekanismi
Kabatsitakseli on syöpälääke, jonka vaikutusmekanismi perustuu solujen mikrotubulusten toiminnan häirintään. Kabatsitakseli sitoutuu tubuliiniin ja edistää tubuliinin kerääntymistä mikrotubuluksiksi samalla, kun se estää niiden hajoamisen. Tämä johtaa mikrotubulusten stabilisoitumiseen johtaen solutoimintojen estoon mitoosissa ja interfaasissa.
Farmakodynaamiset vaikutukset
Kabatsitakseli osoitti laaja-alaista kasvaimen kasvua ehkäisevää tehoa pitkälle edenneissä hiiriin istutetetuissa ihmiskasvaimissa. Kabatsitakselilla on tehoa kasvaimissa, jotka ovat herkkiä dosetakselille. Lisäksi kabatsitakseli osoitti aktiivisuutta kasvainmalleissa, jotka eivät olleet herkkiä kemoterapialle, mukaan lukien dosetakselille.
Kliininen teho ja turvallisuus
JEVTANA-valmisteen teho ja turvallisuus yhdistelmänä prednisonin tai prednisolonin kanssa arvioitiin satunnaistetussa, avoimessa, kansainvälisessä, monikeskus-, faasin III tutkimuksessa (EFC6193) potilaille, joilla oli levinnyt, kastraatioresistentti eturauhassyöpä, joita oli aikaisemmin hoidettu dosetakselilla.
Kokonaiselossaoloaika oli tutkimuksen ensisijainen tehoa mittaava päätetapahtuma.
Toissijaisiin päätetapahtumiin kuuluivat etenemisvapaa elossaoloaika [PFS, progression free survival (aika tutkimukseen satunnaistamisesta taudin etenemiseen), prostataspesifisen antigeenin eli PSA-arvon nousu, kivun voimistuminen tai kuolema mistä syystä tahansa, huolimatta siitä mikä näistä tapahtui ensin], RECIST-kriteerien (Response Evaluation Criteria in Solid Tumours) mukaan arvioitu kasvainvaste, PSA-arvon nousu (määriteltynä ≥ 25 % kasvu tai > 50 % kasvu PSA-arvossa potilailla, joilla ei ollut vastetta hoitoon vs. oli vaste hoitoon), PSA-vaste (lasku seerumin PSA-tasoissa ainakin 50 %), kivun voimistuminen [määriteltynä McGill -Melzack-kysymyslomakkeen Present Pain Intensity (PPI) – kaavakeella ja Analgesic Score (AS)-kipulääkityspisteytyksellä] sekä kipuvaste (määriteltynä kahta pistettä suurempana laskuna lähtötilanteen mediaanin PPI:stä ilman samanaikaista lisäystä kipulääkityspisteytyksessä (AS) tai ≥ 50 % laskuna kipulääkkeiden käytössä verrattuna lähtötilanteen keskimääräiseen kipulääkityspisteytykseen (AS) ilman samanaikaista lisäystä kivussa).
Yhteensä 755 potilasta satunnaistettiin saamaan joko 25 mg/m² JEVTANA-valmistetta laskimoon 3 viikon välein enintään 10 sykliä yhdistettynä prednisoniin tai prednisoloniin 10 mg/vrk päivittäin suun kautta (n=378) tai saamaan 12 mg/m² mitoksantronia laskimoon 3 viikon välein enintään 10 sykliä yhdistettynä prednisoniin tai prednisoloniin 10 mg/vrk päivittäin suun kautta (n=377).
Tähän tutkimukseen kuului yli 18-vuotiaita potilaita, joilla oli levinnyt, kastraatioresistentti eturauhassyöpä, joka oli joko RECIST-kriteereillä mitattavissa tai ei mitattavissa oleva sairaus, johon liittyi nousevat PSA-arvot tai uusien leesioiden ilmaantuminen ja suorituskyky 0-2 Eastern Cooperative Oncology Groupin (ECOG) määritelmän mukaan. Potilaiden neutrofiilitason piti olla >1 500/mm3, verihiutaleiden >100 000/mm3, hemoglobiinin >10 g/dl, kreatiniinin < 1,5 x ULN, kokonaisbilirubiinin <1 x ULN, ASAT ja ALAT <1,5 x ULN.
Tutkimukseen ei otettu potilaita, joilla oli kongestiivinen sydämen vajaatoiminta tai oli ollut sydäninfarkti viimeisten 6 kuukauden aikana tai joiden sydämen rytmihäiriöt, angina pectoris ja/tai verenpaine ei ollut hallinnassa.
Hoitoryhmät olivat samankaltaiset taustatekijöiden, kuten iän, rodun ja suorituskyvyn (0-2 ECOG:n mukaan) kesken. JEVTANA-ryhmässä keski-ikä oli 68 vuotta, vaihteluväli (46-92) ja väestöryhmäjakauma oli 83,9 % kaukaasialaisia, 6,9 % aasialaisia/Kauko-Idästä, 5,3 % tummaihoisia ja 4 % muita.
Mediaani syklien määrä oli 6 JEVTANA-ryhmässä ja 4 mitoksantroniryhmässä. Tutkimuksen loppuun asti (10 sykliä) olleita potilaita oli 29,4 % JEVTANA-ryhmässä ja 13,5 % vertailuryhmässä.
Kokonaiselossaoloaika oli merkitsevästi pidempi JEVTANA-ryhmässä verrattuna mitoksantroniryhmään (15,1 kuukautta vs. 12,7 kuukautta vastaavasti) ja siihen liittyi 30 % kuoleman riskin alenema verrattuna mitoksantroniryhmään (ks. taulukko 3 ja kuva 1).
59 potilaan alaryhmä sai ennen hoitoa kumulatiivisen dosetakseliannoksen < 225 mg/m2 (29 potilasta kuului JEVTANA-ryhmään ja 30 potilasta mitoksantroniryhmään). Kokonaiselossaoloajassa ei ollut merkittävää eroa potilasryhmien välillä (HR (95 % CI) 0,96 (0,49-1,86)).
Taulukko 3 – JEVTANA-valmisteen tehokkuus EFC6193-tutkimuksessa levinnyttä, kastraatioresistenttiä eturauhassyöpää sairastavien potilaiden hoidossa
| - | JEVTANA + prednisoni n=378 | mitoksantroni + prednisoni n=377 |
| Kokonaiselossaoloaika | - | - |
| Kuolleiden potilaiden osuus (%) | 234 (61,9 %) | 279 (74 %) |
| Mediaani elossaoloaika (kuukausina) (95 % CI) | 15,1 (14,1-16,3) | 12,7 (11,6-13,7) |
| Riskisuhde (HR)1 (95 % CI) | 0,70 (0,59-0,83) | |
| p-arvo | <0,0001 | |
1HR (Hazard Ratio) arvioitu Coxin mallin mukaan; riskisuhde alle 1 suosii JEVTANA-valmistetta
Kuva 1: Kokonaiselossaoloaika (EFC6193) Kaplan Meierin mukaan
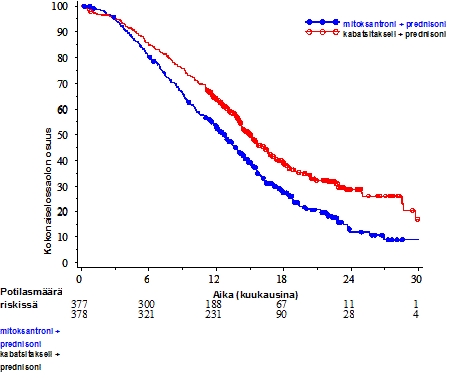
JEVTANA-ryhmässä tautivapaa elossaoloaika (PFS) piteni verrattuna mitoksantroniryhmään, 2,8 (2,4-3,0) kuukautta vs. 1,4 (1,4-1,7) vastaavasti, HR (95% CI) 0,74 (0,64-0,86), p < 0,0001.
Myös hoitovaste kasvaimeen 14,4 % (95 % CI: 9,6-19,3) oli merkitsevästi suurempi JEVTANA-ryhmän potilailla kuin mitoksantroniryhmän 4,4 % (95 % CI: 1,6-7,2), p = 0.0005.
PSA-arvoon liittyvät toissijaiset päätetapahtumat toteutuivat JEVTANA-ryhmässä. Aika PSA-arvon nousuun mediaani oli 6,4 kuukautta (95% CI: 5,1-7,3) JEVTANA-ryhmän potilailla verrattuna mitoksantroniryhmän potilaisiin 3,1 kuukautta (95% CI: 2,2-4,4), HR 0,75 kuukautta (95% CI 0,63-0,90), p=0,0010. PSA-vaste sai 39.2 % JEVTANA-ryhmän potilaista (95 % CI: 33,9-44,5) vs. 17,8 % mitoksantroniryhmän potilaista (95 % CI: 13,7-22,0), p = 0,0002.
Ryhmien välillä ei ollut tilastollista eroa kivun etenemisessä ja kipuvasteessa.
Tehon kliinisesti hyväksyttävää vertailukelpoisuutta tavoittelevassa (non-inferiority), kansainvälisessä, satunnaistetussa, avoimessa, monikeskus-, faasin III tutkimuksessa (EFC11785) 1200 potilasta, joilla oli levinnyt kastraatioresistentti eturauhassyöpä, ja joita oli aiemmin hoidettu dosetakselia sisältävällä hoidolla, satunnaistettiin saamaan kabatsitakselia joko annoksella 25 mg/m2 (n=602) tai 20 mg/m2 (n=598). Kokonaiselossaoloaika (OS) oli ensisijainen tehokkuuden päätetapahtuma.
Tutkimus saavutti ensisijaisen tavoitteensa osoittaessaan kabatsitakseliannoksen 20 mg/m2 olevan yhtä hyvä kuin vertailuannos 25 mg/m2 (ks. taulukko 4). Tilastollisesti merkitsevä suurempi prosenttiosuus potilaista (p < 0,001) sai vasteen PSA-arvoon 25 mg/m2-ryhmässä (42,9 %) verrattuna 20 mg/m2-ryhmään (29,5 %). Tilastollisesti merkitsevä PSA-arvon huononemisen riski oli suurempi potilailla, jotka saivat 20 mg/m2-annoksen verrattuna 25 mg/m2-annoksen saaneisiin (HR 1,195; 95 % CI: 1,025–1,393). Toissijaisissa päätetapahtumissa ei ollut tilastollisesti merkitsevää eroa (tautivapaa elossaoloaika (PFS), kasvaimen ja kivun hoitovaste, kasvaimen kasvun ja kivun eteneminen sekä neljä FACT-P-kyselylomakkeen kohtaa).
Taulukko 4 − Kokonaiselossaoloaika EFC11785-tutkimuksessa kabatsitakseli 25 mg/m2 -ryhmässä verrattuna kabatsitakseli 20 mg/m2 -ryhmään (Intent‑to–treat -analyysi) – tehokkuuden ensisijainen päätepiste
CBZ20+PRED n=598 | CBZ25+PRED n=602 | |
| Kokonaiselossaoloaika | ||
| Kuolemat, lkm (%) | 497 (83,1 %) | 501 (83,2 %) |
| Mediaani elossaoloaika (95 % CI) (kk) | 13,4 (12,19–14,88) | 14,5 (13,47–15,28) |
| Riskisuhdea | ||
| vs. CBZ25+PRED | 1,024 | − |
| 1-suuntainen 98,89 % UCI | 1,184 | − |
| 1-suuntainen 95 % LCI | 0,922 | − |
CBZ20=Kabatsitakseli 20 mg/m2, CBZ25=Kabatsitakseli 25 mg/m2, PRED=Prednisoni/Prednisoloni
CI=luottamusväli, LCI=luottamusvälin alaraja, UCI=luottamusvälin yläraja
a Riskisuhde arvioitiin käyttämällä Coxin suhteellista riskiregressioanalyysimallia. Riskisuhde < 1 osoittaa pienempää riskiä kabatsitakseliannokselle 20 mg/m2 verrattuna annokseen 25 mg/m2.
EFC11785-tutkimuksessa kabatsitakseliannokselle 25 mg/m2 havaittu turvallisuusprofiili oli laadullisesti ja määrällisesti samanlainen kuin EFC6193-tutkimuksessa. EFC11785-tutkimus osoitti, että kabatsitakseliannoksen 20 mg/m2 turvallisuusprofiili oli parempi.
Taulukko 5 − Yhteenveto kabatsitakseli 25 mg/m2 -ryhmän turvallisuustiedoista verrattuna kabatsitakseli 20 mg/m2 -ryhmään EFC11785-tutkimuksessa.
| CBZ20+PRED | CBZ25+PRED | |
| n=580 | n=595 | |
| Syklien lukumäärän mediaani/ hoidon keston mediaani | 6/18 viikkoa | 7/21 viikkoa |
| Niiden potilaiden lukumäärä, joiden annosta laskettiin n (%) | 20–15 mg/m2: 58 (10,0 %) 15–12 mg/m2: 9 (1,6 %) | 25–20 mg/m2: 128 (21,5 %) 20–15 mg/m2: 19 (3,2 %) 15–12 mg/m2: 1 (0,2 %) |
| Haittavaikutukset, kaikki vaikeusasteeta (%) | ||
| Ripuli | 30,7 | 39,8 |
| Pahoinvointi | 24,5 | 32,1 |
| Väsymys | 24,7 | 27,1 |
| Verivirtsaisuus | 14,1 | 20,8 |
| Astenia | 15,3 | 19,7 |
| Vähentynyt ruokahalu | 13,1 | 18,5 |
| Oksentelu | 14,5 | 18,2 |
| Ummetus | 17,6 | 18,0 |
| Selkäkipu | 11,0 | 13,9 |
| Kliininen neutropenia | 3,1 | 10,9 |
| Virtsatietulehdus | 6,9 | 10,8 |
| Perifeerinen sensorinen neuropatia | 6,6 | 10,6 |
| Makuhäiriö | 7,1 | 10,6 |
| Gradus ≥ 3 haittavaikutuksetb (%) | ||
| Kliininen neutropenia | 2,4 | 9,6 |
| Kuumeinen nuetropenia | 2,1 | 9,2 |
| Veriarvojen poikkeamatc (%) | ||
| Gradus ≥ 3 neutropenia | 41,8 | 73,3 |
| Gradus ≥ 3 anemia | 9,9 | 13,7 |
| Gradus ≥ 3 trombosytopenia | 2,6 | 4,2 |
CBZ20=Kabatsitakseli 20 mg/m2, CBZ25=Kabatsitakseli 25 mg/m2, PRED=Prednisoni/Prednisoloni
a Haittavaikutukset (kaikki vaikeusasteet), joiden esiintyvyys on suurempi kuin 10 %
b Gradus ≥ 3 haittavaikutukset, joiden esiintyvyys on suurempi kuin 5 %
c Perustuu laboratorioarvoihin
Prospektiivisessa, kansainvälisessä, satunnaistetussa, aktiivikontrolloidussa ja avoimessa vaiheen IV tutkimuksessa (LPS14201/CARD-tutkimus) 255 potilasta, joilla oli metastaattinen, kastraatioresistentti eturauhassyöpä ja jotka olivat aiemmin saaneet missä tahansa järjestyksessä dosetakselia sisältävää hoitoa ja androgeenireseptoriin vaikuttavaa hoitoa (abirateronia tai entsalutamidia, ja tauti oli edennyt 12 kuukauden kuluessa hoidon aloittamisesta), satunnaistettiin saamaan joko 25 mg/m2 JEVTANA-valmistetta 3 viikon välein yhdistettynä prednisoniin tai prednisoloniin 10 mg/vrk päivittäin suun kautta (n = 129) tai androgeenireseptoriin vaikuttavaa hoitoa (1 000 mg abirateronia kerran vuorokaudessa sekä 5 mg prednisonia tai prednisolonia kaksi kertaa vuorokaudessa tai 160 mg entsalutamidia kerran vuorokaudessa (n = 126). Ensisijainen päätetapahtuma oli PCWG2-kriteerien (Prostate Cancer Working Group-2 ‑kriteerien) mukainen radiologisesti todennettu etenemisvapaa elossaoloaika (rPFS). Toissijaiset päätetapahtumat olivat kokonaiselossaoloaika, etenemisvapaa elossaoloaika, PSA-vaste ja kasvainvaste.
Taustatekijät ja sairauden ominaispiirteet olivat samankaltaiset eri hoitoryhmissä. Lähtötilanteessa kaikkien tutkittavien mediaani-ikä oli 70 vuotta, 95 %:lla tutkittavista ECOG-suorituskykyluokka oli 0 tai 1 ja Gleason-pistemäärän mediaani oli 8. Tutkittavista 61 % oli aiemmin saanut androgeenireseptoriin vaikuttavaa hoitoa dosetakselihoidon jälkeen.
Tutkimuksen ensisijainen päätetapahtuma saavutettiin: JEVTANA-valmistetta saaneilla rPFS oli merkitsevästi pidempi (8,0 kuukautta) kuin androgeenireseptoriin vaikuttavaa hoitoa saaneilla (3,7 kuukautta), ja radiologisesti todennetun etenemisen riski oli 46 % pienempi kuin androgeenireseptoriin vaikuttavaa hoitoa saaneilla (ks. taulukko 6 ja kuva 2).
Taulukko 6 – JEVTANA-hoidon teho CARD-tutkimuksessa metastaattista, kastraatioresistenttiä eturauhassyöpää sairastavilla potilailla (Intent-to-treat-analyysi) – radiologisesti todennettu etenemisvapaa elossaoloaika (rPFS)
| JEVTANA + prednisoni/prednisoloni + G-CSF
n = 129 | Androgeenireseptoriin vaikuttava hoito: abirateroni + prednisoni/prednisoloni tai entsalutamidi n = 126 |
Tapahtumien määrä tiedonkeruun päättymispäivänä (%) | 95 (73,6 %) | 101 (80,2 %) |
rPFS-ajan mediaani (kk) (95 % CI) | 8,0 (5,7–9,2) | 3,7 (2,8–5,1) |
Riskisuhde (HR) (95 % CI) | 0,54 (0,40–0,73) | |
p-arvo1 | < 0,0001 | |
1stratifioitu log rank -testi, merkitsevyyden kynnysarvo = 0,05
Kuva 2 – Ensisijainen päätetapahtuma: Radiologisesti todennettua etenemisvapaata elossaoloaikaa (rPFS) kuvaava Kaplan–Meier-käyrä (ITT-populaatio)
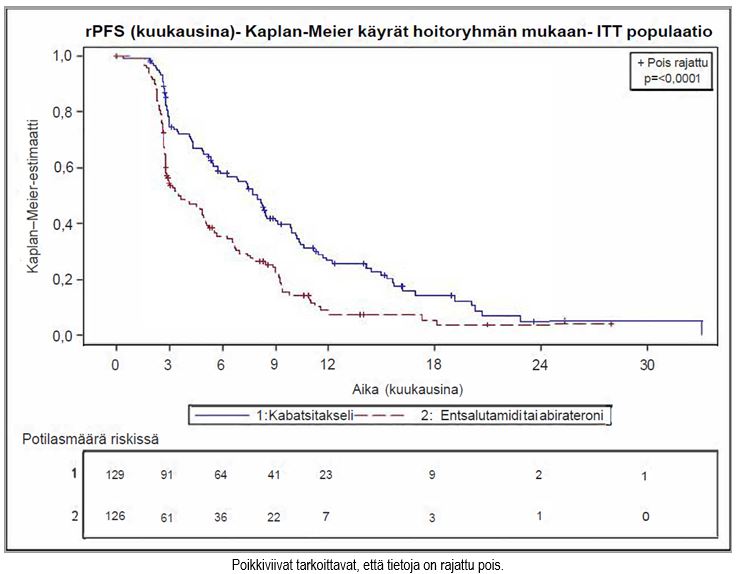
Suunniteltujen, lähtötilanteen mukaisiin stratifiointitekijöihin perustuneiden rPFS-ajan alaryhmäanalyysien perusteella riskisuhteeksi saatiin 0,61 (95 % CI: 0,39–0,96) tutkittavilla, jotka olivat saaneet aiemmin androgeenireseptoriin vaikuttavaa hoitoa ennen dosetakselia, ja 0,48 (95 % CI: 0,32–0,70) potilailla, jotka olivat aiemmin saaneet androgeenireseptoriin vaikuttavaa hoitoa dosetakselin jälkeen.
JEVTANA oli tilastollisesti parempi kuin androgeenireseptoriin vaikuttavat vertailuvalmisteet kaikkien alfavirheeltä suojattujen keskeisten toissijaisten päätetapahtumien suhteen. Näitä olivat kokonaiselossaoloaika (13,6 kuukautta JEVTANA-ryhmässä vs. 11,0 kuukautta androgeenireseptoriin vaikuttavaa hoitoa saaneessa ryhmässä, HR 0,64, 95 % CI: 0,46–0,89; p = 0,008), etenemisvapaa elossaoloaika (4,4 kuukautta JEVTANA-ryhmässä vs. 2,7 kuukautta androgeenireseptoriin vaikuttavaa hoitoa saaneessa ryhmässä, HR 0,52; 95% CI: 0,40–0,68), vahvistettu PSA-vaste (36,3 % JEVTANA-ryhmässä vs. 14,3 % androgeenireseptoriin vaikuttavaa hoitoa saaneessa ryhmässä, p = 0,0003), ja paras kasvainvaste (36,5 % JEVTANA-ryhmässä vs. 11,5 % androgeenireseptoriin vaikuttavaa hoitoa saaneessa ryhmässä, p = 0,0004).
JEVTANA-valmisteen annoksella 25 mg/m2 CARD-tutkimuksessa todettu turvallisuusprofiili oli kaiken kaikkiaan yhdenmukainen TROPIC- ja PROSELICA-tutkimuksissa todetun kanssa (ks. kohta Haittavaikutukset). Gradus ≥ 3 -asteen haittatapahtumien ilmaantuvuus oli 53,2 % JEVTANA-ryhmässä ja 46,0 % androgeenireseptoriin vaikuttavaa hoitoa saaneessa ryhmässä. Gradus ≥ 3 -asteen vakavien haittatapahtumien ilmaantuvuus oli 31,7 % JEVTANA-ryhmässä ja 37,1 % androgeenireseptoriin vaikuttavaa hoitoa saaneessa ryhmässä. Niiden tutkittavien osuus, jotka lopettivat tutkimushoidon kokonaan haittatapahtumien vuoksi, oli 19,8 % JEVTANA-ryhmässä ja 8,1 % androgeenireseptoriin vaikuttavaa hoitoa saaneessa ryhmässä. Niiden tutkittavien osuus, joilla ilmeni kuolemaan johtanut haittatapahtuma, oli 5,6 % JEVTANA-ryhmässä ja 10,5 % androgeenireseptoriin vaikuttavaa hoitoa saaneessa ryhmässä.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset JEVTANA-valmisteen käytöstä kaikkien pediatristen potilasryhmien hoidossa, käyttöaiheessa eturauhassyöpä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
JEVTANA-valmistetta arvioitiin avoimessa, monikeskus-, faasin 1/2 -tutkimuksessa, johon otettiin kaiken kaikkiaan 39 pediatrista potilasta (iältään 4–18 vuotta tutkimuksen osassa faasi 1 ja 3‑16 vuotta osassa faasi 2). Faasin 2 tutkimus ei osoittanut kabatsitakselin tehoa ainoana lääkkeenä pediatrisessa väestössä uusiutuneen tai refraktäärin diffuusin aivosiltagliooman (diffuse intrinsic pontine glioma, DIPG) eikä pahanlaatuisen gliooman (high grade glioma, HGG) hoidossa annoksella 30 mg/m².
Farmakokinetiikka
Populaatiofarmakokineettinen analyysi suoritettiin 170 potilaalla mukaan lukien potilailla, joilla oli edenneitä kiinteitä kasvaimia (n = 69), levinnyt rintasyöpä (n = 34) ja levinnyt eturauhasen syöpä (n = 67). Nämä potilaat saivat kabatsitakselia 10-30 mg/m2 viikoittain tai 3 viikon välein.
Imeytyminen
Levinnyttä eturauhassyöpää sairastavilla potilailla (n=67) Cmax oli 226 ng/ml (variaatiokerroin (Coefficient of Variation, CV): 107 %) ja se saavutettiin yhden tunnin laskimonsisäisen kabatsitakseli 25 mg/m2 infuusion lopussa (Tmax). AUC:n keskiarvo oli 991 ng.h/ml (CV: 34 %).
Suurta hajontaa ei havaittu suhteessa annokseen annettaessa 10 – 30 mg/m2 potilaille, joilla oli edennyt kiinteä kasvain (n = 126).
Jakautuminen
Jakautumistilavuus (Vss) oli 4870 l vakaassa tilassa (2640 l/m2 potilaille, joiden mediaani-BSA oli 1,84 m²).
Kabatsitakseli sitoutui ihmisen seerumin proteiineihin 89-92 % in vitro ja oli saturoitumaton 50 000 ng/ml asti, mikä vastaa maksimipitoisuutta kliinisissä tutkimuksissa. Kabatsitakseli sitoutuu pääasiassa ihmisen seerumin albumiiniin (82,0 %) ja lipoproteiineihin (87,9 % HDL, 69,8 % LDL, ja 55,8 % VLDL). Veri-plasma-pitoisuuksien suhde ihmisveressä vaihteli 0,90-0,99 in vitro osoittaen, että kabatsitakseli jakautui tasaisesti vereen ja plasmaan.
Biotransformaatio
Kabatsitakseli metaboloituu laajasti maksassa (>95 %), lähinnä CYP3A-isoentsyymin kautta (80 - 90 %). Kabatsitakseli on pääasiallinen yhdiste plasmassa ihmisen verenkierrossa. Plasmasta todettiin seitsemän metaboliittia (mukaan lukien 3 aktiivista metaboliittia O-demetylaatioista), joista päämetaboliitille altistuminen vastasi 5 % vaikuttavalle aineelle altistumisesta. Ihmisvirtsaan ja ulosteisiin erittyi noin 20 kabatsitakselin metaboliittia.
In vitro – tutkimusten perusteella on olemassa riski, että kabatsitakselin kliiniset pitoisuudet voivat estää lääkeaineita, jotka ovat CYP3A:n pääasiallisia substraatteja. Kliininen tutkimus on kuitenkin osoittanut, että kabatsitakseli (annosteltuna 25 mg/m2 kerta-annoksena yhden tunnin infuusiona) ei muuttanut midatsolaamin, CYP3A-tutkimussubstraatin, pitoisuutta plasmassa. Siten CYP3A-substraattien terapeuttisten annosten antamisella samaan aikaan kabatsitakselin kanssa ei odoteta olevan mitään kliinistä merkitystä.
Riski lääkeaineineen vaikutuksen estämisestä ei koske lääkeaineita, jotka ovat muiden CYP-entsyymien substraatteja (1A2, 2B6, 2C9, 2C8, 2C19, 2E1 ja 2D6) eikä myöskään ole vaaraa, että kabatsitakseli indusoisi lääkeaineita, jotka ovat CYP1A:n, CYP2C9:n ja CYP3A:n substraatteja. Kabatsitakseli ei estänyt in vitro varfariinin biotransformaatiota 7-hydroksivarfariiniksi CYP2C9:n kautta. Siten kabatsitakselilla ja varfariinilla ei odoteta olevan farmakokineettista yhteisvaikutusta in vivo.
Kabatsitakseli ei estänyt in vitro monilääkeresistenttejä proteiineja (Multidrug-Resistant Proteins (MRP)): MRP1 ja MRP2 tai orgaanisten kationien kuljettajaproteiineja (Organic Cation Transporter (OCT1)). Kabatsitakseli esti P-glykoproteiinin (PgP) (digoksiini, vinblastiini), rintasyövälle resistenttien proteiinien (Breast-Cancer-Resistant-Proteins (BRCP)) (metotreksaatti) ja orgaanisten anionien kuljettajaproteiinipolypeptidien (Organic Anion Transporting Polypeptide (OATP1B3) (CCK8)) kuljetuksen konsentraatioissa, jotka olivat ainakin 15-kertaisia kliinisiin arvoihin verrattuina, kun taas OATP1B1:n (estradioli-17β-glukuronidi) kuljetuksen se esti konsentraatioissa, jotka olivat vain 5-kertaisia kliinisiin arvoihin verrattuina. Siksi yhteisvaikutuksen riski MRP:n, OCT1:n, PgP:n, BCRP:n ja OATP1B3:n substraattien kanssa on epätodennäköinen in vivo annoksella 25 mg/m². Yhteisvaikutuksen riski OATP1B1:n kuljettajaproteiinien kanssa on mahdollista, varsinkin infuusion aikana (1 tunti) ja enintään 20 minuuttia infuusion päättymisen jälkeen (ks. kohta Yhteisvaikutukset).
Eliminaatio
Yhden tunnin aikana potilaille laskimoon annetun 25 mg/m2 [14C]-kabatsitakseli-infuusion jälkeen noin 80 % annetusta annoksesta poistui 2 viikon kuluessa. Kabatsitakseli erittyy pääasiassa ulosteisiin useina metaboliitteina (76 % annoksesta) munuaisten kautta poistuvan kabatsitakselin ja sen metaboliittien vastatessa alle 4 % annoksesta (2,3 % muuttumattomana lääkeaineena virtsassa).
Kabatsitakselin plasmapuhdistuma oli suuri 48,5 l/h (26,4 l/h/m2 potilaalle, jonka mediaani-BSA on 1,84 m2), ja sen terminaalivaiheen puoliintumisaika oli 95 tuntia.
Erityisryhmät
Iäkkäät potilaat
Populaatiofarmakokineettisessä analyysissä 70 potilaalle, jotka olivat 65-vuotiaita tai sitä vanhempia (57 potilasta oli 65-75 -vuotiaita ja 13 yli 75-vuotiaita) ei havaittu iän vaikuttavan kabatsitakselin farmakokinetiikkaan.
Pediatriset potilaat
JEVTANA-valmisteen turvallisuutta ja tehokkuutta lapsille ja alle 18-vuotiaille nuorille ei ole osoitettu.
Maksan vajaatoiminta
Kabatsitakseli poistuu pääasiassa maksan metabolian kautta.
Tutkimus, jossa oli 43 maksan vajaatoimintaa sairastavaa potilasta, ei osoittanut lievän (kokonaisbilirubiini > 1, ≤ 1,5 x normaaliarvon yläraja (ULN) tai ASAT > 1,5 x ULN) tai kohtalaisen (kokonaisbilirubiini > 1,5, ≤ 3,0 x ULN) maksan vajaatoiminnan vaikuttavan kabatsitakselin farmakokinetiikkaan. Suurin siedetty kabatsitakseliannos (MTD) oli lievää maksan vajaatoimintaa sairastavilla 20 mg/m2 ja kohtalaista maksan vajaatoimintaa sairastavilla 15 mg/m2.
Kolmella vaikeaa maksan vajaatoimintaa (kokonaisbilirubiini > 3 x ULN) sairastavalla potilaalla havaittiin 39 %:n alenema puhdistumassa verrattuna lievää maksan vajaatoimintaa sairastaviin potilaisiin, mikä viittaa siihen, että vaikealla maksan vajaatoiminnalla on vaikutusta kabatsitakselin farmakokinetiikkaan. Suurinta siedettyä annosta (MTD) ei pystytty vahvistamaan vaikeaa maksan vajaatoimintaa sairastaville potilaille.
Turvallisuus- ja siedettävyystietojen perusteella kabatsitakseliannosta on pienennettävä potilaille, joilla on lievä maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). JEVTANA on vasta-aiheinen vaikeaa maksan vajaatoimintaa sairastaville potilaille (ks. kohta Vasta-aiheet).
Munuaisten vajaatoiminta
Kabatsitakseli poistuu vain vähän munuaisten kautta (2,3 % annoksesta). Populaatiofarmakokineettinen analyysi 170 potilaalle, joista 14 sairasti kohtalaista munuaisten vajaatoimintaa (kreatiniinipuhdistuma 30−50 ml/min) ja 59 sairasti lievää munuaisten vajaatoimintaa (kreatiniinipuhdistuma 50−80 ml/min), osoitti, että lievä ja kohtalainen munuaisten vajaatoiminta ei vaikuttanut merkittävästi kabatsitakselin farmakokinetiikkaan. Tämä vahvistettiin erillisessä vertailevassa farmakokineettisessä tutkimuksessa, jossa potilailla oli kiinteä kasvain ja normaali munuaisten toiminta (8 potilasta), kohtalainen munuaisten vajaatoiminta (8 potilasta) tai vaikea munuaisten vajaatoiminta (9 potilasta), ja jotka saivat useita kabatsitakselisyklejä laskinmonsisäisinä kertainfuusioina, joissa annos oli enintään 25 mg/m2.
Prekliiniset tiedot turvallisuudesta
Seuraavia haittavaikutuksia ei ole todettu kliinisissä tutkimuksissa, mutta niitä on todettu koirista, joille on annosteltu kerta-annos, 5 päivän ajan tai viikottain kliinisiä pitoisuuksia pienempiä hoitoannoksia vastaavia määriä lääkeainetta. Siksi haitoilla, kuten maksan arteriolaarisella/periarteriolaarisella nekroosilla, sappitiehyiden hyperplasialla ja/tai maksasolujen nekroosilla, voi olla kliinistä merkitystä (ks. kohta Annostus ja antotapa).
Seuraavia haittavaikutuksia ei ole todettu kliinisissä tutkimuksissa, mutta niitä on todettu rotista toistetun annoksen toksisuustutkimuksissa annosteltaessa kliinisiä pitoisuuksia suurempia hoitoannoksia vastaavia määriä lääkeainetta. Siksi silmähaitoilla, joissa linssin subkapsulaariset syyt turposivat/rappeutuivat, voi olla kliinistä merkitystä. Nämä haitat korjaantuivat osittain 8 viikon kuluessa.
Kabatsitakselilla ei ole tehty karsinogeenisuustutkimuksia.
Kabatsitakseli ei indusoinut mutaatioita bakteerien mutaatiokokeessa (Amesin testi). Se ei ollut klastogeeninen in vitro – kokeessa ihmisen lymfosyyteissä (se ei indusoinut rakenteellisia kromosomipoikkeamia, mutta se lisäsi polyploidisten solujen määrää) ja se indusoi mikrotumien määrän kasvua rottien in vivo – testissä. Nämä genotoksisuuslöydökset (jotka perustuvat aneugeeniseen mekanismiin) ovat ominaisia yhdisteen farmakologiselle vaikutukselle (tubuliinin depolymerisaation estäminen).
Kabatsitakseli ei vaikuttanut urosrottien parittelukykyyn tai hedelmällisyyteen. Toistettujen annosten toksisuustutkimuksissa havaittiin kuitenkin rakkularauhasen rappeutumista ja siemenjohtimen surkastumista rotilla ja kivesten rappeutumista (minimaalinen epiteelisolujen nekroosi lisäkiveksessä) koirilla. Eläimet altistettiin ihmisten kliinisiä pitoisuuksia vastaaville tai sitä pienemmille annoksille kabatsitakselia.
Kabatsitakseli oli toksinen alkioille/sikiöille annosteltuna naarasrottien laskimoon kerran päivässä raskauspäivien 6-17 ajan. Tällä oli yhteys toksisuuteen emoille ja se johti sikiöiden kuolemiin ja sikiöiden keskimääräisen painon laskuun liittyneenä luurangon kehityksen viivästymiseen. Eläimet altistettiin ihmisten kliinisiä pitoisuuksia pienemmille annoksille kabatsitakselia. Kabatsitakseli kulkeutui rottien istukan seinämän lävitse.
Kabatsitakseli ja sen metaboliitit erittyivät rottien rintamaitoon 1,5 % pitoisuudeksi asti annetusta annoksesta 24 tunnin aikana.
Ympäristöön kohdistuvien riskien arviointi
Ympäristöön kohdistuvien riskien arvioinnin mukaan JEVTANA-valmisteen käyttö ei aiheuta merkittävää riskiä vesistöille (ks. kohta Käyttö- ja käsittelyohjeet käyttämättömän lääkevalmisteen hävittäminen).
Farmaseuttiset tiedot
Apuaineet
Konsentraatti
Polysorbaatti 80
Sitruunahappo
Liuotin
Etanoli 96 %
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Älä käytä PVC-infuusiopusseja tai polyuretaani-infuusiolaitteita infuusion valmistamisessa tai sen annostelussa.
Kestoaika
Avaamaton injektiopullo
3 vuotta
Avaamisen jälkeen
Konsentraattia ja liuotinta sisältävät injektiopullot on käytettävä välittömästi. Jos sitä ei käytetä välittömästi, käytössä olevan valmisteen säilytysaika ja -olosuhteet ovat käyttäjän vastuulla.
Liuotinlaimennuksen jälkeen
Kemiallinen ja fysikaalinen säilyvyys käytössä on osoitettu 1 tunnin ajan huoneenlämmössä (15 °C-30 °C). Mikrobiologiselta kannalta konsentraatti-liuotinseos pitäisi käyttää välittömästi. Jos sitä ei käytetä välittömästi, käytössä olevan valmisteen säilytysaika ja -olosuhteet ovat käyttäjän vastuulla eivätkä saisi tavallisesti kestää pidempään kuin 24 tuntia 2 °C-8 °C:een lämpötilassa ellei laimennusta ole tehty valvotuissa, validoiduissa, aseptisissa olosuhteissa.
Lopullisen laimennuksen jälkeen infuusiopussissa tai -pullossa
Infuusioliuoksen kemiallinen ja fysikaalinen säilyvyys on osoitettu 8 tunnin ajan huoneenlämmössä (sisältäen 1 tunnin infuusioajan) ja 48 tuntia jääkaapissa (sisältäen 1 tunnin infuusioajan).
Mikrobiologiselta kannalta infuusioneste pitäisi käyttää välittömästi. Jos sitä ei käytetä välittömästi, säilytysaika ja -olosuhteet ovat käyttäjän vastuulla eivätkä saisi tavallisesti kestää pidempään kuin 24 tuntia 2 °C–8 °C:een lämpötilassa ellei laimennusta ole tehty valvotuissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä alle 30 °C.
Älä säilytä kylmässä.
Lääkevalmisteen säilytys avaamisen ja laimentamisen jälkeen, ks. kohta Kestoaika
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
JEVTANA infuusiokonsentraatti ja liuotin, liuosta varten
60 mg (L:ei) 1 pakkaus (5304,12 €)
PF-selosteen tieto
Yksi pakkaus sisältää yhden injektiopullon konsentraattia ja yhden injektiopullon liuotinta:
- Konsentraatti: 1,5 ml konsentraattia 15 ml:n kirkkaassa lasisessa (tyyppi I) injektiopullossa, jonka harmaa klorobutyylikumitulppa on suljettu vaaleanvihreällä muovisella sinetillä suojatulla alumiinikorkilla. Jokainen injektiopullo sisältää 60 mg kabatsitakselia 1,5 ml:aa nimellistilavuutta kohden (täyttötilavuus: 73,2 mg kabatsitakselia/1,83 ml). JEVTANA-valmisteen kehittämisen aikana todettiin, että tämä täyttötilavuus kompensoi liuottamisessa syntyvää nestehävikkiä. Tällä ylitäytöllä varmistetaan, että liuotettua valmistetta saadaan vähintään 6 ml sisältäen 10 mg/ml JEVTANA-valmistetta, mikä vastaa etiketissä ilmoitettua määrää 60 mg/injektiopullo, kun liuottamiseen käytetään pakkauksen mukana tullut liuotin kokonaan.
- Liuotin: 4,5 ml liuotinta 15 ml:n kirkkaassa lasisessa injektiopullossa (tyyppi I), jonka harmaa klorobutyylikumitulppa on suljettu värittömällä muovisella sinetillä suojatulla kullanvärisellä alumiinikorkilla. Jokaisen injektiopullon sisältämä nimellistilavuus on 4,5 ml (täyttötilavuus: 5,67 ml). Tähän täyttötilavuuteen on päädytty kehitystyön aikana ja ylitäytöllä varmistetaan, että saadaan 10 mg/ml pitoisuus liuotettua JEVTANA-valmistetta, kun koko liuotinmäärä lisätään JEVTANA 60 mg konsentraatti-injektiopullon sisältöön.
Valmisteen kuvaus:
Konsentraatti on kirkas, keltainen tai ruskeankeltainen öljymäinen liuos.
Liuotin on kirkas ja väritön liuos.
Käyttö- ja käsittelyohjeet
JEVTANA-valmistetta saa käsitellä ja annostella vain syöpälääkkeiden antoon perehtynyt henkilökunta. Raskaana olevat työntekijät eivät saa käsitellä lääkevalmistetta. Kuten kaikkia syöpälääkkeitä JEVTANA-liuosta on käsiteltävä ja se on valmistettava varoen ottaen huomioon käytettävät välineet, henkilökohtaiset suojavälineet (esim. käsineet) ja valmistukseen liittyvät toimintatavat. Jos JEVTANA-liuos joutuu ihon kanssa kosketuksiin minkä tahansa valmisteluvaiheen aikana, pese kohta välittömästi vedellä ja saippualla. Jos sitä pääsee limakalvoille, pese kohta välittömästi vedellä.
Laimenna infuusiokonsentraatti aina koko mukana toimitetulla liuotinmäärällä ennen sen lisäämistä infuusioliuokseen.
Lue KOKO tämä kohta huolellisesti ennen lääkevalmisteen sekoittamista ja laimentamista. JEVTANA-valmiste vaatii KAKSI laimennuskertaa ennen annostelua. Noudata seuraavia valmistusohjeita.
Huomaa: Sekä JEVTANA 60 mg/1,5 ml konsentraatti-injektiopullo (täyttötilavuus 73,2 mg kabatsitakselia/1,83 ml) että liuotininjektiopullo (täyttötilavuus 5,67 ml) sisältävät ylitäytön kompensoimaan valmistuksen aikana syntyvää nestehävikkiä. Tämä ylitäyttö varmistaa, että kun mukana toimitettu KOKO liuotinmäärä on käytetty laimennukseen, liuos sisältää 10 mg/ml kabatsitakselia.
Seuraava kaksivaiheinen laimennus infuusioliuoksen valmistamista varten on tehtävä aseptisissa olosuhteissa.
Vaihe 1: Ensimmäinen infuusiokonsentraattiliuoksen laimennus mukana toimitetulla liuottimella.
Vaihe 1.1 Tarkista sekä konsentraattipullo että mukana toimitettu liuotinpullo. Konsentraatti- ja liuotinpullossa olevien liuosten pitää olla kirkkaita.
|
| |
Vaihe 1.2 Kallista pulloa ja vedä aseptisesti neulalla varustettuun ruiskuun injektiopullon koko liuotinmäärä.
|
| |
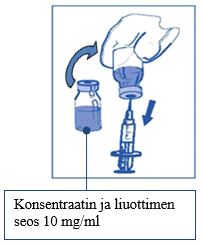
Vaihe 1.3 Injisoi ruiskun koko sisältö konsentraattipulloon. Injisoi liuotin hitaasti konsentraattipullon sisäseinämää pitkin vähentääksesi mahdollisimman tehokkaasti vaahdon syntymistä. Tämän laimennuksen jälkeen saatu liuos sisältää 10 mg/ml kabatsitakselia. |
| |
Vaihe 1.4 Poista ruisku ja neula ja sekoita varovasti kääntelemällä injektiopulloa ylös alas, kunnes liuos on kirkas ja homogeeninen. Tämä kestää noin 45 sekuntia.
|
|



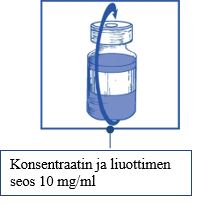
Vaihe 1.5 Anna liuoksen seistä noin 5 minuuttia ja tarkista sen jälkeen, että liuos on kirkas ja homogeeninen. Vaahdon esiintyminen tässä vaiheessa on normaalia.
|
|
Tämä konsentraatin ja liuottimen seos sisältää 10 mg/ml kabatsitakselia (vähintään 6 ml käytettävää liuosta). Toinen laimennus on tehtävä välittömästi (1 tunnin sisällä) vaiheen 2 ohjeiden mukaisesti. Määrätyn annoksen antamiseen saatetaan tarvita enemmän kuin yksi injektiopullollinen konsentraatin ja liuottimen seosta. | |
Vaihe 2: Toinen (viimeinen) infuusioliuoksen laimennus | |
Vaihe 2.1 Vedä aseptisesti neulalla varustettuun mittaruiskuun tarvittava annos konsentraatin ja liuottimen seosta, joka sisältää 10 mg/ml kabatsitakselia. Esimerkiksi 45 mg:n annos JEVTANA-valmistetta vastaa 4,5 ml:aa konsentraatin ja liuottimen seosta, joka on valmistettu vaiheen 1 mukaisesti.
Valmistusvaiheen 1 jälkeen pullon reunoille mahdollisesti jääneen vaahdon takia on suositeltavaa, että liuosta otettaessa neulaa pidetään pullon keskellä.
|
|
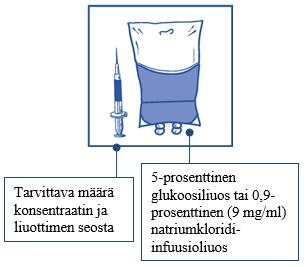
Vaihe 2.2 Injisoi annos steriiliin 5-prosenttista glukoosiliuosta tai 0,9-prosenttista (9 mg/ml) natriumkloridi-infuusioliuosta sisältävään pakkaukseen, joka ei sisällä PVC-muovia. Infuusioliuoksen pitoisuuden on oltava välillä 0,10 mg/ml - 0,26 mg/ml.
|
|
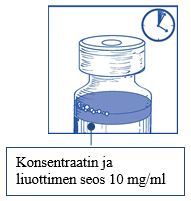
Vaihe 2.3 Poista ruisku ja sekoita infuusiopussin tai -pullon sisältö käsin heiluriliikkeellä.
|  |
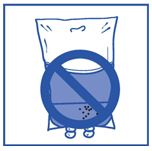
Vaihe 2.4 Kuten kaikki parenteraaliset liuokset, käyttövalmis infuusioliuos on tarkastettava silmämääräisesti ennen käyttöä. Koska infuusioliuos on ylikyllästetty, se voi kiteytyä ajan kuluessa. Tällöin liuosta ei saa käyttää vaan se on hävitettävä.
|  |
Infuusioliuos on käytettävä välittömästi. Käytettäväksi aiotun tuotteen säilytysaika voi kuitenkin olla pidempi kohdassa Kestoaika mainituissa erityistapauksissa.
Annosteluun suositellaan letkunsisäisen 0,22 mikrometrin huokoskoon suodatinta (0,2 mikrometrin huokoskoko on myös hyväksyttävä).
Älä käytä PVC:stä valmistettuja infuusionestepakkauksia tai polyuretaanista valmistettuja infuusiolaitteita JEVTANA-valmisteen valmistukseen tai annosteluun.
JEVTANA-valmistetta ei saa sekoittaa muiden kuin tässä erikseen mainittujen lääkeaineiden kanssa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
JEVTANA infuusiokonsentraatti ja liuotin, liuosta varten
60 mg 1 pakkaus
- Ei korvausta.
ATC-koodi
L01CD04
Valmisteyhteenvedon muuttamispäivämäärä
30.09.2025
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi