
VIDAZA injektiokuiva-aine, suspensiota varten 25 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 100 mg atsasitidiinia. Kun valmiste on saatettu käyttökuntoon, yksi ml suspensiota sisältää 25 mg atsasitidiinia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine, suspensiota varten.
Kliiniset tiedot
Käyttöaiheet
Vidaza on tarkoitettu sellaisten aikuispotilaiden hoitoon, joille ei voi tehdä hematopoieettisten kantasolujen siirtoa (haematopoietic stem cell transplantation, HSCT) ja joilla on:
- keskisuuren-2 tai korkean riskin myelodysplastinen oireyhtymä (myelodysplastic syndromes, MDS) International Prognostic Scoring System (IPSS) -luokituksen mukaan
- krooninen myelomonosyyttileukemia (KMML), luuytimessä blasteja 10–29 % ilman myeloproliferatiivista häiriötä
- akuutti myelooinen leukemia (AML), 20–30 % blasteja ja monilinjainen dysplasia, Maailman terveysjärjestön (WHO) luokituksen mukaan
- AML, luuytimessä blasteja > 30 % WHO:n luokituksen mukaan.
Ehto
Hoito aloitetaan ja toteutetaan kemoterapiaan perehtyneen lääkärin toimesta.
Annostus ja antotapa
Vidaza-hoito tulee aloittaa ja sitä tulee seurata kemoterapeuttisten aineiden käyttöön perehtyneen lääkärin valvonnassa. Potilaille tulee esilääkityksenä antaa antiemeettejä pahoinvointiin ja oksenteluun.
Annostus
Suositeltu aloitusannos ensimmäisellä hoitojaksolla on kaikilla potilailla lähtötason hematologisista laboratorioarvoista riippumatta 75 mg/m2 kehon pinta-alasta ihon alle pistettynä päivittäin 7 vuorokauden ajan, minkä jälkeen seuraa 21 vuorokauden lepojakso (28 vuorokauden hoitojakso).
Suositeltavaa on, että potilaita hoidetaan vähintään 6 jakson ajan. Hoitoa tulee jatkaa niin kauan kuin siitä on potilaalle hyötyä tai kunnes sairaus etenee.
Potilaita tulee tarkkailla hematologisen vasteen/toksisuuden ja munuaistoksisuuden varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet); seuraavan jakson aloittamisen viivästyttäminen tai annoksen pienentäminen jäljempänä kuvatulla tavalla saattaa olla tarpeen.
Vidaza‑valmistetta ei saa käyttää vaihdellen suun kautta otettavan atsasitidiinin kanssa. Altistuserojen takia suun kautta otettavan atsasitidiinin annos- ja hoito-ohjelmasuositukset eivät ole samat kuin injektiona annettavan atsasitidiinin. Terveydenhuollon ammattilaisia suositellaan tarkistamaan lääkevalmisteen nimi, annos ja antoreitti.
Laboratoriokokeet
Maksan toiminta-arvot, seerumin kreatiniini ja seerumin bikarbonaattiarvo tulee määrittää ennen hoidon aloittamista ja ennen jokaista hoitojaksoa. Täydellinen verenkuva on määritettävä ennen hoidon aloittamista ja tarpeen mukaan vasteen ja toksisuuden seuraamiseksi, mutta vähintään ennen jokaisen hoitojakson alkua.
Annoksen sovittaminen hematologisen toksisuuden johdosta
Hematologisella toksisuudella tarkoitetaan tietyssä jaksossa saavutettua alhaisinta verisolujen määrää (nadiiri), jos trombosyyttien määrä on ≤ 50,0 x 109/l ja/tai absoluuttinen neutrofiilimäärä (Absolute Neutrophil Count, ANC) ≤ 1 x 109/l.
Palautuminen määritellään sellais(t)en solulinjan (solulinjojen), joissa hematologista toksisuutta todettiin solumäärän lisääntymisenä, joka oli vähintään puolet nadiirin määrän ja lähtötason määrän absoluuttisesta erotuksesta plus nadiiri määrä (ts. verisolujen määrä palautumisessa ≥ nadiiri määrä + (0,5 x [|lähtötason määrä – nadiiri määrä|]).
Potilaat, joiden lähtötason verisolujen määrä ei ole alentunut (ts. valkosolut ≥ 3,0 x 109/l ja ANC ≥ 1,5 x 109/l ja trombosyytit ≥ 75,0 x 109/l) ennen ensimmäistä hoitoa
Jos Vidaza-hoidon jälkeen huomataan hematologista toksisuutta, seuraavaa hoitojaksoa tulee viivästyttää, kunnes trombosyyttimäärä ja ANC ovat palautuneet. Jos palautuminen saavutetaan 14 vuorokauden kuluessa, annoksen sovittaminen ei ole tarpeen. Jos palautumista ei kuitenkaan saavuteta 14 vuorokauden kuluessa, annosta tulee pienentää seuraavan taulukon mukaan. Annoksen muuttamisen jälkeen jakson kesto tulisi palauttaa 28 vuorokauteen.
| Jakson nadiiri määrä | ||
| ANC (x 109/l) | Trombosyytit (x 109/l) | Annos seuraavassa jaksossa, jos palautumista* ei saavuteta 14 vuorokaudessa (%) |
| ≤ 1,0 | ≤ 50,0 | 50 % |
| > 1,0 | > 50,0 | 100 % |
*Palautuminen = määrät ≥ nadiiri määrä + (0,5 x [lähtötason määrä – nadiiri määrä])
Potilaat, joiden lähtötason verisolujen määrä on alentunut (ts. valkosolut < 3,0 x 109/l tai ANC < 1,5 x 109/l tai trombosyytit < 75,0 x 109/l) ennen ensimmäistä hoitoa
Jos Vidaza-hoidon jälkeen valkosolujen, ANC:n tai trombosyyttien väheneminen hoitoa edeltävään vähenemiseen verrattuna on ≤ 50 %, tai jos se on enemmän kuin 50 %, mutta samalla solulinjojen differentaatiossa on parannusta, seuraavaa jaksoa ei saa viivästyttää eikä annosta sovittaa.
Jos valkosolujen, ANC:n tai trombosyyttien määrän väheneminen on enemmän kuin 50 % hoitoa edeltävään vähenemiseen verrattuna eikä solulinjojen differentaatiossa ole parannusta, seuraavaa Vidaza-hoitojaksoa tulee viivästyttää, kunnes trombosyyttimäärä ja ANC ovat palautuneet. Jos palautuminen saavutetaan 14 vuorokauden kuluessa, annoksen sovittaminen ei ole tarpeen. Jos palautumista ei kuitenkaan saavuteta 14 vuorokauden kuluessa, tulee määrittää luuytimen solukkuus. Jos luuytimen solukkuus on > 50 %, annosta ei tule sovittaa. Jos luuytimen solukkuus on ≤ 50 %, hoitoa tulee viivästyttää ja annosta pienentää seuraavan taulukon mukaan:
| Luuytimen solukkuus | Annos seuraavassa jaksossa, jos palautumista ei saavuteta 14 vuorokaudessa (%) | |
Palautuminen* ≤ 21 vrk | Palautuminen* > 21 vrk | |
| 15-50 % | 100 % | 50 % |
| < 15 % | 100 % | 33 % |
*Palautuminen = määrät ≥ nadiiri määrä + (0,5 x [lähtötason määrä – nadiiri määrä])
Annoksen muuttamisen jälkeen seuraavaan jakson kesto tulisi palauttaa 28 vuorokauteen.
Erityisryhmät
Iäkkäät potilaat
Iäkkäillä potilailla ei suositella erityistä annoksen sovittamista. Koska iäkkäillä potilailla munuaisten toiminta on todennäköisemmin heikentynyttä, munuaisten toiminnan seuranta saattaa olla tarpeen.
Munuaisten vajaatoimintaa sairastavat potilaat
Atsasitidiinia voidaan antaa munuaisten vajaatoimintaa sairastaville potilaille ilman aloitusannoksen säätämistä (ks. kohta Farmakokinetiikka). Jos seerumin bikarbonaattitaso laskee tuntemattomasta syystä alle 20 mmol/l, annosta tulee pienentää 50 %:lla seuraavassa jaksossa. Jos seerumin kreatiniini- tai ureatyppipitoisuus veressä (BUN) nousee tuntemattomasta syystä ≥ 2-kertaiseksi lähtötason arvojen yläpuolelle ja yli normaalin ylärajan (ULN), seuraavaa jaksoa tulee viivästyttää, kunnes arvot palautuvat normaaleiksi tai lähtötasolle, ja annosta tulee pienentää 50 %:lla seuraavassa hoitojaksossa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoimintaa sairastavat potilaat
Maksan vajaatoimintaa sairastavilla potilailla ei ole suoritettu virallisia tutkimuksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Vakavaa maksan vajaatoimintaa sairastavia potilaita tulee seurata tarkasti haittavaikutusten varalta. Maksan vajaatoimintaa sairastavilla potilailla ei suositella erityistä aloitusannoksen muuttamista ennen hoidon aloittamista; myöhemmät annosmuutokset tulee tehdä hematologisiin laboratorioarvoihin perustuen. Vidaza on vasta-aiheinen potilailla, joilla on edenneitä pahanlaatuisia maksakasvaimia (ks. kohdat Vasta-aiheet sekä Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Vidazan-valmisteen turvallisuutta ja tehoa 0–17 vuoden ikäisten lasten hoidossa ei ole vielä osoitettu. Tällä hetkellä saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, ei annossuosituksia voida antaa.
Antotapa
Käyttökuntoon saatettu Vidaza tulee pistää ihon alle käsivarren yläosaan, reiteen tai vatsaan. Pistoskohtia tulee vaihdella. Uudet pistokset tulee antaa vähintään 2,5 cm etäisyydelle aiemmasta pistoskohdasta eikä koskaan alueelle, jossa pistoskohta on arka, mustelmainen, punainen tai kovettunut.
Käyttökuntoon saattamisen jälkeen suspensiota ei saa suodattaa. Ks. kohdasta "Käyttö- ja käsittelyohjeet" ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Edenneet pahanlaatuiset maksakasvaimet (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Hematologinen toksisuus
Atsasitidiinihoitoon on liittynyt anemiaa, neutropeniaa ja trombosytopeniaa, erityisesti kahden ensimmäisen jakson aikana (ks. kohta Haittavaikutukset). Täydellinen verenkuva on määritettävä tarpeen mukaan vasteen ja toksisuuden seuraamiseksi, mutta vähintään ennen jokaisen hoitojakson alkua. Ensimmäisen jakson suositellun annoksen antamisen jälkeen seuraavien jaksojen annosta tulee pienentää tai sen antoa tulee viivästyttää nadiiri määrästä ja hematologisesta vasteesta riippuen (ks. kohta Annostus ja antotapa). Potilaita tulee kehottaa ilmoittamaan heti kuumejaksoista. Potilaita ja lääkäreitä kehotetaan myös tarkkailemaan verenvuodon merkkejä ja oireita.
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastavilla potilailla ei ole suoritettu virallisia tutkimuksia. Potilailla, joilla on etäpesäkkeisestä sairaudesta johtuva huomattava kasvaintaakka, on raportoitu progressiivinen maksakooma ja kuolema atsasitidiinihoidon aikana, erityisesti potilailla, joiden lähtötason seerumin albumiini oli < 30 g/l. Atsasitidiini on vasta-aiheinen potilailla, joilla on edenneitä pahanlaatuisia maksakasvaimia (ks. kohta Vasta-aiheet).
Munuaisten vajaatoiminta
Munuaishäiriöitä, jotka vaihtelivat kohonneista seerumin kreatiniiniarvoista munuaisten vajaatoimintaan ja kuolemaan, raportoitiin potilailla, joita hoidettiin laskimonsisäisellä atsasitidiinilla muihin kemoterapeuttisiin aineisiin yhdistettynä. Lisäksi viidellä kroonista myelooista leukemiaa (KML) sairastavalla sekä atsasitidiinilla ja etoposidilla hoidetulla potilaalla kehittyi renaalinen tubulaarinen asidoosi, joka määriteltiin seerumin bikarbonaattiarvon laskemisella < 20 mmol/l emäksisen virtsan ja hypokalemian (seerumin kaliumarvo < 3 mmol/l) yhteydessä. Jos tuntemattomasta syystä seerumin bikarbonaattiarvo laskee (< 20 mmol/l) tai seerumin kreatiniiniarvo tai BUN nousee, annosta tulee pienentää tai sen antoa tulee viivästyttää (ks. kohta Annostus ja antotapa).
Potilaita tulee kehottaa ilmoittamaan oliguria- ja anuriatapaukset välittömästi terveydenhoidon ammattilaiselle.
Vaikka haittavaikutusten esiintymistiheydessä ei havaittu kliinisesti merkitseviä eroja niiden tutkittavien välillä, joiden munuaisten toiminta oli normaali tai joilla oli munuaisten vajaatoimintaa, munuaisten vajaatoimintaa sairastavia potilaita tulee seurata tarkasti toksisuuden varalta, sillä atsasitidiini ja/tai sen metaboliitit erittyvät pääasiassa munuaisten kautta (ks. kohta Annostus ja antotapa).
Laboratoriokokeet
Maksan toiminta-arvot, seerumin kreatiniini ja seerumin bikarbonaattiarvo tulee määrittää ennen hoidon aloittamista ja ennen jokaista hoitojaksoa. Täydellinen verenkuva on määritettävä ennen hoidon aloittamista ja tarpeen mukaan vasteen ja toksisuuden seuraamiseksi, mutta vähintään ennen jokaisen hoitojakson alkua, ks. myös kohta Haittavaikutukset.
Sydän- ja keuhkosairaus
Potilaat, joilla on aiemmin ollut vakava kongestiivinen sydämen vajaatoiminta, kliinisesti epävakaa sydänsairaus tai keuhkosairaus, suljettiin pois keskeisistä rekisteritutkimuksista (AZA PH GL 2003 CL 001 ja AZA-AML-001), ja sen vuoksi atsasitidiinin turvallisuutta ja tehoa ei ole määritelty näillä potilailla. Kliinisestä tutkimuksesta äskettäin saadut tiedot potilaista, joiden anamneesissa tiedetään olevan sydän- ja verisuonitauti tai keuhkosairaus, osoittivat sydäntapahtumien lisääntyneen huomattavasti atsasitidiinin käytön yhteydessä (ks. kohta Haittavaikutukset). Atsasitidiinin määräämisessä tälle potilasryhmälle kehotetaan sen vuoksi noudattamaan varovaisuutta. Kardiopulmonaalista tutkimusta ennen hoitoa ja hoidon aikana tulee harkita.
Nekrotisoiva faskiitti
Vidaza-hoitoa saaneilla potilailla on raportoitu nekrotisoivaa faskiittia, myös kuolemaan johtaneina tapauksina. Jos potilaalle kehittyy nekrotisoiva faskiitti, Vidaza-hoito on lopetettava ja asianmukainen hoito on aloitettava heti.
Tuumorilyysioireyhtymä
Tuumorilyysioireyhtymän vaara on potilailla, joiden kasvaintaakka on ollut suuri ennen hoitoa. Näiden potilaiden tilaa tulee seurata tarkoin ja asianmukaisia varotoimenpiteitä on noudatettava.
Erilaistumisoireyhtymä
Injektiona annettavaa atsasitidiinihoitoa saavilla potilailla on raportoitu erilaistumisoireyhtymää (tunnetaan myös nimellä retinoiinihappo-oireyhtymä). Erilaistumisoireyhtymä voi johtaa kuolemaan. Sen oireita ja kliinisiä löydöksiä ovat mm. hengitysvaikeus, keuhkoinfiltraatit, kuume, ihottuma, keuhkoedeema, raajojen turvotus, nopea painon nousu, pleuraeffuusiot, perikardiumeffuusiot, hypotensio ja munuaisten toimintahäiriö (ks. kohta Haittavaikutukset). Erilaistumisoireyhtymään viittaavien oireiden ja löydösten ilmaantuessa ensimmäistä kertaa pitää harkita hoitoa suurilla laskimoon annettavilla kortikosteroidiannoksilla ja hemodynaamista seurantaa. Injektiona annettavan atsasitidiinihoidon keskeyttämistä oireiden häviämiseen saakka pitää harkita. Jos hoitoa jatketaan, siinä kehotetaan noudattamaan varovaisuutta.
Yhteisvaikutukset
In vitro -tietoihin perustuen atsasitidiinin metabolia ei vaikuta välittyvän sytokromi P450 -isoentsyymien (CYP:t), UDP-glukuronyylitransferaasien (UGT:t), sulfotransferaasien (SULT:t) ja glutationitransferaasien (GST:t) kautta; näihin metaboloiviin entsyymeihin in vivo liittyviä yhteisvaikutuksia pidetään siten epätodennäköisinä.
Atsasitidiinin kliinisesti merkittävät estävät tai induktiiviset vaikutukset sytokromi P450 -entsyymeihin ovat epätodennäköisiä (ks. kohta Farmakokinetiikka).
Atsasitidiinilla ei ole tehty virallisia kliinisiä lääkkeen yhteisvaikutustutkimuksia.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehille ja naisille
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 6 kuukautta hoidon päättymisen jälkeen. Miehiä on neuvottava olemaan siittämättä lasta hoidon aikana, ja miesten on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 3 kuukautta hoidon päättymisen jälkeen.
Raskaus
Ei ole olemassa tarkkoja tietoja atsasitidiinin käytöstä raskaana oleville naisille. Hiirellä tehdyt kokeet osoittavat reproduktiivista toksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmisille ei tunneta. Eläinkokeiden tuloksiin ja atsasitidiinin vaikutusmekanismiin perustuen atsasitidiinia ei tulisi käyttää raskauden aikana, eikä erityisesti raskauden ensimmäisen kolmanneksen aikana, ellei se ole selvästi välttämätöntä. Hoidon hyötyjä tulee punnita sikiölle mahdollisesti aiheutuvaan riskiin nähden jokaisessa yksittäistapauksessa.
Imetys
Ei tiedetä, erittyykö/erittyvätkö atsasitidiini/metaboliitit ihmisen rintamaitoon. Imetettävälle lapselle mahdollisesti aiheutuvien vakavien haittavaikutusten vuoksi imetys on vasta-aiheinen atsasitidiinihoidon aikana.
Hedelmällisyys
Atsasitidiinin vaikutuksesta ihmisten hedelmällisyyteen ei ole tietoja. Eläimillä on dokumentoitu atsasitidiinin käytöstä aiheutuneita haittavaikutuksia urosten hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta). Ennen hoitoa miespotilaita tulee kehottaa hakeutumaan neuvontaan koskien siittiöiden talteenottoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Atsasitidiinilla on vähäinen tai kohtalainen vaikutus ajokykyyn ja koneiden käyttökykyyn. Atsasitidiinin käytön yhteydessä on raportoitu väsymystä. Sen vuoksi suositellaan varovaisuutta ajettaessa tai käytettäessä koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Aikuispotilaat, joilla on MDS, KMML tai AML (luuytimessä blasteja 20–30 %)
Haittavaikutuksia, joita pidetään mahdollisesti tai todennäköisesti Vidaza-valmisteen antoon liittyvinä, esiintyi 97 %:lla potilaista.
Yleisimmät vakavat haittavaikutukset, jotka havaittiin keskeisessä tutkimuksessa (AZA PH GL 2003 CL 001), olivat kuumeinen neutropenia (8,0 %) ja anemia (2,3 %). Näitä raportoitiin myös tukitutkimuksissa (CALGB 9221 ja CALGB 8921). Muita vakavia haittavaikutuksia näissä kolmessa tutkimuksessa olivat infektiot, kuten neutropeeninen sepsis (0,8 %) ja keuhkokuume (2,5 %) (muutama tapauksista johti kuolemaan), trombosytopenia (3,5 %), yliherkkyysreaktiot (0,25 %) ja hemorragiset tapahtumat (esim. aivoverenvuoto [0,5 %], ruoansulatuselimistön verenvuoto [0,8 %] ja kallonsisäinen verenvuoto [0,5 %]).
Yleisimmät atsasitidiinihoidon yhteydessä raportoidut haittavaikutukset olivat hematologiset reaktiot (71,4 %), mukaan lukien trombosytopenia, neutropenia ja leukopenia (yleensä 3.–4. asteen), ruoansulatuselimistön tapahtumat (60,6 %), mukaan lukien pahoinvointi, oksentelu (yleensä 1.–2. asteen) tai pistoskohdan reaktiot (77,1 %, yleensä 1.–2. asteen).
Vähintään 65-vuotiaat AML-potilaat, joilla on luuytimessä blasteja > 30 %
Yleisimmät vakavat haittavaikutukset (≥ 10 %), jotka havaittiin AZA-AML-001-tutkimuksen atsasitidiinia saavassa hoitoryhmässä, olivat kuumeinen neutropenia (25,0 %), keuhkokuume (20,3 %) ja kuume (10,6 %). Muita, harvemmin raportoituja haittavaikutuksia atsasitidiiniryhmässä olivat sepsis (5,1 %), anemia (4,2 %), neutropeeninen sepsis (3,0 %), virtsatieinfektio (3,0 %), trombosytopenia (2,5 %), neutropenia (2,1 %), selluliitti (2,1 %), heitehuimaus (2,1 %) ja hengenahdistus (2,1 %).
Yleisimmät atsasitidiinihoidon yhteydessä raportoidut haittavaikutukset (≥ 30 %) olivat ruoansulatuselimistön tapahtumat, mukaan lukien ummetus (41,9 %), pahoinvointi (39,8 %) ja ripuli (36,9 %; yleensä 1.–2. asteen); yleisoireet ja antopaikassa todettavat haitat, mukaan lukien kuume (37,7 %, yleensä 1.–2. asteen); ja hematologiset tapahtumat, mukaan lukien kuumeinen neutropenia (32,2 %) ja neutropenia (30,1 %; yleensä 3.–4. asteen).
Haittavaikutustaulukko
Alla olevassa taulukossa 1 esitetään keskeisissä MDS:ää ja AML:ää koskeneissa kliinisissä tutkimuksissa sekä markkinoille tulon jälkeisessä seurannassa havaitut atsasitidiinihoitoon liittyneet haittavaikutukset.
Esiintyvyydet on määritetty seuraavalla tavalla: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Haittavaikutusten yleisyysluokka on ilmoitettu keskeisissä tutkimuksissa todetun suurimman esiintymistiheyden mukaan.
Taulukko 1: Atsasitidiinihoitoa saaneilla MDS- tai AML-potilailla raportoidut haittavaikutukset (kliinisissä tutkimuksissa ja markkinoille tulon jälkeen)
| Elinjärjestelmä | Hyvin yleinen
| Yleinen
| Melko harvinainen
| Harvinainen | Tuntematon |
|---|---|---|---|---|---|
| Infektiot | Keuhkokuume* (mukaan lukien bakteeri-, virus- ja sieni-infektiot), nasofaryngiitti | Sepsis* (mukaan lukien bakteeri-, virus- ja sieni-infektiot) neutropeeninen sepsis*, hengitystie- infektio (mukaan lukien ylähengitystiet ja keuhkoputket), virtsatieinfektio, selluliitti, divertikuliitti, suun sieni-infektio, sinuiitti, faryngiitti, riniitti, herpes simplex, ihoinfektio | Nekrotisoiva faskiitti* | ||
| Hyvän- ja pahan-laatuiset kasvaimet (mukaan lukien kystat ja polyypit) | erilaistumisoire-yhtymä*,a | ||||
| Veri ja imukudos | Kuumeinen neutropenia*, neutropenia, leukopenia, trombosytopenia, anemia | Pansytopenia*, luuytimen vajaatoiminta | |||
| Immuunijärjestelmä | Yliherkkyysreaktiot | ||||
| Aineenvaihdunta ja ravitsemus | Ruokahaluttomuus, ruokahalun heikentyminen, hypokalemia
| Kuivuminen | Tuumorilyysi-oireyhtymä | ||
| Psyykkiset häiriöt | Unettomuus | Sekavuustila, ahdistuneisuus | |||
| Hermosto | Huimaus, päänsärky | Kallonsisäinen verenvuoto*, pyörtyminen, uneliaisuus, letargia | |||
| Silmät | Silmäverenvuoto, sidekalvon verenvuoto | ||||
| Sydän | perikardiaalinen effuusio | sydänpussitulehdus | |||
| Verisuonisto | Hypotensio*, hypertensio, ortostaattinen hypotensio, hematooma | ||||
| Hengityselimet, rintakehä ja välikarsina | Hengenahdistus, nenäverenvuoto | Keuhkopussin nestekertymä, rasitushengenahdistus, nielun ja kurkunpään kipu | Interstitiaalinen keuhkosairaus | ||
Ruoansulatus- elimistö | Ripuli, oksentelu, ummetus, pahoinvointi, vatsakipu (mukaan lukien ylä- ja alavatsavaivat) | Ruoansulatuselimistön verenvuoto* (mukaan lukien suun verenvuoto), pukamavuoto, stomatiitti, ienverenvuoto, ruoansulatushäiriöt | |||
| Maksa ja sappi | Maksan vajaatoiminta*, progressiivinen maksakooma | ||||
| Iho ja ihonalainen kudos | Petekia, kutina (mukaan lukien yleistynyt), ihottuma, mustelma | Purppura, kaljuus, nokkosihottuma, punoitus, täpläihottuma | Sweetin oireyhtymä (akuutti kuumeinen neutrofiilinen dermatoosi), ihon märkäinen kuolio (pyoderma gangraenosum) | Ihovaskuliitti | |
| Luusto, lihakset ja sidekudos | Nivelkipu, tuki- ja liikuntaelimien kipu (mukaan lukien selkä-, luu- ja raajakipu) | Lihasspasmit, lihaskipu | |||
| Munuaiset ja virtsatiet | Munuaisten vajaatoiminta*, verivirtsaisuus, kohonnut seerumin kreatiniiniarvo | Munuaisperäinen asidoosi | |||
| Yleisoireet ja anto-paikassa todettavat haitat | Kuume*, väsymys, voimattomuus, rintakipu, pistoskohdan punoitus, pistoskohdan kipu, pistoskohdan reaktio (määrittelemätön) | Mustelma, hematooma, kovettuma, ihottuma, kutina, tulehdus, värinmuutos, kyhmy ja verenvuoto (pistoskohdassa), huonovointisuus, vilunväreet, katetrointikohdan verenvuoto | Pistoskohdan nekroosi (pistoskohdassa) | ||
| Tutkimukset | Painonlasku |
|
|
|
|
* = kuolemaan johtaneita tapauksia on raportoitu harvoin
a = ks. kohta Varoitukset ja käyttöön liittyvät varotoimet
Valittujen haittavaikutusten kuvaus
Hematologiset haittavaikutukset
Atsasitidiinihoidon yhteydessä yleisimmin raportoidut (≥ 10 %) hematologiset haittavaikutukset ovat anemia, trombosytopenia, neutropenia, kuumeinen neutropenia ja leukopenia, ja ne luokiteltiin yleensä 3. tai 4. asteisiksi. Näiden tapahtumien esiintymisriski on suurempi kahden ensimmäisen jakson aikana, minkä jälkeen niitä esiintyy harvemmin potilailla, joiden hematologinen toiminta on palautunut. Useimpia hematologisia haittavaikutuksia hoidettiin täydellisen verenkuvan rutiininomaisella seurannalla ja viivästyttämällä atsasitidiinin antoa seuraavassa jaksossa, antibioottiprofylaksilla ja/tai kasvutekijätuella (esim. G-CSF) neutropeniassa sekä verensiirroilla anemiassa tai trombosytopeniassa tarpeen mukaan.
Infektiot
Myelosuppressio saattaa johtaa neutropeniaan ja infektioriskin suurenemiseen. Atsasitidiinia saaneilla potilailla raportoitiin vakavia haittavaikutuksia, kuten sepsis, mukaan lukien neutropeeninen sepsis, ja keuhkokuume. Muutamat näistä tapauksista johtivat kuolemaan. Infektioita voidaan hoitaa käyttämällä infektiolääkkeitä ja kasvutekijätukea (esim. G-CSF) neutropeniassa.
Verenvuoto
Verenvuotoa saattaa esiintyä atsasitidiinia saavilla potilailla. Vakavia haittavaikutuksia, kuten ruoansulatuselimistön verenvuotoa ja kallonsisäistä verenvuotoa, on raportoitu. Potilaita tulee seurata verenvuodon merkkien ja oireiden varalta, erityisesti sellaisia potilaita, joilla on aiemmin ollut tai joiden hoidon aikana on esiintynyt trombosytopeniaa.
Yliherkkyys
Atsasitidiinia saaneilla potilailla on raportoitu vakavia yliherkkyysreaktioita. Anafylaktisen kaltaisen reaktion yhteydessä atsasitidiinihoito on välittömästi lopetettava ja sopiva oireenmukainen hoito aloitettava.
Ihon ja ihonalaisen kudoksen haittavaikutukset
Suurin osa ihon ja ihonalaisista haittavaikutuksista liittyi pistoskohtaan. Mikään näistä haittavaikutuksista ei johtanut atsasitidiinihoidon lopettamiseen tai atsasitidiiniannoksen pienentämiseen keskeisissä tutkimuksissa. Suurin osa haittavaikutuksista esiintyi kahden ensimmäisen hoitojakson aikana, ja niillä oli taipumus vähentyä seuraavien jaksojen myötä. Ihonalaiset haittavaikutukset, kuten pistoskohdan ihottuma/tulehdus/kutina, ihottuma, punoitus ja iholeesio saattavat vaatia hoitoa samanaikaisilla lääkevalmisteilla kuten antihistamiineilla, kortikosteroideilla ja muilla tulehduskipulääkkeillä (NSAID). Nämä ihoreaktiot on erotettava pehmytkudosinfektioista, joita voi esiintyä toisinaan pistoskohdassa. Pehmytkudosinfektioita, mukaan lukien selluliittia ja nekrotisoivaa faskiittia, jotka ovat harvinaisissa tapauksissa johtaneet kuolemaan, on raportoitu atsasitidiinin käytössä valmisteen markkinoille tulon jälkeen. Haittavaikutuksina ilmaantuvien infektioiden kliininen hoito, ks. kohta Haittavaikutukset; Infektiot.
Ruoansulatuselimistön haittavaikutukset
Yleisimmin raportoidut atsasitidiinihoitoon liittyvät ruoansulatuselimistön haittavaikutukset sisälsivät ummetuksen, ripulin, pahoinvoinnin ja oksentelun. Näitä haittavaikutuksia hoidettiin oireenmukaisesti antamalla pahoinvointia ja oksentelua vähentäviä lääkkeitä; ripulilääkkeitä sekä laksatiiveja ja/tai ulostuslääkkeitä ummetukseen.
Munuaisiin kohdistuvat haittavaikutukset
Atsasitidiinia saaneilla potilailla raportoitiin munuaishäiriöitä, jotka vaihtelivat kohonneista seerumin kreatiniiniarvoista ja hematuriasta munuaisperäiseen asidoosiin, munuaisten vajaatoimintaan ja kuolemaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksaan kohdistuvat haittavaikutukset
Maksan vajaatoimintaa, progressiivista maksakoomaa ja kuolemantapauksia atsasitidiinihoidon aikana on raportoitu potilailla, joilla on etäpesäkkeisestä sairaudesta johtuva huomattava kasvaintaakka (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sydäntapahtumat
Tiedot kliinisestä tutkimuksesta, johon otettiin mukaan potilaita, joiden anamneesissa tiedettiin olevan sydän- ja verisuonitauti tai keuhkosairaus, osoittivat sydäntapahtumien lisääntyneen potilailla, joilla oli äskettäin diagnosoitu akuutti myelooinen leukemia (AML) ja jotka olivat saaneet atsasitidiinihoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Iäkkäät potilaat
Atsasitidiinin käytöstä vähintään 85-vuotiaiden potilaiden hoitoon on vähän turvallisuutta koskevia tietoja (tutkimuksessa AZA-AML-001 hoidetuista potilaista 14 [5,9 %] oli vähintään 85-vuotiaita).
Pediatriset potilaat
Tutkimuksessa AZA-JMML-001 Vidazalla hoidettiin 28 pediatrista potilasta (yhden kuukauden ikäisistä alle 18-vuotiaisiin). Hoidettavana oli MDS (n = 10) tai juveniili myelomonosyyttinen leukemia (JMML) (n = 18) (ks. kohta Farmakodynamiikka).
Kaikilla 28 potilaalla oli ainakin yksi haittatapahtuma, ja 17:llä (60,7%) oli ainakin yksi hoitoon liittyvä tapahtuma. Yleisimmin raportoidut haittavaikutukset koko pediatrisessa ryhmässä olivat pyreksia, hematologiset tapahtumat, mukaan lukien anemia, trombosytopenia ja kuumeinen neutropenia, sekä ruoansulatuselimistön tapahtumat, mukaan lukien ummetus ja oksentelu.
Kolmella (3) tutkittavalla ilmeni hoidon aikana sellainen uusi haittatapahtuma, jonka johdosta lääkkeen anto lopetettiin (pyreksia, sairauden eteneminen ja vatsakipu).
Tutkimuksessa AZA-AML-004 Vidazalla hoidettiin 7 pediatrista potilasta (2–12-vuotiaisiin), joilla oli todettu AML:n molekulaarinen relapsi ensimmäisen täydellisen remission [CR1] jälkeen (ks. kohta Farmakodynamiikka).
Kaikilla 7 potilaalla oli ainakin yksi hoitoon liittyvä haittatapahtuma. Yleisimmin raportoidut haittatapahtumat olivat neutropenia, pahoinvointi, leukopenia, trombosytopenia, ripuli ja alaniiniaminotransferaasiarvon (ALAT) nousu. Kahdella potilaalla ilmeni hoitoon liittyvä tapahtuma, joka johti hoidon keskeyttämiseen (kuumeinen neutropenia, neutropenia).
Uusia turvallisuuteen liittyviä seikkoja ei tunnistettu kliinisen tutkimuksen aikana tässä Vidazalla hoidettujen pediatristen potilaiden lukumäärältään pienessä ryhmässä. Kokonaisturvallisuusprofiili sopi yhteen aikuispotilashavaintojen kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa lääkevalmisteen myyntiluvan myöntämisen jälkeisistä epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta:
www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisten tutkimusten aikana on raportoitu yksi atsasitidiinin yliannostustapaus. Potilaalla esiintyi ripulia, pahoinvointia ja oksentelua hänen saatuaan laskimonsisäisesti noin 290 mg/m2:n kerta-annos, lähes neljä kertaa suositeltua aloitusannosta enemmän.
Yliannostuksen sattuessa potilaan verenkuvaa tulee tarkkailla asianmukaisesti, ja hänelle on annettava tukihoitoa tarpeen mukaan. Atsasitidiinin yliannostukselle ei tunneta spesifistä vastalääkettä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, pyrimidiinianalogit; ATC-koodi: L01BC07
Vaikutusmekanismi
Atsasitidiinin uskotaan vaikuttavan antineoplastisesti usealla mekanismilla mukaan lukien sytotoksisuus poikkeavia hematopoieettisia soluja kohtaan luuytimessä ja DNA:n hypometylaatio. Atsasitidiinin sytotoksiset vaikutukset voivat johtua useista mekanismeista mukaan lukien DNA:n, RNA:n ja proteiinisynteesin estyminen, liittyminen RNA:han ja DNA:han sekä DNA-vaurion reittien aktivoituminen. Ei-proliferoituvat solut ovat suhteellisen epäherkkiä atsasitidiinille. Atsasitidiinin liittyminen DNA:han johtaa DNA:n metyylitransferaasien inaktivoitumiseen, mikä johtaa DNA:n hypometylaatioon. Normaalissa solukierron säätelyssä, erilaistumisessa ja kuoleman reiteillä osallisten poikkeavasti metyloituneiden geenien DNA:n hypometylaatio voi johtaa geenin uudelleen ilmentymiseen ja syöpäsolujen syöpää estävien toimintojen palautumiseen. DNA:n hypometylaation suhteellista tärkeyttä kliinisiin tuloksiin ei ole määritetty verrattuna sytotoksisuuteen tai muihin atsasitidiinin toimintoihin.
Kliininen teho ja turvallisuus
Aikuispotilaat (MDS, KMML ja AML [luuytimessä blasteja 20–30 %])
Vidaza-valmisteen teho ja turvallisuus tutkittiin kansainvälisessä kontrolloidussa, avoimessa, satunnaistetussa, vaiheen 3 vertailevassa monikeskus- ja rinnakkaisryhmätutkimuksessa (AZA PH GL 2003 CL 001) aikuispotilailla, joilla oli keskisuuren-2 ja korkean riskin MDS International Prognostic Scoring System (IPSS) -luokituksen mukaan, refraktaarinen blastianemia (refractory anaemia with excess blasts, RAEB), refraktaarinen blastianemia transformaatiossa (refractory anaemia with excess blasts in transformation, RAEB-T) ja modifioitunut krooninen myelomonosyyttinen leukemia French American British (FAB) -luokituksen mukaan. RAEB-T-potilaita (blasteja 21-30 %) pidetään nykyisin AML-potilaina tämänhetkisessä WHO:n luokituksessa. Atsasitidiinia ja parasta tukihoitoa (best supportive care, BSC) (n = 179) verrattiin tavanomaisiin hoito-ohjelmiin (conventional care regimens, CCR). CCR koostui pelkästä BSC:stä (n = 105), pienestä sytarabiiniannoksesta ja BSC:stä (n = 49) tai tavanomaisesta induktiokemoterapiasta ja BSC:stä (n = 25). Lääkärit olivat esivalinneet potilaat yhteen kolmesta CCR-hoidosta ennen satunnaistamista. Potilaille noudatettiin tätä esivalittua hoito-ohjelmaa, ellei heitä satunnaistettu saamaan Vidaza-valmistetta. Yhtenä mukaanottokriteerinä potilaiden ECOG-toimintakykyluokan (Eastern Cooperative Oncology Group) oli oltava 0–2. Sekundaarista MDS:ää sairastavat potilaat suljettiin pois tutkimuksesta. Tutkimuksen ensisijainen päätepiste oli eloonjäämisaika. Vidaza-valmistetta annettiin ihonalaisena annoksena 75 mg/m2 päivittäin 7 vuorokautta, minkä jälkeen seuraa 21 vuorokauden lepojakso (28 vuorokauden hoitojakso) mediaanin ollessa 9 jaksoa (vaihteluväli = 1-39) ja keskimäärin 10,2 jaksoa. Hoitoaikomusryhmässä (Intent to Treat, ITT) iän mediaani oli 69 vuotta (vaihteluväli 38–88 vuotta).
358 potilaalla (179 atsasitidiini ja 179 CCR) suoritetussa ITT-analyysissä Vidaza-hoitoon liittyi 24,46 kuukauden eloonjäämisajan mediaani verrattuna 15,02 kuukauteen CCR-hoitoa saavilla potilailla, mikä tarkoittaa 9,4 kuukauden eroa (ositettu log-rank p-arvo 0,0001). Hoitovaikutuksen vaarasuhde (HR) oli 0,58 (95 % CI: 0,43-0,77). Kahden vuoden eloonjäämisluvut olivat 50,8 % atsasitidiinia saavilla potilailla verrattuna 26,2 % CCR-hoitoa saavilla potilailla (p < 0,0001).
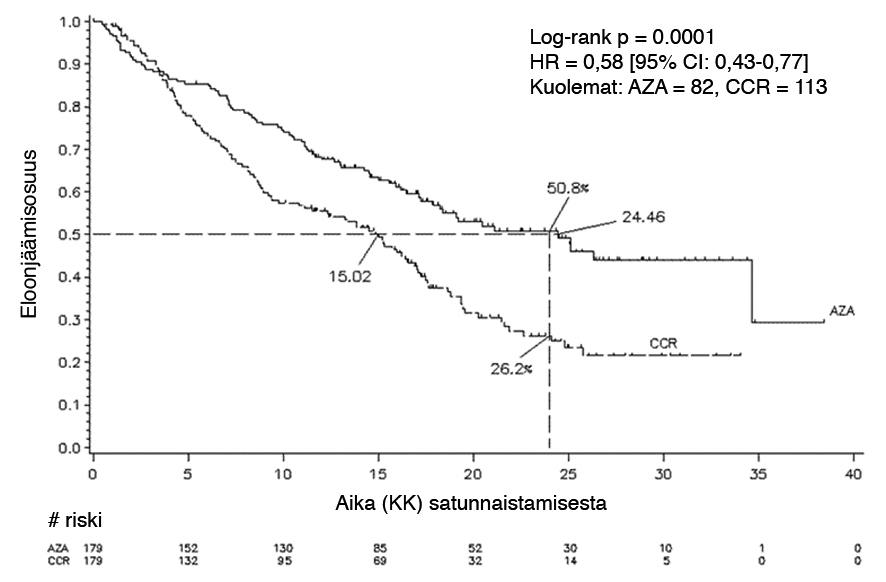
Lyhenteet: AZA=azacitidine (atsasitidiini); CCR=conventional care regimens (tavanomaiset hoito-ohjelmat); CI=confidence interval (luottamusväli); HR=hazard ratio (vaarasuhde).
Vidaza-valmisteen hyödyt eloonjäämiselle olivat yhdenmukaiset kontrolliryhmässä käytetyistä CCR-hoidon vaihtoehdoista (pelkkä BSC, pieni sytarabiiniannos ja BSC tai tavanomainen induktiokemoterapia ja BSC) huolimatta.
Kun IPSS:n sytogeneettisiä alaryhmiä analysoitiin, kaikissa ryhmissä todettiin samanlaisia eloonjäämisajan mediaania koskevia löydöksiä (hyvä, keskisuuri, huono sytogenetiikka, mukaan lukien monosomia 7).
Ikäryhmien analyysissä todettiin eloonjäämisajan mediaanin kohoaminen kaikissa ryhmissä (< 65 vuotta, ≥ 65 vuotta ja ≥ 75 vuotta).
Vidaza-hoitoon liitetty ajan mediaani kuolemaan tai AML:ksi muuttumiseen saakka oli 13,0 kuukautta verrattuna 7,6 kuukauteen CCR-hoitoa saavilla potilailla, mikä tarkoittaa 5,4 kuukauden parannusta ositetun log-rank p-arvon ollessa 0,0025.
Vidaza-hoitoon liittyi myös sytopenioiden ja niihin liittyvien oireiden väheneminen. Vidaza-hoito johti pienempään punasolu- ja trombosyyttisiirtojen tarpeeseen. Lähtötasolla punasolusiirroista riippuvaisista atsasitidiiniryhmän potilaista 45,0 % tuli punasolusiirroista riippumattomiksi hoitojakson aikana, verrattuna 11,4 %:iin yhdistettyjen CCR-ryhmien potilaista (tilastollisesti merkittävä [p < 0,0001] ero 33,6 % [95 % CI: 22,4 - 44,6]). Lähtötasolla punasolusiirroista riippuvaisilla ja sitten riippumattomiksi tulleilla potilailla punasolusiirroista riippumattomuuden keston mediaani oli 13 kuukautta atsasitidiiniryhmässä.
Vasteen arvioi tutkija tai riippumaton arviointitoimikunta (Independent Review Committee, IRC). Tutkijan määrittelemä kokonaisvaste (täydellinen remissio [CR] ja osittainen remissio [PR]) oli 29 % atsasitidiiniryhmässä ja 12 % yhdistetyssä CCR-ryhmässä (p = 0,0001). IRC:n määrittelemä kokonaisvaste (CR + PR) tutkimuksessa AZA PH GL 2003 CL 001 oli 7 % (12/179) atsasitidiiniryhmässä verrattuna 1 %:iin (2/179) yhdistetyssä CCR-ryhmässä (p = 0,0113). IRC:n ja tutkijan vastearvioinnin erot johtuivat International Working Group (IWG) -kriteereistä, joissa vaaditaan verisoluarvojen parantumista ja tämän parantumisen säilymistä vähintään 56 vuorokautta. Eloonjäämiselle aiheutuva hyöty osoitettiin myös potilailla, jotka eivät saavuttaneet täydellistä/osittaista vastetta atsasitidiinihoidon jälkeen. IRC:n määrittelemä hematologinen parannus (suuri tai pieni) saavutettiin 49 %:lla atsasitidiinia saaneista potilaista verrattuna 29 %:iin yhdistetyillä CCR:llä hoidetuista potilaista (p < 0,0001).
Potilailla, joilla oli yksi tai useampi sytogeneettinen poikkeavuus lähtötasolla, suuren sytogeneettisen vasteen osoittaneiden potilaiden prosenttiosuus oli samanlainen atsasitidiiniryhmässä ja yhdistetyssä CCR-ryhmässä. Pieni sytogeneettinen vaste oli tilastollisesti merkittävästi (p = 0,0015) korkeampi atsasitidiiniryhmässä (34 %) yhdistettyyn CCR-ryhmään verrattuna (10 %).
Vähintään 65-vuotiaat AML-potilaat, joilla on luuytimessä blasteja >30 %
Seuraavassa esitetyt tulokset kuvaavat tutkimuksen AZA-AML-001 hoitoaikeen mukaista (intent-to-treat, ITT) potilasjoukkoa (ks. hyväksytty käyttöaihe kohdasta Käyttöaiheet).
Vidaza-valmisteen tehoa ja turvallisuutta arvioitiin vaiheen 3 kansainvälisessä, kontrolloidussa, avoimessa, rinnakkaisryhmillä toteutetussa monikeskustutkimuksessa, johon osallistuneilla vähintään 65-vuotiailla potilailla oli äskettäin todettu de novo- tai sekundaarinen AML ja joilla oli luuytimessä blasteja > 30 % WHO:n luokituksen mukaan ja joille ei voitu tehdä hematopoieettisten kantasolujen siirtoa. Vidaza-valmistetta + BSC-hoitoa (n = 241) verrattiin CCR-hoitoon. CCR-hoito koostui pelkästä BSC-hoidosta (n = 45), pienestä sytarabiiniannoksesta + BSC-hoidosta (n = 158) tai tavanomaisesta intensiivisestä kemoterapiasta, johon kuului sytarabiini ja antrasykliini + BSC-hoito (n = 44). Lääkärit olivat esivalinneet potilaansa yhteen kolmesta CCR-hoidosta ennen satunnaistamista. Potilaat saivat esivalittua hoitoa, ellei heitä satunnaistettu saamaan Vidaza-valmistetta. Mukaanottokriteerien mukaan potilaiden ECOG-toimintakykyluokan oli oltava 0–2 ja sytogeneettisten poikkeavuuksien ennusteeltaan kohtalaisia tai huonoja. Tutkimuksen ensisijainen päätepiste oli eloonjäämisaika.
Vidaza-valmistetta annettiin ihonalaisena annoksena 75 mg/m2 päivittäin 7 vuorokauden ajan, mitä seurasi 21 vuorokauden tauko (28 vuorokauden hoitosykli). Syklien lukumäärän mediaani oli 6 sykliä (vaihteluväli: 1–28). Pelkkää BSC-hoitoa saaneilla potilailla syklien lukumäärän mediaani oli 3 sykliä (vaihteluväli: 1–20), pientä sytarabiiniannosta saaneilla potilailla 4 sykliä (vaihteluväli: 1–25) ja tavanomaista intensiivistä kemoterapiaa saaneilla potilailla 2 sykliä (vaihteluväli: 1–3, induktiosykli + 1–2 vakautussykliä).
Lähtötason yksilölliset parametrit olivat verrannolliset Vidaza- ja CCR-hoitoryhmien välillä. Tutkittavien ikämediaani oli 75,0 vuotta (vaihteluväli: 64–91 vuotta), 75,2 % tutkittavista oli valkoihoisia ja 59,0 % oli miehiä. Lähtötasolla tutkittavien sairaudeksi luokiteltiin WHO:n luokituksen mukaan 60,7 %:lla muutoin määrittämätön AML; 32,4 %:lla AML, johon liittyi myelodysplastisia muutoksia; 4,1 %:lla aikaisempiin hoitoihin liittyvä AML ja 2,9 %:lla AML, johon liittyy toistuva geneettinen poikkeavuus.
488 potilaalla (241 Vidaza ja 247 CCR) suoritetussa ITT-analyysissä Vidaza-hoitoon liittyi 10,4 kuukauden eloonjäämisajan mediaani verrattuna 6,5 kuukauteen CCR-hoitoa saaneilla potilailla: ero oli 3,8 kuukautta (ositetun log-rank-testin p-arvo 0,1009 [kaksisuuntainen]). Hoitovaikutuksen vaarasuhde oli 0,85 (95 % CI: 0,69–1,03). Yhden vuoden eloonjäämisluku oli 46,5 % Vidaza-valmistetta saaneilla potilailla ja 34,3 % CCR-hoitoa saaneilla potilailla.
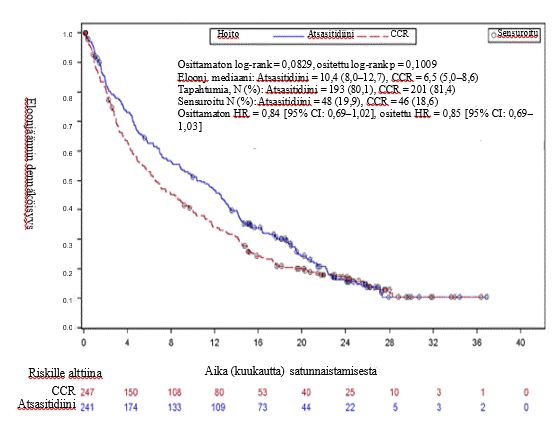
Vidaza- ja CCR-hoitojen välinen vaarasuhde, joka laskettiin etukäteen määriteltyjen lähtötason ennustetekijöiden mukaan korjatulla Coxin suhteellisen vaaran mallilla, oli 0,80 (95 % CI = 0,66, 0,99; p = 0,0355).
Vaikka tutkimuksella ei ollut voimaa osoittaa tilastollisesti merkitsevää eroa, kun atsasitidiinia verrattiin ennalta valittuihin CCR-hoitoryhmiin, Vidaza-hoitoa saaneiden potilaiden elinaika oli pidempi verrattuna CCR-hoitovaihtoehtoja eli pelkkää BSC-hoitoa tai pienen sytarabiiniannoksen ja BSC-hoidon yhdistelmää saaneisiin potilaisiin, ja oli samankaltainen verrattaessa tavanomaiseen intensiiviseen kemoterapiaan + BSC-hoitoon.
Vidaza-valmistetta suosiva trendi eloonjäännin suhteen oli nähtävissä kaikissa ennalta määritetyissä alaryhmissä (ikä [< 75 vuotta ja ≥ 75 vuotta], sukupuoli, rotu, ECOG-toimintakykyluokka [0 tai 1 ja 2], lähtötason sytogeneettisten poikkeavuuksien ennuste (kohtalainen ja huono), maantieteellinen alue, AML:n WHO-luokitus [mukaan lukien AML, johon liittyy myelodysplastisia muutoksia], lähtötason valkosolumäärä [≤ 5 x 109/l ja > 5 x 109/l], luuytimen blastien lähtötaso [≤ 50 % ja > 50 %] sekä anamneesissa MDS). Muutamissa ennalta määritetyissä alaryhmissä eloonjäännin vaarasuhde saavutti tilastollisen merkitsevyyden, mikä koski myös potilaita, joilla oli epäsuotuisa sytogeneettinen riski, AML-potilaita, joilla oli myelodysplastisia muutoksia, alle 75-vuotiaita potilaita, naispotilaita ja valkoihoisia potilaita.
Hematologiset ja sytogeneettiset vasteet olivat samankaltaisia sekä tutkijan että IRC:n arvioimina. IRC:n määrittelemä kokonaisvasteprosentti (täydellinen remissio [CR] + täydellinen remissio ilman verisolujen määrän täydellistä palautumista [CRi]) oli 27,8 % Vidaza-ryhmässä ja 25,1 % yhdistetyssä CCR-ryhmässä (p = 0,5384). CR- tai CRi-vasteen saavuttaneiden potilaiden remission keston mediaani oli 10,4 kuukautta (95 % CI: 7,2–15,2) Vidaza-ryhmässä ja 12,3 kuukautta (95 % CI: 9,0–17,0) yhdistetyssä CCR-ryhmässä. Vidaza-valmisteen hyöty eloonjäämiselle CCR-hoitoon verrattuna osoitettiin myös potilailla, jotka eivät saavuttaneet täydellistä vastetta.
Vidaza-hoito paransi veriarvoja ja vähensi punasolu- ja trombosyyttisiirtojen tarvetta. Potilaan katsottiin olevan riippuvainen punasolu- tai trombosyyttisiirroista lähtötasolla, jos hänelle oli tehty tai tehtiin yksi tai useampi punasolu- tai trombosyyttisiirto satunnaistamista edeltävien tai sen jälkeisten 56 vuorokauden (8 viikon) aikana. Potilaan katsottiin olevan riippumaton punasolu- tai trombosyyttisiirroista hoitojakson aikana, jos hänelle ei tehty punasolu- eikä trombosyyttisiirtoja 56 peräkkäiseen vuorokauteen raportointijakson aikana.
Lähtötasolla punasolusiirroista riippuvaisista Vidaza-ryhmän potilaista 38,5 % (95 % CI: 31,1–46,2) tuli punasolusiirroista riippumattomiksi hoitojakson aikana verrattuna 27,6 %:iin (95 % CI: 20,9–35,1) yhdistettyjen CCR-ryhmien potilaista. Lähtötasolla punasolusiirroista riippuvaisilla ja sitten riippumattomiksi tulleilla potilailla punasolusiirroista riippumattomuuden keston mediaani oli 13,9 kuukautta Vidaza-ryhmässä. CCR-ryhmässä riippumattomuutta ei saavutettu.
Lähtötasolla trombosyyttisiirroista riippuvaisista Vidaza-ryhmän potilaista 40,6 % (95 % CI: 30,9–50,8) tuli trombosyyttisiirroista riippumattomiksi hoitojakson aikana verrattuna 29,3 %:iin (95 % CI: 19,7–40,4) yhdistettyjen CCR-ryhmien potilaista. Lähtötasolla trombosyyttisiirroista riippuvaisilla ja sitten riippumattomiksi tulleilla potilailla trombosyyttisiirroista riippumattomuuden keston mediaani oli 10,8 kuukautta Vidaza-ryhmässä ja 19,2 kuukautta CCR-ryhmässä.
Terveyteen liittyvää elämänlaatua (Health-Related Quality of Life, HRQoL) arvioitiin EORTC QLQ-C30 -kyselylomakkeella (European Organization for Research and Treatment of Cancer Core Quality of Life Questionnaire). HRQoL-tiedot pystyttiin analysoimaan vain osalla koko tutkimusjoukosta. Analyysin rajoituksista huolimatta saatavilla olevat tiedot viittaavat siihen, ettei potilaiden elämänlaatu heikkene merkityksellisesti Vidaza-hoidon aikana.
Pediatriset potilaat
Tutkimus AZA-JMML-001 oli vaiheen 2 kansainvälinen avoin monikeskustutkimus, jossa arvioitiin HSCT:tä edeltävän Vidaza-hoidon farmakokinetiikkaa, farmakodynamiikkaa, turvallisuutta ja aktiivisuutta pediatrisilla potilailla, joilla oli vasta diagnosoitu, pitkälle edennyt MDS tai JMML. Kliinisen tutkimuksen ensisijainen tavoite oli arvioida Vizadan vaikutusta vasteeseen 3. hoitojakson päivänä 28.
Potilaita (MDS n = 10; JMML n = 18; ikä: 3 kk – 15 v.; miehiä 71% ) hoidettiin laskimoon annettavalla Vidazalla, 75 mg/m², 28-päiväisen hoitojakson päivinä 1–7 vähintään kolmen hoitojakson ja korkeintaan kuuden hoitojakson ajan.
Kymmenennen MDS-potilaan jälkeen MDS-tutkimushaaraan ei otettu enempää potilaita hoidon tehottomuudesta johtuen: näillä 10 potilaalla ei havaittu varmistettua hoitovastetta.
JMML-tutkimushaaraan otettiin 18 potilasta (13 potilaalla PTPN11-, 3 potilaalla NRAS-, 1 potilaalla KRAS-somaattinen mutaatio; yhdellä potilaalla kliinisesti diagnosoitu tyypin 1 neurofibromatoosi [NF‑1]). Potilaista 16 sai hoitoa 3 hoitojakson ajan, ja 5:tä heistä hoidettiin 6 hoitojakson ajan. Yhteensä 11 JMML-potilaalla havaittiin kliininen vaste kolmannen hoitojakson 28. päivän kohdalla; näistä yhdeksällä (50% ) oli varmistettu kliininen vaste (kolmella tutkittavalla oli cCR ja kuudella cPR). Koko Vidaza-hoitoa saaneessa JMML-potilasryhmässä seitsemällä potilaalla (43,8% ) havaittiin pitkäkestoinen trombosyyttivaste (määrä ≥ 100 × 109/l), ja seitsemän potilaasta (43,8%) tarvitsi HSCT-hoidon yhteydessä transfuusioita. Potilaista 17/18 eteni HSCT-hoitoon.
Tutkimusasetelmasta johtuen (pieni potilasmäärä ja useita harhaanjohtavia tekijöitä) tämän tutkimuksen pohjalta ei voida päätellä, parantaako ennen HSCT-hoitoa annettu Vidaza-hoito JMML-potilaiden eloonjääntiä.
Tutkimus AZA-AML-004 oli vaiheen 2 avoin monikeskustutkimus, jossa arvioitiin Vidaza-hoidon turvallisuutta, farmakodynamiikkaa ja tehoa lapsilla ja nuorilla aikuisilla, joilla oli todettu AML:n molekulaarinen relapsi CR1:n jälkeen, verrattuna potilaisiin, jotka eivät saaneet syöpähoitoa.
Seitsemän potilasta (iän mediaani 6,7 vuotta [vaihteluväli 2–12 vuotta]; poikia 71,4 %) sai hoitoa laskimoon annettavalla Vidazalla 100 mg/m² päivässä 28-päiväisen hoitojakson päivinä 1–7 korkeintaan kolmen hoitojakson ajan.
Viidelle potilaalle tehtiin minimaalisen jäännöstaudin (MRD) arviointi päivänä 84. Neljä potilasta saavutti joko molekulaarisen vakiintumisen (n = 3) tai molekulaarisen paranemisen (n = 1), ja yhdellä potilaalla todettiin kliininen relapsi. Kuudelle atsasitidiinihoitoa saaneelle potilaalle seitsemästä (90 % [95 % CI = 0,4; 1,0) tehtiin hematopoieettisten kantasolujen siirto (HSCT).
Pienen otoskoon takia Vidazan tehoa pediatristen potilaiden AML:n hoidossa ei voida varmistaa.
Turvallisuustiedot: ks. kohta Haittavaikutukset.
Farmakokinetiikka
Imeytyminen
Atsasitidiini imeytyi ihon alle annettujen kerta-annosten 75 mg/m2 jälkeen nopeasti ja sen huippupitoisuus plasmassa 750 ± 403 ng/ml saavutettiin 0,5 tuntia annon jälkeen (ensimmäinen näytteenottokohta). Atsasitidiinin absoluuttinen hyötyosuus (kerta-annosten 75 g/m2) ihon alle annon jälkeen suhteessa laskimoon antoon oli noin 89 % käyrän alaiseen pinta-alaan (AUC) perustuen.
Ihon alle annetun atsasitidiinin käyrän alainen pinta-ala ja huippupitoisuus plasmassa (Cmax) olivat annosvälillä 25–100 mg/m2 suunnilleen verrannollisia.
Jakautuminen
Laskimoon annon jälkeen keskimääräinen jakautumistilavuus oli 76 ± 26 l ja systeeminen puhdistuma oli 147 ± 47 l/h.
Biotransformaatio
In vitro -tietoihin perustuen atsasitidiinin metabolia ei vaikuta välittyvän sytokromi P450 -isoentsyymien (CYP:t), UDP-glukuronyylitransferaasien (UGT:t), sulfotransferaasien (SULT:t) ja glutationitransferaasien (GST:t) kautta.
Atsasitidiini läpikäy spontaanin hydrolyysin ja sytidiinideaminaasin välittämän deaminaation. Ihmisen maksan S9-fraktioissa metaboliittien muodostuminen oli NADPH:sta riippumatonta, mikä viittaa siihen, ettei atsasitidiinin metabolia ole sytokromi P450:n isoentsyymien välittämää. In vitro -tutkimus, jossa atsasitidiinia tutkittiin viljeltyjen ihmisen hepatosyyttien kanssa, osoitti, että 1,0 µM–100 µM:n pitoisuuksissa (ts. noin 30-kertaisesti suuremmissa kuin kliinisesti saavutettavissa pitoisuuksissa) atsasitidiini ei indusoi CYP 1A2:ta, 2C19:ää tai 3A4:ää tai 3A5:ttä. Tutkimuksissa, joissa arvioitiin erilaisten P450:n isoentsyymien (CYP) (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ja 3A4) inhiboitumista, atsasitidiini aina 100 µM:iin saakka ei kehittänyt inhibitiota. Sen vuoksi on epätodennäköistä, että atsasitidiini indusoisi tai estäisi CYP-entsyymiä kliinisesti saavutettavissa plasmapitoisuuksissa.
Eliminaatio
Atsasitidiini erittyy nopeasti plasmasta keskimääräisen eliminaation puoliintumisajan (t½) ollessa 41 ± 8 minuuttia ihon alle annettuna. Atsasitidiinin annostelu 75 mg/m2 ihon alle kerran vuorokaudessa 7 vuorokauden ajan ei aiheuta kertymää. Atsasitidiini ja/tai sen metaboliitit eliminoituvat pääasiassa erittymällä virtsaan. Kun 14C-atsasitidiinia annettiin laskimoon ja ihon alle, annetusta radioaktiivisuudesta mitattiin virtsassa vastaavasti 85 ja 50 % ja ulosteessa < 1 %.
Erityisryhmät
Maksan vajaatoiminnan (ks. kohta Annostus ja antotapa), sukupuolen, iän tai rodun vaikutuksia atsasitidiinin farmakokinetiikkaan ei ole virallisesti tutkittu.
Pediatriset potilaat
Tutkimuksessa AZA-JMML-001 suoritettiin farmakokineettinen analyysi 10:lle MDS- ja 18:lle JMML-potilaalle ensimmäisen hoitojakson 7. päivänä (ks. kohta Farmakodynamiikka). MDS-potilaiden iän mediaani (vaihteluväli) oli 13,3 (1,9–15) vuotta ja JMML-potilailla 2,1 (0,2–6,9) vuotta.
Kun Vidazaa annettiin laskimoon 75 mg/m2, se saavutti Cmax:n 0,083 tunnissa sekä MDS- että JMML-ryhmässä. Cmax:n geometrinen keskiarvo oli 1797,5 ng/ml (MDS-potilaat) ja 1066,3 ng/ml (JMML-potilaat), ja AUC0-∞:n geometrinen keskiarvo oli 606,9 ng∙h/ml (MDS-potilaat) ja 240,2 ng∙t/ml (JMML-potilaat). Jakautumistilavuuden geometrinen keskiarvo oli MDS-tutkittavilla 103,9 l ja JMML-tutkittavilla 61,1 l. Vaikutti siltä, että kokonaisplasma-altistus Vidazalle olisi ollut MDS-tutkittavilla suurempi; sekä AUC- että Cmax-arvoissa havaittiin kuitenkin kohtalaista tai suurta vaihtelua.
Puoliintumisajan, t½ geometriset keskiarvot olivat 0,4 tuntia (MDS) ja 0,3 tuntia (JMML), ja puhdistumat 166,4 l/h (MDS) ja 148,3 l/h (JMML).
Tutkimuksen AZA-JMML-001 farmakokineettiset tutkimustiedot koottiin yhteen ja niitä verrattiin tutkimuksen AZA-2002-BA-002 kuuteen MDS-aikuispotilaaseen, joille annettiin 75 mg/m2 Vidazaa laskimoon. Laskimoon annetun Vidazan keskimääräiset Cmax ja AUC0-t olivat samankaltaiset aikuispotilaiden ja pediatristen potilaiden välillä (Cmax: 2750 ng/ml vrt. 2841 ng/ml; AUC0-t: 1025 ng∙h/ml vrt. 882.1 ng∙h/ml).
Tutkimuksessa AZA-AML-004 suoritettiin farmakokineettinen analyysi kuudelle niistä seitsemästä pediatrisesta potilaasta, joilta annoksen jälkeinen farmakokineettinen pitoisuus saatiin mitattua vähintään kerran (ks. kohta Farmakodynamiikka). AML-potilaiden iän mediaani (vaihteluväli) oli 6,7 (2–12) vuotta.
Kun atsasitidiinia annettiin useita 100 mg/m2 annoksia, Cmax:n geometrinen keskiarvo oli 1557 ng/ml ja AUC0-tau:n geometrinen keskiarvo oli 899,6 ng∙h/ml ensimmäisen hoitojakson seitsemäntenä päivänä. Tutkittavien välinen vaihtelu oli suurta (CV% 201,6 % ja vastaavasti 87,8 %). Atsasitidiinin Cmax saavutettiin nopeasti mediaaniajassa 0,090 tuntia laskimoon annon jälkeen, ja pitoisuuksien pienenemisen geometrinen keskiarvo t½ oli 0,380 tuntia.
Puhdistuman ja jakautumistilavuuden geometriset keskiarvot olivat 127,2 l/h ja vastaavasti 70,2 l/h.
Farmakokineettinen (atsasitidiinin) altistus lapsilla, joilla todettiin AML:n molekulaarinen relapsi CR1:n jälkeen, oli verrattavissa kymmenestä MDS-lapsipotilaasta ja 18:sta JMML-lapsipotilaasta saatuihin yhdistettyihin altistustietoihin ja myös aikuisten MDS-potilaiden atsasitidiinialtistukseen.
Munuaisten vajaatoiminta
Munuaisten vajaatoiminnalla ei ole merkittävää vaikutusta atsasitidiinin farmakokineettiseen altistukseen ihon alle annettujen kerta-annosten ja toistuvien annosten jälkeen. Ihon alle annetun 75 mg/m2:n kerta-annoksen jälkeen keskimääräiset altistumisarvot (AUC ja Cmax) suurenivat lievää munuaisten vajaatoimintaa sairastavilla 11−21 %, kohtalaista munuaisten vajaatoimintaa sairastavilla 15–27 % ja vaikeaa munuaisten vajaatoimintaa sairastavilla 41–66 % verrattuna tutkittaviin, joiden munuaisten toiminta oli normaali. Altistuminen oli kuitenkin samalla yleisellä altistumisen vaihteluvälillä, joka oli havaittu niillä tutkittavilla, joiden munuaisten toiminta oli normaali. Atsasitidiinia voidaan antaa munuaisten vajaatoimintaa sairastaville potilaille aloitusannosta muuttamatta edellyttäen, että näitä potilaita seurataan toksisuuden havaitsemiseksi, sillä atsasitidiini ja/tai sen metaboliitit erittyvät pääasiassa munuaisten kautta.
Farmakogenomiikka
Tunnetun sytidiinideaminaasin polymorfismin vaikutusta atsasitidiinin metaboliaan ei ole tutkittu virallisesti.
Prekliiniset tiedot turvallisuudesta
Atsasitidiini indusoi sekä geenimutaatioita että kromosomipoikkeavuuksia bakteeri- ja nisäkäslajien solujärjestelmissä in vitro. Atsasitidiinin mahdollista karsinogeenisuutta arvioitiin hiirillä ja rotilla. Atsasitidiini indusoi hematopoieettisen järjestelmän kasvaimia naarashiirissä, kun sitä annettiin vatsakalvonsisäisesti 3 kertaa viikossa 52 viikon ajan. 50 viikkoa vatsakalvonsisäisesti atsasitidiinia saaneilla hiirillä todettiin lymforetikulaarisen järjestelmän, keuhkojen, rintarauhasen ja ihon kasvainten esiintyvyyden lisääntyneen. Rotilla suoritetussa tuumorigeenisuutta koskevassa kokeessa havaittiin kiveskasvainten esiintyvyyden lisääntyneen.
Hiirillä suoritetuissa varhaisen vaiheen sikiötoksisuutta koskevissa kokeissa todettu kohtukuolemien (lisääntynyt imeytyminen) esiintyvyys oli 44 % organogeneesin aikana annetun yksittäisen vatsakalvonsisäisen atsasitidiinipistoksen jälkeen. Aivojen kehitysvaurioita on todettu hiirillä, joille annettiin atsasitidiinia kovan suulaen sulkeutumisen aikana tai ennen sitä. Rotilla atsasitidiini ei aiheuttanut haittavaikutuksia annettaessa ennen implantaatiota, mutta se oli selvästi embryotoksinen annettaessa organogeneesin aikana. Organogeneesin aikaisia sikiövaurioita rotilla olivat: keskushermoston anomaliat (eksenkefalia/enkefaloseele), raajojen anomaliat (mikromelia, kampurajalka, syndaktylia, oligodaktylia) ja muut (mikroftalmia, mikrognatia, vatsahalkio, ödeema ja kylkiluiden epämuodostumat).
Atsasitidiinin anto uroshiirille ennen parittelua naarashiirien kanssa, jotka eivät saaneet atsasitidiinia, johti heikentyneeseen hedelmällisyyteen ja keskenmenoon tai jälkeläisten menetykseen hedelmöitystä seuraavassa embryonaalisessa ja syntymän jälkeisessä kehityksessä. Atsasitidiinin antamisesta urosrotille seurasi kivesten ja lisäkivesten painon pieneneminen, siittiöiden määrän väheneminen, raskauksien väheneminen, epämuodostuneiden alkioiden lisääntyminen ja alkiomenetysten lisääntyminen paritelluilla naarailla (ks. kohta Raskaus ja imetys).
Farmaseuttiset tiedot
Apuaineet
Mannitoli (E421)
Yhteensopimattomuudet
Lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton kuiva-ainetta sisältävä injektiopullo:
4 vuotta
Käyttökuntoon saattamisen jälkeen: Kun Vidaza on saatettu käyttökuntoon käyttämällä injektioita varten tarkoitettua vettä, jota ei ole säilytetty kylmässä, käyttökuntoon saatetun lääkevalmisteen on osoitettu säilyvän käytön aikana kemiallisesti ja fysikaalisesti stabiilina 25 °C:n lämpötilassa 45 minuuttia ja 2°C – 8°C:n lämpötilassa 8 tuntia.
Käyttökuntoon saatetun lääkevalmisteen kestoaikaa voidaan pidentää sekoittamalla se kylmässä (2°C – 8°C:ssa) säilytettyyn injektioveteen. Kun Vidaza on saatettu käyttökuntoon käyttämällä kylmässä (2°C – 8°C:ssa) säilytettyä injektiovettä, käyttökuntoon saatetun lääkevalmisteen on osoitettu säilyvän käytön aikana kemiallisesti ja fysikaalisesti stabiilina 2°C – 8°C:n lämpötilassa 22 tuntia.
Mikrobiologiselta kannalta käyttökuntoon saatettu valmiste tulee käyttää välittömästi. Jos sitä ei käytetä välittömästi, käytön aikaiset säilytysajat ja olosuhteet ennen käyttöä ovat käyttäjän vastuulla, ja ne eivät saa ylittää 8 tuntia 2°C – 8°C:n lämpötilassa kun lääkevalmiste on saatettu käyttökuntoon käyttämällä injektiovettä, jota ei ole säilytetty kylmässä, eivätkä ne saa ylittää 22 tuntia kun lääkevalmiste on saatettu käyttökuntoon käyttämällä kylmässä (2°C – 8°C:ssa) säilytettyä injektiovettä.
Säilytys
Avaamattomat injektiopullot
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Käyttökuntoon saatettu suspensio
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VIDAZA injektiokuiva-aine, suspensiota varten
25 mg/ml (L:ei) 100 mg (515,36 €)
PF-selosteen tieto
Väritön tyypin I lasi-injektiopullo, joka on suljettu butyylielastomeeritulpalla ja polypropyleenimuovipainikkeella varustetulla alumiinikorkilla ja joka sisältää 100 mg atsasitidiinia.
Pakkauskoko: Yksi injektiopullo.
Valmisteen kuvaus:
Valkoinen kylmäkuivattu jauhe.
Käyttö- ja käsittelyohjeet
Turvallista käsittelyä koskevat suositukset
Vidaza on sytotoksinen lääkevalmiste, ja muiden mahdollisesti toksisten aineiden tavoin atsasitidiinisuspensioiden käsittelyssä ja valmistelussa on toimittava varoen. Syöpälääkkeiden asianmukaista käsittelyä ja hävittämistä koskevia toimenpiteitä on noudatettava.
Jos käyttökuntoon saatettu atsasitidiini joutuu kosketukseen ihon kanssa, pese välittömästi ja perusteellisesti vedellä ja saippualla. Jos se pääsee kosketukseen limakalvojen kanssa, huuhtele huolellisesti vedellä.
Ohje käyttökuntoon saattamista varten
Vidaza tulee saattaa käyttökuntoon sekoittamalla se injektioita varten tarkoitettuun veteen. Käyttökuntoon saatetun lääkevalmisteen kestoaikaa voidaan pidentää sekoittamalla se kylmässä (2°C – 8°C:ssa) säilytettyyn injektioveteen. Käyttökuntoon saatetun lääkevalmisteen säilytystä koskevat yksityiskohdat on esitetty alla:
- Ota esille seuraavat tarvikkeet:
- atsasitidiinia sisältävä(t) injektiopullo(t); injektionesteisiin käytettävää vettä sisältävä(t) injektiopullo(t); epästeriilit kirurgiset käsineet; alkoholiin kostutetut puhdistuslaput; 5 ml injektioruisku(t) neuloineen.
- Vedä ruiskuun 4 ml injektionesteisiin käytettävää vettä varmistaen, että tyhjennät ruiskuun jääneen ilman.
- Työnnä 4 ml injektionesteisiin käytettävää vettä sisältävän ruiskun neula atsasitidiinia sisältävän injektiopullon kumisen yläosan läpi, ja ruiskuta injektiopulloon injektionesteisiin käytettävää vettä.
- Kun olet poistanut ruiskun ja neulan, ravista injektiopulloa voimakkaasti, kunnes suspensio on tasainen ja samea. Käyttökuntoon saattamisen jälkeen yksi ml suspensiota sisältää 25 mg atsasitidiinia (100 mg/4 ml). Käyttökuntoon saatettu valmiste on homogeeninen samea suspensio, jossa ei ole agglomeraatteja. Suspensio tulee hävittää, jos se sisältää isoja hiukkasia tai agglomeraatteja. Älä suodata suspensiota käyttökuntoon saattamisen jälkeen, sillä se saattaa poistaa vaikuttavan aineen. Ota huomioon, että suodattimia on joissain sovittimissa, neuloissa ja suljetuissa järjestelmissä. Tällaisia järjestelmiä ei tule käyttää lääkevalmisteen annosteluun
käyttökuntoon saattamisen jälkeen. - Puhdista kuminen yläosa ja aseta uusi ruisku neulan kanssa paikalleen injektiopulloon. Käännä injektiopullo ylösalaisin varmistaen, että neulan kärki on nestetason alapuolella. Vedä sitten asianmukaiseen annokseen vaadittu määrä lääkevalmistetta vetämällä mäntää taaksepäin varmistaen, että tyhjennät ruiskuun jääneen ilman. Vedä ruisku neulan kanssa pois injektiopullosta ja hävitä neula.
- Kiinnitä puhdas ihonalaiseen injektioon tarkoitettu neula (suositellaan 25 gaugea) tiukasti ruiskuun. Neulaa ei saa täyttää ennen injektiota paikallisten pistoskohdan reaktioiden esiintymisen vähentämiseksi.
- Jos annokseen tarvitaan enemmän kuin 1 injektiopullo, toista kaikki edellä mainitut toimenpiteet suspension valmistelussa. Kun annoksen suuruus on enemmän kuin 1 injektiopullo, annos tulee jakaa tasan, esim. annos 150 mg = 6 ml, 2 ruiskua, joissa kummassakin on 3 ml. Neulaan ja injektiopulloon retentoitumisen takia kaiken lääkeaineen vetäminen injektiopullosta ei välttämättä onnistu.
- Annosteluruiskun sisällön tulee antaa suspensoitua uudelleen välittömästi ennen antoa.
Käyttökuntoon saatettua suspensiota sisältävän ruiskun tulee antaa lämmetä tasaisesti enintään 30 minuutin ajan ennen antoa, jotta se saavuttaa noin 20°C - 25°C:n lämpötilan. Jos aikaa kuluu enemmän kuin 30 minuuttia, suspensio tulee hävittää asianmukaisesti ja uusi annos on valmisteltava. Suspensoi sisältö uudelleen pyörittämällä ruiskua voimakkaasti kämmenten välissä, kunnes suspensio on tasainen ja samea. Suspensio tulee hävittää, jos se sisältää isoja hiukkasia tai agglomeraatteja.
Käyttökuntoon saatetun valmisteen säilytys
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Yksilöllisen annoksen laskeminen
Kokonaisannos kehon pinta-alan (body surface area, BSA) mukaan voidaan laskea seuraavalla tavalla:
Kokonaisannos (mg) = Annos (mg/m2) x BSA (m2)
Seuraava taulukko on vain esimerkki siitä, miten yksilölliset atsasitidiiniannokset lasketaan keskimääräiseen BSA-arvoon 1,8 m2 perustuen.
| Annos mg/m2 (% suositellusta aloitusannoksesta) | BSA-arvoon 1,8 m2 perustuva kokonaisannos | Tarvittavien injektiopullojen määrä | Tarvittavan käyttökuntoon saatetun suspension kokonaistilavuus |
| 75 mg/m2 (100 %) | 135 mg | 2 injektiopulloa | 5,4 ml |
| 37,5 mg/m2 (50 %) | 67,5 mg | 1 injektiopullo | 2,7 ml |
| 25 mg/m2 (33 %) | 45 mg | 1 injektiopullo | 1,8 ml |
Antotapa
Käyttökuntoon saatettu Vidaza tulee pistää ihon alle (työnnä neula 45–90°:n kulmassa) 25 gaugen neulaa käyttämällä käsivarren yläosaan, reiteen tai vatsaan.
Yli 4 ml:n annokset tulee pistää kahteen eri kohtaan.
Pistoskohtia tulee vaihdella. Uudet pistokset tulee antaa vähintään 2,5 cm etäisyydelle aiemmasta pistoskohdasta eikä koskaan alueelle, jossa pistoskohta on arka, mustelmainen, punainen tai kovettunut.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VIDAZA injektiokuiva-aine, suspensiota varten
25 mg/ml 100 mg
- Ei korvausta.
ATC-koodi
L01BC07
Valmisteyhteenvedon muuttamispäivämäärä
01.11.2024
Yhteystiedot
09 2512 1244
www.bms.com/fi
medinfo.finland@bms.com