
RETACRIT injektioneste, liuos, esitäytetty ruisku 1000 IU/0,3 ml, 2000 IU/0,6 ml, 3000 IU/0,9 ml, 4000 IU/0,4 ml, 5000 IU/0,5 ml, 6000 IU/0,6 ml, 8000 IU/0,8 ml, 20000 IU/0,5 ml, 30000 IU/0,75 ml, 10000 IU/ml, 40000 IU/ml
Vaikuttavat aineet ja niiden määrät
Retacrit 1 000 IU/0,3 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 1 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 0,3 ml:ssa liuosta. Yksi millilitra liuosta sisältää 3 333 IU epoetiini zeetaa.
Retacrit 2 000 IU/0,6 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 2 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 0,6 ml:ssa liuosta. Yksi millilitra liuosta sisältää 3 333 IU epoetiini zeetaa.
Retacrit 3 000 IU/0,9 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 3 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 0,9 ml:ssa liuosta. Yksi millilitra liuosta sisältää 3 333 IU epoetiini zeetaa.
Retacrit 4 000 IU/0,4 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 4 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 0,4 ml:ssa liuosta. Yksi millilitra liuosta sisältää 10 000 IU epoetiini zeetaa.
Retacrit 5 000 IU/0,5 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 5 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 0,5 ml:ssa liuosta. Yksi millilitra liuosta sisältää 10 000 IU epoetiini zeetaa.
Retacrit 6 000 IU/0,6 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 6 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 0,6 ml:ssa liuosta. Yksi millilitra liuosta sisältää 10 000 IU epoetiini zeetaa.
Retacrit 8 000 IU/0,8 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 8 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 0,8 ml:ssa liuosta. Yksi millilitra liuosta sisältää 10 000 IU epoetiini zeetaa.
Retacrit 10 000 IU/1 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 10 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 1 ml:ssa liuosta. Yksi millilitra liuosta sisältää 10 000 IU epoetiini zeetaa.
Retacrit 20 000 IU/0,5 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 20 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 0,5 ml:ssa liuosta. Yksi millilitra liuosta sisältää 40 000 IU epoetiini zeetaa.
Retacrit 30 000 IU/0,75 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 30 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 0,75 ml:ssa liuosta. Yksi millilitra liuosta sisältää 40 000 IU epoetiini zeetaa.
Retacrit 40 000 IU/1 ml injektioneste, liuos, esitäytetyssä ruiskussa
Yksi esitäytetty ruisku sisältää 40 000 kansainvälistä yksikköä (International Units, IU) epoetiini zeetaa* (yhdistelmä-DNA-tekniikalla valmistettua ihmisen erytropoietiinia) 1 ml:ssa liuosta. Yksi millilitra liuosta sisältää 40 000 IU epoetiini zeetaa.
Apuaineet, joiden vaikutus tunnetaan
Retacrit sisältää fenyylialaniinia 0,5 mg/ml.
Retacrit sisältää polysorbaatti 20:tä (E 432) 0,1 mg/ml.
*Valmistetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa (CHO).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos, esitäytetyssä ruiskussa (injektio).
Kliiniset tiedot
Käyttöaiheet
Retacrit on tarkoitettu krooniseen munuaisten vajaatoimintaan (CRF) liittyvän oireisen anemian hoitoon:
- aikuisille ja 1–18-vuotiaille pediatrisille hemodialyysipotilaille ja aikuisille peritoneaalidialyysipotilaille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- munuaisperäisen vaikean anemian hoitoon aikuisille munuaisten vajaatoimintaa sairastaville potilaille, jotka eivät vielä saa dialyysihoitoa, kun anemiaan liittyy kliinisiä oireita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Retacrit on tarkoitettu anemian hoitoon ja verensiirron tarpeen vähentämiseen solunsalpaajahoitoa saavilla aikuispotilailla, joilla on kiinteitä kasvaimia, pahanlaatuinen lymfooma tai multippeli myelooma ja joilla verensiirron tarve on todennäköinen perustuen arvioon potilaan yleistilasta (esim. kardiovaskulaaritilanne, ennen solunsalpaajahoidon aloittamista esiintynyt anemia).
Retacrit on tarkoitettu aikuisille ennen leikkausta lisäämään autologisesti luovutettavan veren saantoa. Hoitoa tulisi antaa vain kohtalaisesti aneemisille potilaille (hemoglobiinipitoisuus [Hb] 100–130 g/l, [6,2−8,1 mmol/l], ei raudanpuutosta), jos verta säästäviä menetelmiä ei ole käytettävissä tai ne eivät ole riittäviä silloin, kun elektiivisessä leikkauksessa tarvittava verimäärä on suuri (naisilla vähintään 4 ja miehillä vähintään 5 yksikköä).
Retacrit on tarkoitettu aikuisille, joilla ei ole raudanpuutosta, ennen suurta elektiivistä ortopedistä leikkausta, jos on odotettavissa suurentunut verensiirtokomplikaatioiden riski, vähentämään altistusta allogeeniselle verensiirrolle. Käyttö pitää rajata kohtalaisen aneemisiin potilaisiin (hemoglobiinipitoisuus 100–130 g/l eli 6,2–8,1 mmol/l), joille ei ole tarjolla luovutetun autologisen veren siirtomahdollisuutta ja joilla on odotettavissa kohtalainen verenhukka (900–1 800 ml).
Retacrit on tarkoitettu oireisen anemian (hemoglobiinipitoisuus ≤ 100 g/l) hoitoon aikuisille, joilla on pienen tai keskisuuren riskin (riskitaso 1) primaarinen myelodysplastinen oireyhtymä (MDS) ja pieni seerumin erytropoietiinipitoisuus (< 200 mU/ml).
Ehto
Hoidon saavat aloittaa valmisteyhteenvedossa mainittuun indikaatioon perehtyneet lääkärit.
Annostus ja antotapa
Retacrit-hoito tulee aloittaa sellaisen lääkärin valvonnassa, joka on perehtynyt potilaiden hoitoon, joilla lääkevalmistetta käytetään edellä mainittuihin indikaatioihin.
Annostus
Anemian muut syyt (raudan, folaatin tai B12-vitamiinin puutos, alumiinimyrkytys, infektio tai tulehdus, verenhukka, hemolyysi ja mistä tahansa aiheutunut luuydinfibroosi) on tutkittava ja hoidettava ennen epoetiini zeeta -hoidon aloittamista sekä päätettäessä annoksen suurentamisesta. Jotta optimaalinen vaste epoetiini zeetalle voidaan varmistaa, riittävät rautavarannot on varmistettava ja tarvittaessa on annettava rautalisää (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Oireisen anemian hoito kroonista munuaisten vajaatoimintaa sairastavilla aikuispotilailla
Anemian oireet ja seuraukset saattavat vaihdella iän, sukupuolen ja samanaikaisten sairauksien mukaan; lääkärin on arvioitava kunkin potilaan kliininen tila ja hoito yksilöllisesti.
Hemoglobiinin suositeltu pitoisuusväli on 100–120 g/l (6,2–7,5 mmol/l). Retacritia annetaan hemoglobiiniarvon nostamiseksi korkeintaan 120 g/l (7,5 mmol/l). Hemoglobiiniarvon nousua yli 20 g/l (1,25 mmol/l) neljän viikon jakson aikana on vältettävä. Jos näin tapahtuu, annosta on muutettava ohjeiden mukaisesti.
Yksittäisen potilaan hemoglobiiniarvoissa tapahtuvan vaihtelun vuoksi taso voi joskus ylittää tai alittaa halutun hemoglobiinipitoisuusvälin. Hemoglobiiniarvon vaihtelu tulisi hoitaa annosta muuttamalla ja ottamalla huomioon hemoglobiinipitoisuusväli 100–120 g/l (6,2–7,5 mmol/l).
Jatkuvaa 120 g/l (7,5 mmol/l) ylittävää hemoglobiinitasoa tulee välttää. Jos hemoglobiinipitoisuus nousee yli 20 g/l (1,25 mmol/l) kuukautta kohti tai jos hemoglobiinipitoisuus on pitkään yli 120 g/l (7,5 mmol/l), pienennä Retacrit-annosta 25 prosentilla. Jos hemoglobiinipitoisuus on yli 130 g/l (8,1 mmol/l), keskeytä hoito, kunnes pitoisuus laskee alle 120 g/l (7,5 mmol/l), ja aloita Retacrit-hoito sitten uudelleen annoksella, joka on 25 % pienempi kuin edellinen annos.
Potilaita pitää seurata tarkoin, jotta voidaan varmistaa, että käytetään pienintä tehokasta anemian ja sen oireiden riittävään hoitoon hyväksyttyä Retacrit-annosta ja hemoglobiinipitoisuus pysyy samalla edelleen enintään arvossa 120 g/l (7,5 mmol/l).
Kroonista munuaisten vajaatoimintaa sairastaville potilaille annettavan punasolutuotantoa stimuloivan lääkeaineen (erythropoiesis stimulating agent, ESA) annoksen suurentamisessa pitää olla varovainen. Jos potilaan hemoglobiinipitoisuudessa todettava vaste ESA-hoitoon on huono, muut syyt huonoon vasteeseen pitää ottaa huomioon (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Retacrit-hoito jakautuu kahteen vaiheeseen, korjausvaiheeseen ja ylläpitovaiheeseen.
Aikuiset hemodialyysipotilaat
Hemodialyysihoitoa saaville potilaille, joilla laskimoyhteys on helposti avattavissa, valmiste tulisi antaa mieluiten laskimoon.
Korjausvaihe
Aloitusannos on 50 IU/kg kolmesti viikossa.
Annosta on tarvittaessa suurennettava tai pienennettävä 25 IU/kg:n annoksina (kolmesti viikossa), kunnes saavutetaan haluttu hemoglobiinin pitoisuusväli 100–120 g/l (6,2–7,5 mmol/l) (tämä tulisi tehdä vaiheittain niin, että väli on vähintään neljä viikkoa).
Ylläpitovaihe
Suositeltava kokonaisviikkoannos on 75 IU/kg – 300 IU/kg.
Annosta on sovitettava tarkoituksenmukaisesti siten, että hemoglobiiniarvot pysyvät halutussa pitoisuusvälissä 100–120 g/l (6,2–7,5 mmol/l).
Potilaat, joiden hemoglobiinipitoisuus on alussa erittäin pieni (< 60 g/l tai < 3,75 mmol/l), saattavat tarvita suurempia ylläpitoannoksia kuin potilaat, joiden anemia on alussa lievempi (> 80 g/l tai > 5 mmol/l).
Munuaisten vajaatoimintaa sairastavat aikuispotilaat, jotka eivät vielä saa dialyysihoitoa
Kun laskimoyhteys ei ole helposti avattavissa, Retacrit voidaan antaa ihon alle.
Korjausvaihe
Aloitusannos on 50 IU/kg kolmesti viikossa, minkä jälkeen annosta suurennetaan tarvittaessa 25 IU/kg:n kerta-annoksin (kolmesti viikossa), kunnes haluttu tavoite on saavutettu (tämä tulisi tehdä vaiheittain niin, että väli on vähintään neljä viikkoa).
Ylläpitovaihe
Retacrit voidaan antaa ylläpitovaiheessa joko kolme kertaa viikossa tai ihon alle annettaessa kerran viikossa tai kerran joka toinen viikko.
Annos ja antoväli on sovitettava asianmukaisesti hemoglobiiniarvojen säilyttämiseksi halutulla tasolla: Hb 100–120 g/l (6,2–7,5 mmol/l). Antovälin pidentäminen saattaa vaatia annoksen suurentamista.
Enimmäisannostus ei saa olla yli 150 IU/kg kolmesti viikossa, 240 IU/kg (enimmäisannos 20 000 IU) kerran viikossa tai 480 IU/kg (enimmäisannos 40 000 IU) kerran joka toinen viikko.
Peritoneaalidialyysia saavat aikuispotilaat
Kun laskimoyhteys ei ole helposti avattavissa, Retacrit voidaan antaa ihon alle.
Korjausvaihe
Aloitusannos on 50 IU/kg kahdesti viikossa.
Ylläpitovaihe
Suositeltu ylläpitoannos on 25 IU/kg – 50 IU/kg kahdesti viikossa kahtena yhtä suurena injektiona.
Annos on sovitettava asianmukaisesti hemoglobiiniarvojen säilyttämiseksi halutulla tasolla Hb 100–120 g/l (6,2–7,5 mmol/l).
Solunsalpaajahoidon aiheuttamaa anemiaa sairastavat aikuispotilaat
Anemian oireet ja seuraukset saattavat vaihdella iän, sukupuolen ja yleisen tautitaakan mukaan; lääkärin on arvioitava kunkin potilaan kliininen tila ja hoito yksilöllisesti.
Retacrit-valmistetta pitää antaa, jos potilaalla on anemia (esim. hemoglobiinipitoisuus ≤ 100 g/l (6,2 mmol/l)).
Aloitusannos on 150 IU/kg ihon alle kolmesti viikossa.
Retacrit voidaan vaihtoehtoisesti antaa aloitusannoksella 450 IU/kg ihon alle kerran viikossa.
Annos on sovitettava asianmukaisesti hemoglobiiniarvojen säilyttämiseksi halutulla pitoisuusvälillä: Hb 100–120 g/l (6,2–7,5 mmol/l).
Yksittäisen potilaan hemoglobiinipitoisuuksissa tapahtuvan vaihtelun vuoksi pitoisuus voi joskus ylittää tai alittaa halutun hemoglobiinipitoisuusvälin. Hemoglobiinipitoisuuden vaihtelu tulisi hoitaa annosta muuttamalla ja ottamalla huomioon haluttu hemoglobiinipitoisuusväli, joka on 100–120 g/l (6,2–7,5 mmol/l). Jatkuvaa 120 g/l (7,5 mmol/l) ylittävää hemoglobiinipitoisuutta tulee välttää. Ohjeet annoksen muuttamiseksi silloin, kun hemoglobiiniarvo ylittää 120 g/l (7,5 mmol/l) on esitetty seuraavassa.
– Jos hemoglobiinipitoisuus on noussut vähintään 10 g/l (0,62 mmol/l) tai retikulosyyttien määrä on lisääntynyt ≥ 40 000 solua/mikrol yli lähtötason neljän hoitoviikon jälkeen, annos tulisi pitää 150 IU/kg:nä kolmesti viikossa annettuna tai 450 IU/kg:nä kerran viikossa.
– Jos hemoglobiinipitoisuus on noussut < 10 g/l (< 0,62 mmol/l) ja retikulosyyttien määrä on lisääntynyt < 40 000 solua/mikrol yli lähtötason, suurenna annos 300 IU/kg:hen kolmesti viikossa. Jos 300 IU/kg:llä kolmesti viikossa toteutetun neljän lisähoitoviikon jälkeen hemoglobiinipitoisuus on noussut ≥ 10 g/l (≥ 0,62 mmol/l) tai retikulosyyttien määrä on lisääntynyt ≥ 40 000 solua/mikrol, annos tulisi pitää 300 IU/kg:nä kolmesti viikossa.
– Jos hemoglobiinipitoisuus on noussut < 10 g/l (< 0,62 mmol/l) ja retikulosyyttien määrä on lisääntynyt < 40 000 solua/mikrol yli lähtötason, vastetta ei todennäköisesti saavuteta ja hoito tulisi lopettaa.
Annoksen sovittaminen hemoglobiinipitoisuuden pitämiseksi pitoisuusvälillä 100−120 g/l (6,2–7,5 mmol/l)
Jos hemoglobiinipitoisuus nousee enemmän kuin 20 g/l (1,25 mmol/l) kuukaudessa tai jos hemoglobiinipitoisuus ylittää arvon 120 g/l (7,5 mmol/l), pienennä Retacrit-annosta noin 25−50 %:lla.
Jos hemoglobiinipitoisuus ylittää arvon 130 g/l (8,1 mmol/l), keskeytä hoito, kunnes pitoisuus pienenee alle arvon 120 g/l (7,5 mmol/l) ja jatka Retacrit-hoitoa sitten annoksella, joka on 25 % pienempi kuin edellinen annos.
Suositeltava annosteluohjelma on esitetty seuraavassa kaaviossa*:
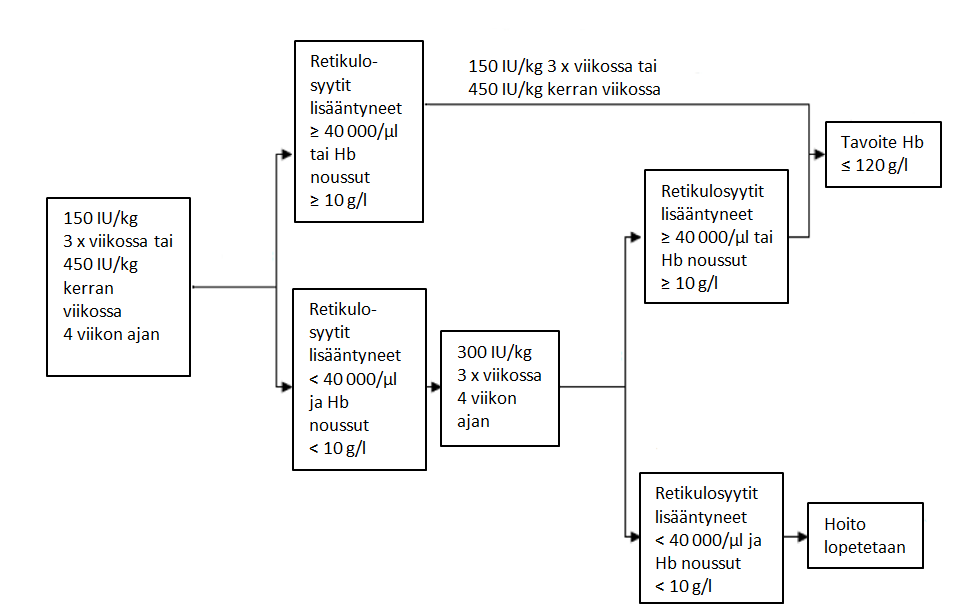
*10 g/l = 0,62 mmol/l; 120 g/l = 7,5 mmol/l
Potilaita tulee seurata tarkoin, jotta voidaan varmistaa, että anemian oireiden hoitoon käytetään pienintä hyväksyttyä annosta punasolutuotantoa stimuloivaa lääkeainetta (ESA).
Retacrit-hoitoa pitää jatkaa kuukauden ajan solunsalpaajahoidon päättymisen jälkeen.
Aikuisten leikkauspotilaiden hoito autologisessa verensiirto-ohjelmassa
Lievästi aneemisille potilaille (B-Hkr 33–39 %), joilta on otettava etukäteen talteen ≥ 4 yksikköä verta, tulisi antaa 600 IU/kg Retacrit-valmistetta laskimoon kahdesti viikossa kolmen viikon ajan ennen leikkausta. Retacrit pitää antaa verenluovutuksen jälkeen.
Aikuispotilaat, joille on suunniteltu suuri elektiivinen ortopedinen leikkaus
Suositeltu Retacrit-annos on 600 IU/kg ihon alle kerran viikossa kolmen viikon ajan (21, 14 ja 7 päivää) ennen leikkausta sekä leikkauspäivänä.
Jos potilaan tila vaatii leikkausta aikaisemmin kuin kolmen viikon kuluttua, annetaan 300 IU/kg Retacrit-valmistetta ihon alle päivittäin kymmenenä perättäisenä päivänä ennen leikkausta, leikkauspäivänä sekä neljänä päivänä heti leikkauksen jälkeen.
Jos hemoglobiinipitoisuus nousee ennen leikkausta arvoon 150 g/l (9,38 mmol/l) tai korkeammalle, Retacrit-valmisteen anto pitää lopettaa eikä lisäannoksia enää anneta.
Aikuispotilaat, joilla on pienen tai keskisuuren riskin (riskitaso 1) myelodysplastinen oireyhtymä (MDS)
Retacrit-valmistetta pitää antaa, jos potilaalla on oireinen anemia (esim. hemoglobiinipitoisuus ≤ 100 g/l [6,2 mmol/l]).
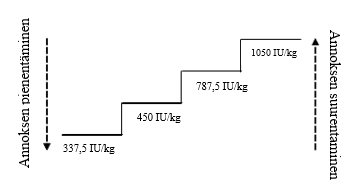
Retacrit-valmisteen suositeltu aloitusannos on 450 IU/kg (kokonaisannos on enintään 40 000 IU) ihon alle kerran viikossa. Annosten välin pitää olla vähintään 5 päivää.
Annosta on sovitettava asianmukaisesti hemoglobiiniarvojen säilyttämiseksi halutulla pitoisuusvälillä: Hb 100–120 g/l (6,2–7,5 mmol/l). Alkuvaiheen erytroidista vastetta suositellaan arvioimaan 8–12 viikon hoidon jälkeen. Annoksen suurentaminen ja pienentäminen pitää tehdä vaiheittain (ks. kuvio jäljempänä). Hemoglobiinipitoisuutta yli 120 g/l (7,5 mmol/l) pitää välttää.
Annoksen suurentaminen
Annosta ei saa suurentaa yli enimmäisannoksen 1 050 IU/kg (kokonaisannos 80 000 IU) viikossa. Jos potilaan vaste annosta pienennettäessä häviää tai hemoglobiinipitoisuus pienenee ≥ 10 g/l, annos pitää suurentaa seuraavalle annostasolle. Annoksen suurentamiskertojen välin pitää olla vähintään 4 viikkoa.
Hoidon keskeyttäminen ja annoksen pienentäminen
Epoetiini zeeta -hoito pitää keskeyttää, jos hemoglobiinipitoisuus ylittää 120 g/l (7,5 mmol/l). Kun hemoglobiinipitoisuus on < 110 g/l, hoitoa voidaan lääkärin harkinnan mukaan jatkaa aiemmalla annoksella tai yhtä annostasoa pienemmällä annoksella. Jos hemoglobiinipitoisuus suurenee nopeasti (> 20 g/l neljän viikon aikana), annoksen pienentämistä yhdellä annostasolla pitää harkita.
Anemian oireet ja seuraukset saattavat vaihdella iän, sukupuolen ja samanaikaisten sairauksien mukaan. Lääkärin on arvioitava potilaan sairaus ja sen kliininen kulku yksilöllisesti.
Pediatriset potilaat
Oireisen anemian hoito kroonista munuaisten vajaatoimintaa sairastavilla hemodialyysipotilailla
Anemian oireet ja seuraukset saattavat vaihdella iän, sukupuolen ja samanaikaisten sairauksien mukaan. Lääkärin on arvioitava potilaan kokonaistila ja sairauden kliininen kulku yksilöllisesti.
Pediatrisille potilaille suositeltu hemoglobiinipitoisuusväli on 95–110 g/l (5,9−6,8 mmol/l). Retacrit annetaan hemoglobiiniarvon nostamiseksi korkeintaan pitoisuuteen 110 g/l (6,8 mmol/l). Hemoglobiiniarvon nousua yli 20 g/l (1,25 mmol/l) neljän viikon jakson aikana on vältettävä. Jos näin tapahtuu, annosta on muutettava ohjeiden mukaisesti.
Potilaita tulee seurata tarkoin, jotta voidaan varmistaa, että anemian ja sen oireiden hoitoon käytetään pienintä hyväksyttyä Retacrit-annosta.
Retacrit-hoito jakautuu kahteen vaiheeseen, korjausvaiheeseen ja ylläpitovaiheeseen.
Hemodialyysihoitoa saaville pediatrisille potilaille, joilla laskimoyhteys on helposti avattavissa, valmiste tulisi antaa mieluiten laskimoon.
Korjausvaihe
Aloitusannos on 50 IU/kg laskimoon kolmesti viikossa.
Annosta on tarvittaessa suurennettava tai pienennettävä 25 IU/kg kerta-annoksin (kolmesti viikossa), kunnes saavutetaan haluttu hemoglobiinin pitoisuusväli 95–110 g/l (5,9–6,8 mmol/l) (tämä tulisi tehdä vaiheittain niin, että väli on vähintään neljä viikkoa).
Ylläpitovaihe
Annosta on sovitettava tarkoituksenmukaisesti siten, että hemoglobiiniarvot pysyvät halutulla hemoglobiinin pitoisuusvälillä 95–110 g/l (5,9–6,8 mmol/l).
Alle 30-kiloiset lapset tarvitsevat yleensä suurempia ylläpitoannoksia kuin yli 30-kiloiset lapset ja aikuiset. Kliinisissä tutkimuksissa havaittiin 6 kuukauden hoidon jälkeen seuraavat ylläpitoannokset:
| Annos (IU/kg kolmesti viikossa) | |
Paino (kg) | Mediaani | Tavanomainen ylläpitoannos |
< 10 | 100 | 75–150 |
10–30 | 75 | 60–150 |
> 30 | 33 | 30–100 |
Pediatriset potilaat, joiden hemoglobiinipitoisuus on alussa erittäin pieni (< 68 g/l tai < 4,25 mmol/l), saattavat tarvita suurempia ylläpitoannoksia kuin ne, joiden hemoglobiinipitoisuus on alussa suurempi (> 68 g/l tai > 4,25 mmol/l).
Kroonista munuaisten vajaatoimintaa sairastavat potilaat, joilla on anemia ennen dialyysihoidon aloittamista tai peritoneaalidialyysihoidon aikana
Retacrit-valmisteen turvallisuutta ja tehoa kroonista munuaisten vajaatoimintaa sairastavien potilaiden hoidossa, jos potilaalla on anemia ennen dialyysihoidon aloittamista tai peritoneaalidialyysihoidon aikana, ei ole varmistettu. Tälle potilasryhmälle ihon alle annettavasta epoetiini alfa ‑hoidosta saatavissa olevan tiedon perusteella, joka on kuvattu kohdassa Farmakodynamiikka, ei voida antaa suosituksia annostuksesta.
Pediatriset potilaat, joilla on solunsalpaajahoidon aiheuttama anemia
Epoetiini alfan turvallisuutta ja tehoa solunsalpaajahoitoa saavien pediatristen potilaiden hoidossa ei ole varmistettu (ks. kohta Farmakodynamiikka).
Pediatristen leikkauspotilaiden hoito autologisessa verensiirto-ohjelmassa
Epoetiini alfan turvallisuutta ja tehoa pediatristen potilaiden hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Pediatriset potilaat, joille on suunniteltu suuri elektiivinen ortopedinen leikkaus
Epoetiini alfan turvallisuutta ja tehoa pediatristen potilaiden hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Varotoimet ennen lääkevalmisteen käsittelyä ja antoa
Retacrit-ruiskun on annettava lämmetä huoneenlämpöiseksi ennen käyttöä. Tämä vie tavallisesti 15–30 minuuttia.
Kroonista munuaisten vajaatoimintaa sairastavat aikuispotilaat, joilla on oireinen anemia
Kroonista munuaisten vajaatoimintaa sairastaville potilaille, joilla laskimoyhteys on avattavissa rutiiniluonteisesti (hemodialyysipotilaat), Retacrit tulisi antaa mieluiten laskimoon.
Jos laskimoyhteyttä ei ole helposti avattavissa (peritoneaalidialyysipotilaat ja potilaat, jotka eivät vielä saa dialyysihoitoa), Retacrit voidaan antaa injektiona ihon alle.
Aikuispotilaat, joilla on solunsalpaajahoidon aiheuttama anemia
Retacrit tulisi antaa injektiona ihon alle.
Aikuisten leikkauspotilaiden hoito autologisessa verensiirto-ohjelmassa
Retacrit tulisi antaa laskimoon.
Aikuispotilaat, joille on suunniteltu suuri elektiivinen ortopedinen leikkaus
Retacrit tulisi antaa injektiona ihon alle.
Aikuispotilaat, joilla on pienen tai keskisuuren riskin (riskitaso 1) myelodysplastinen oireyhtymä
Retacrit tulisi antaa injektiona ihon alle.
Oireisen anemian hoito hemodialyysihoitoa saavilla kroonista munuaisten vajaatoimintaa sairastavilla lapsipotilailla
Kroonista munuaisten vajaatoimintaa sairastaville pediatrisille potilaille, joilla laskimoyhteys on avattavissa rutiiniluonteisesti (hemodialyysipotilaat), Retacrit tulisi antaa mieluiten laskimoon.
Anto laskimoon
Annetaan vähintään 1–5 minuutin kuluessa kokonaisannoksesta riippuen. Hemodialyysipotilaille bolusinjektio voidaan antaa dialyysin aikana dialyysikatetrin laskimoportin kautta. Injektio voidaan vaihtoehtoisesti antaa hoitokerran jälkeen valtimo-laskimoavanteen kanyylin kautta, minkä jälkeen letku huuhdellaan 10 ml:lla isotonista keittosuolaliuosta, jotta injisoitu lääke siirtyy tyydyttävällä tavalla verenkiertoon (ks. Annostus, Aikuiset hemodialyysipotilaat).
Injektio pitää mieluiten antaa hitaammin potilaille, joille hoito aiheuttaa flunssan kaltaisia oireita (ks. kohta Haittavaikutukset).
Retacrit-valmistetta ei saa antaa infuusiona laskimoon eikä yhdistettynä muihin lääkevalmisteliuoksiin (ks. lisätietoja kohdasta Käyttö- ja käsittelyohjeet).
Anto ihon alle
1 ml:n enimmäismäärää yhtä pistoskohtaa kohti ei pitäisi yleensä ylittää. Suurempia tilavuuksia käytettäessä injektio tulisi pistää useaan eri kohtaan.
Injektiot pistetään raajoihin tai vatsaontelon etuseinämään.
Jos lääkäri katsoo, että potilas tai omainen voi turvallisesti ja tehokkaasti antaa Retacrit-valmisteen ihon alle, potilaalle/omaiselle tulee antaa oikeaa annosta ja antotapaa koskevat ohjeet.
Liuos on muiden injektiona annettavien valmisteiden tavoin tarkistettava, ettei siinä ole hiukkasia eikä sen väri ole muuttunut.
Ohjeet Retacrit-pistoksen pistämiseen itse ovat pakkausselosteen lopussa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Jos potilaalle on kehittynyt puhdas punasoluaplasia (pure red cell aplasia, PRCA) minkä tahansa erytropoietiinivalmisteen käytön yhteydessä, häntä ei tule hoitaa Retacrit-valmisteella tai millään muulla erytropoietiinivalmisteella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hallitsematon hypertensio.
Kaikki autologisen veren talteenottoon liittyvät kontraindikaatiot tulisi ottaa huomioon potilailla, jotka saavat Retacrit-lisähoitoa.
Retacrit-valmisteen käyttö on kontraindisoitu potilailla, joilla on suunniteltu suuri elektiivinen ortopedinen leikkaus ja joilla ei käytetä autologista verensiirto-ohjelmaa (oman veren luovuttaminen etukäteen), jos potilaalla on vakava sepelvaltimon, ääreisvaltimoiden, kaulavaltimon tai aivoverisuoniston sairaus tai potilas on saanut äskettäin sydäninfarktin tai aivoinfarktin.
Leikkauspotilaat, joille ei syystä riippumatta voida antaa riittävää tromboosiprofylaksia.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yleistä
Jokaisen epoetiini zeeta ‑hoitoa saavan potilaan verenpainetta on seurattava tarkoin ja sitä on hoidettava tarvittaessa. Varovaisuutta on noudatettava epoetiini zeetan käytössä, jos potilaalla on hoitamaton, riittämättömästi hoidettu tai huonosti hallinnassa oleva verenpainetauti. Potilaalle on ehkä aloitettava lisäksi verenpainetaudin hoito tai hoitoa on lisättävä. Jos verenpainetta ei saada hallintaan, epoetiini zeeta ‑hoito on lopetettava.
Välitöntä lääkärinhoitoa ja tehohoitoa vaatineita hypertensiivisiä kriisejä, joihin on liittynyt enkefalopatiaa ja kouristuskohtauksia, on esiintynyt myös epoetiini zeeta ‑hoidon aikana potilailla, joiden verenpaine on aiemmin ollut normaali tai matala. Pistävään migreenityyppiseen päänsärkyyn on kiinnitettävä erityistä huomiota mahdollisesti varoittavana oireena (ks. kohta Haittavaikutukset).
Varovaisuutta on noudatettava epoetiini zeetan käytössä, jos potilaalla on epilepsia, aiemmin esiintynyt kouristuskohtauksia tai potilaalla on kouristuskohtauksille altistava sairaus, kuten keskushermoston infektio tai aivometastaaseja.
Epoetiini zeetan käytössä on oltava varovainen, jos potilaalla on krooninen maksan vajaatoiminta. Epoetiini zeetan turvallisuutta potilaille, joilla on maksan vajaatoimintaa, ei ole varmistettu.
Punasolutuotantoa stimuloivia lääkeaineita saavilla potilailla on havaittu lisääntynyt tromboottisten verisuonitapahtumien (thrombotic vascular events, TVEs) esiintyvyys (ks. kohta Haittavaikutukset). Tällaisia tapahtumia ovat olleet laskimo- ja valtimotromboosit ja -emboliat (joista osa on johtanut potilaan kuolemaan), kuten syvä laskimotukos, keuhkoembolia, verkkokalvon verisuonitukos ja sydäninfarkti. Lisäksi aivoverisuonitapahtumia (kuten aivoinfarkteja, aivoverenvuotoa ja ohimeneviä aivoverenkiertohäiriöitä) on raportoitu.
Näiden tromboottisten verisuonitapahtumien raportoitua riskiä on punnittava huolellisesti epoetiini zeeta -hoidosta saataviin hyötyihin nähden erityisesti niillä potilailla, joilla on tromboottisten verisuonitapahtumien riskitekijöitä, kuten ylipaino ja aiemmin sairastettu tromboottinen verisuonitapahtuma (esim. syvä laskimotukos, keuhkoembolia tai aivoverisuonitapahtuma).
Kaikkien potilaiden hemoglobiiniarvoja on seurattava tarkoin mahdollisen kohonneen tromboemboliariskin ja kuolemanvaaran vuoksi silloin, kun potilaiden hemoglobiiniarvot ovat suuremmat kuin käyttöaiheen mukainen pitoisuusväli.
Verihiutaleiden määrä saattaa nousta kohtalaisesti normaalin vaihteluvälin rajoissa annoksesta riippuvaisesti epoetiini zeeta ‑hoidon aikana. Verihiutaleiden määrä palautuu ennalleen jatkohoidon aikana. Myös normaalin vaihteluvälin ylittävää trombosytemiaa on raportoitu. Verihiutaleiden määrää suositellaan seurattavaksi säännöllisesti kahdeksan ensimmäisen hoitoviikon aikana.
Kaikki muut anemiaa aiheuttavat tekijät (raudan, folaatin tai B12‑vitamiinin puutos, alumiinimyrkytys, infektio tai tulehdus, verenhukka, hemolyysi ja mistä tahansa aiheutunut luuydinfibroosi) tulisi arvioida ja hoitaa ennen epoetiini zeeta ‑hoidon aloittamista sekä tehtäessä päätöstä annoksen suurentamisesta. Seerumin ferritiiniarvot pienenevät useimmiten samaan aikaan, kun hematokriittiarvo suurenee. Optimaalista epoetiini zeeta ‑vastetta varten on varmistettava potilaan rautavarantojen riittävyys ja tarvittaessa on annettava rautalisää (ks. kohta Annostus ja antotapa):
- rautalisää (elementaarirautaa 200–300 mg/vrk suun kautta aikuisille ja 100–200 mg/vrk suun kautta lapsipotilaille) suositellaan kroonista munuaisten vajaatoimintaa sairastaville potilaille, joiden seerumin ferritiinipitoisuus on alle 100 ng/ml
- rautalisää (elementaarirautaa 200–300 mg/vrk suun kautta) suositellaan syöpäpotilaille, joiden transferriinisaturaatio on alle 20 %
- rautalisää (elementaarirautaa 200 mg/vrk suun kautta) pitää antaa autologisessa verensiirto-ohjelmassa mukana oleville potilaille useita viikkoja ennen autologisen veren talteenoton aloittamista, jotta saavutetaan suuret rautavarannot ennen epoetiini zeeta ‑hoidon aloittamista sekä koko epoetiini zeeta ‑hoidon keston ajaksi
- rautalisää (elementaarirautaa 200 mg/vrk suun kautta) pitää antaa koko epoetiini zeeta ‑hoidon ajan potilaille, joille on suunniteltu suuri elektiivinen ortopedinen leikkaus. Jos mahdollista, rautalisän käyttö pitää aloittaa ennen epoetiini zeeta ‑hoidon aloittamista, jotta saavutetaan riittävät rautavarannot.
Epoetiini zeetalla hoidetuilla potilailla on erittäin harvoin todettu porfyrian kehittymistä tai pahenemista. Epoetiini zeetaa on käytettävä varoen porfyriaa sairastaville potilaille.
Epoetiinihoidon yhteydessä on ilmoitettu vakavia ihoon kohdistuvia haittavaikutuksia, mukaan lukien Stevens–Johnsonin oireyhtymää ja toksista epidermaalista nekrolyysiä, jotka voivat olla hengenvaarallisia tai kuolemaan johtavia. Vakavampia tapauksia on havaittu pitkävaikutteisten epoetiinien yhteydessä.
Lääkettä määrättäessä potilaille on kerrottava oireista ja ihoreaktioita on seurattava tarkasti. Jos näihin reaktioihin viittaavia oireita ilmenee, Retacrit-valmisteen käyttö on lopetettava heti ja vaihtoehtoista hoitoa on harkittava.
Jos potilaalla on vakava ihoon kohdistuva reaktio, kuten Retacrit-valmisteen käytöstä johtuva Stevens–Johnsonin oireyhtymä tai toksinen epidermaalinen nekrolyysi, hoitoa Retacrit-valmisteella ei saa aloittaa kyseisellä potilaalla uudelleen missään vaiheessa.
Potilaille annettava punasolutuotantoa stimuloiva lääkeaine voidaan vaihtaa ainoastaan, jos asianmukainen valvonta on järjestetty.
Puhdas punasoluaplasia
Vasta-ainevälitteistä puhdasta punasoluaplasiaa (pure red cell aplasia, PRCA) on todettu kuukausien tai vuosien mittaisen epoetiinihoidon jälkeen. Tapauksia on raportoitu myös interferonilla ja ribaviriinillä hoidetuilla C-hepatiittia sairastavilla potilailla, jotka ovat saaneet samanaikaisesti punasolutuotantoa stimuloivia lääkeaineita. Epoetiini zeetaa ei ole hyväksytty C-hepatiittiin liittyvän anemian hoitoon.
Potilailta, joille kehittyy hemoglobiiniarvojen heikkenemisellä (10–20 g/l kuukaudessa) osoitettu hoidon tehon äkillinen puuttuminen ja jotka tarvitsevat verensiirtoja tavallista useammin, on tarkistettava retikulosyyttiarvot ja tutkittava tavanomaiset hoitovasteen puuttumiseen johtavat syyt (esim. raudan, folaatin tai B12-vitamiinin puutos, alumiinimyrkytys, infektio tai tulehdus, verenhukka, hemolyysi ja mistä tahansa aiheutunut luuydinfibroosi).
Jos potilaalla esiintyy paradoksaalista hemoglobiiniarvojen laskua ja hänelle kehittyy vaikea anemia, johon liittyy pieni retikulosyyttimäärä, epoetiini zeeta ‑hoito on lopetettava ja potilaalta on tutkittava erytropoietiinivasta-aineet. Luuydinnäytteen tutkimista on myös harkittava puhtaan punasoluaplasian toteamiseksi.
Muiden punasolutuotantoa stimuloivien lääkeaineiden käyttöä ei saa aloittaa ristireaktion riskin vuoksi.
Oireisen anemian hoito kroonista munuaisten vajaatoimintaa sairastavilla aikuis- ja lapsipotilailla
Kroonista munuaisten vajaatoimintaa sairastavien epoetiini zeeta ‑hoitoa saavien potilaiden hemoglobiinipitoisuus pitää mitata säännöllisesti, kunnes pitoisuus tasaantuu, ja vielä ajoittain tämän jälkeenkin.
Kroonisessa munuaisten vajaatoiminnassa hemoglobiinipitoisuuden nousunopeus tulisi olla noin 10 g/l (0,62 mmol/l) kuukautta kohti eikä se saisi ylittää 20 g/l (1,25 mmol/l) kuukautta kohti hypertension pahenemisriskin minimoimiseksi.
Potilailla, joilla on krooninen munuaisten vajaatoiminta, ylläpitohoidon aikainen hemoglobiinipitoisuus ei saa ylittää hemoglobiinin pitoisuusvälin ylärajaa, ks. kohta Annostus ja antotapa. Kliinisissä tutkimuksissa havaittiin kuoleman ja vakavien kardiovaskulaaritapahtumien riskin suurentuminen, kun punasolutuotantoa stimuloivan lääkeaineen tavoitteena oli hemoglobiiniarvon nostaminen yli pitoisuuden 120 g/l (7,5 mmol/l).
Kontrolloiduissa kliinisissä vertailututkimuksissa epoetiinien annosta ei todettu saatavan merkittävää hyötyä silloin, kun hemoglobiiniarvo kohosi suuremmaksi kuin oli tarpeen anemian oireiden hallitsemiseksi ja verensiirron välttämiseksi.
Jos potilaalla on krooninen munuaisten vajaatoiminta, Retacrit-annoksen suurentamisessa pitää olla varovainen, sillä suuriin kumulatiivisiin epoetiiniannoksiin saattaa liittyä lisääntynyt kuolleisuuden, vakavien sydän- ja verisuoni- sekä aivoverisuonitapahtumien riski. Jos potilaan hemoglobiinipitoisuudessa todettava vaste epoetiinihoitoon on huono, muut syyt huonoon vasteeseen pitää ottaa huomioon (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Epoetiini zeeta ‑valmistetta ihon alle saavia kroonista munuaisten vajaatoimintaa sairastavia potilaita on seurattava säännöllisesti lääkkeen vaikutuksen puuttumisen varalta, joka ilmenee epoetiini zeeta ‑hoidon hoitovasteen puuttumisena tai heikkenemisenä potilailla, joilla on aiemmin ilmennyt vaste tällaiseen hoitoon. Tälle on luonteenomaista hemoglobiiniarvojen jatkuva aleneminen epoetiini zeeta ‑hoidon annostuksen suurentamisesta huolimatta (ks. kohta Haittavaikutukset).
Osalla potilaista, joiden epoetiini zeeta ‑annosten antoväliä on pidennetty (pidemmäksi kuin kerran viikossa), hemoglobiinipitoisuus ei välttämättä pysy riittävänä (ks. kohta Farmakodynamiikka), jolloin epoetiini zeeta ‑annosta saattaa olla tarpeen suurentaa. Hemoglobiinipitoisuutta on seurattava säännöllisesti.
Sunttitrombooseja on ilmennyt hemodialyysipotilailla ja erityisesti potilailla, joilla on taipumusta matalaan verenpaineeseen tai joiden valtimo-laskimofistelissä ilmenee häiriöitä (esim. ahtaumia, aneurysmia jne.). Näille potilaille suositellaan suntin varhaista korjaamista ja tromboosin estohoitoa esimerkiksi asetyylisalisyylihapolla.
Hyperkalemiaa on havaittu yksittäistapauksissa, joskaan syy-yhteyttä ei ole osoitettu. Seerumin elektrolyyttitasoja on seurattava potilailla, joilla on krooninen munuaisten vajaatoiminta. Jos seerumin kaliumtason todetaan nousseen tai olevan nousussa, asianmukaisen hyperkalemian hoidon lisäksi on harkittava epoetiini zeeta ‑hoidon keskeyttämistä siihen asti, kunnes seerumin kaliumtaso on korjaantunut.
Hemodialyysin aikana hepariiniannosta on usein suurennettava epoetiini zeeta ‑hoitojakson aikana kohonneen hematokriitin vuoksi. Dialyysilaitteisto voi tukkeutua, jos hepariinihoito ei ole optimaalinen.
Tähänastisten saatavilla olevien tutkimustietojen perusteella anemian korjaaminen epoetiini zeetalla munuaisten vajaatoimintaa sairastavilla aikuispotilailla, jotka eivät vielä saa dialyysihoitoa, ei nopeuta munuaisten vajaatoiminnan pahenemista.
Potilaat, joilla on solunsalpaajahoidon aiheuttama anemia
Epoetiini zeeta ‑hoitoa saavien syöpäpotilaiden hemoglobiinipitoisuus pitää mitata säännöllisesti, kunnes pitoisuus tasaantuu, ja vielä ajoittain tämän jälkeenkin.
Epoetiinit ovat kasvutekijöitä, jotka stimuloivat ensisijaisesti punasolutuotantoa. Erytropoietiinireseptoreja voi esiintyä erilaisten kasvainsolujen pinnalla. Epoetiinien epäillään muiden kasvutekijöiden tavoin voivan stimuloida kasvainten kasvua.
Punasolutuotantoa stimuloivien lääkeaineiden osuutta kasvainten etenemisessä tai taudin etenemättömyysajan lyhenemisessä ei voida sulkea pois. Kontrolloiduissa kliinisissä tutkimuksissa epoetiini zeetan ja muiden punasolutuotantoa stimuloivien lääkeaineiden käyttöön liittyi vähentynyt lokoregionaalisten kasvainten kontrolli ja lyhyempi kokonaiseloonjäämisaika:
- vähensi lokoregionaalista kontrollia sädehoitoa saaneilla pitkälle edennyttä pään ja kaulan alueen syöpää sairastaneilla potilailla, kun hoidon tavoitteena oli hemoglobiinipitoisuuden nostaminen yli 140 g/l:aan (8,7 mmol/l)
- lyhensi kokonaiseloonjäämisaikaa ja lisäsi taudin etenemiseen liittyneitä kuolemia 4 kuukauden kohdalla solunsalpaajahoitoa saaneilla metastasoitunutta rintasyöpää sairastaneilla potilailla, kun hoidon tavoitteena oli hemoglobiinipitoisuuden nostaminen tasolle 120–140 g/l (7,5–8,7 mmol/l)
- lisäsi kuoleman riskiä aktiivista syöpää sairastavilla potilailla, jotka eivät saaneet solunsalpaaja- tai sädehoitoa, kun hoidon tavoitteena oli hemoglobiinipitoisuus 120 g/l (7,5 mmol/l). Punasolutuotantoa stimuloivia lääkeaineita ei ole tarkoitettu tämän potilasryhmän hoitoon.
- epoetiini zeetan ja standardihoidon yhdistelmää saaneessa ryhmässä havaittiin taudin etenemisen tai kuoleman riskin suurentuneen 9 % primaarianalyysista, ja metastasoitunutta rintasyöpää sairastavilla solunsalpaajahoitoa saaneilla potilailla ei voitu tilastollisesti sulkea pois 15 %:n riskin lisäystä silloin, kun he saivat hoitoa hemoglobiinipitoisuuden 100–120 g/l (6,2–7,5 mmol/l) saavuttamiseksi.
Edellä kuvatun perusteella syöpäpotilaiden anemian hoitoon tulisi joissain kliinisissä tilanteissa mieluiten käyttää verensiirtoa. Päätöksen rekombinanttierytropoietiinihoidon antamisesta on perustuttava hyöty-riskiarvioon, jonka tekemiseen potilas itse osallistuu ja päätöksenteossa on otettava huomioon kulloinenkin kliininen kokonaistilanne. Arvioinnissa olisi otettava huomioon mm. kasvaimen tyyppi ja sen levinneisyysaste, anemian vaikeusaste, elinajanodote, hoitoympäristö ja potilaan omat toivomukset (ks. kohta Farmakodynamiikka).
Solunsalpaajahoitoa saavilla syöpäpotilailla tulisi huomioida 2–3 viikon viive punasolutuotantoa stimuloivan lääkeaineen annon ja erytropoietiini-indusoitujen punasolujen ilmaantumisen välillä arvioitaessa epoetiini zeeta ‑hoidon sopivuutta (verensiirtoa todennäköisesti tarvitseva potilas).
Leikkauspotilaat autologisessa verensiirto-ohjelmassa
Kaikkia autologisen veren talteenottoon liittyviä varoituksia ja käyttöön liittyviä varotoimia, etenkin rutiininomaista volyyminkorvausta koskevia varotoimia, on noudatettava.
Potilaat, joille on suunniteltu suuri elektiivinen ortopedinen leikkaus
Leikkausten yhteydessä on aina noudatettava asianmukaisia ohjeita verensiirroista.
Suureen elektiiviseen ortopediseen leikkaukseen tuleville potilaille tulisi antaa riittävä antitromboottinen profylaksi, koska tromboottiset ja vaskulaariset tapahtumat ovat mahdollisia leikkauspotilailla erityisesti, kun potilaalla on ennestään kardiovaskulaarinen sairaus. Lisäksi on noudatettava erityistä varovaisuutta, kun potilaalla on taipumus syvien laskimotukosten muodostumiseen. Ei voida poissulkea myöskään mahdollisuutta, että epoetiini zeeta ‑hoitoon liittyisi lisääntynyt vaara postoperatiivisten tromboottisten tai vaskulaaristen tapahtumien esiintymiseen, kun potilaan hemoglobiinin taso hoidon alussa on > 130 g/l (> 8,1 mmol/l). Siksi epoetiini zeetaa ei tule käyttää potilailla, joiden hemoglobiinin lähtötaso on > 130 g/l (> 8,1 mmol/l).
Apuaineet
Tämä lääkevalmiste sisältää fenyylialaniinia, joka voi olla haitallista fenyyliketonuriaa sairastaville.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Polysorbaatti 20
Tämä lääkevalmiste sisältää enintään 0,1 mg polysorbaatti 20:tä (E 432) per ruisku, joka vastaa pitoisuutta 0,1 mg/ml (ks. kohta Vaikuttavat aineet ja niiden määrät). Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Ei ole näyttöä siitä, että epoetiini zeeta ‑hoito muuttaisi muiden lääkevalmisteiden metaboliaa.
Punasolutuotantoa vähentävät lääkevalmisteet saattavat heikentää vastetta epoetiini zeetalle.
Koska siklosporiini sitoutuu punasoluihin, lääkeinteraktion mahdollisuus on olemassa. Jos epoetiini zeetaa annetaan samanaikaisesti siklosporiinin kanssa, veren siklosporiinitasoa on seurattava ja siklosporiiniannosta on sovitettava hematokriittiarvon noustessa.
Tuumorinäytteiden hematologisen in vitro ‑differentiaation tai ‑proliferaation yhteydessä ei ole esiintynyt viitteitä interaktiosta epoetiini zeetan ja granulosyyttikasvupesäkettä tai granulosyytti-makrofagikantajasoluja stimuloivien tekijöiden (G-CSF ja GM-CSF) välillä.
Metastasoitunutta rintasyöpää sairastavien aikuisten naispotilaiden ihon alle annettu epoetiini alfa ‑annos 40 000 IU/ml yhdessä trastutsumabiannoksen 6 mg/kg kanssa ei vaikuttanut trastutsumabin farmakokinetiikkaan.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja epoetiini zeetan käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Siksi epoetiini zeetaa tulisi käyttää raskausaikana vain, jos mahdollinen hyöty on suurempi kuin sikiölle mahdollisesti aiheutuva riski. Epoetiini zeetan käyttöä ei suositella raskaana oleville leikkauspotilaille, jotka ovat mukana autologisessa verensiirto-ohjelmassa.
Imetys
Ei tiedetä, erittyykö eksogeeninen epoetiini zeeta ihmisen rintamaitoon. Epoetiini zeetaa tulee käyttää varoen imettäville äideille. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Retacrit‑hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Epoetiini zeetan käyttöä ei suositella imettäville leikkauspotilaille, jotka ovat mukana autologisessa verensiirto-ohjelmassa.
Hedelmällisyys
Epoetiini zeetan mahdollista vaikutusta miehen tai naisen hedelmällisyyteen ei ole tutkittu.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia valmisteen vaikutuksesta ajokykyyn tai koneidenkäyttökykyyn ei ole tehty.
Retacrit-valmisteella ei ole merkityksellistä vaikutusta ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin todettu haittavaikutus epoetiini alfa -hoidon aikana oli annosriippuvainen verenpaineen nousu tai aiemmin todetun korkean verenpaineen paheneminen. Potilaiden verenpainetta on seurattava erityisesti hoidon alkuvaiheessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yleisimmin esiintyneitä haittavaikutuksia, joita on havaittu kliinisissä tutkimuksissa epoetiini alfan käytön yhteydessä, ovat ripuli, pahoinvointi, oksentelu, kuume ja päänsärky. Vilustumista muistuttavia oireita voi ilmaantua erityisesti hoidon alussa.
Hengitysteiden tukkoisuutta, ylempien hengitysteiden tukkoisuus, nenän tukkoisuus ja nasofaryngiitti mukaan lukien, on raportoitu tutkimuksissa, joissa munuaisten vajaatoimintaa sairastavien, mutta ei vielä dialyysihoitoa saavien aikuispotilaiden lääkkeenantoväliä oli pidennetty.
Punasolutuotantoa stimuloivia lääkeaineita saavilla potilailla on havaittu lisääntynyt tromboottisten verisuonitapahtumien ilmaantuvuus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Epoetiini alfan yleistä turvallisuusprofiilia tutkittiin 25 satunnaistetussa, kaksoissokkoutetussa, lumehoidolla tai tavallisesti käytettävällä hoidolla kontrolloidussa tutkimuksessa 2 094 aneemisella potilaalla yhteensä 3 417 mukaan otetusta tutkittavasta. Näistä potilaista 228 epoetiini alfa ‑hoitoa saanutta kroonista munuaisten vajaatoimintaa sairastanutta tutkittavaa osallistui neljään kroonista munuaisten vajaatoimintaa koskevaan tutkimukseen (2 tutkimusta potilailla ennen dialyysihoitoa [N = 131 altistettua kroonista munuaisten vajaatoimintaa sairastavaa tutkittavaa] ja 2 dialyysihoitoa saaneilla potilailla [N = 97 altistettua kroonista munuaisten vajaatoimintaa sairastavaa tutkittavaa]; 1 404 altistettua syöpäpotilasta solunsalpaajahoidosta aiheutunutta anemiaa koskevassa 16 tutkimuksessa; 147 altistettua tutkittavaa autologista verenluovutusta koskevissa kahdessa tutkimuksessa; 213 altistettua tutkittavaa leikkauksen yhteydessä tehdyssä yhdessä tutkimuksessa; ja 102 altistettua tutkittavaa kahdessa myelodysplastista oireyhtymää (MDS) koskeneessa tutkimuksessa). Haittavaikutukset, joita raportoitiin näissä tutkimuksissa ≥ 1 %:lla epoetiini alfa ‑hoitoa saaneista tutkittavista, esitetään seuraavassa taulukossa.
Yleisyysluokat: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
MedDRA-elinjärjestelmäluokitus | Haittavaikutus (Preferred Term ‑taso) | Yleisyys |
Veri ja imukudos | Puhdas punasoluaplasia3, | Harvinainen |
trombosytemia | ||
Aineenvaihdunta ja ravitsemus | Hyperkalemia1 | Melko harvinainen |
Immuunijärjestelmä | Yliherkkyys3 | Melko harvinainen |
Anafylaktinen reaktio3 | Harvinainen | |
Hermosto | Päänsärky | Yleinen |
Kouristukset | Melko harvinainen | |
Verisuonisto | Kohonnut verenpaine, laskimo- ja valtimotromboosit2 | Yleinen |
Hypertensiivinen kriisi3 | Tuntematon | |
Hengityselimet, rintakehä ja välikarsina | Yskä | Yleinen |
Hengitystiekongestio | Melko harvinainen | |
Ruoansulatuselimistö | Ripuli, pahoinvointi, oksentelu | Hyvin yleinen |
Iho ja ihonalainen kudos | Ihottuma | Yleinen |
Urtikaria3 | Melko harvinainen | |
Angioneuroottinen edeema3 | Tuntematon | |
Luusto, lihakset ja sidekudos | Nivelsärky, luukipu, lihassärky, raajakipu | Yleinen |
Synnynnäiset ja perinnölliset/geneettiset häiriöt | Akuutti porfyria3 | Harvinainen |
Yleisoireet ja antopaikassa todettavat haitat | Kuume | Hyvin yleinen |
Vilunväristykset, influenssan kaltaiset oireet, pistoskohdan reaktiot, perifeerinen turvotus | Yleinen | |
Lääkkeen tehon puute3 | Tuntematon | |
Tutkimukset | Erytropoietiinin vasta-ainepositiivisuus | Harvinainen |
1 Dialyysihoidon yhteydessä yleinen 2 Sisältää kuolemaan johtaneet ja muut kuin kuolemaan johtaneet valtimo- ja laskimotapahtumat, kuten syvän laskimotukoksen, keuhkoembolian, verkkokalvon verisuonitukoksen, valtimotromboosin (sydäninfarkti mukaan lukien), aivoverisuonitapahtumat (aivoinfarkti ja aivoverenvuoto mukaan lukien), ohimenevät aivoverenkiertohäiriöt ja sunttitromboosin (dialyysilaitteet mukaan lukien) ja aneurysman arteriovenoosisen suntin tukokset. 3 Selitetty seuraavassa alakohdassa ja/tai kohdassa Varoitukset ja käyttöön liittyvät varotoimet | ||
Valittujen haittavaikutusten kuvaus
Yliherkkyysreaktioita, ihottumatapauksia (myös urtikariaa), anafylaktisia reaktioita ja angioedeemaa mukaan lukien, on raportoitu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epoetiinihoidon yhteydessä on ilmoitettu vakavia ihoon kohdistuvia haittavaikutuksia, mukaan lukien Stevens–Johnsonin oireyhtymää ja toksista epidermaalista nekrolyysiä, jotka voivat olla hengenvaarallisia tai kuolemaan johtavia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Välitöntä lääkärinhoitoa ja tehohoitoa vaatineita hypertensiivisiä kriisejä, joihin on liittynyt enkefalopatiaa ja kouristuskohtauksia, on esiintynyt myös epoetiini zeeta ‑hoidon aikana potilailla, joiden verenpaine on aiemmin ollut normaali tai matala. Pistävään migreenityyppiseen päänsärkyyn on kiinnitettävä erityistä huomiota mahdollisesti varoittavana oireena (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vasta-ainevälitteistä puhdasta punasoluaplasiaa on raportoitu hyvin harvoin, < 1/10 000 tapausta potilasvuotta kohden, kuukausien tai vuosien epoetiinihoidon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Tapauksia on raportoitu enemmän ihonalaisen (s.c.) kuin laskimonsisäisen (i.v.) antotavan yhteydessä.
Aikuispotilaat, joilla on pienen tai keskisuuren riskin (riskitaso 1) myelodysplastinen oireyhtymä (MDS)
Satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa, epoetiini alfa -valmisteella tehdyssä monikeskustutkimuksessa neljälle (4,7 %) tutkittavalle ilmaantui tromboottisia verisuonitapahtumia (äkkikuolema, iskeeminen aivohalvaus, embolia ja flebiitti). Kaikki tromboottiset verisuonitapahtumat esiintyivät epoetiini alfa ‑ryhmässä ja tapahtuivat ensimmäisten 24 tutkimusviikon aikana. Näistä kolme varmistui tromboottisiksi verisuonitapahtumiksi, mutta yhdessä tapauksessa (äkkikuolema) tromboembolinen tapahtuma ei varmistunut. Kahdella tutkittavalla oli merkittäviä riskitekijöitä (eteisvärinä, sydämen vajaatoiminta ja tromboflebiitti).
Hemodialyysihoitoa saavat kroonista munuaisten vajaatoimintaa sairastavat pediatriset potilaat
Hemodialyysiä saavista kroonista munuaisten vajaatoimintaa sairastavista pediatrisista potilaista on kliinisistä tutkimuksista ja valmisteen markkinoille tulon jälkeen vähän kokemusta. Tässä potilasryhmässä ei ole raportoitu sellaisia pediatrisille potilaille erityisiä haittavaikutuksia, joita ei ole jo mainittu edellä olevassa taulukossa tai jotka eivät sopineet perussairauteen.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Erytropoietiinin terapeuttinen alue on hyvin laaja. Erytropoietiinin yliannostus saattaa aiheuttaa vaikutuksia, jotka ovat hormonin korostuneita farmakologisia vaikutuksia. Flebotomia voidaan tehdä, jos potilaan hemoglobiinitasot ovat erityisen korkeat. Potilaalle tulee antaa tarvittaessa myös muuta oireenmukaista hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut anemialääkkeet, erytropoietiini, ATC-koodi: B03XA01
Retacrit on ns. biosimilaari lääkevalmiste. Yksityiskohtaisempaa tietoa on saatavilla Euroopan lääkeviraston verkkosivulta: http://www.ema.europa.eu.
Vaikutusmekanismi
Erytropoietiini (EPO) on keskeinen veren punasolutuotantoa säätelevä glykoproteiinihormoni, jota tuotetaan pääasiassa munuaisissa vasteena hypoksiaan. EPO osallistuu erytropoieesin kaikkiin vaiheisiin ja vaikuttaa pääasiassa punasolujen esiasteiden tasolla. Kun EPO on sitoutunut solun pinnalla olevaan reseptoriin, se aktivoi signaalitransduktioreittejä, jotka häiritsevät apoptoosia ja stimuloivat erytrooisten solujen proliferaatiota. Kiinanhamsterin munasarjasoluissa tuotetun rekombinantin humaani-EPOn (epoetiini zeetan) aminohappojärjestys on identtinen ihmisen virtsan EPO:n (165 aminohappoa) kanssa. Näitä kahta ei voi toiminnallisten määritysten perusteella erottaa toisistaan. Erytropoietiinin laskennallinen molekyylipaino on 32 000−40 000 daltonia.
Erytropoietiini on kasvutekijä, joka stimuloi pääasiassa punasolutuotantoa. Erytropoietiinireseptorit saattavat ilmentyä erilaisten kasvainsolujen pinnalla.
Farmakodynaamiset vaikutukset
Terveet vapaaehtoiset
Epoetiini alfa -kerta-annoksen (20 000–160 000 IU ihon alle) jälkeen havaittiin annosriippuvainen vaste tutkituille farmakodynaamisille merkkiaineille, joita olivat retikulosyytit, veren punasolut ja hemoglobiini. Retikulosyyttien prosenttiosuuden muutoksissa havaittiin selkeä pitoisuus-aikaprofiili, jossa oli havaittavissa huippu ja paluu lähtötilanteeseen. Veren punasolujen ja hemoglobiinin osalta profiili ei ollut yhtä selkeä. Kaikki farmakodynaamiset merkkiaineet lisääntyivät yleensä lineaarisesti siten, että maksimivaste saavutettiin suurimmilla annostuksilla.
Farmakodynaamisia tutkimuksia tehtiin lisäksi annoksilla 40 000 IU kerran viikossa verrattuna annokseen 150 IU/kg kolmesti viikossa. Pitoisuus-aikaprofiilin eroista huolimatta farmakodynaaminen vaste (retikulosyyttien, hemoglobiinin ja veren punasolujen kokonaismäärän prosenttimuutoksilla mitattuna) oli samankaltainen näitä annostuksia käytettäessä. Lisäksi tehdyissä tutkimuksissa verrattiin ihon alle annettuja epoetiini alfa -annoksia 40 000 IU kerran viikossa annoksiin 80 000–120 000 IU kerran kahdessa viikossa. Näiden terveillä tutkittavilla tehtyjen farmakodynaamisten tutkimusten tulosten perusteella annostus 40 000 IU kerran viikossa vaikuttaa yleisesti ottaen tehostavan veren punasolutuotantoa enemmän kuin kahden viikon välein annettava hoito huolimatta retikulosyyttituotannossa havaitusta samankaltaisuudesta kerran viikossa ja kahden viikon välein tapahtuvassa annossa.
Krooninen munuaisten vajaatoiminta
Epoetiini alfan on osoitettu stimuloivan erytropoieesia aneemisilla kroonista munuaisten vajaatoimintaa sairastavilla, sekä dialyysihoitoa saavilla ja dialyysillä vielä hoitamattomilla, potilailla. Ensimmäinen osoitus vasteesta epoetiini alfalle on retikulosyyttimäärän suureneminen 10 vuorokauden kuluessa, mitä seuraa veren punasolumäärän, hemoglobiinipitoisuuden ja hematokriitin suureneminen tavallisesti 2–6 viikon kuluessa. Hemoglobiinivaste vaihtelee potilaiden välillä, ja elimistön rautavarannot ja muut samanaikaiset sairaudet saattavat vaikuttaa siihen.
Solunsalpaajahoidosta aiheutuva anemia
Kolme kertaa viikossa tai kerran viikossa annetun epoetiini alfan on osoitettu suurentavan solunsalpaajahoitoa saavien aneemisten syöpäpotilaiden hemoglobiinipitoisuutta ja vähentävän verensiirron tarvetta ensimmäisen hoitokuukauden jälkeen.
Tutkimuksessa, jossa annoksia 150 IU/kg kolmesti viikossa ja 40 000 IU kerran viikossa verrattiin terveillä tutkittavilla ja aneemisilla syöpäpotilailla, retikulosyyttien, hemoglobiinin ja veren punasolujen kokonaismäärän prosenttimuutosten aikaprofiilit olivat näiden kahden annostuksen yhteydessä samankaltaiset sekä terveillä tutkittavilla että aneemisilla syöpäpotilailla. Annosten 150 IU/kg kolmesti viikossa ja 40 000 IU kerran viikossa vastaavien farmakodynaamisten parametrien AUC-arvot olivat terveillä tutkittavilla ja aneemisilla syöpäpotilailla samankaltaiset.
Aikuiset leikkauspotilaat autologisessa verensiirto-ohjelmassa
Epoetiini alfan on osoitettu stimuloivan veren punasolutuotantoa, jotta autologisen veren talteenottoa voidaan lisätä ja vähentää hemoglobiinipitoisuuden laskua aikuispotilailla, joille on suunniteltu suuri elektiivinen leikkaus ja joilta ei odoteta saatavan etukäteen talteen leikkauksen yhteydessä tarvittavaa koko verimäärää. Suurin teho havaitaan potilailla, joiden hemoglobiinipitoisuus on pieni (≤ 130 g/l; 8,1 mmol/l).
Aikuispotilaat, joille on suunniteltu suuri elektiivinen ortopedinen leikkaus
Jos potilaalle suunnitellaan suurta elektiivistä ortopedista leikkausta ja potilaan hemoglobiinipitoisuus ennen hoitoa on > 100 – ≤ 130 g/l, epoetiini alfan on osoitettu pienentävän allogeenisten verensiirtojen saamisen riskiä ja nopeuttavan erytroidista korjautumista (suurentunut hemoglobiinipitoisuus, hematokriitti ja retikulosyyttimäärä).
Kliininen teho ja turvallisuus
Krooninen munuaisten vajaatoiminta
Epoetiini alfaa on tutkittu kliinisissä tutkimuksissa aneemisilla kroonista munuaisten vajaatoimintaa sairastavilla aikuispotilailla, hemodialyysiä saavat ja dialyysillä vielä hoitamattomat potilaat mukaan lukien, anemian hoitoon ja hematokriitin pitämiseen tavoitepitoisuusvälillä 30–36 %.
Kun kliininen tutkimus toteutettiin aloitusannoksilla 50–150 IU/kg kolmesti viikossa, hematokriitti suureni kliinisesti merkitsevästi noin 95 %:lla potilaista. Käytännöllisesti katsoen kaikki potilaat olivat verensiirroista riippumattomia noin kahden kuukauden hoidon jälkeen. Kun tavoitteena ollut hematokriitti oli saavutettu, ylläpitoannos määriteltiin kullekin potilaalle sopivaksi.
Hematokriitin 30–36 %:ssa pitämiseen tarvittavan ylläpitoannoksen mediaani oli kolmessa laajimmassa aikuisilla dialyysipotilailla tehdyssä kliinisessä tutkimuksessa noin 75 IU/kg kolmesti viikossa.
Kaksoissokkoutetussa, lumekontrolloidussa, hemodialyysihoitoa saavien kroonista munuaisten vajaatoimintaa sairastavien potilaiden elämänlaatua selvittävässä monikeskustutkimuksessa osoitettiin epoetiini alfa -hoitoa saaneiden potilaiden elämänlaadun parantuneen kliinisesti ja tilastollisesti merkitsevästi lumeryhmään verrattuna, kun uupumusta, fyysisiä oireita, ihmissuhteita ja masennusta (munuaistautiin liittyvä kysely Kidney Disease Questionnaire) mitattiin kuuden kuukauden hoidon jälkeen. Epoetiini alfa -hoitoa saaneen ryhmän potilaat otettiin mukaan myös avoimeen jatkotutkimukseen, jossa osoitettiin, että heidän elämänlaatunsa osoitettu paraneminen oli säilynyt vielä 12 kuukauden ajan.
Munuaisten vajaatoimintaa sairastavat aikuispotilaat, jotka eivät vielä saa dialyysihoitoa
Hoidon keskimääräinen kesto oli kliinisissä tutkimuksissa epoetiini alfa ‑hoitoa saaneilla kroonista munuaisten vajaatoimintaa sairastavilla potilailla, jotka eivät saaneet dialyysihoitoa, lähes viisi kuukautta. Nämä potilaat saivat vasteen epoetiini alfa -hoitoon vastaavasti kuin dialyysihoitoa saaneilla potilailla oli havaittu. Kroonista munuaisten vajaatoimintaa sairastavilla potilailla, jotka eivät saaneet dialyysihoitoa, todettiin annosriippuvainen ja pitkäkestoisesti suurentunut hematokriitti, kun epoetiini alfa annettiin joko laskimoon tai ihon alle. Vastaavansuuruista hematokriitin nousua huomattiin, kun epoetiini alfa annettiin kumman tahansa näiden antoreittien kautta. Epoetiini alfa ‑annosten 75–150 IU/kg viikossa on lisäksi osoitettu pitävän hematokriitin 36–38 %:ssa enimmillään kuuden kuukauden ajan.
Kahdessa tutkimuksessa, joissa epoetiini alfa ‑hoidon antoväliä oli pidennetty (3 kertaa viikossa, kerran viikossa, kerran joka toinen viikko ja kerran joka neljäs viikko), joidenkin pidemmin antovälein lääkettä saaneiden potilaiden hemoglobiinipitoisuus ei pysynyt riittävänä, ja he täyttivät tutkimussuunnitelmassa määritellyt hemoglobiinipitoisuuteen perustuvat keskeyttämiskriteerit (0 % kerran viikossa, 3,7 % kerran joka toinen viikko ja 3,3 % kerran joka neljäs viikko lääkehoitoa saaneessa ryhmässä).
Satunnaistetussa prospektiivisessa tutkimuksessa (CHOIR) arvioitiin 1 432 aneemista kroonista munuaisten vajaatoimintaa sairastavaa potilasta, jotka eivät saaneet dialyysihoitoa. Potilaat osoitettiin saamaan epoetiini alfa -hoitoa, jolloin hemoglobiinipitoisuuden ylläpitotavoite oli 135 g/l (suositeltua suurempi hemoglobiinipitoisuus) tai 113 g/l. Vakava kardiovaskulaaritapahtuma (kuolema, sydäninfarkti, aivohalvaus tai sairaalahoito kongestiivisen sydämen vajaatoiminnan vuoksi) todettiin 125 potilaalla (18 %) 715 potilaasta, joilla oli suurempi hemoglobiinipitoisuus, verrattuna 97 potilaaseen (14 %) 717 potilaasta, joilla oli pienempi hemoglobiinipitoisuus (riskitiheyksien suhde [hazard ratio, HR] 1,3, 95 %:n luottamusväli: 1,0, 1,7, p = 0,03).
Kliinisten ESA-tutkimusten yhdistetyt post-hoc-analyysit tehtiin kroonista munuaisten vajaatoimintaa sairastavista potilaista (dialyysihoitoa saavat, ei dialyysihoitoa saavat, diabetesta sairastavat ja sairastamattomat potilaat). Suurempiin kumulatiivisiin ESA-annoksiin liittyi yleensä suurempi kokonaiskuolleisuuden sekä sydän- ja verisuoni- sekä aivoverisuonitapahtumien riski riippumatta siitä, sairastiko potilas diabetesta tai saiko hän dialyysihoitoa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Hoito potilaille, joilla on solunsalpaajahoidon aiheuttama anemia
Epoetiini alfaa on tutkittu kliinisissä tutkimuksissa aikuisilla aneemisilla syöpäpotilailla, joilla oli lymfaattisia ja kiinteitä kasvaimia, sekä erilaisia solunsalpaajahoitoja, platinaa ja muuta kuin platinaa sisältävät hoidot mukaan lukien, saavilla potilailla. Näissä tutkimuksissa on osoitettu kolmesti viikossa ja kerran viikossa annetun epoetiini alfan suurentavan aneemisten syöpäpotilaiden hemoglobiinipitoisuutta ja vähentävän verensiirron tarvetta ensimmäisen hoitokuukauden jälkeen. Kaksoissokkoutettua vaihetta seurasi joissakin tutkimuksissa avoin vaihe, jossa kaikki potilaat saivat epoetiini alfaa, ja tehon havaittiin säilyvän.
Saatavissa oleva näyttö viittaa siihen, että vaste epoetiini alfa ‑hoitoon on samanlainen potilailla, joilla on hematologinen pahanlaatuinen sairaus ja kiinteitä kasvaimia, samoin kuin potilailla, joiden kasvain on levinnyt tai ei ole levinnyt luuytimeen. Tutkimuksissa kemoterapian verrannollinen voimakkuus osoitettiin epoetiini alfaa saaneessa ja lumelääkettä saaneessa ryhmässä näiden ryhmien samankaltaisella neutrofiilien AUC-arvolla sekä niiden potilaiden samankaltaisella osuudella epoetiini alfaa ja lumelääkettä saaneissa ryhmissä, joiden absoluuttinen neutrofiilimäärä pieneni alle 1000 solua/mikrol ja 500 solua/mikrol.
Prospektiivisessa satunnaistetussa lumelääkekontrolloidussa kaksoissokkotutkimuksessa, johon osallistui 375 anemiapotilasta, joilla oli erilaisia ei-myeloidisia kasvaimia ja jotka saivat muuta kuin platinapohjaista solunsalpaajahoitoa, nähtiin merkittävä lasku anemiaan liittyvien jälkitilojen (esim. väsymys, alhainen energia- ja aktiivisuustaso) esiintyvyydessä seuraavilla mittareilla ja asteikoilla mitattuna: Functional Assessment of Cancer Therapy-Anemia (FACT-An) ‑yleisasteikko, FACT-An-väsymysasteikko ja Cancer Linear Analogue Scale (CLAS). Kahdessa muussa pienemmässä satunnaistetussa lumelääkekontrolloidussa tutkimuksessa ei voitu osoittaa merkittävää paranemista elämänlaatuparametreissa EORTC-QLQ-C30-asteikolla eikä CLAS-asteikolla.
Eloonjäämistä ja kasvaimen etenemistä on tutkittu viidessä laajassa kontrolloidussa tutkimuksessa, joihin osallistui yhteensä 2 833 potilasta. Tutkimuksista neljä oli kaksoissokkoutettua, lumelääkekontrolloitua tutkimusta ja yksi oli avoin tutkimus. Tutkimuksiin otettiin joko potilaita, jotka saivat solunsalpaajahoitoa (kaksi tutkimusta), tai sellaisia potilasryhmiä, joiden sairauden hoitoon punasolutuotantoa stimuloivat lääkeaineet eivät ole käyttöaiheisia: anemia syöpäpotilailla, jotka eivät saa solunsalpaajahoitoa, ja sädehoitoa saavat pään ja kaulan alueen syöpää sairastavat potilaat. Kahdessa tutkimuksessa hemoglobiinin tavoitearvo oli > 130 g/l (8,1 mmol/l) ja kolmessa muussa tutkimuksessa tavoitearvo oli 120–140 g/l (7,5–8,7 mmol/l). Avoimessa tutkimuksessa ei todettu eroa kokonaiseloonjäämisessä ihmisen rekombinanttierytropoietiinihoitoa saaneiden ja verrokkiryhmän välillä. Neljässä lumelääkekontrolloidussa tutkimuksessa kokonaiseloonjäämisen riskitiheyksien suhde (hazard ratio) vaihteli välillä 1,25–2,47 verrokkiryhmän hyväksi. Näissä tutkimuksissa on todettu säännönmukainen selittämätön tilastollisesti merkitsevä ylikuolleisuus potilailla, joilla oli erilaisiin yleisiin syöpiin liittyvä anemia ja jotka saivat ihmisen rekombinanttierytropoietiinihoitoa, verrattuna verrokkiryhmään. Tutkimuksissa todettua kokonaiseloonjäämistä ei voitu tyydyttävästi selittää tromboosien ja muiden vastaavien komplikaatioiden esiintyvyyden eroilla ihmisen rekombinanttierytropoietiinihoitoa saaneiden ja verrokkiryhmän potilaiden välillä.
Useilla epoetiinivalmisteilla tehtyihin 53 kontrolloituun kliiniseen lääketutkimukseen osallistuneiden yli 13 900 syöpäpotilaan (solunsalpaajahoito, sädehoito, solunsalpaajahoidon ja sädehoidon yhdistelmä ja ei hoitoa) tiedot on myös analysoitu. Kokonaiseloonjääntitietojen meta-analyysistä saatiin riskitiheyksien suhteen piste-estimaatiksi 1,06 verrokkiryhmän hyväksi (95 %:n luottamusväli 1,00, 1,12; 53 tutkimusta ja 13 933 potilasta), ja solunsalpaajahoitoa saaneiden syöpäpotilaiden kokonaiseloonjäämisen riskitiheyksien suhde oli 1,04 (95 %:n luottamusväli 0,97, 1,11; 38 tutkimusta ja 10 441 potilasta). Meta-analyysien tulokset viittaavat myös johdonmukaisesti siihen, että ihmisen rekombinanttierytropoietiinihoitoa saaneilla syöpäpotilailla on tromboembolisten tapahtumien suhteellinen riski huomattavasti lisääntynyt (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Satunnaistettuun, avoimeen monikeskustutkimukseen osallistui 2 098 aneemista naista, jotka sairastivat metastasoitunutta rintasyöpää ja saivat ensilinjan tai toisen linjan solunsalpaajahoitoa. Kyseessä oli vertailukelpoisuustutkimus, jossa oli tarkoitus sulkea pois epoetiini alfan ja standardihoidon yhdistelmän käyttöön liittyvä kasvaimen etenemisen tai kuoleman 15 % riskin lisäys verrattuna pelkkään standardihoitoon. Kliinisten tietojen keruun katkaisuajankohtana tutkijan arvioiman taudin etenemättömyysajan (PFS) mediaani oli kummassakin ryhmässä 7,4 kuukautta (riskitiheyksien suhde [HR] 1,09, 95 %:n luottamusväli: 0,99, 1,20), mikä osoittaa, että tutkimuksen tavoitetta ei saavutettu. Epoetiini alfan ja standardihoidon yhdistelmää saaneen ryhmän potilaista huomattavasti harvempi sai punasolusiirtoja (5,8 % versus 11,4 %), mutta huomattavasti useammalla epoetiini alfan ja standardihoidon yhdistelmää saaneen ryhmän potilaista todettiin tromboottisia verisuonitapahtumia (2,8 % versus 1,4 %). Lopullisessa analyysissa raportoitiin 1 653 kuolemaa. Kokonaiseloonjäämisajan mediaani oli epoetiini alfan ja standardihoidon yhdistelmää saaneessa ryhmässä 17,8 kuukautta verrattuna 18,0 kuukauteen pelkästään standardihoitoa saaneessa ryhmässä (HR 1,07, 95 %:n luottamusväli: 0,97, 1,18). Mediaaniaika taudin etenemiseen (TTP) tutkijan arvioimana oli 7,5 kuukautta epoetiini alfan ja standardihoidon yhdistelmää saaneessa ryhmässä ja 7,5 kuukautta standardihoitoa saaneessa ryhmässä (HR 1,099, 95 %:n luottamusväli: 0,998, 1,210). Mediaani TTP riippumattoman komitean arvioimana oli 8,0 kuukautta epoetiini alfan ja standardihoidon yhdistelmää saaneessa ryhmässä ja 8,3 kuukautta standardihoitoa saaneessa ryhmässä (HR 1,033, 95 %:n luottamusväli: 0,924, 1,156).
Autologinen verensiirto-ohjelma
Epoetiini alfan autologisen veren talteenottoa parantavaa tehoa potilailla, joiden hematokriitti on matala (≤ 39 % eikä anemian taustalla ole raudanpuutosta) ja joille on suunniteltu suurta ortopedista leikkausta, selvitettiin 204 potilaalla tehdyssä kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa sekä 55 potilaalla yksöissokkoutetussa lumekontrolloidussa tutkimuksessa.
Kaksoissokkoutetussa tutkimuksessa potilaat saivat epoetiini alfaa 600 IU/kg tai lumelääkettä laskimoon kerran vuorokaudessa 3–4 vuorokauden välein kolmen viikon ajan (yhteensä 6 annosta). Epoetiini alfalla hoidetut potilaat pystyivät luovuttamaan keskimäärin huomattavasti useampia yksiköitä verta (4,5 yksikköä) kuin lumehoitoa saaneet potilaat (3,0 yksikköä).
Yksöissokkoutetussa tutkimuksessa potilaat saivat epoetiini alfaa 300 IU/kg tai 600 IU/kg tai lumelääkettä laskimoon kerran vuorokaudessa 3–4 vuorokauden välein kolmen viikon ajan (yhteensä 6 annosta). Epoetiini alfalla hoidetut potilaat pystyivät luovuttamaan keskimäärin huomattavasti useampia yksiköitä verta (300 IU/kg epoetiini alfaa = 4,4 yksikköä, 600 IU/kg epoetiini alfaa = 4,7 yksikköä) kuin lumehoitoa saaneet potilaat (2,9 yksikköä).
Epoetiini alfa pienensi allogeeniselle verelle altistumisen riskiä 50 % verrattuna potilaisiin, jotka eivät saaneet epoetiini alfaa.
Suuri elektiivinen ortopedinen leikkaus
Epoetiini alfan (300 IU/kg tai 100 IU/kg) tehoa allogeeniselle verensiirrolle altistumiselle on arvioitu lumekontrolloidussa, kaksoissokkoutetussa kliinisessä tutkimuksessa aikuispotilailla, joilla ei ollut raudanpuutosta ja joille oli suunniteltu suuri elektiivinen ortopedinen lonkka- tai polvileikkaus. Epoetiini alfaa annettiin ihon alle 10 vuorokautta ennen leikkausta, leikkauspäivänä ja neljän päivän ajan leikkauksen jälkeen. Potilaat ositettiin lähtötilanteen hemoglobiinipitoisuuden mukaan (≤ 100 g/l, > 100– ≤ 130 g/l ja > 130 g/l).
Epoetiini alfa -annos 300 IU/kg vähensi allogeenisen verensiirron riskiä merkittävästi, jos potilaan hemoglobiinipitoisuus ennen hoitoa oli > 100 – ≤ 130 g/l. 16 % epoetiini alfa ‑annoksia 300 IU/kg, 23 % epoetiini alfa ‑annoksia 100 IU/kg ja 45 % lumehoitoa saaneista potilaista tarvitsi verensiirron.
Avoimessa rinnakkaisryhmillä toteutetussa tutkimuksessa aikuispotilaille (ei raudan puutosta, hemoglobiinipitoisuus ennen hoitoa ≥ 100 – ≤ 130 g/l, suunniteltu suuri ortopedinen lonkka- tai polvileikkaus) annettuja epoetiini alfa ‑annoksia 300 IU/kg ihon alle päivittäin 10 päivän ajan ennen leikkausta, leikkauspäivänä ja neljän päivän ajan leikkauksen jälkeen verrattiin epoetiini alfa ‑annoksiin 600 IU/kg ihon alle kerran viikossa kolmen viikon ajan ennen leikkausta ja leikkauspäivänä.
Hemoglobiinipitoisuus suureni hoitoa edeltävästä tilanteesta leikkausta edeltävään tilanteeseen annoksia 600 IU/kg viikossa saaneessa ryhmässä kaksinkertaisesti (14,4 g/l) verrattuna pitoisuuksiin, joita todettiin annoksia 300 IU/kg päivittäin saaneessa ryhmässä (7,3 g/l). Keskimääräiset hemoglobiinipitoisuudet olivat kummassakin hoitoryhmässä samankaltaiset koko leikkauksen jälkeisen ajanjakson ajan.
Kummassakin hoitoryhmässä havaittu erytropoieettinen vaste johti verensiirtoon yhtä usein (16 %:lla annoksia 600 IU/kg viikoittain saaneessa ryhmässä ja 20 %:lla annoksia 300 IU/kg päivittäin saaneessa ryhmässä).
Aikuispotilaat, joilla on pienen tai keskisuuren riskin (riskitaso 1) myelodysplastinen oireyhtymä (MDS)
Aikuisilla, joilla oli anemia ja pienen tai keskisuuren riskin (riskitaso 1) myelodysplastinen oireyhtymä (MDS), tehtiin epoetiini alfa -valmisteella satunnaistettu, kaksoissokkoutettu, lumekontrolloitu monikeskustutkimus, jossa selvitettiin epoetiini alfan tehoa ja turvallisuutta.
Tutkittavat ositettiin seulonnassa seerumin erytropoietiinipitoisuuden (sEPO) ja aiemmin saatujen verensiirtojen mukaan. Seuraavassa taulukossa esitetään ositteen < 200 mU/ml keskeiset lähtötilanteen tiedot.
Lähtötilanteen tiedot tutkittavista, joiden sEPO seulonnassa < 200 mU/ml | ||
Satunnaistaminen | ||
Epoetiini alfa | Lume | |
Yhteensä (N)b | 85a | 45 |
Seulonnassa sEPO < 200 mU/ml (N) | 71 | 39 |
Hemoglobiini (g/l) | ||
N | 71 | 39 |
Keskiarvo | 92,1 (8,57) | 92,1 (8,51) |
Mediaani | 94,0 | 96,0 |
Vaihteluväli | (71, 109) | (69, 105) |
Keskiarvon 95 %:n luottamusväli | (90,1, 94,1) | (89,3, 94,9) |
Aiemmat verensiirrot | ||
N | 71 | 39 |
Kyllä | 31 (43,7 %) | 17 (43,6 %) |
≤ 2 punasoluyksikköä | 16 (51,6 %) | 9 (52,9 %) |
˃ 2, mutta ≤ 4 punasoluyksikköä | 14 (45,2 %) | 8 (47,1 %) |
˃ 4 punasoluyksikköä | 1 (3,2 %) | 0 |
Ei | 40 (56,3 %) | 22 (56,4 %) |
a yhdestä tutkittavasta ei ollut sEPO-tietoja b ositteessa ≥ 200 mU/ml epoetiini alfa ‑ryhmässä oli 13 tutkittavaa ja lumeryhmässä oli 6 tutkittavaa | ||
Erytroidinen vaste määriteltiin IWG 2006 ‑kriteerien (International Working Group, IWG) mukaan hemoglobiinipitoisuuden suurenemiseksi ≥ 15 g/l lähtötilanteesta tai siirrettyjen punasoluyksiköiden vähintään neljän yksikön absoluuttiseksi vähenemiseksi kahdeksan viikon välein, kun vertailukohtana oli tilanne kahdeksan viikkoa ennen lähtötilannetta, sekä vasteen säilymiseksi vähintään kahdeksan viikon ajan.
Erytroidinen vaste osoitettiin tutkimuksen ensimmäisten 24 viikon aikana epoetiini alfa ‑ryhmässä 27 tutkittavalla 85 tutkittavasta (31,8 %) verrattuna lumeryhmän 2 tutkittavaan 45 tutkittavasta (4,4 %) (p < 0,001). Kaikki vasteen saaneet tutkittavat olivat seulonnassa ositteessa sEPO < 200 mU/ml. Tässä ositteessa 20 tutkittavaa 40 tutkittavasta (50 %), jotka eivät olleet aiemmin saaneet verensiirtoja, saivat erytroidisen vasteen ensimmäisten 24 hoitoviikon aikana verrattuna 7 tutkittavaan 31 tutkittavasta (22,6 %), jotka olivat aiemmin saaneet verensiirtoja (kaksi verensiirtoja aiemmin saanutta tutkittavaa saavutti ensisijaisen päätetapahtuman, mikä perustui siirrettyjen punasoluyksiköiden vähenemiseen absoluuttisesti vähintään 4 yksiköllä 8 viikon välein verrattuna lähtötilannetta edeltäneisiin 8 viikkoon).
Ajan mediaani lähtötilanteesta ensimmäiseen punasolusiirtoon oli epoetiini alfa ‑ryhmässä tilastollisesti merkitsevästi pidempi verrattuna lumeryhmään (49 päivää vs. 37 päivää; p = 0,046). Aika ensimmäiseen punasolusiirtoon oli pidentynyt epoetiini alfa ‑ryhmässä edelleen neljän viikon hoidon jälkeen (142 päivää vs. 50 päivää, p = 0,007). Niiden tutkittavien prosenttiosuus, jotka saivat punasolusiirron, pieneni epoetiini alfa ‑ryhmässä kahdeksan viikkoa ennen lähtötilannetta olleesta 51,8 %:sta 24,7 %:iin viikkojen 16 ja 24 välillä. Lumeryhmässä punasolusiirtojen määrä samoina ajankohtina lisääntyi 48,9 %:sta 54,1 %:iin.
Pediatriset potilaat
Krooninen munuaisten vajaatoiminta
Epoetiini alfaa tutkittiin avoimessa, satunnaistamattomassa, 52 viikkoa kestäneessä avoimen annosvälin kliinisessä tutkimuksessa, joka toteutettiin hemodialyysihoitoa saavilla kroonista munuaisten vajaatoimintaa sairastavilla pediatrisilla potilailla. Tutkimukseen mukaan otettujen potilaiden iän mediaani oli 11,6 vuotta (vaihteluväli 0,5−20,1 vuotta).
Epoetiini alfaa annettiin dialyysihoidon jälkeen annoksina 75 IU/kg/viikko laskimoon 2−3 annokseen jaettuna, titrattiin annoksina 75 IU/kg/viikko 4 viikon välein (enintään maksimiannokseen 300 IU/kg/viikko), jotta saavutettiin hemoglobiinipitoisuuden suureneminen 10 g/l/kuukausi. Hemoglobiinin tavoitepitoisuus oli 96–112 g/l. Tämän hemoglobiinipitoisuuden saavutti kahdeksankymmentäyksi prosenttia potilaista. Ajan mediaani tavoitteen saavuttamiseen oli 11 viikkoa ja annoksen mediaani tavoitteen saavuttamisajankohtana oli 150 IU/kg/viikko. 90 % tavoitteen saavuttaneista potilaista noudatti hoito-ohjelmaa, jossa epoetiini alfaa annettiin kolme kertaa viikossa.
52 viikon kuluttua tutkimuksessa oli edelleen mukana 57 % potilaista ja annoksen mediaani oli 200 IU/kg/viikko.
Kliinisiä tietoja valmisteen antamisesta lapsille ihon alle on vähän. Lapsille annettiin viidessä suppeassa, avoimessa, kontrolloimattomassa tutkimuksessa (potilaiden lukumäärä oli 9–22, yhteensä N = 72) epoetiini alfaa ihon alle aloitusannoksina 100–150 IU/kg/viikko. Annosta oli mahdollista suurentaa annokseen 300 IU/kg/viikko saakka. Suurin osa näiden tutkimusten potilaista ei vielä saanut dialyysihoitoa (N = 44); 27 potilasta sai peritoneaalidialyysihoitoa ja kaksi sai hemodialyysihoitoa. Potilaat olivat iältään 4 kuukaudesta 17 vuoteen. Näiden tutkimusten menetelmissä oli kaiken kaikkiaan rajoituksensa, mutta hoitoon todettiin yleensä liittyvän hemoglobiinipitoisuuden suurenemista. Odottamattomia haittatapahtumia ei raportoitu (ks. kohta Annostus ja antotapa).
Solunsalpaajahoidon aiheuttama anemia
Epoetiini alfa -annoksia 600 IU/kg (jotka annettiin kerran viikossa laskimoon tai ihon alle) on tutkittu 16 viikkoa kestäneessä satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa sekä 20 viikkoa kestäneessä satunnaistetussa, kontrolloidussa, avoimessa tutkimuksessa pediatrisilla potilailla, joilla oli anemia ja jotka saivat myelosuppressiivista solunsalpaajahoitoa erilaisten lapsuusiän ei-myelooisten syöpien hoitoon.
16 viikkoa kestäneessä tutkimuksessa (n = 222) epoetiini alfa ‑hoitoon ei liittynyt tilastollisesti merkitsevää vaikutusta elämänlaatuun verrattuna lumehoitoa saaneisiin, mitä mitattiin potilaan tai potilaan vanhempien raportoimaa lapsipotilaiden elämänlaatua (Paediatric Quality of Life Inventory) tai syöpää (Cancer Module) koskevilla pisteytyksillä (ensisijainen tehon päätetapahtuma). Myöskään punasolusiirtoja tarvinneiden potilaiden osuudessa ei havaittu tilastollisesti merkitsevää eroa epoetiini alfa ‑hoitoa ja lumehoitoa saaneiden ryhmien välillä.
20 viikkoa kestäneessä tutkimuksessa (n = 225) ei havaittu merkitsevää eroa ensisijaisessa tehoa mitanneessa päätetapahtumassa eli niiden potilaiden osuudessa, jotka tarvitsivat punasolusiirron 28 hoitopäivän jälkeen (62 % epoetiini alfaa saaneista potilaista verrattuna 69 %:iin tavanomaista hoitoa saaneista potilaista).
Farmakokinetiikka
Imeytyminen
Ihon alle injektiona annetun erytropoietiinin huippupitoisuus seerumissa saavutetaan 12−18 tunnissa annoksen annon jälkeen. Ihon alle viikoittain toistuvina annoksina 600 IU/kg annettuna ei tapahtunut kumuloitumista.
Ihon alle injektiona annettavan erytropoietiinin absoluuttinen biologinen hyötyosuus terveillä tutkittavilla on noin 20 %.
Jakautuminen
Keskimääräinen jakautumistilavuus terveille tutkittaville laskimoon annettujen annosten 50 ja 100 IU/kg jälkeen oli 49,3 ml/kg. Kroonista munuaisten vajaatoimintaa sairastaville tutkittaville laskimoon annetun erytropoietiinin jakautumistilavuus oli 57–107 ml/kg kerta-annoksen (12 IU/kg) jälkeen ja 42–64 ml/kg toistettujen annosten (48–192 IU/kg) jälkeen. Jakautumistilavuus on siten hieman suurempi kuin plasmatila.
Eliminaatio
Erytropoietiinin puoliintumisaika on terveille tutkittaville laskimoon toistuvasti annettuna noin 4 tuntia. Ihon alle terveille tutkittaville annettuna puoliintumisajan arvioidaan olevan noin 24 tuntia.
Terveiden tutkittavien keskimääräinen aikaan ja annokseen liittyvä puhdistuma (CL/F) annosten 150 IU/kg kolmesti viikossa annon yhteydessä oli 31,2 ml/h/kg ja annosten 40 000 IU kerran viikossa annon yhteydessä 12,6 ml/h/kg. Aneemisten syöpäpotilaiden keskimääräinen CL/F oli annosten 150 IU/kg kolmesti viikossa annon yhteydessä 45,8 ml/h/kg ja annosten 40 000 IU kerran viikossa yhteydessä 11,3 ml/h/kg. Useimpien syöpää sairastavien solunsalpaajaa sykleinä saavien tutkittavien CL/F oli pienempi ihon alle annettujen annosten 40 000 IU kerran viikossa ja 150 IU/kg kolmesti viikossa yhteydessä verrattuna terveiden tutkittavien arvoihin.
Lineaarisuus/ei-lineaarisuus
Terveillä tutkittavilla havaittiin laskimoon annettujen 150 ja 300 IU/kg kolmesti viikossa annosten jälkeen suhteessa annokseen tapahtuva erytropoietiinipitoisuuden suureneminen seerumissa. Ihon alle annettujen erytropoietiinikerta-annosten 300–2400 IU/kg jälkeen keskimääräisen Cmax-arvon ja annoksen sekä keskimääräisen AUC-arvon ja annoksen välinen suhde oli lineaarinen. Terveillä tutkittavilla havaittiin käänteinen suhde laskennallisen puhdistuman ja annoksen välillä.
Tutkimuksissa, joissa selvitettiin antovälin pidentämistä (40 000 IU kerran viikossa ja 80 000, 100 000 ja 120 000 IU kerran kahdessa viikossa) havaittiin keskimääräisen Cmax-arvon ja annoksen sekä keskimääräisen AUC-arvon ja annoksen välisen suhteen olleen vakaassa tilassa lineaarinen, mutta ei suhteessa annokseen.
Farmakokineettiset/farmakodynaamiset suhteet
Erytropoietiinit vaikuttavat hematologisiin parametreihin antoreitistä riippumattomasti suhteessa annokseen.
Pediatriset potilaat
Kroonista munuaisten vajaatoimintaa sairastavilla pediatrisilla potilailla on raportoitu toistetusti laskimoon annettujen erytropoietiiniannosten jälkeiseksi puoliintumisajaksi noin 6,2–8,7 tuntia. Erytropoietiinien farmakokineettinen profiili vaikuttaa olevan lapsilla ja nuorilla samankaltainen kuin aikuisilla.
Farmakokinetiikasta vastasyntyneillä on vähän tietoja.
Tutkimus, jossa seitsemälle hyvin pienipainoisena syntyneelle vastasyntyneelle keskoselle ja kymmenelle terveelle aikuiselle annettiin erytropoietiinia laskimoon, viittasi siihen, että jakautumistilavuus vastasyntyneillä keskosilla oli noin 1,5–2 kertaa suurempi kuin terveillä aikuisilla ja että puhdistuma vastasyntyneillä keskosilla oli noin 3 kertaa suurempi kuin terveillä aikuisilla.
Munuaisten vajaatoiminta
Kroonista munuaisten vajaatoimintaa sairastaville potilaille laskimoon annetun erytropoietiinin puoliintumisaika on hieman pidentynyt, noin 5 tuntia, verrattuna terveisiin tutkittaviin.
Prekliiniset tiedot turvallisuudesta
Koirilla ja rotilla, mutta ei apinoilla, tehdyissä toistuvien annosten toksisuustutkimuksissa epoetiini alfa ‑hoitoon liittyi subkliinistä luuydinfibroosia. Luuydinfibroosi on kroonisen munuaisten vajaatoiminnan tunnettu komplikaatio ihmisellä ja se saattaa liittyä sekundaariseen lisäkilpirauhasten liikatoimintaan tai tuntemattomiin tekijöihin. Luuydinfibroosin esiintyvyys ei lisääntynyt tutkimuksessa, jossa hemodialyysipotilaita hoidettiin epoetiini alfalla kolme vuotta verrattuna kaltaistettuun hemodialyysipotilaiden verrokkiryhmään, joita ei hoidettu epoetiini alfalla.
Epoetiini alfa ei aiheuta bakteereille geenimutaatioita (Amesin testi), nisäkässoluille kromosomipoikkeavuuksia, hiirelle mikronukleuksia eikä HGPRT-lokuksen geenimutaatioita.
Pitkäkestoisia karsinogeenisuustutkimuksia ei ole tehty. Ihmisen kasvainnäytteistä saatuihin in vitro ‑löydöksiin perustuvat ristiriitaiset raportit kirjallisuudessa viittaavat siihen, että erytropoietiineilla saattaa olla kasvainten kasvua edistäviä vaikutuksia. Tämän merkitys kliiniseltä kannalta ei ole selvä.
Epoetiini alfa stimuloi ihmisen luuydinsolujen soluviljelyssä erityisesti erytropoieesia eikä vaikuta leukopoieesiin. Epoetiini alfalla ei havaittu sytotoksisia vaikutuksia luuydinsoluihin.
Epoetiini alfan on eläinkokeissa osoitettu alentavan sikiön painoa, hidastavan luutumista ja lisäävän sikiökuolleisuutta, kun sitä annettiin viikoittain noin 20 kertaa ihmiselle suositeltua viikkoannosta suurempina annoksina. Näiden muutosten tulkitaan johtuvan emon heikentyneestä painonkehityksestä eikä sen merkitystä ihmiselle terapeuttisia annoksia käytettäessä tiedetä.
Farmaseuttiset tiedot
Apuaineet
Dinatriumfosfaattidihydraatti
Natriumdivetyfosfaattidihydraatti
Natriumkloridi
Kalsiumklorididihydraatti
Polysorbaatti 20 (E 432)
Glysiini
Leusiini
Isoleusiini
Treoniini
Glutamiinihappo
Fenyylialaniini
Injektionesteisiin käytettävä vesi
Natriumhydroksidi (pH:n säätämiseen)
Kloorivetyhappo (pH:n säätämiseen)
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
30 kuukautta
Säilytys
Säilytä jääkaapissa (2 °C - 8 °C). Säilytys tässä lämpötilassa tulee varmistaa aina lääkkeen antoon saakka.
Avohoitokäytössä lääkevalmiste voidaan ottaa jääkaapista ja säilyttää sitä alle 25 °C:ssa yhden kerran enintään kolmen vuorokauden ajan. Jos lääkevalmistetta ei ole käytetty tämän ajanjakson kuluessa, se on hävitettävä.
Ei saa jäätyä. Ei saa ravistaa.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
RETACRIT injektioneste, liuos, esitäytetty ruisku
1000 IU/0,3 ml (L:ei) 6 x 0,3 ml (neulansuojuksella) (48,44 €)
2000 IU/0,6 ml (L:ei) 6 x 0,6 ml (neulansuojuksella) (92,77 €)
3000 IU/0,9 ml (L:ei) 6 x 0,9 ml (neulansuojuksella) (135,90 €)
4000 IU/0,4 ml (L:ei) 6 x 0,4 ml (neulansuojuksella) (178,52 €)
5000 IU/0,5 ml (L:ei) 6 x 0,5 ml (neulansuojuksella) (219,17 €)
6000 IU/0,6 ml (L:ei) 6 x 0,6 ml (neulansuojuksella) (259,80 €)
8000 IU/0,8 ml (L:ei) 6 x 0,8 ml (neulansuojuksella) (341,24 €)
20000 IU/0,5 ml (L:ei) 0,5 ml (neulansuojuksella) (130,28 €)
30000 IU/0,75 ml (L:ei) 0,75 ml (neulansuojuksella) (190,86 €)
10000 IU/ml (L:ei) 6 x 1 ml (neulansuojuksella) (309,72 €)
40000 IU/ml (L:ei) 1 ml (neulansuojuksella) (249,31 €)
PF-selosteen tieto
Retacrit 1 000 IU/0,3 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 0,3 ml liuosta.
Pakkaus sisältää 1 tai 6 esitäytettyä ruiskua.
Retacrit 2 000 IU/0,6 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 0,6 ml liuosta.
Pakkaus sisältää 1 tai 6 esitäytettyä ruiskua.
Retacrit 3 000 IU/0,9 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 0,9 ml liuosta.
Pakkaus sisältää 1 tai 6 esitäytettyä ruiskua.
Retacrit 4 000 IU/0,4 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 0,4 ml liuosta.
Pakkaus sisältää 1 tai 6 esitäytettyä ruiskua.
Retacrit 5 000 IU/0,5 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 0,5 ml liuosta.
Pakkaus sisältää 1 tai 6 esitäytettyä ruiskua.
Retacrit 6 000 IU/0,6 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 0,6 ml liuosta.
Pakkaus sisältää 1 tai 6 esitäytettyä ruiskua.
Retacrit 8 000 IU/0,8 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 0,8 ml liuosta.
Pakkaus sisältää 1 tai 6 esitäytettyä ruiskua.
Retacrit 10 000 IU/1 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 1 ml liuosta.
Pakkaus sisältää 1 tai 6 esitäytettyä ruiskua.
Retacrit 20 000 IU/0,5 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 0,5 ml liuosta.
Pakkaus sisältää 1, 4 tai 6 esitäytettyä ruiskua.
Kerrannaispakkaus sisältää 6 (6 x 1) esitäytettyä ruiskua.
Retacrit 30 000 IU/0,75 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 0,75 ml liuosta.
Pakkaus sisältää 1, 4 tai 6 esitäytettyä ruiskua.
Kerrannaispakkaus sisältää 4 (4 x 1) esitäytettyä ruiskua.
Retacrit 40 000 IU/1 ml injektioneste, liuos, esitäytetyssä ruiskussa
Esitäytetty ruisku (tyypin 1 lasia), jossa on teräsneula ja mäntä (teflonoitua kumia), ruiskussa voi olla myös neulanpistosuoja tai turvaneula.
Yksi esitäytetty ruisku sisältää 1 ml liuosta.
Pakkaus sisältää 1, 4 tai 6 esitäytettyä ruiskua.
Kerrannaispakkaus sisältää 4 (4 x 1) esitäytettyä ruiskua.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas, väritön liuos.
Käyttö- ja käsittelyohjeet
Retacrit-valmistetta ei saa käyttää, vaan se on hävitettävä
- jos sinetti on rikki
- jos neste on värjäytynyttä tai sisältää hiukkasia
- jos esitäytetystä ruiskusta on vuotanut nestettä tai suljetussa kuplassa esiintyy tiivistynyttä kosteutta
- jos tiedät tai epäilet, että valmiste on saattanut epähuomiossa jäätyä tai
- jos jääkaappi on ollut epäkunnossa.
Vain yhtä käyttökertaa varten. Ota kustakin ruiskusta ainoastaan yksi Retacrit-annos.
Ei saa ravistaa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
RETACRIT injektioneste, liuos, esitäytetty ruisku
1000 IU/0,3 ml 6 x 0,3 ml
2000 IU/0,6 ml 6 x 0,6 ml
3000 IU/0,9 ml 6 x 0,9 ml
4000 IU/0,4 ml 6 x 0,4 ml
5000 IU/0,5 ml 6 x 0,5 ml
6000 IU/0,6 ml 6 x 0,6 ml
8000 IU/0,8 ml 6 x 0,8 ml
20000 IU/0,5 ml 0,5 ml
30000 IU/0,75 ml 0,75 ml
10000 IU/ml 6 x 1 ml
40000 IU/ml 1 ml
- Ylempi erityiskorvaus (100 %). Rintasyöpä (115), Eturauhassyöpä (116), Leukemiat, muut pahanlaatuiset veri- ja luuydintaudit sekä pahanlaatuiset imukudostaudit (117), Gynekologiset syövät (128), Pahanlaatuiset kasvaimet, joita ei ole edellä erikseen mainittu (130), Munuaisten vajaatoimintaan liittyvä vaikea anemia (138).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Epoetiini, darbepoetiini alfa ja metoksipolyetyleeniglykoliepoetiini beeta: Krooniseen munuaissairauteen liittyvän anemian hoito erityisin edellytyksin / Epoetiini ja darbepoetiini alfa: Syöpäsairauteen tai solunsalpaajahoitoihin liittyvän anemian hoito sekä valmistauduttaessa autologiseen verensiirtoon (306).
- Oikeus käyttää samaa biologista lääkettä (sama kauppanimi) 6 kk ajan.
ATC-koodi
B03XA01
Valmisteyhteenvedon muuttamispäivämäärä
26.09.2025
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com