
HALAVEN injektioneste, liuos 0,44 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi millilitra sisältää eribuliinimesylaattia, joka vastaa 0,44 mg eribuliinia.
Yksi 2 ml:n injektiopullo sisältää eribuliinimesylaattia, joka vastaa 0,88 mg eribuliinia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste).
Kliiniset tiedot
Käyttöaiheet
Halaven on tarkoitettu niiden aikuispotilaiden hoitoon, jotka sairastavat paikallisesti levinnyttä tai metastasoitunutta rintasyöpää ja joiden sairaus on edennyt sen jälkeen, kun he ovat saaneet vähintään yhden pitkälle edenneen sairauden vuoksi annetun kemoterapiahoitojakson (ks. kohta Farmakodynamiikka). Edeltävään hoitoon on pitänyt kuulua antrasykliini- ja taksaanihoito joko adjuvanttihoitona tai metastaattisessa tilanteessa, paitsi silloin kun nämä hoidot eivät potilaalle sovi.
Halaven on tarkoitettu sellaisten aikuispotilaiden hoitoon, jotka sairastavat ei-resekoitavissa olevaa liposarkoomaa ja jotka ovat aiemmin saaneet antrasykliiniä sisältävää hoitoa (ellei sopimaton potilaalle) pitkälle edenneeseen tai metastasoituneeseen sairauteen (ks. kohta Farmakodynamiikka).
Ehto
Valmistetta saa määrätä vain syöpäpotilaiden hoitoon perehtynyt lääkäri ja sitä saa antaa vain syöpäpotilaiden hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Annostus ja antotapa
HALAVEN-valmisteen saa määrätä vain syövän hoitoon perehtynyt lääkäri. Sen saa antaa vain asianmukaisesti koulutettu terveydenhuollon ammattilainen.
Annostus
Suositeltu eribuliiniannos käyttövalmiina liuoksena on 1,23 mg/m2, joka tulee antaa laskimoon 2–5 minuutin aikana kunkin 21 vrk kestävän hoitojakson päivinä 1 ja 8.
Huom.:
EU:ssa suositeltu annos tarkoittaa vaikuttavan aineen (eribuliinin) emästä. Potilaalle annosteltavan yksilöllisen annoksen laskennan tulee perustua käyttövalmiin liuoksen vahvuuteen, joka sisältää 0,44 mg/ml eribuliinia, ja 1,23 mg/m2:n annossuositukseen. Alla osoitetut suositukset annoksen vähentämisestä on myös esitetty annosteltavana eribuliiniannoksena, joka perustuu käyttövalmiin liuoksen vahvuuteen.
Keskeisissä tutkimuksissa, niitä vastaavissa julkaisuissa ja joillakin muilla alueilla, kuten USA:ssa ja Sveitsissä, suositeltu annos perustuu suolana (eribuliinimesylaattina) annosteltavaan muotoon.
Potilailla saattaa esiintyä pahoinvointia tai oksentelua. Pahoinvointilääkitystä, esim. kortikosteroideja, tulee harkita.
Annon viivästyttäminen hoidon aikana
Halavenin antoa tulee viivästyttää päivinä 1 tai 8 seuraavissa tapauksissa:
- absoluuttinen neutrofiilien määrä (ANC) < 1 x 109/l
- verihiutaleet < 75 x 109/l
- gradus 3 tai 4 ei-hematologinen toksisuus.
Annoksen pienentäminen hoidon aikana
Annoksen pienentämiseen liittyvät suositukset hoitoa uudelleen aloitettaessa on esitetty seuraavassa taulukossa.
Annoksen pienentämistä koskevat suositukset
| Haittavaikutus edellisen Halaven-annon jälkeen | Suositeltu annos eribuliinia |
| Hematologinen: | |
| ANC < 0,5 x 109/l vähintään 7 vuorokauden ajan | 0,97 mg/m2 |
| ANC < 1 x 109/l ja johon on komplikaationa liittynyt kuume tai infektio | |
| Trombosytopenia, jossa verihiutaleet < 25 x 109/l | |
| Trombosytopenia, joka on verenvuodon komplisoima tai vaatii veren- tai verihiutalesiirtoa, verihiutaleet < 50 x 109/l | |
| Ei-hematologinen: | |
| Mikä tahansa gradus 3 tai 4 haittavaikutus edellisen syklin aikana | |
| Minkä tahansa yllä kuvatun hematologisen tai ei-hematologisen haittavaikutuksen uusiutuminen | |
| Huolimatta annoksen pienentämisestä 0,97 mg:aan/m2 | 0,62 mg/m2 |
| Huolimatta annoksen pienentämisestä 0,62 mg:aan/m2 | Harkittava lopettamista |
Eribuliiniannosta ei saa suurentaa uudelleen sen jälkeen, kun sitä on pienennetty.
Maksan vajaatoimintaa sairastavat potilaat
Etäpesäkkeiden aiheuttama maksan vajaatoiminta
Eribuliinin suositettu annos lievää maksan vajaatoimintaa sairastavilla potilailla (Child-Pugh-luokka A) on 0,97 mg/m2 joka annetaan laskimoon 2–5 minuutin aikana kunkin 21 vrk kestävän hoitojakson päivinä 1 ja 8. Keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla (Child-Pugh-luokka B) suositeltava eribuliiniannos on 0,62 mg/m2 joka annetaan laskimoon 2–5 minuutin aikana kunkin 21 vrk kestävän hoitojakson päivinä 1 ja 8.
Vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) ei ole tutkittu, mutta oletetaan, että annosta on välttämätöntä pienentää huomattavasti, jos eribuliinia käytetään näillä potilailla.
Kirroosin aiheuttama maksan vajaatoiminta
Tätä potilasryhmää ei ole tutkittu. Yllä olevia annoksia voidaan käyttää lievää ja keskivaikeaa vajaatoimintaa sairastavilla, mutta huolellista tarkkailua suositellaan, sillä annosten muuttaminen saattaa olla tarpeen.
Munuaisten vajaatoimintaa sairastavat potilaat
Kohtalaista tai vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden (kreatiniinipuhdistuma <50 ml/min) altistuminen eribuliinille saattaa olla lisääntynyt ja heille annettavaa annosta voi olla tarpeen pienentää. Kaikkien munuaisten vajaatoimintaa sairastavien potilaiden kohdalla on noudatettava varovaisuutta ja tarkkailtava turvallisuutta (ks. kohta Farmakokinetiikka).
Iäkkäät potilaat
Erityistä potilaan ikään perustuvaa annoksen muuttamista ei suositella (ks. kohta Haittavaikutukset).
Pediatriset potilaat
Ei ole asianmukaista käyttää HALAVEN-valmistetta pediatrisille ja nuorille potilaille rintasyövän hoitoon.
Ei ole asianmukaista käyttää HALAVEN-valmistetta pediatristen potilaiden pehmytkudossarkooman hoitoon (ks. kohta Farmakodynamiikka).
Antotapa
Halaven annetaan laskimoon. Annos voidaan laimentaa korkeintaan 100 ml:aan 9 mg/ml (0,9 %) natriumkloridi-injektionestettä. Annosta ei saa laimentaa 5-prosenttiseen glukoosi-infuusionesteeseen. Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa. Ennen antoa on varmistettava, että perifeerinen laskimoyhteys on hyvä tai että keskuslaskimoyhteys on avoin. Eribuliinimesylaatin ei ole osoitettu aiheuttavan rakkuloita tai ärsytystä. Mahdollinen ekstravasaatio tulee hoitaa oireenmukaisesti. Katso sytotoksisten lääkevalmisteiden käsittelyyn liittyvät asianmukaiset ohjeet kohdasta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille
- Imettäminen
Varoitukset ja käyttöön liittyvät varotoimet
Hematologia
Luuydinlama on annosriippuvainen ja ilmenee pääasiassa neutropeniana (kohta Haittavaikutukset). Koko verenkuvaa tulee seurata kaikilla potilailla ennen kutakin eribuliiniannosta. Eribuliinihoito tulee aloittaa vain sellaisilla potilailla, joilla absoluuttinen neutrofiilimäärä (ANC) on ≥ 1,5 x 109/l ja verihiutalemäärä > 100 x 109/l.
Kuumeista neutropeniaa esiintyi < 5 %:lla eribuliinihoitoa saaneista potilaista. Kuumeista neutropeniaa, vakavaa neutropeniaa tai trombosytopeniaa sairastavia potilaita tulee hoitaa kohdassa Annostus ja antotapa esitettyjen suositusten mukaisesti.
Potilailla, joiden alaniiniaminotransferaasi (ALAT)- tai aspartaattiaminotransferaasi (ASAT)-arvo oli yli kolminkertaisesti yli normaalin ylärajan (ULN), esiintyi useammin asteen 4 neutropeniaa ja kuumeista neutropeniaa. Potilailla, joiden bilirubiinitaso ylittää normaalin ylärajan 1,5-kertaisesti, esiintyy myös useammin asteen 4 neutropeniaa ja kuumeista neutropeniaa, joskin tätä koskeva aineisto on rajallinen.
Kuumeisen neutropenian, neutropeenisen sepsiksen, sepsiksen ja septisen sokin aiheuttamia kuolemantapauksia on raportoitu.
Vakava neutropenia voidaan hoitaa käyttämällä granulosyyttiryhmiä stimuloivaa kasvutekijää (G-CSF:ää) tai vastaavaa lääkärin harkinnan mukaan asianmukaisia ohjeita noudattaen (ks. kohta Farmakodynamiikka).
Perifeerinen neuropatia
Potilaita tulee seurata huolellisesti perifeerisen motorisen ja sensorisen neuropatian varalta. Jos potilaalle kehittyy vakava perifeerinen neurotoksisuus, annosta tulee viivästyttää tai vähentää (ks. kohta Annostus ja antotapa).
Potilaat joilla oli astetta 2 vaikeampi neuropatia suljettiin pois kliinisistä tutkimuksista. Gradus 1 tai 2 neuropatiaa jo sairastavilla potilailla ei uusien oireiden tai oireiden pahentumisen todennäköisyys ollut suurempi kuin niillä, joilla tutkimukseen mukaan tullessaan ei ollut tätä sairautta.
QT-ajan piteneminen
Kontrolloimattomassa ja avoimessa 26 potilaan EKG-tutkimuksessa eribuliinipitoisuudesta riippumaton QT-ajan pidentyminen havaittiin päivänä 8, mutta QT-ajan pidentymistä ei havaittu päivänä 1. EKG-seurantaa suositellaan, jos hoito aloitetaan kongestiivista sydämen vajaatoimintaa tai bradyarytmiaa sairastavalle potilaalle, potilaalle, jolla on tunnetusti QT-aikaa pidentävä samanaikainen lääkitys, kuten luokan Ia ja III rytmihäiriölääkkeet, tai jos potilaalla on elektrolyyttihäiriö. Hypokalemia, hypokalsemia tai hypomagnesemia tulee korjata ennen Halaven-lääkityksen aloittamista ja näitä elektrolyyttejä tulee seurata määräajoin hoidon aikana. Eribuliinin käyttöä tulee välttää potilaille, joilla on synnynnäinen pitkän QT:n oireyhtymä.
Apuaineet
Tämä lääke sisältää pieniä määriä etanolia (alkoholia), alle 100 mg annosta kohti.
Yhteisvaikutukset
Eribuliini eliminoituu pääasiallisesti (jopa 70-prosenttisesti ) sappierityksen kautta. Tähän prosessiin liittyvää kuljettajaproteiinia ei tunneta. Eribuliini ei ole seuraavien kuljettajaproteiinien substraatti: rintasyövän resistenssiproteiini (BCRP), orgaanisten anionien kuljettajaproteiinit OAT1, OAT3, OATP1B1, OATP1B3, monilääkeresistenssiin liittyvät kuljettajaproteiinit MRP2, MRP4 ja sappisuolapumppu (BSEP).
Lääkkeiden yhteisvaikutuksia ei ole odotettavissa CYP3A4-estäjillä ja indusoijilla. Ketokonatsolilla, joka on CYP3A4- ja P-glykoproteiinin (Pgp) estäjä, ja rifampisiinilla, joka on CYP3A4:n indusoija, ei ollut vaikutusta eribuliinille altistumiseen (AUC ja Cmax).
Eribuliinin vaikutus muiden lääkkeiden farmakokinetiikkaan
In vitro -tiedot osoittavat eribuliinin estävän CYP3A4:tä, joka on tärkeä lääkkeitä metaboloiva entsyymi. In vivo -tietoja ei ole saatavissa. Varovaisuutta ja potilaan huolellista seurantaa haittatapahtumien varalta suositellaan, kun eribuliinia käytetään samanaikaisesti sellaisten lääkeaineiden kanssa, joilla on kapea terapeuttinen leveys ja jotka eliminoituvat pääasiassa CYP3A4-välitteisen metabolian kautta (esim. alfentaniili, siklosporiini, ergotamiini, fentanyyli, pimotsidi, kinidiini, sirolimuusi, takrolimuusi).
Eribuliini ei asianmukaisina kliinisinä pitoisuuksina käytettynä estä CYP-entsyymejä CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 tai 2E1.
Eribuliini ei asianmukaisina kliinisinä pitoisuuksina käytettynä estänyt BCRP-, OCT1-, OCT2-, OAT1-, OAT3-, OATP1B1- ja OATP1B3-kuljettajaproteiinien välittämää toimintaa.
Raskaus ja imetys
Raskaus
Eribuliinin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eribuliini on alkiotoksinen, sikiötoksinen ja teratogeeninen rotilla. Halaven-valmistetta ei pidä käyttää raskauden aikana ellei sen käyttö ole ehdottoman välttämätöntä, ja tällöin vasta äidin tarpeen ja sikiölle koituvan riskin huolellisen arvioinnin jälkeen.
Naisia, jotka voivat tulla raskaaksi, tulee neuvoa välttämään raskaaksi tulemista sinä aikana kun he saavat Halaven-hoitoa, ja heidän tulee käyttää erittäin tehokasta ehkäisyä HALAVEN-hoidon aikana ja 7 kuukauden ajan hoidon jälkeen.
Imetys
Ei tiedetä, erittyvätkö eribuliini ja/tai sen metaboliitit ihmisillä rintamaitoon tai maitoon koe-eläimillä. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois, ja siksi HALAVEN-valmisteen käyttö imetyksen aikana on vasta-aiheista (ks. kohta Vasta-aiheet).
Hedelmällisyys
Kivestoksisuutta on havaittu rotalla ja koiralla (ks. kohta Prekliiniset tiedot turvallisuudesta). Miespotilaiden tulee ennen hoitoa hakeutua neuvontaan saadakseen sperman säilyttämiseen liittyviä ohjeita, sillä Halaven-hoito saattaa aiheuttaa palautumatonta hedelmättömyyttä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Halaven saattaa aiheuttaa haittavaikutuksia, kuten väsymystä ja heitehuimausta, joilla saattaa olla vähäinen tai kohtalainen vaikutus ajokykyyn tai koneiden käyttökykyyn. Potilaita tulee neuvoa välttämään ajamista ja koneiden käyttöä, jos he tuntevat väsymystä tai heitehuimausta.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin raportoitu Halaven-valmisteeseen liittyvä haittavaikutus on luuydinlama, joka ilmenee neutropeniana, leukopeniana, anemiana ja trombosytopeniana sekä näihin liittyvinä infektioina. Myös aiemmin esiintymätöntä perifeeristä neuropatiaa tai olemassa olevan perifeerisen neuropatian pahenemista sekä anoreksiana, pahoinvointina, oksenteluna, ripulina, ummetuksena ja suutulehduksena ilmenevää maha-suolikanavan toksisuutta on raportoitu. Muita haittavaikutuksia ovat väsymys, hiustenlähtö, maksan entsyymiarvojen kohoaminen, sepsis ja muskuloskeletaalinen kipuoireyhtymä.
Taulukkomuotoinen luettelo haittavaikutuksista
Ellei toisin mainita, taulukossa esitetään haittavaikutusten esiintymistiheys rintasyöpää ja pehmytkudossarkoomaa sairastavilla potilailla, joille annettiin suositeltu annos faasin 2 ja faasin 3 tutkimuksissa.
Esiintymisluokat määritetään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, 1/100), harvinainen (≥ 1/10 000, 1/1 000) ja hyvin harvinainen tai tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin) (< 1/10 000). Kussakin esiintymisluokassa haittavaikutukset esitetään vähenevässä järjestyksessä. Haittavaikutuksille, joihin sisältyi graduksen 3 tai 4 vaikutuksia, näytetään varsinainen kokonaisesiintyvyys sekä graduksen 3 tai 4 vaikutusten esiintyvyys.
| Elinjärjestelmä-luokka | Haittavaikutukset - kaikki asteet | |||
|---|---|---|---|---|
Hyvin yleinen (Esiintymistiheys %) | Yleinen (Esiintymistiheys %) | Melko harvinainen (Esiintymistiheys %) | Harvinainen tai tuntematon | |
| Infektiot | Virtsatieinfektio (8,5 %) (G3/4: 0,7 %) Pneumonia (1,6 %) (G3/4: 1,0 %) Suun kandidiaasi Suun herpes Ylähengitystieinfektio Nenän ja nielun tulehdus Riniitti Vyöruusu | Sepsis (0,5 %) (G3/4: 0,5 %)a Neutropeeninen sepsis (0,2 %) (G3/4: 0,2 %)a Septinen sokki (0,2 %) (G3/4: 0,2 %)a
| ||
| Veri ja imukudos | Neutropenia (53,6 %) (G3/4: 46,0 %) Leukopenia (27,9 %) (G3/4: 17,0 %) Anemia (21,8 %) (G3/4: 3,0 %) | Lymfopenia (5,7 %) (G3/4: 2,1 %) Kuumeinen neutropenia (4,5 %) (G3/4: 4,4 %)a Trombosytopenia (4,2 %) (G3/4: 0,7 %) | *Disseminoitunut intra-vaskulaarinen koagulaatiob | |
| Aineenvaihdunta ja ravitsemus | Alentunut ruokahalu (22,5 %) (G3/4: 0,7 %)d | Hypokalemia (6,8 %) (G3/4: 2,0 %) Hypomagnesemia (2,8 %) (G3/4: 0,3 %) Dehydraatio (2,8 %) (G3/4: 0,5 %)d Hyperglykemia Hypofosfatemia Hypokalsemia | ||
| Psyykkiset häiriöt | Unettomuus Masennus | |||
| Hermosto | Perifeerinen neuropatiac (35,9 %) (G3/4: 7,3 %) Päänsärky (17,5 %) (G3/4: 0,7 %) | Makuaistin häiriö Heitehuimaus (9,0 %) (G3/4: 0,4 %)d Hypoestesia Letargia Neurotoksisuus | ||
| Silmät | Kyynelnesteen erityksen lisääntyminen (5,8 %) (G3/4: 0,1 %)d Sidekalvotulehdus | |||
| Kuulo ja tasapainoelin | Kiertohuimaus Tinnitus | |||
| Sydän | Takykardia | |||
| Verisuonisto | Kuumat aallot Keuhkoembolia (1,3 %) (G3/4: 1,1 %)a | Syvä laskimotukos
| ||
| Hengityselimet, rintakehä ja välikarsina | Dyspnea (15,2 %)a (G3/4: 3,5 %)a Yskä (15,0 %) (G3/4: 0,5 %)d | Orofaryngeaalinen kipu Nenäverenvuoto Nuha | Interstitiaalinen keuhkosairaus (0,2 %) (G3/4: 0,1 %) | |
| Ruoansulatus-elimistö | Pahoinvointi (35,7 %) (G3/4: 1,1 %)d Ummetus (22,3 %) (G3/4: 0,7 %)d Ripuli (18,7 %) (G3/4: 0,8 %) Oksentelu (18,1 %) (G3/4: 1,0 %) | Vatsakipu Suutulehdus (11,1 %) (G3/4: 1,0 %)d Suun kuivuminen Dyspepsia (6,5 %) (G3/4: 0,3 %)d Gastroesofageaalinen refluksitauti Vatsan turvotus | Suuhaavauma Pankreatiitti | |
| Maksa ja sappi | Kohonnut aspartaattiaminotransferaasi (7,7 %) (G3/4: 1,4 %)d Kohonnut alaniiniamino-transferaasi (7,6 %) (G3/4: 1,9 %)d Kohonnut gamma- glutamyylitransferaasi (1,7 %) (G3/4: 0,9 %)d Hyperbilirubinemia (1,4 %) (G3/4: 0,4 %) | Maksatoksisuus (0,8 %) (G3/4: 0,6 %) | ||
| Iho ja ihonalainen kudos | Alopesia | Ihottuma (4,9 %) (G3/4: 0,1 %) Kutina (3,9 %) (G3/4: 0,1 %)d Kynsimuutos Yöhikoilu Kuiva iho Eryteema Voimakas hikoilu Kämmenten ja jalkapohjien erytrodysestesia (1,0 %) (G3/4: 0,1 %)d | Angioedeema | **Stevens-Johnsonin oireyhtymä / toksinen epidermaalinen nekrolyysib |
| Luusto, lihakset ja sidekudos | Nivelkipu ja lihaskipu (20,4 %) (G3/4: 1,0 %) Selkäkipu (12,8 %) (G3/4: 1,5 %) Kipu raajassa (10,0 %) (G3/4: 0,7 %)d | Luukipu (6,7 %) (G3/4: 1,2 %) Lihaskrampit (5,3 %) (G3/4: 0,1 %)d Luustolihaskipu Luustolihaskipu rinnassa Lihasheikkous | ||
| Munuaiset ja virtsatiet | Dysuria | Hematuria Proteinuria Munuaisten vajaatoiminta | ||
| Yleisoireet ja antopaikassa todettavat haitat | Uupumus/ Voimattomuus (53,2 %) (G3/4 : 7,7 %) Kuume (21,8 %) (G3/4 : 0,7 %) | Limakalvontulehdus (6,4 %) (G3/4: 0,9 %)d Perifeerinen turvotus Kipu Kylmänväreet Rintakipu Influenssan kaltainen sairaus | ||
| Tutkimukset | Painonlasku (11,4 %) (G3/4: 0,4 %)d |
| ||
a Sisältää graduksen 5 tapaukset
b Spontaanista raportoinnista
c Sisältää alatermit perifeerinen neuropatia, perifeerinen motorinen neuropatia, polyneuropatia, parestesia, perifeerinen sensorinen neuropatia, perifeerinen sensomotorinen neuropatia ja demyelinisoiva polyneuropatia
d Ei graduksen 4 tapauksia
* Harvinainen
** Esiintyvyystiheys tuntematon
Turvallisuusprofiili oli yleisesti ottaen samankaltainen rintasyöpää ja pehmytkudossarkoomaa sairastavissa väestöissä.
Valikoitujen haittavaikutusten kuvaus
Neutropenia
Todettu neutropenia oli palautuvaa eikä se ollut kumulatiivista; keskimääräinen aika, jonka jälkeen pienin arvo oli mitattavissa, oli 13 vuorokautta ja keskimääräinen vakavasta neutropeniasta paranemiseen (< 0,5 x 109/l) kulunut aika oli 8 vuorokautta.
Eribuliinia saaneista rintasyöpäpotilaista 13 %:lla neutrofiiliarvo oli < 0,5 x 109/l pitempään kuin 7 vuorokautta EMBRACE-tutkimuksessa.
Neutropeniaa hoidon aikaisena haittatapahtumana raportoitiin (TEAE) 151/404 potilaalla (37,4 % kaikille graduksille) sarkoomapopulaatiossa ja 902/1 559 potilaalla (57,9 % kaikille graduksille) rintasyöpäpopulaatiossa. Hoidon aikaisten haittatapahtumien ja neutrofiilien poikkeavien laboratoriolöydösten yhteisesiintyvyydet olivat vastaavasti 307/404 potilasta (76,0 %) ja 1 314/1 559 potilasta (84,3 %). Hoidon mediaanikesto oli sarkoomapotilailla 12,0 viikkoa ja rintasyöpäpotilailla 15,9 viikkoa.
Kuumeisen neutropenian, neutropeenisen sepsiksen, sepsiksen ja septisen sokin aiheuttamia kuolemantapauksia on raportoitu. Eribuliinia suositeltuna annoksena kliinisissä tutkimuksissa saaneiden 1 963:n rintasyöpää ja pehmytkudossarkoomaa sairastavan potilaan joukossa neutropeeninen sepsis ja kuumeinen neutropenia johtivat kumpikin yhteen kuolemantapaukseen (0,1 % ja 0,1 %). Lisäksi sepsis aiheutti kolme kuolemantapausta (0,2 %) ja septinen sokki yhden kuolemantapauksen (0,1 %).
Vakavaa neutropeniaa voidaan hoitaa käyttämällä G-CSF:ää tai vastaavaa lääkärin harkinnan mukaan ja asianmukaisia ohjeita noudattamalla. Kahdessa faasin 3 rintasyöpätutkimuksessa (tutkimukset 305 ja 301) eribuliinia saaneista potilaista 18 % ja 13 % sai G-CSF:ää. Faasin 3 sarkoomatutkimuksessa (tutkimus 309) 26 % eribuliinia saaneista potilasta sai G-CSF:ää.
Neutropenia oli hoidon lopettamisen syynä < 1 %:lla eribuliinia saaneista potilaista.
Disseminoitunut intravaskulaarinen koagulaatio
Disseminoituneen intravaskulaarisen koagulaation tapauksia on raportoitu, jotka tavallisesti ovat liittyneet neutropeniaan ja/tai sepsikseen.
Perifeerinen neuropatia
Yleisin 1 559 rintasyöpäpotilaalla todettu eribuliinihoidon lopettamisen aiheuttanut haittavaikutus oli perifeerinen neuropatia (3,4 %). Mediaaniaika graduksen 2 perifeerisen neuropatian kehittymiseen oli 12,6 viikkoa (4 hoitojakson jälkeen). 404 sarkoomapotilaasta 2 lopetti eribuliinihoidon perifeerisen neuropatian takia. Mediaaniaika graduksen 2 perifeerisen neuropatian kehittymiseen oli 18,4 viikkoa.
Graduksen 3 tai 4 perifeerinen neuropatia kehittyi 7,4 %:lla rintasyöpäpotilaista ja 3,5 %:lla sarkoomapotilaista. Kliinisissä tutkimuksissa neuropatiaa jo ennestään sairastaneilla potilailla oli yhtä suuri todennäköisyys saada uusia tai pahempia oireita kuin niillä, joilla tutkimukseen tulleessaan ei ollut tätä sairautta.
Rintasyöpäpotilailla, joilla jo ennestään oli gradus 1 tai 2 perifeerinen neuropatia, hoidon aiheuttaman gradus 3 perifeerisen neuropatian esiintyvyys oli 14 %.
Maksatoksisuus
Maksaentsyymien kohoamista eribuliinihoidon aloittamisen jälkeen on raportoitu joillakin potilailla riippumatta siitä, olivatko hoitoa edeltävät arvot normaaleja vai ei. Useimmilla näistä potilaista maksaentsyymien kohoaminen vaikutti tapahtuneen eribuliinihoidon aikaisessa vaiheessa, hoitojaksoina 1–2. Vaikka arvojen kohoamista pidetään useimmilla potilailla todennäköisenä merkkinä maksan adaptaatiosta eribuliinihoitoon eikä merkkinä merkittävästä maksatoksisuudesta, myös maksatoksisuutta on raportoitu.
Erityispotilasryhmät
Iäkkäät potilaat
Tutkimuksissa hoitoa suositellulla eribuliiniannoksella saaneista 1 559 rintasyöpäpotilaasta 283 (18,2 %) oli iältään ≥ 65 vuotta. Eribuliinia saaneen sarkoomapopulaation 404 potilaasta 90 (22,3 %) oli iältään ≥ 65 vuotta. Eribuliinin turvallisuusprofiili iäkkäillä potilailla (iältään ≥ 65 vuotta) oli samankaltainen kuin < 65 vuoden ikäisillä lukuun ottamatta voimattomuutta/väsymystä, jonka esiintyvyys kasvoi iän myötä. Annoksen säätämistä ei suositella iäkkäälle väestölle.
Maksan vajaatoimintaa sairastavat potilaat
Potilailla, joiden ALAT- tai ASAT-arvo oli yli kolme kertaa yli normaalin ylärajan, esiintyi useammin asteen 4 neutropeniaa ja kuumeista neutropeniaa. Potilailla, joiden bilirubiinitaso on yli 1,5 kertaa yli normaalin ylärajan, on suurempi todennäköisyys sairastua gradus 4 neutropeniaan ja kuumeiseen neutropeniaan (ks. myös kohdat Annostus ja antotapa sekä Farmakokinetiikka), joskin tätä koskeva aineisto on rajallinen.
Pediatriset potilaat
Kolmessa avoimessa tutkimuksessa (tutkimukset 113, 213 ja 223) tutkittiin pediatrisia potilaita, joilla oli hoitoon vastaamattomia tai uusiutuneita kiinteitä kasvaimia ja lymfoomia, pois lukien keskushermoston kasvaimet (ks. kohta Farmakodynamiikka).
Eribuliinimonoterapian turvallisuutta arvioitiin 43 pediatrisella potilaalla, jotka saivat eribuliinia enimmillään 1,58 mg/m2 kunkin 21 vrk kestävän hoitojakson päivinä 1 ja 8 (tutkimukset 113 ja 223). Eribuliinin turvallisuutta arvioitiin myös yhdistelmähoidossa irinotekaanin kanssa 40 pediatrisella potilaalla, jotka saivat kullakin 21 vrk kestävällä hoitojaksolla eribuliinia 1,23 mg/m2 päivinä 1 ja 8 ja irinotekaania 20 tai 40 mg/m2 päivinä 1–5 tai 100 tai 125 mg/m2 päivinä 1 ja 8 (tutkimus 213).
Tutkimuksessa 113 (faasi 1) useimmin raportoidut lääkkeen aiheuttamat haittavaikutukset olivat valkosolumäärän pieneneminen, lymfosyyttimäärän pieneneminen, anemia ja neutrofiilimäärän pieneneminen.
Tutkimuksessa 213 (faasit 1 ja 2) useimmin raportoidut lääkkeen aiheuttamat haittavaikutukset olivat neutropenia (faasi 1) ja ripuli ja neutrofiilimäärän pieneneminen (faasi 2).
Tutkimuksessa 223 (faasi 2) useimmin raportoidut lääkkeen aiheuttamat haittavaikutukset olivat neutrofiilimäärän pieneneminen, anemia ja valkosolumäärän pieneneminen.
Eribuliinin turvallisuusprofiili oli näillä pediatrisilla potilailla sekä monoterapiassa että yhdistelmähoidossa irinotekaanihydrokloridin kanssa yhdenmukainen sen turvallisuusprofiilin kanssa, joka kummallakin tutkimuslääkkeellä tunnetaan aikuispotilailla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
FI-00034 Fimea
Yliannostus
Yhdessä yliannostustapauksessa potilaalle annettiin epähuomiossa 7,6 mg eribuliinia (suunniteltu annos noin nelinkertaisena) ja hänelle kehittyi (gradus 3) yliherkkyysreaktio 3. vuorokautena ja (asteen 3) neutropenia 7. vuorokautena. Molemmat haittavaikutukset hävisivät tukihoidolla.
Eribuliinin yliannokselle ei ole vasta-ainetta. Yliannostustapauksessa potilasta tulee seurata huolellisesti. Yliannostustapauksessa on ilmenneiden kliinisten oireiden hoitamiseksi ryhdyttävä lääkinnällisiin tukitoimiin.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut syöpälääkkeet, ATC-koodi: L01XX41.
Eribuliinimesylaatti on mikrotubulusdynamiikan estäjä ja se kuuluu syöpälääkkeiden halikondriinien luokkaan. Rakenteeltaan se on yksinkertaistettu synteettinen analogi halikondriini B:stä, joka on Halichondria okadai -merisienestä eristetty luonnontuote.
Eribuliini estää mikrotubulusten kasvuvaihetta vaikuttamatta niiden lyhentymisvaiheeseen ja eristää tubuliinin tuottamattomiksi kasaumiksi. Eribuliini vaikuttaa tubuliiniin perustuvalla antimitoottisella mekanismilla, joka johtaa G2/M-solusyklin salpautumiseen, tumasukkuloiden hajoamiseen ja lopulta pitkittyneen, palautumattoman mitoosinsalpauksen jälkeen apoptoottiseen solukuolemaan.
Kliininen teho
Rintasyöpä
Halaven-valmisteen tehoa rintasyövän hoidossa tukee ensisijaisesti kaksi satunnaistettua faasin 3 vertailututkimusta.
Keskeiseen faasin 3 EMBRACE-tutkimukseen (tutkimus 305) osallistuneilla 762 potilaalla oli paikallisesti uusiutunut tai metastasoitunut rintasyöpä, ja heidän hoitoonsa oli aikaisemmin kuulunut vähintään kaksi ja enintään viisi kemoterapiahoitojaksoa, antrasykliini ja taksaani mukaan lukien (elleivät ne olleet vasta-aiheisia). Potilaiden sairauden oli täytynyt edetä 6 kuukauden kuluessa heidän viimeisimmästä kemoterapiahoitojaksostaan. Potilaiden HER2-statukset olivat: 16,1 % positiivisia, 74,2 % negatiivisia ja 9,7 % tuntemattomia. Potilaista 18,9 % oli kolmoisnegatiivisia. Potilaat satunnaistettiin suhteessa 2:1 saamaan joko Halaven-valmistetta tai lääkärin valitsemaa hoitoa (TPC). Lääkärin valitsemista hoidoista 97 % oli kemoterapiaa, 26 % vinorelbiinia, 18 % gemsitabiinia, 18 % kapesitabiinia, 16 % taksaania, 9 % antrasykliiniä, 10 % jotain muuta kemoterapiahoitoa) ja 3 % hormonihoitoa.
Tutkimuksen ensisijaisena päätetapahtumana kokonaiselossaolon tulos oli eribuliiniryhmässä tilastollisesti merkitsevästi parempi kuin lääkärin valitsemaa hoitoa saaneiden ryhmässä (TPC), jossa elossaolo-% oli 55.
Tämä tulos vahvistettiin päivitetyllä kokonaiselossaoloanalyysilla, joka tehtiin 77 %:lla tapauksista.
Tutkimus 305 – Päivitetty kokonaiselossaolo (ITT-väestö)
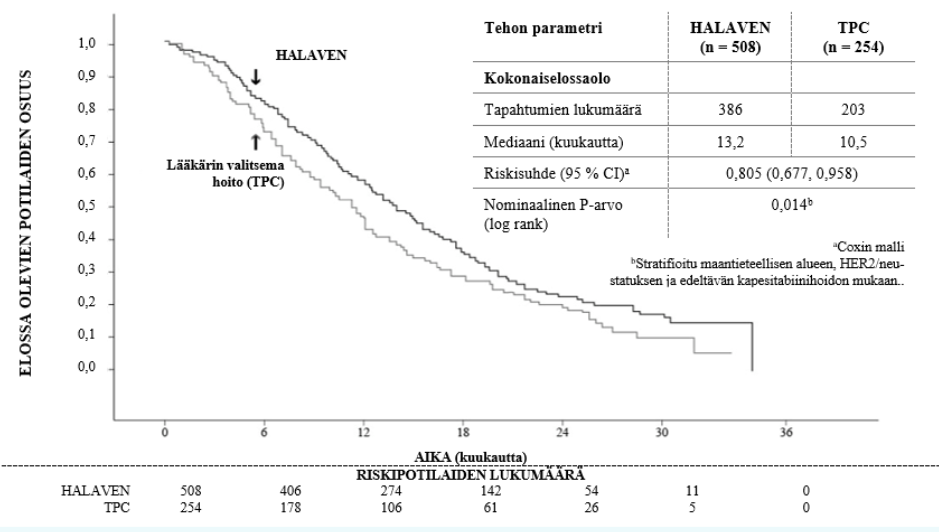
Riippumattoman selvityksen mukaan etenemisvapaan elossaolon mediaani oli 3,7 kuukautta eribuliinille verrattuna TCP-ryhmän 2,2 kuukauteen (riskisuhde 0,865, 95 % CI: 0,714, 1,048, p = 0,137).
Potilailla, joiden vastetta voitiin arvioida, objektiivisen vasteen osuus RECIST-kriteereillä mitattuna oli riippumattoman selvityksen mukaan 12,2 % (95 % CI: 9,4 %, 15,5 %) eribuliiniryhmälle ja 4,7 % (95 % CI: 2,3 %, 8,4 %) TCP-ryhmälle.
Positiivinen vaikutus kokonaiselossaoloon todettiin niin taksaanihoitoon vastaamattomissa kuin siihen vastanneissa potilasryhmissä. Kokonaiselossaolon päivityksessä eribuliinin ja lääkärin valitseman hoidon välinen riskisuhde oli 0,90 (95 % CI: 0,71, 1,14) eribuliinin puolesta taksaanihoitoon vastaamattomilla potilailla ja 0,73 (95 % CI: 0,56, 0,96) taksaanihoitoon vastanneilla potilailla.
Positiivinen vaikutus kokonaiselossaoloon todettiin sekä aiemmin kapesitabiinia saamattomissa että kapesitabiinia edeltävänä hoitona saaneissa potilasryhmissä. Päivitetyn kokonaiselossaolon analyysi osoittaa elossaolon olevan parempi eribuliiniryhmässä verrattuna TPC-ryhmään sekä kapesitabiinia edeltävänä hoitona saaneiden ryhmässä, jossa riskisuhde oli 0,787 (95 % CI: 0,645, 0,961), että kapesitabiinia aikaisemmin saamattomassa potilasryhmässä, jossa vastaava riskisuhde oli 0,865 (95 % CI: 0,606, 1,233).
Toinen faasin 3 tutkimus (tutkimus 301) oli avoin, satunnaistettu tutkimus, joka tehtiin aikaisemman vaiheen metastasoitunutta rintasyöpää sairastavilla potilailla (n = 1 102), joiden syöpä oli paikallisesti pitkälle edennyt tai metastasoitunut. Tutkimuksessa arvioitiin Halaven-monoterapian tehoa kapesitabiinimonoterapiaan verrattuna siten, että kokonaiselossaolo ja etenemisvapaa elossaolo olivat molemmat ensisijaisia päätetapahtumia. Potilaat olivat aiemmin saaneet enintään kolme kemoterapiahoitojaksoa, antrasykliini ja taksaani mukaan lukien, joista enintään kahta oli saatu pitkälle edenneeseen sairauteen. Potilaista 20,0 % ei ollut saanut yhtään kemoterapiahoitojaksoa metastasoituneeseen rintasyöpään, kun taas 52,0 % oli saanut yhden ja 27,2 % kaksi. Potilaiden HER2-statukset olivat: 15,3 % positiivisia, 68,5 % negatiivisia ja 16,2 % tuntemattomia. Potilaista 25,8 % oli kolmoisnegatiivisia.
Tutkimus 301 – Kokonaiselossaolo (ITT-väestö)
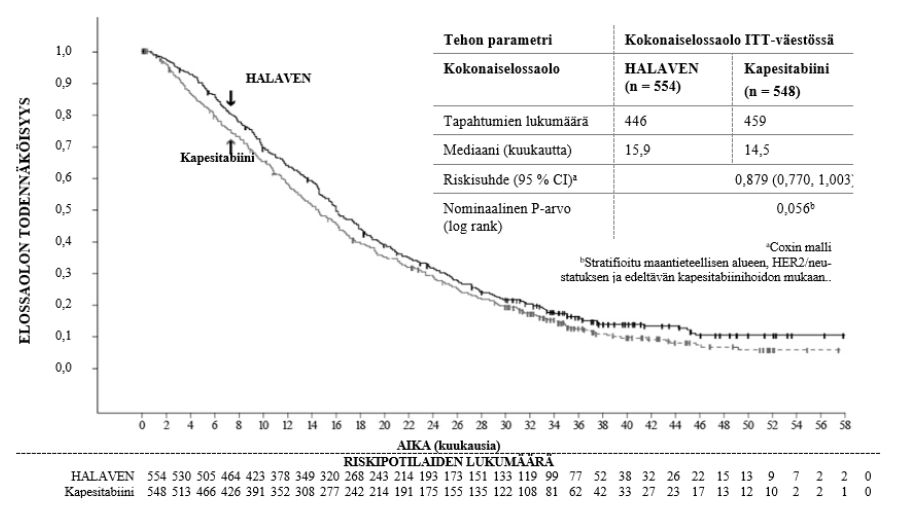
Riippumattoman selvityksen mukainen etenemisvapaa elossaolo oli samankaltainen eribuliinille ja kapesitabiinille; mediaanit olivat 4,1 kuukautta vs. 4,2 kuukautta (riskisuhde 1,08; [95 % CI: 0,932, 1,250]). Riippumattoman selvityksen mukainen objektiivisen vasteen osuus oli myös samankaltainen eribuliinille ja kapesitabiinille; 11,0 % (95 % CI: 8,5, 13,9) eribuliiniryhmässä ja 11,5 % (95 % CI: 8,9, 14,5) kapesitabiiniryhmässä.
Alla esitetään HER2-negatiivisten ja HER2-positiivisten potilaiden kokonaiselossaolo eribuliini- ja vertailuryhmissä tutkimuksissa 305 ja 301:
| Tehon parametri | Tutkimus 305 – Päivitetty kokonaiselossaolo (ITT-väestö) | |||
| HER2-negatiivinen | HER2-positiivinen | |||
| HALAVEN (n = 373) | TPC (n = 192) | HALAVEN (n = 83) | TPC (n = 40) | |
| Tapahtumien lukumäärä | 285 | 151 | 66 | 37 |
| Mediaani (kuukautta) | 13,4 | 10,5 | 11,8 | 8,9 |
| Riskisuhde (95 % CI) | 0,849 (0,695, 1,036) | 0,594 (0,389, 0,907) | ||
| P-arvo (log rank) | 0,106 | 0,015 | ||
| Tehon parametri | Tutkimus 301 – Kokonaiselossaolo (ITT-väestö) | |||
| HER2-negatiivinen | HER2-positiivinen | |||
| HALAVEN (n = 375) | Kapesitabiini (n = 380) | HALAVEN (n = 86) | Kapesitabiini (n = 83) | |
| Tapahtumien lukumäärä | 296 | 316 | 73 | 73 |
| Mediaani (kuukautta) | 15,9 | 13,5 | 14,3 | 17,1 |
| Riskisuhde (95 % CI) | 0,838 (0,715, 0,983) | 0,965 (0,688, 1,355) | ||
| P-arvo (log rank) | 0,030 | 0,837 | ||
Huom. Tutkimuksiin 305 ja 301 ei kuulunut samanaikaista anti-HER2-hoitoa.
Liposarkooma
Eribuliinin tehoa liposarkooman hoidossa tukee keskeinen faasin 3 sarkoomatutkimus (tutkimus 309). Tämän tutkimuksen potilailla (n = 452) oli paikallisesti uusiutunut, ei leikattavissa oleva ja/tai metastasoitunut pehmyskudossarkoomaa, jonka alatyyppi oli joko leiomyosarkooma tai liposarkooma. Potilaat olivat aiemmin saaneet vähintään kaksi kemoterapiahoitojaksoa, joista toisen piti olla antrasykliinillä (ellei vasta-aiheinen).
Potilaiden sairauden oli täytynyt edetä 6 kuukauden kuluessa viimeisimmästä kemoterapiahoitojaksosta. Potilaat satunnaistettiin suhteessa 1:1 saamaan joko eribuliinia 1,23 mg/m2:n annoksena 21 vuorokauden hoitojakson päivinä 1 ja 8 tai dakarbatsiinia 850 mg/m2:n, 1 000 mg/m2:n tai 1 200 mg/m2:n annoksena 21 vuorokauden välein (annokset määräsi tutkija ennen satunnaistamista).
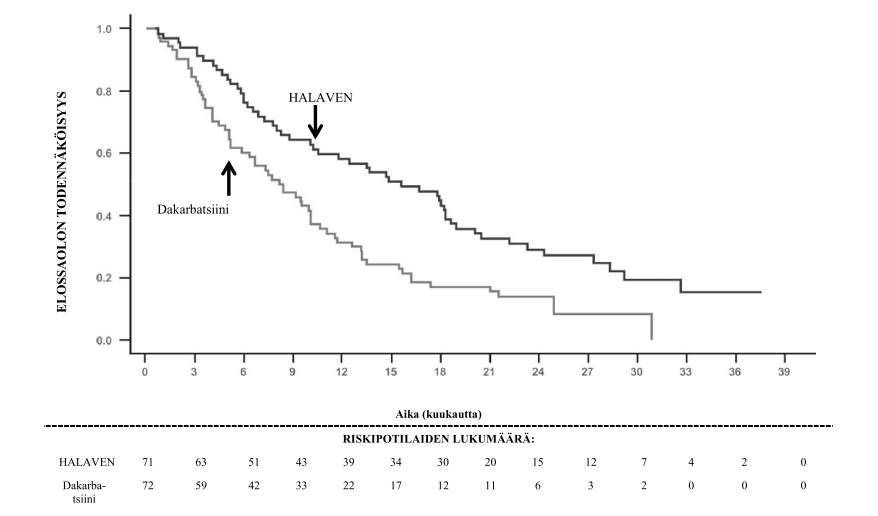
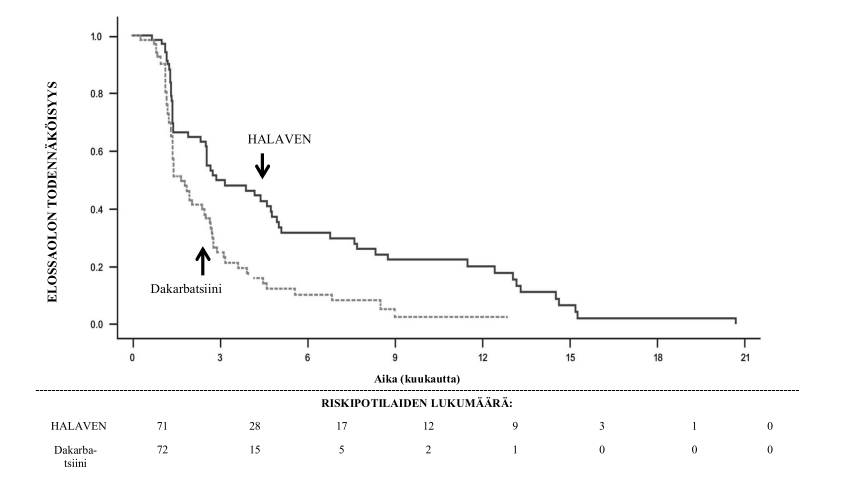
Tutkimuksen 309 eribuliinihaaraan satunnaistetuilla potilailla havaittiin tilastollisesti merkitsevä parannus kokonaiselossaolossa verrokkihaaraan nähden. Ero elossaolon mediaanin parannuksessa oli 2 kuukautta (13,5 kuukautta eribuliinia saaneilla ja 11,5 kuukautta dakarbatsiinia saaneilla potilailla). Elossaolossa ilman taudin etenemistä ja kokonaisvasteessa ei havaittu merkitsevää eroa hoitoryhmien välillä tutkimuksen kokonaisväestössä.
Etukäteen suunniteltujen, kokonaiselossaoloa ja elossaoloa ilman taudin etenemistä koskevien alaryhmäanalyysien perusteella eribuliinin hoitovaikutukset rajoittuivat potilaisiin, joilla oli liposarkooma (dedifferentoitunut 45 %, myksoidinen/pyörösoluinen 37 % ja monimuotoinen 18 % tutkimuksessa 309). Eroa tehossa eribuliinin ja dakarbatsiinin välillä ei havaittu pitkälle edennyttä tai metastasoitunutta leiomykosarkoomaa sairastavilla potilailla.
Tutkimus 309 Liposarkoomaa sairastavien alaryhmä | Tutkimus 309 Leiomyosarkoomaa sairastavien alaryhmä | Tutkimus 309 ITT-väestö | ||||
HALAVEN (n = 71) | Dakarbatsiini (n = 72) | HALAVEN (n = 157) | Dakarbatsiini (n = 152) | HALAVEN (n = 228) | Dakarbatsiini (n = 224) | |
| Kokonaiselossaolo | ||||||
| Tapahtumien lukumäärä | 52 | 63 | 124 | 118 | 176 | 181 |
| Mediaani (kuukautta) | 15,6 | 8,4 | 12,7 | 13,0 | 13,5 | 11,5 |
| Riskisuhde (95 % CI) | 0,511 (0,346, 0,753) | 0,927 (0,714, 1,203) | 0,768 (0,618, 0,954) | |||
| Nominaalinen p‑arvo | 0,0006 | 0,5730 | 0,0169 | |||
| Elossaolo ilman taudin etenemistä | ||||||
| Tapahtumien lukumäärä | 57 | 59 | 140 | 129 | 197 | 188 |
| Mediaani (kuukautta) | 2,9 | 1,7 | 2,2 | 2,6 | 2,6 | 2,6 |
| Riskisuhde (95 % CI) | 0,521 (0,346, 0,784) | 1,072 (0,835, 1,375) | 0,877 (0,710, 1,085) | |||
| Nominaalinen p‑arvo | 0,0015 | 0,5848 | 0,2287 | |||
Tutkimus 309 – Kokonaiselossaolo liposarkoomaa sairastavien alaryhmässä
Tutkimus 309 – Elossaolo ilman taudin etenemistä liposarkoomaa sairastavien alaryhmässä
Pediatriset potilaat
Rintasyöpä
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset eribuliinin käytöstä rintasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Pehmytkudossarkooma
Eribuliinin tehoa on arvioitu mutta ei varmistettu kolmessa avoimessa tutkimuksessa:
Tutkimus 113 oli annoksen määritystä varten tehty faasin 1 avoin monikeskustutkimus, jossa eribuliinia arvioitiin pediatrisilla potilailla, joilla oli hoitoon vastaamattomia tai uusiutuneita kiinteitä kasvaimia ja lymfoomia, pois lukien keskushermoston kasvaimet. Tutkimukseen otettuja ja hoitoa saaneita pediatrisia potilaita (3–17‑vuotiaita) oli yhteensä 22. Potilaille annettiin kunkin 21 vrk kestävän hoitojakson päivinä 1 ja 8 laskimoon jokin seuraavista kolmesta eribuliiniannoksesta: 0,97, 1,23 ja 1,58 mg/m2. Suurimmaksi siedetyksi annokseksi / suositelluksi faasin 2 eribuliiniannokseksi (RP2D) määritettiin 1,23 mg/m2 kunkin 21 vrk kestävän hoitojakson päivinä 1 ja 8.
Tutkimus 223 oli faasin 2 avoin monikeskustutkimus, jossa arvioitiin eribuliinin turvallisuutta ja alustavaa aktiivisuutta pediatrisilla potilailla, joilla oli hoitoon vastaamaton tai uusiutunut rabdomyosarkooma (RMS), jokin muu pehmytkudossarkooma kuin rabdomyosarkooma (NRSTS) tai Ewingin sarkooma (EWS). Tutkimukseen otettiin 21 pediatrista potilasta (2–17‑vuotiaita), joille annettiin laskimoon 1,23 mg/m2 eribuliinia kunkin 21 vrk kestävän hoitojakson päivinä 1 ja 8 (tutkimuksen 113 RP2D). Yhdelläkään potilaalla ei saavutettu vahvistettua osittaista vastetta tai täydellistä vastetta.
Tutkimus 213 oli faasien 1 ja 2 avoin monikeskustutkimus: faasissa 1 arvioitiin eribuliinin turvallisuutta ja tehoa yhdistelmähoidossa irinotekaanihydrokloridin kanssa pediatrisilla potilailla, joilla oli uusiutuneita tai hoitoon vastaamattomia kiinteitä kasvaimia ja lymfoomia, pois lukien keskushermoston kasvaimet, ja faasissa 2 arvioitiin yhdistelmähoidon tehoa pediatrisilla potilailla, joilla oli uusiutunut tai hoitoon vastaamaton RMS, NRSTS tai EWS. Tähän tutkimukseen otettuja ja hoitoa saaneita pediatrisia potilaita oli yhteensä 40. Faasiin 1 otettuja ja hoitoa saaneita pediatrisia potilaita (4–17-vuotiaita) oli 13. Yhdistelmähoidon RP2D-annokseksi määritettiin kullakin 21 vrk kestävällä hoitojaksolla annettu 1,23 mg/m2 eribuliinia päivinä 1 ja 8 ja 40 mg/m2 irinotekaanihydrokloridia päivinä 1–5. Faasiin 2 otettiin 27 pediatrista potilasta (4–17‑vuotiaita), joita hoidettiin RP2D-annoksella. Kolmella potilaalla vahvistettiin osittainen vaste (1 potilaalla kussakin histologisessa RMS-, NRSTS- ja EWS-kohortissa). Objektiivisen vasteen osuus oli 11,1 %.
Näissä kolmessa pediatrisessa tutkimuksessa ei havaittu uusia turvallisuussignaaleja (ks. kohta Haittavaikutukset), mutta potilasryhmien pienen koon vuoksi mitään varmoja johtopäätöksiä ei voida tehdä.
Farmakokinetiikka
Jakautuminen
Eribuliinin farmakokinetiikalle luonteenomaista on nopea jakautumisen vaihe, jota seuraa pitkittynyt eliminaatiovaihe, keskimääräisen lopullisen puoliintumisajan ollessa noin 40 tuntia. Eribuliinilla on suuri jakautumisvolyymi (vaihteluväli keskimäärin 43–114 l/m2).
Eribuliinin sitoutuminen plasman proteiineihin on heikkoa. Eribuliinin (100-1 000 ng/ml) sitoutuminen plasman proteiineihin vaihteli 49 %:n ja 65 %:n välillä ihmisen plasmassa.
Biotransformaatio
Kun potilaille oli annettu 14C-eribuliinia, muuttumaton eribuliini oli plasmassa oleva pääasiallinen muoto. Kanta-aineen < 0,6 %:n metaboliittipitoisuudet osoittavat, että eribuliinin ihmisellä esiintyvät metaboliitit eivät ole huomattavia.
Eliminaatio
Eribuliinin puhdistuma on vähäistä (vaihteluväli keskimäärin 1,16–2,42 l/h/m2). Merkittävää eribuliinin kumuloitumista ei havaita viikoittaisella annostuksella. Farmakokineettiset ominaisuudet eivät ole annoksesta tai ajasta riippuvaisia eribuliiniannoksilla 0,22–3,53 mg/m2.
Eribuliini eliminoituu pääasiallisesti sappierityksen kautta. Erittymiseen liittyvää kuljettajaproteiinia ei tällä hetkellä tunneta. Prekliiniset in vitro -tutkimukset osoittavat, että eribuliini kulkeutuu Pgp:n avulla. On kuitenkin osoitettu, että kliinisesti olennaisin pitoisuuksin eribuliini ei ole Pgp-estäjä in vitro. Lisäksi ketokonatsolin, Pgp-estäjän, samanaikainen in vivo -annostelu ei vaikuta eribuliinille altistumiseen (AUC ja Cmax). In vitro -tutkimukset ovat osoittaneet myös, että eribuliini ei ole OCT1:n substraatti.
Kun 14C-eribuliinia annettiin potilaille, noin 82 % annoksesta eliminoitui ulosteen kautta ja 9 % virtsan kautta, mikä osoittaa, ettei munuaispuhdistuma ole eribuliinin merkitsevä poistumisreitti.
Muuttumaton eribuliini edusti suurinta osaa ulosteessa ja virtsassa havaitusta kokonaisradioaktiivisuudesta.
Maksan vajaatoiminta
Tutkimuksessa arvioitiin eribuliinin farmakokinetiikkaa potilailla, jotka sairastivat maksametastaaseista johtuvaa lievää (Child-Pugh A; n = 7) ja keskivaikeaa (Child-Pugh B; n = 4) maksan vajaatoimintaa. Verrattuna potilaisiin, joilla maksan toiminta oli normaali (n = 6), eribuliinille altistuminen suureni lievää maksan vajaatoimintaa sairastavilla 1,8-kertaiseksi ja keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla 3-kertaiseksi. Halaven-valmisteen anto käyttämällä annosta 0,97 mg/m2 potilaille, jotka sairastivat lievää maksan vajaatoimintaa, ja 0,62 mg/m2 potilaille, jotka sairastivat keskivaikeaa maksan vajaatoimintaa, aikaansai hieman suuremman altistuksen kuin 1,23 mg/m2 -annos potilaille, joilla maksan toiminta oli normaali. Halaven-valmistetta ei tutkittu vakavaa maksan vajaatoimintaa (Child-Pugh C) sairastavilla potilailla. Kirroosin aiheuttamaa maksan vajaatoimintaa sairastavilla potilailla ei ole tehty tutkimusta. Ks. annostussuositus kohdasta Annostus ja antotapa.
Munuaisten vajaatoiminta
Eribuliinille altistumisen lisääntymistä todettiin joillakin munuaisten keskivaikeaa tai vaikeaa vajaatoimintaa sairastavilla potilailla yksilöiden välisten eroavuuksien ollessa suuria. Eribuliinin farmakokinetiikkaa arvioitiin faasin I tutkimuksessa potilailla, joiden munuaisten toiminta oli normaali (kreatiniinipuhdistuma ≥ 80 ml/min, n = 6), keskivaikea (30–50 ml/min, n = 7) tai vaikea (15– < 30 ml/min, n = 6). Kreatiniinipuhdistumaa arvioitiin käyttämällä Cockcroft-Gaultin kaavaa. Annoksen suhteen normalisoidun AUC(0-inf):n havaittiin olevan 1,5 kertaa suurempi (90 % CI: 0,9–2,5) potilailla, joilla munuaisten vajaatoiminta oli keskivaikea tai vaikea. Ks. hoitosuositukset kohdasta Annostus ja antotapa.
Pediatriset potilaat
Eribuliinipitoisuus plasmassa määritettiin 83 pediatriselta potilaalta (2–17‑vuotiaita), joilla oli hoitoon vastaamattomia tai uusiutuneita kiinteitä kasvaimia ja lymfoomia ja jotka saivat eribuliinia tutkimuksissa 113, 213 ja 223. Eribuliinin farmakokinetiikka oli pediatrisissa potilailla vastaavaa kuin aikuisilla, joilla oli pehmytkudossarkooma, ja potilailla, joilla oli jokin muuntyyppinen kasvain. Pediatristen potilaiden altistuminen eribuliinille oli samaa luokkaa kuin aikuispotilailla. Samanaikaisesti annettu irinotekaani ei vaikuttanut eribuliinin farmakokinetiikkaan pediatrisilla potilailla, joilla oli hoitoon vastaamattomia tai uusiutuneita kiinteitä kasvaimia.
Prekliiniset tiedot turvallisuudesta
Eribuliini ei ollut mutageeninen mutageenisuustestissä (Amesin testi) in vitro. Eribuliini oli positiivinen hiiren lymfooma-mutageenisuustestissä ja klastogeeninen rotan mikrotumatestissä in vivo.
Karsinogeenisuustutkimuksia ei ole suoritettu eribuliinilla.
Eribuliinilla ei ole tehty hedelmällisyystutkimusta, mutta toistuvien annosten tutkimuksissa, joissa havaittiin kivestoksisuutta sekä rotilla (siemennestettä muodostavan epiteelin solujen niukkuus, johon liittyy hypospermiaa/aspermiaa) että koirilla, havaittujen muiden kuin kliinisten löydösten perusteella eribuliinihoito saattaa vaikuttaa miehen hedelmällisyyteen. Rotalla tehty alkion ja sikiön kehitystä koskeva tutkimus vahvisti eribuliinin olevan kehitystoksinen ja että sillä on teratogeenista vaikutusta. Tiineille rotille annettiin eribuliinimesylaattia, joka vastasi 0,009, 0,027, 0,088 ja 0,133 mg/kg eribuliinia tiineyden päivinä 8, 10 ja 12. Annokseen liittyvää resorptioiden määrän lisääntymistä ja sikiön painonlaskua havaittiin annoksilla ≥ 0,088 mg/kg ja epämuodostumien (alaleukaluun, kielen, mahalaukun tai pernan puuttumisen) lisääntymistä havaittiin annoksella 0,133 mg/kg.
Farmaseuttiset tiedot
Apuaineet
Etanoli, vedetön
Injektionesteisiin käytettävä vesi
Suolahappo (pH:n säätämistä varten)
Natriumhydroksidi (pH:n säätämistä varten)
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamattomat injektiopullot
5 vuotta.
Käytön aikainen kestoaika
Valmiste tulee mikrobiologisista syistä käyttää välittömästi avaamisen jälkeen. Jos valmistetta ei käytetä välittömästi, käytön aikaiset kestoajat ja olosuhteet ovat käyttäjän vastuulla eivätkä saa normaalisti ylittää 24:ää tuntia 2–8 °C:n lämpötilassa, ellei laimentaminen ole tapahtunut kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Ruiskussa olevan laimentamattoman HALAVEN-liuoksen käytön aikaiseksi kemialliseksi ja fysikaaliseksi stabiliteetiksi on osoitettu enintään 4 tuntia 15–25 °C:n lämpötilassa ja ympäristön vallitsevassa valaistuksessa tai enintään 24 tuntia 2–8 °C:n lämpötilassa.
Laimennetun HALAVEN-liuoksen (0,018 mg/ml – 0,18 mg/ml eribuliinia natriumkloridiliuoksessa 9 mg/ml [0,9 %]) käytön aikaiseksi kemialliseksi ja fysikaaliseksi stabiliteetiksi on osoitettu enintään 72 tuntia 2–8 °C:n lämpötilassa.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Avatun tai laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
HALAVEN injektioneste, liuos
0,44 mg/ml (L:ei) 2 ml (543,55 €)
PF-selosteen tieto
Tyypin I lasista valmistettu 5 ml:n injektiopullo, jossa on teflonpäällystetty butyylikuminen tulppa ja alumiininen repäisysuljin ja joka sisältää 2 ml liuosta.
Pakkauskoot ovat 1 tai 6 injektiopulloa sisältävät rasiat.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas, väritön vesiliuos.
Käyttö- ja käsittelyohjeet
Halaven on sytotoksinen syöpälääke ja muiden myrkyllisten yhdisteiden lailla sen käytössä on noudatettava varovaisuutta. Käsineiden, suojalasien ja suojavaatetuksen käyttämistä suositellaan. Jos iho joutuu kosketuksiin liuoksen kanssa, iho on pestävä välittömästi huolellisesti vedellä ja saippualla. Jos liuos joutuu kosketuksiin limakalvojen kanssa, limakalvot tulee huuhdella perusteellisesti vedellä. Halaven-valmisteen saa valmistaa ja antaa vain henkilökunta, joka on saanut asianmukaisen koulutuksen sytotoksisten aineiden käsittelyssä. Raskaana oleva henkilökunta ei saa käsitellä Halaven-valmistetta.
Aseptista tapaa käyttämällä Halaven voidaan laimentaa korkeintaan 100 ml:aan natriumkloridi-injektioliuosta 9 mg/ml (0,9 %). Annon jälkeen on suositeltavaa huuhdella laskimolinja natriumkloridi-injektioliuoksella 9 mg/ml (0,9 %) sen varmistamiseksi, että koko annos tulee annetuksi. Halaven-valmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa eikä laimentaa 5-prosenttiseen glukoosi-infuusioliuokseen.
Jos käytät valmisteen antoon piikkiä, noudata laitevalmistajan ohjeita. Halaven-injektiopulloissa on 13 mm:n tulppa. Valitun laitteen on oltava yhteensopiva injektiopullojen pienten tulppien kanssa.
Jos käytät valmisteen antoon piikkiä, noudata laitevalmistajan ohjeita. HALAVEN-injektiopulloissa on 13 mm:n tulppa. Valitun laitteen on oltava yhteensopiva injektiopullojen pienten tulppien kanssa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
HALAVEN injektioneste, liuos
0,44 mg/ml 2 ml
- Ei korvausta.
ATC-koodi
L01XX41
Valmisteyhteenvedon muuttamispäivämäärä
26.07.2024
Yhteystiedot
Svärdvägen 15
18233 Danderyd
Ruotsi
+46 8 501 01 60
www.eisai.eu
nordic_medinfo@eisai.net
Edustaja Suomessa: EISAI AB