
WILFACTIN injektiokuiva-aine ja liuotin, liuosta varten 100 IU/ml
not_interestedSaatavuushäiriö
Ei saatavilla
WILFACTIN injektiokuiva-aine ja liuotin, liuosta varten
- 100 IU/ml1000 IU + 10 ml27.07.2026 - 31.01.2027
Saatavilla
Muut samaa lääkeainetta sisältävät valmisteet
VEYVONDI injektiokuiva-aine ja liuotin, liuosta varten
- 650 IU1 kpl
- 1300 IU1 kpl
Vaikuttavat aineet ja niiden määrät
Wilfactin on injektiokuiva-aine ja liuotin, liuosta varten, joka sisältää nimellisesti 500 IU, 1000 IU tai 2000 IU ihmisen von Willebrand -tekijää (VWF) yhtä injektiopulloa kohden.
Valmiste sisältää noin 100 IU/ml ihmisen von Willebrand -tekijää, kun se saatetaan käyttökuntoon 5 ml:aan (500 IU), 10 ml:aan (1000 IU) tai 20 ml:aan (2000 IU) injektionesteisiin käytettävää vettä.
Ennen albumiinin lisäämistä Wilfactin-valmisteen spesifinen aktiivisuus on suurempi tai yhtä suuri kuin 60 IU / VWF:RCo/mg proteiinia.
Von Willebrand -tekijän aktiivisuus (IU) mitataan ristosetiinikofaktorin aktiivisuuden (VWF:RCo) mukaan verrattuna Willebrand-tekijävalmistetta koskevaan kansainväliseen standardiin (WHO).
Ihmisen hyytymistekijän VIII (FVIII) määrä Wilfactin-valmisteessa on ≤ 10 IU/100 IU VWF:RCo.
Tekijä VIII:n aktiivisuus (IU) määritetään Euroopan farmakopean kromogeenisella menetelmällä.
Apuaine, jonka vaikutus tunnetaan:
Tämä lääkevalmiste sisältää natriumia:
- Yksi 5 ml:n injektiopullo (500 IU) sisältää 0,15 mmol (3,4 mg) natriumia.
- Yksi 10 ml:n injektiopullo (1000 IU) sisältää 0,3 mmol (6,9 mg) natriumia.
- Yksi 20 ml:n injektiopullo (2000 IU) sisältää 0,6 mmol (13,8 mg) natriumia
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Wilfactin on tarkoitettu verenvuodon tai leikkaukseen liittyvän vuodon ennaltaehkäisyyn ja hoitoon von Willebrandin tautia (vWD) sairastavilla potilailla, kun pelkkä desmopressiinihoito (DDAVP) yksin on tehoton tai sille on vasta-aiheita.
Wilfactin-valmistetta voidaan käyttää kaikille ikäryhmille.
Wilfactin-valmistetta ei tule käyttää A-hemofilian hoitoon.
Annostus ja antotapa
Veren hyytymishäiriöihin perehtyneen lääkärin tulee valvoa von Willebrandin taudin hoitoa.
Annostus
Yksi IU/kg von Willebrand -tekijää nostaa verenkierron VWF:RCo-tasoa normaalisti 0,02 IU/ml (2 %).
VWF:RCo-tavoitetaso on > 0,6 IU/ml (60 %) ja FVIII:C-tavoitetaso > 0,4 IU/ml (40 %).
Verenvuodon tyrehtyminen edellyttää vähintään hyytymistekijän FVIII aktiivisuuden (FVIII:C) tasoa 0,4 IU/ml (40 %). Yksi von Willebrand ‑tekijäinjektio ei aiheuta FVIII:C-tason enimmäisnousua 6−12 tunnin kuluessa eikä sillä ei voida korjata välittömästi FVIII:C-tasoa. Jos potilaan plasman FVIII:C-perustaso alittaa edellä mainitun kriittisen tason, hyytymistekijä VIII ‑valmistetta tulee antaa von Willebrand ‑tekijän ensimmäisen injektion yhteydessä tilanteissa, joissa vaaditaan verenvuodon nopeaa tyrehdyttämistä (verenvuodon hoitaminen, vakava loukkaantuminen tai hätäleikkaus). Näin saavutetaan plasman hemostaattinen FVIII:C-taso.
Jos FVIII:C-tason välitön nostaminen ei ole tarpeen, esimerkiksi suunnitelluissa leikkauksissa silloin, kun FVIII:C-perustaso riittää varmistamaan verenvuodon tyrehtymisen, FVIII-tekijää ei ehkä tarvitse antaa ensimmäisen VWF-injektion yhteydessä. Lääkäri ratkaisee, annetaanko tekijää.
- Hoidon aloittaminen:
Ensimmäinen annos Wilfactin-valmistetta verenvuodon tai vamman hoidossa on 40–80 IU/kg yhdessä tarvittavan tekijä VIII ‑annoksen kanssa välittömästi ennen toimenpidettä tai mahdollisimman pian vuodon alkamisen tai vakavan loukkaantumisen jälkeen. Riittävän plasman FVIII:C-tason saavuttamiseen tarvittava hyytymistekijä VIII ‑annos lasketaan potilaan plasman FVIII:C-perustason perusteella. Kirurgisissa toimenpiteissä valmiste tulee antaa tunti ennen toimenpiteen alkua.
Wilfactin-valmisteen aloitusannoksena voidaan tarvita 80 IU/kg erityisesti, jos potilaalla on tyypin 3 von Willebrandin tauti, jolloin riittävän tason ylläpitäminen saattaa vaatia suurempia annoksia kuin muissa von Willebrandin tautityypeissä.
Elektiivisessä kirurgiassa Wilfactin-hoito tulee aloittaa 12–24 tuntia ennen toimenpidettä, ja se tulee toistaa tunti ennen toimenpidettä. Tässä tapauksessa tekijä VIII ‑valmistetta ei tarvitse antaa yhdessä valmisteen kanssa, koska endogeeninen FVIII:C-taso on tavallisesti saavuttanut kriittisen tason 0,4 IU/ml (40 %) ennen leikkausta. Tämä tulee kuitenkin varmistaa kunkin potilaan kohdalla.
- Seuraavat injektiot:
Hoitoa tulee tarvittaessa jatkaa sopivalla Wilfactin-annoksella, 40–80 IU/kg päivässä, 1 tai 2 injektiona päivittäin, yhden tai useamman päivän ajan. Annos ja hoidon kesto riippuvat potilaan kliinisestä tilasta, vuodon tyypistä ja vakavuudesta sekä VWF:RCo- ja FVIII:C-pitoisuuksista.
- Pitkäkestoinen estolääkitys
Wilfactin-valmistetta voidaan käyttää pitkäkestoisena estolääkityksenä potilaalle yksilöllisesti määritettyinä annoksina. Wilfactin-annokset 40–60 IU/kg annettuna kahdesta kolmeen kertaa viikossa vähentävät verenvuotokertojen määrää.
- Kotihoito
Kotihoito voidaan aloittaa hoitavan lääkärin hyväksynnällä erityisesti tilanteissa, joissa verenvuoto on vähäistä tai kohtalaista, tai pitkäkestoisessa estolääkityksessä verenvuodon ehkäisyyn. Lääkärin on varmistettava, että potilaalle annetaan asianmukainen koulutus injektion antamiseen, ja hoito on arvioitava uudelleen säännöllisesti määrätyin välein.
Pediatriset potilaat
Kunkin käyttöaiheen mukainen annostus perustuu painoon. Annos ja hoidon kesto on sovitettava potilaan kliiniseen tilaan sekä plasman VWF:RCo- ja FVIII:C-pitoisuuksiin.
-
Hoidon aloitus:
- Alle 6-vuotiailla lapsilla aloitusannos voi perustua potilaan vaiheittaiseen saantoon (IR) tai, jos IR-tietoja ei ole saatavilla, voidaan tarvita aloitusannoksena 60–100 IU/kg tavoitteena nostaa potilaan VWF:RCo-taso arvoon 100 IU/dl.
- Yli 6-vuotiaille lapsille ja nuorille annostus on sama kuin aikuisille potilaille.
-
Seuraavat injektiot:
Lapsilla ja nuorilla myöhemmät annokset on valittava kliinisen tilan ja vWF:RCo-tasojen mukaan ja sovitettava kliiniseen vasteeseen. -
Elektiiviset leikkaukset:
- Alle 6-vuotiaiden lasten elektiivisessä leikkauksessa voidaan antaa uusinta-annos 30 minuuttia ennen toimenpidettä, kun ensimmäinen annos on annettu 12-24 tuntia ennen toimenpidettä.
- Yli 6-vuotiaille lapsille ja nuorille annostus on sama kuin aikuisille potilaille.
-
Profylaksia:
Lapsilla ja nuorilla annos ja uudelleen antamisen tiheys on sovitettava yksilöllisesti potilaan vaiheittaisen toipumisen ja vWF:RCo-tasojen mukaan sekä sovitettava kliiniseen vasteeseen.
Antotapa
Liuota valmiste kohdassa Käyttö- ja käsittelyohjeet kuvatulla tavalla.
Wilfactin-valmiste annetaan laskimoon. Enimmäisantonopeus on 4 ml/minuutti.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille
Varoitukset ja käyttöön liittyvät varotoimet
Vuotaville potilaille suositellaan ensisijaisena hoitona tekijä VIII ‑valmisteen antoa yhdessä sellaisen von Willebrand ‑tekijävalmisteen kanssa, jonka FVIII-pitoisuus on alhainen, erillisellä ruiskulla annettuna.
Yliherkkyys
Kuten muidenkin laskimoon annettavien plasmasta valmistettujen proteiinivalmisteiden kohdalla, yliherkkyysreaktiot ovat mahdollisia. Potilasta tulee seurata tarkasti koko injektion ajan. Potilaalle tulee kertoa yliherkkyysreaktioitten varhaisista merkeistä, joita ovat nokkosihottuma, yleistynyt nokkosihottuma, puristava tunne rinnassa, hengityksen vinkuminen, verenpaineen lasku ja anafylaksia. Jos näitä oireita ilmenee, valmisteen antaminen tulee keskeyttää välittömästi. Anafylaktisen sokin ilmetessä tulee noudattaa normaalia hoitokäytäntöä.
Taudinaiheuttajien siirtyminen
Ihmisen verestä tai plasmasta valmistettujen lääkevalmisteiden käytöstä aiheutuvien infektioiden ehkäisemisessä käytettäviä vakiomenetelmiä ovat luovuttajien valitseminen, luovutetun veren ja plasman infektiovasta-aineiden tutkiminen ja tuotteen valmistaminen siten, että virukset inaktivoituvat tai ne poistetaan. Varotoimenpiteistä huolimatta ihmisen verestä tai plasmasta valmistettujen lääkevalmisteiden antamiseen sisältyvää infektion mahdollisuutta ei voida täysin sulkea pois. Tämä koskee myös tuntemattomia tai uusia viruksia ja muita patogeeneja.
Käytössä olevien varotoimenpiteiden katsotaan tehoavan vaipallisiin viruksiin, kuten ihmisen immuunikatovirukseen (HIV), hepatiitti B -virukseen (HBV) ja hepatiitti C -virukseen (HCV). Näiden toimenpiteiden teho voi olla rajallinen vaipattomiin viruksiin, kuten hepatiitti A -virukseen (HAV) ja parvovirus B19:ään. Parvovirus B19 ‑infektiot saattavat olla vakavia raskaana oleville naisille (sikiön infektio) ja henkilöille, joilla on immuunivaje tai joiden punasolujen muodostus on lisääntynyt (esimerkiksi hemolyyttinen anemia).
Ihmisen plasmasta valmistettua von Willebrand ‑tekijää säännöllisesti saavien potilaiden rokottamista (hepatiitti A ja hepatiitti B) tulee harkita.
On erittäin suositeltavaa kirjata Wilfactin-tuotteen nimi ja eränumero ylös aina, kun valmistetta annetaan potilaalle. Näin potilaan saaman tuotteen erät voidaan jäljittää.
Tromboembolia
Wilfactin on von Willebrand -tekijää sisältävä valmiste, jonka FVIII-pitoisuus on pieni. Valmisteen käyttöön liittyy siitä huolimatta tromboembolisten tapahtumien esiintymisriski, erityisesti potilailla, joilla on tunnettuja kliinisiä tai laboratoriokokein havaittuja riskitekijöitä. Tämän vuoksi riskiryhmän potilaita tulee seurata varhaisten tromboosin oireiden varalta. Laskimoveritulpan estohoito tulee aloittaa voimassa olevien suositusten mukaisesti.
Kun käytetään Wilfactin-valmistetta, hoitavan lääkärin on huomioitava, että pitkäaikainen hoito voi merkittävästi nostaa FVIII:C-pitoisuutta. Sen vuoksi, jos potilas tarvitsee usein Wilfactin-annoksia, erityisesti yhdessä FVIII:aa sisältävän valmisteen kanssa, plasman FVIII:C-pitoisuutta on seurattava, jotta voidaan välttää pitkäaikaiset liian suuret FVIII:C:n pitoisuudet plasmassa, koska ne voivat suurentaa tromboembolisten tapahtumien riskiä.
Immunogeenisuus
Von Willebrandin tautia, erityisesti tyyppiä 3, sairastavilla potilailla saattaa kehittyä neutraloivia vasta-aineita (inhibiittoreita) von Willebrand -tekijälle. Jos odotettua plasman VWF:RCo-aktiivisuustasoa ei saavuteta tai jos verenvuoto ei tyrehdy ohjeen mukaisella annoksella, von Willebrand -tekijän inhibiittorien pitoisuus tulee määrittää asianmukaisilla testeillä. Jos potilaan inhibiittoritasot ovat korkeat, von Willebrand -tekijähoito ei ehkä tehoa. Tällöin tulee harkita muita hoitovaihtoehtoja.
Apuaineen huomioiminen (natriumpitoisuus)
Tämä lääkevalmiste sisältää natriumia.
Potilaiden, joilla on ruokavalion natriumrajoitus, tulee ottaa tämä huomioon, jos annos on suurempi kuin 3300 IU (yli 1 mmol natriumia) (ks. kohta Vaikuttavat aineet ja niiden määrät määrä injektiopulloa kohti).
Yhteisvaikutukset
Ihmisen von Willebrand -tekijävalmisteilla ei ole tunnettuja yhteisvaikutuksia muiden lääkevalmisteiden kanssa.
Raskaus ja imetys
Eläinkokeet eivät ole riittäviä Wilfactin-valmisteen hedelmällisyyttä, lisääntymistä, raskautta, alkion tai sikiön kehitystä tai lapsen syntymän aikaista ja syntymän jälkeistä kehitystä koskevan turvallisuuden arvioimiseksi.
Wilfactin-valmisteen turvallisuutta raskauden ja imetyksen aikana ei ole arvioitu kliinisissä tutkimuksissa.
Tämän vuoksi Wilfactin-valmistetta tulee antaa raskaana oleville ja imettäville von Willebrand ‑tekijän puutosta sairastaville naisille vain, jos siihen on selkeästi aihetta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Vaikutuksia ajokykyyn tai koneiden käyttökykyyn ei ole havaittu.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Seuraavia haittavaikutuksia voi ilmetä Wilfactin-hoidon aikana.
Allergiset reaktiot ja anafylaktiset reaktiot (mukaan lukien harvinaisissa tapauksissa sokki), tromboemboliset tapahtumat (pääasiassa potilailla, joilla on riskitekijöitä), VWF-inhibiittorien muodostuminen ja pistoskohdan reaktiot.
Luettelo haittavaikutuksista
Jäljempänä olevassa taulukossa on yleiskatsaus haittavaikutuksista, jotka on havaittu kuudessa kliinisessä tutkimuksessa ja yhdessä ei-interventionaalisessa myyntiintulon jälkeisessä tutkimuksessa sekä muissa myyntiintulon jälkeisissä lähteissä. Tutkimusten aikana 226 potilasta altistui Wilfactin-valmisteelle yhteensä 16 640 päivän ajan.
Haittavaikutukset on luokiteltu MedDRA-elinluokkajärjestelmän, suositeltavan termin (PT) ja esiintyvyyden mukaan.
Haittavaikutusten esiintyvyys on luokiteltu seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Spontaanisti myyntiluvan myöntämisen jälkeen ilmoitettujen haittavaikutusten esiintyvyydeksi on merkitty tuntematon.
| MedDRAn elinjärjestelmäluokka | Haittavaikutukset (suositeltu termi) | Esiintyvyys |
| Veri ja imukudos | Von Willebrand -tekijän inhibitio* | Tuntematon |
| Immuunijärjestelmä | Yliherkkyys | Melko harvinainen |
| Anafylaktinen sokki* | Tuntematon | |
| Hermosto | Heitehuimaus | Melko harvinainen |
| Parestesia, hypoestesia | Melko harvinainen | |
| Verisuonisto | Kuumat aallot | Melko harvinainen |
| Tromboemboliset tapahtumat* | Tuntematon | |
| Iho ja ihonalainen kudos | Kutina | Melko harvinainen |
| Yleisoireet ja antopaikassa todettavat haitat | Antopaikan reaktiot** (mukaan lukien infuusiokohdan reaktio, infuusiokohdan inflammaatio ja verisuonen pistokohdan inflammaatio) | Yleinen |
| Painostava tunne | Melko harvinainen | |
| Vilunväristykset, palelu | Melko harvinainen | |
| Kuume* | Tuntematon |
* Raportoitu myyntiluvan myöntämisen jälkeisessä käytössä/seurannassa, esiintyvyys tuntematon vakiintuneen käytännön mukaisesti.
** MedDRAn ylätason ryhmätermit.
Valikoitujen haittavaikutusten kuvaus
Yliherkkyysreaktioita tai allergisia reaktioita (näihin saattaa kuulua angioedeemaa, infuusiokohdan polttelua ja pistelyä, vilunväristyksiä, punoitusta, yleistynyttä nokkosihottumaa, päänsärkyä, nokkosihottumaa, verenpaineen laskua, tajunnan menetystä / huonovointisuutta, letargiaa, pahoinvointia, levottomuutta, takykardiaa, puristavaa tunnetta rinnassa, kihelmöintiä, oksentelua, hengityksen vinkumista) on joskus havaittu. Nämä reaktiot voivat joissakin tapauksissa johtaa vaikeaan anafylaksiaan (mukaan lukien sokkiin).
Von Willebrandin tautia, erityisesti tyyppiä 3, sairastavilla potilailla saattaa erittäin harvoin kehittyä neutraloivia vasta-aineita (inhibiittoreita) von Willebrand -tekijälle. Jos inhibiittoreita ilmenee, potilaan hoitovaste on alentunut. Vasta-aineiden esiintyminen saattaa liittyä kiinteästi anafylaktisiin reaktioihin. Tämän vuoksi inhibiittoritasot tulee arvioida potilailla, joilla ilmenee anafylaktisia reaktioita.
Näissä tapauksissa on suositeltavaa ottaa yhteys verenvuotohäiriöihin erikoistuneeseen hoitopaikkaan.
Wilfactin on von Willebrand -tekijää sisältävä valmiste, jonka FVIII-pitoisuus on alhainen. Valmisteen käyttöön liittyy siitä huolimatta tromboembolisten tapahtumien esiintymisriski erityisesti potilailla, joilla on tunnettuja kliinisiä tai laboratoriokokeiden perusteella havaittuja riskitekijöitä. Sen vuoksi riskipotilaita on tarkkailtava.
Valmisteen infektioturvallisuustiedot, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Pediatrinen populaatio
Wilfactin-valmiste arvioitiin 56 alle 18-vuotiaalla potilaalla, joista 23 oli alle 6-vuotiaita, 21 potilasta oli 6–11-vuotiaita ja 12 potilasta yli 11-vuotiaita.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Von Willebrand -tekijän yliannostusoireita ei ole raportoitu. Tromboembolisia tapahtumia saattaa kuitenkin esiintyä suurissa yliannostuksissa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä:
hemostaatit, veren hyytymistekijät, von Willebrand ‑tekijä
ATC-koodi: B02BD10
Vaikutusmekanismi
Wilfactin toimii samalla tavalla kuin endogeeninen von Willebrand ‑tekijä.
Von Willebrand -tekijällä voidaan korjata verenvuodon hyytymiseen liittyviä häiriöitä potilailla, joilla on kyseisen tekijän puutostila (von Willebrandin tauti). Vaikutus tapahtuu kahdella tasolla:
- Von Willebrand -tekijä palauttaa verihiutaleiden kiinnittymiskyvyn verisuonen vauriokohdan endoteeliin (se sitoutuu sekä verisuonen endoteeliin että verihiutaleen kalvoon) ja mahdollistaa primaarin hemostaasin. Tämä näkyy vuotoajan lyhentymisenä. Tämä vaikutus ilmenee heti ja riippuu vaikuttavan aineen multimerisaatiotasosta.
- Von Willebrand -tekijä korjaa viiveellä tapahtumaan liittyvän tekijä VIII ‑vajauksen. Laskimoon annettuna Von Willebrand -tekijä sitoutuu endogeeniseen FVIII-tekijään (jota potilaalla syntyy normaalisti) ja vakauttamalla tämän tekijän estää sen nopean hajoamisen. Tämän vuoksi pelkän VWF-tekijän (VWF-tuote, jonka FVIII-taso on alhainen) antaminen palauttaa sekundaarisena vaikutuksena FVIII:C-tason normaaliksi ensimmäisen infuusion jälkeen. FVIII:C-tekijää sisältävän VWF-valmisteen anto palauttaa FVIII:C-tason normaaliksi välittömästi ensimmäisen infuusion jälkeen.
Farmakokinetiikka
Wilfactin-valmisteen farmakokineettinen tutkimus toteutettiin kahdeksalla tyypin 3 von Willebrandin tautia sairastavalla aikuispotilaalla. Tutkimus osoitti seuraavat tiedot VWF:RCo-pitoisuudesta:
- Keskimääräinen plasman pitoisuuspinta-ala ajasta nolla äärettömään (AUC0-∞) on 3444 IU∙h/dl Wilfactin-valmisteen 100 IU/kg kerta-annoksen jälkeen.
- Keskimääräinen annosvaste on 2,1 IU/dl/IU/kg injektiona.
- Puoliintumisaika on 8–14 tuntia, keskimääräinen puoliintumisaika on 12 tuntia.
- Keskimääräinen puhdistuma on 3,0 ml/h/kg.
Von Willebrand -tekijän huippupitoisuus plasmassa saavutetaan tavallisesti 30–60 minuutin kuluessa injektion jälkeen.
FVIII-tason normalisoituminen on progressiivista ja vaihtelevaa, ja normaalin tason saavuttaminen kestää yleensä 6–12 tuntia. Tämä vaikutus säilyy 2–3 päivän ajan.
FVIII-tason nousu tapahtuu progressiivisesti. Taso palautuu normaaliksi 6–12 tunnin kuluttua. FVIII-tason keskimääräinen nousu on 6 % (IU/dl) tunnissa. Täten myös potilailla, joiden FVIII:C-alkutaso on alle 5 % (IU/dl), FVIII:C-taso nousee noin 40 %:iin (IU/dl) kuuden tunnin kuluttua injektiosta ja säilyy tällä tasolla 24 tunnin ajan.
Pediatriset tiedot
Kattavia farmakokinetiikkatietoja (Cmax, Tmax, AUC, puhdistuma, puoliintumisaika ja keskimääräinen kehossa olon aika) Wilfactin-injektion jälkeen ei ole määritetty pediatrisilla potilailla eli alle 18-vuotiailla.
Seitsemällä alle 6-vuotiaalla lapsella (kaksi lasta iältään 28 vrk–23 kk ja viisi lasta iältään 24 kk–6 v), joilla oli vaikea von Willebrandin taudin muoto (viidellä tyypin 3, yhdellä tyypin 1 ja yhdellä tyypin 2), havaittiin 15 minuutin kuluttua infuusiosta määrällä 101,1 ± 5,0 IU/kg, vaiheittainen VWF:RCo:n saanto 1,75 ± 0,35 (IU/dl)/(IU/kg). Yksilöiden välinen vaihtelu oli suurta (vaihteluväli 1,14–2,03). Vain neljälle näistä lapsista tehtiin sekä alkuvaiheen testaus että 6 kuukauden saannon kontrollitestaus 3–9 hoitopäivän altistuksen jälkeen. Todettu keskimääräinen saantosuhde oli 0,87 ± 0,12 (IU/dl)/(IU/kg) (vaihteluväli 0,7–1,0).
Prekliiniset tiedot turvallisuudesta
Useista eläimillä tehdyistä prekliinisistä tutkimuksista saatujen tietojen perusteella Wilfactin-valmisteen ei ole osoitettu aiheuttavan muita toksisia vaikutuksia kuin niitä, jotka aiheutuvat laboratorioeläinten immunogeenisuudesta ihmisen proteiineille. Toistuvan annoksen toksisuustestejä ei voida tehdä, koska eläimille kehittyy vasta-ainetta vieraaseen proteiiniin.
Prekliiniset tiedot turvallisuudesta eivät osoita, että Wilfactin-valmisteella olisi mutageenisia vaikutuksia.
Farmaseuttiset tiedot
Apuaineet
Injektiokuiva-aine:
ihmisen albumiini
arginiinihydrokloridi
glysiini
natriumsitraatti ja
kalsiumkloridi.
Liuotin:
injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Wilfactin-valmistetta ei saa sekoittaa samassa ruiskussa muiden lääkevalmisteiden kanssa, lukuun ottamatta LFB-BIOMEDICAMENTS-yhtiön valmistamaa plasmasta valmistettua hyytymistekijää VIII, jonka kanssa yhteensopivuustutkimus on tehty. Tätä hyytymistekijä VIII -valmistetta ei kuitenkaan ole myynnissä kaikissa maissa.
Vain hyväksyttyjä polypropeeni-injektiovälineitä tulee käyttää, koska ihmisen von Willebrand ‑tekijän adsorptio joidenkin injektiovälineiden pintaan saattaa aiheuttaa hoidon epäonnistumisen.
Kestoaika
3 vuotta.
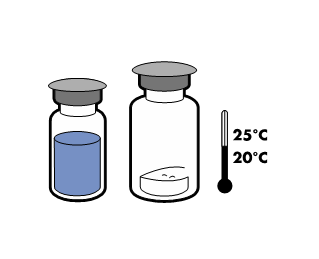
Valmisteen on osoitettu säilyvän kemiallisesti ja fysikaalisesti stabiilina 24 tunnin ajan 25 °C:ssa.
Mikrobiologiselta kannalta valmiste on käytettävä välittömästi.
Säilytys
Säilytä alle 25 °C. Säilytä alkuperäispakkauksessa. Herkkä valolle.
Ei saa jäätyä.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
WILFACTIN injektiokuiva-aine ja liuotin, liuosta varten
100 IU/ml (L:ei) 1000 IU + 10 ml (996,85 €)
PF-selosteen tieto
Injektiokuiva-aine injektiopullossa (tyypin I lasia), jossa on tulppa (bromobutyylikumia) ja suojakorkki + 5 ml liuotinta injektiopullossa (tyypin I tai II lasia), jossa on tulppa (bromobutyylikumia tai klorobutyylikumia) ja suojakorkki; sekä siirtolaite – 1 annoksen pakkauskoko.
Injektiokuiva-aine injektiopullossa (tyypin I lasia), jossa on tulppa (bromobutyylikumia) ja suojakorkki + 10 ml liuotinta injektiopullossa (tyypin I tai II lasia), jossa on tulppa (bromobutyylikumia) ja suojakorkki; sekä siirtolaite – 1 annoksen pakkauskoko.
Injektiokuiva-aine injektiopullossa (tyypin I lasia), jossa on tulppa (bromobutyylikumia) ja suojakorkki + 20 ml liuotinta injektiopullossa (tyypin I tai II lasia), jossa on tulppa (bromobutyylikumia tai klorobutyylikumia) ja suojakorkki; sekä siirtolaite – 1 annoksen pakkauskoko.
Valmisteen kuvaus:
Kuiva-aine: valkoinen tai vaaleankeltainen, kuiva-aine tai mureneva kiinteä kuiva-aine.
Liuotin: kirkas ja väritön.
Käyttö- ja käsittelyohjeet
Käyttövalmiiksi saattaminen
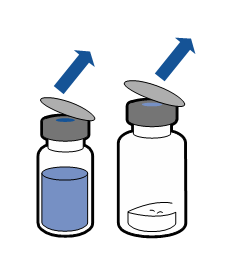
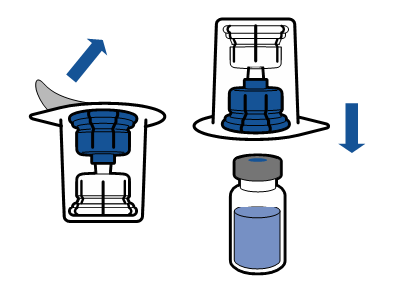
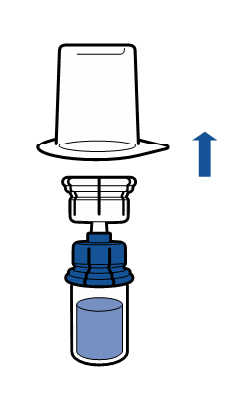
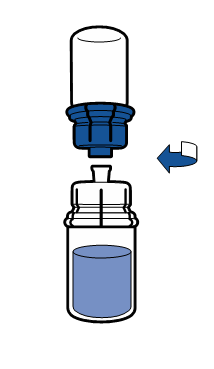
Noudata aseptista toimintatapaa koskevia voimassa olevia ohjeita. Siirtolaite on tarkoitettu ainoastaan lääkevalmisteen käyttökuntoon saattamiseen, kuten jäljempänä on kuvattu. Sitä ei ole tarkoitettu lääkevalmisteen antamiseen potilaalle.
 |
|
 |
|
 |
|
 |
|
|
|
 |
|
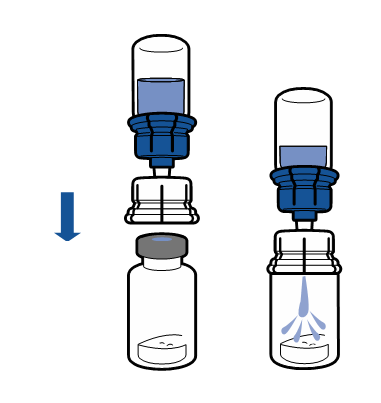
Injektiokuiva-aine liukenee yleensä heti, ja sen pitäisi olla liuennut alle 5 minuutissa.
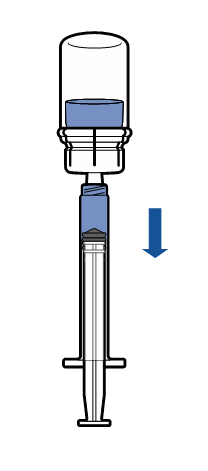
Lääkkeen antaminen
 |
|
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Käyttökuntoon saatettu valmiste pitää tarkistaa ennen antoa silmämääräisesti hiukkasten ja värimuutosten varalta. Liuoksen tulee olla kirkasta tai hieman opalisoivaa, väritöntä tai hieman kellertävää. Älä käytä liuosta, jos se on sameaa tai siinä on sakkaa.
Korvattavuus
WILFACTIN injektiokuiva-aine ja liuotin, liuosta varten
100 IU/ml 1000 IU + 10 ml
- Ylempi erityiskorvaus (100 %). Von Willebrand -tekijävalmiste ja vonikogi alfa: Kroonisten hyytymishäiriöiden hoito erityisin edellytyksin (161).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Von Willebrand -tekijävalmiste ja vonikogi alfa: von Willebrandin taudin hoito erityisin edellytyksin (357).
ATC-koodi
B02BD10
Valmisteyhteenvedon muuttamispäivämäärä
19.01.2024
Yhteystiedot
 PROTHYA BIOSOLUTIONS FINLAND OY
PROTHYA BIOSOLUTIONS FINLAND OY Lars Sonckin kaari 14
02600 Espoo
09 6120 9122
www.prothya.com
info@prothya.fi