
ADCETRIS kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 50 mg
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 50 mg brentuksimabivedotiinia.
Käyttökuntoon saattamisen (ks. kohta Käyttö- ja käsittelyohjeet) jälkeen yksi ml liuosta sisältää 5 mg brentuksimabivedotiinia.
ADCETRIS on vasta‑aineen ja lääkeaineen konjugaatti, joka koostuu CD30‑antigeeniin kohdistuvasta monoklonaalisesta vasta‑aineesta (rekombinantti kimeerinen immunoglobuliini G1 [IgG1], joka valmistetaan yhdistelmä‑DNA‑tekniikalla kiinanhamsterin munasarjasoluissa), joka kytketään kovalenttisesti mikrotubulustoimintaan vaikuttavaan monometyyliauristatiini E:hen (MMAE).
Apuaineet, joiden vaikutus tunnetaan:
Yksi injektiopullo sisältää noin 13,2 mg natriumia.
Yksi injektiopullo sisältää noin 2 mg polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva‑aine välikonsentraatiksi infuusionestettä varten, liuos.
Kliiniset tiedot
Käyttöaiheet
Hodgkinin lymfooma
ADCETRIS on tarkoitettu aiemmin hoitamatonta, CD30‑positiivista levinneisyysasteen III tai IV Hodgkinin lymfoomaa sairastavien aikuispotilaiden hoitoon yhdessä doksorubisiinin, vinblastiinin ja dakarbatsiinin (AVD) kanssa (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
ADCETRIS on tarkoitettu aiemmin hoitamatonta, CD30-positiivista levinneisyysasteen IIB Hodgkinin lymfoomaa, johon liittyy riskitekijöitä, tai levinneisyysasteen III tai IV Hodgkinin lymfoomaa sairastavien aikuispotilaiden hoitoon yhdessä etoposidin, syklofosfamidin, doksorubisiinin, dakarbatsiinin ja deksametasonin (BrECADD) kanssa (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
ADCETRIS on tarkoitettu CD30‑positiivista Hodgkinin lymfoomaa sairastavien aikuispotilaiden hoitoon tilanteissa, joissa taudin uusiutumisen tai etenemisen riski on suurentunut autologisen kantasolusiirron (ASCT) jälkeen (ks. kohta Farmakodynamiikka).
Adcetris on tarkoitettu uusiutunutta tai refraktaarista CD30‑positiivista Hodgkinin lymfoomaa (HL) sairastavien aikuispotilaiden hoitoon:
- autologisen kantasolusiirron (ASCT) jälkeen tai
- vähintään kahden aiemman hoidon jälkeen, jos autologista kantasolusiirtoa (ASCT) tai useammalla lääkeaineella toteutettavaa sytostaattihoitoa ei voida tehdä.
Systeeminen anaplastinen suurisoluinen lymfooma
ADCETRIS on tarkoitettu aiemmin hoitamatonta systeemistä anaplastista suurisoluista lymfoomaa (sALCL) sairastavien aikuispotilaiden hoitoon yhdessä syklofosfamidin, doksorubisiinin ja prednisonin (CHP) kanssa (ks. kohta Farmakodynamiikka).
ADCETRIS on tarkoitettu uusiutunutta tai refraktaarista systeemistä anaplastista suurisoluista lymfoomaa sairastavien aikuispotilaiden hoitoon.
Ihon T‑solulymfooma
ADCETRIS on tarkoitettu CD30‑positiivista ihon T‑solulymfoomaa (CTCL) sairastavien aikuispotilaiden hoitoon vähintään yhden aiemman systeemisen hoidon jälkeen (ks. kohta Farmakodynamiikka).
Ehto
Valmiste on annettava syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
ADCETRIS on annettava syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa.
Annostus
Aiemmin hoitamattoman Hodgkinin lymfooman hoito
ADCETRIS + AVD
Suositusannos yhdistettynä solunsalpaajahoitoon (doksorubisiini [A], vinblastiini [V] ja dakarbatsiini [D] [AVD]) on 1,2 mg/kg 30 minuuttia kestävänä laskimoinfuusiona kunkin 28‑päiväisen hoitojakson päivinä 1 ja 15 yhteensä 6 hoitojakson ajan (ks. kohta Farmakodynamiikka).
Primaariprofylaksia kasvutekijävalmisteella (G‑CSF) ensimmäisestä annoksesta alkaen suositellaan kaikille aiemmin hoitamattomille, yhdistelmähoitoa saaville Hodgkinin lymfoomaa sairastaville aikuispotilaille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tutustu ADCETRIS‑valmisteen kanssa annettavien, aiemmin hoitamattoman Hodgkinin lymfooman hoitoon käytettävien solunsalpaajien valmisteyhteenvetoihin.
BrECADD
Suositusannos yhdistettynä solunsalpaajahoitoon (etoposidi [E], syklofosfamidi [C], doksorubisiini [A], dakarbatsiini [D], deksametasoni [D] [BrECADD]) on 1,8 mg/kg 30 minuuttia kestävänä laskimoinfuusiona kolmen viikon välein yhteensä enintään kuuden hoitojakson ajan (ks. kohta Farmakodynamiikka).
Primaariprofylaksia kasvutekijävalmisteella (G-CSF) on annettava jokaisen hoitojakson päivästä 5 alkaen kaikille aiemmin hoitamattomille, yhdistelmähoitoa saaville Hodgkinin lymfoomaa sairastaville aikuispotilaille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Esihoitoa deksametasonilla neljän päivän ajan ennen ensimmäistä solunsalpaajahoitojaksoa suositellaan > 40-vuotiaille potilaille lääkärin harkinnan mukaan.
Antibioottiprofylaksia on annettava kolme kertaa viikossa koko solunsalpaajahoidon ajan.
Katso ADCETRIS-valmisteen kanssa annettavien, aiemmin hoitamattoman Hodgkinin lymfooman hoitoon käytettävien solunsalpaajien annossuositukset taulukosta 4.
Hodgkinin lymfooma ja suurentunut uusiutumisen tai etenemisen riski
Suositusannos on 1,8 mg/kg 30 minuuttia kestävänä laskimoinfuusiona kolmen viikon välein.
ADCETRIS‑hoito aloitetaan potilaan toivuttua autologisesta kantasolusiirrosta kliinisen arvion mukaan. Näille potilaille annetaan enintään 16 hoitojaksoa (ks. kohta Farmakodynamiikka).
Uusiutunut tai refraktaarinen Hodgkinin lymfooma
Suositusannos on 1,8 mg/kg 30 minuuttia kestävänä laskimoinfuusiona 3 viikon välein.
Suositeltu aloitusannos sellaisten potilaiden uudelleenhoidossa, jotka ovat aiemmin saaneet vasteen ADCETRIS‑hoitoon, on 1,8 mg/kg 30 minuuttia kestävänä laskimoinfuusiona 3 viikon välein. Vaihtoehtoisesti hoito voidaan aloittaa käyttämällä viimeisintä siedettyä annosta (ks. kohta Farmakodynamiikka).
Hoitoa jatketaan, kunnes tauti etenee tai ilmenee sietämätöntä toksisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos hoidolla saavutetaan taudin etenemisen pysähtyminen tai tätä parempi tulos, hoitoa annetaan vähintään 8 hoitojaksoa, mutta enintään 16 hoitojaksoa (noin 1 vuoden ajan) (ks. kohta Farmakodynamiikka).
Aiemmin hoitamattoman systeemisen anaplastisen suurisoluisen lymfooman hoito
Suositusannos yhdistettynä solunsalpaajahoitoon (syklofosfamidi [C], doksorubisiini [H] ja prednisoni [P] [CHP]) on 1,8 mg/kg 30 minuuttia kestävänä laskimoinfuusiona 3 viikon välein yhteensä 6–8 hoitojakson ajan (ks. kohta Farmakodynamiikka).
Primaariprofylaksia kasvutekijävalmisteella (G‑CSF) ensimmäisestä annoksesta alkaen suositellaan kaikille aiemmin hoitamattomille, yhdistelmähoitoa saaville systeemistä anaplastista suurisoluista lymfoomaa sairastaville aikuispotilaille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tutustu ADCETRIS‑valmisteen kanssa annettavien, aiemmin hoitamattoman systeemisen anaplastisen suurisoluisen lymfooman hoitoon käytettävien solunsalpaajien valmisteyhteenvetoihin.
Uusiutunut tai refraktaarinen systeeminen anaplastinen suurisoluinen lymfooma
Suositusannos on 1,8 mg/kg 30 minuuttia kestävänä laskimoinfuusiona kolmen viikon välein.
Suositeltava uusintahoidon aloitusannos potilailla, joilla ADCETRIS‑hoito on aiemmin tuottanut vasteen, on 1,8 mg/kg 30 minuuttia kestävänä laskimoinfuusiona kolmen viikon välein. Vaihtoehtoisesti hoito voidaan aloittaa käyttämällä viimeisintä siedettyä annosta (ks. kohta Farmakodynamiikka).
Hoitoa jatketaan, kunnes tauti etenee tai ilmenee sietämätöntä toksisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilaille, joiden taudin eteneminen pysähtyy tai joilla saavutetaan tätä parempi hoitotulos, annetaan vähintään 8 hoitojaksoa ja enintään 16 hoitojaksoa (noin 1 vuoden hoito) (ks. kohta Farmakodynamiikka).
Ihon T‑solulymfooma
Suositusannos on 1,8 mg/kg 30 minuuttia kestävänä laskimoinfuusiona kolmen viikon välein.
Ihon T‑solulymfoomaa sairastaville potilaille annetaan enintään 16 hoitojaksoa (ks. kohta Farmakodynamiikka).
Yleistä
Jos potilas painaa yli 100 kg, annos lasketaan 100 kg painavalle potilaalle (ks. kohta Käyttö- ja käsittelyohjeet).
Täydellinen verenkuva on tarkistettava ennen jokaista lääkeannosta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilaan vointia on seurattava infuusion aikana ja sen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annoksen muuttaminen
Neutropenia
Jos hoidon aikana kehittyy neutropeniaa, se hoidetaan annosväliä pidentämällä tai muuttamalla annosta seuraavilla hoitojaksoilla. Katso asianmukaiset monoterapian ja yhdistelmähoidon annossuositukset taulukoista 1,2,3 ja 4 (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukko 1: Annossuositukset neutropenian yhteydessä, monoterapia
Neutropenian vaikeusaste (oireet ja löydökset [CTCAE‑kriteeriena lyhyt kuvaus]) | Antoaikataulun muutos |
Aste 1 (< LLN–1 500/mm3 < LLN–1,5 x 109/l) tai Aste 2 (< 1 500–1 000/mm3 < 1,5–1,0 x 109/l) | Jatketaan samalla annoksella ja antoaikataululla. |
Aste 3 (< 1 000–500/mm3 < 1,0–0,5 x 109/l) tai Aste 4 (< 500/mm3 < 0,5 x 109/l) | Lääkkeen anto keskeytetään, kunnes toksisuus palautuu tasolle ≤ Aste 2 tai lähtötasolle, minkä jälkeen hoitoa jatketaan samalla annoksella ja antoaikataulullab. G‑CSF‑ tai GM‑CSF‑valmisteen antamista myöhempien hoitojaksojen yhteydessä on harkittava, jos potilaalle kehittyy asteen 3 tai asteen 4 neutropenia. |
a. Asteikko perustuu Yhdysvaltain kansallisen syöpäinstituutin (NCI) CTCAE‑kriteereihin (Common Terminology Criteria for Adverse Events, versio 3.0); ks. neutrofiilit/granulosyytit; LLN (Lower Limit of Normal) = viitevälin alaraja
b. Jos potilaalle kehittyy asteen 3 tai asteen 4 lymfopenia, hoitoa voidaan jatkaa keskeytyksettä.
Taulukko 2: Annossuositukset neutropenian yhteydessä, AVD/CHP-yhdistelmähoito
Neutropenian vaikeusaste (oireet ja löydökset [CTCAE‑kriteeriena lyhyt kuvaus]) | Antoaikataulun muutos |
Aste 1 (< LLN–1 500/mm3 < LLN–1,5 x 109/l) tai Aste 2 (< 1 500–1 000/mm3 < 1,5–1,0 x 109/l) tai Aste 3 (< 1 000–500/mm3 < 1,0–0,5 x 109/l) tai Aste 4 (< 500/mm3 < 0,5 x 109/l) | G‑CSF‑primaariprofylaksia ensimmäisestä annoksesta alkaen suositellaan kaikille yhdistelmähoitoa saaville aikuispotilaille. Jatketaan samalla annoksella ja antoaikataululla. |
a. Asteikko perustuu Yhdysvaltain kansallisen syöpäinstituutin (NCI) CTCAE‑kriteereihin (Common Terminology Criteria for Adverse Events, versio 4.03); ks. neutrofiilit/granulosyytit; LLN (Lower Limit of Normal) = viitevälin alaraja
Vaikeusaste (oireet ja löydökset [CTCAE‑kriteeriena lyhyt kuvaus]) | Antoaikataulun muutos |
Leukosyytit ≥ 2 500/mm3 Neurofiilit ≥ 1 500/mm3 JA Trombosyytit ≥ 80 000/mm3 | Jatketaan samalla annoksella ja aikataululla. |
Leukosyytit < 2 000–1 000/mm3 Neutrofiilit < 1 000–500/mm3 JA Trombosyytit < 50 000–25 000/mm3 | Lääkkeen anto keskeytetään, kunnes toksisuus palautuu lähtötasolle. Elleivät haittatapahtumat poistu hoitojakson päivään 28 mennessä, voidaan harkita brentuksimabivedotiiniannoksen pienentämistä tasolle 1,2 mg/kg, enintään 120 mg, 3 kolmen viikon välein. |
Leukosyytit < 1 000/mm3 Neutrofiilit < 500/mm3 JA Trombosyytit < 25 000/mm3 | Lääkkeen anto keskeytetään, kunnes toksisuus palautuu lähtötasolle. Brentuksimabivedotiinihoitoa jatketaan sen jälkeen pienennetyllä annoksella 1,2 mg/kg, enintään 120 mg, 3 kolmen viikon välein. |
-
Asteikko perustuu Yhdysvaltain kansallisen syöpäinstituutin (NCI) CTCAE-kriteereihin (Common Terminology Criteria for Adverse Events, versio 4.0); ks. neutrofiilit/granulosyytit; LLN (Lower Limit of Normal) = viitevälin alaraja.
Jos BrECADD-hoitoa saavilla potilailla ilmenee yksi tai useampi haittatapahtuma tietyn hoitojakson aikana, annosta pienennetään yhtä tasoa alemmaksi, jolla jatketaan seuraavien hoitojaksojen ajan.
Jos haittatapahtumia ilmenee kahden peräkkäisen hoitojakson aikana, annos pienennetään lähtötasolle (ks. taulukko 4). Haittatapahtumiin kuuluvat yli neljä päivää kestävä leukopenia, trombosytopenia yhtenä tai useampana päivänä, CTCAE-kriteerien mukaisesti asteen 4 infektio, muut CTCAE-kriteerien mukaiset asteen 4 toksisuudet ja hoidon viivästyminen yli kaksi viikkoa veriarvojen puutteellisen palautumisen vuoksi.
Taulukko 4: Aloitusannos ja annoksen alennupienennystasot, BrECADD-hoito
| Annos-taso | syklofosfamidi (C) | doksorubisiini (A) | etoposidi (E) | dakarbatsiini (D) | deksametasoni (D) |
| 4 (aloitus-annos) | 1 250 mg/m2 | 40 mg/m2 | 150 mg/m2 | 250 mg/m2 | 40 mg |
| 3 | 1 100 mg/m2 | 40 mg/m2 | 125 mg/m2 | 250 mg/m2 | 40 mg |
| 2 | 950 mg/m2 | 40 mg/m2 | 100 mg/m2 | 250 mg/m2 | 40 mg |
| 1 | 800 mg/m2 | 40 mg/m2 | 100 mg/m2 | 250 mg/m2 | 40 mg |
| Lähtötaso (pienin annos) | 650 mg/m2 | 35 mg/m2 | 100 mg/m2 | 250 mg/m2 | 40 mg |
Perifeerinen neuropatia
Jos hoidon aikana ilmenee perifeeristä sensorista tai motorista neuropatiaa tai se pahenee, katso asianmukaiset monoterapian ja yhdistelmähoidon annossuositukset taulukoista 5 ja 6 (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukko 5: Annossuositukset ensimmäistä kertaa ilmenevän tai pahenevan perifeerisen sensorisen tai motorisen neuropatian yhteydessä, monoterapia
Perifeerisen sensorisen tai motorisen neuropatian vaikeusaste (oireet ja löydökset [CTCAE‑kriteeriena lyhyt kuvaus]) | Annoksen ja antoaikataulun muutos |
| Aste 1 (parestesia ja/tai refleksien heikkeneminen; ei vaikutusta toimintakykyyn) | Jatketaan samalla annoksella ja antoaikataululla. |
| Aste 2 (vaikuttaa toimintakykyyn, mutta ei päivittäisistä toimista suoriutumiseen) | Lääkkeen anto keskeytetään, kunnes toksisuus palautuu tasolle ≤ Aste 1 tai lähtötasolle, minkä jälkeen hoito aloitetaan uudelleen pienemmällä annoksella (1,2 mg/kg, enintään 120 mg, kolmen viikon välein). |
| Aste 3 (vaikuttaa päivittäisistä toimista suoriutumiseen) | Lääkkeen anto keskeytetään, kunnes toksisuus palautuu tasolle ≤ Aste 1 tai lähtötasolle, minkä jälkeen hoito aloitetaan uudelleen pienemmällä annoksella (1,2 mg/kg, enintään 120 mg kolmen viikon välein). |
| Aste 4 (toimintakykyä heikentävä sensorinen neuropatia tai henkeä uhkaava tai halvaantumiseen johtava motorinen neuropatia) | Hoito lopetetaan. |
a. Asteikko perustuu Yhdysvaltain kansallisen syöpäinstituutin (NCI) CTCAE‑kriteereihin (Common Terminology Criteria for Adverse Events, versio 3.0); ks. neuropatia: motorinen neuropatia, sensorinen neuropatia ja neuropaattinen kipu.
Taulukko 6: Annossuositukset ensimmäistä kertaa ilmenevän tai pahenevan perifeerisen sensorisen tai motorisen neuropatian yhteydessä, yhdistelmähoito
| Yhdistelmähoito ADCETRIS + AVD ‑yhdistelmällä | Yhdistelmähoito ADCETRIS + CHP ‑yhdistelmällä | Yhdistelmähoito, BrECADD | |
Perifeerisen sensorisen tai motorisen neuropatian vaikeusaste (oireet ja löydökset [CTCAE‑kriteeriena lyhyt kuvaus]) | Annoksen ja antoaikataulun muutos | Annoksen ja antoaikataulun muutos | Annoksen ja antoaikataulun muutos |
| Aste 1 (parestesia ja/tai refleksien heikkeneminen; ei vaikutusta toimintakykyyn) | Jatketaan samalla annoksella ja antoaikataululla. | Jatketaan samalla annoksella ja antoaikataululla. | Jatketaan samalla annoksella ja antoaikataululla. |
Aste 2 (vaikuttaa toimintakykyyn, mutta ei päivittäisistä toimista suoriutumiseen)
| Annos pienennetään tasolle 0,9 mg/kg, enintään 90 mg 2 viikon välein. | Sensorinen neuropatia: Jatketaan hoitoa samalla annoksella.
Motorinen neuropatia: Annos pienennetään tasolle 1,2 mg/kg, enintään 120 mg 3 viikon välein. | Lääkkeen anto keskeytetään, kunnes oireet ovat lieventyneet tasolle ≤ Aste 1 tai lähtötasolle, minkä jälkeen brentuksimabivedotiini-hoitoa jatketaan pienennetyllämmällä annoksella (1,2 mg/kg, enintään 120 mg, 3 kolmen viikon välein).
|
| Aste 3 (vaikuttaa päivittäisistä toimista suoriutumiseen) | ADCETRIS‑valmisteen anto keskeytetään, kunnes toksisuus palautuu tasolle ≤ aste 2, minkä jälkeen hoito aloitetaan uudelleen pienemmällä annoksella (0,9 mg/kg, enintään 90 mg kahden viikon välein). | Sensorinen neuropatia: Annos pienennetään tasolle 1,2 mg/kg, enintään 120 mg 3 viikon välein.
Motorinen neuropatia: Hoito lopetetaan. | |
| Aste 4 (toimintakykyä heikentävä sensorinen neuropatia tai henkeä uhkaava tai halvaantumiseen johtava motorinen neuropatia) | Hoito lopetetaan. | Hoito lopetetaan. | Hoito lopetetaan. |
a. Asteikko perustuu Yhdysvaltain kansallisen syöpäinstituutin (NCI) CTCAE‑kriteereihin (Common Terminology Criteria for Adverse Events, versio 4.03); ks. neuropatia: motorinen neuropatia, sensorinen neuropatia ja neuropaattinen kipu
Erityiset potilasryhmät
Munuaisten ja maksan vajaatoiminta
Yhdistelmähoito
Munuaisten vajaatoimintapotilaita tulee seurata tarkasti haittatapahtumien varalta. Kliinisissä tutkimuksissa ei ole saatu kokemusta ADCETRIS‑valmisteen käytöstä yhdessä solunsalpaajien kanssa munuaisten vajaatoimintapotilaiden hoitoon tilanteessa, jossa seerumin kreatiniinipitoisuus on ≥ 2,0 mg/dl ja/tai kreatiniinipuhdistuma tai laskennallinen kreatiniinipuhdistuma on ≤ 40 ml/min. ADCETRIS‑valmisteen käyttöä yhdessä solunsalpaajien kanssa on vältettävä, jos potilaalla on vaikea munuaisten vajaatoiminta.
Maksan vajaatoimintapotilaita tulee seurata tarkasti haittatapahtumien varalta. Lievää maksan vajaatoimintaa sairastavien potilaiden hoidossa suositeltava aloitusannos on 0,9 mg/kg 30 minuuttia kestävänä laskimoinfuusiona kahden viikon välein, kun potilaat saavat ADCETRIS‑valmistetta yhdessä AVD‑hoidon kanssa. Suositeltava aloitusannos lievää maksan vajaatoimintaa sairastavien potilaiden hoidossa on 1,2 mg/kg 30 minuuttia kestävänä laskimoinfuusiona kolmen viikon välein, kun potilaat saavat ADCETRIS‑valmistetta yhdessä CHP‑hoidon kanssa. Kliinisissä tutkimuksissa ei ole saatu kokemusta ADCETRIS‑valmisteen käytöstä yhdessä solunsalpaajien kanssa maksan vajaatoimintapotilaiden hoitoon tilanteessa, jossa kokonaisbilirubiinipitoisuus on > 1,5 x normaaliarvojen yläraja (ULN) (ellei syynä ole Gilbertin oireyhtymä) tai aspartaattiaminotransferaasi (ASAT) tai alaniiniaminotransferaasi (ALAT) on > 3 x ULN tai, mikäli arvojen nousun voidaan kohtuullisesti olettaa johtuvan Hodgkinin lymfooman maksa‑affisiosta, > 5 x ULN. ADCETRIS‑valmisteen käyttöä yhdessä solunsalpaajien kanssa on vältettävä, jos potilaalla on keskivaikea tai vaikea maksan vajaatoiminta.
Monoterapia
Jos potilas sairastaa vaikeaa munuaisten vajaatoimintaa, suositeltu aloitusannos on 1,2 mg/kg 30 minuuttia kestävänä laskimoinfuusiona 3 viikon välein. Munuaisten vajaatoimintapotilaiden tilaa on seurattava huolellisesti haittavaikutusten varalta (ks. kohta Farmakokinetiikka).
Jos potilas sairastaa maksan vajaatoimintaa, suositeltu aloitusannos on 1,2 mg/kg 30 minuuttia kestävänä laskimoinfuusiona 3 viikon välein. Maksan vajaatoimintapotilaiden tilaa on seurattava huolellisesti haittavaikutusten varalta (ks. kohta Farmakokinetiikka).
Iäkkäät potilaat
Annostussuositukset ovat 65‑vuotiaiden tai sitä vanhempien potilaiden hoidossa samat kuin aikuisillakin. Tällä hetkellä saatavilla olevat tiedot kuvataan kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka. ADCETRIS-valmisteen turvallisuutta ja tehoa 60 vuotiailla ja sitä vanhemmilla potilailla ei ole varmistettu BrECADD-hoidossa.
Pediatriset potilaat
ADCETRIS‑valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tällä hetkellä olevat tiedot kerrotaan kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka mutta annossuosituksia ei voida antaa.
Antotapa
ADCETRIS‑suositusannos annetaan 30 minuuttia kestävänä infuusiona.
Ks. Kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
ADCETRIS‑valmistetta ei saa antaa nopeana laskimoinjektiona eikä bolusinjektiona. ADCETRIS annetaan erillisen infuusioletkun kautta, eikä sitä saa sekoittaa muiden lääkevalmisteiden kanssa (ks. kohta Yhteensopimattomuudet).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Bleomysiinin ja ADCETRIS‑valmisteen samanaikainen käyttö aiheuttaa keuhkotoksisuutta (ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Progressiivinen multifokaalinen leukoenkefalopatia
ADCETRIS‑valmistetta saavilla potilailla voi esiintyä John Cunninghamin viruksen (JC‑viruksen eli papovaviruksen) uudelleenaktivoitumista, joka voi aiheuttaa progressiivisen multifokaalisen leukoenkefalopatian (PML) ja kuoleman. PML:ää on raportoitu potilailla, jotka saivat tätä hoitoa useiden aiempien solunsalpaajahoitojen jälkeen. PML on keskushermoston myeliinikatoa aiheuttava harvinainen sairaus, joka johtuu piilevän JC‑viruksen uudelleenaktivoitumisesta ja johtaa usein kuolemaan.
Potilaiden tilaa on seurattava tarkasti PML:ään mahdollisesti viittaavien uusien tai pahenevien neurologisten, kognitiivisten tai käyttäytymiseen liittyvien oireiden ja löydösten varalta. Jos PML:ää epäillään, ADCETRIS‑valmisteen antaminen on keskeytettävä. PML:ää epäiltäessä suositellaan neurologin konsultoimista, gadoliniumtehosteista aivojen magneettikuvausta ja JC‑viruksen DNA:n analysointia selkäydinnesteestä polymeraasiketjureaktiomenetelmällä (PCR) tai JC‑viruksen toteamista aivobiopsialla. Negatiivinen tulos JC‑viruksen PCR‑tutkimuksesta ei poissulje PML:n mahdollisuutta. Lisäseuranta ja ‑tutkimukset voivat olla aiheellisia, jos mikään vaihtoehtoinen diagnoosi ei tule kysymykseen. Jos potilaalla todetaan PML, ADCETRIS‑hoito on lopetettava pysyvästi.
Lääkärin on tarkkailtava etenkin sellaisia PML:ään viittaavia oireita, joita potilas ei välttämättä huomaa itse (esim. kognitiiviset, neurologiset tai psykiatriset oireet).
Haimatulehdus
Akuuttia haimatulehdusta on todettu ADCETRIS‑valmisteella hoidetuilla potilailla. Kuolemaan johtaneita tapauksia on raportoitu.
Potilaiden tilaa pitää seurata huolellisesti siltä varalta, että heille tulee vatsakipua tai vatsakipu pahenee, mikä saattaa viitata akuuttiin haimatulehdukseen. Potilaiden arvioinnissa voidaan käyttää lääkärintarkastusta, seerumin amylaasin ja seerumin lipaasin selvittämistä laboratoriokokeilla sekä vatsan kuvantamistutkimusta, kuten ultraäänitutkimusta, sekä muita asianmukaisia diagnostisia menetelmiä. ADCETRIS‑valmisteen käyttö pitää keskeyttää, jos epäillään akuuttia haimatulehdusta. ADCETRIS‑valmisteen käyttö pitää lopettaa, jos akuutin haimatulehduksen diagnoosi vahvistuu.
Keuhkotoksisuus
Keuhkotoksisuustapauksia, mukaan lukien pneumoniittia, interstitiaalista keuhkosairautta ja akuuttia hengitysvajausoireyhtymää (ARDS), joista osa on johtanut kuolemaan, on raportoitu ADCETRIS‑valmisteella hoidetuilla potilailla. Vaikka syy‑yhteyttä ADCETRIS‑valmisteen käyttöön ei ole todettu, keuhkotoksisuuden riskiä ei voida sulkea pois. Jos potilaalle tulee keuhko‑oireita (esim. yskä, hengenahdistus) tai hänen keuhko‑oireensa pahenevat, diagnostinen arvio tilanteesta pitää tehdä välittömästi ja potilasta pitää hoitaa asianmukaisesti. ADCETRIS‑hoidon keskeyttämistä arvioinnin ajaksi ja kunnes oireet paranevat on harkittava.
Vakavat infektiot ja opportunistiset infektiot
ADCETRIS‑valmistetta saavilla potilailla on raportoitu vakavia infektioita (mm. keuhkokuume, stafylokokkibakteremia, sepsis / septinen sokki (mukaan lukien kuolemaan johtaneet) ja herpes zoster, sytomegalovirus (CMV (uudelleenaktivoituminen)) ja opportunistisia infektioita (mm. Pneumocystis jiroveci ‑keuhkokuume ja suun kandidiaasi). Potilaiden tilaa on seurattava hoidon aikana huolellisesti mahdollisten vakavien ja opportunististen infektioiden varalta.
Infuusioreaktiot
Välittömästi tai viiveellä kehittyviä infuusioreaktioita ja anafylaktisia reaktioita on raportoitu.
Potilaiden vointia on seurattava huolellisesti infuusion aikana ja sen jälkeen. Jos potilaalle kehittyy anafylaktinen reaktio, ADCETRIS‑valmisteen antaminen on lopetettava välittömästi ja pysyvästi ja potilaalle on annettava asianmukaista lääketieteellistä hoitoa.
Jos potilaalle kehittyy infuusioreaktio, infuusio on keskeytettävä ja tila hoidettava asianmukaisesti. Oireiden hävittyä lääkkeen anto voidaan aloittaa uudelleen hitaammalla infuusionopeudella. Jos infuusioreaktioita on esiintynyt aiemmin, potilaalle on annettava esilääkitystä myöhempien infuusioiden yhteydessä. Esilääkityksenä voidaan antaa esim. parasetamolia, antihistamiinia ja kortikosteroidia.
Infuusioreaktiot ovat yleisempiä ja vaikeampia potilailla, joille on kehittynyt vasta‑aineita brentuksimabivedotiinille (ks. kohta Haittavaikutukset).
Tuumorilyysioireyhtymä
ADCETRIS‑hoidon yhteydessä on raportoitu tuumorilyysioireyhtymää. Tuumorilyysioireyhtymän riski koskee potilaita, joilla on nopeasti kasvava kasvain ja suuri kasvaintaakka. Näiden potilaiden tilaa on seurattava tarkasti ja heitä on hoidettava parhaan kliinisen käytännön mukaisesti. Tuumorilyysioireyhtymän hoitokeinoja voivat olla mm. aggressiivinen nesteytys, munuaistoiminnan seuranta, elektrolyyttihäiriöiden korjaaminen, hyperurikemialääkitys ja tukihoito.
Perifeerinen neuropatia
ADCETRIS voi aiheuttaa sekä sensorista että motorista perifeeristä neuropatiaa. ADCETRIS‑valmisteen aiheuttama perifeerinen neuropatia on yleensä seurausta kumulatiivisesta altistuksesta tälle lääkevalmisteelle ja on useimmissa tapauksissa korjautuvaa. Kliinisissä tutkimuksissa oireet korjautuivat tai lievittyivät valtaosalla potilaista (ks. kohta Haittavaikutukset). Potilaiden tilaa on seurattava neuropatiaoireiden varalta. Oireita voivat olla mm. hypoestesia, hyperestesia, parestesia, epämukavuus, polttelu, neuropaattinen kipu tai heikotus. Jos potilaalla on uusia tai pahenevia perifeerisen neuropatian oireita, on harkittava annosvälin pidentämistä ja ADCETRIS‑annoksen pienentämistä tai hoidon lopettamista (ks. kohta Annostus ja antotapa).
Hematologinen toksisuus
ADCETRIS‑hoidon yhteydessä voi esiintyä asteen 3 tai asteen 4 anemiaa, trombosytopeniaa ja pitkittynyttä (≥ 1 viikko) asteen 3 tai asteen 4 neutropeniaa. Täydellinen verenkuva on tarkistettava ennen jokaista lääkeannosta. Jos potilaalle kehittyy asteen 3 tai asteen 4 neutropenia, katso ohjeet kohdasta Annostus ja antotapa.
Kuumeinen neutropenia
ADCETRIS hoidon yhteydessä on raportoitu kuumeista neutropeniaa (tuntemattomasta syystä johtuvaa kuumetta, johon ei liity mitään kliinisesti eikä mikrobiologisesti dokumentoitua infektiota; absoluuttinen neutrofiiliarvo < 1,0 x 109/l ja kuumetta ≥ 38,5 °C; lähde CTCAE v3). Täydellinen verenkuva on tarkistettava ennen jokaista lääkeannosta. Potilaiden tilaa on seurattava tarkasti kuumeen varalta. Jos kuumeinen neutropenia kehittyy, potilasta on hoidettava parhaan kliinisen käytännön mukaisesti.
Yhdistelmähoidossa AVD tai CHP tai BrECADD-hoidossa korkea ikä oli kuumeisen neutropenian riskitekijä.
Kun ADCETRIS annetaan yhdessä AVD tai CHP hoidon kanssa, kaikille aikuispotilaille iästä riippumatta suositellaan ensimmäisestä annoksesta alkaen primaariprofylaksia G CSF valmisteella.
Kun ADCETRIS annetaan yhdistelmähoitona osana BrECADD-hoitoa, primaariprofylaksia kasvutekijävalmisteella (G-CSF) on annettava jokaisen hoitojakson päivästä 5 alkaen kaikille aikuispotilaille iästä riippumatta.
Vaikeat ihoon liittyvät haittavaikutukset (SCAR)
ADCETRIS‑hoidon yhteydessä on raportoitu vaikeita ihoon liittyviä haittavaikutuksia, mukaan lukien Stevens–Johnsonin oireyhtymää (SJS), toksista epidermaalista nekrolyysiä (TEN) ja yleisoireista eosinofiilistä oireyhtymää (DRESS). Kuolemaan johtaneita Stevens–Johnsonin oireyhtymän ja toksisen epidermaalisen nekrolyysin tapauksia on raportoitu. Jos potilaalle kehittyy Stevens–Johnsonin oireyhtymä, toksinen epidermaalinen nekrolyysi tai yleisoireinen eosinofiilinen oireyhtymä, ADCETRIS‑hoito on lopetettava ja potilaalle on annettava asianmukaista lääketieteellistä hoitoa.
Gastrointestinaaliset komplikaatiot
Gastrointestinaalisia komplikaatioita, mukaan lukien suolitukoksia, ileusta, enterokoliittia, neutropeenistä koliittia, eroosiota, haavaumia, suolen puhkeamista ja verenvuotoa, joista osa on johtanut kuolemaan, on raportoitu ADCETRIS‑valmisteella hoidetuilla potilailla. Jos potilaalle tulee maha‑suolikanavan oireita tai hänen maha‑suolikanavan oireensa pahenevat, diagnostinen arvio tilanteesta pitää tehdä välittömästi ja potilasta pitää hoitaa asianmukaisesti.
Maksatoksisuus
Alaniiniaminotransferaasi (ALT)‑ ja aspartaattiaminotransferaasiarvojen (AST) suurenemisen muodossa ilmenevää maksatoksisuutta on raportoitu ADCETRIS‑hoidon yhteydessä. Myös vakavia maksatoksisuustapauksia, joista osa on johtanut kuolemaan, on esiintynyt. Myös olemassa oleva maksasairaus, muut samanaikaiset sairaudet ja samanaikaiset lääkitykset voivat suurentaa riskiä. ADCETRIS‑valmistetta saavien potilaiden maksan toiminta pitää testata ennen hoidon aloitusta ja sitä pitää seurata rutiininomaisesti. Jos potilaalla ilmenee maksatoksisuutta, on mahdollista, että ADCETRIS‑valmisteen antoväliä pitää pidentää, annosta muuttaa tai hoito lopettaa.
Hyperglykemia
Kliinisissä tutkimuksissa hyperglykemiaa on raportoitu ylipainoisilla potilailla (suuri painoindeksi, BMI) riippumatta siitä, sairastivatko potilaat diabetesta vai eivät. Seerumin glukoosiarvoa on kuitenkin seurattava huolellisesti kaikilta potilailta, joilla on esiintynyt hyperglykemiaa. Diabeteslääkitystä annetaan tarpeen mukaan.
Infuusiokohdan ekstravasaatio
Laskimoinfuusion annon aikana on ilmennyt ekstravasaatiota. Ekstravasaation mahdollisuus huomioon ottaen on suositeltavaa tarkkailla infuusiokohtaa huolellisesti siltä varalta, että lääkettä infiltroituu infuusiokohdasta annon aikana.
Munuaisten ja maksan vajaatoiminta
Munuaisten ja maksan vajaatoimintaa sairastavista potilaista ei ole paljon kokemusta. Käytettävissä olevat tiedot viittaavat siihen, että vaikea munuaisten vajaatoiminta, maksan vajaatoiminta ja pieni seerumin albumiinipitoisuus saattavat vaikuttaa MMAE:n puhdistumaan (ks. kohta Farmakokinetiikka).
CD30‑positiivinen ihon T‑solulymfooma
Mycosis fungoidesta ja ihon primaarista anaplastista suurisoluista lymfoomaa (pcALCL) lukuun ottamatta hoitovaikutuksen suuruus ei ole selvä muissa CD30‑positiivisissa ihon T‑solulymfooma‑alatyypeissä, joiden kohdalla korkeatasoinen näyttö puuttuu. Kahdessa yksiryhmäisessä faasin 2 ADCETRIS‑tutkimuksessa hoidon vaikutus tautiin osoitettiin seuraavissa alatyypeissä: Sézaryn oireyhtymä, lymfomatoidi papuloosi ja histologisesti sekamuotoinen ihon T‑solulymfooma. Tutkimusten tiedot viittaavat siihen, että teho ja turvallisuus voidaan ekstrapoloida koskemaan ihon T‑solulymfooman muita CD30‑positiivisia alatyyppejä. ADCETRIS‑valmistetta on kuitenkin käytettävä varovaisuutta noudattaen muita CD30‑positiivisia ihon T‑solulymfooma‑alatyyppejä sairastavilla potilailla, vasta huolellisen, yksilöllisen hyöty‑riskisuhteen harkinnan jälkeen (ks. kohta Farmakodynamiikka).
Apuaineiden natriumpitoisuus
Tämä lääkevalmiste sisältää 13,2 mg natriumia per injektiopullo, joka vastaa 0,7 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Apuaineiden polysorbaattipitoisuus
Tämä lääkevalmiste sisältää 2 mg polysorbaatti 80:tä per injektiopullo, joka vastaa 0,2 mg/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutukset CYP3A4‑välitteisesti metaboloituvien lääkevalmisteiden kanssa (CYP3A4:n estäjät/indusorit)
Brentuksimabivedotiinin ja ketokonatsolin (voimakas CYP3A4:n ja P‑gp:n estäjä) yhteiskäyttö suurensi altistusta mikrotubulustoimintaan vaikuttavalle MMAE:lle noin 73 %, mutta ei vaikuttanut brentuksimabivedotiinialtistukseen plasmassa. Brentuksimabivedotiinin ja voimakkaiden CYP3A4:n ja P‑gp:n estäjien yhteiskäyttö voi siis suurentaa neutropenian ilmaantuvuutta. Jos potilaalle kehittyy neutropenia, ks. annossuositukset neutropenian yhteydessä taulukoista 1 ja 2 (ks. kohta Annostus ja antotapa).
Brentuksimabivedotiinin ja rifampisiinin (voimakas CYP3A4:n indusori) yhteiskäyttö ei vaikuttanut brentuksimabivedotiinialtistukseen plasmassa. Vaikka farmakokinetiikasta ei ole paljon tietoja, rifampisiinin käyttö samanaikaisesti brentuksimabivedotiinin kanssa näytti pienentävän mitattavissa olevien MMAE‑metaboliittien pitoisuuksia plasmassa.
Brentuksimabivedotiinin ja midatsolaamin (CYP3A4:n substraatti) yhteiskäyttö ei vaikuttanut midatsolaamin metaboliaan, joten brentuksimabivedotiinin ei odoteta vaikuttavan CYP3A4‑entsyymien välityksellä metaboloituvien lääkeaineiden altistukseen.
Doksorubisiini, vinblastiini ja dakarbatsiini (AVD)
Kun tutkittaville annettiin brentuksimabivedotiinia yhdessä AVD‑hoidon kanssa, vasta‑aineen ja lääkeaineen konjugaatin (ADC) farmakokinetiikka seerumissa ja MMAE:n farmakokinetiikka plasmassa olivat samankaltaiset kuin tilanteessa, jossa brentuksimabivedotiinia käytettiin monoterapiana.
Brentuksimabivedotiinin samanaikainen anto ei vaikuttanut plasman AVD‑altistukseen.
Syklofosfamidi, doksorubisiini ja prednisoni (CHP)
Kun tutkittavalle annettiin brentuksimabivedotiinia yhdessä CHP‑hoidon kanssa, ADC:n farmakokinetiikka seerumissa ja MMAE:n farmakokinetiikka plasmassa olivat samankaltaiset kuin tilanteessa, jossa brentuksimabivedotiinia käytettiin monoterapiana.
Brentuksimabivedotiinin samanaikaisen annon ei odoteta vaikuttavan CHP‑altistukseen.
Bleomysiini
Brentuksimabivedotiinia ja bleomysiiniä (B) ei ole arvioitu muodollisissa yhteisvaikutustutkimuksissa. Vaiheen 1 annoshaku‑ ja turvallisuustutkimuksessa (SGN35‑009) todettiin sietämätöntä keuhkotoksisuutta (mukaan lukien 2 kuolemaan johtanutta tapahtumaa) 11 potilaalla (44 %) 25 potilaan ryhmästä, joka sai brentuksimabivedotiinia ja ABVD‑hoitoa. Brentuksimabivedotiinin ja AVD‑hoidon yhdistelmän yhteydessä ei raportoitu keuhkotoksisuutta eikä kuolemaan johtaneita tapahtumia. ADCETRIS‑valmisteen ja bleomysiinin samanaikainen anto on siis vasta‑aiheista (ks. kohta Vasta-aiheet).
Etoposidi, syklofosfamidi, doksorubisiini, dakarbatsiini, deksametasoni (BrECADD-hoito)
ADC:n ja MMAE:n farmakokinetiikkaa ei ole karakterisoitu BrECADD-hoidon yhteydessä. BrECADD-hoidon ei odoteta vaikuttavan brentuksimabivedotiinialtistukseen ja samanaikaiseen solunsalpaaja-altistukseen.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä kahta tehokasta ehkäisymenetelmää ADCETRIS‑hoidon aikana ja 6 kuukauden ajan hoidon päättymisen jälkeen.
Raskaus
ADCETRIS‑valmisteen käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
ADCETRIS‑valmistetta saa käyttää raskauden aikana vain, jos äidille koituva hyöty on suurempi kuin sikiöön mahdollisesti kohdistuva riski. Jos raskaana oleva potilas tarvitsee hoitoa, hänelle on selkeästi kerrottava sikiöön mahdollisesti kohdistuvista riskeistä.
Alla olevassa hedelmällisyyttä käsittelevässä kohdassa on neuvoja naisille, joiden miespuolista kumppania hoidetaan ADCETRIS‑valmisteella.
Imetys
Ei tiedetä, erittyvätkö brentuksimabivedotiini tai sen metaboliitit ihmisillä äidinmaitoon.
Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois.
On päätettävä, lopetetaanko imetys vai pidättäydytäänkö ADCETRIS-hoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Ei‑kliinisissä tutkimuksissa brentuksimabivedotiinihoito on aiheuttanut kivestoksisuutta, joten se saattaa vaikuttaa miehen hedelmällisyyteen. MMAE:llä on todettu aneugeenisia ominaisuuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Tätä lääkettä saavien miesten on hyvä harkita pakastettujen siittiöiden tallettamista spermapankkiin ennen hoidon aloittamista. Tätä lääkettä saavat miehet eivät saa siittää lasta hoidon aikana eivätkä 6 kuukauteen viimeisen annoksen jälkeen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
ADCETRIS‑valmisteella voi olla kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn (esim. huimauksen vuoksi), ks. kohta Haittavaikutukset.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
ADCETRIS‑valmisteen turvallisuusprofiili perustuu käytettävissä oleviin kliinisten tutkimusten tietoihin, Named Patient Program ‑ohjelmaan (NPP) ja tähän mennessä myyntiluvan myöntämisen jälkeen kertyneisiin käyttökokemuksiin. Alla ja taulukossa 5 kuvattujen haittavaikutusten esiintymistiheydet on määritelty kliinisistä tutkimuksista kerättyjen tietojen perusteella.
Monoterapia
Yhdistetyissä tiedoissa tutkimuksista, joissa ADCETRIS‑valmistetta annettiin monoterapiana Hodgkinin lymfoomaa, systeemistä anaplastista suurisoluista lymfoomaa tai ihon T‑solulymfoomaa sairastaville potilaille (SG035‑0003, SG035‑0004, SGN35‑005, SGN35‑006, C25001, C25006 ja C25007, ks. kohta Farmakodynamiikka), yleisimpiä haittavaikutuksia (≥ 10 %) olivat infektiot, perifeerinen sensorinen neuropatia, pahoinvointi, väsymys, ripuli, kuume, neutropenia, ylähengitystieinfektio, nivelkipu, ihottuma, yskä, oksentelu, kutina, perifeerinen motorinen neuropatia, infuusioreaktiot, ummetus, hengenahdistus, lihaskipu, painon lasku ja vatsakipu.
Vakavia haittavaikutuksia esiintyi 12 %:lla potilaista. Kunkin yksittäisen vakavan haittavaikutuksen esiintymistiheys oli ≤ 1 %.
ADCETRIS‑hoitoa saaneista potilaista 24 % keskeytti hoidon haittatapahtumien takia.
ADCETRIS‑hoitoa uudelleen saaneiden potilaiden turvallisuustiedot (SGN35‑006, ks. kohta Farmakodynamiikka) vastasivat yhdistetyissä, keskeisissä faasin 2 tutkimuksissa havaittuja tietoja. Poikkeuksena oli perifeerinen motorinen neuropatia, jonka ilmaantuvuus oli suurempi (28 % vs. keskeisissä faasin 2 tutkimuksissa 9 %) ja jonka aste oli pääasiassa 2. Potilailla esiintyi myös enemmän nivelkipua, asteen 3 anemiaa ja selkäkipua verrattuna yhdistettyjen, keskeisten faasin 2 tutkimusten tietoihin.
Turvallisuustiedot sellaisilta uusiutunutta tai refraktaarista Hodgkinin lymfoomaa sairastavilta potilailta, joille ei ollut tehty autologista kantasolusiirtoa ja joita oli hoidettu suositusannoksella (1,8 mg/kg) kolmen viikon välein yhdellä hoitoryhmällä tehdyssä faasin 4 tutkimuksessa (n = 60), faasin 1 tutkimuksissa (käsittelivät annoksen suurentamista ja kliinistä farmakologiaa, n = 15) ja NPP‑ohjelmassa (n = 26) (ks. kohta Farmakodynamiikka) olivat yhteneväisiä keskeisten kliinisten tutkimusten turvallisuusprofiilin kanssa.
Yhdistelmähoito (AVD/CHP)
Turvallisuustiedot yhdessä ADCETRIS‑valmisteen kanssa annettavista solunsalpaajista (doksorubisiini, vinblastiini ja dakarbatsiini (AVD) tai syklofosfamidi, doksorubisiini ja prednisoni (CHP)), katso kyseisten valmisteiden valmisteyhteenvedot.
Tutkimuksissa, joissa ADCETRIS‑valmistetta annettiin yhdistelmähoitona 662 potilaalle, joilla oli aiemmin hoitamaton, levinnyt Hodgkinin lymfooma (C25003) ja 223 potilaalle, joilla oli aiemmin hoitamaton CD30‑positiivinen perifeerinen T‑solulymfooma (PTCL) (SGN35‑014), yleisimpiä haittavaikutuksia (≥ 10 %) olivat infektiot, neutropenia, perifeerinen sensorinen neuropatia, pahoinvointi, ummetus, oksentelu, ripuli, uupumus, kuume, hiustenlähtö, anemia, painon lasku, stomatiitti, kuumeinen neutropenia, vatsakipu, ruokahalun huononeminen, unettomuus, luustokipu, ihottuma, yskä, hengenahdistus, nivelkipu, lihaskipu, selkäkipu, perifeerinen motorinen neuropatia, ylähengitystieinfektio ja huimaus.
ADCETRIS‑yhdistelmähoitoa saaneessa ryhmässä vakavia haittavaikutuksia esiintyi 34 %:lla potilaista. Vakavia haittavaikutuksia, joita esiintyi ≥ 3 %:lla potilaista, olivat kuumeinen neutropenia (15 %), kuume (5 %) ja neutropenia (3 %).
Haittatapahtumat johtivat hoidon lopettamiseen 10 %:lla potilaista. Haittatapahtumia, jotka johtivat hoidon lopettamiseen ≥ 2 %:lla potilaista, olivat perifeerinen sensorinen neuropatia, ja perifeerinen neuropatia.
Haittavaikutustaulukko
ADCETRIS‑hoidon haittavaikutusten luokittelussa on käytetty MedDRA‑elinjärjestelmäluokitusta ja ‑terminologiaa (ks. taulukko 7). Kussakin elinjärjestelmäluokassa haittavaikutukset on lueteltu esiintymistiheyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 7: ADCETRIS‑valmisteen haittavaikutukset
| Elinjärjestelmäluokitus | Haittavaikutukset (monoterapia) | Haittavaikutukset (yhdistelmähoito) |
| Infektiot | ||
| Hyvin yleinen: | Infektioa, ylähengitystieinfektio | Infektioa, ylähengitystieinfektio |
| Yleinen: | Herpes zoster, keuhkokuume, herpes simplex, suun kandidiaasi | Keuhkokuume, suun kandidiaasi, sepsis / septinen sokki, herpes zoster |
| Melko harvinainen: | Pneumocystis jiroveci ‑keuhkokuume, stafylokokkibakteremia, sytomegalovirusinfektio tai viruksen uudelleenaktivoituminen, sepsis / septinen sokki | Herpes simplex, Pneumocystis jiroveci ‑keuhkokuume |
| Tuntematon: | Progressiivinen multifokaalinen leukoenkefalopatia | |
| Veri ja imukudos | ||
| Hyvin yleinen: | Neutropenia | Neutropeniaa, anemia, kuumeinen neutropenia |
| Yleinen: | Anemia, trombosytopenia | Trombosytopenia |
| Melko harvinainen: | Kuumeinen neutropenia | |
| Immuunijärjestelmä | ||
| Melko harvinainen: | Anafylaktinen reaktio | Anafylaktinen reaktio |
| Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleinen: | Ruokahalun huononeminen | |
| Yleinen: | Hyperglykemia | Hyperglykemia |
| Melko harvinainen: | Tuumorilyysioireyhtymä | Tuumorilyysioireyhtymä |
| Psyykkiset häiriöt | ||
| Hyvin yleinen: | Unettomuus | |
| Hermosto | ||
| Hyvin yleinen: | Perifeerinen sensorinen neuropatia, perifeerinen motorinen neuropatia | Perifeerinen sensorinen neuropatia, perifeerinen motorinen neuropatiaa, huimaus |
| Yleinen: | Huimaus | |
| Melko harvinainen: | Demyelinoiva polyneuropatia | |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleinen: | Yskä, hengenahdistus | Yskä, hengenahdistus |
| Ruoansulatuselimistö | ||
| Hyvin yleinen: | Pahoinvointi, ripuli, oksentelu, ummetus, vatsakipu | Pahoinvointi, ummetus, oksentelu, ripuli, vatsakipu, stomatiitti |
| Melko harvinainen: | Akuutti haimatulehdus | Akuutti haimatulehdus |
| Maksa ja sappi | ||
| Yleinen: | Alaniiniaminotransferaasi-/aspartaattiaminotransferaasiarvojen (ALT/AST) suureneminen | Alaniiniaminotransferaasi-/aspartaattiaminotransferaasiarvojen (ALT/AST) suureneminen |
| Iho ja ihonalainen kudos | ||
| Hyvin yleinen: | Ihottumaa, kutina | Hiustenlähtö, ihottumaa |
| Yleinen: | Hiustenlähtö | Kutina |
| Melko harvinainen: | Stevens–Johnsonin oireyhtymä / toksinen epidermaalinen nekrolyysi | Stevens–Johnsonin oireyhtymäb |
| Tuntematon: | Yleisoireinen eosinofiilinen oireyhtymä (DRESS) | |
| Luusto, lihakset ja sidekudos | ||
| Hyvin yleinen: | Nivelkipu, lihaskipu | Luustokipu, nivelkipu, lihaskipu, selkäkipu |
| Yleinen: | Selkäkipu | |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleinen: | Väsymys, kuume, infuusioreaktiota | Väsymys, kuume, |
| Yleinen: | Vilunväristykset | Infuusioreaktiota, vilunväristykset |
| Tuntematon: | Infuusiokohdan ekstravasaatio c | |
| Tutkimukset | ||
| Hyvin yleinen: | Painon lasku | Painon lasku |
a. Yhdistetty useista haittavaikutustermeistä.
b. Toksista epidermaalista nekrolyysiä ei ilmoitettu yhdistelmähoitoa saaneilla.
c. Ekstravasaatio voi aiheuttaa infuusiokohdan ihon punoitusta, kipua, turvotusta, rakkulointia, hilseilyä tai selluliittia infuusiokohdassa tai sen ympärillä.
Yhdistelmähoito (BrECADD-hoito)
Katso ADCETRIS-valmisteen kanssa annettavien solunsalpaajien (etoposidi, syklofosfamidi, doksorubisiini, dakarbatsiini, deksametasoni [BrECADD]) turvallisuustiedot niiden valmisteyhteenvedoista.
HD21-tutkimuksessa annettiin BrECADD-hoitoa 747 potilaalle ja eBEACOPP-hoitoa (eskaloitu bleomysiini [B], etoposidi [E], doksorubisiini [A], syklofosfamidi [C], vinkristiini [O], prokarbatsiini [P] ja prednisoni [P]) 741 potilaalle. ADCETRIS-valmisteen turvallisuusprofiili oli BrECADD-hoitoa saaneilla potilailla yhdenmukainen muiden yhdistelmähoitojen kanssa (AVD/CHP).
Vakavia haittavaikutuksia esiintyi 39,4 %:lla BrECADD-hoitoa saaneista potilaista ja 36,4 %:lla eBEACOPP-hoitoa saaneista potilaista. BrECADD-hoitoa saaneiden potilaiden yleisimmät vakavat haittavaikutukset (> 3%) olivat kuumeinen neutropenia (19,3 %), pyreksia (3,9 %) ja neutropenia (3,2 %).
Sydämeen liittyviä vakavia haittavaikutuksia esiintyi 2,7 %:lla BrECADD-hoitoa saaneista potilaista ja 1,1 %:lla eBEACOPP-hoitoa saaneista potilaista. Yleisin sydämeen liittyvä vakava haittavaikutus (> 0,5 %) BrECADD-hoitoa saaneilla potilailla oli takykardia (0,9 %).
Vakavat haittatapahtumat johtivat hoidon lopettamiseen 2 %:lla potilaista sekä BrECADD- että eBEACOPP-ryhmässä. Yleisimmät hoidon lopettamiseen johtaneet vakavat haittatapahtumat BrECADD-ryhmässä olivat kuumeinen neutropenia (0,3 %) ja sydämen vajaatoiminta (0,3 %).
Valikoitujen haittavaikutusten kuvaukset
Neutropenia ja kuumeinen neutropenia
Monoterapia
Kliinisissä tutkimuksissa neutropenia johti antovälin pidentämiseen 13 %:lla potilaista. Asteen 3 neutropeniaa ilmoitettiin 13 %:lla potilaista ja asteen 4 neutropeniaa 5 %:lla potilaista. Yhden potilaan annosta oli tarpeen pienentää ja yksi potilas keskeytti hoidon neutropenian vuoksi.
Hoidon yhteydessä voi esiintyä vaikeaa ja pitkittynyttä (≥ 1 viikko) neutropeniaa, joka voi suurentaa vakavien infektioiden kehittymisriskiä. Kuumeista neutropeniaa ilmoitettiin < 1 %:lla potilaista (ks. kohta Annostus ja antotapa).
Keskeisissä faasin 2 tutkimuksissa (SG035‑0003 ja SG035‑0004) asteen 3 tai asteen 4 neutropenian mediaanikesto jäi rajalliseksi (1 viikko); 2 %:lle potilaista kehittyi ≥ 7 vuorokautta kestänyt asteen 4 neutropenia. Alle puolella niistä keskeisten faasin 2 tutkimusten potilaista, joille kehittyi asteen 3 tai asteen 4 neutropenia, esiintyi tähän ajallisesti liittyviä infektioita, ja suurin osa näistä infektioista oli asteen 1 tai asteen 2 infektioita.
Yhdistelmähoito
ADCETRIS‑yhdistelmähoitoa arvioineissa kliinisissä tutkimuksissa C25003 (ADCETRIS + AVD) ja SGN35-014 (ADCETRIS + CHP) neutropenia johti annosvälin pidentämiseen 19 %:lla potilaista. Asteen 3 neutropeniaa ilmoitettiin 17 %:lla potilaista ja asteen 4 neutropeniaa 41 %:lla potilaista. 2 %:lla potilaista annosta oli pienennettävä ja < 1 % lopetti vähintään yhden tutkimuslääkkeen käytön neutropenian vuoksi.
Kuumeista neutropeniaa ilmoitettiin 20 %:lla potilaista, jotka eivät saaneet G‑CSF‑valmistetta primaariprofylaksina (ks. kohta Annostus ja antotapa). Kuumeisen neutropenian esiintymistiheys oli 13 % potilailla, jotka saivat G‑CSF‑valmistetta primaariprofylaksina.
Kliinisessä tutkimuksessa HD21 (BrECADD), jossa ADCETRIS-valmistetta annettiin yhdistelmähoitona, neutropenia johti hoidon viivästymiseen 0,5 %:lla potilaista. Asteen 3 neutropeniaa raportoitiin 0,5 %:lla ja asteen 4 neutropeniaa 9 %:lla potilaista. Annosta jouduttiin pienentämään 1,1 %:lla potilaista, eikä vaikea neutropenia johtanut hoidon lopettamiseen yhdelläkään potilaalla. Kaikki potilaat saivat primaariprofylaksiaa kasvutekijävalmisteella (G-CSF) (ks. kohta Annostus ja antotapa). Kuumeisen neutropenian esiintyvyys oli 26,5 % BrECADD-hoitoa saaneilla potilailla.
Vakavat infektiot ja opportunistiset infektiot
Monoterapia
Kliinisissä tutkimuksissa ilmoitettiin vakavia infektioita ja opportunistisia infektioita 10 %:lla potilaista ja sepsis tai septinen sokki < 1 %:lla potilaista. Yleisimmin ilmoitettuja opportunistisia infektioita olivat herpes zoster ja herpes simplex.
Yhdistelmähoito
ADCETRIS yhdistelmähoitoa arvioineissa kliinisissä tutkimuksissa (C25003 [ADCETRIS + AVD] ja SGN35-014 [ADCETRIS + CHP]) vakavia infektioita, mm. opportunistisia infektioita, esiintyi 15 %:lla potilaista; 4 %:lle potilaista kehittyi sepsis, neutropeeninen sepsis, septinen sokki tai bakteremia. Yleisimmin ilmoitettuja opportunistisia infektioita olivat herpesvirusinfektiot.
Kliinisessä tutkimuksessa (HD21 [BrECADD]), jossa ADCETRIS-valmistetta annettiin yhdistelmähoitona, vaikeita infektioita ja infestaatioita esiintyi 14,3 %:lla potilaista. Yleisimmin raportoituja tapahtumia olivat infektio (2 %), keuhkokuume (1,7 %) ja neutropeeninen infektio (1,2 %).
Perifeerinen neuropatia
Monoterapia
Kliinisissä tutkimuksissa hoidon aikana puhjennutta neuropatiaa oli 57 %:lla tutkimuspotilaista. Perifeeristä motorista neuropatiaa oli 13 %:lla potilaista. Perifeerinen neuropatia johti hoidon keskeyttämiseen 15 %:lla potilaista, annoksen pienentämiseen 15 %:lla potilaista ja antovälin pidentämiseen 16 %:lla potilaista. Niillä potilailla, joilla perifeeristä neuropatiaa esiintyi, haittavaikutuksen puhkeamiseen kuluneen ajan mediaani oli 12 viikkoa. Niillä potilailla, jotka lopettivat hoidon perifeerisen neuropatian vuoksi, hoidon mediaanikesto oli 11 hoitojaksoa.
Potilailla, joilla esiintyi perifeeristä neuropatiaa keskeisissä faasin 2 tutkimuksissa (SG035‑0003 ja SG035‑0004) ja satunnaistetuissa faasin 3 monoterapiatutkimuksissa (SGN35‑005 ja C25001), seurannan mediaanikesto hoidon päättymisestä viimeiseen arviointiin vaihteli 48,9 viikosta 98 viikkoon. Viimeisen arvioinnin hetkellä perifeerisen neuropatian oireet olivat hävinneet tai lievittyneet useimmilla (82–85 %:lla) potilaista, joilla perifeeristä neuropatiaa oli esiintynyt. Kaikkia tapahtumia tarkasteltaessa oireiden ilmaantumisen ja häviämisen tai lievittymisen välinen mediaaniaika vaihteli 16 viikon ja 23,4 viikon välillä.
Perifeerisen neuropatian oireet olivat lievittyneet tai korjautuneet viimeisen arvioinnin hetkellä myös valtaosalla (80 %) uusiutunutta tai refraktaarista Hodgkinin lymfoomaa tai systeemistä anaplastista suurisoluista lymfoomaa sairastavista potilaista, jotka saivat ADCETRIS‑hoitoa uudelleen (SGN35‑006).
Yhdistelmähoito
ADCETRIS‑yhdistelmähoitoa AVD‑hoidon kanssa arvioineessa kliinisessä tutkimuksessa hoidon aikana kehittynyttä neuropatiaa esiintyi 67 %:lla potilaista, ja 11 %:lle potilaista kehittyi perifeerinen motorinen neuropatia. Perifeerinen neuropatia johti hoidon lopettamiseen 7 %:lla potilaista, annoksen pienentämiseen 21 %:lla ja annosvälin pidentämiseen 1 %:lla potilaista. Potilailla, joilla oli perifeeristä neuropatiaa, mediaaniaika perifeerisen neuropatian alkamiseen oli 8 viikkoa. Mediaaniannos potilailla, jotka lopettivat hoidon perifeerisen neuropatian vuoksi, oli 8 annosta ADCETRIS + AVD‑yhdistelmähoitoa (A+AVD) ennen kuin yksi tai useampia yhdistelmän lääkkeistä lopetettiin.
Potilailla, joilla esiintyi perifeeristä neuropatiaa, mediaaniseuranta‑aika hoidon päättymisestä viimeiseen arviointikertaan oli noin 286 viikkoa. Viimeisen arvioinnin ajankohtana perifeerisen neuropatian oireet olivat korjautuneet tai lievittyneet useimmilla perifeeristä neuropatiaa kokeneilla potilailla (86 %). Mediaaniaika perifeeristen neuropatiatapahtumien alusta niiden korjautumiseen tai lievittymiseen oli 17 viikkoa (vaihteluväli 0–283 viikkoa).
ADCETRIS‑yhdistelmähoitoa CHP‑hoidon kanssa arvioineessa kliinisessä tutkimuksessa hoidon aikana kehittynyttä neuropatiaa esiintyi 52 %:lla potilaista, ja 9 %:lle potilaista kehittyi perifeerinen motorinen neuropatia. Perifeerinen neuropatia johti hoidon lopettamiseen 1 %:lla potilaista, annoksen pienentämiseen 7 %:lla ja annosvälin pidentämiseen < 1 %:lla potilaista. Potilailla, joilla oli perifeeristä neuropatiaa, mediaaniaika perifeerisen neuropatian alkamiseen oli 9,1 viikkoa. Mediaaniannos potilailla, jotka lopettivat hoidon perifeerisen neuropatian vuoksi, oli 5 annosta ADCETRIS + CHP‑yhdistelmähoitoa (A + CHP) ennen kuin yksi tai useampia yhdistelmän lääkkeistä lopetettiin.
Potilailla, joilla esiintyi perifeeristä neuropatiaa, mediaaniseuranta‑aika hoidon päättymisestä viimeiseen arviointikertaan oli noin 177 viikkoa. Viimeisen arvioinnin ajankohtana perifeerisen neuropatian oireet olivat korjautuneet tai lievittyneet 64 %:lla perifeeristä neuropatiaa kokeneilla potilailla. Mediaaniaika perifeeristen neuropatiatapahtumien alusta niiden korjautumiseen tai lievittymiseen oli 19 viikkoa (vaihteluväli 0–205 viikkoa).
Kliinisessä tutkimuksessa (HD21 [BrECADD]), jossa ADCETRIS-valmistetta käytettiin yhdistelmähoitona, hoidon aikana ilmaantunutta perifeeristä sensorista neuropatiaa esiintyi 38,8 %:ssa populaatiosta; perifeeristä motorista neuropatiaa esiintyi 3,6 %:lla potilaista. Perifeerinen sensorinen neuropatia ei johtanut vakavana haittatapahtumana hoidon lopettamiseen yhdelläkään potilaalla, mutta se johti annoksen pienentämiseen 4,7 %:lla ja hoidon viivästymiseen 1,5 %:lla potilaista. Perifeerinen motorinen neuropatia ei johtanut vakavana haittatapahtumana hoidon lopettamiseen yhdelläkään potilaalla, mutta se johti annoksen pienentämiseen 0,7 %:lla ja hoidon viivästymiseen 0,1 %:lla potilaista.
Infuusioreaktiot
Monoterapia
Infuusioreaktioita, kuten päänsärky, ihottuma, selkäkipu, oksentelu, vilunväristykset, pahoinvointi, hengenahdistus, kutina ja yskä, ilmoitettiin 12 %:lla potilaista. Anafylaktisia reaktioita on ilmoitettu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Anafylaktisen reaktion oireita voivat olla mm. nokkosihottuma, angioedeema, hypotensio ja bronkospasmi.
Yhdistelmähoito
Kliinisissä tutkimuksissa (C25003 [ADCETRIS + AVD] ja SGN35-014 [ADCETRIS + CHP]) infuusioreaktioita kuten päänsärkyä, ihottumaa, selkäkipua, oksentelua, vilunväristyksiä, pahoinvointia, hengenahdistusta, kutinaa, yskää, infuusiokohdan kipua ja kuumetta ilmoitettiin 8 %:lla potilaista. Anafylaktisia reaktioita on ilmoitettu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Anafylaktisen reaktion oireina voivat olla esimerkiksi nokkosihottuma, angioedeema, hypotensio ja bronkospasmi.
Immunogeenisuus
Kliinisissä tutkimuksissa potilailta tutkittiin määräajoin brentuksimabivedotiini vasta‑aineet herkällä elektrokemiluminesenssimenetelmällä. Potilailla, joilla oli brentuksimabivedotiinivasta‑aineita, havaittiin enemmän infuusioreaktioita kuin potilailla, joilla vasta‑aineita oli ohimenevästi tai ei lainkaan.
Brentuksimabivedotiinivasta‑aineiden ja seerumin brentuksimabivedotiinipitoisuuksien kliinisesti merkittävän pienenemisen välillä ei ollut korrelaatiota, eikä vasta‑aineiden kehittyminen heikentänyt brentuksimabivedotiinin tehoa. Brentuksimabivedotiinivasta‑aineiden kehittyminen ei välttämättä ennakoi infuusioreaktioiden ilmaantumista, mutta pitkäkestoisesti lääkevasta‑ainepositiivisilla potilailla todettiin kuitenkin enemmän infuusioreaktioita kuin ohimenevästi lääkevasta‑ainepositiivisilla potilailla tai potilailla, jotka eivät olleet lääkevasta‑ainepositiivisia missään vaiheessa.
Monoterapiatutkimus C25002
Lääkevasta‑ainepositiivisiksi todetuilla pediatrisilla potilailla havaittiin suuntaus brentuksimabivedotiinin puhdistuman lisääntymiseen. Alle 12‑vuotiaista potilaista yksikään (nolla 11 potilaasta) ei ollut pitkäkestoisesti lääkevasta‑ainepositiivinen, yli 12‑vuotiaissa potilaissa heitä oli 2 (kaksi 23 potilaasta).
Yhdistelmäkäyttötutkimus C25004
Lääkevasta‑ainepositiivisuuden määrä oli vähäinen C25004‑tutkimuksessa; 4 potilasta (ikä ≥ 12 vuotta) 59 potilaasta oli ohimenevästi lääkevasta‑ainepositiivisia, eikä yksikään potilaista ollut pitkäkestoisesti lääkevasta‑ainepositiivinen. Ohimenevästi lääkevasta‑ainepositiivisten potilaiden pienestä määrästä johtuen ei voida varmuudella päätellä, miten lääkevasta‑aineet vaikuttavat lääkkeen tehoon.
Pediatriset potilaat
Monoterapiatutkimus C25002
Turvallisuutta arvioitiin faasin 1/2 tutkimuksessa 7–17‑vuotiailla pediatrisilla potilailla (n = 36), joilla oli uusiutunut tai refraktaarinen Hodgkinin lymfooma tai systeeminen anaplastinen suurisoluinen lymfooma (ks. kohta Farmakodynamiikka). Tässä 36 potilaan tutkimuksessa ei havaittu uusia turvallisuuteen liittyviä huolenaiheita.
Yhdistelmäkäyttötutkimus C25004
Turvallisuutta yhdessä sytostaattihoidon kanssa arvioitiin avoimessa monikeskustutkimuksessa, johon osallistui 59 iältään 6–17‑vuotiasta pediatrista potilasta, joilla oli aiemmin hoitamaton edennyt klassinen CD30‑positiivinen Hodgkinin lymfooma (ks. kohta Farmakodynamiikka). Tässä tutkimuksessa ei raportoitu uusia turvallisuuteen liittyviä huolenaiheita. Yleisin tässä tutkimuksessa raportoitu vakava haittavaikutus oli kuumeinen neutropenia (17 %). Profylaksiaa kasvutekijävalmisteella pohdittiin lääkärin harkinnan mukaan. Perifeeriseen neuropatiaan liittyviä tapahtumia (SMQ‑haun [Standardized MedDRA Query] mukaan) raportoitiin 24 %:lla pediatrisista potilaista tässä tutkimuksessa.
Iäkkäät
Monoterapia
Iäkkäillä potilailla todettu turvallisuusprofiili on yleisesti ottaen vastaava kuin aikuispotilailla todettu. Iäkkäät potilaat saattavat kuitenkin olla alttiimpia haittatapahtumille, kuten keuhkokuumeelle, neutropenialle ja kuumeiselle neutropenialle.
Yhdistelmähoito
Iäkkäillä (≥ 60 vuoden ikäisillä; n = 186 [21 %]) potilailla haittatapahtumien ilmaantuvuus oli kliinisissä tutkimuksissa C25003 (ADCETRIS + AVD) ja SGN35-014 (ADCETRIS + CHP) samaa luokkaa eri hoitoryhmissä. Vakavampia haittatapahtumia ja annosten muutoksia (esim. annosvälin pidentämistä, annoksen pienentämistä ja lääkkeen käytön lopettamista) ilmoitettiin iäkkäillä potilailla useammin kuin tutkimuksen koko populaatiossa. Korkea ikä oli kuumeisen neutropenian riskitekijä molemmissa ryhmissä. Iäkkäillä potilailla, jotka saivat G‑CSF‑valmistetta primaariprofylaksina, neutropenian ja kuumeisen neutropenian ilmaantuvuus oli pienempi kuin potilailla, jotka eivät saaneet G‑CSF‑valmistetta primaariprofylaksina.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyötyhaittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
ADCETRIS‑valmisteelle ei tunneta vastalääkettä. Yliannostustapauksissa potilaan tilaa on seurattava huolellisesti haittavaikutusten (etenkin neutropenian) varalta ja hänelle on annettava tukihoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet; monoklonaaliset vasta‑aineet ja vasta‑ainekonjugoidut lääkkeet, ATC‑koodi: L01FX05
Vaikutusmekanismi
Brentuksimabivedotiini on vasta‑aineen ja lääkeaineen konjugaatti (ADC), josta vapautuva solunsalpaaja aiheuttaa selektiivisesti CD30‑antigeeniä ilmentävien kasvainsolujen apoptoosin. Ei‑kliiniset tiedot viittaavat siihen, että brentuksimabivedotiinin biologinen vaikutus on monivaiheisen prosessin tulos. ADC:n sitoutuminen CD30:een solun pinnalla käynnistää ADC‑CD30‑kompleksin internalisaation ja kulkeutumisen lysosomeihin. Solun sisällä vapautuu proteolyyttisen pilkkoutumisen seurauksena yhtä määriteltyä vaikuttavaa ainetta, monometyyliauristatiini E:tä (MMAE). MMAE:n sitoutuminen tubuliiniin häiritsee solunsisäisen mikrotubulusverkoston toimintaa, pysäyttää solusyklin ja aiheuttaa CD30‑antigeeniä ilmentävän kasvainsolun apoptoosin.
Klassisessa Hodgkinin lymfoomassa, systeemisessä anaplastisessa suurisoluisessa lymfoomassa ja ihon T‑solulymfooman alatyypeissä (kuten mycosis fungoides ja ihon primaarinen anaplastinen suurisoluinen lymfooma) CD30 ilmentyy antigeenina pahanlaatuisten solujen pinnalla. Tämä ilmentymä on riippumaton taudin vaiheesta, hoitotavasta tai siitä, onko kantasolusiirto tehty. Näiden ominaisuuksien ansiosta CD30 on hoidollisen intervention kohde. CD30‑antigeeniin kohdistuvan vaikutusmekanisminsa ansiosta brentuksimabivedotiini tehoaa sytostaattiresistenssissä, sillä CD30 ilmentyy jatkuvasti potilailla, jotka eivät reagoi useammalla lääkeaineella toteutettavaan sytostaattihoitoon, riippumatta siitä, onko aiemmin tehty kantasolusiirto. Brentuksimabivedotiinin CD30‑antigeeniin kohdistuva vaikutusmekanismi, CD30:n jatkuva ilmentymä klassisessa Hodgkinin lymfoomassa, systeemisessä anaplastisessa suurisoluisessa lymfoomassa ja CD30‑positiivisessa ihon T‑solulymfoomassa, sekä hoitokirjot ja kliininen näyttö CD30‑positiivisissa maligniteeteissa useiden hoitotapojen jälkeen ovat biologinen peruste valmisteen käytölle uusiutunutta tai refraktaarista klassista Hodgkinin lymfoomaa ja systeemistä anaplastista suurisoluista lymfoomaa sairastavien hoidossa, aiemmasta autologisesta kantasolusiirrosta riippumatta, sekä CD30‑positiivista ihon T‑solulymfoomaa sairastavien hoidossa vähintään yhden aiemman systeemisen hoidon jälkeen.
Muiden vasta‑aineisiin liittyvien toimintojen mahdollista osuutta vaikutusmekanismiin ei ole poissuljettu.
Farmakodynaamiset vaikutukset
Sydämen elektrofysiologia
46 potilasta, joilla oli CD30‑antigeeniä ilmentäviä hematologisia maligniteetteja, pystyttiin arvioimaan niiden 52 potilaan joukosta, jotka saivat 1,8 mg/kg brentuksimabivedotiinia 3 viikon välein sydänturvallisuutta selvittäneessä, yhdellä hoitoryhmällä tehdyssä avoimessa faasin 1 monikeskus‑tutkimuksessa. Ensisijaisena tavoitteena oli arvioida brentuksimabivedotiinin vaikutusta sydämen kammioiden repolarisaatioon, ja etukäteen määritelty ensisijainen analyysi koski QTc‑ajan muutosta lähtötilanteen ja hoitojakson 1 useiden eri ajankohtien välillä.
Ylempi 90 %:n luottamusväli QTc‑aikaan kohdistuneen keskimääräisen vaikutuksen suhteen oli < 10 msek kaikkina lähtötilanteen jälkeisinä hoitojakson 1 ja hoitojakson 3 mittausajankohtina. Näiden tietojen perusteella brentuksimabivedotiini ei pidennä QT‑aikaa kliinisesti merkitsevästi, kun sitä annetaan 1,8 mg/kg kolmen viikon välein potilaille, joilla on CD30‑antigeeniä ilmentäviä maligniteetteja.
Kliininen teho ja turvallisuus
Hodgkinin lymfooma
Tutkimus C25003
ADCETRIS‑valmisteen tehoa ja turvallisuutta arvioitiin satunnaistetussa, avoimessa, 2‑ryhmäisessä monikeskustutkimuksessa 1 334 potilaalla, joilla oli levinnyt, aiemmin hoitamaton Hodgkinin lymfooma ja jotka saivat ADCETRIS‑valmistetta yhdessä solunsalpaajahoidon kanssa (doksorubisiini [A], vinblastiini [V] ja dakarbatsiini [D] [AVD]). Tutkimukseen ei otettu potilaita, joilla oli nodulaarinen lymfosyyttivaltainen Hodgkinin lymfooma (NLPHL). Kaikilla oli histologisesti vahvistettu CD30‑positiivinen tauti. 62 prosentilla potilaista oli imusolmukealueiden ulkopuolista tautia. Tutkimuksen 1 334 potilaasta 664 potilasta satunnaistettiin ADCETRIS + AVD ‑ryhmään (A+AVD) ja 670 potilasta ABVD‑ryhmään (doksorubisiini [A], bleomysiini [B], vinblastiini [V] ja dakarbatsiini [D]). Potilaat stratifioitiin International Prognostic Factor Project ‑riskitekijöiden (IPFP) lukumäärän ja alueen perusteella. Potilaat saivat hoitoa kunkin 28‑päiväisen hoitojakson päivinä 1 ja 15. Tällöin heille annettiin 1,2 mg/kg ADCETRIS‑valmistetta 30 minuuttia kestävänä laskimoinfuusiona sekä doksorubisiinia (25 mg/m2), vinblastiinia (6 mg/m2) ja dakarbatsiinia (375 mg/m2). Annettujen hoitojaksojen mediaanimäärä oli 6 (vaihteluväli 1–6 hoitojaksoa). Taulukossa 8 esitetään yhteenveto potilaiden ja taudin lähtötasotiedoista. Kahden eri hoitoryhmän potilaiden ja tautien lähtötasotietojen välillä ei ollut merkittäviä eroja.
Taulukko 8: Yhteenveto potilaiden ja taudin lähtötasotiedoista vaiheen 3 tutkimuksessa, jossa arvioitiin aiemmin hoitamattoman Hodgkinin lymfooman hoitoa
| Potilaiden tiedot | ADCETRIS + AVD n = 664 | ABVD n = 670 |
| Iän mediaani (vaihteluväli) | 35 v (18–82) | 37 v (18–83) |
| ≥ 65‑vuotiaat, n (%) | 60 (9) | 62 (9) |
| Sukupuoli, n (%) | 378 miestä (57) 286 naista (43) | 398 miestä (59) 272 naista (41) |
| ECOG‑luokka, n (%) | ||
| 0 | 376 (57) | 378 (57) |
| 1 | 260 (39) | 263 (39) |
| 2 | 28 (4) | 27 (4) |
| Puuttuu | 0 | 2 |
| Taudin tiedot | ||
| Mediaaniaika Hodgkinin lymfooman toteamisesta ensimmäiseen annokseen (vaihteluväli) | 0,92 kk (0,1–21,4) | 0,89 kk (0,0–81,4) |
| Taudin levinneisyysastea Hodgkinin lymfooman toteamishetkellä, n (%) | ||
| III | 237 (36) | 246 (37) |
| IV | 425 (64) | 421 (63) |
| Ei soveltuva | 1 (< 1) | 1 (< 1) |
| Puuttuu | 0 | 2 (< 1) |
| Imusolmukealueiden ulkopuolinen tauti diagnoosihetkellä, n (%) | 411 (62) | 416 (62) |
| IPFPb‑riskitekijät, n (%) | ||
| 0–1 | 141 (21) | 141 (21) |
| 2–3 | 354 (53) | 351 (52) |
| 4–7 | 169 (25) | 178 (27) |
| Luuydinaffisiota diagnoosihetkellä tai tutkimukseenottohetkellä, n (%) | 147 (22) | 151 (23) |
| B‑oireitaa, n (%) | 400 (60) | 381 (57) |
a. Ann Arbor ‑luokituksen mukaisesti
b. IPFP = International Prognostic Factor Project
Tutkimuksen C25003 ensisijainen päätetapahtuma oli riippumattoman arviointielimen (IRF) arvioima muokattu etenemättömyysaika (mPFS), joka määriteltiin ajaksi satunnaistamisesta taudin etenemiseen, kuolemaan tai tilanteeseen, jossa ensilinjan hoidon päättymisen jälkeen todettiin riippumattoman arviointielimen arvion mukaan ei‑täydellinen vaste (non‑CR) ja tämän jälkeen annettiin syöpähoitoa. Muokatun tapahtuman ajankohta oli päivämäärä, jona ensilinjan hoidon päättymisen jälkeen tehty PET‑kuvaus osoitti ensimmäisen kerran, ettei potilaalla ollut täydellistä vastetta (CR); tämä määriteltiin tilanteeksi, jossa Deauville‑pisteet olivat ≥ 3. Riippumattoman arviointielimen arvioimaa mPFS‑mediaania ei saavutettu kummassakaan hoitoryhmässä. Lähtöryhmien mukaisessa populaatiossa (ITT) saavutetut tulokset osoittivat, että muokattu etenemättömyysaika oli ADCETRIS + AVD ‑ryhmässä tilastollisesti merkitsevästi parempi; stratifioitu hasardisuhde (HR) oli 0,770 (95 % lv 0,603; 0,983, p = 0,035) eli mPFS‑tapahtumien riski oli ADCETRIS + AVD ‑ryhmässä 23 % pienempi kuin ABVD‑ryhmässä.
Taulukossa 9 esitetään tehotulokset muokatun etenemättömyysajan ja kokonaiselossaolon (OS) suhteen ITT‑populaatiossa.
Taulukko 9: Tehotulokset aiemmin hoitamattoman Hodgkinin lymfooman hoidossa, kun potilaille annettiin 1,2 mg/kg ADCETRIS + AVD ‑hoito kunkin 28‑päiväisen hoitojakson päivinä 1 ja 15 (ITT‑populaatio)
| Lähtöryhmien mukainen populaatio (ITT) | |||
ADCETRIS + AVD n = 664 | ABVD n = 670 | Stratifioitu hasardisuhde ja p‑arvo | |
| Tapahtumia (%) | 117 (18) | 146 (22) | 0,77 (95 % lv [0,60; 0,98]) p‑arvo = 0,035 |
| Riippumattoman arviointielimen arvioima mPFSa 2 vuoden kohdalla (%) | 82,1 (95 % lv [78,8; 85,0]) | 77,2 (95 % lv [73,7; 80,4]) | |
Kokonaiselossaolob Kuolemantapaukset (%) | 28 (4) | 39 (6) | 0,73 (95 % lv [0,45; 1,18]) p‑arvo = 0,199 |
a Analyysiajankohtana mPFS‑tietojen mediaaniseuranta‑aika oli molemmissa ryhmissä 24,6 kk.
b. Tiedot perustuvat elossaolon välianalyysiin.
Kuva 1: Riippumattoman arviointielimen (IRF) arvioimat mPFS‑tulokset ITT‑populaatiossa (ADCETRIS + AVD vs. ABVD)
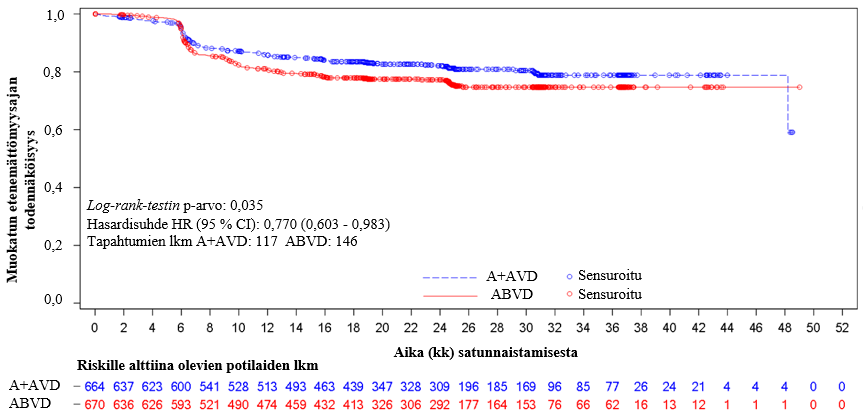
Muut toissijaiset tehon päätetapahtumat, mukaan lukien CR‑vasteen osuus ja ORR satunnaistetun protokollan lopussa, CR‑osuus ensilinjan hoidon lopussa, PET‑negatiivisten potilaiden osuus hoitojakson 2 lopussa, vasteen kesto (DOR), täydellisen remission kesto (DOCR), tauditon elossaoloaika (DFS) ja tapahtumaton elossaolo‑aika (EFS), suosivat kaikki trendinomaisesti ADCETRIS + AVD ‑ryhmää ITT‑populaatiossa.
IRF:n suorittamissa ennalta määritellyissä mPFS-tulosten alaryhmäanalyyseissa ei havaittu kliinisesti merkittävää eroa kahden hoitoryhmän välillä iäkkäillä potilailla (ikä ≥ 60 vuotta [n = 186] [HR = 1,00, 95 % lv (0,58, 1,72)] tai ≥ 65 vuotta [n = 122] [HR = 1,01, 95 % lv (0,53, 1,94)]) eikä potilailla, joilla ei ollut imusolmukealueiden ulkopuolista tautia (n = 445) (HR = 1,04, 95 % lv [0,67, 1,62]).
Kokonaiselossaoloa arvioitiin lopullisessa analyysissa, jossa käytetyissä tiedoissa seurannan mediaanikesto oli kokonaiselossaolon osalta yli 7 vuotta. ITT populaatiossa kuolleiden osuus oli ADCETRIS + AVD ryhmään satunnaistettujen potilaiden joukossa pienempi (46 kuolemantapausta, 7 %) kuin ABVD ryhmään satunnaistettujen potilaiden joukossa (69 kuolemantapausta, 10 %; HR = 0,62, 95 % lv [0,423, 0,899]), katso kuva 2. Levinneisyysasteen III tautia sairastavien potilaiden kohdalla kuolleiden osuus oli ADCETRIS + AVD ryhmään satunnaistettujen joukossa sama (20 kuolemantapausta, 8 %) kuin ABVD ryhmään satunnaistettujen joukossa (20 kuolemantapausta, 8 %; HR = 1,01, 95 % lv [0,542, 1,874]). Levinneisyysasteen IV tautia sairastavien potilaiden kohdalla kuolleiden osuus oli A + AVD ryhmään satunnaistettujen joukossa pienempi (26 kuolemantapausta, 6 %) kuin ABVD ryhmään satunnaistettujen joukossa (48 kuolemantapausta, 11 %; HR = 0,49, 95 % lv [0,303, 0,790]). Kokonaiselossaolon alaryhmän analyysi ei osoittanut kliinisesti merkitsevää eroa kahden hoitoryhmän välillä potilailla, joilla ei ollut imusolmukealueiden ulkopuolista tautia (n = 445) (HR = 1,28, 95 % lv [0,710, 2,303]).
Kuva 2: Kokonaiselossaolon lopullinen analyysi (ADCETRIS + AVD vs. ABVD) (ITT, 7+ vuoden seuranta
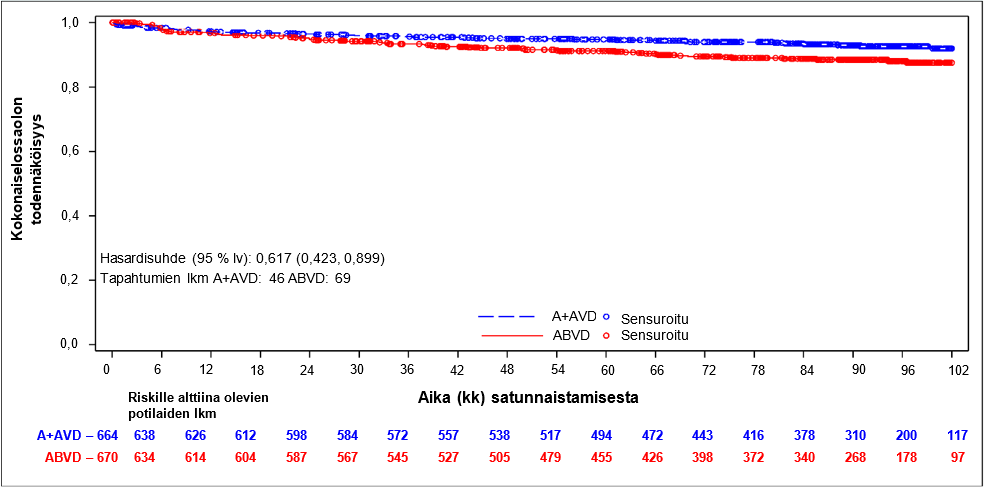
Ensisijaisessa analyysissa ITT‑populaation potilaita, jotka saivat myöhemmin salvage‑hoitoa tai suuriannoksista solunsalpaajahoitoa ja siirteen, oli ADCETRIS + AVD ‑hoitoryhmässä 33 % vähemmän (salvage‑hoidon n = 66; suuriannoksisen solunsalpaajahoidon ja siirteen n = 36) kuin ABVD‑hoitoryhmässä (n = 99 ja n = 54). Levinneisyysasteen IV potilaiden joukossa ADCETRIS + AVD ‑hoitoryhmässä oli 35 % vähemmän potilaita, jotka saivat myöhemmin salvage‑hoitoa (n = 45), kuin ABVD‑hoitoryhmässä (n = 69); ADCETRIS + AVD ‑hoitoryhmässä myös suuriannoksista solunsalpaajahoitoa ja siirteen saaneiden määrä (n = 29) oli 22 % pienempi kuin ABVD‑ryhmässä (n = 37).
Tutkimus HD21
ADCETRIS-valmisteen (brentuksimabivedotiini [Br]) turvallisuutta ja tehoa arvioitiin avoimessa, prospektiivisessa, vaiheen 3 monikeskustutkimuksessa 1 500 potilaalla, joilla oli aiemmin hoitamaton levinneisyysasteen IIB Hodgkinin lymfooma, johon liittyi suuri mediastinaalinen massa ja/tai ekstranodaalisia leesioita, tai levinneisyysasteen III tai IV Hodgkinin lymfooma, yhdessä solunsalpaajahoidon kanssa (etoposidi [E], syklofosfamidi [C], doksorubisiini [A], dakarbatsiini [D], deksametasoni [D] [BrECADD]). Näistä 1 500 potilaasta 751 satunnaistettiin BrECADD-ryhmään ja 749 eBEACOPP-ryhmään (eskaloitu bleomysiini [B], etoposidi [E], doksorubisiini [A], syklofosfamidi [C], vinkristiini [O], prokarbatsiini [P] ja prednisoni [P]). Potilaat ositettiin ryhmiin tutkimukseen ottoalueen, iän, sukupuolen ja IPS (International Prognostic Score) pistemäärän mukaan. BrECADD-ryhmän potilaille annettiin jokaisen 21 päivän hoitojakson päivänä 1 ADCETRIS-valmistetta 1,8 mg/kg 30 minuuttia kestävänä laskimoinfuusiona. Potilaat saivat myös solunsalpaajahoitoa, mukaan lukien syklofosfamidia 1250 mg/m2, doksorubisiinia 40 mg/m2, etoposidia tai etoposidifosfaattia 150 mg/m2, dakarbatsiinia 250 mg/m2 ja deksametasonia 40 mg (ks. kohta Annostus ja antotapa).
Kaikki hoidetut potilaat saivat primaariprofylaksiaa kasvutekijävalmisteella (G-CSF) (ks. kohta Annostus ja antotapa). Potilaille tehtiin PET-kuvantamisella uusi levinneisyysmääritys kahden hoitojakson jälkeen. PET-negatiivisille potilaille annettiin kaikkiaan 4 hoitojaksoa, ja PET-positiivisille potilaille kaikkiaan 6 hoitojaksoa. Hoitojaksojen mediaanilukumäärä oli 4 kummassakin ryhmässä (vaihteluväli 1–6 hoitojaksoa).
Taulukossa 10 esitetään yhteenveto potilaiden ja taudin ominaisuuksista lähtötilanteessa. Potilaiden ja taudin ominaisuuksissa ei ollut olennaisia eroja tutkimuksen kahden ryhmän välillä.
Taulukko 10: Yhteenveto potilaiden ja taudin ominaisuuksista aiemmin hoitamatonta Hodgkinin lymfoomaa koskevassa vaiheen 3 tutkimuksessa
| Potilaiden tiedot | BrECADD n = 751 | eBEACOPP n = 749 |
| Mediaani-ikä (vaihteluväli) | 31 v (18–60) | 31 v (18–60) |
| < 45-vuotiaat, n (%) | 590 (79) | 584 (78) |
| 45–60-vuotiaat, n (%) | 161 (21) | 165 (22) |
| Sukupuoli, n (%) | 419 miestä (56) 332 naista (44) | 419 miestä (56) 330 naista (44) |
| ECOG-luokka, n (%) | ||
| 0 | 514 (68) | 521 (70) |
| 1 | 223 (30) | 205 (27) |
| 2 | 11 (1) | 18 (2) |
| Puuttuu | 3 (< 1)0 | 5 (< 1) |
| Taudin tiedot | ||
| Mediaaniaika Hodgkinin lymfooman toteamisesta satunnaistamiseen (vaihteluväli) | 0,6 kk (0,12) | 0,6 kk (0,10) |
| Taudin levinneisyysastea Hodgkinin lymfooman toteamishetkellä, n (%) | ||
| II | 118 (16) | 117 (16) |
| III | 298 (40) | 293 (39) |
| IV | 332 (44) | 334 (45) |
| Puuttuu | 3 (< 1) | 5 (< 1) |
| IPSb-ryhmät, n (%) | ||
| 0–2 | 394 (52) | 403 (54) |
| 3–7 | 357 (48) | 346 (46) |
| B-oireitaa n (%) | 517 (69) | 501 (67) |
a. Ann Arbor ‑luokituksen mukaisesti
b. IPS = International Prognostic Score
Tutkimuksen HD21 tutkimusasetelmaan kuului kaksi yhdistettyä ensisijaista päätetapahtumaa (hoitoon liittyvä sairastavuus [Treatment Related Morbidity, TRMB] ja etenemisvapaa elossaoloaika [PFS; tutkijan arvioima ja riippumattoman arviointielimen vahvistama]). Tutkimuksen ensimmäinen yhdistetty ensisijainen tavoite oli osoittaa BrECADD-hoidon eBEACOPP-hoitoa vähäisempi toksisuus hoitoon liittyvän sairastavuuden perusteella arvioituna. Jos vähäisempi toksisuus pystyttiin osoittamaan paremmuustestillä, toinen yhdistetty ensisijainen tavoite oli lisäksi osoittaa, että BrECADD-hoidon teho on vähintään samanveroinen kuin eBEACOPP-hoidon teho etenemisvapaan elossaoloajan perusteella arvioituna.
Hoitoon liittyvän sairastavuuden määritelmänä oli mikä tahansa asteen 3 tai asteen 4 CTCAE-kriteerien mukainen elintoksisuus tai asteen 4 hematologinen toksisuus ensisijaisen solunsalpaajahoidon aikana, mukaan lukien enintään 30 päivän jakso viimeisen solunsalpaaja-annoksen jälkeen.
Ensisijaisen analyysin aikaan osoitettiin BrECADD-hoidon paremmuus hoitoon liittyvän sairastavuuden perusteella arvioituna. Riskin absoluuttinen pieneneminen oli -16,7 prosenttiyksikköä, ja suhteellinen riski pieneni tilastollisesti merkitsevästi. Toinen ensisijainen yhdistetty päätetapahtuma, etenemisvapaa elossaoloaika toteutumattomista käynneistä ja uuden syöpähoidon aloittamisesta riippumatta, täytti vähintään samanveroisuutta koskevan kriteerin. Riski oli BrECADD-hoitoryhmässä tilastollisesti merkitsevästi pienempi kuin eBEACOPP-ryhmässä (osittamaton HR = 0,62 [kerrannaisuuden suhteen korjattu 95 %:n luottamusväli: 0,369, 1,040]) (tietojen katkaisupäivä 31.12.2022).
Hoitoon liittyvän sairastavuuden osoitettiin olevan BrECADD-ryhmässä vähäisempää (42 %) kuin eBEACOPP-ryhmässä (58,7 %). Tämä johtui pitkälti asteen 4 hematologisten toksisuuksien vähenemisestä (31,2 % BrECADD-ryhmässä ja 52,1 % eBEACOPP-ryhmässä).
Hoitoon liittyvä sairastavuus on esitetty hoitoryhmittäin taulukossa 11. Päivitetyn etenemisvapaan elossaoloajan (PFS) ja kokonaiseloonjäämisen (OS) analyysin tulokset on esitetty taulukossa 12 (tietojen katkaisupäivä 31.10.2023).
Taulukko 11: Hoitoon liittyvä sairastavuus (TRMB) hoitoryhmittäin (turvallisuuspopulaatio)
BrECADD n = 747 | eBEACOPP n = 741 | |
| Hoitoon liittyvä sairastavuus, potilaiden lukumäärä | 314 (42) | 435 (59) |
| Akuutti hematologinen toksisuus, Aste 4 | 233 (31) | 386 (52) |
| Anemia | 3 (< 1) | 3 (< 1) |
| Trombosytopenia | 227 (30) | 383 (52) |
| Infektio | 13 (2) | 10 (1) |
| Akuutti elintoksisuus; Aste 3 tai Aste 4 | 139 (19) | 129 (17) |
| Sydän | 18 (2) | 10 (1) |
| Ruoansulatuskanava (paitsi oksentelu, pahoinvointi, limakalvotulehdus) | 58 (8) | 32 (4) |
| Maksa ja sappi | 37 (5) | 22 (3) |
| Hermosto | 20 (3) | 40 (5) |
| Perifeerinen sensorinen neuropatia | 9 (1) | 17 (2) |
| Perifeerinen motorinen neuropatia | 2 (< 1) | 1 (< 1) |
| Hermosto, muu kuin neuropatia | 11 (2) | 24 (3) |
| Munuaiset ja virtsatiet | 7 (< 1) | 10 (1) |
| Hengityselimet, rintakehä ja välikarsina | 25 (3) | 35 (5) |
| Ero, % (BrECADD–eBEACOPP) | -16,7 | |
| Täsmällinen 95 %:n luottamusväliLV | -21,7, -11,5 | |
Taulukko 12: Teho aiemmin hoitamattomilla Hodgkinin lymfoomaa sairastavilla potilailla, jotka saivat BrECADD-hoitoa annoksena 1,8 mg/kg 21 päivän hoitojakson ajan (päivitetyn PFSa-analyysin tietojen katkaisupäivä 31.10.2023)
| ITT-populaatio | ||
BrECADD n = 751 | eBEACOPP n = 749 | |
| PFS-tapahtumien määrä (%) | 44 (5,9) | 65 (8,7) |
| PFS-hasardisuhde (95 %:n luottamusväli) | 0,664 (0,453, 0,973) | |
| Arvioitu PFSb,c (95 %:n luottamusväli) | ||
| 3 vuoden aikapisteessä | 95,2 (93,4, 96,6) | 92,4 (90,2, 94,1) |
| 5 vuoden aikapisteessä | 92,8 (90,0, 94,9) | 90,2 (87,5, 92,3) |
| Kokonais-elossaolod Kuolemien määrä (%) | 12 (1,6) | 13 (1,7) |
| OS-hasardisuhde (95 %:n luottamusväli) | 0.919 (0,419, 2,015) | |
| Arvioitu OS-osuus (95 %:n LV) | ||
| 3 vuoden aikapisteessä | 98,9 (97,8, 99,4) | 98,9 (97,8, 99,4) |
| 5 vuoden aikapisteessä | 98,1 (96,5, 98,9) | 97,9 (96,4, 98,8) |
- PFS-analyysissa ei otettu huomioon välitapahtumia eli toteutumatta jääneitä käyntejä ja uuden syöpähoidon aloittamista.
- PFS-osuuksien tutkijakohtaiseen arviointiin käytetään Kaplan–Meierin menetelmää, ja riippumaton arviointielin vahvistaa tiedot.
- Analyysihetkellä PFS:n seurannan mediaaniarvo ITT-populaatiossa oli 50,8 kuukautta.
- Deskriptiivisen analyysin tiedot.
Täydellisen vasteen (CR) keston osalta todettiin BrECADD-ryhmässä kliinisesti merkitsevä hyöty eBEACOPP-ryhmään verrattuna ITT-populaatiossa, kun taas muut toissijaiset tehon päätetapahtumat, kuten objektiivisen vasteen saaneiden osuus (ORR) ja täydellisen vasteen saaneiden osuus solunsalpaajahoidon lopussa, olivat samankaltaiset molemmissa ryhmissä.
Tutkimus SGN35‑005
ADCETRIS‑hoidon tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa 2‑ryhmäisessä monikeskustutkimuksessa, johon osallistuneilla 329:llä Hodgkinin lymfoomaa sairastaneella potilaalla oli taudin uusiutumisen tai etenemisen riski autologisen kantasolusiirron jälkeen. Tutkimuksesta suljettiin pois potilaat, joilla tiedettiin olevan aivo‑/aivokalvosairaus, myös potilaat, joilla oli anamneesissa PML. Potilaiden ominaisuudet, ks. taulukko 8. Koko 329 potilaan ryhmästä 165 potilasta satunnaistettiin hoitoryhmään ja 164 potilasta lumeryhmään. Potilaiden tuli saada ensimmäinen annos autologisesta kantasolusiirrosta toipumisen jälkeen (30‑45 päivän kuluttua autologisesta kantasolusiirrosta). Potilaat saivat 1,8 mg/kg ADCETRIS‑valmistetta tai sitä vastaavaa lumelääkettä laskimoon 30 minuutin kuluessa 3 viikon välein enintään 16 hoitojakson ajan.
Soveltuvilla potilailla tuli olla ainakin yksi seuraavista riskitekijöistä:
- Ensilinjan hoidolle refraktaarinen Hodgkinin lymfooma
- Hodgkinin lymfooma on uusiutunut tai edennyt < 12 kk:n kuluessa ensilinjan hoidon päättymisestä.
- Potilaalla on imusolmukealueiden ulkopuolelle levinnyt tauti, kun tauti uusiutui ennen autologista kantasolusiirtoa; kattaa myös imusolmukemassojen ulottumisen imusolmukkeiden ulkopuolelle niiden viereisiin vitaalielimiin.
Taulukko 13: Yhteenveto potilaiden ja taudin ominaisuuksista lähtötilanteessa faasin 3 tutkimuksessa Hodgkinin lymfoomaa sairastavilla potilailla, joille oli tehty autologinen kantasolusiirto
| Potilaiden tiedot | ADCETRIS n = 165 | Lume n = 164 | |
| Iän mediaani, v (vaihteluväli) | 33 v (18–71) | 32 v (18–76) | |
| Sukupuoli | 76 m (46 %) / 89 n (54 %) | 97 m (59 %) / 67 n (41 %) | |
| ECOG‑luokka | |||
| 0 | 87 (53 %) | 97 (59 %) | |
| 1 | 77 (47 %) | 67 (41 %) | |
| 2 | 1 (1 %) | 0 | |
| Taudin piirteet | |||
| Aiempien solunsalpaajahoitojen määrän mediaani (vaihteluväli) | 2 (2–8) | 2 (2–7) | |
| Mediaaniaika Hodgkinin lymfooman toteamisesta ensimmäiseen annokseen (vaihteluväli) | 18,7 kk (6,1–204,0) | 18,8 kk (7,4–180,8) | |
| Taudin levinneisyysaste Hodgkinin lymfooman toteamishetkellä | |||
| Aste I | 1 (1 %) | 5 (3 %) | |
| Aste II | 73 (44 %) | 61 (37 %) | |
| Aste III | 48 (29 %) | 45 (27 %) | |
| Aste IV | 43 (26 %) | 51 (31 %) | |
| Ei tiedossa | 0 | 2 (1 %) | |
| PET‑kuvausstatus ennen autologista kantasolusiirtoa | |||
| FDG‑POSITIIVINEN | 64 (39 %) | 51 (31 %) | |
| FDG‑NEGATIIVINEN | 56 (34 %) | 57 (35 %) | |
| EI TEHTY | 45 (27 %) | 56 (34 %) | |
| Imusolmukealueiden ulkopuolelle levinnyt tauti, kun tauti uusiutui ennen autologista kantasolusiirtoa | 54 (33 %) | 53 (32 %) | |
| B‑oireitaa | 47 (28 %) | 40 (24 %) | |
| Paras vaste salvage‑hoitoon ennen autologista kantasolusiirtoab | |||
| Täydellinen vaste | 61 (37 %) | 62 (38 %) | |
| Osittainen vaste | 57 (35 %) | 56 (34 %) | |
| Stabiili tauti | 47 (28 %) | 46 (28 %) | |
| Hodgkinin lymfooman status tavanomaisen ensilinjan solunsalpaajahoidon lopussab | |||
| Refraktaarinen | 99 (60 %) | 97 (59 %) | |
| Uusiutuman ajankohta < 12 kk:n kuluessa | 53 (32 %) | 54 (33 %) | |
| Uusiutuman ajankohta ≥ 12 kk:n kuluessa | 13 (8 %) | 13 (8 %) | |
a. Koskee hoitoon vastaamatonta tautia tai taudin etenemistä tai uusiutumista ensilinjan hoidon jälkeen
b. Stratifiointitekijöitä satunnaistamisen yhteydessä
Tehotulokset ensisijaisen päätetapahtuman ensisijaisesta analyysistä lähtien esitetään taulukossa 14. Ensisijainen päätetapahtuma eli etenemisvapaan elossaoloajan piteneminen riippumattoman arviointielimen arvion mukaan saavutettiin, ja etenemisvapaan elossaoloajan mediaani oli hoitoryhmässä 18,8 kk pidempi.
Taulukko 14: Tehotulokset Hodgkinin lymfoomaa sairastavilla potilailla, joilla on suurentunut taudin uusiutumisen tai etenemisen riski autologisen kantasolusiirron jälkeen ja jotka saivat 1,8 mg/kg ADCETRIS‑valmistetta 3 viikon välein (ITT‑populaatio, ensisijainen analyysi)
ADCETRIS n = 165 | Lume n = 164 | Stratifioitu hasardisuhde | |
| Etenemisvapaa elossaoloa | Mediaani riippumattoman arviointielimen mukaan | ||
42,9 kk 95 % lv [30,4; 42,9] | 24,1 kk (95 % lv [11,5; -]) | 0,57 (95 % lv [0,40; 0,81]) Stratifioidun log‑rank‑testin P = 0,001 | |
| Mediaani tutkijan mukaan | |||
Ei saavutettu (95 % lv [26,4; -]) | 15,8 kk (95 % lv [8,5; -]) | 0,5 (95 % lv [0,36; 0,70])b | |
| Kokonaiselossaolo | Kuolemantapauksia (%) | ||
| 28 (17) | 25 (15) | 1,15 (95 % lv [0,67; 1,97]) | |
a. Ensisijaisen analyysin ajankohtana molempien ryhmien mediaaniseuranta‑aika oli 30 kk (vaihteluväli 0–50).
b. Etenemisvapaata elossaoloaikaa ei analysoitu stratifioidulla log‑rank‑testillä tutkijan mukaan.
Riippumattoman arviointielimen mukaan tehdyissä ennalta määrätyissä etenemisvapaan elossaoloajan alaryhmäanalyyseissä otettiin huomioon potilaan paras vaste salvage‑hoitoon ennen autologista kantasolusiirtoa, Hodgkinin lymfooman status ensilinjan hoidon jälkeen, ikä, sukupuoli, lähtöpaino, lähtötilanteen ECOG‑toimintakykyluokka, hoitojen määrä ennen autologista kantasolusiirtoa, maantieteellinen alue, PET‑status ennen autologista kantasolusiirtoa, B‑oireet kun tauti etenee ensilinjan hoidon jälkeen ja taudin tilanne imusolmukealueiden ulkopuolella ennen autologista kantasolusiirtoa. Analyyseissä todettiin johdonmukainen trendi, jossa ADCETRIS‑hoitoa saaneet potilaat saavuttivat suuremman hyödyn kuin lumehoitoa saaneet; poikkeuksena olivat ≥ 65‑vuotiaat potilaat (n = 8).
Hoitoryhmän ja lumeryhmän elämänlaadussa ei todettu eroja. Hodgkinin lymfoomaa sairastaneiden, suurentuneen uusiutumisriskin omaavien potilaiden terveydenhuollon resurssien käyttöä koskeneessa analyysissä todettiin, että ADCETRIS‑hoitoryhmän potilailla oli vähemmän sairaalahoitoja ja poliklinikkakäyntejä ja että tämän ryhmän potilaille ja omaishoitajille kertyi vähemmän poissaolopäiviä töistä ja poissaoloja muista toimista kuin lumeryhmän potilaille.
Päivitetyssä, 3 seurantavuoden jälkeen tehdyssä analyysissä vahvistettiin, että etenemisvapaa elossaoloaika piteni riippumattoman arviointielimen mukaan analysoituna (HR = 0,58 [95 % lv (0,41; 0,81)]).
Tutkimuksen päätyttyä, noin 10 vuotta sen jälkeen, kun ensimmäinen potilas aloitti tutkimuksessa, tutkijan arvioimassa etenemisvapaassa elossaoloajassa nähtävä hyöty säilyi (hasardisuhde = 0,51 [95 % lv (0,37, 0,71)]). Kokonaiselossaoloa koskevat tulokset olivat yhdenmukaiset ensisijaisen analyysin aikaan raportoitujen tulosten kanssa (hasardisuhde = 1,11 [95 % lv (0,72, 1,70)]). Kuvassa 3 esitetään etenemisvapaa elossaoloaika tutkijan mukaan ITT‑populaatiossa tutkimuksen lopussa.
Kuva 3:Etenemisvapaan elossaoloajan Kaplan–Meier‑kuvaaja tutkijan mukaan analysoituna (ITT‑populaatio, tutkimuksen päättyminen)
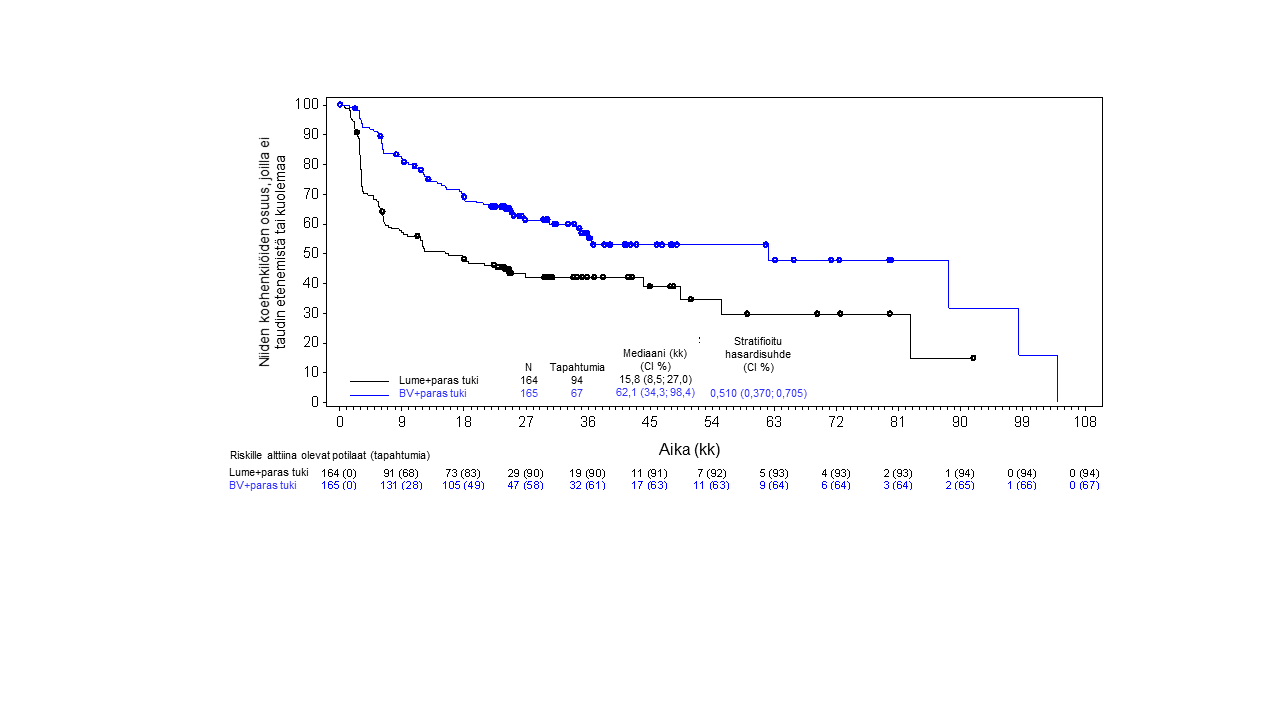
Riskitekijöiden jälkianalyysit (post hoc)
Jälkianalyyseissä arvioitiin ensisijaisen päätetapahtuman ensisijaisen analyysin osalta suurentuneen riskin (riskitekijöiden määrän) vaikutusta kliiniseen hyötyyn (taulukko 10). Näissä analyyseissä arvioituja edustavia riskitekijöitä olivat:
- Hodgkinin lymfooma < 12 kk:n kohdalla tai ensilinjan hoidolle refraktaarinen Hodgkinin lymfooma
- Paras vaste viimeisimmälle salvage‑hoidolle joko osittainen vaste tai taudin etenemisen pysähtyminen, määritetty TT‑ ja/tai PET‑kuvauksella
- Imusolmukealueiden ulkopuolelle levinnyt tauti sen uusiutuessa ennen autologista kantasolusiirtoa
- B‑oireita taudin uusiutuessa ennen autologista kantasolusiirtoa
- Vähintään kaksi aiempaa salvage‑hoitoa.
Näiden jälkianalyysien tulokset viittaavat siihen, että kliininen hyöty on suurempi potilailla, joilla on vähintään kaksi riskitekijää, mutta yksittäisten riskitekijöiden välillä ei ole eroja. Hoidon ei ole todettu parantaneen etenemisvapaata elossaoloaikaa eikä kokonaiselossaoloaikaa potilailla, joilla oli yksi taudin uusiutumisen tai etenemisen riskitekijä.
Taulukko 15: Yhteenveto etenemisvapaan elossaoloajan tiedoista riippumattoman arviointielimen mukaan analysoituna sekä kokonaiselossaoloajan tiedoista riskitekijöiden määrän mukaan analysoituna faasin 3 tutkimuksessa Hodgkinin lymfoomaa sairastavilla potilailla autologisen kantasolusiirron jälkeen (ensisijainen analyysi)
| Etenemisvapaa elossaolo riippumattoman arviointielimen mukaan | ||||||
| Riskitekijöitä = 1 | Riskitekijöitä ≥ 2 | Riskitekijöitä ≥ 3 | ||||
ADCETRIS n = 21 | Lume n = 28 | ADCETRIS n = 144 | Lume n = 136 | ADCETRIS n = 82 | Lume n = 84 | |
| Tapaukset, joissa tauti eteni tai potilas kuolia (%) | 9 (43) | 7 (25) | 51 (35) | 68 (50) | 32 (39) | 49 (58) |
| Stratifioitu hasardisuhde | 1,65 (95 % lv [0,60; 4,55])b | 0,49 (95 % lv [0,34; 0,71]) | 0,43 (95 % lv [0,27; 0,68]) | |||
| Kokonaiselossaolo | ||||||
| Riskitekijöitä = 1 | Riskitekijöitä ≥ 2 | Riskitekijöitä ≥ 3 | ||||
ADCETRIS n = 21 | Lume n = 28 | ADCETRIS n = 144 | Lume n = 136 | ADCETRIS n = 82 | Lume n = 84 | |
| Kuolemantapauksiac (%) | 5 (24) | 1 (4) | 23 (16) | 24 (18) | 15 (18) | 16 (19) |
| Stratifioitu hasardisuhde | 7,94 (95 % lv [0,93; 68,06])b | 0,94 (95 % lv [0,53; 1,67]) | 0,92 (95 % lv [0,45; 1,88]) | |||
a. Kuolema ilman edeltävää taudin etenemistä tai vähintään yksi puuttuva arviointikäynti
b. Viittaa stratifioimattoman analyysin tuloksiin
c. Kaikki kuolemantapaukset syystä riippumatta
Päivitetyn analyysin ajankohtana (3 seurantavuoden jälkeen) potilailla, joilla oli vähintään 2 riski‑tekijää, riippumattoman arviointielimen mukaan analysoitu etenemisvapaan elossaolon hasardisuhde oli 0,49 (95 % lv [0,34; 0,71]) ja tutkijan mukaan analysoitu etenemisvapaan elossaolon hasardisuhde 0,41 (95 % lv [0,29; 0,58]) (ks. kuvat 4 ja 5).
Kuva 4: Etenemisvapaan elossaoloajan Kaplan–Meier‑kuvaaja riippumattoman arviointielimen mukaan analysoituna potilailla, jolla oli ≥ 2 riskitekijää (3 vuoden seuranta)
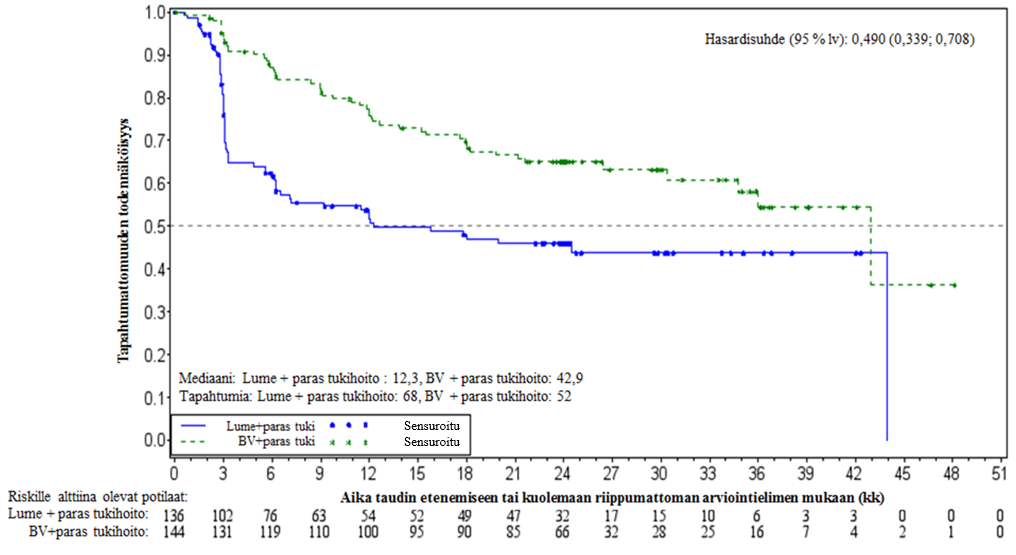
Kuva 5: Etenemisvapaan elossaoloajan Kaplan–Meier‑kuvaaja tutkijan mukaan analysoituna potilailla, jolla oli ≥ 2 riskitekijää (3 vuoden seuranta)
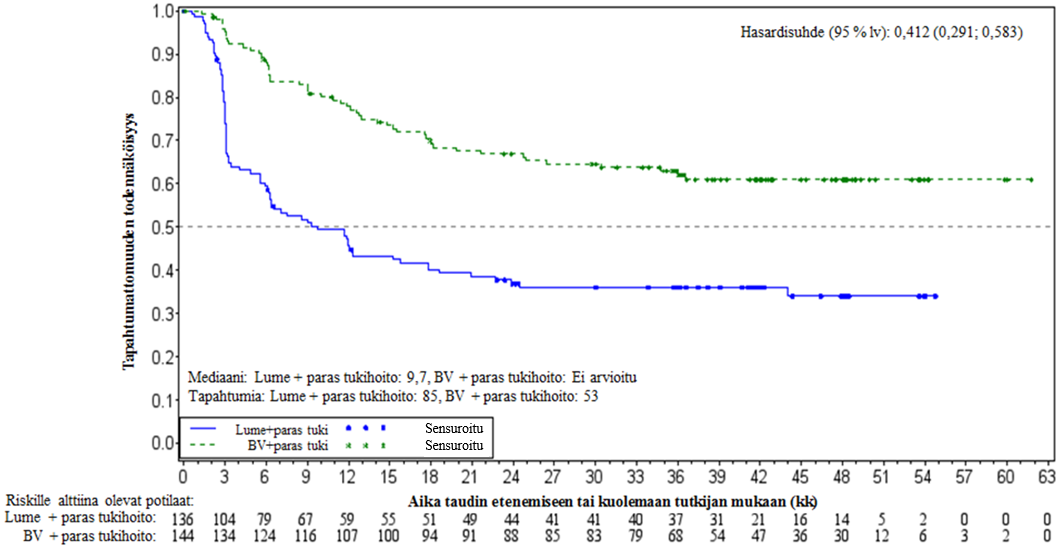
Tutkimuksen päätyttyä, noin 10 vuotta sen jälkeen, kun ensimmäinen potilas aloitti tutkimuksessa, etenemisvapaan elossaoloajan hasardisuhde oli tutkijan arvion mukaan 0,41 (95 % lv [0,29, 0,58]) sellaisilla potilailla, joilla oli 2 tai useampia riskitekijöitä. Etenemisvapaan elossaoloajan hasardisuhde tutkijan arvion mukaan oli 0,38 (95 % lv [0,25, 0,59]) sellaisilla potilailla, joilla oli 3 tai useampia riskitekijöitä. Kokonaiselossaoloa koskevat tulokset olivat yhdenmukaiset ensisijaisen analyysin aikaan raportoitujen tulosten kanssa.
Tutkimus SG035‑0003
ADCETRIS‑monoterapian tehoa ja turvallisuutta arvioitiin yhdellä hoitoryhmällä tehdyssä keskeisessä avoimessa monikeskustutkimuksessa 102:lla uusiutunutta tai refraktaarista Hodgkinin lymfoomaa sairastavalla potilaalla. Yhteenveto lähtötilanteen potilas‑ ja tautitiedoista, ks. taulukko 16 alla.
Taulukko 16: Yhteenveto lähtötilanteen potilas‑ ja tautitiedoista uusiutunutta tai refraktaarista Hodgkinin lymfoomaa koskeneessa faasin 2 tutkimuksessa
| Potilastiedot | n = 102 |
| Mediaani‑ikä, vuotta (vaihteluväli) | 31 vuotta (15–77) |
| Sukupuoli | 48 miestä (47 %) / 54 naista (53 %) |
| ECOG‑toimintakykyluokka | |
| 0 | 42 (41 %) |
| 1 | 60 (59 %) |
| Aiempi ASCT | 102 (100 %) |
| Aiempia solunsalpaajahoitoja | 3,5 (1–13) |
| ASCT:stä taudin ensimmäiseen uusiutumiseen kulunut aika | 6,7 kk (0–131) |
| Histologisesti vahvistettu CD30‑antigeeniä ilmentävä tauti | 102 (100 %) |
| Tautitiedot | |
| Primaaristi refraktaarinen ensilinjan syöpähoidollea | 72 (71 %) |
| Ei vastaa viimeisimpään hoitoon | 43 (42 %) |
| B‑oireita lähtötilanteessa | 35 (33 %) |
| Vaiheen III tauti diagnoosivaiheessa | 27 (26 %) |
| Vaiheen IV tauti diagnoosivaiheessa | 20 (20 %) |
a. Primaaristi refraktaarinen tarkoittaa, ettei HL‑potilaan ensilinjan syöpähoito saanut tautia täydelliseen remissioon tai tauti eteni 3 kuukauden sisällä ensilinjan syöpähoidon päättymisestä.
Kahdeksantoista (18) potilasta (18 %) sai 16 hoitojaksoa ADCETRIS‑valmistetta; hoitojaksojen määrän mediaani oli 9 (vaihteluväli 1–16).
Riippumaton arviointielin (IRF) arvioi vasteen ADCETRIS‑hoitoon päivitettyjen malignin lymfooman vastekriteerien (Cheson, 2007) perusteella. Hoitovaste arvioitiin rintakehän, kaulan, vatsan ja lantion alueen spiraali‑TT‑kuvauksilla, PET‑kuvauksilla ja kliinisten tietojen pohjalta. Vastearvioinnit tehtiin hoitojaksojen 2, 4, 7, 10, 13 ja 16 kohdalla. PET‑kuvaukset tehtiin hoitojaksojen 4 ja 7 kohdalla.
IRF:n arvion mukaan objektiivinen vasteprosentti (ORR) oli 75 % (ITT‑populaatiossa 76 potilasta 102:sta), ja kasvaintaakka pieneni 94 %:lla potilaista. Täydellisen remission (CR) saavutti 33 % potilaista (ITT‑populaatiossa 34 potilasta 102:sta). Kokonaiselossaolon mediaani (OS) oli 40,5 kuukautta (seuranta‑ajan mediaani (ensimmäisestä annoksesta kuolemaan tai viimeiseen yhteydenottoon kulunut aika) oli 35,1 kuukautta (vaihteluväli 1,8–72,9+ kuukautta). Arvioitu kokonaiselossaolo 5 vuoden kohdalla oli 41 % (95 %:n luottamusväli [31 %, 51 %]). Tutkijan tekemät arvioinnit ja kuvien riippumattomat arvioinnit vastasivat yleensä toisiaan. Hoitoa saaneista potilaista 8:lle hoitovasteen saavuttaneelle potilaalle tehtiin myöhemmin allogeeninen kantasolusiirto. Lisää tehotuloksia on taulukossa 17.
Taulukko 17: Tehotulokset uusiutunutta tai refraktaarista Hodgkinin lymfoomaa sairastavilla potilailla, jotka saivat 1,8 mg/kg ADCETRIS‑hoitoa3 viikon välein
| Paras kliininen vaste (n = 102) | IRF n (%) | 95 %:n luottamusväli |
| Objektiivinen vaste (CR + PR) | 76 (75) | 64,9; 82,6 |
| Täydellinen remissio (CR) | 34 (33) | 24,3; 43,4 |
| Osittainen remissio (PR) | 42 (41) | Ei oleellinen |
| Taudin hallinta (CR + PR + SD) | 98 (96) | 90,3; 98,9 |
| Vasteen kesto | IRF:n arvion mukainen mediaani | 95 %:n luottamusväli |
| Objektiivinen vaste (CR + PR) a | 6,7 kuukautta | 3,6; 14,8 |
| Täydellinen remissio (CR) | 27,9 kuukautta | 10,8; EAb |
| Kokonaiselossaoloaika | 95 %:n luottamusväli | |
| Mediaani | 40,5 kuukautta | 28,7; 61,9 |
| Arvioitu kokonaiselossaolo 5 vuoden kohdalla | 41 % | 31 %, 51 % |
a. Vasteen kesto vaihteli 1,2+ kuukaudesta 43+ kuukauteen. Ensimmäisen annoksen jälkeen toteutetun seurannan mediaanikesto oli 9,0 kuukautta potilailla, jotka saavuttivat IRF:n arvion mukaan objektiivisen vasteen (OR).
b. Ei arvioitavissa.
Eksploratiivisessa analyysissä osoitettiin, että noin 64 % ADCETRIS‑hoidetuista Hodgkinin lymfoomaa (kliininen tutkimus SG035‑0003) sairastavista potilaista sai brentuksimabivedotiinista enemmän kliinistä hyötyä kuin edellisestä hoidostaan (mitattuna elinaikana, jolloin tauti ei etene).
Lähtötilanteessa 35 potilaalla (33 %) oli B‑oireita. Näistä potilaista 27:n (77 %) B‑oireet hävisivät täysin 0,7 kuukaudessa (mediaani) ADCETRIS‑hoidon aloittamisen jälkeen.
Tiedot Hodgkinin lymfoomaa sairastavista potilaista, joille ei harkittu kantasolusiirtoa
Tutkimus C25007
Yhden hoitoryhmän faasin 4 tutkimus toteutettiin potilailla, jotka sairastivat uusiutunutta tai refraktaarista Hodgkinin lymfoomaa (n = 60). Potilaat olivat saaneet aiemmin vähintään yhtä sytostaattihoitoa, eikä potilaille harkittu kantasolusiirtoa eikä sytostaattiyhdistelmähoitoa ADCETRIS‑hoidon aloittamishetkellä. Tutkimukseen pääsyn ehtona oli, että potilaille ei ollut aiemmin tehty kantasolusiirtoa. Hoitojaksojen mediaanimäärä oli 7 (vaihteluväli 1–16 hoitojaksoa). Potilaat saivat 1,8 mg/kg ADCETRIS‑valmistetta 3 viikon välein.
Ensisijaisen päätetapahtuman ensisijaisen analyysin aikaan IRF:n arvion mukaan ITT‑populaation objektiivinen vasteprosentti (ORR) oli 50 % (95 % lv, 37; 63 %). Paras CR‑kokonaisvaste raportoitiin 7 potilaalla (12 %); PR raportoitiin 23 potilaalla (38 %). Näillä 30 potilaalla vasteen saavuttamiseen kulunut mediaaniaika (aika ensimmäisestä annoksesta varhaisimpaan osittaiseen remissioon tai täydelliseen remissioon) oli 6 viikkoa (vaihteluväli 5–39 viikkoa). Parhaan kokonaisvasteen saavuttamiseen kulunut mediaaniaika (aika ensimmäisestä annoksesta parhaaseen kliiniseen CR‑ tai PR‑vasteeseen) oli 11 viikkoa (vaihteluväli 5–60 viikkoa). 28 potilasta (47 %) sai kantasolusiirron 7 ADCETRIS‑hoitojakson jälkeen (mediaani; vaihteluväli 4–16 hoitojaksoa). Myös ne 32 potilasta (53 %), jotka eivät saaneet myöhemmin kantasolusiirtoa, saivat ADCETRIS‑valmistetta 7 hoitojakson ajan (mediaani; vaihteluväli 1–16 hoitojaksoa).
Tutkimukseen osallistuneista 60 potilaasta 49 potilasta (82 %) oli saanut aiemmin useampaa kuin yhtä syöpään liittyvää hoitoa ja 11 potilasta (18 %) oli saanut yhtä syöpään liittyvää hoitoa. IRF:n arvion mukaan ORR oli 51 % (95 % lv [36 %, 66 %]) potilailla, jotka olivat saaneet aiemmin useampaa kuin yhtä syöpään liittyvää hoitoa, ja 45 % (95 % lv [17 %, 77 %]) potilailla, jotka olivat saaneet aiemmin yhtä syöpään liittyvää hoitoa. Useampaa kuin yhtä aiempaa syöpään liittyvää hoitoa saaneilla potilailla paras CR‑kokonaisvaste raportoitiin 6 potilaalla (12 %); PR raportoitiin 19 potilaalla (39 %). Yhtä aiempaa syöpään liittyvää hoitoa saaneilla potilailla CR raportoitiin 1 potilaalla (9 %) ja PR 4 potilaalla (36 %). 49:stä useampaa kuin yhtä aiempaa hoitolinjaa saaneesta potilaasta 22 potilasta (45 %) sai myöhemmin kantasolusiirron. 11:stä yhtä aiempaa hoitoa saaneesta potilaasta 6 potilasta (55 %) sai myöhemmin kantasolusiirron.
Tietoja kerättiin myös potilailta (n = 15) annosten suurentamista ja kliinistä farmakologiaa tarkastelleista faasin 1 tutkimuksista sekä NPP‑ohjelman potilailta (n = 26), jotka sairastivat uusiutunutta tai refraktaarista Hodgkinin lymfoomaa ja joille ei ollut tehty autologista kantasolusiirtoa. Potilaiden saama hoito oli 1,8 mg/kg ADCETRIS‑valmistetta kolmen viikon välein.
Lähtötilanteessa potilaiden ominaisuutena oli, että he eivät olleet reagoineet useisiin aiempiin sytostaattihoitojaksoihin (mediaani 3 hoitojaksoa, vaihtelu 1–7) ennen ADCETRIS‑valmisteen antamista. Potilaista 59 %:lla tauti oli edennyt pitkälle (vaihe III tai IV) diagnosointihetkellä.
Faasin 1 tutkimusten tulokset ja NPP‑ohjelmasta saadut kokemukset osoittivat, että uusiutunutta tai refraktaarista Hodgkinin lymfoomaa sairastavilla potilailla, joille ei ollut tehty autologista kantasolusiirtoa, voidaan saavuttaa merkittävä vaste, mikä ilmenee tutkijan arvioimasta, objektiivisesta 54 %:n vasteesta ja 22 %:n täydellisestä remissiosta 5 ADCETRIS‑hoitojakson (mediaani) jälkeen.
Tutkimus SGN35‑006 (uudelleenhoitotutkimus)
Uudelleenhoidon tehoa sellaisilla potilailla, jotka olivat aiemmin saaneet vasteen (CR tai PR) ADCETRIS‑hoitoon, arvioitiin avoimessa faasin 2 monikeskustutkimuksessa. Kaksikymmentä uusiutunutta tai refraktaarista Hodgkinin lymfoomaa sairastavaa potilasta sai aloitusannoksena 1,8 mg/kg ja yksi potilas 1,2 mg/kg ADCETRIS‑valmistetta 30 minuuttia kestävänä laskimoinfuusiona 3 viikon välein. Hoitojaksojen mediaani oli 7 (vaihteluväli 2–37 jaksoa). Niistä 20 Hodgkinin lymfoomaa sairastavasta potilaasta, jotka pystyttiin arvioimaan, 6 potilasta (30 %) saavutti täydellisen remission ja 6 potilasta (30 %) saavutti osittaisen remission uudelleen annetulla ADCETRIS‑hoidolla. Objektiivinen vasteprosentti oli 60 %. Vasteen keston mediaani oli 9,2 kuukautta niillä potilailla, jotka saavuttivat objektiivisen vasteen (CR + PR) ja 9,4 kuukautta niillä potilailla, jotka saavuttivat täydellisen remission.
Systeeminen anaplastinen suurisoluinen lymfooma
Tutkimus SGN35‑014
ADCETRIS‑valmisteen tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa, kaksoislumekontrolloidussa, aktiivikontrolloidussa monikeskustutkimuksessa, johon osallistui 452 aiemmin hoitamatonta CD30‑positiivista perifeeristä T‑solulymfoomaa sairastavaa potilasta. ADCETRIS‑ tai lumevalmisteiden lisäksi potilaat saivat syklofosfamidia ([C], doksorubisiinia [H] ja prednisonia [P] (CHP). Tutkimukseen osallistumiseen vaadittiin ≥ 10 %:n immunohistokemiallisesti määritelty CD30‑positiivisten solujen ilmentymä. Vain ne potilaat, joilla oli CD30‑positiivinen perifeerinen T‑solulymfooma ja jotka voivat käyttää syklofosfamidin [C], doksorubisiinin [H], vinkristiinin [O] ja prednisonin [P] (CHOP) yhdistelmähoitoa, otettiin mukaan tutkimukseen. ADCETRIS + CHP ‑yhdistelmää ei ole tutkittu kaikissa perifeerisen T‑solulymfooman alatyypeissä. Ks. taulukosta 18 mukaan otetut perifeerisen T‑solulymfooman alatyypit. Tutkimuksen 452 potilaasta 226 satunnaistettiin saamaan ADCETRIS + CHP ‑hoitoa ja 226 satunnaistettiin saamaan CHOP‑hoitoa. Satunnaistamisen stratifiointi tapahtui perifeerisen T‑solulymfooman alatyypin (ALK‑positiivinen systeeminen anaplastinen suurisoluinen lymfooma / muut alatyypit) ja International Prognostic Index (IPI) ‑luokituksen mukaan. Potilaat saivat hoitona 1,8 mg/kg ADCETRIS‑valmistetta 30 minuuttia kestävänä laskimoinfuusiona kunkin 21‑päiväisen hoitojakson päivänä 1 sekä CHP‑hoitoa (syklofosfamidia 750 mg/m2 joka 3. viikko laskimoinfuusiona; doksorubisiinia 50 mg/m2 joka 3. viikko laskimoinfuusiona; ja prednisonia 100 mg kunkin 3‑viikkoisen hoitojakson päivinä 1–5 suun kautta) yhteensä 6–8 hoitojakson ajan. Annettujen hoitojaksojen mediaanimäärä oli 6 (vaihteluväli 1–8 hoitojaksoa); 70 % potilaista sai 6 hoitojaksoa ja 18 % sai 8 hoitojaksoa. Taulukossa 18 esitetään yhteenveto lähtötilanteen potilas‑ ja tautitiedoista.
Taulukko 18: Yhteenveto lähtötilanteen potilas‑ ja tautitiedoista aiemmin hoitamatonta perifeeristä T‑solulymfoomaa koskeneessa faasin 3 tutkimuksessa (ITT ja sALCL)
| ITT populaatio | sALCL populaatio b | |||
| Potilastiedot | ADCETRIS + CHP n = 226 | CHOP n = 226 | ADCETRIS + CHP n = 162 | CHOP n = 154 |
| Mediaani‑ikä (vaihteluväli) | 58.0 (18‑85) | 58.0 (18‑83) | 55.0 (18‑85) | 54.0 (18‑83) |
| ≥ 65‑vuotiaat potilaat (%) | 69 (31) | 70 (31) | 38 (23) | 36 (23) |
| Miessukupuoli, n (%) | 133 (59) | 151 (67) | 95 (59) | 110 (71) |
| ECOG‑toimintakykyluokka, n (%) | ||||
| 0 | 84 (37) | 93 (41) | 58 (36) | 53 (34) |
| 1 | 90 (40) | 86 (38) | 62 (38) | 61 (40) |
| 2 | 51 (23) | 47 (21) | 41 (25) | 40 (26) |
| Tautitiedot | ||||
| Diagnoosi, paikallisen arvioinnin mukaan, n (%)a | ||||
| sALCL | 162 (72) | 154 (68) | 162 (100) | 154 (100) |
| ALK‑positiivinen | 49 (22) | 49 (22) | 49 (30) | 49 (32) |
| ALK‑negatiivinen | 113 (50) | 105 (46) | 113 (70) | 105 (68) |
| Perifeerinen T‑solulymfooma (PTCL‑NOS) | 29 (13) | 43 (19) | NA | NA |
| Angioimmunoblastinen T‑solulymfooma (AITL) | 30 (13) | 24 (11) | NA | NA |
| Aikuisen T‑soluleukemia/lymfooma (ATLL) | 4 (2) | 3 (1) | NA | NA |
Enteropaattinen T‑solulymfooma (EATL) Mediaaniaika diagnoosista ensimmäiseen annokseen, | 1 (0) | 2 (1) | NA | NA |
| kk (vaihteluväli) | 0.8 (0, 19) | 0.9 (0, 10) | 0.8 (0, 19) | 0.9 (0, 10) |
| Taudin levinneisyysaste PTCL:n toteamishetkellä, n (%) | ||||
| Aste I | 12 (5) | 9 (4) | 12 (7) | 7 (5) |
| Aste II | 30 (13) | 37 (16) | 22 (14) | 27 (18) |
| Aste III | 57 (25) | 67 (30) | 29 (18) | 46 (30) |
| Aste IV | 127 (56) | 113 (50) | 99 (61) | 74 (48) |
| IPI ‑ luokitus | ||||
| 0 | 8 (4) | 16 (7) | 7 (4) | 14 (9) |
| 1 | 45 (20) | 32 (14) | 34 (21) | 18 (12) |
| 2 | 74 (33) | 78 (35) | 58 (36) | 60 (39) |
| 3 | 66 (29) | 66 (29) | 37 (23) | 40 (26) |
| 4 | 29 (13) | 25 (11) | 22 (14) | 16 (10) |
| 5 | 4 (2) | 9 (4) | 4 (2) | 6 (4) |
| Imusolmukealueiden ulkopuolinen tauti diagnoosihetkellä, n (%) | ||||
| ≤ 1 kohta | 142 (63) | 146 (65) | 94 (58) | 95 (62) |
| > 1 kohta | 84 (37) | 80 (35) | 68 (42) | 59 (38) |
| Luuydinbiopsia lähtötilanteessa, lymfoomaa, n (%) | ||||
| Kyllä | 30 (13) | 34 (15) | 15 (9) | 13 (8) |
| Ei | 196 (87) | 192 (85) | 147 (91) | 141 (92) |
a. WHO:n vuoden 2008 luokituksen mukaisesti
b. Potilaat, joilla on paikallisesti diagnosoitu sALCL
SGN35‑014‑tutkimuksen ensisijainen päätetapahtuma oli etenemisvapaa elossaoloaika riippumattoman arviointielimen (IRF) mukaan, minkä määritelmä on satunnaistamispäivästä taudin ensimmäisen etenemisen dokumentointipäivään kulunut aika, mistä tahansa syystä aiheutunut kuolema tai myöhempi syövän hoitoon tarkoitetun sytostaattihoito jäännöstaudin tai etenevän taudin hoitamiseksi, sen mukaan mikä ilmenee ensin. Hoidon jälkeen konsolidaatiohoitona annettavaa sädehoitoa, hoidon jälkeen perifeerisen veren kantasolujen mobilisoimiseksi annettua sytostaattihoitoa tai konsolidaatiohoitona tehtyä autologista tai allogeenista kantasolusiirtoa ei katsottu taudin etenemiseksi tai uuden syöpähoidon aloittamiseksi.
Tärkeimpiä toissijaisia päätetapahtumia olivat etenemisvapaa elossaoloaika riippumattoman arviointielimen mukaan kun potilailla oli keskuslaboratoriossa vahvistettu systeeminen anaplastinen suurisoluinen lymfooma, täydellinen remissio riippumattoman arviointielimen mukaan tutkimushoidon päättymisen jälkeen, kokonaiselossaolo ja objektiivinen vasteprosentti riippumattoman arviointielimen mukaan tutkimushoidon päättymisen jälkeen, ja nämä testattiin käyttämällä kiinteää testausmenetelmää, joka noudattelee riippumattoman arviointielimen mukaisen etenemisvapaan elossaoloajan tilastollista merkitsevyyttä.
Ensisijainen päätetapahtuma ja alfasuojatut tärkeimmät toissijaiset päätetapahtumat, jotka arvioitiin hierarkkisesti, saavutettiin. Riippumattoman arviointielimen mukaisen etenemisvapaan elossaoloajan (PFS) mediaani ITT‑populaatiossa oli 48,2 kuukautta ADCETRIS + CHP ‑hoitoryhmässä verrattuna CHOP‑ryhmän 20,8 kuukauteen. Stratifioitu hasardisuhde oli 0,71 (95 % lv: 0,54, 0,93, p = 0,011), mikä osoittaa 29 %:n pienenemisen PFS‑tapahtumien riskissä ADCETRIS + CHP ‑hoidolla verrattuna CHOP‑hoitoon. Kokonaiselossaolon stratifioitu hasardisuhde oli 0,66 (95 % lv: 0,46; 0,95, p = 0,024) ja kokonaiselossaoloon liittyvä kuolemanriski oli 34 % pienempi ADCETRIS + CHP ‑hoidolla verrattuna CHOP‑hoitoon.
Etenemisvapaa elossaoloaika riippumattoman arviointielimen mukaan potilailla, joilla oli keskuslaboratoriossa vahvistettu systeeminen anaplastinen suurisoluinen lymfooma, oli ennalta määritetty keskeinen toissijainen päätetapahtuma. Etenemisvapaan elossaoloajan mediaani riippumattoman arviointielimen mukaan oli 55,7 kuukautta ADCETRIS+CHP ‑ryhmässä verrattuna 54,2 kuukauteen CHOP‑ryhmässä. Stratifioitu hasardisuhde oli 0,59 (95 % lv, 0,42; 0,84), mikä on yhdenmukainen PFS‑tapahtumien riskissä havaitun tilastollisesti merkitsevän 41 %:n pienenemän kanssa ADCETRIS + CHP ‑ryhmässä verrattuna CHOP‑ryhmään (p‑arvo = 0,003), ks. kuva 6 ja taulukko 19.
Alaryhmien analyysejä tehtiin potilailla, joilla oli paikallisesti diagnosoitu systeeminen anaplastinen suurisoluinen lymfooma. Kokonaiselossaolon stratifioitu hasardisuhde oli 0,54 (95 % lv: 0,34; 0,87) ja kuolemanriski oli 46 % pienempi ADCETRIS + CHP ‑hoidolla verrattuna CHOP‑hoitoon, ks. kuva 7. Hoidon lopussa IRF:n arvion mukaisen täydellisen remission osuus oli 71,0 % ADCETRIS + CHP‑ryhmän potilailla verrattuna 53,2 %:iin CHOP‑ryhmän potilailla, jolloin ero oli 17,7 % (95 % lv: 7,2 %; 28,3 %). Hoidon lopussa IRF:n arvion mukainen objektiivinen vasteprosentti oli 87,7 % ADCETRIS + CHP‑ryhmän potilailla verrattuna 70,8 %:iin CHOP‑ryhmän potilailla, jolloin ero oli 16,9 % (95 % lv: 8,1 %; 25,7 %). Alaryhmässä, johon kuuluivat ALK‑positiivista systeemistä anaplastista suurisoluista lymfoomaa sairastavat potilaat, IRF:n arvion mukaisen etenemisvapaan elossaoloajan stratifioitu hasardisuhde oli 0,29 (95 % lv: 0,11; 0,79), ALK‑negatiivisen systeemisen anaplastisen suurisoluisen lymfooman alaryhmässä se oli 0,65 (95 % lv: 0,44; 0.95).
Taulukko 19. Tehotulokset aiemmin hoitamattoman systeemisen anaplastisen suurisoluisen lymfooman hoidossa, kun potilaille annettiin 1,8 mg/kg ADCETRIS‑valmistetta 3‑viikkoisen hoitojakson päivänä 1 (ensisijainen analyysi)
ADCETRIS + CHP n = 162a | CHOP n = 154a | ||
| Etenemisvapaa elossaoloaika IRF:n mukaan | |||
Tapaukset, joissa potilaalla oli PFS‑tapahtuma, n (%) | 56 (34) | 73 (48) | |
| PFS:n mediaani, kk (95 % lv) | 55,66 (48,20, EA) | 54,18 (13,44, EA) | |
| Hasardisuhde (95 % CI)b | 0,59 (0,42, 0,84) | ||
| p‑arvoc | 0,0031 | ||
| Arvioitu PFS (95 % lv)d: | |||
| 6 kk:n kohdalla | 88,0 % (81,8 %, 92,2 %) | 68,4 % (60,3 %, 75,2 %) | |
| 12 kk:n kohdalla | 78,7 % (71,4 %, 84,4 %) | 60,3 % (51,9 %, 67,6 %) | |
| 24 kk:n kohdalla | 68,4 % (60,4 %, 75,2 %) | 53,9 % (45,5 %, 61,5 %) | |
| 36 kk:n kohdalla | 65,5 % (57,1 %, 72,7 %) | 50,2 % (41,6 %, 58,1 %) | |
| Kokonaiselossaolo (OS)e | |||
| Kuolemantapausten lukumäärä (%) | 29 (18) | 44 (29) | |
| Kokonaiselossaolon mediaani, kk (95 % lv) | EA (EA, EA) | EA (EA, EA) | |
| Hasardisuhde (95 % lv)b | 0,54 (0,34, 0,87) | ||
| p‑arvoc,f | 0,0096 | ||
| Täydellinen remissio (CR)g | |||
| % (95 % lv) | 71 % (63,3 %, 77,8 %) | 53 % (45,0 %, 61,3 %) | |
| p‑arvof,h | 0.0004 | ||
| Objektiivinen vasteprosentti (ORR)g | |||
| % (95 % lv) | 88 % (81,6 %, 92,3 %) | 71 % (62,9 %, 77,8 %) | |
| p‑arvof,h | < 0,0001 | ||
CR = täydellinen remissio; IRF = riippumaton arviointielin (Independent Review Facility); EA = ei arvioitavissa; ORR = objektiivinen vasteprosentti; PFS = etenemisvapaa elossaoloaika.
a. IRF:n arvion mukainen PFS lasketaan käyttämällä potilaita, joilla on keskuslaboratoriossa vahvistettu sALCL (n = 163 A + CHP‑ryhmässä ja n = 151 CHOP‑ryhmässä). OS, CR ja ORR lasketaan käyttämällä potilaita, joilla on paikallisesti diagnosoitu sALCL).
b. Hasardisuhde (A + CHP/CHOP) ja 95 %:n luottamusvälit perustuvat stratifioituun Coxin suhteellisen vaaran regressiomalliin stratifiointitekijöiden kanssa (ALK‑positiivinen sALCL vs. kaikki muut ja lähtötilanteen IPI‑luokitus [International Prognostic Index]). Hasardisuhde < 1 suosii A + CHP‑ryhmää.
c. P‑arvo lasketaan käyttämällä stratifioitua log‑rank‑testiä.
d. PFS arvioidaan käyttämällä Kaplan–Meier‑menetelmiä ja 95 %:n luottamusväli lasketaan käyttämällä täydentävää log‑log‑muunnosmenetelmää.
e. OS:n seuranta‑ajan mediaani A + CHP‑ryhmässä oli 38,5 kuukautta ja CHOP‑ryhmässä 41,0 kuukautta.
f. P‑arvoa ei ole muokattua kerrannaisuuden mukaan.
g. Vaste 2007 International Working Group Criteria ‑kriteerien mukaan hoidon lopussa.
h. P‑arvo lasketaan käyttämällä stratifioitua Cochran–Mantel–Haenszel‑testiä.
Kuva 6: Etenemisvapaa elossaoloaika riippumattoman arviointielimen mukaan systeemistä anaplastista suurisoluista lymfoomaa sairastavassa sALCL‑populaatiossa (ADCETRIC + CHP vs. CHOP) (ensisijainen analyysi)
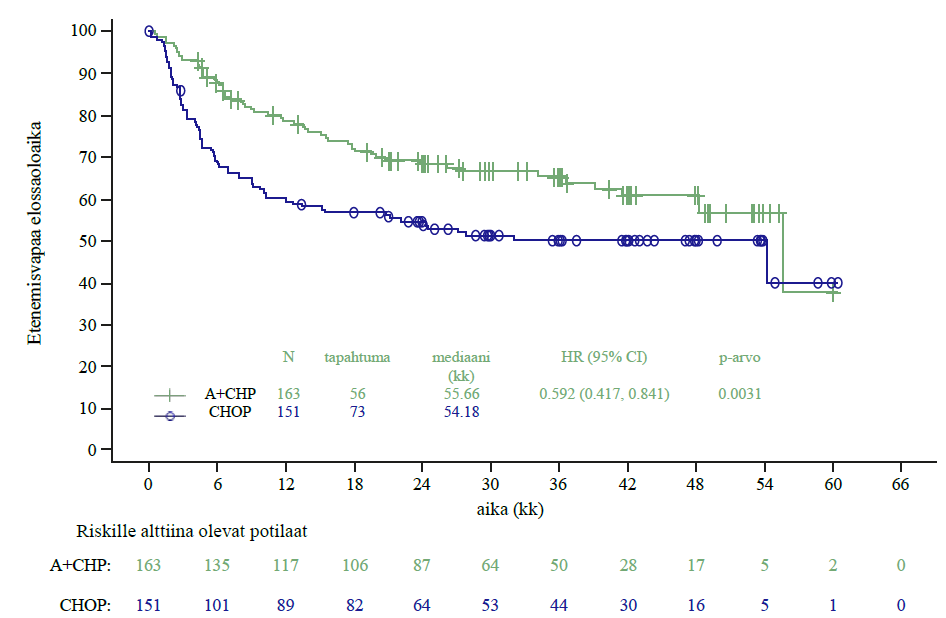
Kuva 7: Kokonaiselossaolo sALCL‑populaatiossa (ADCETRIS + CHP vs. CHOP) (ensisijainen analyysi)
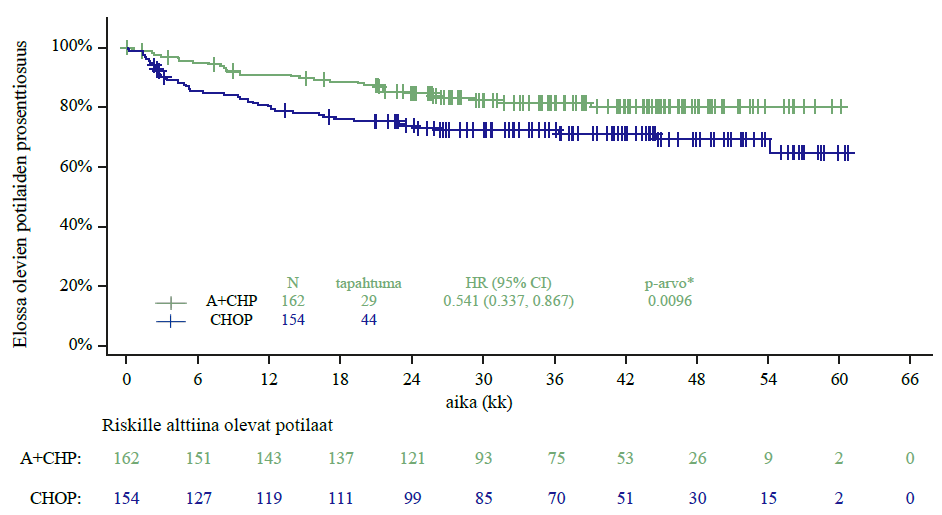
* P‑arvoa ei ole muokattua kerrannaisuuden mukaan.
Tutkimuksen päätyttyä, yli 7 vuotta sen jälkeen, kun ensimmäinen potilas aloitti tutkimuksessa, tutkijan arvioima etenemisvapaa elossaoloaika ITT‑populaatiossa osoitti, että PFS‑tapahtumien riski oli 30 % pienempi ADCETRIS+CHP‑tutkimushaarassa verrattuna CHOP‑hoitoa saaneisiin potilaisiin (HR = 0,70 [95 % lv (0,53, 0.91)]). Tutkijan arvioima etenemisvapaa elossaoloaika sALCL‑populaatiossa osoitti, että PFS‑tapahtumien riski oli 45 % pienempi ADCETRIS+CHP‑tutkimushaarassa verrattuna CHOP‑hoitoa saaneisiin potilaisiin (HR = 0,55 [95 % lv (0,39, 0,79)]).
Tutkimuksen päätyttyä kokonaiselossaoloa osoittavissa tuloksissa nähtävä hyöty säilyi ja tulokset olivat yhdenmukaiset ensisijaisen analyysin aikaan raportoitujen tulosten kanssa. Kokonaiselossaoloa osoittavat tulokset ITT‑populaatiossa osoittivat, että kuoleman riski oli 28 % pienempi ADCETRIS+CHP‑tutkimushaarassa verrattuna CHOP‑hoitoa saaneisiin potilaisiin (HR = 0,72 [95 % lv (0,53–0,99)]). Kokonaiselossaoloa osoittavat tulokset sALCL‑populaatiossa osoittivat, että kuoleman riski oli 34 % pienempi ADCETRIS+CHP‑tutkimushaarassa verrattuna CHOP‑hoitoa saaneisiin potilaisiin (HR = 0,66 [95 % lv (0,43, 1,01)]), ks. kuva 8.
Kuva 8: Kokonaiselossaolo sALCL‑populaatiossa (ADCETRIS + CHP vs. CHOP) (tutkimuksen päätyttyä)
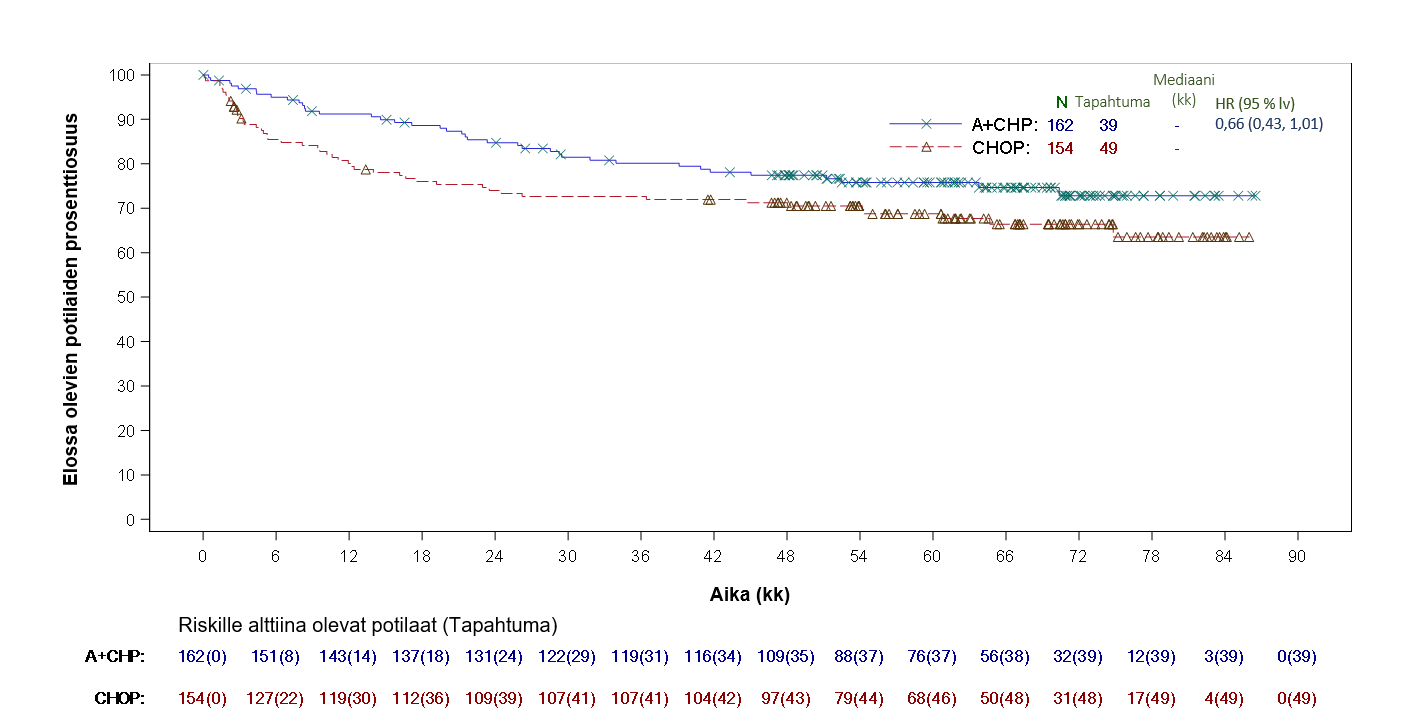
Tutkimus SG035‑0004
ADCETRIS‑monoterapian tehoa ja turvallisuutta arvioitiin yhdellä hoitoryhmällä tehdyssä avoimessa monikeskustutkimuksessa 58:lla uusiutunutta tai refraktaarista systeemistä anaplastista suurisoluista lymfoomaa sairastavalla potilaalla. Yhteenveto lähtötilanteen potilas‑ ja tautitiedoista, ks. taulukko 20 alla.
Taulukko 20: Yhteenveto lähtötilanteen potilas‑ ja tautitiedoista uusiutunutta tai refraktaarista systeemistä anaplastista suurisoluista lymfoomaa koskeneessa faasin 2 tutkimuksessa
| Potilastiedot | n = 58 |
| Mediaani‑ikä, vuotta (vaihteluväli) | 52 vuotta (14–76) |
| Sukupuoli | 33 miestä (57 %) / 25 naista (43 %) |
| ECOG‑toimintakykyluokkaa | |
| 0 | 19 (33 %) |
| 1 | 38 (66 %) |
| Aiempi autologinen kantasolusiirto | 15 (26 %) |
| Aiempia solunsalpaajahoitoja (vaihteluväli) | 2 (1–6) |
| Histologisesti vahvistettu CD30‑antigeeniä ilmentävä tauti | 57 (98 %) |
| Anaplastinen lymfoomakinaasi (ALK) ‑negatiivinen tauti | 42 (72 %) |
| Tautitiedot | |
| Primaaristi refraktaarinen ensilinjan syöpähoidolleb | 36 (62 %) |
| Ei vastaa viimeisimpään hoitoon | 29 (50 %) |
| Tauti uusiutunut viimeisimmän hoidon yhteydessä | 29 (50 %) |
| B‑oireita lähtötilanteessa | 17 (29 %) |
| Vaiheen III tauti diagnoosivaiheessa | 8 (14 %) |
| Vaiheen IV tauti diagnoosivaiheessa | 21 (36 %) |
a. Yhden potilaan ECOG‑toimintakykyluokka oli lähtötilanteessa 2. Arvo oli tutkimussuunnitelman vastainen, joten kyseisen potilaan ei katsottu täyttävän tutkimuksen sisäänottokriteerejä.
b. Primaaristi refraktaarinen tarkoittaa, ettei sALCL‑potilaan ensilinjan syöpähoito ollut saanut tautia täydelliseen remissioon tai tauti oli edennyt 3 kuukauden sisällä ensilinjan syöpähoidon päättymisestä.
Mediaaniaika potilaan alkuperäisestä sALCL‑diagnoosista ensimmäiseen ADCETRIS‑annokseen oli 16,8 kuukautta.
10 potilasta (17 %) sai 16 hoitojaksoa ADCETRIS‑valmistetta; hoitojaksojen määrän mediaani oli 7 (vaihteluväli 1–16).
Riippumaton arviointielin (IRF) arvioi vasteen ADCETRIS‑hoitoon päivitettyjen malignin lymfooman vastekriteerien (Cheson, 2007) perusteella. Hoitovaste arvioitiin rintakehän, kaulan, vatsan ja lantion alueen spiraali‑TT‑kuvauksilla, PET‑kuvauksilla ja kliinisten tietojen pohjalta. Vastearvioinnit tehtiin hoitojaksojen 2, 4, 7, 10, 13 ja 16 kohdalla. PET‑kuvaukset tehtiin hoitojaksojen 4 ja 7 kohdalla.
IRF:n arvion mukaan ORR oli 86 % (ITT‑populaatiossa 50 potilasta 58:sta). CR oli 59 % (ITT‑populaatiossa 34 potilasta 58:sta) ja kasvaintaakka pieneni (kaikki pieneneminen mukaan lukien) 97 %:lla potilaista. Arvioitu kokonaiselossaolo oli 5 vuoden kohdalla 60 % (95 % lv [47 %, 73 %]). Seuranta‑ajan mediaani (ensimmäisestä annoksesta kuolemaan tai viimeiseen yhteydenottoon kulunut aika) oli 71,4 kuukautta. Tutkijan tekemät arvioinnit ja kuvien riippumattomat arvioinnit vastasivat yleensä toisiaan. Hoitoa saaneista potilaista 9:lle hoitovasteen saavuttaneelle tehtiin myöhemmin allogeeninen kantasolusiirto ja 9:lle hoitovasteen saavuttaneelle tehtiin autologinen kantasolusiirto. Lisää tehotuloksia on taulukossa 21 ja kuvassa 9.
Taulukko 21: Tehotulokset uusiutunutta tai refraktaarista systeemistä anaplastista suurisoluista lymfoomaa sairastavilla potilailla, jotka saivat 1,8 mg/kg ADCETRIS‑valmistetta3 viikon välein
| Paras kliininen vaste (n = 58) | IRF n (%) | 95 %:n luottamusväli |
| Objektiivinen vaste (CR + PR) | 50 (86) | 74,6; 93,9 |
| Täydellinen remissio (CR) | 34 (59) | 44,9; 71,4 |
| Osittainen remissio (PR) | 16 (28) | Ei oleellinen |
| Taudin hallinta (CR + PR + SD) | 52 (90) | 78,8; 96,1 |
| Vasteen kesto | IRF:n arvion mukainen mediaani | 95 %:n luottamusväli |
| Objektiivinen vaste (CR + PR)a | 13,2 | 5,7; 26,3 |
| Täydellinen remissio (CR) | 26,3 | 13,2; EAb |
| Etenemisvapaa elossaolo | IRF:n arvion mukainen mediaani | 95 %:n luottamusväli |
| Mediaani | 14,6 | 6,9; 20,6 |
| Kokonaiselinaika | Mediaani | 95 %:n luottamusväli |
| Mediaani | Ei saavutettu | 21,3, EAb |
a. Vasteen kesto vaihteli 0,1 kuukaudesta 39,1+ kuukauteen. Ensimmäisen annoksen jälkeen toteutetun seurannan mediaanikesto oli 15,5 kuukautta potilailla, jotka saavuttivat IRF:n arvion mukaan objektiivisen vasteen (OR).
b. Ei arvioitavissa.
Kuva 9: Kokonaiselossaolon Kaplan–Meier‑kuvaaja
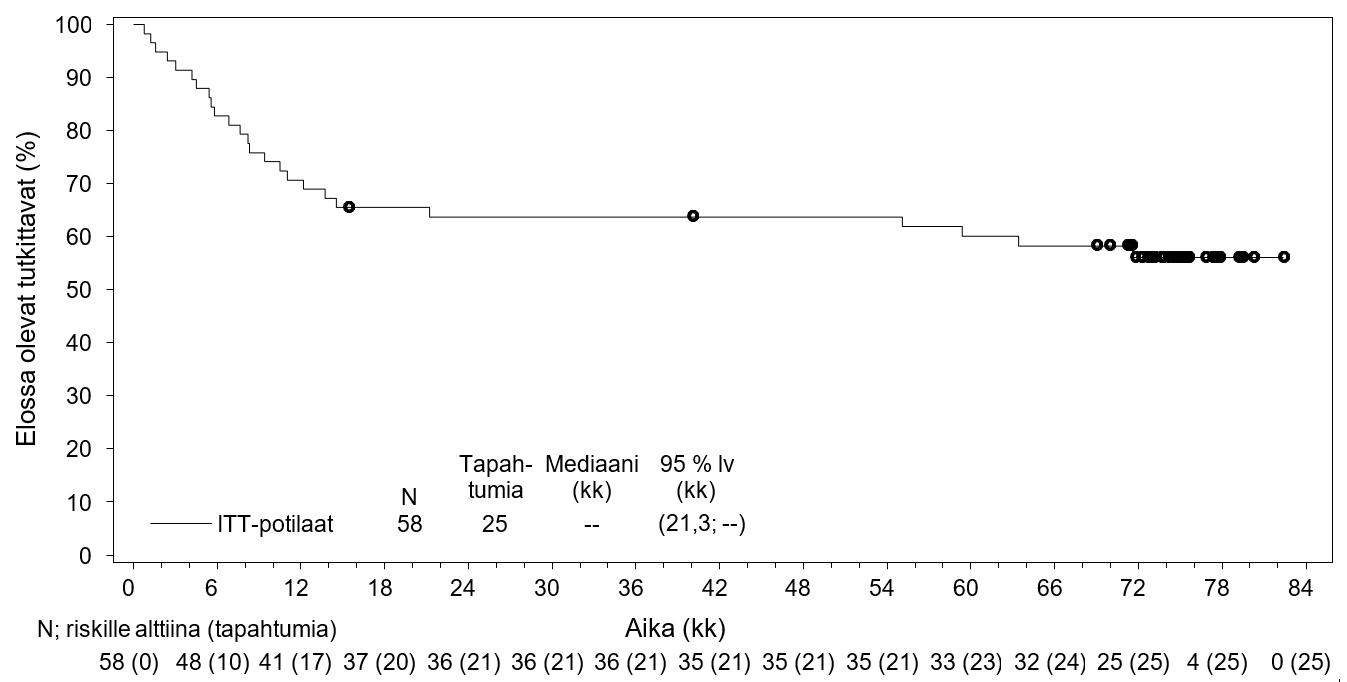
Eksploratiivisessa analyysissä osoitettiin, että noin 69 % brentuksimabivedotiinilla hoidetuista systeemistä anaplastista suurisoluista lymfoomaa (kliininen tutkimus SG0035‑0004) sairastavista potilaista sai ADCETRIS‑valmisteesta enemmän kliinistä hyötyä kuin edellisestä hoidostaan (mitattuna elinaikana, jolloin tauti ei etene).
Lähtötilanteessa 17 potilaalla (29 %) oli B‑oireita. Näistä potilaista 14:n (82 %) B‑oireet hävisivät täysin 0,7 kuukaudessa (mediaani) ADCETRIS‑hoidon aloittamisen jälkeen.
Tutkimus C25006
ADCETRIS‑monoterapian tehoa ja turvallisuutta arvioitiin myös avoimessa faasin 4 yhden tutkimushaaran monikeskustutkimuksessa 50 potilaalla, joilla oli uusiutunut tai refraktaarinen systeeminen anaplastinen suurisoluinen lymfooma (sALCL). IRF:n arvion mukainen ORR oli 64 % (ITT‑populaatiossa 32 potilasta 50:stä). IRF:n arvion mukaista vasteen keston (DOR) mediaania ei saavutettu (95 % lv 19,71 kuukautta, EA). CR‑vasteen saavutti 30 % (ITT‑populaatiossa 15 potilasta 50:stä) ja kasvaintaakka pieneni (kaikki pieneneminen mukaan lukien) 93 %:lla arvioitavissa olevista potilaista. IRF:n arvion mukaista täydellisen remission keston (DOCR) mediaania ei saavutettu (95 % lv 10,61 kuukautta, EA). IRF:n ja tutkijan arviot vasteesta olivat yleisesti ottaen yhdenmukaiset. Hoitoa saaneista potilaista 13 potilaan hoito jatkui kantasolusiirrolla.
Tutkimusten C25006 ja SG035‑0004 (n = 108) yhdistetyissä tiedoissa IRF:n arvion mukainen ORR oli 76 % (ITT‑populaatiossa 82 potilasta 108:sta). IRF:n arvion mukainen mediaani DOR oli 17,0 kuukautta (95 % lv 12,62, 32,46). CR‑vasteen saavutti 45 % (ITT‑populaatiossa 49 potilasta 108:sta) ja kasvaintaakka pieneni (kaikki pieneneminen mukaan lukien) 96 %:lla arvioitavissa olevista potilaista. IRF:n arvion mukainen mediaani DOCR oli 26,3 kuukautta (95 % lv 16,16, EA). IRF:n ja tutkijan arviot vasteesta olivat yleisesti ottaen yhdenmukaiset.
Tutkimus SGN35‑006 (uudelleenhoitotutkimus)
Uudelleenhoidon tehoa sellaisilla potilailla, jotka olivat aiemmin saaneet vasteen (CR tai PR) ADCETRIS‑hoitoon, arvioitiin avoimessa faasin 2 monikeskustutkimuksessa. Seitsemän uusiutunutta systeemistä anaplastista suurisoluista lymfoomaa sairastavaa potilasta sai aloitusannoksena 1,8 mg/kg ja yksi potilas 1,2 mg/kg ADCETRIS‑valmistetta 30 minuuttia kestävänä laskimoinfuusiona 3 viikon välein. Hoitojaksojen mediaani oli 8,5 (vaihteluväli 2–30 jaksoa). Kahdeksasta systeemistä anaplastista suurisoluista lymfoomaa sairastavasta potilaasta kolmea hoidettiin kahdesti. Uudelleenhoitokertoja oli siis yhteensä 11. Uudelleenhoito ADCETRIS‑valmisteella sai aikaan 6 täydellistä remissiota (55 %) ja 4 osittaista remissiota (36 %). Objektiivinen vasteprosentti oli 91 %. Vasteen keston mediaani oli 8,8 kuukautta niillä potilailla, jotka saavuttivat objektiivisen vasteen (CR + PR) ja 12,3 kuukautta niillä potilailla, jotka saavuttivat täydellisen remission.
Ihon T‑solulymfooma
Tutkimus C25001
ADCETRIS‑valmisteen tehoa ja turvallisuutta ainoana lääkkeenä arvioitiin keskeisessä faasin 3 avoimessa, satunnaistetussa monikeskustutkimuksessa 128 potilaalla, joilla oli histologisesti vahvistettu CD30‑positiivinen ihon T‑solulymfooma. CD30‑positiviivisuus määriteltiin seuraavasti: immunohistokemiallisessa määrityksessä (Ventana anti‑CD30 [Ber‑H2]) ≥ 10 %:ssa imukudoksen soluista näkyi solukalvon, soluliman ja/tai Golgin laitteiden värjäytymistä. Tutkimukseen soveltuviksi katsottiin potilaat, joilla oli todettu mycosis fungoides (MF) tai ihon primaarinen anaplastinen suurisoluinen lymfooma (pcALCL). Potilaat stratifioitiin näiden syöpätyyppien perusteella ja satunnaistettiin suhteessa 1:1 saamaan joko ADCETRIS‑valmistetta tai lääkärin valinnan mukaan joko metotreksaattia tai beksaroteenia. Ihon primaarista anaplastista suurisoluista lymfoomaa sairastavat potilaat olivat saaneet joko aiempaa sädehoitoa tai vähintään yhtä aiempaa systeemistä hoitoa, ja mycosis fungoidesta sairastavat potilaat olivat saaneet vähintään yhtä aiempaa systeemistä hoitoa. Potilaat, joilla oli samanaikainen systeeminen anaplastinen suurisoluinen lymfooma, Sézaryn oireyhtymä tai jokin muu non‑Hodgkin‑lymfooma (paitsi lymfomatoidi papuloosi), suljettiin pois tästä tutkimuksesta. Potilaat saivat 1,8 mg/kg ADCETRIS‑valmistetta laskimoon 30 minuutin aikana 3 viikon välein enintään 16 hoitojakson ajan tai lääkärin valitsemaa hoitoa enimmillään 48 viikon ajan. ADCETRIS‑ryhmässä hoitojaksojen mediaanimäärä oli noin 12. Lääkärin valitseman hoidon ryhmässä hoidon mediaanikesto (hoitojaksojen määrä) oli beksaroteenia saaneilla noin 16 viikkoa (5,5 hoitojaksoa) ja metotreksaattia saaneilla 11 viikkoa (3 hoitojaksoa). Taulukossa 22 on yhteenveto potilaiden ja taudin tiedoista lähtötilanteessa.
Taulukko 22: Yhteenveto potilaiden ja taudin tiedoista lähtötilanteessa faasin 3 tutkimuksessa ihon T‑solulymfoomaa sairastavilla potilailla (ITT‑populaatio)
| Potilaan ominaisuudet | ADCETRIS n = 64 | Lääkärin valinta (metotreksaatti tai beksaroteeni) n = 64 |
| Iän mediaani (vaihteluväli) | 62 v (22–83) | 58,5 v (22–83) |
≥ 65‑vuotiaat potilaat, n (%) Sukupuoli, n (%) | 28 (44 %) 33 M (52 %) / 31 N (48 %) | 24 (38 %) 37 M (58 %) / 27 N (42 %) |
| ECOG‑luokka, n (%) | ||
| 0 | 43 (67) | 46 (72) |
1 2 | 18 (28) 3 (5) | 16 (25) 2 (3) |
| Taudin piirteet | ||
| Aiempien hoitojen mediaanimäärä (vaihteluväli) | 4 (0–13) | 3,5 (1–15) |
| Aiempien ihoon kohdennettujen hoitojen mediaanimäärä (vaihteluväli) | 1 (0–6) | 1 (0–9) |
| Aiempien systeemisten hoitojen mediaanimäärä (vaihteluväli) | 2 (0–11) | 2 (1–8) |
| Mycosis fungoides, n (%) | 48 (75) | 49 (77) |
| Varhaisvaihe (IA–IIA) | 15 (31) | 18 (37) |
| Pitkälle edennyt (IIB–IVBa) | 32 (67) | 30 (61) |
| pcALCL, n (%) | 16 (25) | 15 (23) |
| Ihoon rajoittuva | 9 (56) | 11 (73) |
| Tautiaffisiota muualla kuin ihossa | 7 (44) | 4 (27) |
a. Kummassakin ryhmässä yhden potilaan levinneisyysastetiedot olivat puutteelliset, eikä niitä otettu mukaan taulukkoon
Yleisimmät aiemmat ihoon kohdennetut hoidot ITT‑populaatiossa olivat sädehoito (64 %), valohoito (48 %) ja paikallinen steroidihoito (17 %). Yleisimmät aiemmat systeemiset hoidot ITT‑populaatiossa olivat sytostaattihoito (71 %), immunoterapia (43 %) ja beksaroteeni (38 %).
Ensisijainen päätetapahtuma oli objektiivinen vaste, joka säilyi vähintään 4 kuukauden ajan (ORR4) (kesto ensimmäisestä vasteesta viimeiseen vasteeseen ≥ 4 kuukautta), GRS‑pistemäärän (Global Response Score) riippumattoman arvioinnin perusteella. Tähän kuului ihon arviointi (mSWAT‑pisteet [modified severity weighted assessment tool] tutkijan suorittamana), nodaalinen ja viskeraalinen radiologinen arviointi sekä verenkierron Sézaryn solujen määritys (Olsen 2011). Taulukossa 23 esitetään ORR4‑tulokset ja muut tärkeimmät toissijaiset päätetapahtumat.
Taulukko 23: Tehotulokset ihon T‑solulymfoomaa sairastavilla potilailla, jotka saivat 1,8 mg/kg ADCETRIS‑valmistetta 3 viikon välein (ITT‑populaatio)
ADCETRIS (n = 64) | Lääkärin valinta (metotreksaatti tai beksaroteeni) n = 64 | |||
| Objektiivinen vaste, kesto vähintään 4 kk (ORR4) riippumattoman arviointielimen mukaan | ||||
n (%) Prosentuaalinen ero (95 % lv) | 36 (56,3) | 43,8 (29,1; 58,4) | 8 (12,5) | |
| p‑arvo | < 0,001 | |||
| Täydellinen vaste riippumattoman arviointielimen mukaan | ||||
n (%) Prosentuaalinen ero (95 % lv) | 10 (15,6) | 14,1 (‑4,0; 31,5) | 1 (1,6) | |
| Korjattu p‑arvoa | 0,0046 | |||
| Etenemisvapaa elossaolo riippumattoman arviointielimen mukaan | ||||
| Mediaani (kk) | 16,7 | 3,5 | ||
| Hasardisuhde | 0,270 | |||
| 95 % lv | (0,17; 0,43) | |||
| Korjattu p‑arvoa | < 0,001 | |||
a. Laskutapa: painotettu Holmin menetelmä
Ennalta määritetyt ORR4:n alaryhmäanalyysit teki riippumaton arviointielin potilaiden ihon T‑solulymfooman alatyypin, lääkärin valitseman hoidon, lähtötilanteen ECOG‑luokan, iän, sukupuolen ja maantieteellisen alueen perusteella. Analyyseissä todettiin johdonmukainen trendi, jossa ADCETRIS‑valmistetta saaneet potilaat saavuttivat paremmat tulokset kuin lääkärin valitsemaa hoitoa saaneet. ORR4 oli ADCETRIS‑ryhmässä 50 % (mycosis fungoides ‑potilaat) ja 75 % (pcALCL‑potilaat), ja lääkärin valitseman hoidon ryhmässä 10,2 % (mycosis fungoides potilaat) ja 20 % (pcALCL‑potilaat).
Hoitoryhmien välillä ei havaittu merkittäviä eroja elämänlaadussa (EuroQoL 5 Dimensions kyselylomakkeella [EQ‑5D] ja Functional Assessment of Cancer Therapy – General‑kyselylomakkeella [FACT‑G] mitattuna).
ADCETRIS‑hoidon tehoa ja turvallisuutta arvioitiin lisäksi kahdessa avoimessa tutkimuksessa 108 potilaalla, joilla oli uusiutunut CD30‑positiivinen ihon T‑solulymfooma (mukaan lukien mycosis fungoides ja pcALCL sekä Sézaryn oireyhtymä, lymfomatoidi papuloosi ja histologisesti sekamuotoinen ihon T‑solulymfooma), riippumatta CD30‑ekspressiotasosta. Potilaat saivat 1,8 mg/kg ADCETRIS‑valmistetta laskimoon 30 minuutin aikana 3 viikon välein enintään 16 hoitojakson ajan. Näiden tutkimusten turvallisuus‑ ja tehotulokset vastasivat C25001‑tutkimuksen tuloksia. Kokonaisvasteet eri potilasryhmillä olivat seuraavat: mycosis fungoides 54–66 %, pcALCL 67 %, Sézaryn oireyhtymä 50 %, lymfomatoidi papuloosi 92 % ja histologisesti sekamuotoinen ihon T‑solulymfooma 82–85 %.
Pediatriset potilaat
Yhdistelmähoito
C25004
ADCETRIS‑hoidon turvallisuutta ja tuumorin kasvua ehkäisevää vaikutusta arvioitiin yhdessä sytostaattihoidon kanssa (doksorubisiini [A], vinblastiini [V] ja dakarbatsiini [D] [AVD]) avoimessa monikeskustutkimuksessa, johon osallistui 59 pediatrista potilasta (ikä 6–17 vuotta), joilla oli aiemmin hoitamaton edennyt klassinen CD30‑positiivinen Hodgkinin lymfooma. Kaikilla potilailla oli histologisesti vahvistettu CD30‑positiivinen tauti. Potilaista 59 %:lla (n = 35) oli imusolmukealueiden ulkopuolista tautia. Kaikkia 59 pediatrista potilasta hoidettiin kunkin 28‑päiväisen hoitojakson päivinä 1 ja 15 antamalla 48 mg/m2 ADCETRIS‑valmistetta 30 minuuttia kestävänä infuusiona laskimoon sekä lisäksi 25 mg/m2 doksorubisiinia, 6 mg/m2 vinblastiinia ja 375 mg/m2 dakarbatsiinia. Kehon pinta‑alaan perustuva ADCETRIS‑annos valittiin niin, että se vastasi aikuisilla tutkimuksessa C25003 havaittuja farmakokineettisiä altistuksia. Pediatristen potilaiden maksimaalista siedettyä annosta ei saavutettu. Suurin osa (88 %) potilaista saavutti objektiivisen vasteen riippumattoman arviointielimen arvion mukaan tutkimuksen lopussa, ja 67 % saavutti täydellisen vasteen (CR). Yksikään potilaista ei kuollut. Kaikkiaan 13 turvallisuuspopulaatioon kuuluneen potilaan (22 %) raportoitiin saaneen sädetystä hoitojakson 6 jälkeen.
Monoterapia
C25002
ADCETRIS‑hoidon turvallisuutta, farmakokinetiikkaa ja tuumorin kasvua ehkäisevää vaikutusta 36:lla uusiutunutta tai refraktaarista Hodgkinin lymfoomaa (HL) tai systeemistä anaplastista suurisoluista lymfoomaa (sALCL) sairastavalla pediatrisella potilaalla (7–17-vuotiailla; 7–11-vuotiaat lapset, n = 12 ja 12–17-vuotiaat nuoret, n = 24) arvioitiin faasin 1/2 avoimessa, yhden vaikuttavan aineen, suurenevan annoksen monikeskustutkimuksessa (C25002). Tutkimuksen faasi 1 ‑osassa arvioitiin turvallisuusprofiilia (ks. kohta Haittavaikutukset), määritettiin suurin siedetty annos pediatrisilla potilailla ja/tai suositeltu annos faasi 2 ‑osaan sekä arvioitiin ADCETRIS‑valmisteen farmakokinetiikkaa (ks. kohta Farmakokinetiikka). Faasiin 1 osallistui 3 uusiutunutta tai refraktaarista HL:ää sairastavaa potilaista, joita hoidettiin annoksella 1,4 mg/kg, ja 9 potilasta (7:llä uusiutunut tai refraktaarinen HL ja 2:lla sALCL), joita hoidettiin annoksella 1,8 mg/kg. Suurinta siedettyä annosta ei saavutettu. Faasin 2 suositelluksi annokseksi määritettiin 1,8 mg/kg. Koko tutkimuksen aikana kaikkiaan 16 potilasta, joilla oli uusiutunut tai refraktaarinen HL ja 17 potilasta, joilla oli uusiutunut tai refraktaarinen sALCL (joista 10:llä oli ensimmäinen uusiutuma), hoidettiin ADCETRIS‑annoksella 1,8 mg/kg. Riippumattoman arviointielimen mukaan kokonaisvaste arvioitiin kummankin faasin koko ajalta faasin 2 suositellulla annoksella. Näistä 33 potilaasta, jotka saivat faasin 2 suositeltua annosta, vaste pystyttiin arvioimaan 32 potilaalta. Niillä potilailla, joiden vaste pystyttiin arvioimaan, kokonaisvaste oli 47 % uusiutunutta tai refraktaarista HL:ää, 53 % uusiutunutta tai refraktaarista sALCL:ää ja 60 % sALCL:ää sairastavilla, joilla oli ensimmäinen uusiutuma. Kahdeksalle Hodgkinin lymfoomaa sairastavalle ja yhdeksälle systeemistä anaplastista suurisoluista lymfoomaa sairastavalle potilaalle tehtiin kantasolusiirto ADCETRIS‑hoidon jälkeen.
Farmakokinetiikka
Monoterapia
Brentuksimabivedotiinin farmakokinetiikkaa arvioitiin faasin 1 tutkimuksissa ja populaatiofarmakokineettisessä analyysissä, jossa oli mukana 314 potilaan tiedot. Brentuksimabivedotiini annettiin kaikissa kliinisissä tutkimuksissa laskimoinfuusiona.
Brentuksimabivedotiinin vasta‑aine/lääkekonjugaatin (ADC) maksimipitoisuudet havaittiin yleensä joko infuusion lopussa tai infuusion päättymistä lähimpänä olevana mittausajankohtana. Seerumin ADC‑pitoisuuksien todettiin pienenevän multieksponentiaalisesti, ja terminaalinen puoliintumisaika oli noin 4–6 vuorokautta. Altistukset olivat melko riippuvaisia annoksesta. ADC‑pitoisuuksissa ei todettu lainkaan tai juuri lainkaan kumulaatiota, kun potilaille annettiin toistuvia annoksia 3 viikon välein. Tämä löydös tukee arviota valmisteen terminaalisesta puoliintumisajasta. Faasin 1 tutkimuksessa ADC:n tyypilliset Cmax‑ ja AUC‑arvot yhden kerta‑annoksen (1,8 mg/kg) jälkeen olivat noin 31,98 μg/ml ja 79,41 μg/ml x vrk.
MMAE on brentuksimabivedotiinin tärkein metaboliitti. Faasin 1 tutkimuksessa MMAE:n Cmax‑arvon mediaani oli ADC‑kerta‑annoksen (1,8 mg/kg) jälkeen noin 4,97 ng/ml, AUC‑arvon noin 37,03 ng/ml x vrk ja Tmax‑arvon noin 2,09 vrk. MMAE‑altistus pieneni toistuvien brentuksimabivedotiiniannosten jälkeen noin 50–80 %:iin ensimmäisen annoksen jälkeen todetusta altistuksesta. MMAE metaboloituu edelleen pääasiassa yhtä voimakkaaksi metaboliitiksi; sen altistus on kuitenkin suuruusluokkaa pienempi kuin MMAE:n. Se ei siis todennäköisesti myötävaikuta MMAE:n systeemisiin vaikutuksiin oleellisesti.
Ensimmäisen hoitojakson aikana suurempaan MMAE‑altistukseen liittyi neutrofiilimäärän absoluuttista pienenemistä.
Yhdistelmähoito
ADCETRIS‑valmisteen farmakokinetiikkaa yhdessä AVD‑hoidon kanssa arvioitiin yhdessä vaiheen 3 tutkimuksessa 661 potilaan joukossa. Populaatiofarmakokinetiikan analyysi viittasi siihen, että ADCETRIS‑valmisteen farmakokinetiikka AVD‑hoidon yhteydessä vastasi sen farmakokinetiikkaa monoterapiakäytössä.
Kun tutkittaville annettiin toistuvia 1,2 mg/kg brentuksimabivedotiiniannoksia laskimoinfuusiona kahden viikon välein, ADC:n maksimipitoisuudet seerumissa todettiin lähellä infuusion päättymistä. Eliminaatiovaiheessa pitoisuudet pienenivät multieksponentiaalisesti, ja t1/2z‑aika oli noin 4–5 vrk. MMAE:n maksimipitoisuudet plasmassa todettiin noin 2 päivän kuluttua infuusion päättymisestä. Pitoisuudet pienenivät monoeksponentiaalisesti, ja t1/2z‑aika oli noin 3–4 vrk.
Kun tutkittaville annettiin toistuvia 1,2 mg/kg brentuksimabivedotiiniannoksia laskimoinfuusiona kahden viikon välein, ADC:n ja MMAE:n vakaan tilan minimipitoisuudet saavutettiin hoitojaksoon 3 mennessä. Kun vakaa tila oli saavutettu, ADC:n farmakokinetiikka ei vaikuttanut muuttuvan ajan mittaan. ADC:n kumuloituminen (arvioituna AUC14D‑arvon perusteella hoitojakson 1 ja hoitojakson 3 välillä) oli 1,27‑kertaista. MMAE‑altistus (arvioituna AUC14D‑arvon perusteella hoitojakson 1 ja hoitojakson 3 välillä) vaikutti pienenevän ajan mittaan noin 50 %.
ADCETRIS‑valmisteen farmakokinetiikkaa yhdessä CHP‑hoidon kanssa arvioitiin yhdessä vaiheen 3 tutkimuksessa 223 potilaan joukossa (SGN35‑014). Kun potilaille oli annettu useita ADCETRIS‑annoksia (1,8 mg/kg) laskimoinfuusiona 3 viikon välein, ADC:n ja MMAE:n farmakokinetiikka oli samanlaista kuin monoterapian jälkeen.
ADCETRIS-valmisteen farmakokinetiikkaa ei ole tutkittu BrECADD-hoidon yhteydessä.
Jakautuminen
In vitro MMAE sitoutui 68–82‑prosenttisesti ihmisen plasman proteiineihin. MMAE ei todennäköisesti syrjäytä voimakkaasti proteiineihin sitoutuvia lääkeaineita eikä syrjäydy niiden vaikutuksesta. In vitro MMAE oli P‑gp:n substraatti. Kliinisinä pitoisuuksina se ei estänyt P‑gp:n toimintaa.
Ihmisellä ADC:n keskimääräinen vakaan tilan jakautumistilavuus oli noin 6–10 l. Populaatiofarmakokineettisten arvioiden perusteella MMAE:n tyypillinen näennäinen sentraalinen jakautumistilavuus oli 35,5 l.
Biotransformaatio
ADC todennäköisesti kataboloituu proteiinina, ja sen aminohappokomponentit joko kierrättyvät tai poistuvat elimistöstä.
Invivo ‑tiedot eläimistä ja ihmisistä viittaavat siihen, että vain pieni osa brentuksimabivedotiinista vapautuvasta MMAE:stä metaboloituu. MMAE:n metaboliittien pitoisuuksia ihmisen plasmassa ei ole mitattu. Ainakin yhden MMAE:n metaboliitin on todettu olevan aktiivinen in vitro.
MMAE on CYP3A4:n ja mahdollisesti myös CYP2D6:n substraatti. In vitro‑tietojen perusteella MMAE metaboloituu pääasiassa CYP3A4/5‑välitteisen oksidaation kautta. Ihmisen maksan mikrosomeilla tehtyjen in vitro‑tutkimusten perusteella MMAE estää vain CYP3A4/5‑entsyymejä pitoisuuksina, jotka ovat huomattavasti kliinisessä käytössä saavutettavia pitoisuuksia suurempia. MMAE ei estä muiden isoformien toimintaa.
MMAE ei indusoinut mitään merkittäviä CYP450‑entsyymejä ihmisen hepatosyyteillä tehdyissä primaariviljelyissä.
Eliminaatio
ADC eliminoituu katabolian kautta. Valmisteen tyypillinen arvioitu puhdistuma on 1,5 l/vrk ja puoliintumisaika 4–6 vuorokautta.
MMAE:n eliminaatiota rajoitti sen vapautumisnopeus ADC:stä. Sen tyypillinen näennäinen puhdistuma oli 19,99 l/vrk ja puoliintumisaika 3–4 vuorokautta.
Valmisteen erittymistä tutkittiin potilailla, jotka saivat brentuksimabivedotiinia annoksena 1,8 mg/kg. Noin 24 % ADC:hen sitoutuneesta MMAE‑kokonaismäärästä, jonka potilaat saivat brentuksimabivedotiini‑infuusion aikana, erittyi virtsaan ja ulosteeseen 1 viikon kuluessa. Noin 72 % erittyneestä MMAE:sta erittyi ulosteeseen. MMAE erittyi virtsaan vähäisemmässä määrin (28 %).
Farmakokinetiikka erityisryhmissä
Populaatiofarmakokineettisessä analyysissä todettiin, että lähtötilanteessa mitattu seerumin albumiinipitoisuus oli merkitsevä MMAE:n puhdistuman kovariaatti. Analyysin perusteella MMAE:n puhdistuma oli kaksi kertaa pienempi potilailla, joiden seerumin albumiinipitoisuus oli pieni (< 3,0 g/dl) verrattuna potilaisiin, joiden seerumin albumiinipitoisuus oli normaaliarvojen rajoissa.
Maksan vajaatoiminta
Tutkimuksessa arvioitiin brentuksimabivedotiinin ja MMAE:n farmakokinetiikkaa sen jälkeen, kun lievää (Child–Pugh‑luokka A; n = 1), keskivaikeaa (Child–Pugh‑luokka B; n = 5) ja vaikeaa (Child–Pugh‑luokka C; n = 1) maksan vajaatoimintaa sairastaville potilaille oli annettu 1,2 mg/kg ADCETRIS‑valmistetta. MMAE‑altistus suureni maksan vajaatoimintapotilailla noin 2,3‑kertaisesti (90 %:n luottamusväli 1,27‑4,12‑kertainen) verrattuna potilaisiin, joiden maksan toiminta oli normaalia.
Munuaisten vajaatoiminta
Tutkimuksessa arvioitiin brentuksimabivedotiinin ja MMAE:n farmakokinetiikkaa sen jälkeen, kun lievää (n = 4), keskivaikeaa (n = 3) ja vaikeaa (n = 3) munuaisten vajaatoimintaa sairastaville potilaille oli annettu 1,2 mg/kg ADCETRIS‑valmistetta. MMAE‑altistus suureni vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min) sairastavilla noin 1,9‑kertaisesti (90 %:n luottamusväli 0,85‑4,21‑kertainen) verrattuna potilaisiin, joiden munuaisten toiminta oli normaalia. Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla ei havaittu vaikutusta.
Iäkkäät potilaat
Brentuksimabivedotiinin populaatiofarmakokinetiikkaa arvioitiin useissa tutkimuksissa, mukaan lukien 380:n enintään 87‑vuotiaan potilaan tiedot (34 potilasta oli ≥ 65 – < 75‑vuotiaita ja 17 potilasta ≥ 75‑vuotiaita). Lisäksi arvioitiin brentuksimabivedotiinin ja AVD‑hoidon yhdistelmän populaatiofarmakokinetiikkaa. Mukana oli tietoja 661 potilaasta, joiden ikä oli enintään 82 v (42 potilaan ikä oli ≥ 65 – < 75 v ja 17 potilaan ikä ≥ 75 v). Iän merkitystä farmakokinetiikkaan arvioitiin kussakin analyysissä, eikä se ollut merkitsevä kovariaatti.
Pediatriset potilaat
Monoterapia
C25002
Brentuksimabivedotiinin vasta‑aine/lääkekonjugaatin (ADC) ja MMAE:n farmakokinetiikkaa sen jälkeen kun brentuksimabivedotiinia oli annettu 30 minuutin infuusiona laskimoon annoksella 1,4 mg/kg tai 1,8 mg/kg kolmen viikon välein arvioitiin faasin 1/2 kliinisessä tutkimuksessa, johon osallistui 36 uusiutunutta tai refraktaarista Hodgkinin lymfoomaa tai systeemistä anaplastista suurisoluista lymfoomaa sairastavaa pediatrista potilasta (7–17‑vuotiaita; 7–11‑vuotiaat lapset, n = 12 ja 12–17‑vuotiaat nuoret, n = 24) (ks. kohta Farmakodynamiikka). ADC:n Cmax‑arvo havaittiin tyypillisesti infuusion lopussa tai infuusion loppua lähinnä otettavista näytteistä. ADC:n pitoisuuksissa seerumissa havaittiin multieksponentiaalinen pienenemä; terminaalinen puoliintumisaika oli noin 4–5 vuorokautta. Altistukset olivat suunnilleen suhteessa annokseen, ja havaittiin suuntaus, että altistus ADC:lle oli vähäisempi nuoremmilla / vähemmän painavilla tutkimushenkilöillä. ADC:n AUC‑arvon mediaani tämän tutkimuksen lapsilla oli noin 14 % ja nuorilla noin 3 % pienempi kuin aikuispotilailla, kun taas MMAE‑altistus oli lapsilla noin 53 % pienempi ja nuorilla noin 13 % suurempi kuin aikuispotilailla. ADC:n Cmax‑ ja AUC‑arvojen mediaanit kerta‑annoksen (1,8 mg/kg) jälkeen olivat alle 12‑vuotiailla potilailla 29,8 µg/ml (Cmax) ja 67,9 µg*vrk/ml (AUC), ja 12-vuotiailla ja sitä vanhemmilla 34,4 µg/ml (Cmax) ja 77,8 µg*vrk/ml (AUC). MMAE:n Cmax‑, AUC‑ ja Tmax‑arvojen mediaanit kerta‑annoksen (1,8 mg/kg) jälkeen olivat alle 12‑vuotiailla potilailla 3,73 ng/ml (Cmax), 17,3 ng*vrk/ml (AUC) ja 1,92 päivää (Tmax), ja 12‑vuotiailla ja sitä vanhemmilla 6,33 ng/ml (Cmax), 42,3 ng*vrk/ml (AUC) ja 1,82 päivää (Tmax). Lääkevasta‑ainepositiivisiksi todetuilla pediatrisilla potilailla havaittiin suuntaus brentuksimabivedotiinin puhdistuman lisääntymiseen. Alle 12‑vuotiaista potilaista yksikään (nolla 11 potilaasta) ei ollut pitkäkestoisesti lääkevasta‑ainepositiivinen, yli 12‑vuotiaissa potilaissa heitä oli kaksi (kaksi 23 potilaasta).
Yhdistelmähoito
C25004
Brentuksimabivedotiinin vasta‑aine/lääkekonjugaatin (ADC) ja MMAE:n farmakokinetiikkaa sen jälkeen, kun brentuksimabivedotiinia oli annettu 30 minuutin infuusiona laskimoon annoksella 48 mg/m2 kahden viikon välein yhdessä doksorubisiinin, vinblastiinin ja dakarbatsiinin (AVD) kanssa, arvioitiin faasin 1/2 kliinisessä tutkimuksessa, johon osallistui 59 edennyttä, äskettäin diagnosoitua CD30‑positiivista klassista Hodgkinin lymfoomaa sairastavaa pediatrista potilasta (6–17 vuotiaita; 6–11‑vuotiaat lapset, n = 11 ja 12–17‑vuotiaat nuoret, n = 48). ADC:n huippupitoisuus (Cmax)‑ havaittiin seerumissa noin infuusion lopussa ja se pieneni multieksponentiaalisesti. Terminaalinen puoliintumisaika oli noin 4 vuorokautta. MMAE:n huippupitoisuus (Cmax)‑ havaittiin plasmassa noin 2 vuorokautta brentuksimabivedotiinin annon jälkeen, ja puoliintumisaika oli noin 2 vuorokautta. ADC:n geometrinen keskimääräinen Cmax‑arvo oli 22,5 µg/ml ja AUC‑arvo oli 46,7 µg*vrk/ml kerta‑annoksen (48 mg/m2) jälkeen. MMAE:n geometrinen keskimääräinen Cmax‑arvo oli 4,9 ng/ml ja AUC‑arvo oli 27,2 ng*vrk/ml kerta‑annoksen (48 mg/m2) jälkeen. Samankaltaiset ADC:n altistukset saavutettiin pediatrisissa ikäryhmissä (< 12 vuotta, 12–16 vuotta ja > 16 vuotta) sen jälkeen, kun brentuksimabivedotiinia oli annettu kehon pinta‑alaan perustuvan annostelun mukaan käyttämällä annosta 48 mg/m2 yhdessä AVD:n kanssa.
Prekliiniset tiedot turvallisuudesta
MMAE:llä on todettu aneugeenisia ominaisuuksia in vivo rotilla tehdyssä luuytimen mikrotumatutkimuksessa. Tulokset olivat yhtäpitäviä solujen sukkularihmastoon kohdistuvan MMAE:n farmakologisen vaikutuksen (mikrotubulusverkoston toiminnan häiritseminen) kanssa.
Brentuksimabivedotiinin vaikutuksia miehen ja naisen hedelmällisyyteen ei ole tutkittu. Rotilla tehtyjen, toistuvan altistuksen aiheuttamaa toksisuutta selvittäneiden tutkimusten tulokset viittaavat kuitenkin siihen, että brentuksimabivedotiini saattaa heikentää miesten suvunjatkamiskykyä ja hedelmällisyyttä. Kivesten atrofia ja degeneraatio korjaantuivat osittain 16 viikon pituisen hoidottoman jakson jälkeen.
Brentuksimabivedotiini aiheutti alkio‑ ja sikiökuolemia tiineille naarasrotille.
Ei‑kliinisissä tutkimuksissa todettiin imukudoskatoa ja kateenkorvan painon laskua, mikä on johdonmukaista brentuksimabivedotiinista vapautuvan MMAE:n aiheuttaman farmakologisen mikrotubulusten toiminnan eston kanssa.
Farmaseuttiset tiedot
Apuaineet
Sitruunahappomonohydraatti (pH:n säätöön) (E330)
Natriumsitraattidihydraatti (pH:n säätöön) (E331)
α,α‑trehaloosidihydraatti
Polysorbaatti 80 (E433)
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
4 vuotta
Käyttökuntoon saattamisen / laimentamisen jälkeen valmiste on mikrobiologiselta kannalta käytettävä heti. Valmisteen kemiallinen ja fysikaalinen stabiliteetti on kuitenkin osoitettu 24 tunnin ajalta 2–8 ºC:n lämpötilassa.
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Ei saa jäätyä.
Säilytä injektiopullo alkuperäispakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ADCETRIS kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
50 mg (L:ei) 50 mg (5 mg/ml) (3926,10 €)
PF-selosteen tieto
Tyypin I lasista valmistettu, 50 mg kuiva‑ainetta sisältävä injektiopullo, jossa butyylikumitulppa ja alumiinista/muovista valmistettu repäisysinetti.
Pakkaus sisältää yhden injektiopullon.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen kakku tai kuiva-aine.
Käyttö- ja käsittelyohjeet
Yleiset varotoimet
Syöpälääkevalmisteiden asianmukaista käsittelyä ja hävittämistä koskevia ohjeita on noudatettava.
Tämän lääkevalmisteen käsittelyssä on noudatettava asianmukaista aseptista tekniikkaa.
Ohjeet käyttökuntoon saattamisesta
Kertakäyttöinen injektiopullo saatetaan käyttökuntoon 10,5 ml:lla injektionesteisiin käytettävää vettä, jolloin lopulliseksi pitoisuudeksi tulee 5 mg/ml. Yhdessä injektiopullossa on 10 %:n ylimäärä, eli yhdessä injektiopullossa on 55 mg ADCETRIS‑valmistetta, ja käyttökuntoon saatetun valmisteen kokonaistilavuus on 11 ml.
- Suuntaa vesi injektiopullon seinämää kohti, ei siis suoraan kuiva‑ainetta/kakkua päin.
- Pyörittele injektiopulloa varovasti liukenemisen helpottamiseksi. EI SAA RAVISTAA.
- Injektiopulloon muodostuva käyttökuntoon saatettu liuos on kirkasta tai hieman opaalinhohtoista ja väritöntä, ja sen lopullinen pH on 6,6.
- Käyttökuntoon saatettu liuos on tarkistettava silmämääräisesti hiukkasten ja värimuutosten varalta. Jos jompaakumpaa havaitaan, lääkevalmiste pitää hävittää.
Infuusionesteen valmistus
Vedä asianmukainen määrä käyttökuntoon saatettua ADCETRIS‑valmistetta injektiopullo(i)sta ja lisää se 0,9‑prosenttista (9 mg/ml) NaCl‑injektionestettä sisältävään infuusiopussiin siten, että lopulliseksi ADCETRIS‑pitoisuudeksi tulee 0,4–1,2 mg/ml. Suositeltava laimentimen määrä on 150 ml. Käyttökuntoon saatettu ADCETRIS voidaan laimentaa myös 5‑prosenttisella glukoosi‑injektionesteellä tai Ringerin laktaattiliuoksella.
Sekoita ADCETRIS‑valmisteen sisältävä liuos kääntelemällä pussia varovasti. EI SAA RAVISTAA.
Jos injektiopulloon jää lääkeainetta sen jälkeen, kun laimennettava määrä on poistettu, se pitää hävittää paikallisten ohjeiden mukaisesti.
Käyttövalmiiseen ADCETRIS‑infuusionesteeseen tai infuusiojärjestelmään ei saa lisätä mitään muita lääkevalmisteita. Lääkkeenannon jälkeen infuusioletku on huuhdeltava 0,9‑prosenttisella (9 mg/ml) NaCl‑injektionesteellä, 5‑prosenttisella glukoosi‑injektionesteellä tai Ringerin laktaattiliuoksella.
Laimennuksen jälkeen ADCETRIS‑liuos annetaan välittömästi suositellulla infuusionopeudella.
Liuoksen yhteenlaskettu säilytysaika käyttökuntoon saattamisesta potilaalle antamiseen ei saa olla yli 24 tuntia.
Annoksen määrittäminen:
Jatkolaimennettavan ADCETRIS‑kokonaisannoksen (ml) laskukaava (ks. kohta Annostus ja antotapa):
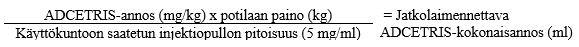
Huom. Jos potilas painaa yli 100 kg, annos lasketaan 100 kg painavalle potilaalle. Suositeltu enimmäisannos on 180 mg.
Tarvittavien ADCETRIS‑injektiopullojen kokonaismäärän laskukaava:

Taulukko 24: Esimerkki annoksen laskemisesta 60–120 kg painaville potilaille, jotka saavat ADCETRIS‑suositusannosta 1,8 mg/kg, 1,2 mg/kg tai 0,9 mg/kga, b
| Suositeltu annos | Potilaan paino (kg) | Kokonaisannos = potilaan paino kerrotaan suositusannoksella | Laimennettava kokonaistilavuusc = kokonaisannos jaetaan käyttökuntoon saatetun injektiopullon pitoisuudella (5 mg/ml) | Tarvittavien injektiopullojen lukumäärä = laimennettava kokonaistilavuus jaetaan injektiopullokohtaisella kokonaistilavuudella (10 ml/injektiopullo) |
| 1,8 mg/kg (enintään 180 mg) | 60 kg | 108 mg | 21,6 ml | 2,16 injektiopulloa |
| 80 kg | 144 mg | 28,8 ml | 2,88 injektiopulloa | |
| 100 kg | 180 mg | 36 ml | 3,6 injektiopulloa | |
| 120 kgd | 180 mg | 36 ml | 3,6 injektiopulloa | |
| 1,2 mg/kg (enintään 120 mg) | 60 kg | 72 mg | 14,4 ml | 1,44 injektiopulloa |
| 80 kg | 96 mg | 19,2 ml | 1,92 injektiopulloa | |
| 100 kg | 120 mg | 24 ml | 2,4 injektiopulloa | |
| 120 kgd | 120 mg | 24 ml | 2,4 injektiopulloa | |
| 0,9 mg/kg (enintään 90 mg) | 60 kg | 54 mg | 10,8 ml | 1,08 injektiopulloa |
| 80 kg | 72 mg | 14,4 ml | 1,44 injektiopulloa | |
| 100 kg | 90 mg | 18 ml | 1,8 injektiopulloa | |
| 120 kgd | 90 mg | 18 ml | 1,8 injektiopulloa |
a. Tässä taulukossa esitetään esimerkkejä annoslaskelmista aikuispotilaille.
b. Kliinisissä tutkimuksissa tutkituilla pediatrisilla potilailla (ikä 6–17 vuotta) kehon pinta‑alaan perustuvan annoksen laskettiin olevan 48 mg/m2 joka toinen viikko yhdessä AVD:n kanssa 28‑päiväisenä hoitojaksona tai 72 mg/m2 joka kolmas viikko monoterapiana. (Ks. kohdista Farmakodynamiikka ja Farmakokinetiikka tiedot pediatrisilla potilailla tehdyistä kliinisistä tutkimuksista.)
c. Laimennetaan 150 ml:aan laimenninta ja annetaan 30 minuutin laskimoinfuusiona.
d. Jos potilas painaa yli 100 kg, annos lasketaan 100 kg painavalle potilaalle.
Hävittäminen
ADCETRIS on tarkoitettu vain yhtä käyttökertaa varten.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ADCETRIS kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
50 mg 50 mg
- Ei korvausta.
ATC-koodi
L01FX05
Valmisteyhteenvedon muuttamispäivämäärä
18.02.2026
Yhteystiedot
 TAKEDA OY
TAKEDA OY PL 1406, Ilmalankuja 3
00101 Helsinki
0800 774 051
www.takeda.fi
etunimi.sukunimi@takeda.com