
KEPPRA infuusiokonsentraatti, liuosta varten 100 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi ml sisältää 100 mg levetirasetaamia.
Yksi 5 ml:n injektiopullo sisältää 500 mg levetirasetaamia.
Apuaine, jonka vaikutus tunnetaan:
Yksi injektiopullo sisältää 19 mg natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Keppra on tarkoitettu ainoaksi epilepsialääkkeeksi paikallisalkuisten (sekundaarisesti yleistyvien tai yleistymättömien) kohtausten hoitoon aikuisille ja vähintään 16‑vuotiaille nuorille, joilla on äskettäin diagnosoitu epilepsia.
Keppra on tarkoitettu lisälääkkeeksi
- epilepsiapotilaiden paikallisalkuisten (sekundaarisesti yleistyvien tai yleistymättömien) kohtausten hoitoon aikuisille, nuorille ja vähintään 4‑vuotiaille lapsille.
- nuoruusiän myoklonista epilepsiaa sairastavien potilaiden myoklonisten kohtausten hoitoon aikuisille ja vähintään 12‑vuotiaille nuorille.
- idiopaattista yleistynyttä epilepsiaa sairastavien potilaiden primaarisesti yleistyvien toonis-kloonisten kohtausten hoitoon aikuisille ja vähintään 12‑vuotiaille nuorille.
Keppra-konsentraatti on vaihtoehto potilaille, kun suun kautta antaminen ei ole tilapäisesti mahdollista.
Annostus ja antotapa
Annostus
Keppra-hoito voidaan aloittaa joko laskimoon tai suun kautta annettuna.
Siirtyminen suun kautta annettavasta hoidosta laskimoon annettavaan hoitoon tai päinvastoin voidaan toteuttaa suoraan ilman titrausta.
Kokonaisvuorokausiannos ja antotiheys on pidettävä samana.
Paikallisalkuiset kohtaukset
Suositeltu annostus ainoana lääkkeenä (vähintään 16‑vuotiaille) ja lisälääkkeenä on sama, kuten jäljempänä esitetään.
Kaikki käyttöaiheet
Aikuiset (≥ 18‑vuotiaat) ja 12–17‑vuotiaat nuoret (≥ 50 kg)
Aloitusannos on 500 mg kaksi kertaa päivässä. Lääkityksen voi aloittaa tällä annoksella jo ensimmäisestä hoitopäivästä lähtien. Aloitusannos voi kuitenkin olla pienempi 250 mg kaksi kertaa päivässä, jos lääkäri katsoo sen aiheelliseksi arvioituaan kohtausten vähenemistä mahdollisiin haittavaikutuksiin nähden. Annos voidaan kahden viikon jälkeen nostaa 500 mg:aan kaksi kertaa päivässä.
Kliinisestä vasteesta ja siedettävyydestä riippuen vuorokausiannos voidaan nostaa annokseen 1500 mg kaksi kertaa päivässä. Annosta voidaan muuttaa lisäämällä tai vähentämällä vuorokausiannosta 250 mg tai 500 mg kaksi kertaa päivässä 2-4 viikon välein.
12–17-vuotiaat nuoret (< 50 kg) ja vähintään 4 vuoden ikäiset lapset
Lääkärin on määrättävä potilaalle hänen painonsa, ikänsä ja annoksensa perusteella sopivin lääkemuoto, pakkauskoko ja vahvuus. Katso painoon perustuvat annostusohjeet kohdasta Pediatriset potilaat.
Hoidon kesto
Levetirasetaamin annosta laskimonsisäisesti pidempään kuin 4 vuorokauden ajan ei ole kokemusta.
Hoidon lopettaminen
Jos levetirasetaamihoito on lopetettava, lääkitystä on suositeltavaa vähentää asteittain (esim. yli 50 kg:n painoisilla aikuisilla ja nuorilla vähentämällä 500 mg kaksi kertaa päivässä 2−4 viikon välein; alle 50 kg:n painoisilla lapsilla ja nuorilla annosta ei saa laskea enempää kuin 10 mg/kg kaksi kertaa päivässä kahden viikon välein).
Erityispotilasryhmät
Iäkkäät (vähintään 65‑vuotiaat)
Iäkkäiden potilaiden annos suositellaan määritettäväksi munuaisten toimintakyvyn perusteella (ks. Munuaisten vajaatoiminta).
Munuaisten vajaatoiminta
Vuorokausiannos on yksilöitävä munuaisten toiminnan mukaan.
Aikuisille potilaille annos säädetään seuraavan taulukon mukaisesti. Annostaulukkoa varten tarvitaan arvio potilaan kreatiniinipuhdistumasta (CLcr) ml/min. Aikuisten ja vähintään 50 kg:n painoisten nuorten CLcr voidaan arvioida määrittämällä seerumin kreatiniinipitoisuus (mg/dl) ja sijoittamalla se seuraavaan kaavaan:

Kreatiniinipuhdistuma suhteutetaan tämän jälkeen kehon pinta-alaan (BSA = body surface area) seuraavasti:
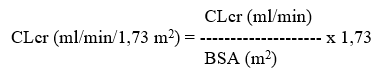
Annosmuutos munuaisten vajaatoiminnassa aikuisilla ja yli 50 kg:n painoisilla nuorilla potilailla:
| Ryhmä | Kreatiniinipuhdistuma (ml/min/1,73 m2) | Annos ja antotiheys |
| Normaali | ≥ 80 | 500-1500 mg kahdesti päivässä |
| Lievä | 50-79 | 500-1000 mg kahdesti päivässä |
| Keskivaikea | 30-49 | 250-750 mg kahdesti päivässä |
| Vaikea | < 30 | 250-500 mg kahdesti päivässä |
| Myöhäisvaiheen munuaissairaus –dialyysipotilas (1) | - | 500-1000 mg kerran päivässä (2) |
(1) 750 mg levetirasetaamia on suositeltava aloitusannos ensimmäisenä hoitopäivänä.
(2) Dialyysin jälkeen suositellaan 250−500 mg:n lisäannosta.
Lapsille, joilla on munuaisten vajaatoiminta, levetirasetaamin annos täytyy määrittää munuaisten toiminnan mukaisesti, sillä levetirasetaamin puhdistuma riippuu munuaisten toiminnasta. Suositus perustuu tutkimukseen aikuisilla munuaisten vajaatoimintapotilailla.
Nuorten ja lasten CLcr (ml/min/1,73 m2) voidaan arvioida määrittämällä seerumin kreatiniinipitoisuus (mg/dl) ja sijoittamalla se seuraavaan kaavaan (Schwartzin laskukaava):

ks = 0,55 alle 13‑vuotiaat lapset ja nuoret tytöt; ks = 0,7 nuoret pojat
Annosmuutos munuaisten vajaatoiminnassa lapsilla ja alle 50 kg:n painoisilla nuorilla:
| Ryhmä | Kreatiniini-puhdistuma (ml/min/1,73 m2) | Annos ja antotiheys |
| 50 kg:n painoiset nuoret ja vähintään 4‑vuotiaat lapset | ||
| Normaali | ≥ 80 | 10–30 mg/kg (0,10–0,30 ml/kg) kahdesti päivässä |
| Lievä | 50–79 | 10–20 mg/kg (0,10–0,20 ml/kg) kahdesti päivässä |
| Keskivaikea | 30–49 | 5–15 mg/kg (0,05–0,15 ml/kg) kahdesti päivässä |
| Vaikea | < 30 | 5–10 mg/kg (0,05–0,10 ml/kg) kahdesti päivässä |
| Myöhäisvaiheen munuaissairaus –dialyysipotilas | - | 10–20 mg/kg (0,10–0,20 ml/kg) kerran päivässä (1) (2) |
(1) Kyllästysannosta 15 mg/kg (0,15 ml/kg) levetirasetaamia suositellaan ensimmäisenä hoitopäivänä.
(2) Dialyysin jälkeen suositellaan lisäannosta 5−10 mg/kg (0,05–0,10 ml/kg).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa lievässä tai keskivaikeassa maksan vajaatoiminnassa. Vaikeassa maksan vajaatoiminnassa kreatiniinipuhdistuma ei välttämättä anna todellista kuvaa munuaisten vajaatoiminnan asteesta. Tämän vuoksi suositellaan päivittäisen ylläpitoannoksen pienentämistä 50 %:lla, jos kreatiniinipuhdistuma on < 60 ml/min/1,73 m2.
Pediatriset potilaat
Lääkärin on määrättävä potilaalle hänen ikänsä, painonsa ja annoksensa perusteella sopivin lääkemuoto, pakkauskoko ja vahvuus.
Ainoana lääkkeenä
Keppran turvallisuutta ja tehoa lasten ja alle 16‑vuotiaiden nuorten hoidossa ainoana lääkkeenä ei ole varmistettu.
Tietoja ei ole saatavilla.
16–17-vuotiaat nuoret (≥ 50 kg), joilla on paikallisalkuisia (sekundaarisesti yleistyviä tai yleistymättömiä) kohtauksia ja äskettäin diagnosoitu epilepsia.
Katso edellä kohta Aikuiset (≥ 18-vuotiaat) ja 12–17-vuotiaat nuoret (≥ 50 kg).
Lisälääkkeenä 4−11‑vuotiaille lapsille ja 12−17‑vuotiaille nuorille (< 50 kg)
Aloitusannos on 10 mg/kg kaksi kertaa päivässä.
Kliinisestä vasteesta ja siedettävyydestä riippuen vuorokausiannos voidaan nostaa annokseen 30 mg/kg kaksi kertaa päivässä. Annosta ei saa muuttaa enempää kuin lisäämällä tai vähentämällä vuorokausiannosta 10 mg/kg kaksi kertaa päivässä kahden viikon välein. Kaikkiin käyttöaiheisiin tulee käyttää matalinta tehokasta annosta.
Kaikissa käyttöaiheissa annos lapsille, jotka painavat 50 kg tai enemmän, on sama kuin aikuisille.
Katso tiedot kaikista käyttöaiheista edellä kohdasta Aikuiset (≥ 18-vuotiaat) ja 12–17-vuotiaat nuoret (≥ 50 kg).
Annossuositukset lapsille ja nuorille:
| Paino | Aloitusannos: 10 mg/kg kaksi kertaa päivässä | Enimmäisannos: 30 mg/kg kaksi kertaa päivässä |
| 15 kg (1) | 150 mg kaksi kertaa päivässä | 450 mg kaksi kertaa päivässä |
| 20 kg (1) | 200 mg kaksi kertaa päivässä | 600 mg kaksi kertaa päivässä |
| 25 kg | 250 mg kaksi kertaa päivässä | 750 mg kaksi kertaa päivässä |
| Yli 50 kg (2) | 500 mg kaksi kertaa päivässä | 1500 mg kaksi kertaa päivässä |
(1) Lapsilla, jotka painavat ≤ 25 kg, hoito tulisi mieluiten aloittaa Keppra 100 mg/ml oraaliliuoksella.
(2) Annos lapsille ja nuorille, jotka painavat 50 kg tai enemmän, on sama kuin aikuisille.
Liitännäishoito alle 4‑vuotiaille imeväisille ja lapsille
Keppra-infuusiokonsentraatin turvallisuutta ja tehoa alle 4‑vuotiaiden imeväisten ja lasten hoidossa ei ole varmistettu.
Tällä hetkellä käytettävissä olevat tiedot on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, mutta annossuosituksia ei voida antaa.
Antotapa
Keppra-infuusiokonsentraatti on tarkoitettu vain laskimoon annettavaksi ja suositeltu annos on laimennettava vähintään 100 ml:aan yhteensopivaa laimenninta ja annettava laskimoon 15 minuuttia kestävänä infuusiona (ks. kohta Käyttö- ja käsittelyohjeet).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle, muille pyrrolidonijohdoksille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Munuaisten vajaatoiminta
Levetirasetaamin käyttö munuaisten vajaatoiminnan yhteydessä saattaa edellyttää annoksen pienentämistä. Potilailla, joilla on vaikea maksan vajaatoiminta, suositellaan munuaisten toimintakyvyn arviointia ennen annoksen valintaa (ks. kohta Annostus ja antotapa).
Akuutti munuaisvaurio
Levetirasetaamin käyttöön on joissakin hyvin harvinaisissa tapauksissa yhdistetty akuutti munuaisvaurio, jonka ilmenemisajankohta on vaihdellut muutamasta päivästä useisiin kuukausiin hoidon aloittamisesta.
Verisolumäärät
Levetirasetaamin annon yhteydessä, yleensä hoidon alussa, on joissakin harvinaisissa tapauksissa kuvattu verisolumäärien pienenemistä (neutropeniaa, agranulosytoosia, leukopeniaa, trombosytopeniaa ja pansytopeniaa). Täydellinen verenkuva tulisi määrittää, jos potilaalla ilmenee huomattavaa heikkoutta, kuumetta, uusiutuvia infektioita tai hyytymishäiriöitä (ks. kohta Haittavaikutukset).
Itsemurha
Epilepsialääkkeiden (myös levetirasetaamin) käyttäjillä on raportoitu itsemurhia, itsemurhayrityksiä, itsetuhoajatuksia ja -käyttäytymistä. Satunnaistettujen, lumekontrolloitujen epilepsialääketutkimusten meta-analyysi osoitti itsetuhoajatusten ja -käyttäytymisen riskin vähäistä lisääntymistä. Riskin kasvun mekanismia ei tunneta.
Potilaita tulee seurata masennuksen ja/tai itsetuhoajatusten ja -käyttäytymisen varalta, ja asianmukaisen hoidon tarvetta tulee harkita. Potilaita (ja heidän omaisiaan) tulee neuvoa ottamaan yhteyttä lääkäriin, mikäli masennusta ja/tai itsetuhoajatuksia tai -käyttäytymistä esiintyy.
Poikkeava ja aggressiivinen käyttäytyminen
Levetirasetaami voi aiheuttaa psykoottisia oireita ja poikkeavaa käyttäytymistä, kuten ärtyneisyyttä ja aggressiivisuutta. Levetirasetaamihoitoa saavia potilaita on seurattava merkittäviin mielialan ja/tai persoonallisuuden muutoksiin viittaavien psykiatristen oireiden varalta. Jos tällaista käyttäytymistä havaitaan, on harkittava hoidon mukauttamista tai asteittaista lopettamista. Jos harkitaan hoidon lopettamista, ks. kohta Annostus ja antotapa.
Kohtausten paheneminen
Levetirasetaami voi muiden epilepsialääkkeiden tavoin harvinaisissa tapauksissa lisätä kouristusten määrää tai niiden vaikeusastetta. Tätä paradoksaalista vaikutusta on raportoitu lähinnä ensimmäisen kuukauden kuluessa levetirasetaamin aloittamisesta tai annoksen suurentamisesta, ja se korjautuu lääkkeen lopettamisen tai annoksen pienentämisen jälkeen. Potilaita on neuvottava ottamaan välittömästi yhteyttä lääkäriinsä, jos epilepsia pahenee.
Tehon puutteesta tai kohtausten pahenemisesta on raportoitu esimerkiksi potilailla, joiden epilepsiaan liittyy jänniteherkän natriumkanavan alfa-alayksikön 8 (SCN8A) mutaatioita.
Sydänsähkökäyrässä todettava QT-ajan pidentyminen
Markkinoille tulon jälkeisessä seurannassa on havaittu harvinaisina tapauksina sydänsähkökäyrässä (EKG) todettavaa QT-ajan pidentymistä. Jos potilaalla on pidentynyt QTc-aika, jos potilasta hoidetaan samanaikaisesti QTc-aikaan vaikuttavilla lääkkeillä tai jos potilaalla on ennestään oleellinen sydänsairaus tai elektrolyyttihäiriöitä, levetirasetaamin käytössä on oltava varovainen.
Pediatriset potilaat
Saatavilla olevan tiedon perusteella vaikutuksia kasvuun ja puberteettiin lapsilla ei ole. Pitkäaikaiset vaikutukset lasten oppimiseen, älykkyyteen, kasvuun, umpieritykseen, puberteettiin ja lisääntymiskykyyn eivät kuitenkaan ole selvillä.
Apuaineet
Tämä lääkevalmiste sisältää 2,5 mmol (tai 57 mg) natriumia/enimmäiskerta-annos [0,8 mmol (tai 19 mg)/injektiopullo)], mikä vastaa 2,85 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille. Potilaiden, jotka ruokavaliossaan pyrkivät rajoittamaan natriumin saantia, tulee huomioida tämä.
Yhteisvaikutukset
Epilepsialääkkeet
Kliinisten tutkimusten tulokset aikuisilla osoittavat, että levetirasetaami ei vaikuta seuraavien epilepsialääkkeiden pitoisuuksiin seerumissa: fenytoiini, karbamatsepiini, valproaatti, fenobarbitaali, lamotrigiini, gabapentiini, primidoni ja että mainitut epilepsialääkkeet eivät vaikuta levetirasetaamin farmakokinetiikkaan.
Kliinisesti merkittäviä yhteisvaikutuksia muiden lääkeaineiden kanssa ei havaittu lapsipotilailla, joille annettiin levetirasetaamia jopa 60 mg/kg/vrk ja tämä tulos vastaa havaintoja aikuisilla.
Retrospektiivinen arvio farmakokineettisistä yhteisvaikutuksista 4−17‑vuotiailla epilepsiaa sairastavilla lapsilla ja nuorilla vahvisti, että suun kautta annetun levetirasetaamin käyttö lisälääkityksenä ei vaikuttanut samanaikaisesti annettujen karbamatsepiinin ja valproaatin vakaan tilan pitoisuuksiin seerumissa. Kuitenkin entsyymejä indusoivia epilepsialääkkeitä käyttävillä lapsilla levetirasetaamin puhdistuma kasvaa 20 %:lla. Annoksen muuttaminen ei ole tarpeen.
Probenesidi
Probenesidin (aine, joka estää eritystä munuaistiehyistä) on osoitettu annoksella 500 mg neljästi päivässä pienentävän päämetaboliitin, mutta ei levetirasetaamin, munuaispuhdistumaa. Metaboliitin pitoisuudet jäävät kuitenkin pieniksi.
Metotreksaatti
Levetirasetaamin ja metotreksaatin samanaikaisen annon on raportoitu vähentävän metotreksaatin puhdistumaa, jolloin metotreksaatin pitoisuus veressä suurenee tai metotreksaatti säilyy veressä tavallista pitempään ja voi aiheuttaa toksisuutta. Veren metotreksaatti- ja levetirasetaamipitoisuuksia on seurattava tarkoin, jos potilas saa samanaikaista hoitoa näillä kahdella lääkeaineella.
Oraaliset ehkäisyvalmisteet ja muut farmakokineettiset yhteisvaikutukset
Levetirasetaami (1000 mg päivässä) ei vaikuttanut oraalisten ehkäisyvalmisteiden (etinyyliestradioli ja levonorgestreeli) farmakokinetiikkaan, eikä endokriinisiin muuttujiin (luteinisoiva hormoni ja progesteroni). Levetirasetaami (2000 mg päivässä) ei vaikuttanut digoksiinin eikä varfariinin farmakokinetiikkaan, protrombiiniajat pysyivät muuttumattomina. Yhteiskäyttö digoksiinin, oraalisten ehkäisyvalmisteiden ja varfariinin kanssa ei vaikuttanut levetirasetaamin farmakokinetiikkaan.
Alkoholi
Tietoja levetirasetaamin ja alkoholin yhteisvaikutuksesta ei ole.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisille, jotka voivat tulla raskaaksi, tulee antaa asiantuntijan neuvontaa. Levetirasetaamihoitoa on arvioitava, kun nainen suunnittelee raskautta. Kaikkien epilepsialääkkeiden tavoin myös levetirasetaamin äkillistä keskeyttämistä on vältettävä, sillä se voi aiheuttaa läpilyöntikohtauksia (breakthrough), joilla voi puolestaan olla vakavia seurauksia naiselle ja syntymättömälle lapselle. Monoterapiaa on suosittava aina kuin mahdollista, koska hoitoon useilla epilepsialääkkeillä voi käytetyistä epilepsialääkkeistä riippuen liittyä monoterapiaan verrattuna korkeampi synnynnäisten epämuodostumien riski.
Raskaus
Huomattava määrä markkinoille tulon jälkeistä tietoa raskaana olevista naisista, jotka altistuivat levetirasetaamimonoterapialle (yli 1800, joista yli 1500 altistui raskauden ensimmäisen kolmanneksen aikana), ei viittaa vakavien synnynnäisten epämuodostumien riskin lisääntymiseen. Levetirasetaamimonoterapialle kohdussa altistuneiden lasten neurologisesta kehityksestä on saatavilla vain vähän tietoa. Tiedot kahdesta ei-kokeellisesta väestöpohjaisesta rekisteritutkimuksesta, jotka tehtiin pääosin samasta pohjoismaisesta tietoaineistosta ja johon kuului yli 1 000 epilepsiaa sairastavalle äidille syntynyttä lasta, jotka olivat raskauden aikana altistuneet levetirasetaamimonoterapialle, eivät viittaa autismikirjon häiriöiden tai älyllisen kehitysvammaisuuden riskin lisääntymiseen verrattuna epilepsiaa sairastaville äideille syntyneisiin lapsiin, jotka eivät olleet altistuneet epilepsialääkkeille kohdussa. Levetirasetaamiryhmän lasten keskimääräinen seuranta-aika oli lyhyempi kuin epilepsialääkkeille altistumattomien lasten seuranta-aika (esim. 4,4 vuotta vs. 6,8 vuotta yhdessä tutkimuksessa).
Levetirasetaamia voi käyttää raskauden aikana, jos sitä huolellisen arvioinnin jälkeen pidetään kliinisesti tarpeellisena. Tällaisessa tapauksessa suositellaan pienintä tehokasta annosta.
Raskaudenaikaiset fysiologiset muutokset voivat vaikuttaa levetirasetaamin pitoisuuteen. Levetirasetaamipitoisuuden pienenemistä plasmassa on havaittu raskauden aikana. Pieneneminen on voimakkaampaa raskauden viimeisen kolmanneksen aikana (enimmillään 60 % pitoisuudesta ennen raskautta). Levetirasetaamia saavien raskaana olevien naisten asianmukainen hoito tulee taata.
Imetys
Levetirasetaami erittyy äidinmaitoon, joten imettämistä ei suositella. Mikäli imetyksen aikana tarvitaan levetirasetaamihoitoa, tulisi hoidon hyödyt ja riskit punnita rintaruokinnan tärkeys huomioiden.
Hedelmällisyys
Eläinkokeissa ei havaittu vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta). Kliinisiä tietoja ei ole, joten mahdollista riskiä ihmiselle ei tiedetä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Levetirasetaamilla on vähäinen tai kohtalainen vaikutus ajokykyyn ja koneiden käyttökykyyn. Koska yksilöllinen herkkyys vaihtelee, jotkut potilaat saattavat olla etenkin hoidon alussa tai annosta suurennettaessa uneliaita tai kokea muita keskushermostoon liittyviä oireita. Tämän vuoksi näille potilaille suositellaan varovaisuutta tarkkuutta vaativissa tehtävissä, esim. autolla ajamisen ja koneiden käytön yhteydessä. Potilaita kehotetaan välttämään autolla ajoa ja koneiden käyttämistä kunnes on todettu, että heidän kykynsä tehdä näitä tehtäviä ei ole heikentynyt.
Haittavaikutukset
Turvallisuustietojen yhteenveto
Yleisimmin raportoidut haittavaikutukset olivat nenänielun tulehdus, uneliaisuus, päänsärky, väsymys ja heitehuimaus. Seuraava haittavaikutusprofiili perustuu yhdistettyihin tutkimustuloksiin lumekontrolloiduista kliinisistä tutkimuksista, jotka kattoivat valmisteen kaikki käyttöaiheet. Yhteensä 3416 potilasta sai levetirasetaamihoitoa. Näitä tietoja on täydennetty levetirasetaamihoitoa koskevilla tuloksilla vastaavista avoimista jatkotutkimuksista sekä valmisteen markkinoilletulon jälkeisillä kokemuksilla. Levetirasetaamin turvallisuusprofiili on yleisesti ottaen samankaltainen kaikissa ikäryhmissä (aikuis- ja lapsipotilailla) sekä kaikissa valmisteelle hyväksytyissä epilepsian käyttöaiheissa. Koska altistus laskimoon annetulle Keppralle oli vähäistä ja koska suun kautta ja laskimoon annettavat lääkemuodot ovat biologisesti samanarvoisia, turvallisuustiedot laskimoon annettavasta Kepprasta perustuvat suun kautta käytettävään Keppraan.
Haittavaikutustaulukko
Kliinisistä tutkimuksista (aikuisilla, nuorilla, lapsilla ja yli 1 kuukauden ikäisillä imeväisillä) ja markkinoille tulon jälkeisestä seurannasta kertyneet tiedot haittavaikutuksista on lueteltu ohessa kohde-elimien ja haittavaikutuksen yleisyyden mukaisesti. Haittavaikutukset on esitetty vakavuudeltaan alenevassa järjestyksessä, ja niiden yleisyys on ilmaistu seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000) ja hyvin harvinainen (< 1/10 000).
| Elinjärjestelmä (MedDRA) | Yleisyysluokka | ||||
| Hyvin yleiset | Yleiset | Melko harvinaiset | Harvinaiset | Hyvin harvinaiset | |
| Infektiot | Nenänielun tulehdus | Infektio | |||
| Veri ja imukudos | Trombosytopenia, leukopenia | Pansytopenia, neutropenia, agranulosytoosi | |||
| Immuunijärjestelmä | Lääkeaineihottuma, johon liittyy eosinofiliaa ja systeemioireita (DRESS)(1), yliherkkyys (myös angioedeema ja anafylaksia) | ||||
| Aineenvaihdunta ja ravitsemus | Syömishäiriö | Painonlasku, painonnousu | Hyponatremia | ||
| Psyykkiset häiriöt | Masennus, vihamielisyys/ aggressiivisuus, ahdistuneisuus, unettomuus, hermostuneisuus/ ärtyneisyys | Itsemurhayritys, itsemurha-ajatukset, psykoottinen häiriö, poikkeava käyttäytyminen, aistiharha, vihantunne, sekavuustila, paniikkikohtaus, affektilabiilius/ mielialanvaihtelut, agitaatio | Itsemurha, persoonallisuushäiriö, poikkeavat ajatukset, delirium | Pakko-oireinen häiriö(2) | |
| Hermosto | Uneliaisuus, päänsärky | Kouristus, tasapainohäiriö, heitehuimaus, letargia, vapina | Muistinmenetys, muistin heikkeneminen, koordinaatiohäiriö/ ataksia, tuntoharhat, tarkkaavaisuuden häiriintyminen | Koreoatetoosi, dyskinesia, hyperkinesia, kävelyn häiriö, enkefalopatia, kohtausten paheneminen, pahanlaatuinen neuroleptioireyhtymä(3) | |
| Silmät | Kaksoiskuvat, näön hämärtyminen | ||||
| Kuulo ja tasapainoelin | Kiertohuimaus | ||||
| Sydän | Sydänsähkökäyrässä todettava pidentynyt QT-aika | ||||
| Hengityselimet, rintakehä ja välikarsina | Yskä | ||||
| Ruoansulatus-elimistö | Vatsakipu, ripuli, dyspepsia, oksentelu, pahoinvointi | Haimatulehdus | |||
| Maksa ja sappi | Poikkeavat tulokset maksan toimintakokeista | Maksan vajaatoiminta, maksatulehdus | |||
| Iho ja ihonalainen kudos | Ihottuma | Hiustenlähtö, ekseema, kutina | Toksinen epidermaalinen nekrolyysi, Stevens‑Johnsonin oireyhtymä, erythema multiforme | ||
| Luusto, lihakset ja sidekudos | Lihasheikkous, lihaskipu | Rabdomyolyysi ja veren kreatiinikinaasipitoisuuden suureneminen(3) | |||
| Munuaiset ja virtsatiet | Akuutti munuaisvaurio | ||||
| Yleisoireet ja antopaikassa todettavat haitat | Voimattomuus/ väsymys | ||||
| Vammat ja myrkytykset | Vamma | ||||
(1) Katso Kuvaus joistakin haittavaikutuksista.
(2) Hyvin harvinaisissa tapauksissa pakko-oireinen häiriö (OCD) on kehittynyt potilaille, joilla on ollut anamneesissa pakko-oireinen häiriö tai muita psyykkisiä häiriöitä. Näitä tapauksia on havaittu markkinoilletulon jälkeisessä seurannassa.
(3) Merkitsevästi yleisempi japanilaispotilailla kuin muilla potilailla.
Kuvaus joistakin haittavaikutuksista
Useisiin elimiin vaikuttavat yliherkkyysreaktiot
Levetirasetaamilla hoidetuilla potilailla on harvoin raportoitu useisiin elimiin vaikuttavia yliherkkyysreaktioita (tunnetaan myös nimellä yleisoireinen eosinofiilinen oireyhtymä [DRESS]), joiden kliiniset ilmenemismuodot voivat kehittyä 2–8 viikkoa hoidon aloittamisen jälkeen. Reaktioiden ilmenemismuoto vaihtelee, mutta tyypillisesti niihin liittyy kuumetta, ihottumaa, kasvojen turvotusta, lymfadenopatioita ja hematologisia poikkeavuuksia, ja näihin reaktioihin voi liittyä eri elinjärjestelmiä, enimmäkseen maksa. Jos useisiin elimiin vaikuttavaa yliherkkyysreaktiota epäillään, levetirasetaamin käyttö on keskeytettävä.
Syömishäiriön riski on suurempi, kun levetirasetaamia annetaan yhdessä topiramaatin kanssa.
Useissa alopesiatapauksissa hiukset kasvoivat takaisin, kun levetirasetaamin käyttö keskeytettiin.
Joissakin pansytopeniatapauksissa todettiin luuydinlama.
Enkefalopatiatapauksia ilmeni yleensä hoidon alussa (muutamasta päivästä joihinkin kuukausiin hoidon aloittamisesta), ja ne hävisivät hoidon lopettamisen jälkeen.
Pediatriset potilaat
Levetirasetaamihoitoa on annettu sekä lumekontrolloiduissa että avoimissa jatkotutkimuksissa yhteensä 190:lle 1 kk–≤ 4‑vuotiaalle lapsipotilaalle. Näistä potilaista 60 sai levetirasetaamihoitoa lumekontrolloiduissa tutkimuksissa. Ikäryhmässä 4–16 vuotta levetirasetaamihoitoa on saanut yhteensä 645 lapsipotilasta sekä lumekontrolloiduissa että avoimissa jatkotutkimuksissa. Näistä potilaista 233 sai levetirasetaamihoitoa lumekontrolloiduissa tutkimuksissa. Markkinoilletulon jälkeen saadut kokemukset levetirasetaamin käytöstä täydentävät näiden molempien pediatristen ikäryhmien tutkimustuloksia.
Lisäksi valmisteen myyntiluvan saamisen jälkeen on tehty tutkimus, jossa levetirasetaamille altistettiin 101 alle 1-vuotiasta imeväistä. Levetirasetaamilla ei tunnistettu mitään uusia turvallisuusseikkoja epilepsiaa sairastaneilla alle 1-vuotiailla imeväisillä.
Levetirasetaamin haittavaikutusprofiili on yleensä samankaltainen kaikissa ikäryhmissä ja kaikissa valmisteelle hyväksytyissä epilepsian käyttöaiheissa. Lapsipotilaita koskeneet turvallisuustulokset lumekontrolloiduista kliinisistä tutkimuksista olivat yhdenmukaiset aikuisten turvallisuusprofiilin kanssa lukuun ottamatta käyttäytymiseen ja psyykeen kohdistuneita haittavaikutuksia, jotka olivat yleisempiä lapsilla kuin aikuisilla. 4–16‑vuotiailla lapsilla ja nuorilla raportoitiin muita ikäryhmiä tai kokonaisturvallisuusprofiilia useammin oksentelua (hyvin yleinen, 11,2 %), agitaatiota (yleinen, 3,4 %), mielialanvaihteluja (yleinen, 2,1 %), affektilabiiliutta (yleinen, 1,7 %), aggressiivisuutta (yleinen, 8,2 %), poikkeavaa käyttäytymistä (yleinen, 5,6 %) ja letargiaa (yleinen, 3,9 %). Pikkulapsilla ja lapsilla ikäryhmässä 1 kk–≤ 4 v raportoitiin muita ikäryhmiä tai kokonaisturvallisuusprofiilia enemmän ärtyneisyyttä (hyvin yleinen, 11,7 %) ja koordinaatiohäiriöitä (yleinen, 3,3 %).
Lapsipotilailla tehdyssä kaksoissokkoutetussa, lumekontrolloidussa turvallisuustutkimuksessa, jonka oli tarkoitus osoittaa valmisteen yhdenvertaisuus (non-inferiority), arvioitiin levetirasetaamin kognitiivisia ja neuropsykologisia vaikutuksia paikallisalkuisia kohtauksia saavilla 4–16‑vuotiailla lapsilla. Siinä todettiin, että Keppra ei eronnut (oli yhdenvertainen) lumelääkkeestä lähtöryhmissä pysyneiden Leiter-R Attention and Memory, Memory Screen Composite ‑pisteiden muutoksessa lähtötilanteeseen nähden. Käytökselliseen ja emotionaaliseen toimintakykyyn liittyvät tulokset osoittivat aggressiivisen käyttäytymisen pahentuneen levetirasetaamihoitoa saaneilla potilailla, mikä mitattiin standardoidusti ja systemaattisesti validoitua menetelmää (CBCL – Achenbach Child Behavior Checklist) käyttäen. Levetirasetaamihoitoa avoimessa pitkäkestoisessa jatkotutkimuksessa saaneilla potilailla ei kuitenkaan keskimääräisesti esiintynyt käytöksellisen ja emotionaalisen toimintakyvyn huononemista, etenkään heidän aggressiivista käyttäytymistä osoittavat mittarinsa eivät olleet huonontuneet lähtötilanteeseen nähden.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet
Keppran yliannoksilla on ilmaantunut uneliaisuutta, kiihtymystä, vihamielisyyttä, tajunnantason laskua, hengityslamaa ja koomaa.
Yliannostuksen hoito
Levetirasetaamille ei ole spesifistä vastalääkettä. Yliannoksen hoito on oireenmukaista ja hemodialyysia voidaan käyttää. Dialyysin hyötyosuus on 60 % levetirasetaamille ja 74 % päämetaboliitille.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: epilepsialääkkeet, muut epilepsialääkkeet, ATC-koodi: N03AX14.
Vaikuttava aine, levetirasetaami, on pyrrolidonijohdos (α-etyyli-2-okso-1-pyrrolidiiniasetamidin S‑enantiomeeri). Se ei ole kemiallisesti sukua muille tunnetuille antiepileptisesti vaikuttaville lääkeaineille.
Vaikutusmekanismi
Levetirasetaamin vaikutusmekanismia ei ole vielä tarkalleen selvitetty. In vitro ja in vivo tehdyt kokeet viittaavat siihen, että levetirasetaami ei vaikuta solun perusominaisuuksiin eikä normaaliin neurotransmissioon.
In vitro tehdyt kokeet osoittavat, että levetirasetaami vaikuttaa neuronien Ca2+-ionikonsentraatioon estämällä osittain N‑tyypin Ca2+-virtausta ja vähentämällä Ca2+-ionien vapautumista neuronivarastoista. Lisäksi se kumoaa osittain sinkin ja ß‑karboliinien aikaansaamaa GABA- ja glysiinivälitteisen virtauksen vähenemistä. Levetirasetaamin on todettu in vitro tehdyissä kokeissa sitoutuvan spesifiseen kohtaan jyrsijän aivokudoksessa. Tämä sitoutumiskohta on synapsirakkulan proteiini 2A, jonka uskotaan osallistuvan vesikkelifuusioon ja neurotransmitterin eksosytoosiin. Levetirasetaamilla ja samantyyppisillä analogeilla on todettu saman luokan affiniteetti sitoutumisessa synaptiseen rakkulaproteiiniin 2A, mikä korreloi niiden kouristuksenestotehoon hiiren audiogeenisessä epilepsiamallissa. Tämä havainto viittaa siihen, että levetirasetaamin ja rakkulaproteiinin 2A vuorovaikutus näyttää olevan osallisena tämän lääkkeen antiepileptisessa vaikutuksessa.
Farmakodynaamiset vaikutukset
Levetirasetaami estää useissa eläinmalleissa paikallisalkuisia ja primaaristi yleistyviä kohtauksia, mutta sillä ei ole kouristuksia edistävää vaikutusta. Päämetaboliitti on inaktiivinen. Ihmisellä tehokkuus sekä paikallisalkuisissa että yleistyvissä epileptisissä tiloissa (epilepsian kaltaiset purkaukset / vilkkuvalon aiheuttama vaste) vahvistaa levetirasetaamin laajan farmakologisen tehon.
Kliininen teho ja turvallisuus
Lisälääkkeenä epilepsiapotilaiden paikallisalkuisten (sekundaarisesti yleistyvien tai yleistymättömien) kohtausten hoidossa aikuisilla, nuorilla ja vähintään 4‑vuotiailla lapsilla
Levetirasetaamin teho on osoitettu aikuisilla kolmessa kaksoissokkoutetussa lumekontrolloidussa tutkimuksessa, joissa levetirasetaamia annettiin 1000 mg, 2000 mg tai 3000 mg päivässä jaettuna kahteen osa-annokseen ja hoidon kesto oli jopa 18 viikkoa. Yhdistetyn analyysin mukaan prosenttiosuus potilaista, joilla paikallisalkuisten kohtausten määrä viikossa väheni vähintään 50 % lähtöarvosta, kun käytettiin vakaata annosta (12/14 viikkoa) oli 27,7 % levetirasetaamiannoksen ollessa 1000 mg, 31,6 % levetirasetaamiannoksen ollessa 2000 mg ja 41,3 % levetirasetaamiannoksen ollessa 3000 mg. Lumelääkettä saaneiden ryhmässä vastaava prosenttiosuus oli 12,6 %.
Pediatriset potilaat
Levetirasetaamin teho on osoitettu lapsilla (4−16‑vuotiailla) kaksoissokkoutetussa lumekontrolloidussa tutkimuksessa, johon osallistui 198 potilasta ja hoidon kesto oli 14 viikkoa. Tässä tutkimuksessa potilaat saivat levetirasetaamia vakaalla annoksella 60 mg/kg päivässä (kahdesti päivässä annosteltuna).
44,6 %:lla potilaista levetirasetaamiryhmässä ja 19,6 %:lla potilaista lumelääkeryhmässä paikallisalkuisten kohtausten määrä viikossa väheni vähintään 50 % lähtöarvosta.
Hoidon jatkuessa pitkäkestoisesti 11,4 %:lla potilaista ei ilmennyt kohtauksia laisinkaan ainakaan 6 kuukauteen ja 7,2 %:lla potilaista ei ilmennyt kohtauksia laisinkaan ainakaan 1 vuoteen.
Lumekontrolloiduissa kliinisissä tutkimuksissa levetirasetaamille on altistettu 35 alle 1‑vuotiasta imeväistä, joilla oli paikallisalkuisia kohtauksia. Näistä potilaista vain 13 oli alle 6 kuukauden ikäisiä.
Ainoana epilepsialääkkeenä paikallisalkuisten (sekundaarisesti yleistyvien tai yleistymättömien) kohtausten hoidossa vähintään 16‑vuotiailla potilailla, joilla on äskettäin diagnosoitu epilepsia
Levetirasetaamin teho ainoana epilepsialääkkeenä osoitettiin kaksoissokkoutetussa, rinnakkaisryhmillä toteutetussa tutkimuksessa, jossa levetirasetaamia verrattiin säädellysti karbamatsepiinia vapauttavaan lääkevalmisteeseen (ei huonompi kuin vertailu) 576:lla vähintään 16‑vuotiaalla potilaalla, joilla on äskettäin diagnosoitu epilepsia. Potilailla piti esiintyä provosoimattomia paikallisalkuisia kohtauksia tai pelkästään yleistyneitä toonis-kloonisia kohtauksia. Potilaat satunnaistettiin saamaan joko säädellysti karbamatsepiinia vapauttavaa lääkevalmistetta 400−1200 mg päivässä tai levetirasetaamia 1000−3000 mg päivässä ja vasteesta riippuen hoidon kesto oli jopa 121 viikkoa.
6 kuukauden kohtaukseton jakso saatiin 73,0 %:lla potilaista levetirasetaamiryhmässä ja 72,8 %:lla potilaista karbamatsepiiniryhmässä; absoluuttinen ero hoitojen välillä oli 0,2 % (95 % CI: −7,8; 8,2). Yli puolella tutkimushenkilöistä kohtauksia ei esiintynyt 12 kuukauden aikana (56,6 %:lla potilaista levetirasetaamiryhmässä ja 58,5 %:lla potilaista karbamatsepiiniryhmässä).
Kliinistä hoitokäytäntöä noudattavassa tutkimuksessa voitiin osalta potilaista, joilla levetirasetaamilla saatiin lisälääkkeenä hyvä vaste, purkaa muu samanaikainen epilepsialääkitys (36 aikuista potilasta 69:stä).
Lisälääkkeenä nuoruusiän myoklonista epilepsiaa sairastavien potilaiden myoklonisten kohtausten hoidossa aikuisilla ja vähintään 12‑vuotiailla nuorilla
Levetirasetaamin teho osoitettiin 16 viikkoa kestäneessä kaksoissokkoutetussa lumekontrolloidussa tutkimuksessa vähintään 12‑vuotiailla potilailla, joilla on idiopaattinen yleistynyt epilepsia ja myoklonisia kohtauksia erilaisissa oireyhtymissä. Suurimmalla osalla potilaista oli nuoruusiän myokloninen epilepsia.
Tässä tutkimuksessa 58,3 %:lla potilaista levetirasetaamiryhmässä (annos 3000 mg päivässä jaettuna kahteen osa-annokseen) ja 23,3 %:lla potilaista lumelääkeryhmässä myoklonisten kohtausten määrä viikossa väheni vähintään 50 %. Hoidon jatkuessa pitkäkestoisesti 28,6 %:lla potilaista ei ilmennyt kohtauksia laisinkaan ainakaan 6 kuukauteen ja 21,0 %:lla potilaista ei ilmennyt kohtauksia laisinkaan ainakaan 1 vuoteen.
Lisälääkkeenä idiopaattista yleistynyttä epilepsiaa sairastavien potilaiden primaarisesti yleistyvien toonis-kloonisten kohtausten hoidossa aikuisilla ja vähintään 12‑vuotiailla nuorilla
Levetirasetaamin teho osoitettiin 24 viikkoa kestäneessä kaksoissokkoutetussa lumekontrolloidussa tutkimuksessa aikuisilla, nuorilla ja pienellä määrällä lapsia, joilla on idiopaattinen yleistynyt epilepsia ja primaarisesti yleistyviä toonis-kloonisia kohtauksia erilaisissa oireyhtymissä (nuoruusiän myokloninen epilepsia, nuoruusiän poissaoloepilepsia, lapsuusiän poissaoloepilepsia tai epilepsia jossa grand mal -kohtauksia esiintyy herätessä). Tässä tutkimuksessa levetirasetaamia annettiin aikuisille ja nuorille 3000 mg päivässä ja lapsille 60 mg/kg päivässä jaettuna kahteen osa-annokseen.
72,2 %:lla potilaista levetirasetaamiryhmässä ja 45,2 %:lla potilaista lumelääkeryhmässä primaarisesti yleistyvien toonis-kloonisten kohtausten määrä viikossa väheni vähintään 50 %. Hoidon jatkuessa pitkäkestoisesti 47,4 %:lla potilaista ei ilmennyt kohtauksia laisinkaan ainakaan 6 kuukauteen ja 31,5 %:lla potilaista ei ilmennyt kohtauksia laisinkaan ainakaan 1 vuoteen.
Farmakokinetiikka
Levetirasetaamin farmakokineettinen profiili on karakterisoitu oraalisen annon jälkeen. 1500 mg:n kerta-annos levetirasetaamia laimennettuna 100 ml:aan yhteensopivaa laimenninta ja infusoituna laskimoon 15 minuutin kuluessa on bioekvivalentti 1500 mg:n levetirasetaamiannokseen otettuna suun kautta kolmena 500 mg:n tablettina.
Levetirasetaamia annettiin laskimoon infuusiona annoksella jopa 4000 mg laimennettuna 100 ml:aan 0,9 % natriumkloridiliuosta 15 minuutin ajan sekä jopa 2500 mg laimennettuna 100 ml:aan 0,9 % natriumkloridia 5 minuutin ajan ja tulokset arvioitiin. Farmakokinetiikka ja turvallisuusprofiilit eivät viitanneet mihinkään turvallisuusriskiin.
Levetirasetaami on hyvin liukeneva ja hyvin läpäisevä yhdiste. Farmakokinetiikka on lineaarinen ja sekä yksilöiden välinen että yksilöllinen vaihtelu on vähäistä. Puhdistuma ei muutu toistuvassa annostelussa. Levetirasetaamin ajasta riippumaton farmakokineettinen profiili todettiin myös annettaessa 1500 mg infuusiona laskimoon 4 päivän ajan kahdesti päivässä annosteltuna.
Sukupuolen, rodun tai vuorokausirytmin aiheuttamaa vaihtelua ei ole todettu. Farmakokinetiikka on samanlainen terveillä vapaaehtoisilla ja epilepsiaa sairastavilla potilailla.
Aikuiset ja nuoret
Jakautuminen
Huippupitoisuus plasmassa (Cmax) oli 51 ± 19 µg/ml (aritmeettinen keskiarvo ± keskihajonta), kun 17 koehenkilölle annettiin kerta-annoksena 1500 mg levetirasetaamia laskimoon infuusiona 15 minuutin ajan.
Tietoa lääkkeen jakautumisesta ihmisen kudoksiin ei ole.
Levetirasetaami ja sen päämetaboliitti eivät sitoudu merkittävästi plasman proteiineihin (< 10 %).
Levetirasetaamin jakaantumistilavuus on noin 0,5−0,7 l/kg, mikä vastaa elimistön koko nestetilavuutta.
Biotransformaatio
Levetirasetaamista metaboloituu ihmisessä vain pieni osuus. Päämetaboliareitti (24 % annoksesta) on asetamidiryhmän entsymaattinen hydrolyysi. Päämetaboliitin, ucb L057, muodostuminen ei tapahdu maksan sytokromi P450-isoentsyymien avulla. Asetamidiryhmän hydrolyysi tapahtuu useissa eri kudoksissa, kuten verisoluissa. Metaboliitti ucb L057 on farmakologisesti inaktiivinen.
Lisäksi on identifioitu kaksi muuta metaboliittia. Toinen muodostuu pyrrolidiinirenkaan hydrolysoituessa (1,6 % annoksesta) ja toinen pyrrolidiinirenkaan avautuessa (0,9 % annoksesta).
Muiden tunnistamattomien metaboliittien osuus on vain 0,6 % annoksesta.
Enantiomeerin muuttumista toiseen muotoon ei tapahdu levetirasetaamille tai sen päämetaboliitille in vivo.
In vitro -interaktiotutkimukset ovat osoittaneet, että levetirasetaami ja sen päämetaboliitti eivät estä tärkeimpiä maksan sytokromi P450-isoentsyymejä (CYP3A4, 2A6, 2C9, 2C19, 2D6, 2E1 ja 1A2), glukuronyylitransferaaseja (UGT1A1 ja UGT1A6) eivätkä vaikuta ihmisen epoksidihydroksylaasin aktiivisuuteen. Levetirasetaami ei vaikuta myöskään valproaatin glukuronidaatioon in vitro. Levetirasetaamilla oli vähän tai ei lainkaan vaikutusta CYP1A2-, SULT1E1- tai UGT1A1-maksaentsyymeihin ihmisen maksasoluviljelmässä. Levetirasetaami indusoi lievästi CYP2B6- ja CYP3A4-entsyymejä. In vitro ja in vivo -yhteisvaikutustutkimukset oraalisten ehkäisyvalmisteiden, digoksiinin ja varfariinin kanssa osoittavat, ettei merkittävää entsyymi-induktiota in vivo ole odotettavissa. Tästä johtuen Keppran interaktiot muiden lääkeaineiden kanssa (tai päinvastoin) ovat epätodennäköisiä.
Eliminaatio
Puoliintumisaika plasmassa on aikuisilla 7±1 tuntia eikä se muutu annoksen tai antotavan muuttuessa eikä toistuvan annostelun yhteydessä. Kokonaispuhdistuman keskiarvo on 0,96 ml/min/kg.
Eritys tapahtuu pääosin virtsaan, keskimäärin 95 % annoksesta (noin 93 % annoksesta on erittynyt 48 tunnin aikana). Ulosteeseen erittyy vain 0,3 % annoksesta.
Ensimmäisen 48 tunnin aikana levetirasetaamista erittyy virtsaan kumulatiivisesti 66 % annoksesta ja vastaavasti päämetaboliitista 24 %.
Levetirasetaamin munuaispuhdistuma on 0,6 ml/min/kg ja ucb L057:n 4,2 ml/min/kg. Tämä osoittaa, että levetirasetaami erittyy suodattumalla munuaiskeräsistä, mutta imeytyy takaisin munuaistiehyistä ja että päämetaboliitti erittyy samoin munuaiskeräsistä suodattumalla mutta sen lisäksi myös aktiivisesti munuaistiehyistä. Levetirasetaamin eritys on suhteessa kreatiniinipuhdistumaan.
Iäkkäät
Iäkkäillä puoliintumisaika on pidentynyt noin 40 % (10−11 tuntia). Tämä johtuu iäkkäiden munuaistoiminnan heikkenemisestä (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Levetirasetaamin ja sen päämetaboliitin kokonaispuhdistuma on verrannollinen kreatiniinipuhdistumaan. Sen vuoksi potilaille, joilla on keskivaikea tai vaikea munuaisten vajaatoiminta, suositellaan Keppran päivittäisen ylläpitoannoksen pienentämistä potilaan kreatiniinipuhdistuman mukaisesti (ks. kohta Annostus ja antotapa).
Vastaavasti aikuisilla myöhäisvaiheen munuaispotilailla, joilla oli anuria, puoliintumisaika oli noin 25 tuntia dialyysien välisenä aikana ja 3,1 tuntia dialyysin aikana.
Tavanomaisen 4 tuntia kestävän dialyysin aikana poistui 51 % levetirasetaamista.
Maksan vajaatoiminta
Lievä ja keskivaikea maksan vajaatoiminta eivät vaikuta merkitsevästi levetirasetaamin puhdistumaan. Useimmilla vaikeaa maksan vajaatoimintaa sairastavilla potilailla levetirasetaamin puhdistuma on pienentynyt yli 50 %, mikä johtuu samanaikaisesta munuaisten vajaatoiminnasta (ks. kohta Annostus ja antotapa).
Pediatriset potilaat
Lapset (4−12‑vuotiaat)
Levetirasetaamin farmakokinetiikkaa lapsipotilailla laskimonsisäisen annon jälkeen ei ole tutkittu. Levetirasetaamin farmakokineettisten ominaisuuksien, farmakokinetiikan aikuisilla laskimonsisäisen annon jälkeen ja lapsilla suun kautta annon jälkeen perusteella altistuksen (AUC) levetirasetaamille oletetaan olevan samanlainen 4−12‑vuotiailla lapsilla laskimonsisäisen ja suun kautta annon jälkeen.
Levetirasetaamin puoliintumisaika epilepsiaa sairastavilla lapsilla (6−12‑vuotiailla) oli 6,0 tuntia suun kautta annetun kerta-annoksen (20 mg/kg) jälkeen. Potilaan painoon suhteutettu kokonaispuhdistuma oli 30 % suurempi kuin aikuisilla epilepsiapotilailla.
Levetirasetaami imeytyi nopeasti epilepsiaa sairastavilla lapsilla (4−12‑vuotiailla) toistuvan oraalisen annostelun jälkeen (20−60 mg/kg/vrk). Huippupitoisuus plasmassa havaittiin 0,5−1 tunnin kuluttua annostelun jälkeen. Huippupitoisuus plasmassa ja AUC-arvo kasvoivat lineaarisesti ja annoksesta riippuvaisesti. Eliminaation puoliintumisaika oli keskimäärin 5 tuntia. Näennäinen puhdistuma oli 1,1 ml/min/kg.
Prekliiniset tiedot turvallisuudesta
Ei-kliinisissä tutkimuksissa ei ole ilmaantunut erityisiä ihmiseen kohdistuvia riskejä tavanomaisten farmakologisten turvallisuustutkimusten eikä genotoksisuus- tai karsinogeenisuuskokeiden perusteella.
Haittavaikutukset, joita ei havaittu kliinisissä tutkimuksissa, mutta joita todettiin rotilla ja vähäisemmässä määrin myös hiirillä, olivat adaptaatioon viittaavia maksamuutoksia, kuten maksan painon nousu ja keskilohkon hypertrofia sekä rasvakertymät ja kohonneet plasman maksaentsyymiarvot; eläinten altistus oli samaa luokkaa kuin ihmisellä ja muutoksilla saattaa olla merkitystä myös kliinisessä käytössä.
Rotilla annoksiin 1800 mg/kg/vrk saakka (kuusinkertainen annos ihmiselle suurimpaan suositeltuun annokseen nähden kehon pinta-alan (mg/m2) tai altistuksen perusteella) vanhemmilla ja F1-sukupolvella ei havaittu haitallisia reaktioita urosten tai naaraiden hedelmällisyyteen eikä lisääntymiseen liittyviin toimintoihin.
Rotilla tehtiin kaksi alkion ja sikiön kehitystä selvittävää tutkimusta annoksilla 400 mg/kg/vrk, 1200 mg/kg/vrk ja 3600 mg/kg/vrk. Annoksella 3600 mg/kg/vrk vain toisessa näistä kahdesta alkion ja sikiön kehitystä selvittävästä tutkimuksesta sikiön painon vähäiseen vähenemiseen liittyi luuston muutosten/lievien poikkeavuuksien marginaalista lisääntymistä. Alkiokuolleisuuteen kohdistuvaa vaikutusta ei todettu eikä epämuodostumien esiintyvyys ollut lisääntynyt. Tiineille naarasrotille haitaton annos (NOAEL, No Observed Adverse Effect Level) oli 3600 mg/kg/vrk (12 kertaa ihmiselle suositeltu suurin vuorokausiannos kehon pinta-alan perusteella laskettuna) ja sikiöille 1200 mg/kg/vrk.
Kaniineilla tehtiin neljä alkion ja sikiön kehitystä selvittävää tutkimusta annoksilla 200 mg/kg/vrk, 600 mg/kg/vrk, 800 mg/kg/vrk, 1200 mg/kg/vrk ja 1800 mg/kg/vrk. Annos 1800 mg/kg/vrk aiheutti emolle huomattavaa toksisuutta, ja sikiön painon laskuun liittyi suurentunutta sikiön sydämen ja verisuoniston/luuston poikkeavuuksien esiintyvyyttä. Haitaton annos (NOAEL) oli emoille < 200 mg/kg/vrk ja sikiöille 200 mg/kg/vrk (vastaa suurinta ihmiselle suositeltua vuorokausiannosta kehon pinta-alan perusteella laskettuna).
Peri- ja postnataalista kehitystä tutkittiin rotilla levetirasetaamiannoksilla 70 mg/kg/vrk, 350 mg/kg/vrk ja 1800 mg/kg/vrk. Haitaton annos (NOAEL) F0-naaraille, samoin kuin F1-jälkeläisten eloonjäännin, kasvun ja kehityksen kannalta vieroitukseen saakka, oli ≥ 1800 mg/kg/vrk (6 kertaa ihmiselle suositeltu suurin vuorokausiannos kehon pinta-alan perusteella laskettuna).
Vastasyntyneillä ja nuorilla rotilla ja koirilla tehdyt eläintutkimukset osoittivat, että haitallisia vaikutuksia ei havaittu missään tavallisissa kehittymisen tai henkisen kypsymisen loppuarvoissa annoksilla aina 1800 mg/kg/vrk saakka (6–17 kertaa ihmiselle suositeltu suurin vuorokausiannos kehon pinta-alan perusteella laskettuna).
Farmaseuttiset tiedot
Apuaineet
Natriumasetaatti
Etikkahappo, väkevä
Natriumkloridi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
3 vuotta.
Mikrobiologisesti katsoen tuote tulee käyttää välittömästi laimentamisen jälkeen. Jos tuotetta ei käytetä välittömästi, säilytysaika ja säilytysolosuhteet ennen käyttöä ovat käyttäjän vastuulla. Ne eivät normaalisti ole enempää kuin 24 tuntia 2−8 °C:ssa, jollei laimentaminen ole tapahtunut kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Laimennetun lääkevalmisteen säilytysolosuhteet: ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
KEPPRA infuusiokonsentraatti, liuosta varten
100 mg/ml (L:ei) 10 x 5 ml (403,30 €)
PF-selosteen tieto
5 ml:n lasipullo (tyyppi I), joka on suljettu päällystämättömällä harmaalla klorobutyylikumitulpalla ja alumiini/polypropeeni-sulkimella. Yksi kotelo sisältää 10 pulloa.
Valmisteen kuvaus:
Kirkas, väritön neste.
Käyttö- ja käsittelyohjeet
Katso taulukosta 1 Keppra infuusiokonsentraatti, liuosta varten ‑valmisteen suositeltu valmistus ja annostelu, jotta kokonaisvuorokausiannokseksi saadaan 500 mg, 1000 mg, 2000 mg tai 3000 mg levetirasetaamia kahtena annoksena.
Taulukko 1. Keppra-infuusiokonsentraatti, liuosta varten ‑valmisteen valmistus ja annostelu
| Annos | Tarvittava määrä | Laimentimen määrä | Infuusioaika | Antotiheys | Kokonaisvuoro-kausiannos |
| 250 mg | 2,5 ml (puolet 5 ml:n injektiopullosta) | 100 ml | 15 minuuttia | 2 kertaa päivässä | 500 mg/vrk |
| 500 mg | 5 ml (yksi 5 ml:n injektiopullo) | 100 ml | 15 minuuttia | 2 kertaa päivässä | 1000 mg/vrk |
| 1000 mg | 10 ml (kaksi 5 ml:n injektiopulloa) | 100 ml | 15 minuuttia | 2 kertaa päivässä | 2000 mg/vrk |
| 1500 mg | 15 ml (kolme 5 ml:n injektiopulloa) | 100 ml | 15 minuuttia | 2 kertaa päivässä | 3000 mg/vrk |
Tämä lääkevalmiste on tarkoitettu vain kertakäyttöön, ja kaikki käyttämätön liuos on hävitettävä.
Keppra infuusiokonsentraatti, liuosta varten oli fysikaalisesti yhteensopiva ja kemiallisesti stabiili, ainakin 24 tunnin ajan, kun sitä sekoitettiin seuraavien laimentimien kanssa, ja säilytettin PVC-pusseissa kontrolloidussa huoneenlämpötilassa 15−25 °C.
Laimentimet:
- 9 mg/ml (0,9 %) natriumkloridi-injektioneste
- laktatoitu Ringerin injektioneste
- 50 mg/ml (5 %) glukoosi-injektioneste
Lääkevalmistetta, jossa on havaittavissa hiukkasia tai värinmuutosta, ei tule käyttää.
Käyttämätön valmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
KEPPRA infuusiokonsentraatti, liuosta varten
100 mg/ml 10 x 5 ml
- Ei korvausta.
- Epilepsian hoidossa lääkevaihto vain saman kauppanimen valmisteeseen.
ATC-koodi
N03AX14
Valmisteyhteenvedon muuttamispäivämäärä
13.10.2025
Yhteystiedot
Bertel Jungin aukio 5, 6 krs.
02600 Espoo
+358 9 2514 4221
etunimi.sukunimi@ucb.com