
ZELBORAF tabletti, kalvopäällysteinen 240 mg
Vaikuttavat aineet ja niiden määrät
Yksi tabletti sisältää 240 mg vemurafenibia (vemurafenibin ja hypromelloosiasetaattisuksinaatin presipitaattina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Vemurafenibi on tarkoitettu yksinään käytettynä leikkaukseen soveltumattoman tai etäpesäkkeisen melanooman hoitoon aikuisille potilaille, joiden kasvaimessa on BRAF V600 ‑mutaatio (ks. kohta Farmakodynamiikka).
Ehto
Valmistetta tulee käyttää vain syövän hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Syövän hoitoon perehtyneen lääkärin on aloitettava vemurafenibihoito ja valvottava sen toteuttamista.
Ennen vemurafenibihoidon aloittamista kasvaimen BRAF V600 ‑mutaatiostatus on vahvistettava validoidulla testillä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Annostus
Suositeltu vemurafenibiannos on 960 mg (neljä 240 mg:n tablettia) kahdesti vuorokaudessa (vastaa 1920 mg:n kokonaisvuorokausiannosta). Vemurafenibi voidaan ottaa joko aterian yhteydessä tai ilman ateriaa, mutta molempien annosten jatkuvaa ottamista tyhjään mahaan on vältettävä (ks. kohta Farmakokinetiikka).
Hoidon kesto
Vemurafenibihoitoa jatketaan, kunnes tauti etenee tai ilmaantuu kestämättömiä haittavaikutuksia (ks. taulukot 1 ja 2 jäljempänä).
Annoksen unohtuminen
Jos annos unohtuu, se voidaan ottaa viimeistään neljä tuntia ennen seuraavaa annosta, jotta annostelu kahdesti vuorokaudessa voi toteutua. Molempia annoksia ei saa ottaa yhtaikaa.
Oksentelu
Jos potilas oksentaa vemurafenibiannoksen jälkeen, ylimääräistä annosta ei pidä ottaa, vaan hoitoa on jatkettava normaaliin tapaan.
Annoksen muuttaminen
Haittavaikutukset tai QTc-ajan piteneminen saattavat vaatia annoksen pienentämistä tai hoidon keskeyttämistä tai lopettamista (ks. taulukot 1ja 2). Pienennetyn annoksen pitäisi kuitenkin olla vähintään 480 mg kahdesti vuorokaudessa.
Jos potilaalle kehittyy ihon okasolusyöpä (cuSCC), vemurafenibihoitoa tulisi jatkaa annostusta muuttamatta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Taulukko 1: Annostuksen säätäminen haittatapahtumien vaikeusasteen mukaan
Vaikeusaste (CTC-AE)(a) | Suositeltu annostuksen muutos |
1. tai 2. aste (siedettävä) | Vemurafenibihoitoa jatketaan annostuksella 960 mg kahdesti vuorokaudessa. |
2. aste (sietämätön) tai 3. aste | |
Mikä tahansa 2. tai 3. asteen haittatapahtuma esiintyy 1. kerran | Hoito keskeytetään, kunnes vaikeusaste on laskenut tasolle 0–1. Hoito aloitetaan uudelleen annostuksella 720 mg kahdesti vuorokaudessa (tai 480 mg kahdesti vuorokaudessa, jos annosta on jo pienennetty). |
Mikä tahansa 2. tai 3. asteen haittatapahtuma esiintyy 2. kerran tai jatkuu hoidon keskeyttämisen jälkeen | Hoito keskeytetään, kunnes vaikeusaste on laskenut tasolle 0–1. Hoito aloitetaan uudelleen annostuksella 480 mg kahdesti vuorokaudessa (tai lopetetaan pysyvästi, jos annos on jo pienennetty tasolle 480 mg kahdesti vuorokaudessa). |
Mikä tahansa 2. tai 3. asteen haittatapahtuma esiintyy 3. kerran tai jatkuu toisen annosmuutoksen jälkeen | Hoito lopetetaan pysyvästi. |
4. aste | |
Mikä tahansa 4. asteen haittatapahtuma esiintyy 1. kerran | Vemurafenibihoito lopetetaan pysyvästi tai keskeytetään, kunnes vaikeusaste on laskenut tasolle 0–1. Hoito aloitetaan uudelleen annostuksella 480 mg kahdesti vuorokaudessa (tai lopetetaan pysyvästi, jos annos on jo pienennetty tasolle 480 mg kahdesti vuorokaudessa). |
Mikä tahansa 4. asteen haittatapahtuma esiintyy 2. kerran tai mikä tahansa 4. asteen haittatapahtuma jatkuu ensimmäisen annosmuutoksen jälkeen | Hoito lopetetaan pysyvästi. |
(a) Kliinisten haittatapahtumien vaikeusaste on määritetty CTC-AE-kriteerien version 4 mukaan (Common Terminology Criteria for Adverse Events v4.0).
Avoimessa ei-vertailevassa toisen vaiheen tutkimuksessa, johon osallistui etäpesäkkeistä melanoomaa sairastavia aikaisemmin hoitoa saaneita potilaita, todettiin altistuksesta riippuvaa QT-ajan pitenemistä. QTc-ajan piteneminen saattaa vaatia erityisiä seurantatoimenpiteitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukko 2: Annostuksen säätäminen QT-ajan pitenemisen mukaan
QTc-aika | Suositeltu annostuksen muutos |
QTc > 500 ms lähtötilanteessa | Hoitoa ei suositella. |
QTc pidentynyt > 500 ms:iin ja muutos hoitoa edeltäneestä arvosta > 60 ms | Hoito lopetetaan pysyvästi. |
QTc > 500 ms 1. kerran hoidon aikana ja muutos hoitoa edeltäneestä arvosta < 60 ms | Hoito keskeytetään, kunnes QTc laskee 500 ms:n alapuolelle. Ks. seurantatoimenpiteet kohdasta Varoitukset ja käyttöön liittyvät varotoimet. Hoito aloitetaan uudelleen annostuksella 720 mg kahdesti vuorokaudessa (tai 480 mg kahdesti vuorokaudessa, jos annosta on jo pienennetty). |
QTc > 500 ms 2. kerran hoidon aikana ja muutos hoitoa edeltäneestä arvosta < 60 ms | Hoito keskeytetään, kunnes QTc laskee 500 ms:n alapuolelle. Ks. seurantatoimenpiteet kohdasta Varoitukset ja käyttöön liittyvät varotoimet. Hoito aloitetaan uudelleen annostuksella 480 mg kahdesti vuorokaudessa (tai lopetetaan pysyvästi, jos annos on jo pienennetty tasolle 480 mg kahdesti vuorokaudessa). |
QTc > 500 ms 3. kerran hoidon aikana ja muutos hoitoa edeltäneestä arvosta < 60 ms | Hoito lopetetaan pysyvästi. |
Erityisryhmät
Iäkkäät potilaat
Erityiset annosmuutokset eivät ole tarpeen yli 65-vuotiaita potilaita hoidettaessa.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavien potilaiden hoidosta on vain vähän tietoja. Lääkeainealtistuksen suurenemisen riskiä ei voida sulkea pois, jos potilaalla on vaikea munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden tilaa on seurattava tarkoin (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastavien potilaiden hoidosta on vain vähän tietoja. Koska vemurafenibi poistuu maksan kautta, lääkeainealtistus saattaa olla normaalia suurempi, jos potilaalla on kohtalainen tai vaikea maksan vajaatoiminta, ja näiden potilaiden tilaa on seurattava tarkoin (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Vemurafenibin turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tällä hetkellä saatavissa olevat tiedot kuvataan kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, mutta annostussuosituksia ei voida antaa.
Muut kuin valkoihoiset potilaat
Vemurafenibin turvallisuutta ja tehoa ei ole varmistettu muiden kuin valkoihoisten potilaiden hoidossa. Tietoja ei ole saatavilla.
Antotapa
Vemurafenibi otetaan suun kautta. Tabletit niellään kokonaisina veden kera. Niitä ei saa pureskella eikä murskata.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Kasvaimen BRAF V600 -mutaatiostatus on vahvistettava validoidulla testillä ennen vemurafenibihoidon aloittamista. Vemurafenibihoidon tehoa ja turvallisuutta ei ole osoitettu vakuuttavasti potilailla, joiden kasvaimessa on muu harvinainen BRAF–mutaatio kuin V600E- tai V600K-mutaatio (ks. kohta Farmakodynamiikka). Vemurafenibihoitoa ei pidä antaa potilaille, joilla on villin tyypin BRAF-geeniä ilmentävä maligni melanooma.
Yliherkkyysreaktio
Vemurafenibihoidon yhteydessä on raportoitu vakavia yliherkkyysreaktioita, myös anafylaksiaa (ks. kohdat Vasta-aiheet ja Haittavaikutukset). Vaikeita yliherkkyysreaktioita voivat olla Stevens-Johnsonin oireyhtymä, yleistynyt ihottuma, ihon punoitus tai verenpaineen lasku. Vemurafenibihoito on lopetettava pysyvästi, jos potilaalla todetaan vaikea yliherkkyysreaktio.
Ihoreaktiot
Keskeisessä kliinisessä tutkimuksessa vemurafenibia saaneilla potilailla on raportoitu vaikeita ihoreaktioita, harvinaisina tapauksina myös Stevens-Johnsonin oireyhtymää ja toksista epidermaalista nekrolyysiä. Vemurafenibin käytön yhteydessä on sen markkinoille tulon jälkeen raportoitu lääkeainereaktioita, joihin liittyy eosinofiliaa ja systeemioireita (DRESS) (ks. kohta Haittavaikutukset). Vemurafenibihoito on lopetettava pysyvästi, jos potilaalla todetaan vaikea ihoreaktio.
Sädehoidon toksisuuden voimistuminen
Sädehoitoa joko ennen vemurafenibihoitoa tai sen aikana tai jälkeen saaneilla potilailla on raportoitu sädehoitoihottumaa ja herkistymistä sädehoidolle. Tällaiset vaikutukset liittyivät useimmiten ihoon, mutta joissakin tapauksissa ne kohdistuivat sisäelimiin ja johtivat potilaan kuolemaan (ks. kohdat Yhteisvaikutukset ja Haittavaikutukset).
Vemurafenibin käytössä samanaikaisesti tai peräkkäin sädehoidon kanssa pitää olla varovainen.
QT-ajan piteneminen
Avoimessa ei-vertailevassa toisen vaiheen tutkimuksessa, johon osallistui etäpesäkkeistä melanoomaa sairastavia aikaisemmin hoitoa saaneita potilaita, todettiin altistuksesta riippuvaa QT-ajan pitenemistä (ks. kohta Haittavaikutukset). QT-ajan piteneminen voi lisätä kammioperäisten rytmihäiriöiden, myös kääntyvien kärkien takykardian, riskiä. Vemurafenibihoitoa ei suositella, jos potilaalla on korjautumattomia poikkeavia elektrolyyttiarvoja (magnesium mukaan lukien) tai pitkä QT ‑oireyhtymä tai jos potilas käyttää lääkkeitä, joiden tiedetään pidentävän QT-aikaa.
Kaikilta potilailta on otettava elektrokardiografia (EKG) ja määritettävä elektrolyyttiarvot (myös magnesium) ennen vemurafenibihoidon aloittamista, kuukauden kuluttua hoidon alkamisesta ja annoksen muuttamisen jälkeen. Seurantaa tulisi jatkaa kerran kuukaudessa kolmen ensimmäisen hoitokuukauden aikana ja sen jälkeen kolmen kuukauden välein tai useammin, jos se on kliinisesti perusteltua, varsinkin jos potilaalla on kohtalainen tai vaikea maksan vajaatoiminta. Vemurafenibihoidon aloittamista ei suositella, jos QTc on > 500 millisekuntia (ms). Jos QTc on vemurafenibihoidon aikana yli 500 ms, hoito on keskeytettävä, poikkeavat elektrolyyttiarvot (myös magnesium) on korjattava ja QT-ajan pitenemisen sydänperäiset riskitekijät (esim. kongestiivinen sydämen vajaatoiminta, bradyarytmiat) on saatava hallintaan. Kun QTc on laskenut 500 ms:n alapuolelle, hoito aloitetaan uudelleen pienemmällä annoksella taulukossa 2 annettuja ohjeita noudattaen. Jos QTc-aika on yli 500 ms ja muutos hoitoa edeltäneestä arvosta on yli 60 ms, vemurafenibihoito tulisi lopettaa pysyvästi.
Silmäoireet
Vakavia silmäoireita, kuten uveiittia, iriittiä ja verkkokalvon laskimotukoksia, on raportoitu. Potilaiden silmät on tutkittava säännöllisin välein silmäoireiden havaitsemiseksi.
Ihon okasolusyöpä
Vemurafenibia saaneilla potilailla on raportoitu joitakin ihon okasolusyöpätapauksia (joihin kuuluvat myös keratoakantooman tai sekamuotoisen keratoakantooman alatyyppiin luokitellut tapaukset) (ks. kohta Haittavaikutukset).
Kaikille potilaille tulisi tehdä dermatologinen tutkimus ennen hoidon aloittamista, ja tilannetta on seurattava säännöllisin välein hoidon aikana. Kaikki epäilyttävät ihomuutokset on poistettava, lähetettävä ihopatologiseen tutkimukseen ja hoidettava normaalin käytännön mukaisesti. Hoitavan lääkärin on tutkittava potilas ihon okasolusyövän varalta kuukauden välein hoidon aikana ja enintään kuuden kuukauden ajan hoidon päättymisen jälkeen. Jos potilaalle kehittyy ihon okasolusyöpä, suositellaan hoidon jatkamista annostusta muuttamatta. Seurantaa on jatkettava kuuden kuukauden ajan vemurafenibihoidon lopettamisen jälkeen tai kunnes aloitetaan jokin muu syöpälääkitys. Potilaita on kehotettava kertomaan kaikista ihomuutoksista heti lääkärille.
Muu levyepiteelisyöpä kuin ihon okasolusyöpä
Vemurafenibia kliinisissä tutkimuksissa saaneilla potilailla on raportoitu muuta levyepiteelisyöpää kuin ihon okasolusyöpää. Potilaille on tehtävä pään ja kaulan alueen tutkimus, johon kuuluu ainakin suun limakalvon silmämääräinen tarkastelu ja imusolmukkeiden tunnustelu, ennen hoidon aloittamista ja kolmen kuukauden välein hoidon aikana.
Lisäksi on tehtävä rintakehän tietokonetomografiatutkimus (TT) ennen hoidon aloittamista ja kuuden kuukauden välein hoidon aikana.
Peräaukon tunnustelu ja silmämääräinen tarkastelu sekä gynekologinen sisätutkimus (naisille) tulisi tehdä ennen hoidon aloittamista ja hoidon päättyessä tai aina, kun se on kliinisesti perusteltua.
Seurantaa muun levyepiteelisyövän kuin ihon okasolusyövän havaitsemiseksi on jatkettava enintään kuuden kuukauden ajan vemurafenibihoidon lopettamisen jälkeen tai kunnes jokin muu syöpälääkitys aloitetaan. Poikkeavat löydökset on hoidettava hoitosuositusten mukaisesti.
Uusi primaarimelanooma
Kliinisissä tutkimuksissa on raportoitu uusia primaarisia melanoomia. Näissä tapauksissa melanooma poistettiin kirurgisesti, ja potilaat jatkoivat hoitoa samalla annostuksella. Ihomuutosten ilmaantumista on seurattava samalla tavoin kuin edellä ihon okasolusyövän yhteydessä on kuvattu.
Muut syövät
Vemurafenibi voi vaikutusmekanisminsa perusteella aiheuttaa RAS-mutaatioihin liittyvien syöpien etenemistä (ks. kohta Haittavaikutukset). Vemurafenibi-hoidon hyödyt ja riskit on arvioitava tarkoin, jos sitä saavalla potilaalla on aiemmin ollut tai parhaillaan on RAS-mutaatioon liittyvä syöpä.
Pankreatiitti
Vemurafenibihoitoa saaneilla tutkittavilla on raportoitu pankreatiittejä. Selittämätön vatsakipu pitää tutkia viipymättä (seerumin amylaasi- ja lipaasimääritykset mukaan lukien). Potilaita tulee seurata huolellisesti jatkettaessa vemurafenibihoitoa pankreatiittiepisodin jälkeen.
Maksavaurio
Vemurafenibia käytettäessä on raportoitu maksavaurioita, mukaan lukien vaikea-asteisia maksavaurioita (ks. kohta Haittavaikutukset). Maksaentsyymit (aminotransferaasit ja alkalinen fosfataasi) ja bilirubiiniarvo on määritettävä ennen hoidon aloittamista ja niitä on seurattava kuukauden välein hoidon aikana tai kliinisen tarpeen mukaan. Jos poikkeavia laboratorioarvoja havaitaan, annosta on pienennettävä tai hoito on keskeytettävä tai lopetettava (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Munuaistoksisuus
Vemurafenibin käytön yhteydessä on raportoitu munuaistoksisuutta, joka vaihtelee kohonneista seerumin kreatiniinipitoisuuksista akuuttiin interstitiaalinefriittiin ja akuuttiin tubulusnekroosiin. Seerumin kreatiniinipitoisuus pitää määrittää ennen hoidon aloittamista ja sitä on seurattava hoidon aikana kliinisen tarpeen mukaan (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Maksan vajaatoiminta
Aloitusannosta ei tarvitse muuttaa maksan vajaatoiminnan vuoksi. Maksan etäpesäkkeiden aiheuttamaa lievää maksan vajaatoimintaa, johon ei liity hyperbilirubinemiaa, voidaan seurata yleisten suositusten mukaisesti. Kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavien potilaiden hoidosta on vain hyvin vähän tietoja. Kohtalainen tai vaikea maksan vajaatoiminta saattaa suurentaa lääkeainealtistusta (ks. kohta Farmakokinetiikka). Siksi potilaiden tilaa on syytä seurata tarkoin, varsinkin ensimmäisten hoitoviikkojen jälkeen, sillä kumuloitumista saattaa tapahtua pitemmän ajan (useiden viikkojen) kuluessa. Myös EKG-rekisteröinti tulisi tehdä kerran kuukaudessa kolmen ensimmäisen kuukauden aikana.
Munuaisten vajaatoiminta
Aloitusannosta ei tarvitse muuttaa lievän tai kohtalaisen munuaisten vajaatoiminnan vuoksi. Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden hoidosta on vain hyvin vähän tietoja (ks. kohta Farmakokinetiikka). Vemurafenibin käytössä on noudatettava varovaisuutta, jos potilaalla on vaikea munuaisten vajaatoiminta, ja potilaan tilaa on seurattava tarkoin.
Valoyliherkkyys
Eriasteista valoyliherkkyyttä on raportoitu potilailla, jotka ovat saaneet vemurafenibia kliinisissä tutkimuksissa (ks. kohta Haittavaikutukset). Kaikkia potilaita on kehotettava välttämään auringonvaloaltistusta vemurafenibihoidon aikana. Potilaita on neuvottava suojaamaan iho palamiselta hoidon aikana käyttämällä peittäviä vaatteita ja laajakirjoista ultravioletti A (UVA) / ultravioletti B (UVB) ‑auringonsuojavoidetta ja huulivoidetta (suojakerroin ≥ 30) ulkona ollessaan.
Annostuksen muuttamista suositellaan, jos valoyliherkkyyden vaikeusaste on vähintään 2 (sietämätön) (ks. kohta Annostus ja antotapa).
Dupuytrenin kontraktuura ja jalkapohjan kalvojänteen fibromatoosi
Vemurafenibin käytön yhteydessä on raportoitu Dupuytrenin kontraktuuraa ja jalkapohjan kalvojänteen fibromatoosia. Valtaosa oli 1. tai 2. asteen tapauksia, mutta myös vaikea-asteista, invalidisoivaa Dupuytrenin kontraktuuraa on raportoitu (ks. kohta Haittavaikutukset).
Tapahtumat pitää hoitaa pienentämällä annosta tai keskeyttämällä tai lopettamalla hoito (ks. kohta Annostus ja antotapa).
Vemurafenibin vaikutukset muihin lääkevalmisteisiin
Vemurafenibi saattaa suurentaa pääasiassa CYP1A2-entsyymin välityksellä metaboloituvien ja pienentää pääasiassa CYP3A4:n välityksellä metaboloituvien lääkevalmisteiden pitoisuutta plasmassa. Vemurafenibin samanaikaista käyttöä ei suositella sellaisten CYP1A2:n ja CYP3A4:n välityksellä metaboloituvien lääkeaineiden kanssa, joiden terapeuttinen ikkuna on kapea. Pääasiassa CYP1A2:n tai CYP3A4:n välityksellä metaboloituvien lääkevalmisteiden annoksen muuttamista on harkittava niiden terapeuttisten pitoisuusalueiden perusteella ennen kuin niitä annetaan yhdessä vemurafenibin kanssa (ks. kohdat Yhteisvaikutukset ja Raskaus ja imetys).
Varovaisuutta on noudatettava, jos vemurafenibia annetaan yhtaikaa varfariinin kanssa, ja ylimääräisiä INR (International Normalised Ratio) ‑mittauksia on harkittava.
Vemurafenibi saattaa suurentaa sellaisten lääkevalmisteiden altistusta plasmassa, jotka ovat P-gp:n substraatteja. Jos vemurafenibia käytetään samanaikaisesti P-gp:n substraattien kanssa, hoidossa on oltava varovainen. Jos P-gp:n substraatin terapeuttinen indeksi on kapea (esim. digoksiini, dabigatraanieteksilaatti, aliskireeni), hoidossa on oltava varovainen ja annoksen pienentämistä ja/tai lääkepitoisuuksien lisäseurantaa saattaa olla syytä harkita (ks. kohta Yhteisvaikutukset).
Muiden lääkkeiden vaikutus vemurafenibiin
Voimakkaiden CYP3A4:n, P-gp:n ja glukuronidaation induktorien (esim. rifampisiinin, rifabutiinin, karbamatsepiinin, fenytoiinin tai mäkikuisman [hyperisiinin]) samanaikainen käyttö saattaa johtaa vemurafenibialtistuksen vähenemiseen. Samanaikaista käyttöä pitää välttää, mikäli mahdollista (ks. kohta Yhteisvaikutukset). Vaihtoehtoisia hoitomuotoja, joiden indusoiva vaikutus on heikompi, on harkittava vemurafenibin tehon säilyttämiseksi. Vemurafenibin käytössä on noudatettava varovaisuutta, jos sitä annetaan yhdessä voimakkaiden CYP3A4:n/P-gp:n estäjien kanssa. Hoidon turvallisuutta potilaalle on seurattava tarkoin, ja annosta on muutettava, jos se on kliinisesti aiheellista (ks. taulukko 1 kohdassa Annostus ja antotapa).
Samanaikainen ipilimumabihoito
Ensimmäisen vaiheen tutkimuksessa raportoitiin ipilimumabia (3 mg/kg) ja vemurafenibia (960 mg kaksi kertaa vuorokaudessa tai 720 mg kaksi kertaa vuorokaudessa) samanaikaisesti käytettäessä transaminaasipitoisuuden (ALAT/ASAT >5 x viitealueen yläraja) ja bilirubiinipitoisuuden (kokonaisbilirubiini >3x viitealueen yläraja) oireetonta 3. asteen suurenemista. Ipilimumabin ja vemurafenibin samanaikaista käyttöä ei näiden alustavien tietojen perusteella suositella.
Yhteisvaikutukset
Vemurafenibin vaikutukset lääkkeitä metaboloiviin entsyymeihin
Metastasoitunutta melanoomaa sairastavilla potilailla tehdyn lääkkeiden yhteisvaikutustutkimuksen in vivo -tulokset osoittivat, että vemurafenibi on CYP1A2:n kohtalainen estäjä ja CYP3A4:n induktori.
Vemurafenibin samanaikaista käyttöä ei suositella CYP1A2:n välityksellä metaboloituvien aineiden kanssa, joiden terapeuttinen ikkuna on kapea (esim. agomelatiini, alosetroni, duloksetiini, melatoniini, ramelteoni, takriini, titsanidiini, teofylliini). Jos samanaikaista käyttöä ei voida välttää, hoidossa on oltava varovainen, koska vemurafenibi saattaa suurentaa CYP1A2:n substraattien pitoisuutta plasmassa. Samanaikaisesti käytettävän CYP1A2:n substraatin annoksen pienentämistä saattaa olla syytä harkita, jos se on kliinisesti aiheellista.
Vemurafenibin samanaikainen käyttö suurensi eräässä kliinisessä tutkimuksessa kofeiinialtistuksen (CYP1A2:n substraatti) plasmassa (AUC-arvon) 2,6-kertaiseksi. Toisessa kliinisessä tutkimuksessa vemurafenibi suurensi 2 mg:n titsanidiinikerta-annoksen (titsanidiini on CYP1A2:n substraatti) Cmax-arvon noin 2,2-kertaiseksi ja AUC-arvon 4,7-kertaiseksi.
Vemurafenibin samanaikaista käyttöä ei suositella CYP3A4:n välityksellä metaboloituvien aineiden kanssa, joiden terapeuttinen ikkuna on kapea. Jos samanaikaista käyttöä ei voida välttää, on otettava huomioon, että vemurafenibi voi pienentää CYP3A4:n substraattien pitoisuutta plasmassa ja niiden teho voi siksi heikentyä. Tämän perusteella CYP3A4:n välityksellä metaboloituvien ehkäisytablettien teho voi heikentyä, jos niitä käytetään yhtä aikaa vemurafenibin kanssa. Jos CYP3A4:n substraattien terapeuttinen ikkuna on kapea, niiden annoksen muuttamista saattaa olla syytä harkita, jos se on kliinisesti aiheellista (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys). Kliinisessä tutkimuksessa vemurafenibin samanaikainen anto pienensi midatsolaamin (CYP3A4:n substraatti) AUC-arvoa keskimäärin 39 % (pienenemä enintään 80 %).
Vemurafenibin havaittiin indusoivan heikosti CYP2B6-entsyymin toimintaa in vitro, kun vemurafenibipitoisuus oli 10 µM. Toistaiseksi ei tiedetä, pienentävätkö potilaiden plasmasta vakaan tilan aikana mitatut 100 µM:n (noin 50 µg/ml) vemurafenibipitoisuudet samanaikaisesti annettujen CYP2B6-substraattien, kuten bupropionin, pitoisuutta plasmassa.
Vemurafenibin samanaikainen käyttö suurensi S-varfariinin (CYP2C9:n substraatti) AUC-arvoa 18 %. Hoidossa pitää olla varovainen ja INR-arvon (international normalised ratio) seurantaa pitää harkita, jos vemurafenibia annetaan samanaikaisesti varfariinin kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vemurafenibi inhiboi kohtalaisesti CYP2C8 -entsyymiä in vitro. Löydöksen in vivo-merkitystä ei tunneta, mutta kliinisesti merkitsevän vaikutuksen riskiä CYP2C8-substraatin toimintaan ei voida poissulkea. CYP2C8:n substraattien, joiden terapeuttinen ikkuna on kapea, samanaikaisessa käytössä pitää olla varovainen, koska vemurafenibi saattaa suurentaa niiden pitoisuutta.
Koska vemurafenibin puoliintumisaika on pitkä, muiden samanaikaisesti annettujen lääkkeiden toimintaa estävä vaikutus saattaa tulla täydellisenä esiin vasta 8 vuorokautta jatkuneen vemurafenibihoidon jälkeen. Kahdeksan vuorokauden puhdistumisjakso (”wash out”) saattaa olla tarpeen vemurafenibihoidon lopettamisen jälkeen, jotta vältetään mahdolliset yhteisvaikutukset myöhemmän lääkehoidon kanssa.
Sädehoito
Vemurafenibihoitoa saaneilla potilailla on raportoitu sädehoidon toksisuuden voimistumista (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Potilaan saama sädehoitoannos oli useimmiten 2 Gy/vrk tai suurempi (hypofraktioitu sädehoito).
Vemurafenibin vaikutus lääkeaineiden kuljetusjärjestelmiin
In vitro ‑tutkimukset ovat osoittaneet, että vemurafenibi on kuljetusproteiinien P-glykoproteiini (P-gp) ja rintasyövän resistenssiproteiinin (BCRP) estäjä.
Kliininen yhteisvaikutustutkimus osoitti, että suun kautta toistuvasti otetut vemurafenibiannokset (960 mg kaksi kertaa vuorokaudessa) suurentavat suun kautta otetun digoksiinikerta-annoksen aikaansaamaa altistusta siten, että digoksiinin (P-gp:n substraatti) AUClast-arvo suurenee noin 1,8‑kertaiseksi ja Cmax-arvo noin 1,5-kertaiseksi.
Vemurafenibin samanaikaisessa käytössä P-gp:n substraattien (esim. aliskireeni, ambrisentaani, kolkisiini, dabigatraanieteksilaatti, digoksiini, everolimuusi, feksofenadiini, lapatinibi, maraviroki, nilotinibi, posakonatsoli, ranolatsiini, sirolimuusi, sitagliptiini, talinololi, topotekaani) kanssa pitää olla varovainen. Samanaikaisesti käytettävän P-gp:n substraatin annoksen pienentämistä saattaa olla syytä harkita, jos se on kliinisesti aiheellista. Jos vemurafenibia käytetään samanaikaisesti sellaisen lääkevalmisteen kanssa, joka on P-gp:n substraatti ja jonka terapeuttinen indeksi on kapea (esim. digoksiini, dabigatraanieteksilaatti, aliskireeni), harkitse P-gp:n substraatin lääkeainepitoisuuksien lisäseurantaa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vemurafenibin vaikutusta sellaisiin lääkkeisiin, jotka ovat BCRP:n substraatteja, ei tiedetä. Ei voida sulkea pois sitä, että vemurafenibi saattaa lisätä altistusta BCPR:n kuljettamille lääkeaineille (esim. metotreksaatti, mitoksantroni, rosuvastatiini).
Monet syöpälääkkeet ovat BCPR:n substraatteja, joten yhteisvaikutusten riski vemurafenibin kanssa on teoreettisesti olemassa.
Vemurafenibin mahdollisia vaikutuksia muihin kuljetusproteiineihin ei toistaiseksi tunneta.
Muiden samanaikaisesti käytettyjen lääkkeiden vaikutukset vemurafenibiin
In vitro ‑tutkimukset viittaavat siihen, että vemurafenibi metaboloituu CYP3A4-entsyymin ja glukuronidaation välityksellä. Erittyminen sappeen näyttää olevan toinen merkittävä eliminoitumistie. In vitro ‑tutkimukset ovat osoittaneet, että vemurafenibi on kuljetusproteiinien P-gp ja BCRP substraatti. Toistaiseksi ei tiedetä, onko vemurafenibi myös muiden kuljetusproteiinien substraatti. CYP3A4:n voimakkaiden estäjien tai induktorien tai kuljetusproteiinien toiminnan estäjien/induktorien samanaikainen käyttö saattaa muuttaa vemurafenibipitoisuutta.
Itrakonatsolin, joka on voimakas CYP3A4:n/P-gp:n estäjä, samanaikainen anto suurensi vemurafenibin vakaan tilan AUC-arvoa noin 40 %. Vemurafenibin käytössä on noudatettava varovaisuutta, jos sitä annetaan yhdessä CYP3A4:n, glukuronidaation ja/tai kuljetusproteiinien voimakkaiden estäjien (esim. ritonaviirin, sakinaviirin, telitromysiinin, ketokonatsolin, itrakonatsolin, vorikonatsolin, posakonatsolin, nefatsodonin, atatsanaviirin) kanssa. Tällaisia lääkeaineita samanaikaisesti käyttävien potilaiden hoidon turvallisuutta on seurattava tarkoin, ja annosta on muutettava, jos se on kliinisesti aiheellista (ks. taulukko 1 kohdassa Annostus ja antotapa).
Kliinisessä tutkimuksessa 960 mg:n vemurafenibikerta-annoksen antaminen samaan aikaan rifampisiinin kanssa pienensi vemurafenibialtistusta plasmassa huomattavasti, noin 40 %.
Vemurafenibin yhteiskäyttö P-gp:n, glukuronidaation ja/tai kuljetusproteiinien voimakkaiden induktorien (esim. rifampisiinin, rifabutiinin, karbamatsepiinin, fenytoiinin tai mäkikuisman [Hypericum perforatum]) kanssa voi johtaa suboptimaaliseen vemurafenibialtistukseen, ja sitä on vältettävä.
SellaistenP-gp:n ja BCRP:n estäjien, jotka eivät ole myös voimakkaita CYP3A4:n estäjiä, vaikutuksia ei tunneta. Sitä ei voida sulkea pois, että tällaiset lääkeaineet voivat vaikuttaa vemurafenibin farmakokinetiikkaan P-gp:n toimintaan aiheutuvien vaikutusten välityksellä (esim. verapamiili, siklosporiini, kinidiini) tai BCRP:n toimintaan aiheutuvien vaikutusten välityksellä (esim. siklosporiini, gefitinibi).
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset / raskauden ehkäisy
Hedelmällisessä iässä olevien naisten on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 6 kuukauden ajan hoidon päättymisen jälkeen.
Vemurafenibi saattaa heikentää hormonaalisten ehkäisymenetelmien tehoa (ks. kohta Yhteisvaikutukset).
Raskaus
Ei ole olemassa tietoja vemurafenibin käytöstä raskaana oleville naisille.
Vemurafenibin ei havaittu aiheuttavan epämuodostumia rottien eikä kaniinien alkioille tai sikiöille (ks. kohta Prekliiniset tiedot turvallisuudesta). Eläinkokeissa vemurafenibin on havaittu läpäisevän istukan. Jos vemurafenibia annetaan raskaana olevalle naiselle, se voi vaikutusmekanisminsa perusteella vahingoittaa sikiötä. Vemurafenibia ei pidä antaa raskaana oleville naisille, paitsi jos hoidon odotettu hyöty äidille on suurempi kuin sikiölle mahdollisesti aiheutuva vaara.
Imetys
Ei tiedetä, erittyykö vemurafenibi ihmisen rintamaitoon. Vastasyntyneisiin/imeväisiin kohdistuvaa riskiä ei voida sulkea pois. Kun harkitaan imetyksen tai vemurafenibihoidon lopettamista, on otettava huomioon toisaalta imetyksestä koituva hyöty lapselle ja toisaalta hoidon hyöty äidille.
Hedelmällisyys
Vemurafenibin vaikutuksia hedelmällisyyteen ei ole tutkittu spesifisillä eläinkokeilla. Uros- ja naarasrottien ja ‑koirien lisääntymiselimissä ei kuitenkaan havaittu histopatologisia muutoksia toistuvilla annoksilla tehdyissä toksisuustutkimuksissa (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Vemurafenibilla on vähäinen vaikutus ajokykyyn ja koneiden käyttökykyyn. Potilaita on varoitettava mahdollisesta väsymyksestä ja silmäoireista, joiden vuoksi voi olla syytä välttää ajamista.
Haittavaikutukset
Tiivistelmä turvallisuustiedoista
Yleisimmin raportoituja (> 30 %) vemurafenibin kaikkien vaikeusasteiden haittavaikutuksia ovat nivelkipu, väsymys, ihottuma, valoyliherkkyysreaktio, hiustenlähtö, pahoinvointi, ripuli, päänsärky, kutina, oksentelu, ihon papillooma ja hyperkeratoosi. Yleisimpiä (≥ 5 %) vaikeusasteen 3 haittavaikutuksia olivat ihon okasolusyöpä, keratoakantooma, ihottuma, nivelkipu ja suurentunut gammaglutamyylitransferaasipitoisuus (GGT). Ihon okasolusyövän hoitona oli useimmiten kasvaimen paikallinen poisto.
Haittavaikutusten yhteenveto
Alla luetellaan raportoidut melanoomapotilailla esiintyneet haittavaikutukset MedDRA-elinjärjestelmän, yleisyyden ja vaikeusasteen mukaan. Yleisyysluokituksessa on noudatettu seuraavaa käytäntöä:
Hyvin yleinen ≥ 1/10
Yleinen ≥ 1/100, < 1/10
Melko harvinainen ≥ 1/1000, < 1/100
Harvinainen ≥ 1/10 000, < 1/1000
Hyvin harvinainen < 1/10 000
Tässä luetellut haittavaikutukset perustuvat 468 potilaan tuloksiin kolmannen vaiheen avoimessa satunnaistetussa tutkimuksessa, johon osallistuneilla aikuisilla potilailla oli BRAF V600 -mutatoitunut leikkaukseen soveltumaton tai levinneisyysasteen IV melanooma, ja toisen vaiheen yhden hoitohaaran tutkimukseen, johon osallistuneilla potilailla oli BRAF V600 ‑mutatoitunut levinneisyysasteen IV melanooma ja joiden vähintään yksi aikaisempi systeeminen hoito oli osoittautunut tehottomaksi (ks. kohta Farmakodynamiikka). Lisäksi kaikkien kliinisten tutkimusten ja valmisteen markkinoille tulon jälkeisistä turvallisuusselvityksistä peräisin olevat haittavaikutukset on raportoitu. Kaikki käytetyt termit perustuvat suurimpaan prosentuaaliseen esiintyvyyteen toisen ja kolmannen vaiheen kliinisissä tutkimuksissa. Haittavaikutukset on esitetty kussakin yleisyysluokassa vaikeusasteen mukaan alenevassa järjestyksessä, ja ne on raportoitu käyttäen toksisuuden arvioinnissa yleisten toksisuuskriteerien versiota 4.0 (NCI-CTCAE v 4.0).
Taulukko 3: Vemurafenibia saaneilla potilailla esiintyneet haittavaikutukset toisen tai kolmannen vaiheen kliinisessä tutkimuksessa sekä kaikkien kliinisten tutkimusten(1) ja valmisteen markkinoille tulon jälkeisistä(2) turvallisuusselvityksistä peräisin olevat tapahtumat
| Elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen |
| Infektiot | Karvatupen-tulehdus | |||
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Ihon okasolusyöpä(d), keratoakantooma, seborrooinen keratoosi, ihon papillooma | Tyvisolusyöpä, uusi primaari-melanooma(3) | Muu levyepiteelisyöpä kuin ihon okasolusyöpä(1)(3) | Krooninen myelomonosyytti-leukemia(2)(4), haiman adenokarsinooma(5) |
| Veri ja imukudos | Neutropenia, trombosytopenia(6) | |||
| Immuunijärjestelmä | Sarkoidoosi(1)(2)(j) | |||
| Aineenvaihdunta ja ravitsemus | Heikentynyt ruokahalu | |||
| Hermosto | Päänsärky, makuhäiriö, huimaus | Kasvohermo-halvaus, perifeerinen neuropatia | ||
| Silmät | Uveiitti | Verkkokalvon laskimotukos, iridosykliitti | ||
| Verisuonisto | Vaskuliitti | |||
| Hengityselimet, rintakehä ja välikarsina | Yskä | |||
| Ruoansulatuselimistö | Ripuli, oksentelu, pahoinvointi, ummetus | Stomatiitti | Pankreatiitti(2) | |
| Maksa ja sappi | Maksavaurio(1)(2)(g) | |||
| Iho ja ihonalainen kudos | Valoyliherkkyysreaktio, aktiininen keratoosi, ihottuma, makulopapulaarinen ihottuma, kutina, hyperkeratoosi, punoitus, käsi-jalkaoireyhtymä, hiustenlähtö, kuiva iho, auringonpolttama | Papulaarinen ihottuma, pannikuliitti (kyhmyruusu mukaan lukien), keratosis pilaris | Toksinen epidermaalinen nekrolyysi(e), Stevens-Johnsonin oireyhtymä(f) | Lääkeainereaktio, johon liittyy eosinofiliaa ja systeemioireita (DRESS)(1)(2) |
| Luusto, lihakset ja sidekudos | Nivelkipu, lihaskipu, raajakipu, lihas- ja luustokipu, selkäkipu | Niveltulehdus | Jalkapohjan kalvojänteen fibromatoosi(1)(2), Dupuytrenin kontraktuura(1)(2) | |
| Munuaiset ja virtsatiet | Akuutti interstitiaali-nefriitti(1)(2)(h), akuutti tubulusnekroosi(1)(2)(h) | |||
| Yleisoireet ja antopaikassa todettavat haitat | Väsymys, kuume, perifeerinen edeema, voimattomuus | |||
| Tutkimukset | ALAT-arvon nousu(c), alkalisen fosfataasiarvon nousu(c), ASAT-arvon nousu(c), bilirubiiniarvon nousu(c), GGT-arvon nousu(c), painon lasku, sydän-sähkökäyrässä todettava QT-ajan piteneminen, suurentunut veren kreatiniini-pitoisuus(1)(2)(h) | |||
| Vammat ja myrkytykset | Sädehoidon toksisuuden voimistuminen (1)(2)(i) |
(1) Kaikkien kliinisten tutkimusten turvallisuusselvityksistä peräisin olevat tapahtumat.
(2) Valmisteen markkinoille tulon jälkeisestä raportoinnista peräisin olevat tapahtumat.
(3) Syy-yhteys lääkevalmisteen ja haittatapahtuman välillä on mahdollinen
(4) Potilaan jo ennestään sairastaman NRAS-mutaatioon liittyvän kroonisen myelomonosyyttileukemian eteneminen.
(5) Potilaan jo ennestään sairastaman KRAS-mutaatioon liittyvän haiman adenokarsinooman eteneminen.
(6) Laskettu toisen vaiheen ja kolmannen vaiheen tutkimusten perusteella.
Tärkeimpien haittavaikutusten kuvaus
Maksaentsyymiarvojen kohoaminen(c)
Kolmannen vaiheen kliinisessä tutkimuksessa raportoidut maksaentsyymiarvojen muutokset ilmoitetaan seuraavassa niiden potilaiden osuuden perusteella, joilla todettiin 3. tai 4. asteen muutos lähtöarvoihin verrattuna:
- Hyvin yleiset: GGT
- Yleiset: ALAT, alkalinen fosfataasi, bilirubiini
- Melko harvinaiset: ASAT
ALAT, alkalinen fosfataasi ja bilirubiini eivät nousseet yhdessäkään tapauksessa 4. asteen muutosta vastaavalle tasolle.
Maksavaurio(g)
Kansainvälisen (lääkäreistä ja tutkijoista koostuvan) asiantuntijaryhmän kehittämien kriteereiden perusteella, potilaalla on lääkkeen aiheuttama maksavaurio, mikäli hänellä on todettu poikkeama jossakin seuraavista laboratorioarvoista:
- ALAT ≥ 5x ULN
- AFOS ≥ 2x ULN (ilman muuta syytä AFOS-arvon nousulle)
- ALAT ≥ 3x ULN ja samanaikainen bilirubiinipitoisuuden nousu > 2x ULN
Ihon okasolusyöpä (d)
Vemurafenibihoitoa saaneilla potilailla on raportoitu ihon okasolusyöpää. Ihon okasolusyövän ilmaantuvuus oli vemurafenibia saaneilla potilailla noin 20 % kaikissa tutkimuksissa. Poistetut ihomuutokset tutkittiin riippumattomassa ihopatologian keskuslaboratoriossa, ja niistä suurin osa luokiteltiin alatyyppiin okasolusyöpä-keratoakantooma tai niihin liittyi sekamuotoisen keratoakantooman piirteitä (52 %). Luokkaan ”muut” sijoitetuista muutoksista suurin osa (43 %) oli hyvänlaatuisia ihomuutoksia (esim. tavallinen syylä, aktiininen keratoosi, hyvänlaatuinen keratoosi, kysta / hyvänlaatuinen kysta). Ihon okasolusyöpä ilmaantui yleensä hoidon alkuvaiheessa, ja mediaaniaika sen ensimmäiseen ilmaantumiseen oli 7–8 viikkoa. Noin 33 prosentille niistä potilaista, joilla todettiin ihon okasolusyöpä, se ilmaantui useammin kuin kerran, ja ilmaantumiskertojen välisen ajan mediaani oli 6 viikkoa. Ihon okasolusyövän tyypillinen hoitomuoto oli kirurginen poisto, ja potilaat jatkoivat yleensä hoitoa ilman annosmuutoksia (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Muu levyepiteelisyöpä kuin ihon okasolusyöpä
Vemurafenibia saaneilla kliinisiin tutkimuksiin osallistuneilla potilailla on raportoitu muuta levyepiteelisyöpää kuin ihon okasolusyöpää. Siksi seurantaa on jatkettava kohdassa Varoitukset ja käyttöön liittyvät varotoimet. kuvatulla tavalla.
Uusi primaarimelanooma
Kliinisissä tutkimuksissa on raportoitu uusia primaarisia melanoomia. Näissä tapauksissa melanooma poistettiin kirurgisesti, ja potilaat jatkoivat hoitoa samalla annostuksella. Ihomuutosten ilmaantumista on seurattava kohdassa Varoitukset ja käyttöön liittyvät varotoimet kuvatulla tavalla.
Sädehoidon toksisuuden voimistuminen (i)
Raportoituja tapauksia ovat olleet sädehoitoihottuma, sädehoidosta aiheutuva ihovaurio, sädepneumoniitti, sädehoidon aiheuttama esofagiitti, sädehoidon aiheuttama peräsuolitulehdus, sädehoidon aiheuttama hepatiitti, sädehoidon aiheuttama virtsarakkotulehdus ja sädenekroosi.
Sädehoidon toksisuuden voimistumista raportoitiin vaiheen III kliinisessä tutkimuksessa (MO25515, N = 3219) yleisemmin vemurafenibipotilailla, jotka saivat sädehoitoa ennen vemurafenibihoitoa ja sen aikana (9,1 %), verrattuna potilaisiin, jotka saivat sädehoitoa ja vemurafenibia samanaikaisesti (5,2 %) tai jotka saivat sädehoitoa ennen vemurafenibihoitoa (1,5 %).
Yliherkkyysreaktiot (e)
Vemurafenibihoidon yhteydessä on raportoitu vakavia yliherkkyysreaktioita, myös anafylaksiaa. Vaikeita yliherkkyysreaktioita voivat olla Stevens-Johnsonin oireyhtymä, yleistynyt ihottuma, ihon punoitus tai verenpaineen lasku. Vemurafenibihoito on lopetettava pysyvästi, jos potilaalla todetaan vaikea yliherkkyysreaktio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ihoreaktiot (f)
Keskeisessä kliinisessä tutkimuksessa vemurafenibia saaneilla potilailla on raportoitu vaikeita ihoreaktioita, harvinaisina tapauksina myös Stevens-Johnsonin oireyhtymää ja toksista epidermaalista nekrolyysiä. Vemurafenibihoito on lopetettava pysyvästi, jos potilaalla todetaan vaikea ihoreaktio.
QT-ajan piteneminen
Keskitettyjen EKG-tulosten analyysi toisen vaiheen avoimen ei-vertailevan tutkimuksen QT-alatutkimuksesta, jossa 132 potilaalle annettiin vemurafenibia 960 mg kahdesti vuorokaudessa (NP22657), osoitti lääkeainealtistuksesta riippuvan QTc-ajan pitenemisen. QTc-vaikutus pysyi stabiilina, keskimäärin 12–15 ms:n tasolla, ensimmäisen hoitokuukauden jälkeen, ja suurin keskimääräinen QTc-ajan piteneminen (15,1 ms; 95 %:n CI:n yläraja: 17,7 ms) havaittiin kuuden ensimmäisen hoitokuukauden aikana (n = 90 potilasta). Kahden potilaan (1,5 %) absoluuttinen QTc-arvo nousi hoidon aikana yli 500 ms:iin (3. aste CTC-kriteerien mukaan), ja vain yhdellä potilaalla (0,8 %) QTc-ajan muutos lähtöarvosta oli > 60 ms (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Akuutti munuaisvaurio(h)
Vemurafenibin käytössä on raportoitu munuaistoksisuutta, joka on vaihdellut kohonneista kreatiniinipitoisuuksista akuuttiin interstitiaalinefriittiin ja akuuttiin tubulusnekroosiin. Osa tapauksista on havaittu dehydraation yhteydessä. Seerumin kreatiniinipitoisuuksien kohoaminen oli lähinnä lievää (> 1–1,5x ULN) tai keskivaikeaa (> 1,5–3x ULN) ja sen havaittiin olevan luonteeltaan korjautuvaa (ks. taulukko 4).
Taulukko 4: Kreatiniinipitoisuuden muutokset lähtötilanteesta kolmannen vaiheen tutkimuksessa
| Vemurafenibi (%) | Dakarbatsiini (%) | |
| Muutos ≥ 1 aste lähtötilanteesta kaikkiin vaikeusasteisiin | 27,9 | 6,1 |
| Muutos ≥ 1 aste lähtötilanteesta 3. asteeseen tai sitä suurempaan vaikeusasteeseen | 1,2 | 1,1 |
| • 3. asteeseen | 0,3 | 0,4 |
| • 4. asteeseen | 0,9 | 0,8 |
Taulukko 5: Akuutit munuaisvauriot kolmannen vaiheen tutkimuksessa
| Vemurafenibi (%) | Dakarbatsiini (%) | |
| Akuutit munuaisvauriot* | 10,0 | 1,4 |
| Dehydraatioon liittyneet akuutit munuaisvauriot | 5,5 | 1,0 |
| Annosmuutokset akuutin munuaisvaurion vuoksi | 2,1 | 0 |
Kaikki prosenttiosuudet on ilmaistu kummankin lääkevalmisteen osalta tapauksina koko altistetusta potilasjoukosta.
* Sisältää akuutin munuaisvaurion, munuaisten vajaatoiminnan ja akuuttiin munuaisvaurioon sopivat laboratorioarvojen muutokset.
Sarkoidoosi (j)
Vemurafenibihoitoa saaneilla potilailla on raportoitu sarkoidoosia lähinnä ihossa, keuhkoissa ja silmissä. Valtaosassa tapauksista vemurafenibihoitoa jatkettiin, ja sarkoidoosi joko hävisi tai pitkittyi.
Erityisryhmät
Iäkkäät potilaat
Kolmannen vaiheen tutkimuksessa vemurafenibia annettiin 336 potilaalle, joilla oli leikkaukseen soveltumaton tai etäpesäkkeinen melanooma, ja heistä 94 (28 %) oli yli 65-vuotiaita. Iäkkäät (≥ 65-vuotiaat) potilaat saattavat olla alttiimpia haittavaikutusten, kuten ihon okasolusyövän, ruokahalun heikkenemisen ja sydänoireiden, kehittymiselle.
Sukupuoli
Vemurafenibin kliinisissä tutkimuksissa 3. asteen haittavaikutuksia, joita raportoitiin useammin naisilla kuin miehillä, olivat ihottuma, nivelkipu ja valoyliherkkyys.
Pediatriset potilaat
Vemurafenibin turvallisuutta lapsille ja nuorille ei ole varmistettu. Kuudella nuorella potilaalla tehdyssä kliinisessä tutkimuksessa ei havaittu uusia turvallisuussignaaleja.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 Fimea
Yliannostus
Vemurafenibin yliannostukseen ei ole spesifistä vastalääkettä. Jos haittavaikutuksia ilmaantuu, on annettava oireenmukaista hoitoa tarpeen mukaan. Vemurafenibin yliannostustapauksia ei ole havaittu kliinisissä tutkimuksissa. Yliannostusta epäiltäessä vemurafenibin antamista on lykättävä ja aloitettava tukihoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Solunsalpaajat, proteiinikinaasin estäjät, ATC-koodi: L01EC01
Vaikutusmekanismi ja farmakodynaamiset vaikutukset
Vemurafenibi on BRAF-seriinitreoniinikinaasin estäjä. BRAF-geenin mutaatiot johtavat BRAF-proteiinien konstitutiiviseen aktivaatioon, joka voi aiheuttaa solujen jakautumisen ilman siihen liittyviä kasvutekijöitä.
Biokemiallisista analyyseistä saadut prekliiniset tulokset osoittivat, että vemurafenibi pystyy estämään voimakkaasti sellaisten BRAF-kinaasien toimintaa, joissa on aktivoivia mutaatioita kodonissa 600 (taulukko 6).
Taulukko 6: Vemurafenibin eri BRAF-kinaaseja estävä teho
Kinaasi | Oletettu esiintymistiheys V600-mutaatiopositiivisissa melanoomissa (t) | Estävä pitoisuus (IC50) (nM) |
BRAFV600E | 87,3 % | 10 |
BRAFV600K | 7,9 % | 7 |
BRAFV600R | 1 % | 9 |
BRAFV600D | < 0,2 % | 7 |
BRAFV600G | < 0,1 % | 8 |
BRAFV600M | < 0,1 % | 7 |
BRAFV600A | < 0,1 % | 14 |
BRAFWT | N/A | 39 |
(t) Arvioitu yleisen COSMIC-tietokannan 16 403 melanoomasta, joihin oli ilmoitettu liittyvän BRAF-mutaatio kodonissa 600 (release 71, November 2014).
Tämä estovaikutus vahvistettiin ERK:n fosforylaatiotestillä ja solujen antiproliferaatiotestillä käytettävissä olleissa melanoomasolulinjoissa, jotka ilmensivät V600-mutatoitunutta BRAF-proteiinia. Solujen antiproliferaatiotesteissä puolet V600-mutatoituneista solulinjoista (V600E-, V600R-, V600D- ja V600K-mutatoituneita solulinjoja) estävä pitoisuus (IC50) oli 0,016–1,131 μM, kun taas villin tyypin BRAF-geeniä ilmentävien solulinjojen IC50 oli 12,06 ja 14,32 μM.
BRAF-mutaatiostatuksen määrittäminen
BRAF V600 -mutaation esiintyminen kasvaimessa on vahvistettava validoidulla testillä ennen vemurafenibihoidon aloittamista. Toisen ja kolmannen vaiheen kliinisissä tutkimuksissa tutkimukseen soveltuvat potilaat valittiin reaaliaikaisella polymeraasiketjureaktiomenetelmällä (PCR) (Cobas 4800 BRAF V600 ‑mutaatiotesti). Tällä testillä on CE-hyväksyntä, ja sitä käytetään BRAF-mutaatiostatuksen määrittämiseen DNA:sta, joka on eristetty formaliinilla kiinnitetystä parafiiniin valetusta (FFPE) kasvainkudoksesta. Se on suunniteltu tunnistamaan vallitseva BRAF V600E ‑mutaatio erittäin suurella tarkkuudella (vain 5 %:n V600E-jakson FFPE-näytteestä eristetyn DNA:n villin tyypin jaksosta). Prekliiniset ja kliiniset tutkimukset, joissa on käytetty retrospektiivisiä sekvensointianalyysejä, ovat osoittaneet, että testi tunnistaa heikommalla tarkkuudella myös harvinaisemmat BRAF V600D- ja V600K ‑mutaatiot. Prekliinisistä ja kliinisistä tutkimuksista saaduista näytteistä (n = 920), jotka todettiin mutatoituneiksi Cobas-testillä ja jotka analysoitiin lisäksi sekvensoimalla, yhdessäkään näytteessä ei todettu villiä tyyppiä Sangerin menetelmällä eikä 454-sekvensointia käyttäen.
Kliininen teho ja turvallisuus
Vemurafenibin tehoa on arvioitu vaiheen kolme kliinisessä tutkimuksessa (NO25026), jossa oli mukana 336 potilasta, ja kahdessa vaiheen kaksi kliinisessä tutkimuksessa (NP22657 ja MO25743), joissa oli mukana 278 potilasta. Kaikilla potilailla tuli olla levinnyt melanooma, jossa esiintyi BRAF V600 ‑mutaatioita Cobas 4800 BRAF V600 ‑mutaatiotestin perusteella.
Vaiheen kolme tutkimuksen (NO25026) tulokset aikaisemmin hoitamattomilla potilailla
Kansainvälisen avoimen satunnaistetun vaiheen kolme monikeskustutkimuksen tulosten perusteella vemurafenibi soveltuu aikaisemmin hoitamattomille potilaille, joilla on leikkaukseen soveltumaton tai etäpesäkkeinen BRAF V600E ‑mutaatiopositiivinen melanooma. Potilaat saivat satunnaistetusti joko vemurafenibia (960 mg kahdesti vuorokaudessa) tai dakarbatsiinia (1000 mg/m2 joka kolmannen viikon 1. päivänä).
Yhteensä 675 potilasta jaettiin satunnaistetusti vemurafenibia (n = 337) tai dakarbatsiinia (n = 338) saavaan ryhmään. Suurin osa potilaista oli miehiä (56 %) ja valkoihoisia (99 %), mediaani-ikä oli 54 vuotta (24 % oli yli 65-vuotiaita), kaikkien potilaiden ECOG-toimintakykyluokka oli 0 tai 1 ja suurimmalla osalla (65 %) oli levinneisyysluokan M1c tauti. Tutkimuksen ensisijaiset tehoa mittaavat päätetapahtumat olivat kokonaiselinaika (OS) ja aika ilman taudin etenemistä (PFS).
Etukäteen määritetyssä välianalyysissä (tiedonkeräyksen katkaisukohta (data cut-off) 30.12.2010), havaittiin merkitsevää paranemista ensisijaisissa päätetapahtumissa eli kokonaiselinajassa (p < 0,0001) ja ajassa ilman taudin etenemistä (p < 0,0001) (stratifioimaton logrank-testi). Nämä tulokset julkaistiin tammikuussa 2011 puolueettoman valvontakomitean (Data and Safety Monitoring Board (DSMB) suosituksesta, ja tutkimusta muutettiin siten, että dakarbatsiinia saaneille potilaille annettiin mahdollisuus siirtyä vemurafenibihoitoon. Post hoc ‑elossaoloanalyysit tehtiin tämän jälkeen taulukossa 7 kuvatulla tavalla.
Taulukko 7: Kokonaiselossaolo-osuus tutkimuksen tiedonkeräyksen katkaisukohdassa aikaisemmin hoitamattomilla melanoomapotilailla, joiden kasvaimessa oli BRAF V600 ‑mutaatio (N = 338 dakarbatsiini, N = 337 vemurafenibi)
Tiedonkeräyksen katkaisukohdat | Hoito | Kuolemantapauksia (%) | Riskisuhde (HR) (95 % CI) | Hoitoa vaihtaneet potilaat (%) |
30.12.2010 | dakarbatsiini | 75 (22) | 0,37 (0,26, 0,55) | 0 (ei sovelleta) |
vemurafenibi | 43 (13) | |||
31.3.2011 | dakarbatsiini | 122 (36) | 0,44 (0,33, 0,59) (w) | 50 (15 %) |
vemurafenibi | 78 (23) | |||
3.10.2011 | dakarbatsiini | 175 (52) | 0,62 (0,49, 0,77) (w) | 81 (24 %) |
vemurafenibi | 159 (47) | |||
1.2.2012 | dakarbatsiini | 200 (59) | 0,70 (0,57, 0,87) (w) | 83 (25 %) |
vemurafenibi | 199 (59) | |||
20.12.2012 | dakarbatsiini | 236 (70) | 0,78 (0,64, 0,94) (w) | 84 (25 %) |
vemurafenibi | 242 (72) |
(w) Tulokset, jotka on laskettu sensuroiduista havaintoarvoista hoidon vaihtamisen ajankohtana
Tulokset, jotka on laskettu sensuroimattomista havaintoarvoista hoidon vaihtamisen ajankohtana: 31. maaliskuuta 2011: HR (95 % CI) = 0,47 (0,35, 0,62); 3. lokakuuta, 2011: HR (95 % CI) = 0,67 (0,54, 0,84) ); 1. helmikuuta, 2012: HR (95 % CI)= 0,76 (0,63, 0,93); 20. joulukuuta, 2012 HR (95 % CI) = 0,79 (0.66, 0.95).
Kuva 1: Kaplan-Meierin kuvaajat kokonaiselinajasta – aikaisemmin hoitamattomat potilaat (tiedonkeräyksen katkaisukohdassa 20.12.2012)
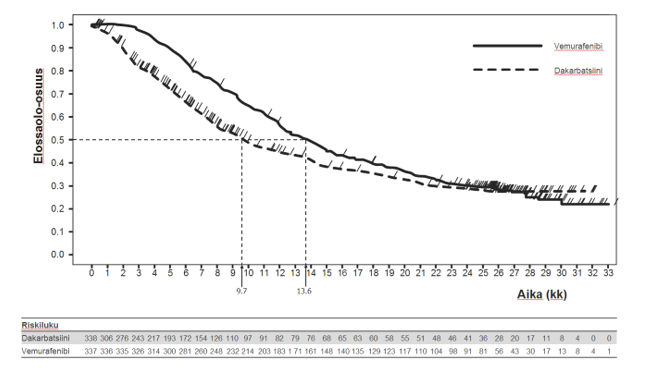
Taulukko 8 kuvaa hoitotehoa kaikkien ennustetekijöiksi määriteltyjen, etukäteen valittujen stratifiointimuuttujien osalta.
Taulukko 8: Kokonaiselinaika aikaisemmin hoitamattomilla melanoomapotilailla, joiden kasvaimessa oli BRAF V600 ‑mutaatio, LDH-arvon, kasvaimen levinneisyysasteen ja ECOG-toimintakykyluokan mukaan (post-hoc-analyysi tiedonkeräyksen katkaisukohdassa 20.12.2012, sensuroiduista havaintoarvoista hoidon vaihtamisen ajankohtana)
Stratifiointimuuttuja | N | Riskisuhde (HR) | 95 %:n luottamusväli |
LDH normaali | 391 | 0,88 | 0,67; 1,16 |
LDH > ULN | 284 | 0,57 | 0,44; 0,76 |
Levinneisyysaste IIIC/M1a/M1b | 234 | 1,05 | 0,73; 1,52 |
Levinneisyysaste M1c | 441 | 0,64 | 0,51; 0,81 |
ECOG-luokka = 0 | 459 | 0,86 | 0,67; 1,10 |
ECOG-luokka = 1 | 216 | 0,58 | 0,42; 0,9 |
LDH: laktaattidehydrogenaasi, ECOG: Eastern Cooperative Oncology Group
Taulukossa 9 on esitetty kokonaisvaste ja aika ilman taudin etenemistä aikaisemmin hoitamattomilla melanoomapotilailla, joiden kasvaimessa oli BRAF V600 ‑mutaatio.
Taulukko 9: Kokonaisvaste ja aika ilman taudin etenemistä aikaisemmin hoitamattomilla melanoomapotilailla, joiden kasvaimessa oli BRAF V600 ‑mutaatio
vemurafenibi | dakarbatsiini | p-arvo (x) | |
Tiedonkeräyksen katkaisukohta 30.12.2010 (y) | |||
Kokonaisvaste (95 % CI) | 48,4 % (41,6 %, 55,2 %) | 5,5% (2,8 %, 9,3 %) | < 0,0001 |
Elinaika ilman taudin etenemistä Riskisuhde (95 % CI) | 0,26 (0,20, 0,33) | < 0,0001 | |
Tapahtumien lukumäärä (%) | 104 (38 %) | 182 (66 %) | |
PFS-mediaani (kk) (95 % CI) | 5,32 (4,86, 6,57) | 1,61 (1,58, 1,74) | |
Tiedonkeräyksen katkaisukohta 1.2.2012 (z) | |||
Elinaika ilman taudin etenemistä Riskisuhde (95 % CI) | 0,38 (0,32, 0,46) | < 0,0001 | |
Tapahtumien lukumäärä (%) | 277 (82 %) | 273 (81 %) | |
PFS-mediaani (kk) (95 % CI) | 6,87 (6,14, 6,97) | 1,64 (1,58, 2,07) | |
(x) PFS-vertailussa käytettiin stratifioimatonta logrank-testiä ja kokonaishoitovasteiden vertailussa khii2-testiä.
(y) Elinaika ilman taudin etenemistä (PFS) voitiin arvioida 549 potilaalta ja kokonaishoitovaste 439 potilaalta (tiedonkeräyksen katkaisukohta 30.12.2010).
(z) Elinaika ilman taudin etenemistä (PFS) voitiin arvioida 675 potilaalta post hoc ‑analyysissä (tiedonkeräyksen katkaisukohta 1.2.2012).
NO25026-tutkimuksessa 673 melanoomapotilaan kasvaimesta tehtiin retrospektiivisesti sekvensointi, ja 57:llä näistä potilaista todettiin BRAF V600K ‑mutaatio. Vaikka potilasmäärä oli pieni, tehoa mittaava analyysi näistä V600K-positiivisista potilaista viittasi siihen, että vemurafenibihoito vaikutti suotuisasti kokonaiselinaikaan, aikaan ilman taudin etenemistä ja vahvistettuun parhaaseen kokonaisvasteeseen. Melanoomapotilaista, joilla olisi todettu jokin muu harvinainen BRAF V600 ‑mutaatio kuin V600E tai V600K, ei ole tutkimustietoa.
Vaiheen kaksi tutkimuksen tulokset (NP22657) potilailla, joiden vähintään yksi aikaisempi hoito oli osoittautunut tehottomaksi
Vaiheen kaksi kansainvälisessä yhden hoitohaaran monikeskustutkimuksessa oli mukana 132 potilasta, joilla oli BRAF V600E ‑mutatoitunut etäpesäkkeinen melanooma Cobas 4800 BRAF V600 ‑mutaatiotestin perusteella. Potilaat olivat saaneet aikaisemmin vähintään yhtä hoitoa. Mediaani-ikä oli 52 vuotta, ja 19 % oli yli 65-vuotiaita. Potilaiden enemmistö oli miehiä (61 %) ja valkoihoisia (99 %) ja suurimmalla osalla oli levinneisyysluokan M1c tauti (61 %). Vähintään kaksi aikaisempaa hoitoa oli osoittautunut tehottomaksi 49 prosentilla potilaista.
Kun seuranta-ajan mediaani oli 12,9 kuukautta (vaihteluväli 0,6–20,1), ensisijainen päätetapahtuma eli riippumattoman arviointikomitean (IRC) arvioon perustuva vahvistettu paras kokonaisvaste (täydellinen vaste + osittainen vaste), oli 53 % (95 % CI: 44 %, 62 %). Kokonaiselinajan mediaani oli 15,9 kuukautta (95 % CI: 11,6, 18,3). Kokonaiselossaolo-osuus oli 6 kuukauden kuluttua 77 % (95 % CI: 70 %, 85 %) ja 12 kuukauden kuluttua 58 % (95 % CI: 49 %, 67 %).
Yhdeksällä NP22657-tutkimukseen otetuista 132 potilaasta oli Sangerin menetelmällä tehdyn retrospektiivisen sekvensoinnin mukaan V600K-mutaatiopositiivinen kasvain. Näistä potilaista kolmella oli osittainen hoitovaste, kolmella oli stabiili tauti, kahdella oli etenevä tauti ja yhtä ei voitu arvioida.
Vaiheen kaksi tutkimuksen tulokset (MO25743) potilailla, joilla oli etäpesäkkeitä aivoissa
Yhden hoitohaaran monikeskustutkimuksessa (N = 146) tutkittiin vemurafenibihoitoa aikuispotilailla, joilla oli histologisesti varmistettu BRAF V600‑mutatoitunut (cobas 4800 BRAF V600 mutaatiotestin mukaan) etäpesäkkeinen melanooma ja etäpesäkkeitä aivoissa. Tutkimuksessa oli kaksi samaan aikaan mukaan otettua kohorttia:
- kohortti 1 koostui aiemmin hoitamattomista potilaista (N = 90): potilaat eivät olleet aiemmin saaneet hoitoa etäpesäkkeisiin aivoissa; etäpesäkkeisen melanooman aiempi systeeminen hoito oli BRAF:n estäjiä ja MEK:n estäjiä lukuun ottamatta sallittu
- kohortti 2 koostui aiempaa hoitoa saaneista potilaista (N = 56): potilaat olivat aiemmin saaneet hoitoa etäpesäkkeisiin aivoissa ja sairaus oli edennyt tämän hoidon jälkeen. Jos potilas oli saanut stereotaktista sädehoitoa tai kasvain oli hoidettu leikkauksella, potilaalle piti olla kehittynyt uusi, RECIST-kriteerein arvioitavissa oleva aivoleesio ennen tätä hoitoa.
Tutkimukseen otettiin mukaan yhteensä 146 potilasta. Potilaiden enemmistö oli miehiä (61,6 %) ja valkoihoisia (92,5 %), ja iän mediaani oli 54 vuotta (vaihteluväli 26–83 vuotta). Jakauma oli kummassakin kohortissa samankaltainen. Aivoissa lähtötilanteessa olleiden kohdeleesioiden lukumäärän mediaani oli kummassakin kohortissa kaksi (vaihteluväli 1–5).
Tutkimuksen ensisijainen tehoa koskeva tavoite oli riippumattoman arviointikomitean (IRC) arvioima paras kokonaisvaste potilailla, joilla oli melanooman etäpesäkkeitä aivoissa ja jotka eivät olleet aiemmin saaneet hoitoa aivoissa oleviin etäpesäkkeisiin.
Toissijaisia tavoitteita olivat aivoissa todettuun parhaaseen kokonaisvasteeseen perustuva arvio vemurafenibin tehosta aiempaa hoitoa saaneilla potilailla, vasteen kesto, aika ilman taudin etenemistä ja kokonaiselinaika potilailla, joilla oli melanooman etäpesäkkeitä aivoissa (ks. taulukko 10).
Taulukko 10: Vemurafenibin teho potilailla, joilla oli etäpesäkkeitä aivoissa
Kohortti 1 Ei aiempaa hoitoa saaneet n = 90 | Kohortti 2 Aiempaa hoitoa saaneet n = 56 | Yhteensä n = 146 | |
Paras kokonaisvastea aivoissa Vasteen saaneiden n (%) (95 % CI)b | 16 (17,8 %) (10,5, 27,3) | 10 (17,9 %) (8,9, 30,4) | 26 (17,8 %) (12,0, 25,0) |
Vasteen kestoc aivoissa (n) Mediaani (kk) (95 % CI)d | (n = 16) 4,6 (2,9, 6,2) | (n = 10) 6,6 (2,8, 10,7) | (n = 26) 5,0 (3,7, 6,6) |
Paras kallonulkoinen kokonaisvaste n (%)a | 26 (32,9 %) | 9 (22,5 %) | 35 (29,4 %) |
Aika ilman taudin etenemistä, yhteensä Mediaani (kk)e (95 % CI)d | 3,7 (3,6, 3,7) | 3,7 (3,6, 5,5) | 3,7 (3,6, 3,7) |
Aika ilman taudin etenemistä, pelkästään aivoissa Mediaani (kk)e (95 % CI)d | 3,7 (3,6, 4,0) | 4,0 (3,6, 5,5) | 3,7 (3,6, 4,2) |
Kokonaiselinaika Mediaani (kk) (95 % CI)d | 8,9 (6,1, 11,5) | 9,6 (6,4, 13,9) | 9,6 (6,9, 11,5) |
a Riippumattoman arviointikomitean arvio parhaasta varmistetusta kokonaisvasteesta, vasteen saaneiden lukumäärä n (%)
b Kaksitahoinen Clopper-Pearsonin 95 % luottamusväli (Confidence Interval, CI)
c Riippumattoman arviointikomitean arvio vasteen kestosta
d Kaplan-Meierin estimaatti
e Tutkijan arvio
Pediatriset potilaat
Pediatrisilla potilailla tehdyn vaiheen I tutkimuksen (NO25390) tulokset
Vaiheen I annoseskalaatiotutkimuksessa arvioitiin vemurafenibin käyttöä kuudelle nuorelle potilaalle, joilla oli levinneisyysasteen IIIC tai IV BRAF V600 ‑mutatoitunut melanooma. Kaikki hoidetut potilaat olivat vähintään 15-vuotiaita ja painoivat vähintään 45 kg. Kolme potilasta sai hoitona 720 mg vemurafenibia kaksi kertaa vuorokaudessa, ja kolme potilasta sai hoitona 960 mg vemurafenibia kaksi kertaa vuorokaudessa. Suurinta siedettyä annosta ei pystytty määrittämään. Vaikka ohimenevää kasvaimen regressiota havaittiin, varmistettuihin vasteisiin perustuva paras kokonaisvasteluku (best overall response rate, BORR) oli 0 % (95 %:n luottamusväli: 0 %, 46 %). Tutkimus lopetettiin, koska mukaan otettuja potilaita oli vähän. Tiedot valmisteen käytöstä pediatrisille potilaille, ks. kohta Annostus ja antotapa.
Farmakokinetiikka
Vemurafenibi on luokan IV lääkeaine (vähäinen liukoisuus ja läpäisevyys) biofarmaseuttisen luokittelujärjestelmän (Biopharmaceutics Classification System) kriteerien mukaan. Vemurafenibin farmakokineettiset parametrit määritettiin tilamalleista riippumattomalla analyysillä ensimmäisen ja kolmannen vaiheen tutkimuksissa (20 potilaalta, kun hoitoa oli jatkettu 15 vuorokautta annostuksella 960 mg kahdesti vuorokaudessa, ja 204 potilaalta vakaan tilan aikana 22. päivänä) sekä populaatiofarmakokineettisellä analyysillä, joka tehtiin 458 potilaan yhdistetyistä tiedoista. Näistä potilaista 457 oli valkoihoisia.
Imeytyminen
Vaiheen I tutkimuksessa, jossa lääkevalmisteen ottamisen ja ruokailun välistä yhteyttä ei seurattu, oli mukana neljä BRAF V600 ‑positiivista syöpää sairastavaa potilasta. Näiden potilaiden vakaan tilan biologinen hyötyosuus oli 32–115 % (keskiarvo 64 %) suhteessa laskimoon annettuun mikroannokseen.
Aika vemurafenibin huippupitoisuuden saavuttamiseen (Tmax) 960 mg:n kerta-annoksen (neljä 240 mg:n tablettia) jälkeen on noin 4 tuntia (mediaani). Potilaiden väliset erot ovat suuria. Toisen vaiheen tutkimuksessa 1. päivän AUC0-8h oli 22,1 ± 12,7 µg⋅h/ml ja Cmax 4,1 ± 2,3 µg/ml. Kumuloitumista esiintyy, kun vemurafenibia annetaan toistuvina annoksina kahdesti vuorokaudessa. Tilamalleista riippumattomassa analyysissä kahdesti vuorokaudessa annetun 960 mg:n vemurafenibiannoksen jälkeen 15. päivän ja 1. päivän välinen suhde oli AUC-arvon osalta 15–17-kertainen ja Cmax-arvon osalta 13–14-kertainen, joten AUC0-8h oli 380,2 ± 143,6 µg⋅h/m ja Cmax 56,7 ± 21,8 µg/m vakaan tilan aikana.
Ruoka (runsasrasvainen ateria) suurentaa 960 mg:n vemurafenibikerta-annoksen suhteellista hyötyosuutta. Ruokailun jälkeen ja paastotilassa mitattujen huippupitoisuuksien (Cmax) välisen geometrisen suhteen keskiarvo oli 2,5-kertainen ja AUC:n geometrisen suhteen keskiarvo oli 4,6–5,1-kertainen. Aika huippupitoisuuden saavuttamiseen (Tmax-arvon mediaani) piteni 4 tunnista 7,5 tuntiin, kun vemurafenibikerta-annos otettiin ruokailun yhteydessä.
Ruoan vaikutusta vakaan tilan vemurafenibialtistukseen ei toistaiseksi tiedetä. Vemurafenibin jatkuva ottaminen tyhjään mahaan voi johtaa merkitsevästi alhaisempaan altistukseen vakaassa tilassa verrattuna vemurafenibin jatkuvaan ottamiseen aterian yhteydessä tai heti sen jälkeen. Vemurafenibin satunnaisella ottamisella tyhjään vastaan oletetaan olevan mitätön vaikutus vakaan tilan altistukseen johtuen vemurafenibin korkeasta kertymisestä vakaassa tilassa. Pivotaalitutkimusten tehoa ja turvallisuutta koskevat tiedot kerättiin potilaista, jotka ottivat vemurafebinia ruokailun yhteydessä tai ilman ruokailua.
Lääkeainealtistuksessa saattaa myös esiintyä vaihteluita, jotka johtuvat maha-suolikanavan nestesisällön, nestemäärien, pH:n, motiliteetin ja läpikulkuaikojen sekä sapen koostumuksen eroista.
Vakaan tilan aikana vemurafenibialtistuksen keskiarvo plasmassa pysyy stabiilina 24 tunnin jakson ajan, kuten ennen aamuannosta ja 2–4 tuntia aamuannoksen jälkeen mitattujen plasman lääkeainepitoisuuksien suhde 1,13 (keskiarvo) osoittaa. Etäpesäkkeistä melanoomaa sairastavilla potilailla imeytymisvakion arvioidaan olevan suun kautta annetun annoksen jälkeen 0,19 h‑1 (potilaiden välinen vaihtelu 101 %).
Jakautuminen
Vemurafenibin lasketun jakautumistilavuuden potilaspopulaatiossa arvioidaan olevan etäpesäkkeistä melanoomaa sairastavilla potilailla 91 litraa (potilaiden välinen vaihtelu 64,8 %). Lääkeaine sitoutuu voimakkaasti (> 99 %) ihmisen plasman proteiineihin in vitro.
Biotransformaatio
Vemurafenibin ja sen metaboliittien suhteellisia osuuksia ihmiselimistössä tutkittiin ihmisillä tehdyn massatasapainotutkimuksen avulla antamalla 14C-merkittyä vemurafenibia kerta-annoksena suun kautta. CYP3A4 on tärkein vemurafenibin metaboloitumisesta vastaava entsyymi in vitro. Ihmiselimistössä todettiin myös konjugoitumalla (glukuronidaatio ja glykosylaatio) syntyneitä metaboliitteja. Lähtöaine oli kuitenkin vallitseva komponentti (95 %) plasmassa. Vaikka metaboliittien määrä plasmassa ei näytä nousevan merkittävälle tasolle metabolian seurauksena, metabolian merkitystä lääkeaineen erittymisessä ei voida sulkea pois.
Eliminaatio
Vemurafenibin lasketun puhdistuman potilaspopulaatiossa arvioidaan olevan etäpesäkkeistä melanoomaa sairastavilla potilailla 29,3 l/vrk (potilaiden välinen vaihtelu 31,9 %). Populaatiofarmakokineettisen analyysin perusteella arvioitu vemurafenibin eliminoitumisen puoliintumisaika potilaspopulaatiossa on 51,6 tuntia (yksittäisten puoliintumisaikojen estimaattien 5. ja 95. persentiilin vaihteluväli on 29,8–119,5 tuntia).
Suun kautta annetulla vemurafenibilla tehdyssä massatasapainotutkimuksessa keskimäärin 95 % annoksesta erittyi 18 vuorokauden kuluessa. Suurin osa vemurafenibista peräisin olevasta materiaalista (94 %) erittyi ulosteeseen ja < 1 % virtsaan. Eliminaatiolla munuaisten kautta ei vaikuta olevan merkitystä vemurafenibin eliminaation kannalta, kun taas erittyminen sappeen saattaa olla tärkeä eliminoitumistie. Vemurafenibi on P-gp:n substraatti ja estäjä in vitro.
Erityisryhmät
Iäkkäät potilaat
Populaatiofarmakokineettisen analyysin perusteella iällä ei ole tilastollisesti merkitsevää vaikutusta vemurafenibin farmakokinetiikkaan.
Sukupuoli
Populaatiofarmakokineettinen analyysi osoitti, että laskettu puhdistuma (CL/F) oli 17 % suurempi ja laskettu jakautumistilavuus (V/F) 48 % suurempi miehillä kuin naisilla.On epäselvää, johtuuko tämä vaikutus sukupuolesta vai koosta. Lääkeainealtistuksen erot eivät ole kuitenkaan niin suuria, että annosta tarvitsisi muuttaa koon tai sukupuolen perusteella.
Munuaisten vajaatoiminta
Populaatiofarmakokineettinen analyysi etäpesäkkeistä melanoomaa sairastavien potilaiden kliinisten tutkimusten tuloksista osoitti, ettei lievä eikä kohtalainen munuaisten vajaatoiminta vaikuttanut vemurafenibin laskettuun puhdistumaan (kreatiniinipuhdistuma > 40 ml/min). Vaikeaa munuaisten vajaatoimintaa sairastavista potilaista ei ole tutkimustietoa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoiminta
Prekliinisten tietojen ja ihmisillä tehdyn massatasapainotutkimuksen perusteella suurin osa vemurafenibista eliminoituu maksan kautta. Populaatiofarmakokineettinen analyysi etäpesäkkeistä melanoomaa sairastavien potilaiden kliinisten tutkimusten tuloksista osoitti, ettei ASAT- eikä ALAT-arvon kohoaminen enintään kolminkertaiseksi normaalialueen ylärajaan (ULN) verrattuna vaikuttanut vemurafenibin laskettuun puhdistumaan. Tiedot ovat riittämättömät, jotta voitaisiin arvioida maksan aineenvaihdunta- tai eritystoimintojen heikkenemisen vaikutuksia vemurafenibin farmakokinetiikkaan (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Kuudesta nuoresta iältään 15–17-vuotiaasta potilaasta, jotka sairastivat levinneisyysasteen IIIC tai IV BRAF V600 ‑mutatoitunutta melanoomaa, on saatu suppeita farmakokineettisiä tietoja. Nämä tiedot viittaavat siihen, että vemurafenibin farmakokineettiset ominaisuudet ovat nuorilla yleisesti samankaltaiset kuin aikuisilla. Tiedot valmisteen käytöstä pediatrisille potilaille, ks. kohta Annostus ja antotapa.
Prekliiniset tiedot turvallisuudesta
Vemurafenibin prekliinistä turvallisuusprofiilia arvioitiin rotilla, koirilla ja kaniineilla.
Toistuvien annosten toksisuustutkimukset osoittivat, että koiralla kohde-elimiä ovat maksa ja luuydin. Koirien 13 viikon tutkimuksissa havaittiin korjautuvia toksisia vaikutuksia maksassa (maksasolujen nekroosia ja degeneraatiota), kun altistus oli pienempi kuin odotettavissa oleva kliininen altistus (AUC-vertailujen perusteella). Yhdellä koiralla havaittiin fokaalista luuydinnekroosia ennenaikaisesti lopetetussa 39 viikon tutkimuksessa, jossa lääke annettiin kahdesti vuorokaudessa, altistuksen ollessa samanlainen kuin odotettu kliininen altistus (AUC-vertailujen perusteella). Luuytimen sytotoksisuustutkimuksessa in vitro havaittiin vähäistä sytotoksisuutta joissakin rotan, koiran ja ihmisen lymfo-hematopoieettisissa solupopulaatioissa kliinisesti merkittävillä pitoisuusalueilla.
Vemurafenibi todettiin fototoksiseksi in vitro hiiren fibroblastiviljelmässä UVA-säteilytyksen jälkeen, mutta ei in vivo rotilla tehdyssä tutkimuksessa, kun annostus oli enintään 450 mg/kg/vrk (odotettua kliinistä altistusta pienempi altistus (AUC- vertailun perusteella). Vemurafenibin vaikutuksia hedelmällisyyteen ei ole tutkittu spesifisillä eläinkokeilla. Toistuvien annosten toksisuustutkimuksissa rotilla ja koirilla ei kuitenkaan havaittu histopatologisia muutoksia urosten eikä naaraiden lisääntymiselimissä, kun annostus oli enintään 450 mg/kg/vrk (odotettua kliinistä altistusta pienempi altistus AUC- vertailun perusteella). Alkion- ja sikiönkehitystutkimuksissa ei havaittu epämuodostumia rotilla, kun annostus oli enintään 250 mg/kg/vrk, eikä kaniineilla, kun annostus oli enintään 450 mg/kg/vrk. Näiden annosten aikaansaama altistus oli pienempi kuin odotettu kliininen altistus (AUC-vertailun perusteella). Koska altistukset olivat alkion- ja sikiönkehitystutkimuksissa pienempiä kuin kliininen altistus AUC-vertailun perusteella, on vaikeaa arvioida, missä määrin näitä tuloksia voidaan soveltaa ihmisiin. Siksi vemurafenibin vaikutusta sikiöön ei voida sulkea pois. Pre- ja postnataalista kehitystä koskevia tutkimuksia ei ole tehty.
Viitteitä vemurafenibin genotoksisuudesta ei havaittu in vitro ‑testeissä (bakteerimutaatio [Amesin testi], ihmisen lymfosyyttien kromosomipoikkeavuus) eikä in vivo rotan luuytimen mikrotumatestissä.
Vemurafenibilla ei ole tehty karsinogeenisuustutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Kroskarmelloosinatrium
Vedetön kolloidinen piidioksidi
Magnesiumstearaatti
Hydroksipropyyliselluloosa
Kalvopäällyste
Polyvinyylialkoholi
Titaanidioksidi (E171)
Makrogoli 3350
Talkki
Punainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ZELBORAF tabletti, kalvopäällysteinen
240 mg (L:ei) 56 x 1 fol (1520,55 €)
PF-selosteen tieto
Yksittäispakatut alumiini–alumiini-läpipainolevyt, joissa on repäisykohdat.
Pakkauskoko: 56 x 1 kalvopäällysteistä tablettia (7 läpipainolevyä, joissa kussakin 8 x 1 tablettia).
Valmisteen kuvaus:
Vaaleanpunavalkoisia tai oranssinvalkoisia, soikeita, kaksoiskuperia kalvopäällysteisiä, noin 19 mm:n pituisia tabletteja, joissa on toisella puolella merkintä VEM.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ZELBORAF tabletti, kalvopäällysteinen
240 mg 56 x 1 fol
- Ylempi erityiskorvaus (100 %). Vemurafenibi: BRAF V600 -mutaatiopositiivisen melanooman hoito aikuispotilailla erityisin edellytyksin (167).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Vemurafenibi: BRAF V600 -mutaatiopositiivisen melanooman hoito aikuispotilailla erityisin edellytyksin (362).
ATC-koodi
L01EC01
Valmisteyhteenvedon muuttamispäivämäärä
16.06.2025
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com