
ROACTEMRA infuusiokonsentraatti, liuosta varten 20 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Tämä potilaskortti sisältää tärkeitä turvallisuutta koskevia tietoja, joista potilaan on oltava tietoinen ennen RoActemra-hoidon aloittamista ja hoidon aikana.
Vaikuttavat aineet ja niiden määrät
Yksi ml konsentraattia sisältää 20 mg tosilitsumabia*.
Yksi 4 ml:n injektiopullo sisältää 80 mg tosilitsumabia* (20 mg/ml).
Yksi 10 ml:n injektiopullo sisältää 200 mg tosilitsumabia* (20 mg/ml).
Yksi 20 ml:n injektiopullo sisältää 400 mg tosilitsumabia* (20 mg/ml).
*yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa (CHO) tuotettu humanisoitu monoklonaalinen IgG1-vasta-aine.
Apuaineet, joiden vaikutus tunnetaan:
Yksi 80 mg:n injektiopullo sisältää 0,10 mmol (2,21 mg) natriumia ja 2 mg (0,5 mg/ml) polysorbaattia 80.
Yksi 200 mg:n injektiopullo sisältää 0,20 mmol (4,43 mg) natriumia ja 5 mg (0,5 mg/ml) polysorbaattia 80.
Yksi 400 mg injektiopullo sisältää 0,39 mmol (8,85 mg) natriumia ja 10 mg (0,5 mg/ml) polysorbaattia 80.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Nivelreuma
RoActemra on tarkoitettu yhdessä metotreksaatin (MTX) kanssa:
- vaikean, aktiivisen ja etenevän nivelreuman hoitoon aikuisille, joita ei aiemmin ole hoidettu MTX:lla
- aikuispotilaiden kohtalaisen tai vaikean aktiivisen nivelreuman hoitoon, kun aikaisempi hoito yhdellä tai useammalla tautiprosessia hidastavalla reumalääkkeellä (disease modifying anti-rheumatic drugs, DMARD) tai tuumorinekroositekijän (TNF) estäjällä ei ole tuottanut riittävää hoitovastetta tai potilas ei siedä niitä.
RoActemraa voidaan antaa monoterapiana näille potilaille, jos he eivät siedä MTX:a tai jos jatkuva MTX-hoito ei sovi heille.
RoActemran on röntgentutkimuksissa havaittu vähentävän nivelvaurioiden etenemistä ja parantavan fyysistä toimintakykyä, kun sitä annetaan yhdessä MTX:n kanssa.
Koronavirustauti 2019 (COVID-19)
RoActemra on tarkoitettu COVID-19-taudin hoitoon aikuisille, jotka saavat systeemistä kortikosteroidihoitoa ja jotka tarvitsevat lisähappea tai hengityskonehoitoa.
Yleisoireinen lastenreuma
RoActemra on tarkoitettu 2-vuotiaiden ja sitä vanhempien lasten aktiivisen yleisoireisen lastenreuman hoitoon, kun aikaisempi hoito tulehduskipulääkkeillä (NSAID-lääkkeillä) ja systeemisillä kortikosteroideilla ei ole tuottanut riittävää hoitovastetta. RoActemraa voidaan antaa monoterapiana (jos potilas ei siedä MTX:a tai MTX-hoito ei sovi hänelle) tai yhdessä MTX:n kanssa.
Lasten polyartriitti
RoActemra on tarkoitettu yhdessä MTX:n kanssa 2-vuotiaiden ja sitä vanhempien lasten polyartriitin hoitoon (reumatekijä positiivinen tai negatiivinen ja laajentunut oligoartriitti), kun aikaisempi hoito MTX:lla ei ole tuottanut riittävää hoitovastetta. RoActemraa voidaan antaa monoterapiana, jos potilas ei siedä MTX:a tai jos jatkuva MTX-hoito ei sovi hänelle.
Sytokiinioireyhtymä
RoActemra on tarkoitettu aikuisille sekä 2-vuotiaille ja sitä vanhemmille pediatrisille potilaille kimeeristä antigeenireseptoria (CAR) ilmentävien T-solujen aikaansaaman vaikea-asteisen tai hengenvaarallisen sytokiinioireyhtymän hoitoon.
Ehto
Hoidon saavat aloittaa valmisteyhteenvedossa mainittuun indikaatioon perehtyneet lääkärit.
Annostus ja antotapa
Hoidon saa aloittaa nivelreuman, COVID-19-taudin, yleisoireisen lastenreuman, lasten polyartriitin tai sytokiinioireyhtymän diagnosointiin ja hoitoon perehtynyt lääkäri.
Kaikille RoActemra-hoitoa saaville potilaille on annettava potilaskortti.
Annostus
Nivelreumaa sairastavat potilaat
Suositeltu annostus on 8 mg/kg. Lääke annostellaan neljän viikon välein.
Yli 100 kg painaville potilaille ei suositella 800 mg:aa ylittäviä kerta-annoksia (ks. kohta Farmakokinetiikka).
Kliinisissä tutkimuksissa ei ole arvioitu yli 1,2 g:n annoksia (ks. kohta Farmakodynamiikka).
Annoksen muuttaminen poikkeavien laboratorioarvojen takia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Poikkeavat maksaentsyymit
| Laboratorioarvo | Toimenpide |
> 1–3 × viitevälin yläraja (ULN) (Upper Limit of Normal = viitevälin yläraja) | Muuta samanaikaisesti annettu MTX-annos, jos tarkoituksenmukaista. Jos arvon nousu on pitkäaikaista, vähennä tosilitsumabiannosta 4 mg:aan/kg tai keskeytä hoito, kunnes ALAT- (alaniiniaminotransferaasi) ja ASAT (aspartaattiaminotransferaasi)-arvot ovat normalisoituneet. Aloita uudelleen annoksella 4 mg/kg tai 8 mg/kg, kliinisen tilan mukaan. |
> 3–5 × viitevälin yläraja (varmistetaan toistetulla mittauksella, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). | Keskeytä tosilitsumabihoito, kunnes arvo on < 3 × viitevälin yläraja ja noudata yllä annettuja suosituksia arvolle > 1–3 × viitevälin yläraja. Jos arvot nousevat uudestaan > 3 × viitevälin yläraja, lopeta hoito. |
| > 5 × viitevälin yläraja | Lopeta hoito. |
- Matala absoluuttinen neutrofiilien määrä (ANC)
Aiemmin tosilitsumabilla hoitamattomille potilaille ei suositella hoidon aloitusta, jos absoluuttinen neutrofiilien määrä (ANC) on < 2 × 109/l.
| Laboratorioarvo (solumäärä × 109/l) | Toimenpide |
| ANC > 1 | Jatka samalla annoksella. |
| ANC 0,5–1 | Keskeytä tosilitsumabihoito. Kun ANC nousee > 1 × 109/l, aloita hoito uudelleen annoksella 4 mg/kg ja nosta annokseen 8 mg/kg kliinisen tilan mukaan. |
| ANC < 0,5 | Lopeta hoito. |
- Pieni trombosyyttien määrä
| Laboratorioarvo (solumäärä × 103/μl) | Toimenpide |
| 50–100 | Keskeytä tosilitsumabihoito. Kun trombosyyttien määrä nousee > 100 × 103/μl, aloita hoito uudelleen annoksella 4 mg/kg ja nosta annokseen 8 mg/kg kliinisen tilan mukaan. |
| < 50 | Lopeta hoito. |
COVID-19-potilaat
COVID-19-taudin hoitoon suositeltu annostus potilaille, jotka saavat systeemistä kortikosteroidihoitoa ja tarvitsevat lisähappea tai hengityskonehoitoa, on 60 minuuttia kestävä 8 mg/kg kertainfuusio laskimoon, ks. kohta Farmakodynamiikka. Jos kliiniset oireet tai löydökset pahenevat tai eivät lievene ensimmäisen annoksen jälkeen, voidaan antaa yksi 8 mg/kg tosilitsumabi-lisäinfuusio. Kahden infuusion välisen ajan on oltava vähintään 8 tuntia.
Yli 100 kg painaville potilaille ei suositella 800 mg:aa ylittäviä kerta-annoksia (ks. kohta Farmakokinetiikka).
Tosilitsumabin antoa ei suositella COVID-19-potilaille, joilla on jokin seuraavista poikkeavista laboratorioarvoista:
| Laboratoriotestityyppi | Laboratorioarvo | Toimenpide |
| Maksaentsyymit | > 10 × viitevälin yläraja | Tosilitsumabin antoa ei suositella |
| Absoluuttinen neutrofiilimäärä | < 1 × 109/l | |
| Trombosyyttien määrä | < 50 × 103/μl |
Sytokiinioireyhtymä (aikuiset ja pediatriset potilaat)
Suositeltu annostus sytokiinioireyhtymän hoitoon on 8 mg/kg vähintään 30 kg:n painoisille potilaille ja 12 mg/kg alle 30 kg:n painoisille potilaille. Annos annetaan 60 minuutin kestoisena infuusiona laskimoon. Tosilitsumabi voidaan antaa yksinään tai yhdistelmänä kortikosteroidien kanssa.
Jos ensimmäisen annoksen jälkeen ei havaita sytokiinioireyhtymän oireiden ja löydösten kliinistä lievenemistä, tosilitsumabia voidaan antaa enintään 3 lisäannosta. Seuraavien annosten välillä on oltava vähintään 8 tuntia. Potilaille, joilla on sytokiinioireyhtymä, ei suositella yli 800 mg:n annoksia infuusiota kohden.
Potilailla, joilla on vaikea-asteinen tai hengenvaarallinen sytokiinioireyhtymä, on usein sytopenioita tai kohonnut ALAT- tai ASAT-arvo perussairautena sairastetun syövän, aiemman lymfosyyttivajetta aiheuttaneen solunsalpaajahoidon tai sytokiinioireyhtymän vuoksi.
Erityisryhmät
Iäkkäät
Annoksen muuttaminen ei ole tarpeen yli 65-vuotiaille potilaille.
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen lievää munuaisten vajaatoimintaa sairastaville potilaille. Tosilitsumabin käyttöä ei ole tutkittu kohtalaista tai vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka). Munuaisten toimintaa on seurattava tarkasti näillä potilailla.
Maksan vajaatoiminta
Tosilitsumabin käyttöä ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla. Siksi annossuosituksia ei voida antaa.
Pediatriset potilaat
Yleisoireista lastenreumaa sairastavat potilaat
Suositeltu annostus yli 2-vuotiaille potilaille on 8 mg/kg joka toinen viikko 30 kg tai sitä enemmän painaville potilaille, tai 12 mg/kg joka toinen viikko alle 30 kg painaville potilaille. Annos on laskettava potilaan painon mukaan joka antokerralla. Annosmuutoksia tehdään vain, jos potilaan paino ajan mittaan muuttuu pysyvästi.
Laskimoon annosteltavan tosilitsumabin turvallisuutta ja tehoa alle 2 vuoden ikäisten lasten hoidossa ei ole varmistettu. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Tosilitsumabihoidon keskeyttämistä suositellaan, jos yleisoireista lastenreumaa sairastavilla potilailla todetaan seuraavassa taulukossa esitettyjä poikkeavia laboratorioarvoja. Samanaikaisesti annetun MTX:n ja/tai muiden lääkevalmisteiden annoksia on tarvittaessa muutettava tai annostus on lopetettava ja tosilitsumabin antaminen on keskeytettävä, kunnes potilaan kliininen tila on selvitetty. Koska monet samanaikaiset sairaudet voivat vaikuttaa yleisoireista lastenreumaa sairastavien potilaiden laboratorioarvoihin, päätöksen tosilitsumabihoidon lopettamisesta poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin.
- Poikkeavat maksaentsyymit
| Laboratorioarvo | Toimenpide |
| > 1–3 × viitevälin yläraja | Muuta samanaikaisesti annettu MTX-annos, jos tarkoituksenmukaista. Jos arvon nousu on pitkäaikaista, keskeytä tosilitsumabihoito, kunnes ALAT- (alaniiniaminotransferaasi) ja ASAT (aspartaattiaminotransferaasi)-arvot ovat normalisoituneet. |
| > 3–5 × viitevälin yläraja | Muuta samanaikaisesti annettu MTX-annos, jos tarkoituksenmukaista. Keskeytä tosilitsumabihoito, kunnes arvo on < 3 × viitevälin yläraja ja noudata yllä annettuja suosituksia arvolle > 1–3 × viitevälin yläraja. |
| > 5 × viitevälin yläraja | Lopeta tosilitsumabihoito. Päätöksen hoidon lopettamisesta yleisoireista lastenreumaa sairastavilla potilailla poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin. |
- Matala absoluuttinen neutrofiilien määrä (ANC)
| Laboratorioarvo (solumäärä × 109/l) | Toimenpide |
| ANC > 1 | Jatka samalla annoksella. |
| ANC 0,5–1 | Keskeytä tosilitsumabihoito. Kun ANC nousee > 1 × 109/l, aloita hoito uudelleen. |
| ANC < 0,5 | Lopeta tosilitsumabihoito. Päätöksen hoidon lopettamisesta yleisoireista lastenreumaa sairastavilla potilailla poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin. |
- Pieni trombosyyttien määrä
| Laboratorioarvo (solumäärä × 103/μl) | Toimenpide |
| 50–100 | Muuta samanaikaisesti annettu MTX-annos, jos tarkoituksenmukaista. Keskeytä tosilitsumabihoito. Kun trombosyyttien määrä nousee > 100 × 103/μl, aloita hoito uudelleen. |
| < 50 | Lopeta tosilitsumabihoito. Päätöksen hoidon lopettamisesta yleisoireista lastenreumaa sairastavilla potilailla poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin. |
Ei ole riittävästi kliinisiä tietoja, jotta voitaisiin arvioida tosilitsumabiannoksen pienentämisen vaikutukset yleisoireista lastenreumaa sairastavilla potilailla, joiden laboratorioarvot ovat olleet poikkeavia.
Saatavilla olevat tiedot viittaavat siihen, että kliininen vaste on havaittavissa 6 viikon kuluessa tosilitsumabihoidon aloittamisesta. Ellei potilaan tilassa tapahdu paranemista tänä aikana, hoidon jatkamista on arvioitava huolellisesti uudelleen.
Lasten polyartriittia sairastavat potilaat
Suositeltu annostus yli 2-vuotiaille potilaille on 8 mg/kg kerran joka 4. viikko 30 kg tai sitä enemmän painaville potilaille, tai 10 mg/kg kerran joka 4. viikko alle 30 kg painaville potilaille. Annos on laskettava potilaan painon mukaan joka antokerralla. Annosmuutoksia tehdään vain, jos potilaan paino ajan mittaan muuttuu pysyvästi.
Laskimoon annosteltavan tosilitsumabin turvallisuutta ja tehoa alle 2 vuoden ikäisten lasten hoidossa ei ole varmistettu. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Tosilitsumabihoidon keskeyttämistä suositellaan, jos lasten polyartriittia sairastavilla potilailla todetaan seuraavassa taulukossa esitettyjä poikkeavia laboratorioarvoja. Samanaikaisesti annetun MTX:n ja/tai muiden lääkevalmisteiden annoksia on tarvittaessa muutettava tai annostus on lopetettava ja tosilitsumabin antaminen on keskeytettävä, kunnes potilaan kliininen tila on selvitetty. Koska monet samanaikaiset sairaudet voivat vaikuttaa lasten polyartriittia sairastavien potilaiden laboratorioarvoihin, päätöksen tosilitsumabihoidon lopettamisesta poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin.
- Poikkeavat maksaentsyymit
| Laboratorioarvo | Toimenpide |
| > 1–3 × viitevälin yläraja | Muuta samanaikaisesti annettu MTX-annos, jos tarkoituksenmukaista Jos arvon nousu on pitkäaikaista, keskeytä tosilitsumabihoito, kunnes ALAT- (alaniiniaminotransferaasi) ja ASAT (aspartaattiaminotransferaasi)-arvot ovat normalisoituneet. |
| > 3–5 × viitevälin yläraja | Muuta samanaikaisesti annettu MTX-annos, jos tarkoituksenmukaista Keskeytä tosilitsumabihoito, kunnes arvo on < 3 × viitevälin yläraja ja noudata yllä annettuja suosituksia arvolle > 1–3 × viitevälin yläraja |
| > 5 × viitevälin yläraja | Lopeta tosilitsumabihoito. Päätöksen hoidon lopettamisesta lasten polyartriittia sairastavilla potilailla poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin. |
- Matala absoluuttinen neutrofiilien määrä (ANC)
| Laboratorioarvo (solumäärä × 109/ l) | Toimenpide |
| ANC > 1 | Jatka samalla annoksella |
| ANC 0,5–1 | Keskeytä tosilitsumabihoito Kun ANC nousee > 1 × 109/l, aloita hoito uudelleen |
| ANC < 0,5 | Lopeta tosilitsumabihoito Päätöksen hoidon lopettamisesta lasten polyartriittia sairastavilla potilailla poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin. |
- Pieni trombosyyttien määrä
| Laboratorioarvo (solumäärä × 103/μl) | Toimenpide |
| 50–100 | Muuta samanaikaisesti annettu MTX-annos, jos tarkoituksenmukaista Keskeytä tosilitsumabihoito Kun trombosyyttien määrä nousee > 100 × 103/μl, aloita hoito uudelleen |
| < 50 | Lopeta tosilitsumabihoito. Päätöksen hoidon lopettamisesta lasten polyartriittia sairastavilla potilailla poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin. |
Tosilitsumabiannoksen pienentämistä poikkeavien laboratorioarvojen vuoksi ei ole tutkittu lasten polyartriittia sairastavilla potilailla.
Saatavilla olevat tiedot viittaavat siihen, että kliininen vaste on havaittavissa 12 viikon kuluessa tosilitsumabihoidon aloittamisesta. Ellei potilaan tilassa tapahdu paranemista tänä aikana, hoidon jatkamista on arvioitava huolellisesti uudelleen.
Sytokiinioireyhtymä
Tosilitsumabia voidaan käyttää pediatrisille potilaille (2-vuotiaille ja sitä vanhemmille), joilla on sytokiinioireyhtymä, samalla annostuksella kuin aikuisille. Ks. kohta Annostus ja antotapa Annostus ja antotapa, sytokiinioireyhtymä (aikuiset ja pediatriset potilaat).
Antotapa
Laimentamisen jälkeen tämä lääkevalmiste annetaan laskimoon tunnin kestävänä infuusiona. Jos infuusioon liittyviä oireita ja löydöksiä ilmaantuu, infuusiota pitää hidastaa tai sen anto lopettaa ja sopivaa lääkevalmistetta/tukihoitoa on annettava välittömästi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Nivelreumaa, yleisoireista lastenreumaa, lasten polyartriittia ja COVID-19-tautia sairastavat potilaat sekä potilaat, joilla on sytokiinioireyhtymä (≥ 30 kg)
Tämä lääkevalmiste pitää laimentaa steriilillä, pyrogeenittomalla 0,9-prosenttisella (9 mg/ml) natriumkloridiliuoksella 100 ml:n tilavuuteen aseptista tekniikkaa käyttäen.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Yleisoireista lastenreumaa ja lasten polyartriittia sairastavat potilaat sekä potilaat, joilla on sytokiinioireyhtymä (< 30 kg)
Tämä lääkevalmiste pitää laimentaa steriilillä, pyrogeenittomalla 0,9-prosenttisella (9 mg/ml) natriumkloridiliuoksella 50 ml:n tilavuuteen aseptista tekniikkaa käyttäen.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiiviset vaikeat infektiot, COVID-19-infektiota lukuun ottamatta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Nivelreumaa, lasten polyartriittia ja yleisoireista lastenreumaa sairastavat potilaat
Infektiot
Vakavia ja joskus kuolemaan johtaneita infektioita on raportoitu potilailla, jotka ovat saaneet immunosuppressiivisia lääkkeitä mukaan lukien tosilitsumabia (ks. kohta Haittavaikutukset). Hoitoa ei saa aloittaa, jos potilaalla on aktiivinen infektio (ks. kohta Vasta-aiheet). Jos potilaalle kehittyy vakava infektio, tosilitsumabihoito on keskeytettävä, kunnes infektio on saatu hallintaan (ks. kohta Haittavaikutukset). Tämän lääkevalmisteen käyttöä harkitessaan lääkärin on noudatettava varovaisuutta, jos potilaalla on aikaisemmin esiintynyt uusiutuvia tai kroonisia infektioita tai jos hänellä on infektioalttiutta lisäävä perussairaus (esim. divertikuliitti, diabetes ja interstitiaalinen keuhkosairaus).
Biologisia lääkkeitä saavien potilaiden tilaa on seurattava tarkoin, jotta mahdolliset vakavat infektiot havaitaan ajoissa, sillä akuutin vaiheen reaktion estyminen voi heikentää akuutin tulehduksen oireita. Potilaan mahdollista infektiota arvioitaessa on huomioitava tosilitsumabin vaikutukset C‑reaktiiviseen proteiiniin (CRP), neutrofiileihin ja infektion oireisiin. Potilaita (myös yleisoireista lastenreumaa tai lasten polyartriittia sairastavia pienempiä lapsia, jotka eivät ehkä itse kykene kertomaan oireistaan) ja yleisoireista lastenreumaa tai lasten polyartriittia sairastavien potilaiden vanhempia/hoitajia pitää ohjeistaa ottamaan heti yhteyttä terveydenhoitohenkilöstöön, jos he havaitsevat mitä tahansa infektioon viittaavia oireita, jotta tutkimukset ja hoito voidaan aloittaa mahdollisimman nopeasti.
Tuberkuloosi
Kuten muitakin biologisia nivelreumalääkkeitä käytettäessä latentin tuberkuloosin poissulkemista seulontatutkimusten avulla suositellaan nivelreumaa, lasten polyartriittia ja yleisoireista lastenreumaa sairastavilla potilailla ennen tosilitsumabihoidon aloittamista. Jos latentti tuberkuloosi todetaan, se on hoidettava tavanomaisilla mykobakteerilääkkeillä ennen kuin hoito aloitetaan. Lääkettä määrääviä lääkäreitä muistutetaan tuberkuliinikokeen ja verinäytteestä määritettävän IGRA-testin väärän negatiivisen tuloksen riskistä, varsinkin vaikeasti sairailla tai immuunipuutteisilla potilailla.
Potilaita tulisi kehottaa ottamaan yhteyttä lääkäriin, jos tällä lääkevalmisteella annettavan hoidon aikana tai sen jälkeen ilmenee tuberkuloosi-infektioon viittaavia merkkejä tai oireita (esim. itsepintainen yskä, kuihtuminen/painon lasku, lievä lämmönnousu).
Virustautien uudelleen aktivoituminen
Virusten uudelleen aktivoitumista (esim. hepatiitti B -virus) on raportoitu biologisilla nivelreumalääkkeillä hoidetuilla potilailla. Tosilitsumabin kliinisiin tutkimuksiin ei ole hyväksytty hepatiittipositiivisia potilaita.
Divertikuliitin komplikaatiot
Melko harvinaisena divertikuliitin komplikaationa on raportoitu divertikkelien perforaatioita tosilitsumabihoidon yhteydessä nivelreumapotilailla (ks. kohta Haittavaikutukset). Tätä lääkevalmistetta tulisi antaa varoen potilaille, joilla on aikaisemmin esiintynyt suoliston haavaumia tai divertikuliittia. Jos potilaalle ilmaantuu komplisoituneeseen divertikuliittiin viittaavia oireita, kuten vatsakipua, verenvuotoa ja/tai selittämättömiä muutoksia suolen toiminnassa ja kuumetta, potilas on tutkittava heti, jotta divertikuliitti ja siihen mahdollisesti liittyvä suolen puhkeaminen havaitaan mahdollisimman varhaisessa vaiheessa.
Yliherkkyysreaktiot
Vakavia yliherkkyysreaktioita on raportoitu tosilitsumabi-infuusion yhteydessä (ks. kohta Haittavaikutukset). Reaktiot voivat olla vaikeampia ja mahdollisesti fataaleja niillä potilailla, jotka ovat kokeneet yliherkkyysreaktioita aikaisempien infuusioiden yhteydessä, vaikka heille annetaan esilääkityksenä steroideja ja antihistamiineja. Anafylaktisen reaktion hoidossa tarvittavien välineiden on oltava välittömästi saatavilla hoidon aikana. Anafylaktisen tai muun vakavan yliherkkyys-/infuusioreaktion ilmaantuessa, tosilitsumabin anto on heti keskeytettävä ja hoito lopetettava pysyvästi.
Aktiivinen maksasairaus ja maksan vajaatoiminta
Tosilitsumabihoitoon voi liittyä kohonneita aminotransferaasiarvoja, varsinkin jos sitä annetaan yhdessä MTX:n kanssa. Siksi on noudatettava varovaisuutta, jos harkitaan hoidon aloittamista potilaille, joilla on aktiivinen maksasairaus tai maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Maksatoksisuus
Ohimenevää tai jaksottaista lievää tai kohtalaista maksan aminotransferaasiarvojen nousua on raportoitu yleisesti tosilitsumabihoidon yhteydessä (ks. kohta Haittavaikutukset). Kohonneita arvoja havaittiin enemmän, kun tosilitsumabihoitoon yhdistettiin jokin mahdollisesti maksatoksinen lääkevalmiste (esim. MTX). Muita maksantoimintakokeissa ilmenevien yhdisteiden määritystä, kuten bilirubiini, on harkittava kliinisen tarpeen mukaan.
Tosilitsumabihoitoa saaneilla potilailla on havaittu vakavia lääkkeestä aiheutuneita maksavaurioita, mukaan lukien akuutti maksan vajaatoiminta, hepatiitti ja ikterus (ks. kohta Haittavaikutukset). Vakava maksavaurio ilmeni kahdesta viikosta yli viiteen vuoteen hoidon aloittamisesta. Maksansiirron vaativia maksan vajaatoimintatapauksia on raportoitu. Potilaita on neuvottava ottamaan yhteyttä lääkäriin heti, jos maksavaurion oireita ilmaantuu.
Varovaisuutta on noudatettava harkittaessa hoidon aloittamista potilaille, joiden kohonneet ALAT- tai ASAT-arvot ovat > 1,5 × viitevälin yläraja. Hoitoa ei suositella nivelreumaa, lasten polyartriittia ja yleisoireista lastenreumaa sairastaville potilaille, joiden ALAT- tai ASAT-arvot hoidon alussa ovat > 5 × viitevälin yläraja.
Nivelreumapotilaiden sekä yleisoireista lastenreumaa ja lasten polyartriittia sairastavien potilaiden ALAT- ja ASAT-arvoja on seurattava joka 4.–8. viikko hoidon kuuden ensimmäisen kuukauden aikana ja sen jälkeen joka 12. viikko. Suositukset annoksen muuttamisesta aminotransferaasiarvojen perusteella, mukaan lukien tosilitsumabihoidon lopettamista, ks. kohta Annostus ja antotapa. Jos ASAT- tai ALAT-arvojen nousu on > 3–5 × viitevälin yläraja, ja se on varmistettu toistetulla mittauksella, hoito on keskeytettävä.
Hematologiset poikkeamat
Neutrofiili- ja trombosyyttiarvon laskua on esiintynyt, kun tosilitsumabia on annettu 8 mg/kg yhdessä MTX:n kanssa (ks. kohta Haittavaikutukset). Neutropenian riski voi olla suurentunut, jos potilasta on aikaisemmin hoidettu TNF-estäjällä.
Aiemmin tosilitsumabilla hoitamattomille potilaille ei suositella hoidon aloitusta, jos absoluuttinen neutrofiilien määrä (ANC) on < 2 × 109/l. Varovaisuutta on noudatettava harkittaessa hoidon aloittamista potilaille, joiden trombosyyttiarvo on alentunut (trombosyyttiarvo alle 100 × 103/μl). Nivelreumaa, lasten polyartriittia ja yleisoireista lastenreumaa sairastavien potilaiden hoidon jatkamista ei suositella, jos potilaan ANC on < 0,5 × 109/l tai trombosyyttiarvo < 50 × 103/μl.
Vaikeaan neutropeniaan voi liittyä vakavien infektioiden kohonnut riski. Tosilitsumabin kliinisissä tutkimuksissa ei ole tähän mennessä ilmennyt selvää yhteyttä alentuneiden neutrofiiliarvojen ja vakavien infektiotapausten välillä.
Nivelreumapotilailla neutrofiili- ja trombosyyttiarvoja on seurattava 4–8 viikon ajan hoidon alkamisesta ja myöhemmin normaalin kliinisen käytännön mukaan. Suositukset annoksen muuttamisesta ANC- ja trombosyyttiarvojen mukaan, ks. kohta Annostus ja antotapa.
Lasten polyartriittia ja yleisoireista lastenreumaa sairastavilla potilailla neutrofiili- ja trombosyyttiarvoja on seurattava toisen infuusion yhteydessä ja sen jälkeen hyvän hoitokäytännön mukaan, ks. kohta Annostus ja antotapa.
Veren rasva-arvot
Veren rasva-arvojen, kuten kokonaiskolesterolin, LDL- ja HDL-kolesterolin sekä triglyseridien, nousua havaittiin tosilitsumabilla hoidetuilla potilailla (ks. kohta Haittavaikutukset). Suurimmalla osalla potilaista ei havaittu aterogeenisen vaikutuksen lisääntymistä. Kohonneet kokonaiskolesteroliarvot saatiin yleensä hallintaan kolesterolia alentavilla lääkkeillä.
Nivelreumaa, lasten polyartriittia ja yleisoireista lastenreumaa sairastavilla potilailla rasva-arvot on määritettävä 4–8 viikon kuluttua hoidon aloittamisesta. Potilaita tulisi hoitaa hyperlipidemian paikallisten hoitosuositusten mukaisesti.
Neurologiset häiriöt
Lääkäreiden tulisi kiinnittää erityistä huomiota oireisiin, jotka voivat viitata keskushermoston myeliinikadon puhkeamiseen. Toistaiseksi ei tiedetä, liittyykö tosilitsumabihoitoon keskushermoston myeliinikatoa.
Maligniteetti
Nivelreumapotilailla on lisääntynyt pahanlaatuisten kasvainten riski. Immunomodulatoriset lääkkeet voivat suurentaa tätä riskiä. Kliiniset tiedot eivät ole riittäviä, jotta voitaisiin arvioida pahanlaatuisten kasvainten mahdollista ilmaantuvuutta tosilitsumabialtistuksen jälkeen. Pitkäaikainen turvallisuusarviointi on meneillään.
Rokotukset
Eläviä tai eläviä heikennettyjä taudinaiheuttajia sisältäviä rokotteita ei tulisi antaa tällä lääkevalmisteella annettavan hoidon aikana, sillä niiden kliinistä turvallisuutta ei ole osoitettu. Avoimessa satunnaistetussa tutkimuksessa tosilitsumabin ja MTX:n yhdistelmällä hoidetut aikuiset nivelreumapotilaat saavuttivat tehokkaan vasteen sekä 23-valenttiselle pneumokokkipolysakkaridi- että jäykkäkouristusrokotteelle. Vaste oli verrattavissa pelkkää MTX-hoitoa saaneiden potilaiden vasteeseen. Kaikille potilaille, erityisesti lasten polyartriittia ja yleisoireista lastenreumaa sairastaville potilaille, suositellaan kaikkien ajantasaisten rokotusten antamista voimassa olevien rokotussuositusten mukaisesti ennen hoidon aloittamista. Eläviä taudinaiheuttajia sisältävien rokotteiden antamisen ja hoidon aloittamisen välillä on pidettävä tauko, jonka kesto on nykyisten immunosuppressantteja koskevien rokotussuositusten mukainen.
Sydän- ja verisuonitautiriski
Nivelreumapotilailla on suurentunut sydän- ja verisuonisairauksien riski, ja riskitekijöiden (esim. hypertensio, hyperlipidemia) hallinnan on oltava osa näiden potilaiden tavanomaista perushoitoa.
Yhteiskäyttö TNF-estäjien kanssa
Tosilitsumabin käytöstä yhdessä TNF-estäjien tai muiden biologisten reumalääkkeiden kanssa ei ole kokemuksia nivelreumaa, lasten polyartriittia eikä yleisoireista lastenreumaa sairastavilla potilailla. Tämän lääkevalmisteen käyttöä yhdessä muiden biologisten reumalääkkeiden kanssa ei suositella.
COVID-19-potilaat
- Tämän lääkevalmisteen tehoa ei ole varmistettu niiden COVID-19-potilaiden hoidossa, joiden CRP-arvo ei ole koholla, ks. kohta Farmakodynamiikka.
- Tätä lääkevalmistetta ei saa antaa COVID-19-potilaille, jotka eivät saa systeemistä kortikosteroidihoitoa, koska kuolleisuuden kasvua ei voida poissulkea tässä alaryhmässä, ks. kohta Farmakodynamiikka.
Infektiot
Tätä lääkevalmistetta ei pidä antaa COVID-19-potilaalle, jolla on jokin toinen samanaikainen vaikea-asteinen aktiivinen infektio. Tosilitsumabin käyttöä harkitessaan lääkärin on noudatettava varovaisuutta, jos potilaalla on aikaisemmin esiintynyt uusiutuvia tai kroonisia infektioita tai jos hänellä on infektioalttiutta lisäävä perussairaus (esim. divertikuliitti, diabetes tai interstitiaalinen keuhkosairaus).
Maksatoksisuus
Sairaalahoidossa olevien COVID-19-potilaiden ALAT- tai ASAT-arvot voivat olla koholla. Maksaan vaikuttava monielinhäiriö on vaikea-asteisen COVID-19-taudin tunnettu komplikaatio. Tosilitsumabin antamisesta päätettäessä pitää arvioida COVID-19-taudin hoidon mahdolliset hyödyt ja akuuttihoitona annetun tosilitsumabin mahdolliset riskit. Tosilitsumabihoitoa ei suositella COVID-19-potilaille, joiden ALAT- tai AST-arvo on kohonnut tasolle yli 10 × viitevälin yläraja. COVID-19-potilaiden ALAT- tai ASAT-arvoa pitää seurata voimassa olevien tavanomaisten kliinisten käytäntöjen mukaisesti.
Hematologiset poikkeamat
Hoitoa ei suositella COVID-19-potilaille, joiden ANC on < 1 × 109/l tai trombosyyttiarvo on < 50 × 103/μl. Neutrofiili- ja trombosyyttiarvoja on seurattava voimassa olevien tavanomaisten kliinisten käytäntöjen mukaisesti, ks. kohta Annostus ja antotapa.
Pediatriset potilaat
Yleisoireista lastenreumaa sairastavat potilaat
Makrofagiaktivaatio-oireyhtymä (MAS) on vakava henkeä uhkaava tila, joka voi kehittyä yleisoireista lastenreumaa sairastaville potilaille. Kliinisissä tutkimuksissa tosilitsumabia ei ole tutkittu aktiivisen makrofagiaktivaatio-oireyhtymän oireita saaneilla potilailla.
Natrium
0,9-prosenttisella natriumkloridiliuoksella laimentamisen jälkeen tämä lääkevalmiste sisältää 230,6 mg natriumia per 800 mg:n enimmäisannos, mikä vastaa 11,5 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Polysorbaatti 80 (E 433)
Tämä lääkevalmiste sisältää 2 mg polysorbaattia 80 per 80 mg:n injektiopullo, 5 mg polysorbaattia 80 per 200 mg:n injektiopullo ja 10 mg polysorbaattia 80 per 400 mg:n injektiopullo, mikä vastaa 0,5 mg:aa/ml.
Polysorbaatit saattavat aiheuttaa allergisia reaktioita. Tiedossa olevat potilaan allergiat on otettava huomioon.
Yhteisvaikutukset
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Tosilitsumabi ei vaikuttanut kliinisesti merkitsevästi MTX-altistukseen, kun sitä annettiin 10 mg/kg kerta-annoksena MTX-hoidon (10–25 mg kerran viikossa) aikana.
Populaatiofarmakokineettisessä analyysissä havaittiin, etteivät MTX, steroideihin kuulumattomat tulehduskipulääkkeet (NSAID-lääkkeet) eivätkä kortikosteroidit vaikuttaneet tosilitsumabin puhdistumaan.
Kroonista tulehdusta vahvistavat sytokiinit, kuten IL-6, estävät maksan CYP450-entsyymien ilmentymistä. CYP450-entsyymien ilmentyminen voi siis palautua, kun aloitetaan voimakas sytokiinia estävä lääkitys, esimerkiksi tosilitsumabi.
In vitro ‑tutkimukset viljellyillä ihmisen maksasoluilla osoittivat, että IL-6 pienensi CYP1A2-, CYP2C9-, CYP2C19- ja CYP3A4-entsyymien ilmentymää. Tosilitsumabi normalisoi näiden entsyymien ilmentymää.
Kliinisessä tutkimuksessa nivelreumapotilailla simvastatiinipitoisuudet (CYP3A4) olivat laskeneet 57 % viikon kuluttua tosilitsumabin kerta-annoksen antamisesta ja olivat vastaavat tai hieman korkeammat kuin terveillä koehenkilöillä mitatut pitoisuudet.
Potilaita on seurattava tarkoin tosilitsumabihoitoa aloitettaessa tai lopetettaessa, jos he saavat CYP450-entsyymien 3A4, 1A2 tai 2C9 välityksellä metaboloituvia lääkkeitä (esim. metyyliprednisoloni, deksametasoni [oraalisen glukokortikoidihoidon lopettamisoireiden mahdollisuus], atorvastatiini, kalsiuminestäjät, teofylliini, varfariini, fenprokumoni, fenytoiini, siklosporiini tai bentsodiatsepiinit). Näiden lääkkeiden annokset määritellään yksilöllisesti ja annosten suurentaminen saattaa olla tarpeen hoitotehon säilyttämiseksi. Eliminaation pitkän puoliintumisajan (t1/2) takia tosilitsumabin vaikutus CYP450-entsyymin toimintaan saattaa jatkua vielä useita viikkoja hoidon päättymisen jälkeen.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä luotettavaa ehkäisyä hoidon aikana ja vähintään kolmen kuukauden ajan hoidon päättymisen jälkeen.
Raskaus
Ei ole riittävästi tietoja tosilitsumabin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu spontaanien keskenmenojen / alkio- ja sikiökuolemien lisääntymistä suuria annoksia annettaessa (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmisille ei tunneta.
RoActemraa ei pidä käyttää raskauden aikana, ellei se ole ehdottoman välttämätöntä.
Imetys
Ei tiedetä, erittyykö tosilitsumabi ihmisillä äidinmaitoon. Tosilitsumabin erittymistä äidinmaitoon ei ole tutkittu eläimillä. On päätettävä, lopetetaanko imetys vai pidättäydytäänkö RoActemra‑hoidosta ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Käytettävissä olevat ei-kliiniset tiedot eivät viittaa tosilitsumabihoidon vaikuttavan hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
RoActemralla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn, esim. huimaus (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Nivelreuma, yleisoireinen lastenreuma, lasten polyartriitti ja sytokiinioireyhtymä
Yleisimmät haittavaikutukset ovat ylähengitystieinfektiot, nasofaryngiitti, päänsärky, hypertensio ja ALAT-arvon nousu.
Vakavimmat haittavaikutukset ovat vakavat infektiot, divertikuliitin komplikaatiot ja yliherkkyysreaktiot.
COVID-19-tauti
Yleisimmin raportoidut haittavaikutukset ovat maksan transaminaasiarvojen nousu, ummetus ja virtsatieinfektio.
Taulukoitu lista haittavaikutuksista
Taulukossa 1 luetellaan haittavaikutukset, jotka on todettu kliinisissä tutkimuksissa ja/tai tosilitsumabin markkinoille tulon jälkeisessä käytössä spontaanien tapausselostusten, kirjallisuudessa raportoitujen tapausten ja ei-interventiotutkimusohjelmissa todettujen tapausten perusteella, ja taulukossa 2 haittavaikutukset luetellaan MedDRA-elinjärjestelmän (SOC) mukaan. Kunkin haittavaikutuksen vastaava yleisyysluokka perustuu seuraavaan esitystapaan: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Nivelreumapotilaat
Taulukko 1. Lista nivelreumapotilailla esiintyneistä haittavaikutuksista, kun tosilitsumabia annettiin monoterapiana tai yhdessä MTX:n tai muiden DMARDien kanssa kaksoissokkoutetussa kontrollivaiheessa tai valmisteen markkinoille tulon jälkeisessä käytössä
| MedDRA-elinjärjestelmä | Yleisyysluokat suositelluin termein | ||||
| Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Hyvin harvinainen | |
| Infektiot | Ylähengitystie-infektiot | Selluliitti, keuhkokuume, suun herpes simplex -infektio, vyöruusu | Divertikuliitti | ||
| Veri ja imukudos | Leukopenia, neutropenia, hypofibrinogenemia | ||||
| Immuunijärjestelmä | Anafylaksia (fataali)1, 2, 3 | ||||
| Umpieritys | Kilpirauhasen vajaatoiminta | ||||
| Aineenvaihdunta ja ravitsemus | Hyperkoleste-rolemia* | Hypertriglyse-ridemia | |||
| Hermosto | Päänsärky, huimaus | ||||
| Silmät | Sidekalvotulehdus | ||||
| Verisuonisto | Hypertensio | ||||
| Hengityselimet, rintakehä ja välikarsina | Yskä, hengenahdistus | ||||
| Ruoansulatuselimistö | Vatsakipu, suun haavaumat, gastriitti | Suutulehdus, mahahaava | |||
| Maksa ja sappi | Lääkkeestä aiheutunut maksavaurio, hepatiitti ja ikterus. | Maksan vajaatoiminta | |||
| Iho ja ihonalainen kudos | Ihottuma, kutina, nokkosihottuma | Stevens-Johnsonin oireyhtymä3 | |||
| Munuaiset ja virtsatiet | Munuaiskivitauti | ||||
| Yleisoireet ja antopaikassa todettavat haitat | Perifeerinen ödeema, yliherkkyysreaktiot | ||||
| Tutkimukset | Maksan aminotransferaasi-arvojen nousu, painon nousu, kokonaisbilirubiiniarvon nousu* | ||||
*Sisältää tavanomaisessa laboratorioseurannassa kerätyt kohonneet arvot (ks. alempana oleva teksti)
1 Katso kohta Vasta-aiheet
2 Katso kohta Varoitukset ja käyttöön liittyvät varotoimet
3 Tämä haittavaikutus havaittiin markkinoille tulon jälkeen, mutta sitä ei ole raportoitu kontrolloiduissa kliinisissä tutkimuksissa. Yleisyysluokka arvioitiin käyttämällä 95 prosentin luottamusvälin ylärajaa ja se perustuu kliinisissä tutkimuksissa tosilitsumabille altistuneiden potilaiden kokonaismäärään.
COVID-19-potilaat
Tämän lääkevalmisteen turvallisuuden arviointi COVID-19-taudin hoidossa perustui kolmeen satunnaistettuun, kaksoissokkoutettuun, lumekontrolloituun tutkimukseen (tutkimukset ML42528, WA42380 ja WA42511). Näissä tutkimuksissa yhteensä 974 potilasta altistui tosilitsumabille. RECOVERY-tutkimuksesta kerätyt turvallisuutta koskevat tiedot olivat suppeita, eikä niitä esitetä tässä.
Seuraavat MedDRA-elinjärjestelmien mukaisesti taulukossa 2 luetellut haittavaikutukset on varmistettu tapahtumista, joita esiintyi kliinisten tutkimusten ML42528, WA42380 ja WA42511 turvallisuuden osalta arvioitavissa olleessa yhdistetyssä potilasjoukossa vähintään 3 %:lla tosilitsumabihoitoa saaneista potilaista ja yleisemmin kuin lumelääkettä saaneilla potilailla.
Taulukko 2. Luettelo haittavaikutuksista1, jotka tunnistettiin COVID-19-potilailla kliinisten tosilitsumabitutkimusten turvallisuuden osalta arvioitavissa olleessa yhdistetyssä potilasjoukossa2
| MedDRA-elinjärjestelmä | Suositellut termit ja esiintyvyys Yleinen |
| Infektiot | Virtsatieinfektio |
| Aineenvaihdunta ja ravitsemus | Hypokalemia |
| Psyykkiset häiriöt | Ahdistuneisuus, unettomuus |
| Verisuonisto | Hypertensio |
| Ruoansulatuselimistö | Ummetus, ripuli, pahoinvointi |
| Maksa ja sappi | Maksan transaminaasiarvojen nousu |
1 Potilaat on laskettu kussakin luokassa kerran reaktioiden lukumäärästä riippumatta
2 Sisältää tutkimuksissa WA42511, WA42380 ja ML42528 raportoidut varmistetut reaktiot
Yleisoireista lastenreumaa tai lasten polyartriittia sairastavat potilaat
Yleisoireista lastenreumaa ja lasten polyartriittia sairastavilla tosilitsumabihoitoa saaneilla potilailla todetut haittavaikutukset luetellaan taulukossa 3 MedDRA-elinjärjestelmän mukaan. Kunkin haittavaikutuksen vastaava yleisyysluokka perustuu seuraavaan esitystapaan: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10) ja melko harvinainen (≥ 1/1000, < 1/100).
Taulukko 3: Lista yleisoireista lastenreumaa (sJIA) tai lasten polyartriittia (pJIA) sairastavilla potilailla kliinisissä tutkimuksissa esiintyneistä haittavaikutuksista, kun tosilitsumabia annettiin monoterapiana tai yhdessä MTX:n kanssa
| MedDRA-elinjärjestelmä | Suositeltu termi | Yleisyys | ||
| Infektiot | Hyvin yleinen | Yleinen | Melko harvinainen | |
| Ylähengitystieinfektiot | pJIA, sJIA | |||
| Nasofaryngiitti | pJIA, sJIA | |||
| Hermosto | ||||
| Päänsärky | pJIA | sJIA | ||
| Ruoansulatuselimistö | ||||
| Pahoinvointi | pJIA | |||
| Ripuli | pJIA, sJIA | |||
| Yleisoireet ja antopaikassa todettavat haitat | ||||
| Infuusioon liittyvät reaktiot | pJIA1, sJIA2 | |||
| Tutkimukset | ||||
| Maksan aminotransferaasiarvojen nousu | pJIA | |||
| Neutrofiiliarvojen lasku | sJIA | pJIA | ||
| Trombosyyttiarvojen lasku | sJIA | pJIA | ||
| Kohonnut kolesteroli | sJIA | pJIA | ||
1. Lasten polyartriittia sairastavilla potilailla raportoituihin infuusioreaktiotapauksiin sisältyi mm. päänsärkyä, pahoinvointia ja hypotensiota
2. Yleisoireista lastenreumaa sairastavilla potilailla raportoituihin infuusioreaktiotapauksiin sisältyi mm. ihottumaa, nokkosihottumaa, ripulia, epigastrista kipua, nivelkipua ja päänsärkyä
Valikoitujen haittavaikutusten kuvaus
Nivelreumaa sairastavat potilaat
Infektiot
Kuusi kuukautta kestäneissä vertailututkimuksissa infektioiden esiintyvyys oli 127 tapahtumaa 100 potilasvuotta kohti potilailla, jotka saivat 8 mg/kg tosilitsumabia yhdessä DMARDien kanssa, ja 112 tapahtumaa 100 potilasvuotta kohti potilailla, jotka saivat lumevalmistetta ja DMARDeja. Populaatiossa, jossa altistuminen tosilitsumabille oli pitkäaikaista, infektioiden kokonaisesiintyvyys oli 108 tapahtumaa 100 potilasvuotta kohti.
Kuusi kuukautta kestäneissä kliinisissä vertailututkimuksissa vakavien infektioiden esiintyvyys oli 5,3 tapahtumaa 100 potilasvuotta kohti potilailla, jotka saivat 8 mg/kg tosilitsumabia yhdessä DMARDien kanssa ja 3,9 tapahtumaa 100 potilasvuotta kohti potilailla, jotka saivat lumevalmistetta ja DMARDeja. Monoterapiatutkimuksessa vakavien infektioiden esiintyvyys oli 3,6 tapahtumaa 100 potilasvuotta kohti tosilitsumabiryhmässä ja 1,5 tapahtumaa 100 potilasvuotta kohti MTX-ryhmässä.
Populaatiossa, jossa altistuminen oli pitkäaikaista, vakavien infektioiden (bakteeri-, virus- tai sieni-infektio) kokonaisesiintyvyys oli 4,7 tapahtumaa 100 potilasvuotta kohti. Raportoituja vakavia infektioita, joista jotkut johtivat kuolemaan, olivat aktiivinen tuberkuloosi (intra- tai ekstrapulmonaalinen), invasiiviset keuhkoinfektiot, joihin kuuluvat kandidiaasi, aspergilloosi, koksidioidomykoosi ja pneumocystis jirovecii ‑infektio, keuhkokuume, selluliitti, vyöruusu, gastroenteriitti, divertikuliitti, sepsis ja bakteeriartriitti. Opportunistisia infektioita on raportoitu.
Interstitiaalinen keuhkosairaus
Keuhkojen heikentynyt toimintakyky voi lisätä infektioriskiä. Interstitiaalista keuhkosairautta on raportoitu markkinoille tulon jälkeen (mukaan lukien keuhkotulehdus ja keuhkofibroosi). Näistä tapauksista muutama on ollut kuolemaan johtava.
Ruoansulatuskanavan perforaatiot
Kuusi kuukautta kestäneissä kontrolloiduissa kliinisissä tutkimuksissa ruoansulatuskanavan perforaatioiden kokonaisesiintyvyys tosilitsumabihoidon aikana oli 0,26 tapahtumaa 100 potilasvuotta kohti. Populaatiossa, jossa altistuminen oli pitkäaikaista, kokonaisesiintyvyys oli 0,28 tapahtumaa 100 potilasvuotta kohti. Ruoansulatuskanavan perforaatioita hoidon yhteydessä raportoitiin ensisijaisesti divertikuliitin komplikaatioina mukaan lukien yleistynyt märkäinen peritoniitti, ruoansulatuskanavan alaosan perforaatio, fistelit ja absessi.
Infuusioon liittyvät reaktiot
Kuusi kuukautta kestäneissä tutkimuksissa infuusioon liittyviä haittatapahtumia (valikoidut tapahtumat, jotka ilmaantuivat infuusion aikana tai 24 tunnin kuluessa sen päättymisestä) esiintyi 6,9 %:lla potilaista, jotka saivat tosilitsumabia 8 mg/kg yhdessä DMARDien kanssa, ja 5,1 %:lla potilaista, jotka saivat lumevalmistetta ja DMARDeja. Infuusion aikana raportoidut tapahtumat olivat pääasiassa hypertensioepisodeja. Tapahtumia, joita raportoitiin 24 tunnin aikana infuusion päättymisen jälkeen, olivat päänsärky ja ihoreaktiot (ihottuma, nokkosihottuma). Nämä tapahtumat eivät olleet hoitoa rajoittavia.
Anafylaktisten reaktioiden esiintyvyys (yhteensä 8 tapausta / 4 009 potilasta, 0,2 %) oli moninkertainen annoksen ollessa 4 mg/kg verrattuna annokseen 8 mg/kg. Hoidon keskeyttämistä vaatineita kliinisesti merkitseviä tosilitsumabihoitoon liittyneitä yliherkkyysreaktioita todettiin yhteensä 56:lla (1,4 %) niistä 4 009 potilaasta, jotka saivat hoitoa vertailututkimuksissa ja avoimissa kliinisissä tutkimuksissa. Nämä reaktiot ilmaantuivat yleensä 2.–5. tosilitsumabi-infuusion aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Myyntiluvan myöntämisen jälkeen on raportoitu yksi fataali anafylaksiatapaus tosilitsumabihoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Immunogeenisuus
Anti-tosilitsumabivasta-aineet määritettiin kuusi kuukautta kestäneissä kliinisissä vertailututkimuksissa yhteensä 2 876 potilaalta. Näistä 46 potilaalle (1,6 %) kehittyi anti-tosilitsumabivasta-aineita, ja heistä kuudella esiintyi lääketieteellisesti merkitsevä yliherkkyysreaktio, jonka seurauksena viidellä potilaalla hoito lopetettiin pysyvästi. Neutraloivia vasta-aineita kehittyi 30 potilaalla (1,1 %).
Neutrofiilit
Kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa neutrofiiliarvo laski tason 1 × 109/l alapuolelle 3,4 %:lla potilaista, jotka saivat tosilitsumabia 8 mg/kg ja DMARDeja verrattuna < 0,1 %:iin potilaista, jotka saivat lumevalmistetta ja DMARDeja. Potilaista, joiden absoluuttinen neutrofiiliarvo laski tasolle < 1 × 109/l, noin puolella lasku todettiin kahdeksan viikon kuluessa hoidon alkamisesta. Tason 0,5 × 109/l alapuolelle laskeneita arvoja todettiin 0,3 %:lla potilaista, jotka saivat tosilitsumabia 8 mg/kg yhdessä DMARDien kanssa. Infektioita, joihin liittyi neutropenia, on raportoitu.
Kaksoissokkoutetussa kontrollivaiheessa ja pitkäaikaisessa altistumisessa havaittiin neutrofiiliarvojen laskeneen vastaavalla tavalla ja ilmaantuvuudella kuin kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa.
Trombosyytit
Kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa trombosyyttiarvo laski tason 100 × 103/μl alapuolelle 1,7 %:lla tosilitsumabia 8 mg/kg ja DMARDeja saaneista potilaista verrattuna < 1 %:lla lumevalmistetta ja DMARDeja saaneisiin potilaisiin. Näihin muutoksiin ei liittynyt verenvuototapahtumia.
Kaksoissokkoutetussa kontrollivaiheessa ja pitkäaikaisessa altistumisessa havaittiin trombosyyttiarvojen laskeneen vastaavalla tavalla ja ilmaantuvuudella kuin kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa.
Myyntiluvan myöntämisen jälkeisessä seurannassa on hyvin harvoin raportoitu pansytopeniaa.
Maksan aminotransferaasiarvojen nousu
Kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa ohimenevä ALAT-/ASAT-arvojen nousu > 3 × viitevälin yläraja todettiin 2,1 %:lla tosilitsumabia 8 mg/kg saaneista ja 4,9 %:lla MTX:a saaneista potilaista sekä 6,5 %:lla potilaista, jotka saivat tosilitsumabia 8 mg/kg yhdessä DMARDien kanssa, sekä 1,5 %:lla lumevalmistetta ja DMARDeja saaneista potilaista.
Kohonneet arvot yleistyivät, kun tosilitsumabimonoterapiahoitoon lisättiin jokin mahdollisesti maksatoksinen lääkevalmiste (esim. MTX). ALAT-/ASAT-arvojen nousu > 5 × viitevälin yläraja todettiin 0,7 %:lla pelkkää tosilitsumabia saaneista potilaista ja 1,4 %:lla potilaista, jotka saivat tosilitsumabia yhdessä DMARDien kanssa. Suurimmalla osalla näistä potilaista tosilitsumabihoito keskeytettiin pysyvästi. Kaksoissokkoutetun kontrollivaiheen tavanomaisessa laboratorioseurannassa 6,2 %:lla tosilitsumabia 8 mg/kg ja DMARDeja saaneista potilaista todettiin konjugoitumattoman bilirubiinin pitoisuuksia, jotka olivat korkeammat kuin viitevälin yläraja. Konjugoitumattoman bilirubiinin pitoisuus nousi tasolle > 1–2 × viitevälin yläraja yhteensä 5,8 %:lla potilaista ja 0,4 %:lla nousu oli > 2 × viitevälin yläraja.
Kaksoissokkoutetussa kontrollivaiheessa ja pitkäaikaisessa altistumisessa havaittiin ALAT-/ASAT-arvojen nousseen vastaavalla tavalla ja ilmaantuvuudella kuin kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa.
Veren rasva-arvot
Kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa on raportoitu yleisesti veren rasva-arvojen, kuten kokonaiskolesterolin, triglyseridien, LDL- ja/tai HDL-kolesterolin, nousua. Tavanomaisessa laboratorioseurannassa noin 24 %:lla potilaista, jotka saivat tosilitsumabia kliinisissä tutkimuksissa, todettiin pysyvästi kohonneita kokonaiskolesteroliarvoja (≥ 6,2 mmol/l). 15 %:lla potilaista havaittiin pysyvästi kohonneita LDL-kolesteroliarvoja (≥ 4,1 mmol/l). Kohonneet veren rasva-arvot saatiin hallintaan kolesterolia alentavilla lääkkeillä.
Kaksoissokkoutetussa kontrollivaiheessa ja pitkäaikaisessa altistumisessa havaittiin veren rasva-arvojen kohonneen vastaavalla tavalla ja ilmaantuvuudella kuin kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa.
Ihoreaktiot
Myyntiluvan myöntämisen jälkeisessä seurannassa on harvoin raportoitu Stevens-Johnsonin oireyhtymää.
COVID-19-potilaat
Infektiot
Infektioiden / vakavien infektioon liittyvien tapahtumien esiintyvyys tutkimusten ML42528, WA42380 ja WA42511 turvallisuuden osalta arvioitavissa olleessa yhdistetyssä potilasjoukossa oli tasapainossa tosilitsumabia saaneiden (30,3 % / 18,6 %, n = 974) ja lumelääkettä saaneiden (32,1 % / 22,8 %, n = 483) COVID-19-potilaiden välillä.
Lähtötilanteessa systeemistä kortikosteroidihoitoa saaneen ryhmän havaittu turvallisuusprofiili oli yhdenmukainen koko potilasjoukon tosilitsumabia koskevan turvallisuusprofiilin kanssa, kuten taulukossa 2 esitetään. Tässä alaryhmässä tosilitsumabia laskimoon saaneista potilaista 27,8 %:lla esiintyi infektioita ja 18,1 %:lla vakavia infektioita ja kun taas lumehoitoa saaneista potilaista 30,5 %:lla esiintyi infektioita ja 22,9 %:lla vakavia infektioita.
Poikkeavat laboratorioarvot
Poikkeavien laboratorioarvojen ilmaantuvuus satunnaistetuissa, kaksoissokkoutetuissa lumekontrolloiduissa tutkimuksissa oli muutamaa poikkeusta lukuun ottamatta yleensä samankaltainen COVID-19-potilailla, jotka saivat yhden tai kaksi annosta tosilitsumabia laskimoon, verrattuna lumelääkettä saaneisiin potilaisiin. Trombosyyttien ja neutrofiilien väheneminen ja ALAT- ja ASAT-arvojen kohoaminen olivat yleisempiä tosilitsumabia laskimoon saaneilla kuin lumelääkettä saaneilla potilailla (ks. kohta Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Lasten polyartriittia ja yleisoireista lastenreumaa sairastavilla potilailla todetut haittavaikutukset olivat yleensä luonteeltaan samanlaisia kuin nivelreumapotilailla raportoidut, ks. kohta Haittavaikutukset.
Lasten polyartriittia sairastavien potilaiden valikoitujen haittavaikutusten kuvaus
Laskimoon annosteltavan tosilitsumabin turvallisuusprofiilia lasten polyartriitin hoidossa on tutkittu 188 potilaalla, iältään 2–17-vuotiaita. Potilasvuosien määrä oli yhteensä 184,4. Haittavaikutusten yleisyys lasten polyartriittia sairastavilla potilailla on esitetty taulukossa 3. Lasten polyartriittia sairastavilla potilailla todetut haittavaikutukset olivat luonteeltaan samanlaisia kuin nivelreumapotilailla ja yleisoireista lastenreumaa sairastavilla potilailla. Aikuisiin nivelreumapotilaisiin verrattuna lasten polyartriittia sairastavilla potilailla raportoitiin yleisemmin nasofaryngiittia, päänsärkyä, pahoinvointia ja neutrofiiliarvojen laskua. Kolesteroliarvojen nousua raportoitiin harvemmin lasten polyartriittia sairastavilla potilailla kuin aikuisilla nivelreumapotilailla.
Infektiot
Infektioiden esiintyvyys kaikilla tosilitsumabia saaneilla potilailla oli 163,7 tapausta 100 potilasvuotta kohti. Yleisimmät havaitut tapahtumat olivat nasofaryngiitti ja ylempien hengitysteiden infektiot. Vakavien infektioiden esiintyvyys oli suurempi < 30 kg:n painoisilla 10 mg/kg tosilitsumabiannoksia saaneilla potilailla (12,2 tapausta 100 potilasvuotta kohden) verrattuna ≥ 30 kg:n painoisiin 8 mg/kg tosilitsumabiannoksia saaneisiin potilaisiin (4,0 tapausta 100 potilasvuotta kohden). Hoidon keskeyttämiseen johtaneiden infektioiden ilmaantuvuus oli myös suurempi < 30 kg:n painoisilla 10 mg/kg tosilitsumabiannoksia saaneilla potilailla (21,4 %) verrattuna ≥ 30 kg:n painoisiin 8 mg/kg tosilitsumabiannoksia saaneisiin potilaisiin (7,6 %).
Infuusioon liittyvät reaktiot
Infuusioon liittyviksi reaktioiksi määritellään lasten polyartriittia sairastavilla potilailla kaikki haittatapahtumat, jotka ilmaantuvat infuusion aikana tai 24 tunnin kuluessa sen antamisesta. Kaikista tosilitsumabia saaneista potilaista 11 potilaalla (5,9 %) esiintyi infuusioon liittynyt reaktio infuusion aikana ja 38 potilaalla (20,2 %) infuusioon liittynyt reaktio ilmaantui 24 tunnin kuluessa infuusion antamisesta. Yleisimpiä infuusion aikana esiintyneitä tapahtumia olivat päänsärky, pahoinvointi ja hypotensio. Yleisimpiä tapahtumia 24 tunnin kuluessa infuusion antamisesta olivat huimaus ja hypotensio. Infuusion annon aikana tai 24 tunnin kuluessa infuusion antamisesta esiintyneet haittavaikutukset olivat yleensä luonteeltaan samankaltaisia kuin nivelreumaa tai yleisoireista lastenreumaa sairastavilla potilailla, ks. kohta Haittavaikutukset.
Tosilitsumabiin liittyviä ja hoidon keskeyttämistä vaativia kliinisesti merkittäviä yliherkkyysreaktioita ei esiintynyt.
Immunogeenisuus
Yhdelle < 30 kg:n painoisten 10 mg/kg tosilitsumabiannoksia saaneiden ryhmän potilaalle kehittyi positiivisia anti-tosilitsumabivasta-aineita, mihin ei liittynyt yliherkkyysreaktion kehittymistä. Potilas keskeytti tämän jälkeen osallistumisensa tutkimukseen.
Neutrofiilit
Kaikkien tosilitsumabia saaneiden potilaiden tavanomaisessa laboratorioseurannassa 3,7 %:lla potilaista esiintyi neutrofiiliarvojen laskua tason 1 × 109/l alapuolelle.
Trombosyytit
Kaikkien tosilitsumabia saaneiden potilaiden tavanomaisissa laboratorioseurannoissa 1 %:lla potilaista esiintyi trombosyyttiarvon laskua tasoon ≤ 50 × 103/μl, mutta tähän ei liittynyt verenvuototapahtumia.
Maksan aminotransferaasiarvojen nousu
Kaikkien tosilitsumabia saaneiden potilaiden tavanomaisissa laboratorioseurannoissa todettiin maksan aminotransferaasiarvojen nousua tasolle ≥ 3 × viitevälin yläraja 3,7 %:lla (ALAT) ja < 1 %:lla (ASAT) potilaista.
Veren rasva-arvot
Laskimoon annosteltavaa tosilitsumabia koskeneen tutkimuksen WA19977 tavanomaisissa laboratorioseurannoissa veren LDL-kolesterolipitoisuus oli jossakin vaiheessa tutkimushoidon aikana kohonnut arvoon ≥ 130 mg/dl 3,4 %:lla potilaista ja kokonaiskolesterolipitoisuus oli kohonnut arvoon ≥ 200 mg/dl 10,4 %:lla potilaista.
Yleisoireista lastenreumaa sairastavien potilaiden valikoitujen haittavaikutusten kuvaus
Laskimoon annosteltavan tosilitsumabin turvallisuusprofiilia yleisoireisessa lastenreumassa on tutkittu 112 potilaalla, iältään 2–17-vuotiaita. Tutkimuksen 12 viikkoa kestäneessä kaksoissokkoutetussa, kontrollivaiheessa 75 potilasta sai tosilitsumabihoitoa (8 mg/kg tai 12 mg/kg painon mukaan). 12 viikon jälkeen tai vaihdettuaan lumelääkkeestä tosilitsumabiin taudin pahenemisen vuoksi potilaat hoidettiin avoimessa jatkotutkimuksessa.
Yleisoireista lastenreumaa sairastavilla potilailla todetut haittavaikutukset olivat yleensä luonteeltaan samanlaisia kuin nivelreumapotilailla. Haittavaikutusten yleisyys yleisoireista lastenreumaa sairastavilla potilailla on esitetty taulukossa 3. Verrattuna aikuisiin nivelreumapotilaisiin yleisoireista lastenreumaa sairastavilla potilailla raportoitiin yleisemmin nasofaryngiittia, neutrofiiliarvojen laskua, maksan aminotransferaasiarvojen nousua ja ripulia. Kolesteroliarvojen nousua raportoitiin harvemmin yleisoireista lastenreumaa sairastavilla potilailla kuin aikuisilla nivelreumapotilailla.
Infektiot
12 viikkoa kestäneessä kontrollivaiheessa infektioiden kokonaisesiintyvyys laskimoon annosteltavaa tosilitsumabia saaneessa ryhmässä oli 344,7 tapausta 100 potilasvuotta kohti. Lumelääkeryhmässä esiintyvyys oli 287,0 tapausta 100 potilasvuotta kohti. Tutkimuksen avoimessa jatkovaiheessa infektioiden kokonaisesiintyvyys on pysynyt samalla tasolla, 306,6 tapausta 100 potilasvuotta kohti.
12 viikkoa kestäneessä kontrollivaiheessa vakavien infektioiden esiintyvyys laskimoon annosteltavaa tosilitsumabia saaneessa ryhmässä oli 11,5 tapausta 100 potilasvuotta kohti. Avoimessa jatkotutkimuksessa vakavien infektioiden kokonaismäärä oli vuoden kuluttua samalla tasolla eli 11,3 infektiota 100 potilasvuotta kohti. Raportoidut vakavat infektiot olivat samankaltaisia kuin nivelreumassa raportoidut, lisäksi raportoitiin vesirokkoa ja välikorvantulehdusta.
Infuusioon liittyvät reaktiot
Infuusioon liittyviksi reaktioiksi määritellään kaikki haittatapahtumat, jotka ilmaantuvat infuusion aikana tai 24 tunnin kuluessa sen antamisesta. 12 viikkoa kestäneessä kontrollivaiheessa 4 % tosilitsumabiryhmän potilaista koki reaktion infuusion aikana. Yhtä tapahtumaa (angioedeema) pidettiin vakavana ja henkeä uhkaavana, ja potilas keskeytti tutkimushoidon.
12 viikkoa kestäneessä kontrollivaiheessa 16 % tosilitsumabiryhmän potilaista ja 5,4 % lumelääkeryhmän potilaista sai infuusioon liittyvän reaktion 24 tunnin kuluessa infuusiosta. Tosilitsumabiryhmässä infuusioreaktio-oireisiin sisältyi mm. ihottumaa, nokkosihottumaa, ripulia, epigastrista kipua, nivelkipua ja päänsärkyä. Yhtä näistä oireista, nokkosihottumaa, pidettiin vakavana. Oireet eivät rajoittuneet vain edellä mainittuihin.
Tutkimuksen kontrollivaiheessa ja sen avoimessa seurantavaiheessa raportoitiin yhdellä tosilitsumabia saaneella potilaalla 112:sta (< 1 %) tosilitsumabiin liittyviä ja hoidon keskeyttämistä vaativia kliinisesti merkittäviä allergisia reaktioita.
Immunogeenisuus
Anti-tosilitsumabi-vasta-aineet määritettiin kaikilta 112 potilaalta ennen hoidon aloittamista. Kahdelle potilaalle kehittyi positiivisia anti-tosilitsumabivasta-aineita. Toinen näistä potilaista sai yliherkkyysreaktion, joka johti hoidon keskeytykseen. Anti-tosilitsumabi-vasta-ainemuodostuksen ilmaantuvuutta on saatettu aliarvioida, koska tosilitsumabi vaikuttaa määritystulokseen ja koska tosilitsumabipitoisuuksien on havaittu olevan lapsilla korkeammat kuin aikuisilla.
Neutrofiilit
Kontrollivaiheen 12 viikon aikana tehdyssä tavanomaisessa laboratorioseurannassa 7 %:lla tosilitsumabiryhmän potilaista esiintyi neutrofiiliarvojen laskua tason 1 × 109/l alapuolelle. Lumelääkeryhmässä ei havaittu neutrofiiliarvojen alenemista.
Avoimessa jatkotutkimuksessa neutrofiiliarvot laskivat tason 1 × 109/l alapuolelle 15 %:lla tosilitsumabiryhmän potilaista.
Trombosyytit
Kontrollivaiheen 12 viikon aikana tehdyssä tavanomaisessa laboratorioseurannassa 3 %:lla lumelääkeryhmän potilaista ja 1 %:lla tosilitsumabiryhmän potilaista esiintyi trombosyyttiarvon laskua tason 100 × 103/μl alapuolelle.
Avoimessa jatkotutkimuksessa trombosyyttiarvot laskivat tason 100 × 103/μl alapuolelle 3 %:lla tosilitsumabia saaneista potilaista. Trombosyyttiarvon laskuun ei liittynyt verenvuototapahtumia.
Maksan aminotransferaasiarvojen nousu
Kontrollivaiheen 12 viikon aikana tehdyssä tavanomaisessa laboratorioseurannassa maksan aminotransferaasiarvojen nousua tasolle ≥ 3 × viitevälin yläraja todettiin 5 %:lla (ALAT) ja 3 %:lla (ASAT) tosilitsumabia saaneista potilaista. Lumelääkeryhmässä maksan aminotransferaasiarvot eivät nousseet (0 %).
Avoimessa jatkotutkimuksessa aminotransferaasiarvojen nousua tasolle ≥ 3 × viitevälin yläraja esiintyi 12 %:lla (ALAT) ja 4 %:lla (ASAT) tosilitsumabia saaneista potilaista.
Immunoglobuliini G
IgG-pitoisuudet laskevat hoidon aikana. Laskua viitevälin alapuolelle havaittiin 15 potilaalla jossakin vaiheessa tutkimuksen aikana.
Veren rasva-arvot
Kontrollivaiheen 12 viikon aikana (tutkimus WA18221) tehdyssä tavanomaisessa laboratorioseurannassa veren LDL-kolesterolipitoisuus oli jossakin vaiheessa tutkimushoidon aikana kohonnut arvoon ≥ 130 mg/dl 13,4 %:lla potilaista ja kokonaiskolesterolipitoisuus oli kohonnut arvoon ≥ 200 mg/dl 33,3 %:lla potilaista.
Avoimessa jatkotutkimuksessa (tutkimus WA18221) veren LDL-kolesterolipitoisuus oli jossakin vaiheessa tutkimushoidon aikana kohonnut arvoon ≥ 130 mg/dl 13,2 %:lla potilaista ja kokonaiskolesterolipitoisuus oli kohonnut arvoon ≥ 200 mg/dl 27,7 %:lla potilaista.
Potilaat, joilla on sytokiinioireyhtymä
Tosilitsumabin turvallisuutta sytokiinioireyhtymän hoitoon on selvitetty kliinisten tutkimustietojen retrospektiivisessä analyysissä. Kyseisissä kliinisissä tutkimuksissa 51 potilasta sai kimeeristä antigeenireseptoria ilmentävien T-solujen (CAR-T-solujen) aikaansaaman vaikea-asteisen tai hengenvaarallisen sytokiinioireyhtymän hoitoon tosilitsumabiannoksia 8 mg/kg (12 mg/kg, jos potilas painoi alle 30 kg) laskimoon, minkä lisäksi joko annettiin tai ei annettu suuria kortikosteroidiannoksia. Annettujen tosilitsumabiannosten lukumäärän mediaani oli 1 annos (vaihteluväli 1–4 annosta).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tietoja tosilitsumabin yliannostuksesta on rajoitetusti. Yksi tahaton yliannostustapaus on raportoitu. Siinä multippelia myeloomaa sairastava potilas sai kerta-annoksena 40 mg/kg. Haittavaikutuksia ei havaittu.
Vakavia haittavaikutuksia ei havaittu, kun terveille koehenkilöille annettiin kerta-annoksena enintään 28 mg/kg, mutta annostusta rajoittavaa neutropeniaa todettiin.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressiiviset lääkeaineet, interleukiinin estäjät, ATC-koodi: L04AC07.
Vaikutusmekanismi
Tosilitsumabi sitoutuu spesifisesti sekä liukoisiin että kalvoon sitoutuneisiin IL‑6-reseptoreihin (sIL‑6 ja mIL‑6R). Tosilitsumabin on osoitettu estävän sIL‑6R- ja mIL‑6R-reseptorien kautta tapahtuvaa signaalinvälitystä. IL-6 on pleiotrooppinen tulehdusta vahvistava sytokiini, jota tuottavat useat eri solutyypit, kuten T- ja B-solut, monosyytit ja fibroblastit. IL-6 osallistuu erilaisiin fysiologisiin tapahtumaketjuihin, joita ovat esimerkiksi T-solun aktivoituminen, immunoglobuliinierityksen käynnistyminen, akuutin vaiheen proteiinisynteesin käynnistyminen maksassa ja hematopoieesin stimuloituminen. IL-6 on yhdistetty erilaisten sairauksien, kuten tulehdussairauksien, osteoporoosin ja kasvainten, patogeneesiin.
Farmakodynaamiset vaikutukset
Nivelreumapotilailla tehdyissä kliinisissä tosilitsumabitutkimuksissa havaittiin CRP:n (C-reaktiivisen proteiinin), laskon (La), seerumin amyloidi A:n (SAA) ja fibrinogeenin nopea lasku. Akuutin vaiheen proteiineihin kohdistuvan vaikutuksen mukaisesti tosilitsumabihoitoon liittyi myös trombosyyttiarvon lasku normaalialueen sisällä. Hemoglobiiniarvon nousua havaittiin, sillä tosilitsumabi heikentää IL-6:n vaikutuksia hepsidiinin tuotantoon, mikä lisää raudan saatavuutta. Hoitoa saaneilla potilailla CRP-arvon lasku normaalialueelle havaittiin jo toisella hoitoviikolla, ja lasku säilyi koko hoidon ajan.
Kun tosilitsumabia annettiin annoksilla 2–28 mg/kg terveille vapaaehtoisille koehenkilöille, heidän neutrofiiliarvonsa laskivat alimmalle tasolleen 3–5 päivän kuluttua infuusiosta. Sen jälkeen neutrofiiliarvot palautuivat hoitoa edeltävälle tasolle annosriippuvaisesti. Nivelreumapotilailla havaittiin neutrofiiliarvojen muuttuvan vastaavalla tavalla tosilitsumabin annon jälkeen (ks. kohta Haittavaikutukset).
COVID-19-potilailla, joille annettiin yksi 8 mg/kg tosilitsumabiannos laskimoon, CRP-arvon havaittiin laskeneen normaaleihin viitearvoihin jo 7. päivänä.
Nivelreumaa sairastavat potilaat
Kliininen teho ja turvallisuus
Tosilitsumabin tehoa arvioitiin viidessä satunnaistetussa kaksoissokkoutetussa monikeskustutkimuksessa nivelreuman oireiden lievittymisen perusteella. Tutkimuksiin I–V otettiin ≥ 18-vuotiaita potilaita, joilla oli ACR-kriteerien (American College of Rheumatology) mukaan diagnosoitu aktiivinen nivelreuma ja lähtötilanteessa vähintään kahdeksan aristavaa ja kuusi turvonnutta niveltä.
Tutkimuksessa I tosilitsumabi annettiin laskimoon neljän viikon välein monoterapiana. Tutkimuksissa II, III ja V tosilitsumabi annettiin laskimoon neljän viikon välein yhdessä MTX:n kanssa. Vertailuna käytettiin lumevalmisteen ja MTX:n yhdistelmää. Tutkimuksessa IV tosilitsumabi annettiin laskimoon neljän viikon välein yhdessä muiden DMARDien kanssa. Vertailuna käytettiin lumevalmisteen ja muiden DMARDien yhdistelmää. Kaikissa viidessä tutkimuksessa ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat ACR 20‑vasteen viikkoon 24 mennessä.
Tutkimuksessa I arvioitiin 673 potilasta, jotka eivät olleet saaneet MTX:a satunnaistamista edeltäneiden kuuden kuukauden aikana ja joiden aikaisempaa MTX-hoitoa ei oltu keskeytetty kliinisesti merkittävien toksisten vaikutusten tai hoidon tehottomuuden vuoksi. Valtaosa (67 %) potilaista ei ollut aikaisemmin saanut MTX:a. Tosilitsumabia annettiin 8 mg/kg neljän viikon välein monoterapiana. Vertailuryhmä sai MTX:a viikon välein (viikkoannos titrattiin 7,5 mg:sta enintään 20 mg:aan kahdeksan viikon jakson aikana).
Tutkimus II kesti kaksi vuotta, ja siihen kuului suunniteltu analyysi viikkojen 24, 52 ja 104 kohdalla. Tutkimuksessa arvioitiin 1 196 potilasta, joille MTX ei ollut tuottanut riittävää hoitovastetta. Tosilitsumabiannos 4 mg/kg tai 8 mg/kg tai lumevalmiste annettiin sokkoutetusti neljän viikon välein 52 viikon ajan yhdistettynä vakaaseen MTX-annokseen (10–25 mg viikossa). Avoimessa vaiheessa, viikon 52 jälkeen, kaikilla potilailla oli mahdollisuus saada tosilitsumabihoitoa 8 mg/kg. Niistä potilaista, jotka olivat mukana tutkimuksen loppuun asti ja jotka oli satunnaistettu saamaan lumevalmistetta ja MTX:a, 86 % sai avoimessa vaiheessa tosilitsumabia 8 mg/kg tutkimuksen toisena vuonna. Ensisijainen päätetapahtuma 24 viikon kohdalla tehdyssä analyysissä oli ACR 20‑vasteen kriteerit täyttävien potilaiden suhteellinen osuus. Viikoilla 52 ja 104 muut ensisijaiset päätetapahtumat olivat nivelvaurioiden estyminen ja fyysisen toimintakyvyn paraneminen.
Tutkimuksessa III arvioitiin 623 potilasta, joille MTX ei ollut tuonut riittävää hoitovastetta. Tosilitsumabiannos 4 mg/kg tai 8 mg/kg tai lumevalmiste annettiin neljän viikon välein yhdistettynä vakaaseen MTX-annokseen (10–25 mg viikossa).
Tutkimuksessa IV arvioitiin 1 220 potilasta, jotka eivät olleet saavuttaneet riittävää hoitovastetta nykyisellä reumalääkityksellään, johon kuului vähintään yksi DMARD. Tosilitsumabiannos 8 mg/kg tai lumevalmiste annettiin neljän viikon välein yhdistettynä DMARDien vakaaseen annokseen.
Tutkimuksessa V arvioitiin 499 potilasta, joille yksi tai useampi TNF-estäjä ei ollut tuonut riittävää hoitovastetta tai jotka eivät sietäneet näitä lääkkeitä. Hoito TNF-estäjillä lopetettiin ennen satunnaistamista. Tosilitsumabiannos 4 mg/kg tai 8 mg/kg tai lumevalmiste annettiin neljän viikon välein yhdistettynä vakaaseen MTX-annokseen (10–25 mg viikossa).
Kliininen vaste
Kaikissa tutkimuksissa kuuden kuukauden ACR 20-, ACR 50- ja ACR 70-vasteet olivat tilastollisesti merkitsevästi suuremmat tosilitsumabia 8 mg/kg saaneiden potilaiden ryhmissä kuin vertailuryhmissä (taulukko 4). Tutkimuksessa I tosilitsumabi 8 mg/kg todettiin tehokkaammaksi kuin vaikuttava vertailuaine MTX.
Hoitoteho oli sama riippumatta potilaiden reumatekijästatuksesta, iästä, sukupuolesta, etnisestä taustasta, aikaisempien hoitojen lukumäärästä tai taudin tilasta. Vaikutus alkoi nopeasti (jo toisella hoitoviikolla), ja vaste suureni jatkuvasti hoidon keston myötä. Avoimissa jatkotutkimuksissa I–V vasteen on todettu kestävän yli 3 vuotta.
Kaikissa tutkimuksissa tosilitsumabiannosta 8 mg/kg saaneilla potilailla todettiin merkitsevää paranemista ACR-vasteen kaikissa yksittäisissä osatekijöissä (aristavien ja turvonneiden nivelten lukumäärä, potilaan ja lääkärin yleisarvio, toimintakykyä mittaava pistearvo, kivun arviointi ja CRP-arvo) verrattuna potilaisiin, jotka saivat lumevalmistetta yhdessä MTX:in tai muiden DMARDien kanssa.
Tutkimuksissa I–V potilaiden keskimääräinen DAS28 (Disease Activity Score) -indeksi hoidon alussa oli 6,5–6,8. DAS28-indeksin merkitsevää laskua (keskimääräinen parannus) havaittiin tosilitsumabilla hoidetuilla potilailla verrattuna kontrolliryhmän potilaisiin. Tosilitsumabilla hoidetuilla potilailla DAS28 laski 3,1–3,4 yksikköä ja kontrolliryhmässä 1,3–2,1 yksikköä. Tosilitsumabihoitoa saaneista potilaista 28–34 % saavutti DAS28-remission viikolla 24 (DAS28 < 2,6). Osuus oli huomattavasti suurempi kuin kontrolliryhmässä, jossa vastaava luku oli 1–12 %. Tutkimuksessa II, 65 % potilaista saavutti DAS28 < 2,6 viikolla 104. Vastaava luku viikolla 52 oli 48 % ja viikolla 24 33 %.
Tutkimusten II, III ja IV yhdistetyssä analyysissä ACR 20-, ACR 50- ja ACR 70-vasteiden saavuttaneiden potilaiden osuus oli merkitsevästi suurempi ryhmässä, joka sai tosilitsumabia 8 mg/kg yhdessä DMARDien kanssa (59 %, 37 % ja 18 %), kuin ryhmässä, joka sai tosilitsumabia 4 mg/kg yhdessä DMARDien kanssa (50 %, 27 % ja 11 %; p < 0,03). Vastaavasti DAS28-remission (DAS28 < 2,6) saavuttaneiden potilaiden osuus oli merkitsevästi suurempi tosilitsumabia 8 mg/kg ja DMARDeja saaneessa ryhmässä (31 %) kuin tosilitsumabia 4 mg/kg ja DMARDeja saaneessa ryhmässä (16 %, p < 0,0001).
Taulukko 4. ACR-vasteet kontrolloiduissa tutkimuksissa, joissa vertailuaineina olivat lumevalmiste / MTX / DMARDit, (% potilaista)
Tutkimus I AMBITION | Tutkimus II LITHE | Tutkimus III OPTION | Tutkimus IV TOWARD | Tutkimus V RADIATE | ||||||
| Vko | TCZ 8 mg/kg | MTX | TCZ 8 mg/kg + MTX | Lume + MTX | TCZ 8 mg/kg + MTX | Lume + MTX | TCZ 8 mg/kg + DMARD | Lume+ DMARD | TCZ 8 mg/kg + MTX | Lume + MTX |
n = 286 | n = 284 | n = 398 | n = 393 | n = 205 | n = 204 | n = 803 | n = 413 | n = 170 | n = 158 | |
| ACR 20 | ||||||||||
| 24 | 70 %*** | 52 % | 56 %*** | 27 % | 59 %*** | 26 % | 61 %*** | 24 % | 50 %*** | 10 % |
| 52 | 56 %*** | 25 % | ||||||||
| ACR 50 | ||||||||||
| 24 | 44 %** | 33 % | 32 %*** | 10 % | 44 %*** | 11 % | 38 %*** | 9 % | 29 %*** | 4 % |
| 52 | 36 %*** | 10 % | ||||||||
| ACR 70 | ||||||||||
| 24 | 28 %** | 15 % | 13 %*** | 2 % | 22 %*** | 2 % | 21 %*** | 3 % | 12 %** | 1 % |
| 52 | 20 %*** | 4 % | ||||||||
ACR - American College of Rheumatology (ACR) ‑kriteerit
TCZ - tosilitsumabi
MTX - metotreksaatti
DMARD - tautiprosessia hidastava reumalääke (disease modifying anti-rheumatic drug)
** - p < 0,01, tosilitsumabi vs. lumevalmiste + MTX / DMARD
*** - p < 0,0001, tosilitsumabi vs. lumevalmiste + MTX / DMARD
Merkittävä kliininen vaste
Tosilitsumabia yhdessä MTX:n kanssa saaneista potilaista 14 % saavutti merkittävän kliinisen vasteen, kun hoitoa oli annettu kaksi vuotta (ACR 70-vaste säilyi 24 viikkoa tai pitempään).
Radiologinen vaste
Tutkimukseen II osallistui potilaita, joiden aikaisempi hoito MTX:lla ei tuonut riittävää hoitovastetta. Rakenteellisten nivelvaurioiden estymistä arvioitiin radiologisesti ja tulos ilmaistiin modifioidun Sharp-indeksin ja sen osatekijöiden, eroosioasteen ja nivelraon kaventumisen muutoksena. Rakenteellisen nivelvaurion estyminen näkyi merkitsevästi hitaampana radiologisena etenemisenä tosilitsumabihoitoa saaneilla potilailla kuin vertailuryhmän potilailla (taulukko 5).
Avoimessa jatkotutkimuksessa (tutkimus II) niveltuhon etenemistä estävä vaikutus säilyi hoidon toisena vuonna tosilitsumabia yhdessä MTX:n kanssa saaneilla potilailla. Sharp-Genant-kokonaispistearvon keskimääräinen muutos lähtötilanteesta viikolla 104 oli huomattavasti pienempi potilailla, jotka oli satunnaistettu saamaan tosilitsumabia 8 mg/kg yhdessä MTX:n kanssa (p < 0,0001) kuin niillä potilailla, jotka oli satunnaistettu saamaan lumevalmistetta yhdessä MTX:n kanssa.
Taulukko 5. Radiologiset muutokset (keskiarvo) 52 viikon aikana tutkimuksessa II
Lume + MTX (+TCZ viikosta 24 alkaen) n = 393 | TCZ 8 mg/kg + MTX n = 398 | |
| Sharp-Genant-kokonaispistearvo | 1,13 | 0,29* |
| Eroosioindeksi | 0,71 | 0,17* |
| Nivelraon kaventuminen | 0,42 | 0,12** |
MTX - metotreksaatti
TCZ - tosilitsumabi
* - p ≤ 0,0001, kun tosilitsumabia verrattiin lumevalmisteen + MTX:n yhdistelmään.
** - p ≤ 0,005, kun tosilitsumabia verrattiin lumevalmisteen + MTX:n yhdistelmään.
Vuoden kestäneen hoidon jälkeen tosilitsumabia yhdessä MTX:n kanssa saaneista potilaista 85 %:lla (n = 348) ei havaittu niveltuhon etenemistä Sharp-Genant-kokonaispistearvon muutoksena mitattuna (muutos 0 tai vähemmän). Vastaava tulos lumevalmistetta yhdessä MTX:n kanssa saaneilla potilailla oli 67 % (n = 290) (p ≤ 0,001). Tulos säilyi samanlaisena kaksi vuotta kestäneen hoidon jälkeen (83 %; n = 353). Viikkojen 52 ja 104 välillä 93 %:lla potilaista (n = 271) ei havaittu niveltuhon etenemistä.
Terveydentilaa ja elämänlaatua kuvaavat tulokset
Raportit tosilitsumabihoitoa saaneilta potilailta osoittivat paranemista kaikissa potilaan arviointiin perustuvissa mittareissa (HAQ-DI = Health Assessment Questionnaire Disability Index, Short Form ‑36- ja FACIT = Functional Assessment of Chronic Illness Therapy -kyselylomakkeet). Fyysistä toimintakykyä mittaava HAQ-DI-tulos parani tilastollisesti merkitsevästi tosilitsumabihoitoa saaneiden potilaiden ryhmässä DMARDeja saaneisiin potilaisiin verrattuna. Tutkimuksen II avoimessa vaiheessa havaittu parannus fyysisessä toimintakyvyssä säilyi jopa kahden vuoden ajan. Viikolla 52 keskimääräinen muutos HAQ-DI:ssä oli -0,58 niillä potilailla, jotka saivat tosilitsumabia 8 mg/kg yhdessä MTX:n kanssa. Lumevalmistetta yhdessä MTX:n kanssa saaneilla potilailla vastaava tulos oli 0,39. Keskimääräinen muutos HAQ-DI:ssä säilyi viikon 104 loppuun asti tosilitsumabia 8 mg/kg yhdessä MTX:n kanssa saaneilla potilailla (-0,61).
Hemoglobiiniarvot
Tosilitsumabihoitoa saaneiden potilaiden hemoglobiiniarvot olivat parantuneet tilastollisesti merkitsevästi viikolla 24 verrattuna DMARDeja saaneiden potilaiden arvoihin (p < 0,0001). Hemoglobiiniarvojen keskiarvot nousivat viikkoon kaksi mennessä ja pysyivät normaalialueella viikkoon 24 asti.
Tosilitsumabin vertailu adalimumabiin monoterapiassa
Tutkimuksessa VI (WA19924), joka oli 24 viikon pituinen kaksoissokkoutettu tosilitsumabimonoterapiaa ja adalimumabimonoterapiaa vertaileva tutkimus, oli mukana 326 nivelreumapotilasta, jotka eivät sietäneet MTX-hoitoa tai joille MTX-hoidon jatkamista ei katsottu tarkoituksenmukaiseksi (mukaan lukien potilaat, jotka eivät saaneet riittävää vastetta MTX-hoitoon). Tosilitsumabiryhmän potilaat saivat tosilitsumabia (8 mg/kg) infuusiona laskimoon neljän viikon välein ja lumevalmistetta ihon alle kahden viikon välein. Adalimumabiryhmän potilaat saivat adalimumabia (40 mg) injektiona ihon alle kahden viikon välein sekä lumevalmistetta infuusiona laskimoon neljän viikon välein.
Tosilitsumabihoidon osoitettiin olevan adalimumabia tilastollisesti merkitsevästi tehokkaampi tautiaktiivisuuden hallinnassa sekä ensisijaisen päätetapahtuman osalta (DAS28-indeksin muutos lähtötilanteesta viikolle 24) että kaikkien toissijaisten päätetapahtumien osalta (taulukko 6).
Taulukko 6. Tutkimuksen VI (WA19924) tehon tulokset
ADA + lume (i.v.) n = 162 | TCZ + lume (s.c.) n = 163 | p-arvo(a) | |
| Ensisijainen päätetapahtuma: keskimuutos lähtötilanteesta viikkoon 24 | |||
| DAS28 (korjattu keskiarvo) | -1,8 | -3,3 | |
| Korjatun keskiarvon ero (95 %:n luottamusväli) | -1,5 (-1,8, -1,1) | < 0,0001 | |
| Toissijaiset päätetapahtumat: vasteen saaneiden prosenttiosuus viikolla 24 (b) | |||
| DAS28 < 2,6, n (%) | 17 (10,5) | 65 (39,9) | < 0,0001 |
| DAS28 ≤ 3,2, n (%) | 32 (19,8) | 84 (51,5) | < 0,0001 |
| ACR 20-vaste, n (%) | 80 (49,4) | 106 (65,0) | 0,0038 |
| ACR 50-vaste, n (%) | 45 (27,8) | 77 (47,2) | 0,0002 |
| ACR 70-vaste, n (%) | 29 (17,9) | 53 (32,5) | 0,0023 |
a p-arvo on korjattu potilaiden maantieteellisen sijainnin ja nivelreuman kestoajan suhteen kaikkien päätetapahtumien osalta sekä lisäksi kaikkien jatkuvien päätetapahtumien lähtötilanteen arvon osalta.b Hoitoon vastaamattomien potilaiden puuttuvien tietojen paikkaus. Monivertailun hallintaan käytetty Bonferroni-Holmin menetelmää
i.v. = laskimoon
s.c. = ihon alle
TCZ = tosilitsumabi
ADA = adalimumabi
Tosilitsumabin ja adalimumabin kliiniset haittatapahtumaprofiilit olivat yleisesti ottaen samankaltaiset. Niiden potilaiden osuus, joilla esiintyi vakavia haittatapahtumia, oli samankaltainen kummassakin hoitoryhmässä (tosilitsumabi 11,7 % vs. adalimumabi 9,9 %). Tosilitsumabihaarassa havaitut haittavaikutukset olivat luonteeltaan yhdenmukaisia tosilitsumabin tunnetun turvallisuusprofiilin kanssa ja ne ilmenivät vastaavalla frekvenssillä kuin taulukossa 1 luetellut haittavaikutukset. Infektioita ja infestaatioita ilmeni enemmän tosilitsumabihaarassa (48 % vs. 42 %), vakavien infektioiden ilmaantuvuus oli samanlainen (3,1 %). Kumpikin tutkimushoito aiheutti samantyyppisiä turvallisuuteen liittyvien laboratorioarvojen muutoksia (neutrofiili- ja trombosyyttimäärän vähenemistä, ALAT- ja ASAT- sekä lipidiarvojen suurenemista), mutta muutosten suuruus ja huomattavien poikkeavuuksien esiintymistiheys oli tosilitsumabiryhmässä suurempi kuin adalimumabiryhmässä. Neljällä (2,5 %) potilaalla tosilitsumabiryhmässä ja kahdella (1,2 %) potilaalla adalimumabiryhmässä esiintyi CTC-luokituksen mukaista gradus 3 tai 4 neutrofiilimäärän vähenemistä. Yhdellätoista (6,8 %) potilaalla tosilitsumabiryhmässä ja viidellä (3,1 %) potilaalla adalimumabiryhmässä esiintyi CTC-luokituksen mukaista gradus 2 tai vaikeampiasteista ALAT-arvon suurenemista. LDL-arvo suureni lähtötilanteesta tosilitsumabiryhmässä keskimäärin 0,64 mmol/l (25 mg/dl) ja adalimumabiryhmässä 0,19 mmol/l (7 mg/dl). Turvallisuuden havaittiin olleen tosilitsumabiryhmässä yhdenmukainen tosilitsumabin tunnetun turvallisuusprofiilin kanssa eikä uusia tai odottamattomia haittavaikutuksia havaittu (ks. taulukko 1).
Tuoretta nivelreumaa sairastavat potilaat, jotka eivät ole aikaisemmin saaneet MTX-hoitoa
Tutkimus VII (WA19926) kesti kaksi vuotta, ja sen ensisijainen analyysi tehtiin viikon 52 kohdalla. Tutkimuksessa arvioitiin 1 162 aikuispotilasta, joilla oli keskivaikea tai vaikea aktiivinen tuore nivelreuma (keskimääräinen sairauden kesto ≤ 6 kuukautta) ja jotka eivät olleet aikaisemmin saaneet MTX:a. Noin 20 % potilaista oli saanut aikaisemmin jotakin muuta DMARD-lääkettä kuin MTX:a. Tutkimuksessa arvioitiin miten tehokkaasti laskimoon annettu tosilitsumabi 4 tai 8 mg/kg neljän viikon välein yhdessä MTX:n kanssa, laskimoon annettu tosilitsumabi 8 mg/kg monoterapiana ja MTX-monoterapia vähensivät nivelvaurioiden löydöksiä ja oireita sekä etenemisnopeutta 104 viikon aikana. Ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat DAS28-remission viikolla 24 (DAS28 < 2,6). Ensisijaisen päätetapahtuman saavuttaneiden potilaiden osuus oli merkitsevästi suurempi ryhmässä, joka sai tosilitsumabia 8 mg/kg yhdessä MTX:n kanssa tai tosilitsumabia monoterapiana, kuin pelkkää MTX:ia saaneiden ryhmässä. Lisäksi ryhmässä, joka sai tosilitsumabia 8 mg/kg yhdessä MTX:n kanssa, kaikkia tärkeimpiä toissijaisia päätetapahtumia koskevat tulokset olivat tilastollisesti merkitseviä. Ryhmässä, joka sai tosilitsumabia 8 mg/kg monoterapiana, vasteet olivat numeerisesti suurempia kaikkien toissijaisten päätetapahtumien osalta radiologiset päätetapahtumat mukaan lukien, kuin pelkkää MTX:ia saaneessa ryhmässä. Tässä tutkimuksessa ACR/EULAR-remissio (Boolen kriteereiden ja indeksin perusteella) analysoitiin myös ennalta määritettyinä eksploratiivisina päätetapahtumina, ja havaitut vasteet olivat suurempia tosilitsumabiryhmissä. Tutkimuksen VII tulokset esitetään taulukossa 7.
Taulukko 7. Tutkimuksen VII (WA19926) tehon tulokset tuoretta nivelreumaa sairastavilla potilailla, jotka eivät olleet aiemmin saaneet MTX:a
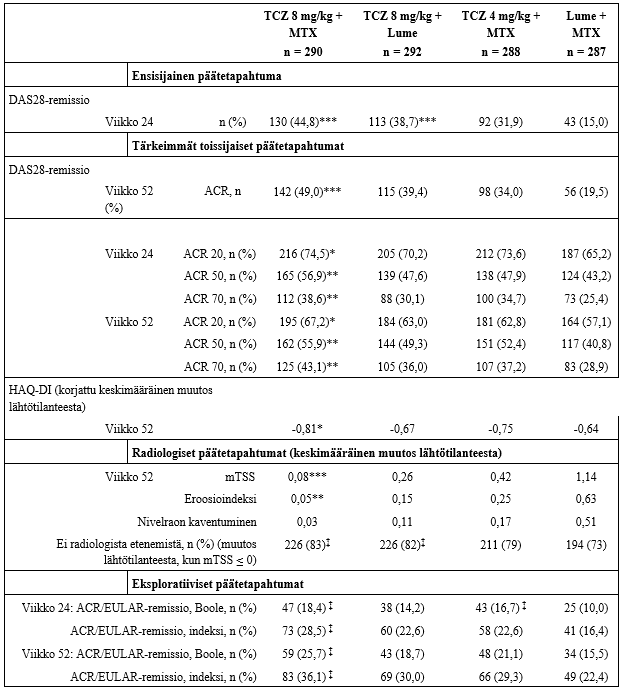
mTSS - modified Total Sharp Score
TCZ - tosilitsumabi
MTX - metotreksaatti
ACR - American College of Rheumatology (ACR) ‑kriteerit
Kaikki tehoa koskevat vertailut vs. lumevalmiste + MTX. ***p≤0,0001; **p<0,001; *p<0,05;
‡p-arvo < 0,05 vs. lumevalmiste + MTX, mutta päätetapahtuma oli eksploratiivinen (ei mukana tilastollisen testauksen hierarkiassa eikä monivertailua ole siksi kontrolloitu)
COVID-19
Kliininen teho
RECOVERY (Randomised Evaluation of COVID-19 Therapy) ‑työryhmätutkimus sairaalahoidossa olevilla COVID-19-diagnoosin saaneilla aikuisilla
RECOVERY oli laaja, satunnaistettu, kontrolloitu, avoin, monikeskusalustatutkimus, joka tehtiin Isossa-Britanniassa mahdollisten hoitojen tehon ja turvallisuuden arvioimiseksi sairaalahoidossa olevilla vaikeaa COVID-19-tautia sairastavilla aikuispotilailla. Kaikki mukaan soveltuneet potilaat saivat tavanomaista hoitoa ja alkuvaiheessa (päävaiheessa) heidät satunnaistettiin. Tutkimukseen mukaan soveltuneilla potilailla oli kliinisesti epäilty tai laboratoriotestein varmistettu SARS-CoV-2-infektio, eikä heillä ollut vasta-aiheita millekään hoidolle. Potilaat, joilla oli kliinisesti havaittu etenevä COVID-19-tauti (määritelty happisaturaatioksi < 92 % huoneilmassa tai happihoito ja CRP-arvo ≥ 75 mg/l), soveltuivat toiseen satunnaistamiseen saamaan joko tosilitsumabia laskimoon tai pelkästään tavanomaista hoitoa.
Tehon analyysit tehtiin hoitoaikeen mukaisesta (ITT) potilasjoukosta, johon kuului 4116 potilasta, joista 2022 potilasta satunnaistettiin tosilitsumabin ja tavanomaisen hoidon yhdistelmää saaneeseen haaraan ja 2 094 potilasta satunnaistettiin pelkästään tavanomaista hoitoa saaneeseen haaraan. Hoitoaikeen mukaisen potilasjoukon demografiset ja taudin ominaisuudet lähtötilanteessa olivat hyvin tasapainossa hoitohaarojen kesken. Osallistujien ikä oli keskimäärin 63,6 vuotta (keskihajonta 13,6 vuotta). Valtaosa potilaista oli miehiä (67 %) ja valkoihoisia (76 %). CRP-arvon mediaani (vaihteluväli) oli 143 mg/l (75–982).
Lähtötilanteessa 0,2 % (n = 9) potilaista ei saanut lisähappea, 45 % potilaista tarvitsi pienivirtauksista happihoitoa, 41 % potilaista tarvitsi kajoamatonta hengityskonehoitoa tai suurivirtauksista happihoitoa ja 14 % potilaista tarvitsi invasiivista hengityskonehoitoa; 82 %:n raportoitiin saaneen systeemistä kortikosteroidihoitoa (määriteltiin potilaiksi, jotka aloittivat systeemisen kortikosteroidihoidon joko ennen satunnaistamista tai satunnaistamisajankohtana). Yleisimmät samanaikaiset sairaudet olivat diabetes (28,4 %), sydänsairaus (22,6 %) ja krooninen keuhkosairaus (23,3 %).
Ensisijainen hoitotulos oli kuolleisuus päivän 28 kohdalla. Tosilitsumabin ja tavanomaisen hoidon yhdistelmää saaneen haaran ja pelkästään tavanomaista hoitoa saaneen haaran vertailussa riskitiheyksien suhde oli 0,85 (95 %:n luottamusväli: 0,76–0,94), joka oli tilastollisesti merkitsevä tulos (p = 0,0028). Kuoleman todennäköisyyden päivään 28 mennessä arvioitiin olleen tosilitsumabihaarassa 30,7 % ja tavanomaisen hoidon haarassa 34,9 %. Riskin eroksi arvioitiin ‑4,1 % (95 %:n luottamusväli: -7,0 – ‑1,3 %), mikä on yhdenmukainen ensisijaisen analyysin kanssa. Ennalta määritellyssä alaryhmässä, jossa potilaat saivat lähtötilanteessa systeemistä kortikosteroidihoitoa, riskitiheyssuhde oli 0,79 (95 %:n luottamusväli: 0,70–0,89), ja ennalta määritellyssä alaryhmässä, jossa potilaat eivät saaneet lähtötilanteessa systeemistä kortikosteroidihoitoa, se oli 1,16 (95 %:n luottamusväli: 0,91–1,48).
Sairaalasta kotiutumiseen kuluneen ajan mediaani oli tosilitsumabin ja tavanomaisen hoidon yhdistelmää saaneessa haarassa 19 päivää ja tavanomaista hoitoa saaneessa haarassa > 28 päivää (riskitiheyksien suhde [95 %:n luottamusväli] = 1,22 [1,12–1,33]).
Niistä potilaista, jotka eivät tarvinneet lähtötilanteessa invasiivista hengityskonehoitoa, päivään 28 mennessä hengityskonehoitoa tarvinneiden tai kuolleiden osuus oli tosilitsumabin ja tavanomaisen hoidon yhdistelmää saaneessa haarassa 35 % (619/1754) ja pelkästään tavanomaista hoitoa saaneessa haarassa 42 % (754/1800) (riskisuhde [95 %:n luottamusväli] = 0,84, [0,77–0,92] p < 0,0001).
Yleisoireista lastenreumaa sairastavat pediatriset potilaat
Kliininen teho
Tosilitsumabin tehoa aktiivisen yleisoireisen lastenreuman hoidossa on selvitetty 12 viikkoa kestäneessä satunnaistetussa kaksoissokkoutetussa lumelääkekontrolloidussa tutkimuksessa, jossa oli kaksi rinnakkaista ryhmää. Tutkimukseen hyväksytyillä potilailla oli aktiivinen tauti ja he olivat sairastaneet yleisoireista nivelreumaa vähintään kuusi kuukautta. Akuutissa pahenemisvaiheessa olevia potilaita, jotka tarvitsivat > 0,5 mg/kg prednisoniannoksia, ei hyväksytty tutkimukseen. Vaikutusta makrofagiaktivaatio-oireyhtymän (MAS) hoidossa ei ole tutkittu.
Potilaat (MTX-hoidossa tai ilman) satunnaistettiin (tosilitsumabi:lumelääke = 2:1) jompaankumpaan kahdesta hoitoryhmästä. Toisessa ryhmässä 75 potilasta sai tosilitsumabi-infuusion joka toinen viikko, joko annoksella 8 mg/kg (≥ 30 kg painavat potilaat) tai annoksella 12 mg/kg (< 30 kg painavat potilaat) ja toisessa ryhmässä 37 potilaalle annettiin lumelääkeinfuusio joka toinen viikko. Kortikosteroidiannoksen asteittainen pienentäminen oli sallittua kuudennelta viikolta alkaen niille potilaille, jotka saavuttivat JIA ACR 70-vasteen (JIA = juveniili idiopaattinen artriitti). 12 viikon jälkeen tai vaihdettuaan hoitoa taudin pahenemisen vuoksi potilaita siirrettiin tutkimuksen avoimeen vaiheeseen, jossa he saivat painonmukaista jatkohoitoa.
Kliininen vaste
Tutkimuksen ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka viikkoon 12 mennessä saavuttivat JIA ACR-kriteerien perusteella vähintään 30 % paremman vasteen (JIA ACR 30-vaste) ja jotka olivat kuumeettomia (ei ≥ 37,5 °C:n mittaustuloksia seitsemän edellisen vuorokauden aikana). 85 % (64/75) tosilitsumabihoitoa ja 24,3 % (9/37) lumelääkettä saaneista potilaista saavutti tämän päätetapahtuman. Ryhmien välinen ero oli erittäin merkitsevä (p < 0,0001).
Potilaiden jakaumat saavutettujen JIA ACR 30, 50, 70 ja 90 -vasteiden mukaan esitetään taulukossa 8.
Taulukko 8. JIA ACR-vasteosuudet viikolla 12 (% potilaista)
| Vaste | Tosilitsumabi n = 75 | Lume n = 37 |
| JIA ACR 30 | 90,7 %1 | 24,3 % |
| JIA ACR 50 | 85,3 %1 | 10,8 % |
| JIA ACR 70 | 70,7 %1 | 8,1 % |
| JIA ACR 90 | 37,3 %1 | 5,4 % |
1p < 0.0001, tosilitsumabi vs. lumelääke
Systeemiset vaikutukset
Tosilitsumabiryhmän potilaista, joilla oli yleisoireisesta lastenreumasta johtuvaa kuumetta hoidon alussa, 85 % oli kuumeettomia viikolla 12 (ei ≥ 37,5 °C:n mittaustuloksia 14 edellisen vuorokauden aikana). Lumelääkeryhmässä kuumeettomien potilaiden osuus oli vain 21 % (p < 0,0001).
12 viikon tosilitsumabihoidon jälkeen vakioitu keskimääräinen muutos VAS-kipuindeksissä oli 41 VAS-pisteen vähennys (asteikolla 0–100 pistettä) verrattuna yhden VAS-pisteen vähennykseen lumelääkeryhmässä (p < 0,0001).
Kortikosteroidiannoksen asteittainen pienentäminen
Kortikosteroidiannoksen pienentäminen sallittiin JIA ACR 70 ‑vasteen saavuttaneille potilaille. Tosilitsumabiryhmässä 17 potilasta (24 %) ja lumelääkeryhmässä yksi (3 %) potilas pystyi pienentämään kortikosteroidiannostaan vähintään 20 % ilman JIA ACR 30 ‑vasteen heikkenemistä tai systeemisten oireiden ilmaantumista viikkoon 12 mennessä (p = 0,028). Kortikosteroidiannoksen pienentämistä jatkettiin, ja tutkimusviikolla 44 oraalisten kortikosteroidien ottamisen oli lopettanut 44 potilasta. Samaan aikaan JIA ACR-vasteet pysyivät ennallaan.
Terveydentilaa ja elämänlaatua kuvaavat tulokset
Niiden potilaiden osuus, jotka osoittivat pienintä kliinisesti merkittävää paranemista kaikissa lapsipotilaan toimintakyvyn arviointiin perustuvissa mittareissa (yksilöllisen kokonais-HAQ-DI-indeksin pieneneminen ≥ 0,13 yksikköä), oli viikolla 12 huomattavasti suurempi tosilitsumabiryhmässä kuin lumelääkeryhmässä, 77 % vs 19 % (p < 0,0001).
Laboratorioarvot
Tosilitsumabiryhmän potilaista 67 prosentilla (50/75) hemoglobiiniarvo oli hoidon alkaessa viitealueen alarajan alapuolella. Viikolla 12 hemoglobiiniarvo oli noussut viitealueelle 80 prosentilla näistä potilaista (40/50), kun lumeryhmässä vastaava nousu todettiin vain 7 prosentilla (2/29) potilaista, joiden hemoglobiini oli hoidon alkaessa viitealueen alapuolella (p < 0,0001).
Lasten polyartriittia sairastavat pediatriset potilaat
Kliininen teho
Tosilitsumabin tehoa aktiivista lasten polyartriittia sairastavien lasten hoidossa on selvitetty kolmiosaisessa tutkimuksessa WA19977, johon kuului avoin jatkotutkimus. Osa I koostui 16 viikkoa kestäneestä aktiivisesta tosilitsumabihoidon aloitusjaksosta (n = 188), jonka jälkeinen osa II oli 24 viikkoa kestänyt satunnaistettu kaksoissokkoutettu lumekontrolloitu hoidon lopettamisjakso (n = 163). Tämän jälkeen seurasi osa III, joka oli 64 viikkoa kestänyt avoin jakso. Osassa I tutkimukseen hyväksytyt ≥ 30 kg:n painoiset potilaat saivat neljä 8 mg/kg tosilitsumabiannosta laskimoon neljän viikon välein, kun taas < 30 kg:n painoiset potilaat satunnaistettiin suhteessa 1:1 saamaan laskimoon neljä tosilitsumabiannosta (joko 8 mg/kg tai 10 mg/kg) neljän viikon välein. Jos potilas oli tutkimuksessa mukana osan I päättymiseen saakka ja sai viikolla 16 vähintään JIA ACR 30 -vasteen verrattuna lähtötilanteeseen, potilas soveltui jatkamaan tutkimusta sokkoutetussa hoidon lopettamisjaksossa (osa II). Osassa II potilaat satunnaistettiin joko tosilitsumabihoitoon (samoina annoksina kuin osassa I) tai lumelääkkeeseen suhteessa 1:1 samanaikaisen MTX:n ja kortikosteroidien käytön perusteella stratifioituna. Potilaat olivat mukana osassa II viikkoon 40 saakka tai kunnes potilas täytti JIA ACR 30 ‑pahenemisvaiheen kriteerit (viikkoon 16 verrattuna), mikä johti potilaan siirtymiseen tosilitsumabihoitoon (osassa I annetulla annostuksella).
Kliininen vaste
Tutkimuksen ensisijainen päätetapahtuma oli niiden potilaiden osuus, joiden sairaus oli pahentunut ACR 30 ‑kriteerien mukaisesti viikolta 16 viikolle 40. Sairaus paheni 48 %:lla (48,1 %, 39/81) lumelääkettä saaneista ja 25,6 %:lla (21/82) tosilitsumabia saaneista potilaista. Nämä osuuksien väliset erot olivat tilastollisesti merkitseviä (p = 0,0024).
Osan I päättyessä JIA ACR 30 -vasteen saaneita oli 89,4 %, JIA ACR 50 -vasteen saaneita 83,0 %, JIA ACR 70 -vasteen saaneita 62,2 % ja JIA ACR 90 -vasteen saaneita oli 26,1 %.
Hoidon lopettamisjakson aikana (osa II) JIA ACR 30 -,50 - ja 70 -vasteen saaneiden potilaiden prosenttiosuudet viikolla 40 verrattuna lähtötilanteeseen on esitetty taulukossa 9. Jos potilaalla oli pahenemisvaihe osan II aikana tai potilas vetäytyi tutkimuksesta, hänet luokiteltiin tässä tilastollisessa analyysissä ryhmään ei-vastetta. JIA ACR -vasteiden lisäanalyysi, osoitti, että niistä potilaista, joilla tosilitsumabihoito oli jatkuvaa viikolle 40 asti, 95,1 % saavutti vähintään JIA ACR 30 -vasteen. Tässä lisäanalyysissa huomioitiin havaintoarvot viikolla 40 riippumatta potilaalla esiintyneistä pahenemisvaiheista.
Taulukko 9. JIA ACR -vasteosuudet viikolla 40 verrattuna lähtötilanteeseen (potilaiden prosenttiosuudet)
| Vaste | Tosilitsumabi n = 82 | Lume n = 81 |
| ACR 30 | 74,4 %* | 54,3 %* |
| ACR 50 | 73,2 %* | 51,9 %* |
| ACR 70 | 64,6 %* | 42,0 %* |
*p<0,01, tosilitsumabi vs. lumelääke
Taudin aktiivisuutta osoittavien nivelten lukumäärä väheni merkitsevästi lähtötilanteesta tosilitsumabihoitoa saaneilla potilailla verrattuna lumelääkettä saaneisiin potilaisiin (korjattu keskimuutos -14,3 vs -11,4, p = 0,0435). Lääkärin yleisarvio taudin aktiivisuudesta asteikolla 0−100 mm mitattuna osoitti, että tosilitsumabi vähensi taudin aktiivisuutta enemmän kuin lumelääke (korjattu keskimuutos ‑45,2 mm vs ‑35,2 mm, p = 0,0031).
VAS-kipuindeksin korjattu keskimuutos viikon 40 jälkeen oli tosilitsumabihoitoa saaneilla 32,4 mm (asteikolla 0–100 mm) ja 22,3 mm lumelääkettä saaneilla potilailla (tilastollisesti erittäin merkitsevä; p = 0,0076).
ACR-vasteita saavuttaneiden potilaiden osuudet olivat numeerisesti pienempiä niillä potilailla, jotka olivat aiemmin saaneet biologista lääkehoitoa, kuten taulukossa 10 esitetään.
Taulukko 10. Niiden potilaiden määrä ja prosenttiosuus, joiden tauti paheni JIA ACR -kriteerien perusteella vähintään 30 % sekä niiden potilaiden prosenttiosuus, jotka saivat JIA ACR 30/50/70/90 ‑vasteen viikolla 40, jaoteltuna aikaisemman biologisen lääkityksen mukaan (ITT populaatio -tutkimuksen osa II)
| Lume | Tosilitsumabi | |||
| Biologinen käyttö | Kyllä (n = 23) | Ei (n = 58) | Kyllä (n = 27) | Ei (n = 55) |
| ACR 30 ‑kriteerien mukainen taudin paheneminen | 18 (78,3) | 21 (36,2) | 12 (44,4) | 9 (16,4) |
| JIA ACR 30 -vaste | 6 (26,1) | 38 (65,5) | 15 (55,6) | 46 (83,6) |
| JIA ACR 50 -vaste | 5 (21,7) | 37 (63,8) | 14 (51,9) | 46 (83,6) |
| JIA ACR 70 -vaste | 2 (8,7) | 32 (55,2) | 13 (48,1) | 40 (72,7) |
| JIA ACR 90 -vaste | 2 (8,7) | 17 (29,3) | 5 (18,5) | 32 (58,2) |
Tosilitsumabiryhmään satunnaistetuilla potilailla oli vähemmän ACR 30 ‑kriteerien mukaisia taudin pahenemisia ja paremmat ACR-kokonaisvasteet kuin plaseboryhmän potilailla, riippumatta aiemmasta biologisesta lääkehoidosta.
Sytokiinioireyhtymä
Tosilitsumabin tehoa sytokiinioireyhtymän hoitoon selvitettiin hematologisten syöpien hoitoon käytettyjä CAR-T-soluhoitoja (tisagenlekleuseeli ja aksikabtageenisiloleuseeli) koskevien kliinisten tutkimustietojen retrospektiivisessä analyysissä. Arvioitavissa olleet potilaat olivat saaneet vaikea-asteisen tai hengenvaarallisen sytokiinioireyhtymän hoitoon tosilitsumabia annoksina 8 mg/kg (12 mg/kg, jos potilaan paino oli < 30 kg), jonka lisäksi annettiin tai ei annettu suuria kortikosteroidiannoksia. Vain ensimmäinen sytokiinioireyhtymäepisodi otettiin mukaan analyysiin. Tisagenlekleuseeli-kohortin tehoa koskevassa potilasjoukossa oli 28 miestä ja 23 naista (yhteensä 51 potilasta), joiden iän mediaani oli 17 vuotta (vaihteluväli 3−68 vuotta). Ajan mediaani sytokiinioireyhtymän ilmenemisestä ensimmäiseen tosilitsumabiannokseen oli 3 vuorokautta (vaihteluväli 0−18 vuorokautta). Sytokiinioireyhtymän häviämiseksi määriteltiin, ettei potilaalla ollut kuumetta eikä hän käyttänyt vasopressoreita vähintään 24 tuntiin. Potilaan katsottiin saaneen vasteen, jos sytokiinioireyhtymä hävisi 14 vuorokauden kuluessa ensimmäisestä tosilitsumabiannoksesta, jos tarvittiin enintään kaksi annosta eikä hoitoon käytetty muita lääkevalmisteita kuin tosilitsumabia ja kortikosteroideja. Kolmekymmentäyhdeksän potilasta (76,5 %; 95 %:n luottamusväli: 62,5–87,2 %) sai vasteen. 15 potilaan (vaihteluväli: 9−75 vuotta) riippumattomassa kohortissa potilailla oli aksikabtageenisiloleuseelista aiheutunut sytokiinioireyhtymä, ja 53 % sai vasteen.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset tosilitsumabin käytöstä kaikkien pediatristen potilasryhmien hoidossa CAR-T-soluhoidon aikaansaamassa sytokiinioireyhtymässä.
COVID-19
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset tosilitsumabin käytöstä COVID-19-taudin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Nivelreumaa sairastavat potilaat
Tosilitsumabin farmakokinetiikka määritettiin populaatiofarmakokineettisellä analyysillä 3 552 nivelreumapotilaan tietokannasta. Potilaat saivat tosilitsumabia 4 mg/kg tai 8 mg/kg tunnin kestävänä infuusiona neljän viikon välein 24 viikon ajan, tai 162 mg ihon alle joko kerran viikossa tai kerran kahdessa viikossa 24 viikon ajan.
Seuraavat parametrit (odotettu keskiarvo ± SD) arvioitiin tosilitsumabiannokselle 8 mg/kg, joka annettiin neljän viikon välein: vakaan tilan AUC = 38 000 ± 13 000 h·µg/ml, minimipitoisuus (Cmin) = 15,9 ± 13,1 μg/ml ja maksimipitoisuus (Cmax) = 182 ± 50,4 µg/ml. AUC- ja Cmax-arvojen kumulaatiosuhteet olivat pienet: AUC 1,32 ja Cmax 1,09. Kumulaatiosuhde oli suurempi Cmin-arvojen osalta (2,49), mikä oli odotettua johtuen epälineaarisesta puhdistumasta alhaisilla pitoisuustasoilla. Cmax-arvon osalta vakaa tila saavutettiin ensimmäisen annoksen jälkeen, AUC:n osalta 8 viikon kuluttua ja Cmin-arvon osalta 20 viikon kuluttua. Tosilitsumabin AUC-, Cmin- ja Cmax-arvot nousivat potilaan painon mukaan. Jos potilas painoi 100 kg tai enemmän, tosilitsumabin vakaan tilan AUC-arvo (odotettu keskiarvo ± SD) oli 50 000 ± 16 800 μg × h/ml, Cmin-arvo 24,4 ± 17,5 μg/ml, ja Cmax-arvo 226 ± 50,3 μg/ml. Arvot ovat korkeammat kuin yllä analysoidun potilaspopulaation keskimääräiset arvot (ts. kaikki painoryhmät). Tosilitsumabin annosvastekäyrä tasaantuu suuremmilla altistuksilla siten, että lisäykset tosilitsumabin pitoisuudessa eivät samassa suhteessa anna lisähyötyä tehossa. Kliinisesti merkittävää parannusta tehossa ei havaittu potilailla, joita hoidettiin yli 800 mg:n tosilitsumabiannoksilla. Siksi ei suositella tosilitsumabin kerta-annoksia, jotka ylittävät 800 mg:aa (ks. kohta Annostus ja antotapa).
COVID-19-potilaat
Tosilitsumabin farmakokinetiikkaa selvitettiin WA42380-tutkimuksessa (COVACTA) ja CA42481-tutkimuksessa (MARIPOSA) tekemällä populaatiofarmakokineettinen analyysi tietokannasta, joka koostuu 380 aikuisen COVID-19-potilaan tiedoista. Nämä potilaat saivat 8 mg/kg tosilitsumabia kertainfuusiona tai vähintään 8 tunnin välein annettuina kahtena infuusiona. Tosilitsumabiannoksen 8 mg/kg osalta arvioitiin seuraavat parametrit (ennustettu keskiarvo ± keskihajonta): käyrän alle jäävä pinta-ala 28 päivän aikana (AUC0-28) = 18 312 (5 184) tuntia × μg/ml, pitoisuus päivänä 28 (Cday28) = 0,934 (1,93) μg/ml ja huippupitoisuus (Cmax) = 154 (34,9) μg/ml. AUC0-28-, Cday28- ja Cmax-arvot arvioitiin myös, kun 8 mg/kg tosilitsumabia annettiin kahtena annoksena 8 tunnin välein (ennustettu keskiarvo ± keskihajonta): AUC0-28 42 240 (11 520) tuntia × μg/ml ja Cday28 8,94 (8,5) μg/ml ja Cmax 296 (64,7) μg/ml.
Jakautuminen
Nivelreumapotilailla sentraalinen jakautumistilavuus oli 3,72 l ja perifeerinen jakautumistilavuus 3,35 l, joten jakautumistilavuus oli vakaan tilan aikana 7,07 l.
Aikuisilla COVID-19-potilailla sentraalinen jakautumistilavuus oli 4,52 l ja perifeerinen jakautumistilavuus 4,23 l, joten jakautumistilavuus oli 8,75 l.
Eliminaatio
Laskimoon annettu tosilitsumabi poistuu verenkierrosta kaksivaiheisesti, joista toinen vaihe on lineaarinen puhdistuma ja toinen noudattaa pitoisuudesta riippuvaista epälineaarista puhdistumaa. Nivelreumapotilailla lineaarinen puhdistuma oli 9,5 ml/h. Aikuisilla COVID-19-potilailla lineaarinen puhdistuma oli seuraava: 17,6 ml/h lähtötilanteen järjestysasteikon (ordinal scale, OS) arvon ollessa 3 (OS 3, lisähappea tarvitsevat potilaat), 22,5 ml/h lähtötilanteessa OS 4 ‑potilailla (suurivirtauksista happihoitoa tai kajoamatonta hengityskonehoitoa tarvitsevat potilaat), 29 ml/h lähtötilanteessa OS 5 ‑potilailla (hengityskonehoitoa tarvitsevat potilaat) ja 35,4 ml/h lähtötilanteessa OS 6 ‑potilailla (ECMO-hoitoa eli veren kehonulkoista happeuttamista tai hengityskonehoitoa ja lisähappitukea tarvitsevat potilaat). Pitoisuudesta riippuva epälineaarinen puhdistuma on ratkaisevassa asemassa, kun tosilitsumabipitoisuus on alhainen. Tosilitsumabipitoisuuden suurentuessa epälineaarinen puhdistumareitti kyllästyy, minkä jälkeen puhdistuma määräytyy pääasiassa lineaarisen puhdistuman perusteella.
Nivelreumapotilailla tosilitsumabin puoliintumisaika (t1/2) riippui pitoisuudesta. Kun vakaa tila oli saavutettu annoksen ollessa 8 mg/kg neljän viikon välein, efektiivinen t1/2 lyheni pitoisuuden pienentyessä annosvälin aikana 18 vuorokaudesta 6 vuorokauteen.
COVID-19-potilailla pitoisuudet seerumissa olivat määritysrajan alapuolella keskimäärin 35 päivää yhden laskimoon annetun 8 mg/kg tosilitsumabi-infuusion jälkeen.
Lineaarisuus
Tosilitsumabin farmakokineettiset parametrit eivät muuttuneet ajan myötä. AUC- ja Cmin-arvojen havaittiin suurenevan annossuhdetta enemmän annoksen ollessa 4 mg/kg tai 8 mg/kg neljän viikon välein. Cmax nousi samassa suhteessa kuin annos. Annoksen ollessa 8 mg/kg arvioitu vakaan tilan AUC-arvo oli 3,2-kertainen verrattuna annokseen 4 mg/kg. Vastaava vakaan tilan Cmin-arvo oli 30-kertainen.
Erityisryhmät
Munuaisten vajaatoiminta
Varsinaista tutkimusta munuaisten vajaatoiminnan vaikutuksesta tosilitsumabin farmakokinetiikkaan ei ole tehty. Useimmilla populaatiofarmakokineettisessä analyysissä mukana olleilla potilailla oli normaali munuaistoiminta tai lievä munuaisten vajaatoiminta. Lievä munuaisten vajaatoiminta (Cockcroft-Gaultin kaavalla laskettu kreatiniinipuhdistuma < 80 ml/min ja ≥ 50 ml/min) ei vaikuttanut tosilitsumabin farmakokinetiikkaan.
Maksan vajaatoiminta
Varsinaista tutkimusta maksan vajaatoiminnan vaikutuksista tosilitsumabin farmakokinetiikkaan ei ole tehty.
Ikä, sukupuoli ja etninen tausta
Aikuisten nivelreuma- ja COVID-19-potilaiden populaatiofarmakokineettiset analyysit osoittivat, etteivät ikä, sukupuoli ja etninen tausta vaikuttaneet tosilitsumabin farmakokinetiikkaan.
COVID-19-potilaiden populaatiofarmakokineettisen analyysin tulokset varmistivat, että sekä paino että taudin vaikeusaste ovat kovariaatteja, joilla on merkittävä vaikutus tosilitsumabin lineaariseen puhdistumaan.
Yleisoireista lastenreumaa sairastavat potilaat
Tosilitsumabin farmakokinetiikka määritettiin populaatiofarmakokineettisellä analyysillä 140 yleisoireista lastenreumaa sairastavan potilaan tietokannasta. Potilaita oli hoidettu tosilitsumabilla joko 8 mg/kg laskimoon joka toinen viikko (≥ 30 kg painavat potilaat), 12 mg/kg laskimoon joka toinen viikko (< 30 kg painavat potilaat), 162 mg ihon alle joka viikko (≥ 30 kg painavat potilaat), 162 mg ihon alle 10 päivän välein tai joka toinen viikko (< 30 kg painavat potilaat).
Taulukko 11. Yleisoireista lastenreumaa sairastaville potilaille laskimoon annetun hoidon farmakokineettisten parametrien ennakoidut vakaan tilan keskiarvot ± keskihajonnat
Tosilitsumabin farmakokineettiset parametrit | 8 mg/kg joka 2. viikko ≥ 30 kg | 12 mg/kg joka 2. viikko alle 30 kg |
Cmax (µg/ml) | 256 ± 60,8 | 274 ± 63,8 |
Ctrough (µg/ml) | 69,7 ± 29,1 | 68,4 ± 30,0 |
Cmean (µg/ml) | 119 ± 36,0 | 123 ± 36,0 |
Kumuloitumisen Cmax | 1,42 | 1,37 |
Kumuloitumisen Ctrough | 3,20 | 3,41 |
Kumuloitumisen Cmean tai AUCτ* | 2,01 | 1,95 |
*τ = hoito-ohjelma 2 viikon ajan laskimoon
Noin 90 % potilaista saavutti vakaan tilan laskimoon annettujen annosten 12 mg/kg (< 30 kg painavat potilaat) ja 8 mg/kg joka toinen viikko (≥ 30 kg painavat potilaat) yhteydessä viikkoon 8 mennessä.
Yleisoireista lastenreumaa sairastavilla potilailla sentraalinen jakautumistilavuus oli 1,87 l ja perifeerinen jakautumistilavuus 2,14 l, joten jakautumistilavuus oli vakaan tilan aikana 4,01 l. Lineaarinen puhdistuma arvioituna populaatiofarmakokineettisen analyysin parametrina oli 5,7 ml/h.
Viikolla 12 tosilitsumabin puoliintumisaika yleisoireista lastenreumaa sairastavilla potilailla on jopa 16 vuorokautta molemmissa painoryhmissä (8 mg/kg ≥ 30 kg painaville ja 12 mg/kg < 30 kg painaville).
Lasten polyartriittia sairastavat potilaat:
Tosilitsumabin farmakokinetiikka määritettiin lasten polyartriittia sairastavilla potilailla populaatiofarmakokineettisellä analyysillä, jossa oli mukana 237 potilaan tiedot. Potilaat olivat saaneet hoitona annoksia 8 mg/kg laskimoon joka neljäs viikko (≥ 30 kg painavat potilaat), 10 mg/kg laskimoon joka neljäs viikko (< 30 kg painavat potilaat), 162 mg ihon alle joka toinen viikko (≥ 30 kg painavat potilaat) tai 162 mg ihon alle joka kolmas viikko (< 30 kg painavat potilaat).
Taulukko 12. Lasten polyartriittia sairastaville potilaille laskimoon annetun hoidon farmakokineettisten parametrien ennakoidut vakaan tilan keskiarvot ± keskihajonnat
Tosilitsumabin farmakokineettiset parametrit | 8 mg/kg joka 4. viikko ≥ 30 kg | 10 mg/kg joka 4. viikko alle 30 kg |
Cmax (µg/ml) | 183 ± 42,3 | 168 ± 24,8 |
Ctrough (µg/ml) | 6,55 ± 7,93 | 1,47 ± 2,44 |
Cmean (µg/ml) | 42,2 ± 13,4 | 31,6 ± 7,84 |
Kumuloitumisen Cmax | 1,04 | 1,01 |
Kumuloitumisen Ctrough | 2,22 | 1,43 |
Kumuloitumisen Cmean tai AUCτ* | 1,16 | 1,05 |
*τ = hoito-ohjelmat 4 viikon ajan laskimoon
Noin 90 % potilaista saavutti vakaan tilan laskimoon annettujen annosten 10 mg/kg (paino < 30 kg) yhteydessä viikkoon 12 mennessä ja annosten 8 mg/kg (paino ≥ 30 kg) yhteydessä viikkoon 16 mennessä.
Tosilitsumabin puoliintumisaika lasten polyartriittia sairastavilla potilailla on jopa 16 vuorokautta molemmissa painoryhmissä (8 mg/kg ≥ 30 kg painaville ja 10 mg/kg < 30 kg painaville) (vakaa tila antovälin aikana).
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta ja genotoksisuutta koskevien konventionaalisten prekliinisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Karsinogeenisuustutkimuksia ei ole tehty, koska monoklonaalisia IgG1-vasta-aineita ei pidetä luontaisina karsinogeeneinä.
Käytettävissä olevat ei-kliiniset tiedot osoittivat, että IL-6 vaikuttaa tiettyjen syöpätyyppien etenemiseen ja apoptoosiresistenssiin. Nämä tiedot eivät viittaa siihen, että tosilitsumabihoitoon liittyisi merkittävää syövän kehittymisen ja etenemisen riskiä. Pitkäaikaisessa kuuden kuukauden toksisuustutkimuksessa ei havaittu proliferoivia leesioita jaavanmakakeilla (cynomolgus-apinoilla) eikä IL-6-defisienteillä hiirillä.
Käytettävissä olevat ei-kliiniset tiedot eivät viittaa hedelmällisyyteen kohdistuviin vaikutuksiin tosilitsumabihoidon aikana. Jaavanmakakeilla tehdyssä toksisuustutkimuksessa ei havaittu endokriinisesti aktiivisiin elimiin eikä lisääntymiselimiin kohdistuneita vaikutuksia. Lisääntymistoimintoihin kohdistuvia vaikutuksia ei havaittu myöskään IL-6-defisienteillä hiirillä.
Tosilitsumabilla ei havaittu olevan suoraa eikä epäsuoraa haitallista vaikutusta tiineyteen eikä alkion- tai sikiönkehitykseen, kun sitä annettiin jaavanmakakeille tiineyden alkuvaiheessa. Keskenmenojen / alkio- ja sikiökuolemien vähäistä lisääntymistä havaittiin kuitenkin suurta annostusta 50 mg/kg/vrk saaneiden ryhmässä, jossa systeeminen altistus oli suuri (> 100-kertainen ihmisen altistukseen verrattuna), lumeryhmään ja muihin pientä annosta saaneiden ryhmiin verrattuna. Vaikka IL-6 ei näytä olevan ratkaisevan tärkeä sytokiini sikiön kasvun kannalta eikä emon ja sikiön immunologisessa vuorovaikutuksen säätelyssä, tämän löydöksen yhteyttä tosilitsumabihoitoon ei voida sulkea pois.
Hoito hiiren analogeilla ei aiheuttanut lisääntynyttä toksisuutta nuorilla hiirillä. Häiriöitä ei havaittu etenkään luuston kehityksessä, immuniteetissä eikä seksuaalisessa kypsymisessä.
Farmaseuttiset tiedot
Apuaineet
Sakkaroosi
Polysorbaatti 80 (E 433)
Dinatriumfosfaattidodekahydraatti (pH:n säätöön)
Natriumdivetyfosfaattidihydraatti (pH:n säätöön)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta
Valmis liuos
Infuusiokonsentraatti laimennetaan 0,9-prosenttiseen natriumkloridiliuokseen (9 mg/ml), ja valmis infuusioliuos on kemiallisesti ja fysikaalisesti stabiili. Sitä voidaan säilyttää 24 tuntia 30 ºC:ssa ja enintään 2 viikkoa jääkaapissa (2–8 °C).
Mikrobiologiselta kannalta valmis infuusioliuos on käytettävä välittömästi. Ellei liuosta käytetä heti, säilytysajat ja olosuhteet ennen käyttöä ovat käyttäjän vastuulla. Valmista liuosta ei tulisi yleensä säilyttää yli 24 tuntia 2–8 °C:ssa, ellei laimentaminen ole tapahtunut kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä injektiopullo(t) jääkaapissa (2 °C–8 °C). Ei saa jäätyä.
Pidä injektiopullot ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ROACTEMRA infuusiokonsentraatti, liuosta varten
20 mg/ml (L:ei) 4 ml (194,56 €), 10 ml (462,35 €), 20 ml (897,57 €)
PF-selosteen tieto
RoActemra on pakattu injektiopulloon (kirkas tyypin I lasi, butyylikumitulppa). Sisältää 4 ml, 10 ml tai 20 ml konsentraattia. Pakkauskoot: 1 ja 4 injektiopulloa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai opalisoiva, väritön tai vaaleankeltainen liuos, jonka pH on 6,3–6,7 ja jonka osmolaalisuus on 172–229 mosm/kg.
Käyttö- ja käsittelyohjeet
Laimentamisohjeet (ennen annostelua)
Parenteraalisesti annettavat lääkkeet on tarkastettava silmämääräisesti ennen annostelua hiukkasten ja värimuutosten havaitsemiseksi. Laimennettavan liuoksen on oltava kirkasta tai opalisoivaa ja väritöntä tai vaaleankeltaista, eikä siinä saa olla näkyviä hiukkasia. Käytä steriiliä neulaa ja ruiskua valmisteen käyttökuntoon saattamista varten.
Aikuiset nivelreumapotilaat ja potilaat, joilla on sytokiinioireyhtymä (≥ 30 kg), sekä COVID-19-potilaat
Vedä aseptisissa olosuhteissa ruiskuun steriiliä, pyrogeenitonta 0,9-prosenttista (9 mg/ml) natriumkloridiliuosta 100 ml:n infuusiopussista. Määrän tulee vastata potilaan annosta varten tarvittavaa tilavuutta konsentraattia. Tarvittava määrä konsentraattia (0,4 ml/kg) vedetään injektiopullosta ja siirretään 100 ml:n infuusiopussiin. Lopullisen tilavuuden pitää olla 100 ml. Sekoita kääntämällä pussia varovasti ylösalaisin, jotta vältetään vaahdon muodostuminen.
Pediatriset potilaat
Yleisoireista lastenreumaa ja lasten polyartriittia sairastavat potilaat sekä potilaat, joilla on sytokiinioireyhtymä (≥ 30 kg)
Vedä aseptisissa olosuhteissa ruiskuun steriiliä, pyrogeenitonta 0,9-prosenttista (9 mg/ml) natriumkloridiliuosta 100 ml:n infuusiopussista. Määrän tulee vastata potilaan annosta varten tarvittavaa tilavuutta konsentraattia. Tarvittava määrä konsentraattia (0,4 ml/kg) vedetään injektiopullosta ja siirretään 100 ml:n infuusiopussiin. Lopullisen tilavuuden pitää olla 100 ml. Sekoita kääntämällä pussia varovasti ylösalaisin, jotta vältetään vaahdon muodostuminen.
Yleisoireista lastenreumaa sairastavat potilaat sekä potilaat, joilla on sytokiinioireyhtymä (< 30 kg)
Vedä aseptisissa olosuhteissa ruiskuun steriiliä, pyrogeenitonta 0,9-prosenttista (9 mg/ml) natriumkloridiliuosta 50 ml:n infuusiopussista. Määrän tulee vastata potilaan annosta varten tarvittavaa tilavuutta konsentraattia. Tarvittava määrä konsentraattia (0,6 ml/kg) vedetään injektiopullosta ja siirretään 50 ml:n infuusiopussiin. Lopullisen tilavuuden pitää olla 50 ml. Sekoita kääntämällä pussia varovasti ylösalaisin, jotta vältetään vaahdon muodostuminen.
Lasten polyartriittia sairastavat potilaat (< 30 kg)
Vedä aseptisissa olosuhteissa ruiskuun steriiliä, pyrogeenitonta 0,9-prosenttista (9 mg/ml) natriumkloridiliuosta 50 ml:n infuusiopussista. Määrän tulee vastata potilaan annosta varten tarvittavaa tilavuutta konsentraattia. Tarvittava määrä konsentraattia (0,5 ml/kg) vedetään injektiopullosta ja siirretään 50 ml:n infuusiopussiin. Lopullisen tilavuuden pitää olla 50 ml. Sekoita kääntämällä pussia varovasti ylösalaisin, jotta vältetään vaahdon muodostuminen.
9 mg/ml (= 0,9 %) natriumkloridiliuoksella laimennettu RoActemra on yhteensopiva polyvinyylikloridista (PVC), polyeteenistä (PE) ja polypropeenista (PP) valmistettujen infuusiopussien kanssa.
RoActemra on tarkoitettu vain kertakäyttöön.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ROACTEMRA infuusiokonsentraatti, liuosta varten
20 mg/ml 4 ml, 10 ml, 20 ml
- Ei korvausta.
- Oikeus käyttää samaa biologista lääkettä (sama kauppanimi) 6 kk ajan.
ATC-koodi
L04AC07
Valmisteyhteenvedon muuttamispäivämäärä
12.02.2026
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com