
ECALTA kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 100 mg
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 100 mg anidulafungiinia.
Käyttökuntoon saatettu infuusiokonsentraattiliuos sisältää anidulafungiinia 3,33 mg/ml ja laimennettu liuos 0,77 mg/ml.
Apuaineet, joiden vaikutus tunnetaan:
ECALTA sisältää 119 mg fruktoosia per injektiopullo.
ECALTA sisältää 250 mg polysorbaattia 80 per 100 mg:n injektiopullon annos, mikä vastaa 8,33 mg:aa/ml polysorbaattia 80 käyttökuntoon saatetussa liuoksessa ja 1,92 mg:aa/ml polysorbaattia 80 laimennetussa liuoksessa.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos.
Kliiniset tiedot
Käyttöaiheet
Invasiivisen kandidiaasin hoitoon aikuisille ja pediatrisille potilaille (iältään 1 kuukaudesta < 18 vuotialle) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Ehto
Invasiivisten sieni-infektioiden hoitoon perehtyneen lääkärin tulisi aloittaa hoito.
Annostus ja antotapa
ECALTA-hoito on aloitettava sellaisen lääkärin toimesta, jolla on kokemusta invasiivisten sieni-infektioiden hoidosta.
Annostus
Näytteet sieniviljelyä varten on otettava ennen hoidon aloittamista. Hoidon voi aloittaa jo ennen viljelytuloksia, joiden valmistuttua hoitoa voi tarvittaessa muuttaa.
Aikuispotilaat (annostus ja hoidon kesto)
Ensimmäisenä päivänä annetaan yksi 200 mg:n kyllästysannos ja sen jälkeen 100 mg päivittäin. Hoidon kesto riippuu potilaan kliinisestä vasteesta.
Sienilääkitystä annetaan yleensä vähintään 14 päivän ajan viimeisen positiivisen viljelytuloksen jälkeen.
Tiedot pidemmästä kuin 35 päivän hoidosta 100 mg:n annoksella ovat riittämättömiä.
Potilaat, joilla on heikentynyt munuaisten ja maksan toiminta
Annostusta ei tarvitse muuttaa potilaille, joiden maksan toiminta on heikentynyt lievästi, kohtalaisesti tai vaikeasti.
Annostusta ei tarvitse muuttaa munuaisten vajaatoimintapotilaille vajaatoiminnan asteesta riippumatta, ei myöskään dialyysihoitoa saaville potilaille. ECALTAN voi antaa hemodialyysin ajoituksesta riippumatta (ks. Farmakokinetiikka).
Muut erityisryhmät
Annostusta ei tarvitse muuttaa aikuispotilaille sukupuolen, ruumiinpainon, rodun, HIV-positiivisuuden tai korkean iän vuoksi (ks. Farmakokinetiikka).
Pediatriset potilaat (iältään 1 kuukaudesta < 18 vuoteen)(annostus ja hoidon kesto)
Ensimmäisenä päivänä annetaan yksi 3,0 mg/kg (enintään 200 mg) kyllästysannos ja sen jälkeen ylläpitoannos 1,5 mg/kg (enintään 100 mg) päivittäin.
Hoidon kesto riippuu potilaan kliinisestä vasteesta.
Sienilääkitystä annetaan yleensä vähintään 14 päivän ajan viimeisen positiivisen viljelytuloksen jälkeen.
ECALTAN turvallisuutta ja tehoa vastasyntyneiden vauvojen (< 1 kuukauden ikäisten) hoidossa ei ole varmistettu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Antotapa
Vain laskimoon.
ECALTA saatetaan käyttövalmiiksi liuottamalla kuiva-aine injektionesteisiin käytettävällä vedellä pitoisuuteen 3,33 mg/ml ja saatu välikonsentraattiliuos laimennetaan vielä lopulliseksi infuusioliuokseksi pitoisuuteen 0,77 mg/ml. Pediatrisille potilaille annoksen antamiseksi tarvittava infuusioliuostilavuus riippuu lapsen painosta. Ks. kohdasta Käyttö- ja käsittelyohjeet. ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
ECALTA-infuusion suositeltava enimmäisnopeus on 1,1 mg/min (vastaa 1,4 ml:aa/min kun kuiva-aine on liuotettu ja laimennettu ohjeen mukaan). Kun anidulafungiini-infuusion enimmäisnopeus on 1,1 mg/min, infuusioon liittyviä reaktioita esiintyy harvemmin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
ECALTAA ei saa antaa bolusinjektiona.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Yliherkkyys muille ekinokandiiniryhmän lääkevalmisteille.
Varoitukset ja käyttöön liittyvät varotoimet
ECALTAN käyttöä ei ole tutkittu potilailla, joilla on kandidaendokardiitti, osteomyeliitti tai meningiitti.
ECALTAn teho on arvioitu vain pienellä määrällä neutropeniapotilaita (ks. kohta Farmakodynamiikka).
Pediatriset potilaat
ECALTA-hoitoa ei suositella vastasyntyneille vauvoille (< 1 kuukauden ikäisille). Vastasyntyneiden vauvojen hoito edellyttää, että se kattaa disseminoituneen kandidiaasin, myös keskushermoston kandidiaasin; ei-kliiniset infektiomallit osoittavat, että riittävä keskushermostoon penetroituminen edellyttää suurempia anidulafungiiniannoksia (ks. kohta Prekliiniset tiedot turvallisuudesta), jolloin valmisteen apuaineena sisältämän polysorbaatti 80:n annos on suurempi. Kirjallisuudessa on raportoitu, että suuriin polysorbaattiannoksiin on vastasyntyneillä liittynyt mahdollisesti hengenvaarallista toksisuutta.
Kohdan Annostus ja antotapa suosituksia suurempien anidulafungiiniannosten tehon ja turvallisuuden tueksi ei ole kliinisiä tietoja.
Maksavaikutukset
Anidulafungiinia saaneilla terveillä tutkimushenkilöillä ja potilailla on todettu kohonneita maksaentsyymitasoja. Joillakin potilailla, joilla oli jokin vakava lääketieteellinen perussairaus ja jotka saivat anidulafungiinin lisäksi monia lääkkeitä samanaikaisesti, on ilmennyt kliinisesti merkittäviä maksan poikkeavuuksia. Merkittävä maksan toimintahäiriö, maksatulehdus ja maksan vajaatoiminta olivat melko harvinaisia kliinisissä tutkimuksissa. Jos potilaan maksaentsyymitasot ovat koholla anidulafungiinihoidon aikana, on potilasta seurattava maksan toiminnan heikkenemisen varalta ja arvioitava anidulafungiinihoidon jatkamisen hyöty-/riskisuhde.
Yliherkkyys-/anafylaktiset reaktiot
Anidulafungiinihoidon aikana on raportoitu ilmenneen anafylaktisia reaktioita, mukaan lukien anafylaktista sokkia. Jos näitä rekatioita ilmaantuu anidulafungiinihoito tulee lopettaa ja tarkoituksenmukainen hoito on aloitettava.
Infuusioon liittyvät reaktiot
Anidulafungiinihoidon yhteydessä on havaittu infuusioon liittyviä reaktiota mukaan lukien ihottuma, urtikaria, punoitus, kutina, hengenahdistus, bronkospasmi ja hypotensio. Kun anidulafungiini-infuusion enimmäisnopeus on 1,1 mg/min, infuusioon liittyviä reaktioita esiintyy harvemmin (ks. kohta Haittavaikutukset).
Ei-kliinisessä (rotta)tutkimuksessa on todettu, että anesteettien samanaikainen anto pahentaa infuusioon liittyviä reaktioita (ks. Prekliiniset tiedot turvallisuudesta). Tämän kliinistä merkitystä ei tiedetä. Varovaisuutta on kuitenkin noudatettava anidulafungiinin ja anesteettien samanaikaisessa annossa.
Apuaineet, joiden vaikutus tunnetaan
Fruktoosi
ECALTA sisältää fruktoosia.
Potilaille, joilla on perinnöllinen fruktoosi-intoleranssi (HFI) ei saa antaa tätä lääkevalmistetta, ellei se ole ehdottoman välttämätöntä.
Vauvoilla ja pienillä lapsilla (alle 2-vuotiaat) ei vielä välttämättä ole diagnosoitu perinnöllistä fruktoosi-intoleranssia (HFI). Laskimoon annettavat lääkevalmisteet (fruktoosia sisältävät) saattavat olla hengenvaarallisia, eikä niitä pidä antaa tälle väestöryhmälle, paitsi tilanteissa, joissa lääkevalmisteen käytölle on ehdoton kliininen tarve eikä muita hoitovaihtoehtoja ole saatavilla.
Kunkin potilaan yksityiskohtaiset HFI-oireisiin liittyvät esitiedot on selvitettävä ennen tämän lääkevalmisteen antoa.
Natrium
ECALTA sisältää alle 1 mmol natriumia (23 mg) per injektiopullo. Potilaille, joilla on ruokavalion natriumrajoitus, voidaan sanoa, että tämä lääkevalmiste on ”natriumiton”.
ECALTA voidaan laimentaa natriumia sisältäviin liuoksiin (ks. kohta Käyttö- ja käsittelyohjeet), mikä pitää ottaa huomioon potilaan kaikista lähteistä saamassa natriumin kokonaismäärässä.
Polysorbaatti
ECALTA sisältää polysorbaattia 80. Polysorbaatit saattavat aiheuttaa allergisia reaktioita ja vaikuttaa sydämeen ja verenkiertoon (esim. epäsäännöllinen tai poikkeava sydämensyke tai matala verenpaine). Sydämen minuuttitilavuuden pienenemisen ja hypotension riskin pienentämiseksi on harkittava tavanomaista hitaampaa infuusionopeutta.
Yhteisvaikutukset
Anidulafungiini ei ole sytokromi P450 -isoentsyymien (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A) kliinisesti merkittävä substraatti, induktori tai estäjä. On kuitenkin huomattava, että in vitro
-tutkimukset eivät poissulje kokonaan yhteisvaikutuksia in vivo.
Lääkeyhteisvaikutustutkimukset on tehty anidulafungiinilla ja sellaisilla muilla lääkevalmisteilla, joita todennäköisesti käytetään anidulafungiinin kanssa samanaikaisesti. Sen paremmin anidulafungiinin kuin sen kanssa samanaikaisesti annettavan siklosporiinin, vorikonatsolin tai takrolimuusin annostusta ei suositella muutettavaksi. Anidulafungiini-annostuksen muuttamista ei suositella myöskään silloin, kun potilas saa samanaikaisesti amfoterisiini B:tä tai rifampisiinia.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja anidulafungiinin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
ECALTAN käyttöä ei suositella raskauden aikana, ellei lääkkeestä mahdollisesti saatava hyöty äidille ole suurempi kuin mahdollinen sikiölle koituva haitta.
Imetys
Ei tiedetä, erittyykö anidulafungiini ihmisen rintamaitoon. Olemassa olevat farmakodynaamiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet anidulafungiinin erittyvän rintamaitoon.
Imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko ECALTA-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Anidulafungiinilla uros- ja naarasrotilla tehdyissä tutkimuksissa ei todettu vaikutuksia fertiliteettiin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ei oleellinen.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Anidulafungiinilla on ilmoitettu infuusioon liittyviä haittavaikutuksia kliinisissä tutkimuksissa: tällaisia vaikutuksia olivat mm. ihottuma, kutina, dyspnea, bronkospasmi, hypotensio (yleisiä tapahtumia), punoitus, kuumat aallot ja nokkosihottuma (melko harvinaisia tapahtumia). Yhteenveto näistä haittavaikutuksista on esitetty taulukossa 1 (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Seuraavassa taulukossa on esitetty lääkkeeseen liittyvät, kaikista syistä johtuvat haittavaikutukset (MedDRA termit) 840 tutkittavalla, jotka saivat 100 mg:n annoksen anidulafungiinia yleisyysluokissa hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1000) tai hyvin harvinainen (< 10 000) , sekä yleisyysluokassa tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin) spontaaniraporteissa ilmoitetut haittavaikutukset. Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Haittavaikutukset
| Elinjärjestelmä | Hyvin yleinen ≥ 1/10 | Yleinen ≥ 1/100, < 1/10 | Melko harvinainen ≥ 1/1 000, < 1/100 | Harvinainen ≥ 1/10 000, < 1/1 000 | Hyvin harvinainen < 1/10 000 | Tuntematon |
| Veri ja imukudos | Hyytymishäiriöt | |||||
| Immuunijärjestelmä | Anafylaktinen sokki, anafylaktinen reaktio* | |||||
Aineenvaihdunta ja ravitsemus | Hypokalemia | Hyperglykemia | ||||
| Hermosto | Kouristus, päänsärky | |||||
| Verisuonisto | Hypotensio, hypertensio | Kasvojen punoitus ja kuumotus, kuumat aallot | ||||
| Hengityselimet, rintakehä ja välikarsina | Bronkospasmi, hengenahdistus | |||||
| Ruoansulatus-elimistö | Ripuli, pahoinvointi | Oksentelu | Ylävatsakipu | |||
| Maksa ja sappi | Kohonnut alaniiniamino-transferaasi, kohonnut veren alkalinen fosfataasi, kohonnut aspartaatti-aminotrans-feraasi, kohonnut veren bilirubiini, kolestaasi | Kohonnut gammagluta-myylitrans-feraasi | ||||
| Iho ja ihonalainen kudos | Ihottuma, kutina | Nokkosihottuma | ||||
| Munuaiset ja virtsatiet | Kohonnut veren kreatiniini | |||||
| Yleisoireet ja antopaikassa todettavat haitat | Kipu infuusiokohdassa |
*ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Pediatriset potilaat
Anidulafungiinin turvallisuutta tutkittiin prospektiivisesssa, avoimessa, vertailemattomassa pediatrisessa tutkimuksessa 68 pediatrisella potilaalla (iältään 1 kuukaudesta < 18 vuoteen), joilla oli invasiivinen kandidiaasi, kandidemia mukaan lukien (ks. kohta Farmakodynamiikka). Tiettyjen maksan ja sapen haittavaikutusten, mukaan lukien suurentuneen alaniiniaminotransferaasipitoisuuden (ALAT) ja suurentuneen aspartaattiaminotransferaasipitoisuuden (ASAT), esiintyvyyden havaittiin suurentuneen yleisemmin näillä pediatrisilla potilailla (7–10 %) kuin aikuisilla (2 %). Perussairauden vaikeusasteen mahdollisuus tai erot ovat saattaneet osaltaan vaikuttaa tähän, mutta sitä ei voida sulkea pois, että maksan ja sapen haittavaikutuksia esiintyy pediatrisilla potilailla paljon useammin kuin aikuisilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kuten aina yliannostuksessa, elintoimintoja tukevaa hoitoa on annettava tarpeen mukaan. Yliannostuksen sattuessa voi ilmetä kohdassa Haittavaikutukset mainittuja haittavaikutuksia.
Kliinisissä tutkimuksissa annettiin tahattomasti yksi 400 mg:n kerta-annos anidulafungiinia kyllästysannoksena: mitään kliinisiä haittareaktioita ei ilmoitettu. Tutkimuksessa, jossa 10 terveelle tutkimushenkilölle annettiin 260 mg:n kyllästysannos ja sen jälkeen 130 mg päivittäin, ei ilmoitettu annosta rajoittavaa toksisuutta. Näistä kymmenestä tutkimushenkilöstä kolmella transaminaasiarvot kohosivat (≤ 3 x normaalin vaihteluvälin yläraja-arvo) ohimenevästi ja oireettomasti.
Pediatrisilla potilailla tehdyn kliinisen tutkimuksen aikana yksi tutkittava sai kaksi anidulafungiiniannosta, jotka olivat 143 % tarkoitetusta annoksesta. Kliinisiä haittavaikutuksia ei raportoitu.
ECALTA ei ole dialysoitavissa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Systeemiset sienilääkkeet, muut systeemiset sienilääkkeet, ATC-koodi: J02AX06
Vaikutusmekanismi
Anidulafungiini on semisynteettinen ekinokandiini, lipopeptidi, joka on syntetisoitu Aspergillus nidulans -homesienen käymistuotteesta.
Anidulafungiini estää selektiivisesti 1,3-β-D-glukaanisyntaasi-entsyymiä, jota on sienisoluissa mutta ei nisäkässoluissa. Tämän seurauksena sienen soluseinämälle oleellisen aineosan, 1,3-β-D-glukaanin, muodostuminen estyy. Anidulafungiinilla on osoitettu fungisidista aktiivisuutta Candida-lajeja vastaan ja Aspergillus fumigatus -sienirihmojen kasvukohdissa.
Herkkyystestauksen raja-arvot
European Committee on Antimicrobial Susceptibility Testing (EUCAST) on vahvistanut mikrobilääkeherkkyyden testausta koskevat MIC-arvon (pienin bakteerin kasvun estävä pitoisuus) tulkintakriteerit anidulafungiinille, ja ne luetellaan täällä: https://www.ema.europa.eu/documents/other/minimum-inhibitory-concentration-mic-breakpoints_en.xlsx
Aktiivisuus in vitro
Anidulafungiinilla on osoitettu tehoa in vitroC. albicans-, C. glabrata-,C. parapsilosis-, C. krusei- ja C. tropicalis -lajeihin. Näiden löydösten kliinistä merkitystä on käsitelty kohdassa ”Kliininen teho ja turvallisuus”.
Kohdegeenin tietyillä alueilla esiintyvät mutaatioita sisältävät isolaatit on yhdistetty kliinisiin epäonnistumisiin tai infektioiden uusiutumisiin. Suurin osa kliinisista tapauksista on koskenut kaspofungiinihoitoa. Eläinkokeissa nämä mutaatiot ilmentävät kuitenkin ristiresistenssiä kaikille kolmelle ekinokandiinille, minkä vuoksi nämä isolaatit luokitellaan ekinokandiineille resistenteiksi, kunnes saadaan anidulafungiinia koskevia kliinisiä lisäkokemuksia.
Anidulafungiinilla osoitettu teho in vitro Candida-lajeihin ei ole yhtenäistä. Erityisesti C. parapsilosis -lajia estävät anidulafungiinin pienimmät pitoisuudet (MIC) ovat korkeampia kuin muiden Candida-lajien kohdalla.
Aktiivisuus in vivo
Parenteraalisesti annettu anidulafungiini tehosi Candida-lajeihin hiiri- ja rottamalleissa, joissa osalla koe-eläimistä vastustuskyky oli normaali ja osalla heikennetty. Anidulafungiinihoito pidensi elossaoloaikaa ja myös vähensi elimen Candida-kuormaa määrityksissä, jotka tehtiin 24–96 tunnin kuluttua viimeisen hoitokerran jälkeen.
Koetarkoituksessa aiheutettuja infektioita olivat mm. disseminoitunut C. albicans -infektio neutropeenisille kaniineille, esofageaalinen/orofaryngeaalinen infektio neutropeenisille kaniineille, joilla oli flukonatsolille vastustuskykyinen C. albicans, ja disseminoitunut infektio neutropeenisille hiirille, joilla oli flukonatsolille vastustuskykyinen C. glabrata.
Kliininen teho ja turvallisuus
Kandidemia ja muut invasiivisen kandidiaasin muodot
Anidulafungiinin turvallisuutta ja tehoa on arvioitu kolmannen vaiheen keskeisessä monikansallisessa monikeskustutkimuksessa, joka oli satunnaistettu ja kaksoissokkoutettu. Suurimmalla osalla tutkimukseen osallistuneista potilaista oli kandidemia, johon ei liittynyt neutropeniaa, ja pienellä osalla potilaista oli jokin syvän kudoksen Candida-infektio tai märkäpesäkkeitä muodostava sairaus. Tutkimuksesta poissuljettiin erityisesti sellaiset potilaat, joilla oli Candidan aiheuttama endokardiitti, osteomyeliitti tai meningiitti, tai joilla oli C. krusein aiheuttama infektio. Potilaat satunnaistettiin saamaan joko anidulafungiinia (200 mg:n kyllästysannos laskimoon ja sen jälkeen 100 mg laskimoon päivittäin) tai flukonatsolia (800 mg:n kyllästysannos laskimoon ja sen jälkeen 400 mg laskimoon päivittäin). Potilaat ositettiin APACHE II -pisteiden (≤ 20 ja > 20) ja neutropenian esiintymisen tai esiintymättömyyden mukaan. Hoitoa annettiin vähintään 14 päivää ja enintään 42 päivää. Molempien tutkimusryhmien potilaat saivat vaihtaa suun kautta annettavaan flukonatsoliin aikaisintaan 10 päivän laskimonsisäisen hoidon jälkeen edellyttäen, että he sietivät suun kautta annettuja lääkevalmisteita, olivat olleet kuumeettomia vähintään 24 tunnin ajan ja että viimeisimmät veriviljelyt olivat negatiivisia Candida-lajien suhteen.
Ne potilaat, jotka saivat vähintään yhden annoksen tutkimuslääkevalmisteita ja joilta oli ennen tutkimukseen ottamista saatu positiivinen Candida-viljelytulos normaalisti steriilistä kohdasta, otettiin mukaan modifioituun intent-to-treat (MITT) -populaatioon. MITT-populaatioiden ensisijaisessa tehoanalyysissa (kokonaisvaste laskimonsisäisen hoidon loputtua) anidulafungiinia verrattiin flukonatsoliin ennalta määritellyssä kaksivaiheisessa tilastollisessa vertailussa (sama teho ja parempi teho). Kokonaisvaste määriteltiin onnistuneeksi, silloin kun potilaan kliininen tila parani ja taudinaiheuttajan eradikaatio osoitettiin mikrobiologisesti. Potilaita seurattiin kuuden viikon ajan hoidon päättymisen jälkeen.
Hoitoryhmiin satunnaistettiin 256 iältään 16–91-vuotiasta potilasta, jotka saivat vähintään yhden annoksen tutkimuslääkettä. Lähtötilanteessa useimmiten eristetyt lajit olivat C. albicans (63,8 %:lla anidulafungiiniryhmässä, 59,3 %:lla flukonatsoliryhmässä) ja sitten C. glabrata (15,7 %, 25,4 %), C. parapsilosis (10,2 %, 13,6 %) ja C. tropicalis (11,8 %, 9,3 %) – anidulafungiiniryhmässä oli kolmea viimeksi mainittua lajia kutakin 20, 13 ja 15 isolaattia. Suurimmalla osalla potilaista APACHE II -pisteet olivat ≤ 20, ja vain muutamalla oli neutropenia.
Taulukossa 2 on esitetty tehotiedot sekä yhdistettyinä että alaryhmittäin.
| Taulukko 2. Hoidon kokonaisonnistuminen MITT-populaatiossa: ensi- ja toissijaiset päätemuuttujat | |||
| Anidulafungiini | Flukonatsoli | Ryhmien välinen ero a ( 95 % CI) | |
| Laskimonsisäisen hoidon loputtua (ensisijainen päätemuuttuja) | 96/127 (75,6 %) | 71/118 (60,2 %) | 15,42 (3,9, 27,0) |
| Vain kandidemia | 88/116 (75,9 %) | 63/103 (61,2 %) | 14,7 (2,5; 26,9) |
| Muut steriilit paikatb | 8/11 (72,7 %) | 8/15 (53,3 %) | - |
| Peritoneaalineste/IAc-märkäpesäke | 6/8 | 5/8 | |
| Muu | 2/3 | 3/7 | |
| C. albicansd | 60/74 (81,1 %) | 38/61 (62,3 %) | - |
| Muu kuin albicans-lajid | 32/45 (71,1 %) | 27/45 (60,0 %) | - |
| Apache II -pisteet ≤ 20 | 82/101 (81,2 %) | 60/98 (61,2 %) | - |
| Apache II -pisteet > 20 | 14/26 (53,8 %) | 11/20 (55,0 %) | - |
| Ei neutropeniaa (ANC, soluja/mm3 > 500) | 94/124 (75,8 %) | 69/114 (60,5 %) | - |
| Neutropenia (ANC, soluja/mm3 ≤ 500) | 2/3 | 2/4 | - |
| Toissijaiset päätemuuttujat | |||
| Hoidon loputtua | 94/127 (74,0 %) | 67/118 (56,8 %) | 17,24 (2,9, 31,6)e |
| 2 viikon seuranta | 82/127 (64,6 %) | 58/118 (49,2 %) | 15,41 (0,4, 30,4)e |
| 6 viikon seuranta | 71/127 (55,9 %) | 52/118 (44,1 %) | 11,84 (-3,4, 27,0)e |
a Laskettu: anidulafungiini miinus flukonatsoli
bJoko samanaikainen kandidemia tai ei kandidemiaa
cIntra-abdominaalinen
d Tiedot potilaista, joilla oli lähtötilanteessa vain yksi taudinaiheuttaja.
e 98,3 %:n luottamusvälit, sovitettu post hoc toissijaisten päätemuuttujien toistuvia vertailuja varten.
Taulukossa 3 on kuolleisuusluvut sekä anidulafungiini- että flukonatsoliryhmässä.
| Taulukko 3. Kuolleisuus | ||
| Anidulafungiini | Flukonatsoli | |
| Kokonaiskuolleisuus tutkimuksessa | 29/127 (22,8 %) | 37/118 (31,4 %) |
| Kuolleisuus hoidon aikana | 10/127 (7,9 %) | 17/118 (14,4 %) |
| Candida-infektiosta johtunut kuolleisuus | 2/127 (1,6 %) | 5/118 (4,2 %) |
Neutropeenisiä potilaita koskevat lisätiedot
Anidulafungiinin tehoa (200 mg:n kyllästysannos laskimoon ja sen jälkeen 100 mg laskimoon päivittäin) aikuisilla neutropeenisillä (neutrofiilimäärä ≤ 0,5x109/l, leukosyyttimäärä ≤ 0,5x109/l tai tutkija on luokitellut potilaan neutropeeniseksi jo lähtötilanteessa) potilailla, joilla oli mikrobiologisesti varmistettu invasiivinen kandidiaasi, on arvioitu viidestä prospektiivisesta tutkimuksesta yhdistetyistä tiedoista (1 vertailututkimus vs. kaspofungiini ja 4 avointa ei-vertailevaa tutkimusta). Hoitoa annettiin vähintään 14 päivää. Kliinisesti stabiilit potilaat saivat vaihtaa suun kautta annettavaan atsolihoitoon aikaisintaan 5–10 päivän anidulafungiinihoidon jälkeen. Analyysissa oli mukana kaikkiaan 46 potilasta. Suurimmalla osalla potilaista oli ainoastaan kandidemia (84,8 %; 39/46). Lähtötilanteessa yleisimmin eristetyt patogeenit olivat C. tropicalis (34,8 %; 16/46), C. krusei (19,6 %; 9/46), C. parapsilosis (17,4 %; 8/46), C. albicans (15,2 %; 7/46) ja C. glabrata (15,2 %; 7/46). Onnistunut kokonaisvaste laskimonsisäisen hoidon loputtua (ensisijainen päätepiste) oli 26/46 (56,5 %) ja koko hoidon loputtua 24/46 (52,2 %). Kaikista syistä johtuva kuolleisuus tutkimuksen loppumiseen saakka (6 viikon seurantakäynti) oli 21/46 (45,7 %).
Anidulafungiinin tehoa aikuisilla neutropeenisillä (absoluuttinen neutrofiilimäärä ≤ 0,5x109/l lähtötilanteessa) potilailla, joilla oli invasiivinen kandidiaasi, on arvioitu prospektiivisessa, kaksoissokkoutetussa ja satunnaistetussa kontrolloidussa tutkimuksessa. Soveltuvat potilaat saivat joko anidulafungiinia (200 mg:n kyllästysannos laskimoon ja sen jälkeen 100 mg laskimoon päivittäin) tai kaspofungiinia (70 mg:n kyllästysannos laskimoon ja sen jälkeen 50 mg laskimoon päivittäin) (2:1 satunnaistaminen). Hoitoa annettiin vähintään 14 päivää. Kliinisesti stabiilit potilaat saivat vaihtaa suun kautta annettavaan atsolihoitoon aikaisintaan 10 päivää kestäneen tutkimushoidon jälkeen. Tutkimuksessa oli mukana 14 neutropeenistä potilasta (anidulafungiini 11; kaspofungiini 3), joilla oli mikrobiologisesti varmistettu invasiivinen kandidiaasi (MITT-populaatio). Suurimmalla osalla potilaista oli ainoastaan kandidemia. Lähtötilanteessa yleisimmin eristetyt patogeenit olivat C. Tropicalis (anidulafungiini 4, kaspofungiini 0), C. parapsilosis (anidulafungiini 2, kaspofungiini 1), C. krusei (anidulafungiini 2, kaspofungiini 1), C. ciferrii (anidulafungiini 2, kaspofungiini 0). Onnistunut kokonaisvaste laskimonsisäisen hoidon loputtua (ensisijainen päätepiste) oli 8/11 (72,7 %) anidulafungiin kohdalla ja 3/3 (100,0 %) kaspofungiinin kohdalla (ero -27,3, 95 %:n CI -80,9, 40,3) sekä vastaavasti koko hoidon loputtua. Kaikista syistä johtuva kuolleisuus 6 viikon seurantakäyntiin saakka oli 4/11 (36,4 %) anidulafungiinin kohdalla (MITT populaatio) ja 2/3 (66,7 %) kaspofungiinin kohdalla.
Potilaat, joilla oli mikrobiologisesti vahvistettu invasiivinen kandidiaasi (MITT-populaatio) ja neutropenia, tunnistettiin neljästä samalla tavoin suunnitellusta prospektiivisesta, avoimesta, ei vertailevasta tutkimuksesta yhdistetyistä tiedoista. Anidulafungiinin tehoa (200 mg:n kyllästysannos laskimoon ja sen jälkeen 100 mg laskimoon päivittäin) arvioitiin 35 aikuisella neutropeenisellä potilaalla, joiden neutrofiilimäärä oli ≤0,5x109/l tai 22 potilaalla, joiden leukosyyttimäärä oli ≤0,5x109/l tai 13 potilaalla, jotka tutkija oli luokitellut neutropeenisiksi jo lähtötilanteessa. Kaikkia potilaita hoidettiin vähintään 14 päivää. Kliinisesti stabiilit potilaat saivat vaihtaa suun kautta annettavaan atsolihoitoon aikaisintaan 5–10 päivää kestäneen anidulafungiinihoidon jälkeen. Suurimmmalla osalla (85,7 %:lla) potilaista oli ainoastaan kandidemia. Lähtötilanteessa yleisimmin eristetyt patogeenit olivat C. tropicalis (12 potilasta), C. albicans (7 potilasta), C. glabrata (7 potilasta), C. krusei (7 potilasta) ja C. parapsilosis (6 potilasta). Onnistunut kokonaisvaste laskimonsisäisen hoidon loputtua (ensisijainen päätepiste) oli 18/35 (51,4 %) ja koko hoidon loputtua 16/35 (45,7 %). Kaikista syistä johtuva kuolleisuus päivään 28 mennessä oli 10/35 (28,6 %). Tutkijoiden lähtötilanteessa neutropeeniseksi arvioimilla 13 potilaalla onnistunut kokonaisvaste laskimonsisäisen hoidon loputtua ja koko hoidon loputtua oli molemmissa tapauksissa 7/13 (53,8 %).
Lisätiedot potilaista, joilla oli syvän kudoksen infektio
Anidulafungiinin tehoa (200 mg:n kyllästysannos laskimoon ja sen jälkeen 100 mg laskimoon päivittäin) aikuisilla potilailla, joilla on mikrobiologisesti varmistettu syvän kudoksen kandidiaasi, on arvioitu viidestä prospektiivisesta (1 vertailevasta ja 4 avoimesta) tutkimuksesta yhdistetyistä tiedoista. Hoitoa annettiin vähintään 14 päivää. Neljässä avoimessa tutkimuksessa mukana olleet potilaat saivat vaihtaa suun kautta annettavaan atsolihoitoon aikaisintaan 5–10 päivää kestäneen anidulafungiinihoidon jälkeen. Analyysissa oli mukana kaikkiaan 129 potilasta. 21 (16,3 %) potilaalla oli samanaikainen kandidemia. APACHE II -pisteiden keskiarvo oli 14,9 (vaihteluväli 2–44). Yleisimmin infektoita esiintyi vatsakalvonontelossa (54,3 %; 70/129), maksa-sappiteissä (7,0 %; 9/129), keuhkopussinontelossa (5,4 %; 7/129) ja munuaisissa (3,1 %; 4/129). Lähtötilanteessa yleisimmin eristetyt patogeenit olivat C. albicans (64,3 %; 83/129), C. glabrata (31,0 %; 40/129), C. tropicalis (11,6 %; 15/129) ja C. krusei (5,4 %; 7/129). Onnistunut kokonaisvaste laskimonsisäisen hoidon loputtua (ensisijainen päätepiste) ja koko hoidon loputtua sekä kaikista syistä johtuva kuolleisuus 6 viikon seurantakäyntiin saakka esitetään taulukossa 4.
| Taulukko 4. Onnistunut kokonaisvastea ja kaikista syistä johtuva kuolleisuus potilailla, joilla oli syvän kudoksen kandidiaasi – Yhdistetty analyysi | |
MITT-Populaatio n/N (%) | |
| Onnistunut kokonaisvaste IV-hoidon loputtua | |
| Kaikkiaan | 102/129 (79,1) |
| Vatsakalvonontelo | 51/70 (72,9) |
| Maksa-sappitiet | 7/9 (77,8) |
| Keuhkopussinontelo | 6/7 (85,7) |
| Munuainen | 3/4 (75,0) |
| Onnistunut kokonaisvaste koko hoidon loputtua | 94/129 (72,9) |
| Kaikista syistä johtuva kuolleisuus | 40/129 (31,0) |
| a Onnistunut kokonaisvaste määriteltiin sekä kliinisenä että mikrobiologisena onnistumisena | |
Pediatriset potilaat
Prospektiivisessa, avoimessa, vertailemattomassa, monikansallisessa tutkimuksessa arvioitiin anidulafungiinin turvallisuutta ja tehoa 68 pediatrisella potilaalla (iältään 1 kuukaudesta < 18 vuoteen). Potilaat ositettiin iän mukaan (1 kuukaudesta < 2 vuoteen, 2 vuodesta < 5 vuoteen ja 5 vuodesta < 18 vuoteen), ja he saivat yhden anidulafungiiniannoksen laskimoon vuorokaudessa (ensimmäisenä päivänä yksi 3,0 mg/kg kyllästysannos ja sen jälkeen päivittäin ylläpitoannos 1,5 mg/kg) enintään 35 päivän ajan, minkä jälkeen heillä oli mahdollisuus siirtyä suun kautta otettavaan flukonatsolihoitoon (6–12 mg/kg/vrk, enintään 800 mg/vrk). Potilaiden seuranta toteutettiin 2 viikkoa ja 6 viikkoa sen jälkeen, kun hoito oli päättynyt.
Niistä 68 potilaasta, jotka saivat anidulafungiinia, 64 potilaalla oli mikrobiologisesti varmistettu Candida-infektio, ja he olivat mukana modifioidun intent-to-treat (MITT) ‑populaation tehoarviossa. Kaikkiaan 61 potilaalta (92,2 %) oli eristetty Candida vain verestä. Yleisimmin eristettyjä taudinaiheuttajia olivat Candida albicans (25 [39,1 %] potilaalla), ja sen jälkeen Candida parapsilosis (17 [26,6 %] potilaalla) ja Candida tropicalis (9 [14,1 %] potilaalla). Onnistuneeksi kokonaisvasteeksi määriteltiin sekä onnistunut kliininen vaste (paraneminen tai infektion lieveneminen) että onnistunut mikrobiologinen vaste (eradikaatio tai oletettu eradikaatio). MITT-populaation onnistuneen kokonaisvasteen kokonaisosuudet esitetään taulukossa 5.
| Taulukko 5. Yhteenveto onnistuneesta kokonaisvasteesta ikäryhmittäin, MITT-populaatio | |||||
| Onnistunut kokonaisvaste, n (%) | |||||
| Ajankohta | Kokonaisvaste | 1 kuukaudesta < 2 vuoteen (N = 16) n (n/N, %) | 2 vuodesta < 5 vuoteen (N = 18) n (n/N, %) | 5 vuodesta < 18 vuoteen (N = 30) n (n/N, %) | Yhteensä (N = 64) n (n/N, %) |
| IV-hoidon lopussa | Onnistunut | 11 (68,8) | 14 (77,8) | 20 (66,7) | 45 (70,3) |
| 95 % CI | (41,3; 89,0) | (52,4; 93,6) | (47,2; 82,7) | (57,6; 81,1) | |
| Koko hoidon lopussa | Onnistunut | 11 (68,8) | 14 (77,8) | 21 (70,0) | 46 (71,9) |
| 95 % CI | (41,3; 89,0) | (52,4; 93,6) | (50,6; 85,3) | (59,2; 82,4) | |
| 2 viikon seuranta | Onnistunut | 11 (68,8) | 13 (72,2) | 22 (73,3) | 46 (71,9) |
| 95 % CI | (41,3; 89,0) | (46,5; 90,3) | (54,1; 87,7) | (59,2; 82,4) | |
| 6 viikon seuranta | Onnistunut | 11 (68,8) | 12 (66,7) | 20 (66,7) | 43 (67,2) |
| 95 % CI | (41,3; 89,0) | (41,0; 86,7) | (47,2; 82,7) | (54,3; 78,4) | |
95 % CI = binomiosuuksien eksakti 95 %:n luottamusväli Clopper–Pearsonin menetelmällä; MITT = modifioitu intent-to-treat ‑populaatio; N = tutkittavien lukumäärä populaatiossa; n = niiden tutkittavien lukumäärä, jotka saivat vasteen
Farmakokinetiikka
Yleiset farmakokineettiset ominaisuudet
Anidulafungiinin farmakokinetiikka on karakterisoitu terveillä tutkimushenkilöillä, erityisryhmillä ja potilailla. Systeemisen altistuksen (variaatiokerroin ∼25 %) todettiin vaihtelevan yksilöiden välillä vain vähän. Vakaa tila saavutettiin kyllästysannoksen (2 x päivittäinen ylläpitoannos) annostelua seuraavana päivänä.
Jakautuminen
Anidulafungiinin farmakokinetiikalle on tyypillistä jakautumisen lyhyt puoliintumisaika
(0,5–1 h) ja 30–50 litran jakautumistilavuus, joka on samaa luokkaa kuin elimistön koko nestetilavuus. Anidulafungiini sitoutuu suuressa määrin (> 99 %) ihmisen plasman proteiineihin. Anidulafungiinin kudoksiin jakautumista ei ole tutkittu ihmisellä. Siksi ei ole tietoa anidulafungiinin kulkeutumisesta aivo-selkäydinnesteeseen ja/tai veri-aivoesteen läpi.
Biotransformaatio
Anidulafungiinilla ei ole todettu metaboliaa maksassa. Anidulafungiini ei ole kliinisesti merkittävä sytokromi P450 -isoentsyymien substraatti, induktori tai estäjä. Anidulafungiinilla ei todennäköisesti ole kliinisesti merkittäviä vaikutuksia sellaisten lääkkeiden metaboliaan, jotka metaboloituvat sytokromi P450 -isoentsyymien välityksellä.
Anidulafungiini hajoaa fysiologisessa lämpötilassa ja pH:ssa hitaasti ja kemiallisesti avorenkaiseksi peptidiksi, joka ei tehoa sieniin. Anidulafungiinin hajoamisen puoliintumisaika on fysiologisissa in vitro -olosuhteissa noin 24 tuntia. Avorenkainen yhdiste muuntuu in vivo peptidin hajoamistuotteiksi ja eliminoituu pääasiassa erittymällä sappeen.
Eliminaatio
Anidulafungiinin puhdistuma on noin 1 l/h. Anidulafungiinin eliminaation puoliintumisaika on noin 24 tuntia (karakterisoi suurinta osaa plasman pitoisuus-aika-profiilista) ja terminaalinen puoliintumisaika 40–50 tuntia (karakterisoi profiilin terminaalisen eliminaation vaihetta).
Terveille tutkimushenkilöille annettiin kliinisessä kerta-annostutkimuksessa radioaktiivisesti (14C) merkittyä anidulafungiinia (∼88 mg). Tästä radioaktiivisesta annoksesta eliminoitui 9 päivän kuluessa ulosteeseen noin 30 %, josta alle 10 % muuttumattomana lääkeaineena. Annetusta radioaktiivisesta annoksesta erittyi virtsaan alle 1 %, mikä viittaa häviävän pieneen munuaispuhdistumaan. Anidulafungiinipitoisuudet pienenivät alle alimpien mittausrajojen 6 päivän kuluttua annostelusta. Lääkeperäistä radioaktiivisuutta löytyi häviävän pieniä määriä verestä, virtsasta ja ulosteesta 8 viikon kuluttua annostelusta.
Lineaarisuus
Anidulafungiinin farmakokinetiikka on lineaarinen kerran vuorokaudessa annettavien annosten laajalla vaihteluvälillä (15–130 mg).
Erityisryhmät
Potilaat, joilla on sieni-infektioita
Farmakokineettisten populaatioanalyysien mukaan anidulafungiinin farmakokinetiikka on samankaltainen sieni-infektioita sairastavilla potilailla ja terveillä tutkimushenkilöillä. Kun vuorokausiannos oli 200/100 mg ja infuusionopeus oli 1,1 mg/min, vakaan tilan suurin pitoisuus (Cmax) oli noin 7 mg/l ja pienin pitoisuus (Cmin) 3 mg/l. Vakaan tilan keskimääräinen AUC-arvo oli 110 mg⋅h/l.
Paino
Vaikka farmakokineettisessä populaatioanalyysissa puhdistumavaihtelun lähteeksi tunnistettiin paino, tämän kliininen merkitys anidulafungiinin farmakokinetiikan kannalta on pieni.
Sukupuoli
Anidulafungiinin pitoisuudet terveiden miesten ja naisten plasmassa ovat samaa luokkaa. Toistuvan annon potilastutkimuksissa anidulafungiini puhdistui hieman nopeammin (noin 22 %) miehillä.
Iäkkäät potilaat
Farmakokineettinen populaatioanalyysi osoitti, että mediaanipuhdistuma erosi hieman iäkkäiden (potilaat ≥ 65 v, mediaani CL = 1,07 l/h) ja muunikäisten (potilaat < 65 v, mediaani CL = 1,22 l/h) välillä. Puhdistuman vaihteluväli oli kuitenkin samaa luokkaa.
Rotu
Anidulafungiinin farmakokinetiikka oli samankaltainen valkoihoisilla, mustaihoisilla, aasialaisilla ja latinoilla.
HIV-positiivisuus
Annostusta ei tarvitse muuttaa potilaan HIV-positiivisuuden ja samanaikaisen antiretroviraalisen lääkehoidon perusteella.
Maksan vajaatoiminta
Anidulafungiini ei metaboloidu maksassa. Sen farmakokinetiikkaa on tutkittu tutkimushenkilöillä, joiden maksan vajaatoiminta oli Child-Pugh-asteikolla arvioituna luokkaa A, B tai C. Anidulafungiinipitoisuudet eivät suurentuneet minkäänasteista maksan vajaatoimintaa sairastaneilla tutkimushenkilöillä. Vaikka AUC-arvon todettiin pienenevän hieman potilailla, joilla maksan vajaatoiminnan Child-Pugh-luokka oli C, arvon pienenemä pysyi vaihteluvälillä, joka on todettu terveitä tutkimushenkilöitä sisältäneissä populaatioarvioissa.
Munuaisten vajaatoiminta
Anidulafungiinin munuaispuhdistuma on häviävän pieni (< 1 %). Kliinisessä tutkimuksessa, jonka tutkimushenkilöillä oli lievä, kohtalainen, vaikea tai loppuvaiheen (dialyysihoitoa vaativa) munuaisten vajaatoiminta, anidulafungiinin farmakokinetiikka oli samankaltainen kuin tutkimushenkilöillä, joiden munuaiset toimivat normaalisti. Anidulafungiini ei ole dialysoitavissa, ja sen voi antaa hemodialyysin ajoituksesta riippumatta.
Pediatriset potilaat
Anidulafungiinin farmakokinetiikkaa vähintään 5 vuorokausiannoksen jälkeen on tutkittu 24:llä neutropeenisellä immuniteetiltaan heikentyneellä lapsella (2–11 v) ja nuorella (12–17 v). Vakaa tila saavutettiin kyllästysannoksen (2 x ylläpitoannos) annostelua seuraavana päivänä, ja vakaan tilan Cmax- ja AUCss-arvot suurenivat suhteessa annokseen. Päivittäisten ylläpitoannosten (0,75 mg/kg/vrk ja 1,5 mg/kg/vrk) tuottama systeeminen altistus oli tässä populaatiossa verrattavissa aikuisilla todettuun systeemiseen altistukseen annoksilla 50 mg/vrk ja 100 mg/vrk. Molemmat annokset olivat näillä potilailla hyvin siedettyjä.
Anidulafungiinin farmakokinetiikkaa tutkittiin prospektiivisessa, avoimessa, vertailemattomassa, pediatrisessa tutkimuksessa 66 pediatrisella potilaalla (1 kuukaudesta < 18 vuoteen), joilla oli invasiivinen kandidiaasi, kandidemia mukaan lukien, ja jotka saivat kyllästysannoksen 3,0 mg/kg ja ylläpitoannoksen 1,5 mg/kg/vrk (ks. kohta Farmakodynamiikka). Invasiivista kandidiaasia, kandidemia mukaan lukien, sairastavien aikuisten ja pediatristen potilaiden yhdistettyjen tietojen populaatiofarmakokineettisen analyysin perusteella keskimääräistä altistusta vakaassa tilassa kuvaavat parametrit (AUC0-24,ss ja Cmin,ss) olivat pediatristen potilaiden kaikissa ikäryhmissä (1 kuukaudesta < 2 vuoteen, 2 vuodesta < 5 vuoteen ja 5 vuodesta < 18 vuoteen) kaikkiaan verrattavissa niihin aikuisiin nähden, jotka saivat kyllästysannoksen 200 mg ja ylläpitoannoksen 100 mg/vrk. Painon suhteen vakioitu puhdistuma (l/h/kg) ja jakautumistilavuus vakaassa tilassa (l/kg) olivat kaikissa ikäryhmissä samankaltaiset.
Prekliiniset tiedot turvallisuudesta
Kolmen kuukauden tutkimuksissa, joissa rotille ja apinoille annetut annokset olivat 4–6 kertaa suurempia kuin odotettavissa oleva kliininen hoitoaltistus, saatiin näyttöä maksatoksisuudesta, johon kuului entsyymiarvojen suurenemista ja morfologisia muutoksia. Anidulafungiinin in vitro- ja in vivo -geenitoksisuustutkimuksista ei saatu näyttöä geenitoksisuudesta. Eläimillä ei ole tehty pitkäaikaistutkimuksia, joissa olisi arvioitu anidulafungiinin karsinogeenisuutta.
Anidulafungiinin anto rotille ei osoittanut vaikutuksia lisääntymiseen, ei myöskään urosten tai naaraiden hedelmällisyyteen.
Anidulafungiini läpäisi rottien istukan, ja sitä löytyi sikiöiden plasmasta.
Alkioiden ja sikiöiden kehitystutkimuksissa käytetyt annokset olivat 0,2 ja 2 kertaa (rotta) sekä 1 ja 4 kertaa suurempia (kaniinit) kuin hoidon ylläpitoannokseksi ehdotettu 100 mg/vrk. Anidulafungiini ei aiheuttanut rotille lääkkeeseen liittyvää kehitystoksisuutta suurimmalla testatulla annoksella. Kaniineilla havaittuja kehitysvaikutuksia (sikiöiden painon lievä aleneminen) ilmeni vain suurimmalla testatulla annoksella, joka oli myös toksinen emolle.
Terveillä aikuisilla ja vastasyntyneillä rotilla anidulafungiinipitoisuus aivoissa oli pieni (aivo/plasma-suhde noin 0,2) kerta-annoksen jälkeen. Pitoisuus aivoissa kuitenkin suureni terveillä vastasyntyneillä rotilla viiden päivittäisen annoksen jälkeen (aivo/plasma-suhde noin 0,7). Anidulafungiinin on osoitettu vähentävän aivojen sienikuormaa moniannostutkimuksissa, jotka tehtiin disseminoitunutta kandidaasia sairastavilla kaniineilla ja keskushermoston kandidainfektiota sairastavilla hiirillä. Disseminoituneen kandidiaasin ja hematogeenisen Candida-peräisen meningoenkefaliitin farmakokineettis-farmakodynaamisten tutkimusten kaniinimallien tulokset osoittivat, että keskushermoston kudosten infektioiden optimaaliseen hoitoon tarvittiin suurempia annoksia muihin kudoksiin verrattuna (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Rotille annettiin kolmea erisuuruista anidulafungiiniannosta, ja ne nukutettiin ketamiinin ja ksylatsiinin yhdistelmällä yhden tunnin sisällä anidulafungiinin annostelusta. Suurta annosta saaneilla rotilla ilmeni infuusioon liittyneitä reaktioita, joita anestesia pahensi. Joillakin keskisuurta annosta saaneilla rotilla ilmeni samankaltaisia reaktioita mutta vasta anesteettien annon jälkeen. Pientä annosta saaneilla rotilla ei ilmennyt haittavaikutuksia anesteettien kanssa eikä ilman niitä. Keskisuurta annosta saaneilla rotilla ei ilmennyt infuusioon liittyneitä reaktioita ilman anesteetteja.
Nuorilla rotilla tehdyt tutkimukset eivät osoittaneet aikuisiin eläimiin verrattuna suurempaa alttiutta anidulafungiinin maksatoksisuudelle.
Farmaseuttiset tiedot
Apuaineet
Fruktoosi
Mannitoli
Polysorbaatti 80
Viinihappo
Natriumhydroksidi (happamuuden säätöön)
Vetykloridihappo (happamuuden säätöön)
Yhteensopimattomuudet
Lääkevalmistetta ei saa sekoittaa eikä antaa muiden lääkevalmisteiden tai elektrolyyttien kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
3 vuotta.
Säilytyslämpötilan ylittyminen voi olla enintään 96 tuntia korkeintaan 25 °C:n lämpötilassa, minkä jälkeen infuusiokuiva-aineen voi palauttaa jääkaappiin.
Käyttövalmiiksi saatettu infuusiokonsentraattiliuos
Käyttövalmiiksi saatetun infuusiokonsentraattiliuoksen kemiallinen ja fysikaalinen stabiliteetti käytön aikana on 24 tuntia 25 °C:ssa.
Kun noudatetaan hyvää aseptista toimintatapaa, infuusiokonsentraattiliuos on mikrobiologiselta kannalta käyttökelpoista enintään 24 tuntia 25 °C:ssa säilytettynä.
Infuusioliuos
Ei saa jäätyä.
Infuusioliuoksen kemiallinen ja fysikaalinen stabiliteetti käytön aikana on 48 tuntia 25 °C:ssa.
Kun noudatetaan hyvää aseptista toimintatapaa, infuusioliuos on mikrobiologiselta kannalta käyttökelpoista enintään 48 tuntia valmistuksesta 25 °C:ssa säilytettynä.
Säilytys
Säilytä jääkaapissa (2 °C–8 °C ).
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ECALTA kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
100 mg (L:ei) 100 mg (736,98 €)
PF-selosteen tieto
Tyypin 1 lasista tehty injektiopullo (30 ml), jossa on elastomeerinen suljin (butyylikumisuljin, jonka sisäpinnalla on inertti polymeeripäällyste ja ulkopinnalla liukastusaine käsittelyn helpottamiseksi tai vaihtoehtoisesti bromobutyylikumisuljin ja liukastusaine) ja alumiininen repäisysinetti.
Pakkauskoko: 1 injektiopullo.
Valmisteen kuvaus:
Valkoinen tai lähes valkoinen kiinteä aine.
Käyttövalmiiksi saatetun infuusiokonsentraattiliuoksen pH on 3,5‑5,5.
Käyttö- ja käsittelyohjeet
Ei erityisvaatimuksia hävittämisen suhteen.
ECALTA on liuotettava injektionesteisiin käytettävällä vedellä ja laimennettava sen jälkeen VAIN joko 9 mg/ml (0,9 %) natriumkloridi-infuusioliuoksella tai 50 mg/ml (5 %) glukoosi-infuusioliuoksella. Käyttövalmiiksi saatetun ECALTAN yhteensopivuutta muiden laskimonsisäisesti annosteltavien aineiden, infuusioliuosten tai lääkkeiden kanssa kuin 9 mg/ml (0,9 %) natriumkloridi-infuusioliuoksen tai 50 mg/ml (5 %) glukoosi-infuusioliuoksen kanssa ei ole vahvistettu. Infuusioliuos ei saa jäätyä.
Käyttövalmiiksi saattaminen
Saata jokainen injektiopullo aseptisesti käyttövalmiiksi 30 ml:lla injektionesteisiin käytettävää vettä, jolloin pitoisuudeksi tulee 3,33 mg/ml. Käyttövalmiiksi saattaminen voi kestää jopa 5 minuuttia. Jos infuusiokonsentraattiliuoksessa näkyy hiukkasia tai värjääntymistä seuraavaksi tehtävän laimentamisen jälkeen, liuos on hävitettävä.
Laimentaminen ja infuusio
Parenteraalista lääkevalmistetta sisältävä liuos ja säiliö on mahdollisuuksien mukaan aina tarkastettava silmämääräisesti ennen antoa hiukkasten ja värjäytymien varalta. Liuos on hävitettävä, jos siinä näkyy hiukkasia tai värjäytymiä.
Aikuispotilaat
Siirrä käyttövalmiiksi saatettu infuusiokonsentraattiliuos aseptisesti injektiopullosta/-pulloista infuusiopussiin (tai -pulloon), jossa on joko 9 mg/ml (0,9 %) natriumkloridi-infuusioliuosta tai 50 mg/ml (5 %) glukoosi-infuusioliuosta saavuttaaksesi sopivan ECALTA-pitoisuuden. Seuraavassa taulukossa on esitetty laimentaminen siten, että infuusioliuoksen lopullinen pitoisuus on 0,77 mg/ml, sekä kunkin annoksen infuusio-ohjeet.
ECALTAN laimentaminen annostelua varten
Annos | Kuiva-ainepullo-ja | Infuusio-konsen-traattiliuosta yhteensä | Laimenti-mena käytettävää infuusio-liuostaA | Infusoitavaa liuosta yhteensäB | Infuusio-nopeus | Infuusion minimi-kesto |
100 mg | 1 | 30 ml | 100 ml | 130 ml | 1,4 ml/min tai 84 ml/h | 90 min |
200 mg | 2 | 60 ml | 200 ml | 260 ml | 1,4 ml/min tai 84 ml/h | 180 min |
A Joko 9 mg/ml (0,9 %) natriumkloridi-infuusioliuosta tai 50 mg/ml (5%) glukoosi-infuusioliuosta.
B Infuusioliuoksen pitoisuus on 0,77 mg/ml.
Infuusionopeus ei saa ylittää 1,1 mg/min (vastaa 1,4 ml/min tai 84 ml/h, kun liuottaminen ja laimentaminen on tehty ohjeen mukaan) (ks. kohta Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Pediatriset potilaat
Pediatristen potilaiden (iältään 1 kuukaudesta alle 18 vuoteen) annokseen tarvittava infuusiotilavuus vaihtelee potilaiden painon perusteella. Käyttövalmiiksi saatettu liuos on laimennettava edelleen siten, että lopullisen infuusioliuoksen pitoisuus on 0,77 mg/ml. Ohjelmoitavan ruisku- tai infuusiopumpun käyttöä suositellaan. Infuusionopeus ei saa ylittää 1,1 mg:aa/min (vastaa 1,4 ml:aa/min tai 84 ml:aa/h, kun käyttövalmiiksi saattaminen ja laimentaminen on tehty ohjeiden mukaan) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
1. Laske potilaan annos ja saata tarvittava(t) injektiopullo(t) käyttövalmiiksi ohjeiden mukaan siten, että pitoisuus on 3,33 mg/ml (ks. kohdat Vaikuttavat aineet ja niiden määrät ja Annostus ja antotapa).
2. Laske tarvittava tilavuus (ml) käyttövalmiiksi saatettua anidulafungiinia:

3. Laske annettavan liuoksen kokonaistilavuus (ml) siten, että lopullinen pitoisuus on 0,77 mg/ml:
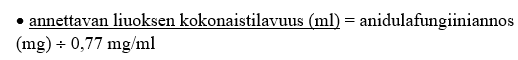
4. Laske annettavaa liuosta varten tarvittavan laimentimen tilavuus [5 %:n dekstroosi-injektioliuos (USP) tai 0,9 %:n natriumkloridi-injektioliuos (USP, normaali keittosuolaliuos)]:
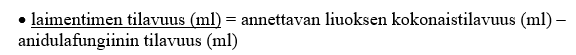
5. Siirrä tarvittava määrä (ml) anidulafungiinia ja 5 %:n dekstroosi-injektioliuosta (USP) tai 0,9 %:n natriumkloridi-injektioliuosta (USP, normaali keittosuolaliuos) aseptisesti antoa varten tarvittavaan infuusioruiskuun tai i.v. infuusiopussiin.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ECALTA kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
100 mg 100 mg
- Ei korvausta.
ATC-koodi
J02AX06
Valmisteyhteenvedon muuttamispäivämäärä
09.09.2025
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com